

Abstract
In hepatocellular carcinoma (HCC), dysregulated expression of microRNA-224 (miR-224) and impaired autophagy have been reported separately. However, the relationship between them has not been explored. In this study we determined that autophagy is down-regulated and inversely correlated with miR-224 expression in hepatitis B virus (HBV)-associated HCC patient specimens. These results were confirmed in liver tumors of HBV X gene transgenic mice. Furthermore, miR-224 was preferentially recruited and degraded during autophagic progression demonstrated by real-time polymerase chain reaction and miRNA in situ hybridization electron microscopy after extraction of autophagosomes. Our in vitro study demonstrated that miR-224 played an oncogenic role in hepatoma cell migration and tumor formation through silencing its target gene Smad4. In HCC patients, the expression of low-Atg5, high-miR-224, and low-Smad4 showed significant correlation with HBV infection and a poor overall survival rate. Autophagy-mediated miR-224 degradation and liver tumor suppression were further confirmed by the autophagy inducer amiodarone and miR-224 antagonist using an orthotopic SD rat model. Conclusion: A noncanonical pathway links autophagy, miR-224, Smad4, and HBV-associated HCC. These findings open a new avenue for the treatment of HCC. (Hepatology 2014;59:505–517)
Hepatocellar carcinoma (HCC) is the fifth most common cancer worldwide and is the leading cause of cancer-related death. Moreover, HCC is often diagnosed at a late stage and the prognosis is poor.1 Recent studies highlight new molecular mechanisms involved in HCC progression, including autophagy and microRNAs (miRNAs).2 Autophagy induced by metabolic stresses is a complicated process to balance cellular energy metabolites involving degradation, renewal of energy, quality control of intracellular organelles and proteins, embryonic development, and presentation of antigens for immunity. Adequate autophagic responses protect cells from different kinds of stress and maintain cell survival mechanisms.
A group of autophagy-related genes (Atg), including Atg5 and Atg7 genes are essential for the autophagic process, as demonstrated by gene silencing studies. Impaired autophagy causes diverse pathologic conditions in humans, including liver dysfunction and tumorigenesis.3 For example, mice with mosaic deletion of Atg5 and liver-specific Atg7(−/−) developed multiple liver tumors. In addition, autophagic gene Beclin 1 (Atg6 in yeast) expression was decreased in HCC tissues compared with adjacent nontumor tissues.4,5 The polyubiquitin-binding protein p62/SQSTM1 interacts with the ubiquitinated cargo protein followed by binding with LC3 (Atg8 in yeast), and then is transported into the autophagosomes for degradation. Suppression of autophagy causes p62 accumulation, as identified in various cancers.6 Although accumulating evidence indicates that autophagy suppresses tumorigenesis to preserve cellular fitness and genome integrity,7 the molecular mechanisms whereby autophagy inhibits tumorigenesis remain unclear.
MiRNAs are small noncoding RNAs that are initially transcribed as primary miRNAs, and then undergo sequential processing to precursor miRNAs by Drosha. Precursor miRNAs are then transported into the cytoplasm followed by Dicer processing to become mature miRNAs.8 MiRNAs suppress their target-gene expression either by transcriptional degradation or by translational inhibition, depending on sequence homology between the miRNA and the target gene.9 MiR-216a and miR-224 are highly expressed in HCC.10 Overexpression of miR-224 promotes liver tumorigenesis.11 Recent studies have identified the Atg members (Atg4, Beclin 1, and LC3) as the targets of miRNAs (miR-376b, miR-30a, and miR-204).12 These studies indicate that the autophagic process could be regulated by miRNAs, but little is known as to how autophagy regulates miRNA to affect specific functions especially tumorigenesis. In this study we aimed to identify the miRNA in HCC tumorigenesis, which is regulated by autophagy and to clarify the underlying mechanism.
Materials and Methods
Clinical Specimens
HCC tissue array and specimens were purchased from the Taiwan Liver Cancer Network (Zhunan, Taiwan). Informed consent was signed by the patients with approval from the Institutional Review Board, National Cheng Kung University Hospital (Tainan, Taiwan).
