

Abstract
We hypothesized that GTI-2040, a 20-mer oligonucleotide complementary to the R2 subunit mRNA of ribonucleotide reductase, combined with high dose cytarabine (HiDAC) would result in enhanced cytotoxicity by favoring Ara-CTP DNA incorporation. In a phase I dose escalation trial, adults (≥60 years) with refractory or relapsed acute myeloid leukemia (AML) received daily HiDAC plus infusional GTI-2040. Using a novel assay, evidence of intracellular drug accumulation and target R2 down-regulation was observed. GTI-2040/HiDAC can be administered safely. However, with no complete remissions observed, alternative doses and schedules may need to be investigated to achieve clinical activity in older patients with AML.
Keywords: Acute myeloid leukemia, antisense therapy, phase I study, GTI-2040, ribonucleotide reductase
Introduction
Therapeutic options for patients older than 60 years remain dismal, with inferior 5-year survival rates compared to younger adults (4.3% vs. 34.4%) [1]. Given that this population, particularly in the relapsed setting, is also more likely to have additional comorbid illness, experience treatment-related death and have salvage response rates as low as 4% to cytarabine based regimens [2], many physicians are reluctant to consider treatment [3,4]. However, the potential for curative therapy remains in patients who can achieve adequate disease control and are able to proceed to allogeneic stem cell transplant [5], and investigation into new therapies for this disease are warranted.
Nucleoside analog drugs induce inhibition and chain termination of newly synthesized nucleic acid, ultimately leading to apoptosis by mimicking endogenous nucleosides [6]. One such pyrimidine analog, cytarabine (AraC), constitutes the backbone of several primary and salvage chemotherapy regimens for acute myeloid leukemia (AML) [7,8]. Differing from endogenous cytidine by the substitution of a ribose sugar with arabinoside, AraC is phosphorlyated into Ara-CTP, the active metabolite which is incorporated into newly synthesized DNA. Intracellular levels of the active metabolite have been directly correlated with the anti-leukemic properties of AraC, and attempts to increase levels of Ara-CTP hold promise to overcome resistance [9–13].
One such strategy is to inhibit ribonucleotide reductase (RNR), an enzyme required for the reductive conversion of ribonucleotides to deoxynucleotides, a crucial rate-limiting step during DNA synthesis and repair [14,15]. One potential mechanism of nucleoside analog chemoresistance is over-expression of RNR, frequently observed in malignant cells, leading to increased pools of endogenous deoxynucleoside triphosphates (dNTPs) that compete for DNA incorporation [16–21]. Therefore, AraC cytotoxicity may potentially be enhanced by RNR inhibition.
GTI-2040 is a 20-mer oligonucleotide that is complementary to the mRNA of the RNR subunit R2. These antisense oligodeoxynucleotides (ODN) obstruct gene expression by forming duplexes with complementary sequences of target mRNAs [22–24]. Therefore, we hypothesized that treatment with GTI-2040 would reduce expression of RNR and decrease the levels of endogenous dNTPs, favoring DNA incorporation of Ara-CTP and enhanced cytotoxicity. We initiated a phase I study of GTI-2040 in combination with high dose cytarabine (HiDAC) in patients with AML aged ≥60 years with goals to define the safety and tolerability of the combination and validate R2 as an in vivo target in this elderly population of AML.
Patients and methods
Eligibility criteria and study design
The dose escalation schema of this National Cancer Institute (NCI)/Cancer Therapy Evaluation Program-sponsored phase I study in patients over the age of 60 with refractory/relapsed AML is detailed in Table I. GTI-2040 was administered intravenously via continuous infusion, and AraC was infused over 4 h each day. Adequate performance status (Eastern Cooperative Oncology Group [ECOG] ≤2), cardiac function (left ventricular ejection fraction ≥50%) and normal hepatic/renal function were necessary for enrollment. Informed consent was obtained from all subjects before enrollment, and the local Institutional Review Board approved the study.
Table I.