MiRNA In Situ Hybridization (miRNA ISH)
MiRNA ISH was performed as described.13 Briefly, slides were hybridized with 200 nM of 5′-digoxigenin (DIG) LNA-modified-miR-224 (Blossom Biotechnologies, Hyderabad, India) using the IsHyb In Situ Hybridization kit (Biochain, Newark, NJ). After washing and blocking, slides were incubated with anti-DIG-horseradish peroxidase (HRP) (Jackson ImmunoResearch, West Grove, PA) for 1 hour. MiR-224 signal was amplified by the TSA Plus DNP system (Perkin Elmer, Waltham, MA) and then incubated with anti-DNP-HRP for 1 hour. Slides were treated with an AEC solution and hematoxylin.
Fluorescent MiRNA ISH and Immunofluorescence Staining
The cells were fixed with 4% formaldehyde (Thermo Scientific, Santa Clara, CA) followed by ISH. The hybridization probes used were: 200 nM of 5′-DIG LNA-modified-miR-224 and 5′-DIG LNA-modified-let-7a (Blossom Biotechnologies). The miRNA signal was amplified by TSA Plus Cyanine 5 system (Perkin Elmer). Anti-LC3 (Medical and Biological Laboratories, Nagoya, Japan) and anti-Lamp1 antibodies (Abcam, Cambridge, UK) were incubated with miRNA solution for 1 hour. Alexa Flour 488 (Invitrogen, Boston, MA) was added for another 1 hour. Fluorescent change of the cells was investigated under a confocal microscope (FV-1000; Olympus, Tokyo, Japan).
Autophagosome Extraction
The cells were suspended in 0.4 mL 10% sucrose and mixed with 0.5 mL 1 M Hepes/0.1 M EDTA and homogenized by a Dounce homogenizer. This homogenate was diluted with homogenization buffer (HB; 0.25 M sucrose, 10 mM Hepes, 1 mM EDTA, pH 7.3) containing 1.5 mM glycyl-l-phenylalanine 2-naphthylamide and 1% dimethyl sulfoxide (DMSO). After incubation for 7 minutes at 37°C to destroy the lysosomes, the homogenate was cooled to 4°C. The extraction was performed as previously reported.14
MiRNA ISH-Electron Microscopy
MiRNA ISH-electron microscopy was conducted as previously described with modification.15 Briefly, the ultrathin sections of autophagosome fraction (AP) on the nickel grids were treated with 10% H2O2 for 10 minutes, prehybridized at 50°C for 30 minutes, and hybridized at 50°C for 2 hours with 200 nM 5′-DIG LNA-miRNA probes. Grids were then washed in SSC gradient and blocked for 30 minutes followed by anti-DIG antibody (Jackson ImmunoResearch) treatment. Grids were incubated with anti-mouse IgG H&L (18 nm Gold) secondary antibody (Abcam) for 1 hour. The sections on the grids were stained with saturated uranyl acetate and lead citrate and investigated under the electron microscope (HITACHI-7000, Tokyo, Japan).
Orthotopic Rat Model of Liver Tumor Formation
SD rats were obtained from the Laboratory Animal Center of National Cheng Kung University (Tainan, Taiwan). The experimental protocol complied with Taiwan’s Animal Protection Act and was approved by the Laboratory Animal Care and Use Committee of the National Cheng Kung University. Four-week-old male SD rats were anesthetized by intraperitoneal injection of sodium pentobarbital (MTC Pharmaceuticals, Hamilton, Canada) at a concentration of 6 mg/100 g of body weight. The liver was exposed by subxiphoid laparotomy. The NS1 cells (1.5 × 106) in 250 μL were injected slowly into the right hepatic lobe under the liver capsule. The puncture site was gently compressed and the wound was closed in layers. The cells grew successfully and formed tumors in the orthotopic rat model 1 week after inoculation.
Hepatitis B Virus X Protein (HBx) Transgenic Mice
HBx transgenic mice (C57B/6 background) were generated as described.16 Six- and 17.5-month-old HBx transgenic mice were sacrificed and RNA extracts and paraffin sections were prepared.