Dose-escalation of GTI-2040 in combination with high-dose AraC.
| Dose level | GTI-2040 CIVI days 1–6 (mg/kg/day) | AraC IV q24 h days 2–6 (g/m2/dose) | Cummulative dose AraC (g/m2) | Number of patients treated |
|---|---|---|---|---|
| 1 | 3.5 | 1.5 | 7.5 | 3 |
| 2 | 5 | 1.5 | 7.5 | 3 |
| 3 | 5 | 2 | 10 | 6 |
| 4 | 7 | 1.5 | 7.5 | 4 |
AraC, cytarabine; CIVI, continuous intravenous infusion.
Patients were monitored closely for adverse events, including thorough neurologic examination before AraC doses. Toxicities were graded according to NCI Common Toxicity Criteria (version 3.0). Myeloid growth factors were permitted according to American Society of Clinical Oncology guidelines. Grade 3 or 4 non-hematologic toxicity related to GTI-2040 and grade 3 or 4 hematologic toxicity at day 42 in patients without evidence of persistent AML defined dose limiting toxicity (DLT). AML responses were assessed according to published NCI criteria [25].
Analysis of plasma and intracellular levels of GTI-2040
A novel hybridization-ligation based enzyme-linked immunosorbent assay (ELISA) was developed and validated in our laboratory for the measurement of GTI-2040 levels in plasma and lysates from bone marrow (BM) mononuclear cells (MNCs) [26,27]. The sample was mixed with a capture ODN (5′-TAACTAGTGCTTGGTGGAGCGATTTAGCC/3 biotin/3′), diluted in buffer (60 mmol/L phosphate buffer [pH 7.4], 1.0 mol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid [EDTA] and 0.2% Tween 20] and heated at 95°C. Samples were added to NeutrAvidin-coated 96-well plates (Pierce Co., Rockford, IL), incubated at 42°C for 2 h and subsequently washed with buffer (Tris-buffered saline [TBS] in 0.1% Tween 20). Next, a detection ODN probe (5′-CACTAGTTA-3′) with phosphate at the 5′-end and digoxigenin at the 3′-end was diluted in ligation buffer (66 mmol/L Tris-HCl [pH 7.6], 10 mmol/L MgCl2, 10 mmol/L dithiothreitol [DTT], 1 mmol/L adenosine triphosphate [ATP]) containing 5 U/mL T4 DNA ligase (Amersham Biosciences, Piscataway, NJ) and incubated overnight at 18°C. Excess probe bound to capture ODN was removed by incubation in 60 units of S1 nuclease (Invitrogen, Carlsbad, CA) in 3 mmol/L sodium acetate (pH 4.6), 1 mmol/L zinc acetate, 100 mmol/L NaCl and 5% glycerol for 2 h at 37°C. The plate was washed with buffer, and anti-digoxigenin-alkaline phosphatase (1:2500 with bovine serum albumin block buffer in TBS; Roche, Indianapolis, IN) was added. After 30 min incubation at room temperature, the plate was washed and AttoPhos substrate (Promega, Madison, WI) in diethanolamine buffer, prepared as recommended by the manufacturer, was added. Fluorescence intensity was measured at excitation 430/emission 560 (filter = 550 nm) using a Gemini XS plate reader (Molecular Devices, Sunnyvale, CA).
Plasma was collected before treatment; at 2, 4, 6, 12, 24, 48 and 72 h from the beginning of GTI-2040 infusion; at the end of GTI-2040 infusion; and 0.25, 0.5, 1, 2, 4, 6, 12, 24 and 48 h after drug discontinuation.
BM samples for the quantification of GTI-2040 intracellular concentrations were collected at 24 and 120 h after initiation of the infusions. BM MNCs were pelleted and treated with 0.1 μmol/L of phosphorothioate 28-mer polycytidine. Lysis buffer (10 mmol/L Tris-HCl [pH 8.5], 0.5 mmol/L EDTA and 1% Triton X-100) was added and incubated on ice for 10 min. The cells were then mechanically lysed, the homogenate centrifuged and the supernatant collected for use in the assay described above. Intracellular concentrations (ICs) were obtained by dividing the GTI-2040 amount by the mononuclear cell volume as measured on a Samba Image Analyzer 4000 (Imaging Products International, Inc., Chantilly, VA) and using a conversion factor of 0.035 μg protein equal to 1 μL cell volume or 2 × 106 cell number equal to 1 μL cell volume.
Quantification of R2 expression
Using BM lysates collected before treatment and at 24 and 120 h after initiation of GTI-2040, standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting were performed for the quantification of R2 (Santa Cruz Biotechnology, Santa Cruz, CA) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology). Protein levels were quantified by scanning immunoblots with ImageQuant software (Molecular Dynamics, Sunnyvale, CA). R2 levels were normalized to GAPDH.
Statistical considerations
Descriptive statistics including means, medians when large variability in the biologic samples was noted and standard deviations were calculated. Mann–Whitney U-tests and analysis of variance for continuous data were used to compare groups, and the Wilcoxon matched pairs signed test was used to analyze paired data. Plasma concentration–time profiles were fit and pharmacokinetics (PK) parameters were calculated using WinNonlin (Version 4.0; Pharsight, Mountain View, CA).
Results
Patient characteristics
Demographic and pretreatment features of the 16 patients with AML enrolled are detailed in Table II. Ten patients had relapsed disease, and complete remission (CR) durations for six of the relapsed patients were <6 months (median CR duration 4.1 months). Six patients had primary refractory disease. No patient had core binding factor (CBF)-positive AML. Five patients had a complex karyotype (≥5 abnormalities), two patients had a normal karyotype and the remaining patients had other cytogenetic abnormalities. Five had received prior HiDAC.
Table II.
Patient clinical characteristics.
| Characteristic | (n = 16) |
|---|---|
| Age, years | |
| Median | 68 |
| Range | 60–79 |
| Sex | |
| Female, % | 44 |
| Hemoglobin, g/dL | |
| Median | 9.1 |
| Range | 7.4–13.0 |
| Platelets, ×109/L | |
| Median | 50 |
| Range | 8–250 |
| WBC count, ×109/L | |
| Median | 3.7 |
| Range | 1.1–20.3 |
| PB blasts, % | |
| Median | 32 |
| Range | 0–85 |
| BM blasts, % | |
| Median | 63 |
| Range | 6–95 |
| Cytogenetics, n | |
| CBF | 0 |
| Normal | 2 |
| Complex (≥5 abnormalities) | 5 |
| Other | 9 |
| Disease status, n | |
| Refractory | 6 |
| Relapsed | 10 |
| CR duration (relapse patients), n | |
| 0–6 months | 6 |
| >6 months | 4 |
| No. of prior therapies | |
| Median | 1 |
| Range | 1–3 |
| Prior therapy, n | |
| HiDAC | 5 |
| Antisense | 3 |
WBC, white blood cell; PB, peripheral blood; BM, bone marrow; CBF, core binding factor; CR, complete remission; HiDAC, high-dose cytarabine.
Toxicity and disease response
No toxicity was dose-limiting. Compiled grades 3 and 4 non-hematologic toxicities observed are listed in Table III. One death occurred due to central nervous system (CNS) hemorrhage on day 24 in a patient with transfusion-refractory thrombocytopenia and persistent leukemia. One patient developed fevers and confusion 5 h into GTI-2040 infusion attributed to sepsis. The infusion was stopped, and the patient did not receive any additional therapy on the study. Other toxicities were reversible and previously observed with HiDAC alone. All 16 patients in the older cohort failed to achieve a CR. Although a maximum tolerated dose (MTD) was not defined, accrual was closed due to lack of clinical responses.
Table III.