Results
Level of Autophagy Is Inversely Correlated With MiR-224 Expression in Hepatitis B Virus (HBV)-Associated HCC
MiR-224 overexpression and impaired autophagy have been reported in HCC separately, but their relationship remains unclear.4,10 The levels of miR-224 expression in the two autophagy-deficient cell lines, MEF-Atg5(−/−) and MEF-Atg7(−/−), were higher than that in the wild-type MEF cells (Supporting Fig. 1), indicating a negative correlation between autophagy and miR-224 expression. This study focused on their relationship and the underlying mechanism in HCC. Expression of the autophagy related genes (Atg5 and Beclin 1) generally reflects their corresponding autophagic activity17 and accumulation of p62 represents impaired autophagy.18 Therefore, the expression of Atg5, Beclin 1, as well as p62 accumulation were determined to evaluate the level of autophagy. A total of 93 paired HCC specimens (tumor and adjacent nontumor tissue) in the tissue array were analyzed (Supporting Table 1). The protein expression of Atg5, Beclin 1, and p62 was evaluated by immunohistochemistry (IHC), and miR-224 RNA expression was determined by miRNA ISH. Following analysis of a number of clinical-pathological factors using univariate and multivariate analysis, only virus status (HBV compared with hepatitis C virus [HCV]) showed significant correlation with Atg5 and/or miR-224 expression (Supporting Table 2, Rows 1, 2, and 4). In HBV-associated HCC specimens, Atg5 and Beclin 1 expression levels were low. Accordingly, p62 accumulation and miR-224 expression were higher in the tumors compared with the adjacent nontumor tissues (Fig. 1A). Further analysis showed that miR-224 was negatively correlated with Atg5 expression and positively correlated with p62 expression in the HBV-associated HCC specimens (Fig. 1B). In contrast, the expression levels of Atg5, Beclin 1, p62, and miR-224 were not changed and miR-224 was not correlated with Atg5 or p62 in HCV-associated HCC (Supporting Fig. 2). In summary, the level of autophagy was low and inversely correlated with miR-224 expression only in HBV-associated HCC.
Figure 1.
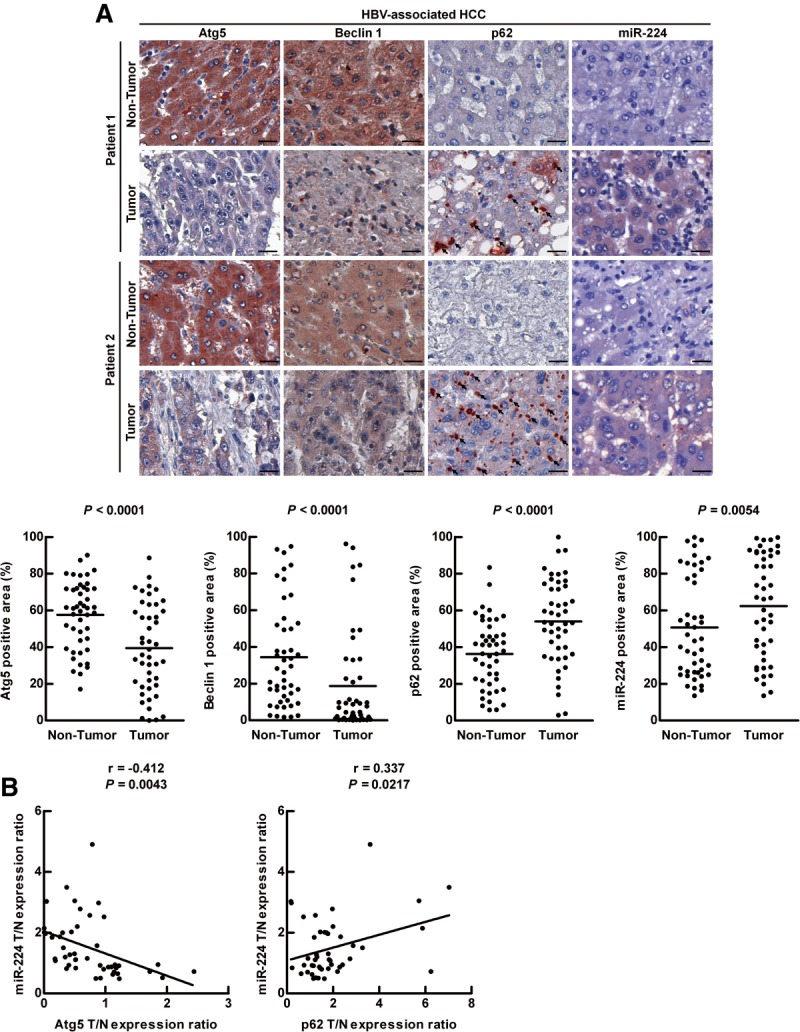
Low autophagic level and high miR-224 expression correlate with HBV-associated HCC. (A) Representative IHC of two paired HBV-associated HCC specimens showing the staining of Atg5, Beclin 1, and p62. MiR-224 staining was shown by miRNA in situ hybridization. Protein and miR-224 expression were determined by defining regions of interest (ROIs) using automated cell acquisition and quantification software (Histoquest). Arrow points to p62 accumulation. Scale bar = 20 μm. Quantification of the expression levels of Atg5, Beclin 1, p62, and miR-224 in 46 paired HBV-associated HCC specimens is shown as graphs. (B) Correlation of miR-224 expression with Atg5 and p62 was analyzed in 46 paired HBV-associated HCC specimens and linear regression coefficient and statistical significance are indicated. N, adjacent nontumor tissue; T, tumor tissue.