Grades 3 and 4 non-hematologic toxicities without regard to attribution.
| Event | Grade 3 | Grade 4 |
|---|---|---|
| Constitutional | ||
| Fatigue | 5 | 0 |
| Dermatologic | ||
| Rash/erythema | 2 | 0 |
| Gastrointestinal | ||
| Anorexia | 1 | 0 |
| Diarrhea | 3 | 0 |
| Infection | ||
| Febrile neutropenia | 5 | 1 |
| Infection w/neutropenia | 9 | 0 |
| Metabolic | ||
| ALT | 2 | 0 |
| Bilirubin | 1 | 0 |
| Neurology | ||
| Confusion | 1 | 0 |
w/, with; ALT, alanine aminotransferase.
Pharmacokinetic studies
Fifteen patients had samples collected for PK analyses. Similar to observations in the younger cohort [28], the steady-state concentrations (Css) were achieved within 4 h and remained so until the end of continuous intravenous infusion (CIVI) with measurable GTI-2040 up to 48 h post-infusion, as demonstrated in the concentration–time profiles shown in Figure 1(A). The PK profiles were fitted to a two-compartment infusion model and the relevant parameters were computed. No statistical differences in dose-normalized PK parameters (dose-normalized area under the curve [AUC] and Css, and elimination half-life [t½β]) among different dose levels were observed (Table IV).
Figure 1.
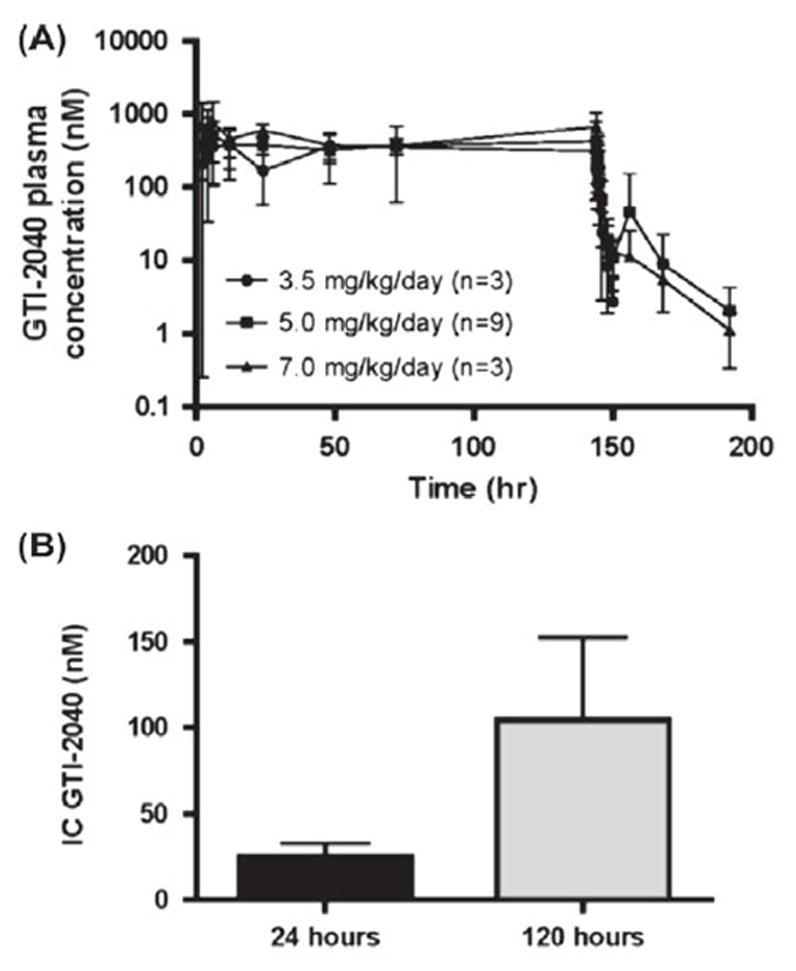
Mean (± SD) plasma GTI-2040 concentration vs. time in patients (A) and mean (with SEM) IC GTI-2040 after 24 h and 120 h of GTI-2040 exposure (B).
Table IV.
Relevant PK parameters of GTI-2040 (mean ± SD).
| Parameter | 3.5 mg/kg/day* (n = 3) | 5.0 mg/kg/day (n = 9) | 7.0 mg/kg/day (n = 3) |
|---|---|---|---|
| Css (nM) | 269±111 | 396±200 | 430±44.4 |
| AUC 0–∞ (μM×h) | 46.1±20.5 | 57.3±30.5 | 76.9±10.9 |
| CL (L/h) | 9.62±7.61 | 10.2±6.80 | 8.93±1.40 |
| Vss (L) | 8.71±5.69 | 27.7±22.2 | 14.6±4.02 |
| t1/2α (h) | 0.670±0.150 | 0.900±0.290 | 0.773±0.162 |
| t1/2β (h) | 32.1±16.2 | 16.7±4.42 |
Css, steady-state concentration; AUC, area under the curve; CL, clearance; Vss, steady-state volume; t1/2α, redistribution half-life; t1/2β, elimination half-life.
Samples of three patients were only collected to 4 h after the end of infusion; data were fitted by one-compartment model.
We next examined GTI-2040 uptake by measuring ICs in BM MNCs collected from treated patients at 24 and 120 h following initiation of GTI-2040, since target down-regulation is likely dependent on intracellular drug levels. Ten patients had adequate samples at 24 h and nine patients at 120 h. The median IC of GTI-2040 in BM was 12.8 nM (n = 10 patients; range 1.4–76 nM) at 24 h, and 23 nM (n = 9 patients; range 3.9–380 nM) at 120 h. BM ICs at 120 h were significantly higher than ICs at 24 h of drug exposure (p = 0.04), suggesting intracellular accumulation of the drug over time [Figure 1(B)]. Among different doses of GTI-2040, there was a difference in median IC achieved (i.e. 22 and 104 nM in 5 mg/kg, and 7 mg/kg, respectively), but this did not achieve statistical significance.
Pharmacodynamics studies
Eight patients had samples assessable for the R2 target protein in BM collected before treatment and at 24 and 120 h after initiation of GTI-2040 infusion. A >50% reduction in R2 protein at 24 and 120 h was seen in one and five patients, respectively. R2 protein reductions in pretreatment versus treated samples were noted, but the difference was not statistically significant (Figure 2).
Figure 2.
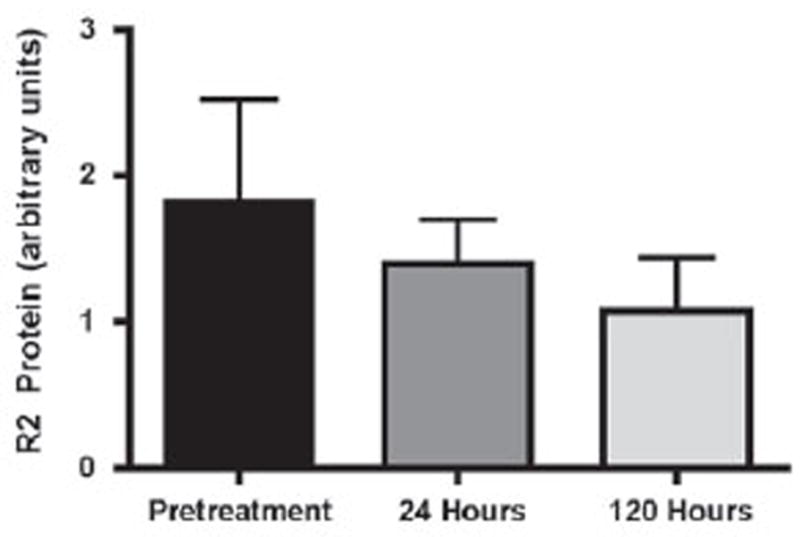
R2 protein expression (with range) changes in patients treated with GTI-2040 and HiDAC.