Low Level of Autophagy and High MiR-224 Expression Are Detected in the Liver Tumors of HBx-Transgenic Mice
An HBx transgenic model was used to confirm the relationship between autophagy and miR-224 expression in HBV-induced tumorigenesis. HBx gene participates in carcinogenesis of HCC, and the transgenic mice harboring the liver-specific albumin promoter-driven HBx gene develop liver tumors at 16 months after birth.16 The liver specimens from the 6-month (liver tissue without tumor) and 17.5-month (tumor and adjacent nontumor tissue) old transgenic mice were analyzed (Fig. 2A). Low Atg5 expression accompanied with high p62 accumulation (Fig. 2B) as well as high miR-224 expression (Fig. 2C) was detected in the tumors compared to adjacent nontumor tissues of 17.5-month-old transgenic mice and the liver tissues without tumor of the 6-month-old transgenic mice. Furthermore, the expression of Atg5, p62, and miR-224 was not changed in the livers of 6-month- and 17.5-month-old non-HBx transgenic mice (Supporting Fig. 3), excluding the age effect on autophagy-related genes and miR-224 expression. In summary, our data confirmed that a low level of autophagy together with high miR-224 expression is involved in the liver tumorigenesis of HBx transgenic mice.
Figure 2.

Liver tumor formation in HBx transgenic mice is correlated with low autophagic level and high miR-224 expression. (A) Liver without tumor was from 6-month-old transgenic mice (6 M), and liver with tumor was from 17.5-month-old transgenic mice (17.5 M). The arrow points to the tumor. Scale bar = 1 cm. (B) Representative IHC showing the staining of Atg5 and p62 in paraffin section including liver tissue without tumor formation of 6-month-old transgenic mice, liver tumor tissue and adjacent nontumor tissue of 17.5-month-old transgenic mice. Arrow points to p62 accumulation. Scale bar = 20 μm. Atg5 and p62 expression levels were determined by ROI followed by HistoQuest analysis. Quantification of the expression levels of Atg5 and p62 in transgenic mice are shown as bar graphs. (C) Quantification of miR-224 expression in HBx transgenic mice by real-time PCR and data are shown as mean ± SEM (n = 5). The relative expression fold of miR-224 is normalized to snoRNA-55. **P < 0.01 and ***P < 0.001. N, adjacent nontumor tissue; T, tumor tissue.