Discussion
In this article, we report the first trial in older patients with refractory/relapsed AML treated with a combination of GTI-2040 and HiDAC. This study was based on the rationale that the cytotoxic activity of Ara-C is enhanced by GTI-2040-induced RNR down-regulation when this target mediates chemoresistance and is supported by encouraging results in younger patients [28]. The results of the present phase I dose-escalating trial demonstrate that this combination can be administered safely in an older population with a toxicity profile similar to that observed with HiDAC alone. In particular, none of the patients experienced cerebellar toxicity, and the MTD was not reached.
This study included extensive PK and pharmacodynamics studies of this combination. Although we were able to demonstrate intracellular accumulation of GTI-2040, no clinical responses were observed. Notably, the intracellular levels of GTI-2040 were approximately half those observed in the younger cohort, which may partially explain clinical response differences observed [28]. Furthermore, it is possible that optimal pharmacologic activity of antisense compounds may not depend solely on absolute ICs. Perhaps drug levels achievable in distinct subcellular compartments, such as the nucleus or cytoplasm, may more accurately predict efficient target down-regulation by the antisense molecule [26]. In a younger cohort, although there was no difference in intracellular levels of GTI-2040 between patients achieving a CR and non-responders, there was a trend toward higher levels in the nuclear compared to cytoplasmic compartment in patients achieving a CR [28]. Future studies should include measures of ICs in different subcellular fractions to identify specific patterns of intracellular distribution of the parent drug and its metabolites that may correlate with disease response as well as explore the impact of age and other factors on intracellular drug levels.
In contrast to observations in younger patients where reductions in R2 protein correlated with clinical response [28], no correlation between reductions in target expression and clinical response was observed. Additionally, increases in R2 expression were also noted, suggesting overexpression of R2 as a potential mechanism of resistance with treatment. Indeed, up-regulation of the target has been observed with other antisense compounds if the antisense–target ratios are not optimal and potential feedback mechanisms of gene transcription become operative [4]. Thus, differences between the two age-based groups may be related to distinct mechanisms of chemoresistance (e.g. higher expression of anti-apoptotic protein in elderly patients, overexpression of R2 or induction of p53R2), biology of the disease or schedule and dosing of the combination.
We hypothesized that GTI-2040 should modulate the dNTP pools, in favor of Ara-CTP incorporation during combination therapy. We recently, in a separate preclinical study, demonstrated that GTI-2040 decreased dCTP pools 1–2-fold in K562 cells following treatment with 5 μM GTI-2040 for 24 h using our newly developed liquid chromatography–tandem mass spectrometry (LC-MS/MS) method which is more suitable for use in clinical samples [29]. Failure to decrease these dNTP pools in a clinical setting may explain the disappointing disease activity observed in this trial. Although limited sample material prohibited completion of these studies in this population, these assays should be incorporated into future clinical trials with GTI-2040 or a similar rationale.
In summary, we conclude that GTI-2040 in combination with HiDAC is feasible in an older population and is active against its target R2. However, an improved understanding of the impact of the R2 target in elderly AML and GTI-2040 on its subcellular distribution and dNTP pools will be necessary to evaluate its role in older patients with AML.
Acknowledgments
This study was supported by NCI U01 CA 076576-10 and NCI R21 CA 105879.
Footnotes
Potential conflict of interest: Disclosure forms provided by the authors are available with the full text of this article at www.informahealthcare.com/lal.