Mature MiR-224 Is Preferentially Recruited and Degraded Through the Autophagosome-Lysosome Pathway
To clarify that autophagy regulates miR-224 at the transcriptional or posttranscriptional level, specific primers were designed to measure the expression of precursor and mature forms of miR-224 in HCC specimens and HBx transgenic mice (Supporting Table 3). Our data showed that only the mature miR-224 was overexpressed in the tumors of the HCC patients and HBx transgenic mice (Fig. 3A; Supporting Fig. 4A), indicating that mature miR-224 is regulated at the posttranscriptional level. Lee et al.19 reported that a fraction of miRNA-loaded AGO2 complex is sorted into lysosomes for secretion or lysosomal degradation. Therefore, we hypothesize that mature miR-224 in the cytoplasm is recruited to the autophagosome and degraded in the autolysosome (fusion of autophagosome and lysosome) during autophagic flux.
Figure 3.

Autophagic flux participates in the degradation of mature miR-224 expression. (A) Quantification of the expression of precursor and mature forms miR-224 in 75 paired HCC specimens by real-time PCR. (B) Hep 3B cells were treated with amiodarone (5 μM) for various times. The expression of LC3 and p62 was determined by western blotting. Beta-actin was used as an internal control. (C) MiR-224 expression in cells in (B) was measured by real-time PCR. (D) Amiodarone-induced autophagic flux was blocked by CQ. Hep 3B cells were treated with amiodarone (5 μM) for 48 hours. In the CQ group, CQ (50 μM) was added at 24 hours in the presence or absence of amiodarone treatment for another 24 hours. LC3 expression was determined by western blotting. (E) MiR-224 expression in (D) was determined by real-time PCR. The data are shown as mean ± SEM (n = 3). The relative expression fold of miR-224 is normalized to U54. P values were obtained using a paired Student t test. *P < 0.05 and **P < 0.01. Pre-miR 224, precursor miR-224; CQ, chloroquine.
To confirm our speculation, amiodarone (an autophagy inducer)20 was used in the human hepatoma Hep 3B cells (with HBV DNA integration). Our data showed that amiodarone induced the autophagic flux (Fig. 3B), and the mature miR-224 expression was decreased in a time-dependent manner (Fig. 3C). The expression of precursor miR-224 was not changed after amiodarone treatment (Supporting Fig. 4B). To confirm that the autophagic flux proceeds with degradation, chloroquine (CQ. a blocker of autophagosome and lysosome fusion) was used to block autophagic progression and degradation. The accumulation of LC3-type II in the presence of CQ indicates that the autophagic flux was blocked (Fig. 3D). Under such conditions, only mature miR-224 expression was reversed after CQ treatment (Fig. 3E; Supporting Fig. 4C). To further confirm our investigation, another autophagy inducer, rapamycin, was used. Similarly, our data showed that mature but not precursor miR-224 was degraded during autophagic progression (Supporting Fig. 5). To clarify whether miR-224 degradation through autophagic pathway is HBV-related, an Huh7 derivate, Con-1, which stably expresses HCV RNA, was used. Similarly, our data showed that only mature miR-224 was degraded during autophagic flux in Con-1 cells (Supporting Fig. 6). Collectively, we reveal that mature miR-224 degradation by autophagic flux is a general event which can be induced by various inducers and not dependent on the viral status of the hepatoma cells.
Because the expression kinetics of intracellular and extracellular miR-224 were similar (Fig. 3E versus Supporting Fig. 7), it indicates that miR-224 expression is not affected by secretion. To clarify whether miR-224 expression affects autophagy, synthetic miR-224 and miR-224 antagonist (anti-miR-224) were transfected into Hep 3B cells. The expression of LC3-type II was not affected by miR-224 with or without CQ treatment (Supporting Fig. 8), indicating that miR-224 does not affect autophagic progression.
To clarify whether miR-224 is preferentially degraded by autophagy, the distribution of miR-224 and a control miRNA let-7a in the cells was investigated by fluorescent miRNA ISH. Let-7a, a widely expressed miRNA in diverse tissue cells and not affected by autophagy, was used as a control (Supporting Fig. 9A,B). Colocalization of miR-224 and autophagosomes (represented by LC3 puncta) was induced after amiodarone treatment and this phenomenon was further enhanced by CQ in Hep 3B cells (Fig. 4A). In contrast, colocalization of let-7a and autophagosomes was not induced by the same treatment (Fig. 4B). Similarly, colocalization of miR-224 and autolysosomes (represented by LAMP1) was also increased by NH4Cl treatment (to neutralize acidic pH and block degradation in autolysosomes) after autophagy induction (Supporting Fig. 10A). Again, colocalization of let-7a and autolysosome was not changed by the same treatment (Supporting Fig. 10B). Taken together, we reveal that miR-224 is preferentially recruited to autophagosomes followed by autophagic degradation in autolysosomes.
Figure 4.
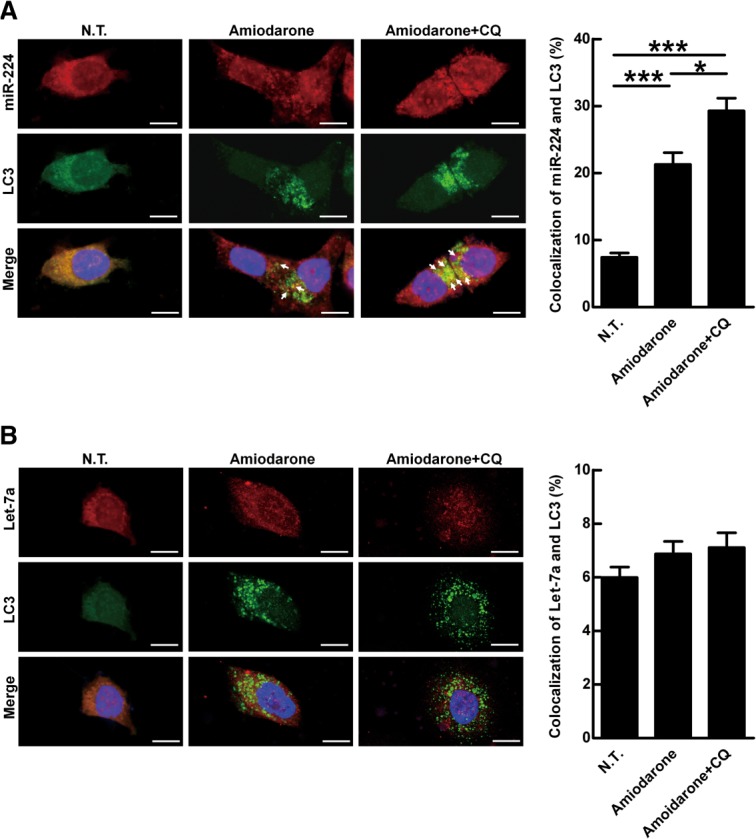
MiR-224 is accumulated in the autophagosome but not let-7a. MiR-224 accumulation in the autophagosome was demonstrated by CQ blockage. Hep 3B cells were treated with amiodarone (5 μM) for 24 hours followed by CQ (50 μM) blockage for another 24 hours. The LC3 protein was labeled with green fluorescent tag. MiR-224 and let-7a were labeled with red fluorescent tags. The arrows point to colocalization of LC3 and miR-224 (A) or LC3 and let-7a (B). Scale bar = 10 μm. Quantification of colocalization was performed by counting 30 cells. *P < 0.05 and ***P < 0.001. N.T., nontreatment; CQ, chloroquine.
MiR-224 Is Accumulated in the Autophagosomes
To further confirm the preferential accumulation of miR-224 in the autophagosomes, density gradient centrifugation of the autophagosomes was conducted.14 The successful purification of AP was confirmed by enriched LC3-type II expression and low background expression of mitochondria and endoplasmic reticulum (ER) markers (Fig. 5A). Furthermore, the ultrastructure of the double membrane autophagosomes was detected under transmission electron microscopy (TEM) (Supporting Fig. 11). The expression level of miR-224 was about 50-fold increased in AP (Fig. 5B). In contrast, the amount of let-7a in AP was very low compared with whole cell lysate (Fig. 5B). The preferential accumulation of miR-224 in autophagosome was further confirmed by miRNA ISH-electron microscopy using immunogold-labeled miR-224 and let-7a. The amount of the immunogold-labeled miR-224 was about 6-fold higher than that of let-7a in the autophagosomes (Fig. 5C-E). Collectively, we reveal the preferential accumulation of miR-224 in the autophagosomes.