References
- 1.Dai G, Chan KK, Liu S, et al. Cellular uptake and intracellular levels of the bcl-2 antisense g3139 in cultured cells and treated patients with acute myeloid leukemia. Clin Cancer Res. 2005;11:2998–3008. doi: 10.1158/1078-0432.CCR-04-1505. [DOI] [PubMed] [Google Scholar]
- 2.Litzow MR, Othus M, Cripe LD, et al. Failure of three novel regimens to improve outcome for patients with relapsed or refractory acute myeloid leukaemia: a report from the Eastern Cooperative Oncology Group. Br J Haematol. 2010;148:217–225. doi: 10.1111/j.1365-2141.2009.07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marcucci G, Stock W, Dai G, et al. G3139, a BCL-2 antisense oligonucleotide, in AML. Ann Hematol. 2004;83(Suppl 1):S93–S94. doi: 10.1007/s00277-004-0850-2. [DOI] [PubMed] [Google Scholar]
- 4.Marcucci G, Byrd JC, Dai G, et al. Phase 1 and pharmacodynamic studies of G3139, a Bcl-2 antisense oligonucleotide, in combination with chemotherapy in refractory or relapsed acute leukemia. Blood. 2003;101:425–432. doi: 10.1182/blood-2002-06-1899. [DOI] [PubMed] [Google Scholar]
- 5.Chemnitz JM, von Lilienfeld-Toal M, Holtick U, et al. Intermediate intensity conditioning regimen containing FLAMSA, treosulfan, cyclophosphamide, and ATG for allogeneic stem cell transplantation in elderly patients with relapsed or high-risk acute myeloid leukemia. Ann Hematol. 2012;91:47–55. doi: 10.1007/s00277-011-1253-9. [DOI] [PubMed] [Google Scholar]
- 6.Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia. 2001;15:875–890. doi: 10.1038/sj.leu.2402114. [DOI] [PubMed] [Google Scholar]
- 7.Estey EH. Treatment of relapsed and refractory acute myelogenous leukemia. Leukemia. 2000;14:476–479. doi: 10.1038/sj.leu.2401568. [DOI] [PubMed] [Google Scholar]
- 8.Kimby E, Nygren P, Glimelius B. A systematic overview of chemotherapy effects in acute myeloid leukaemia. Acta Oncol. 2001;40:231–252. doi: 10.1080/02841860116883. [DOI] [PubMed] [Google Scholar]
- 9.Estey E, Thall P, Andreeff M, et al. Use of granulocyte colony-stimulating factor before, during, and after fludarabine plus cytarabine induction therapy of newly diagnosed acute myelogenous leukemia or myelodysplastic syndromes:comparison with fludarabine plus cytarabine without granulocyte colony-stimulating factor. J Clin Oncol. 1994;12:671–678. doi: 10.1200/JCO.1994.12.4.671. [DOI] [PubMed] [Google Scholar]
- 10.Gandhi V, Plunkett W. Modulation of arabinosylnucleoside metabolism by arabinosylnucleotides in human leukemia cells. Cancer Res. 1988;48:329–334. [PubMed] [Google Scholar]
- 11.Gandhi V, Estey E, Keating MJ, et al. Fludarabine potentiates metabolism of cytarabine in patients with acute myelogenous leukemia during therapy. J Clin Oncol. 1993;11:116–124. doi: 10.1200/JCO.1993.11.1.116. [DOI] [PubMed] [Google Scholar]
- 12.Gandhi V, Estey E, Du M, et al. Modulation of the cellular metabolism of cytarabine and fludarabine by granulocyte-colony-stimulating factor during therapy of acute myelogenous leukemia. Clin Cancer Res. 1995;1:169–178. [PubMed] [Google Scholar]
- 13.Yee KW, Cortes J, Ferrajoli A, et al. Triapine and cytarabine is an active combination in patients with acute leukemia or myelodysplastic syndrome. Leuk Res. 2006;30:813–822. doi: 10.1016/j.leukres.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 14.Fan H, Villegas C, Wright JA. Ribonucleotide reductase R2 component is a novel malignancy determinant that cooperates with activated oncogenes to determine transformation and malignant potential. Proc Natl Acad Sci USA. 1996;93:14036–14040. doi: 10.1073/pnas.93.24.14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan H, Villegas C, Huang A, et al. The mammalian ribonucleotide reductase R2 component cooperates with a variety of oncogenes in mechanisms of cellular transformation. Cancer Res. 1998;58:1650–1653. [PubMed] [Google Scholar]
- 16.Fukushima M, Fujioka A, Uchida J, et al. Thymidylate synthase (TS) and ribonucleotide reductase (RNR) may be involved in acquired resistance to 5-fluorouracil (5-FU) in human cancer xenografts in vivo. Eur J Cancer. 2001;37:1681–1687. doi: 10.1016/s0959-8049(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 17.Iwasaki H, Huang P, Keating MJ, et al. Differential incorporation of ara-C, gemcitabine, and fludarabine into replicating and repairing DNA in proliferating human leukemia cells. Blood. 1997;90:270–278. [PubMed] [Google Scholar]
- 18.Kubota T, Watanabe M, Otani Y, et al. Different pathways of 5-fluorouracil metabolism after continuous venous or bolus injection in patients with colon carcinoma: possible predictive value of thymidylate synthetase mRNA and ribonucleotide reductase for 5-fluorouracil sensitivity. Anticancer Res. 2002;22:3537–3540. [PubMed] [Google Scholar]
- 19.Mansson E, Spasokoukotskaja T, Sallstrom J, et al. Molecular and biochemical mechanisms of fludarabine and cladribine resistance in a human promyelocytic cell line. Cancer Res. 1999;59:5956–5963. [PubMed] [Google Scholar]
- 20.Matsusaka S, Yamasaki H, Fukushima M, et al. Upregulation of enzymes metabolizing 5-fluorouracil in colorectal cancer. Chemotherapy. 2007;53:36–41. doi: 10.1159/000098249. [DOI] [PubMed] [Google Scholar]
- 21.Okamura H, Kamei T, Sakuma N, et al. Ribonucleotide reductase immunoreactivity in adenocarcinoma cells and malignant or reactive mesothelial cells in serous effusions. Acta Cytol. 2003;47:209–215. doi: 10.1159/000326506. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal N, Gewirtz AM. Oligonucleotide therapeutics for hematologic disorders. Biochim Biophys Acta. 1999;1489:85–96. doi: 10.1016/s0167-4781(99)00142-6. [DOI] [PubMed] [Google Scholar]
- 23.Gewirtz AM. Antisense oligonucleotide therapeutics for human leukemia. Curr Opin Hematol. 1998;5:59–71. doi: 10.1097/00062752-199801000-00011. [DOI] [PubMed] [Google Scholar]
- 24.Marcucci G, Caligiuri MA. Antisense and gene transfer as therapeutic strategies in acute and chronic leukemia. In: Henderson EA, editor. Leukemia. 7. New York: WB Saunders; 2002. pp. 449–457. [Google Scholar]
- 25.Cheson BD, Cassileth PA, Head DR, et al. Report of the National Cancer Institute-sponsored workshop on definitions of diagnosis and response in acute myeloid leukemia. J Clin Oncol. 1990;8:813–819. doi: 10.1200/JCO.1990.8.5.813. [DOI] [PubMed] [Google Scholar]
- 26.Marcucci G, Stock W, Dai G, et al. Phase I study of oblimersen sodium, an antisense to Bcl-2, in untreated older patients with acute myeloid leukemia: pharmacokinetics, pharmacodynamics, and clinical activity. J Clin Oncol. 2005;23:3404–3411. doi: 10.1200/JCO.2005.09.118. [DOI] [PubMed] [Google Scholar]
- 27.Wei X, Dai G, Marcucci G, et al. A specific picomolar hybridization-based ELISA assay for the determination of phosphorothioate oligonucleotides in plasma and cellular matrices. Pharm Res. 2006;23:1251–1264. doi: 10.1007/s11095-006-0082-3. [DOI] [PubMed] [Google Scholar]
- 28.Klisovic RB, Blum W, Wei X, et al. Phase I study of GTI-2040, an antisense to ribonucleotide reductase, in combination with high-dose cytarabine in patients with acute myeloid leukemia. Clin Cancer Res. 2008;14:3889–3895. doi: 10.1158/1078-0432.CCR-08-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen P, Liu Z, Liu S, et al. A LC-MS/MS method for the analysis of intracellular nucleoside triphosphate levels. Pharm Res. 2009;26:1504–1515. doi: 10.1007/s11095-009-9863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]