Figure 5.
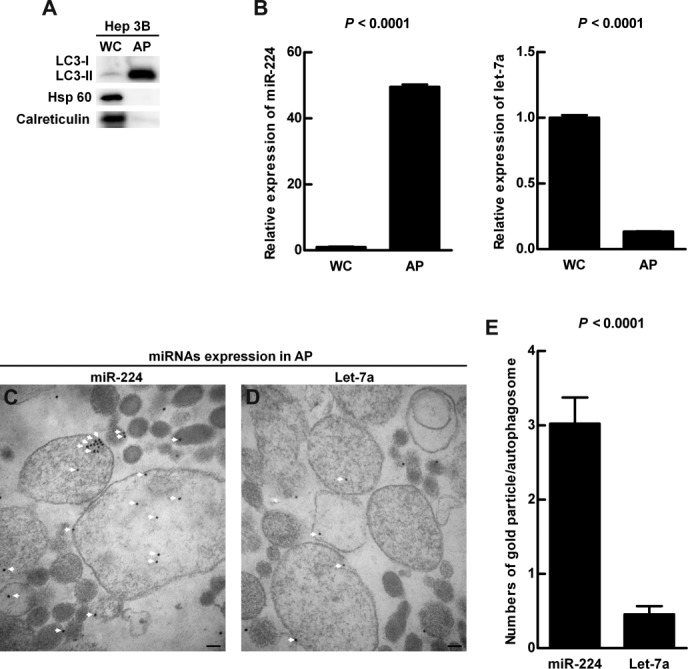
Preferential accumulation of miR-224 was demonstrated in the autophagosome. Hep 3B cells were treated with amiodarone and chloroquine (CQ) as in Fig. 4A. Protein and RNA were extracted from the whole cell lysate and followed by autophagosome purification. (A) Protein was analyzed by western blotting for LC3, Hsp 60 (mitochondria marker), and calreticulin (endoplasmic reticulum marker) expression. (B) The quantification of miR-224 and let-7a conducted by real-time PCR is shown as mean ± SEM (n = 5). (C) Accumulation of immune-gold labeled miR-224 (18 nm gold beads) in the purified double membrane autophagosomes was detected by miRNA in situ hybridization under TEM. (D) Accumulation of immune-gold labeled let-7a (18 nm gold beads) in the autophagosomes. The arrows highlight gold labeled miR-224 in (C) and let-7a in (D). Scale bar = 100 nm. (E) Quantification was performed by counting the gold particle labeled miR-224 and let-7a in 50 autophagosomes. WC, whole cell lysate; AP, autophagosome fraction.
MiR-224 Promotes Tumorigenesis Through Silencing Its Target Gene Smad4
The target gene of miR-224 was determined by overlapping the target genes predicted with three softwares: TargetScan, PicTar, and miRBase (data not shown). Among the predicated genes, Smad4 was chosen because it contains two miR-224-binding sites in the 3′ untranslated region. To confirm that miR-224 targets Smad4, luciferase reporter plasmids containing wild- or mutant-type miR-224 targeting sequence of Smad4 in the 3′ untranslated region (Supporting Fig. 12A) were constructed. In the cells transfected with the wild-type plasmid, the luciferase activity was repressed by synthetic miR-224 and enhanced by anti-miR-224; however, synthetic miR-224 and anti-miR-224 showed no effect on the luciferase activity of the mutant-type plasmid (Fig. 6A). This indicates miR-224 specifically targets Smad4 and blocks its translation. Furthermore, autophagy was induced accompanied with decreased miR-224 expression (Fig. 3C) and increased Smad4 expression (Supporting Fig. 12B). These results indicate that increased the level of autophagy is related to down-regulation of miR-224 and increased Smad4 expression.
Figure 6.

MiR-224 promotes tumor formation through silencing Smad4, and together with impaired autophagy correlates with poor overall survival rate. (A) HEK293T cells were transfected with wild-type or mutant-type pMIR-Report-Smad4 plasmid and cotransfected with miR-224, miR-224 antagonist (anti-miR-224) or their negative controls (N.C. and anti-N.C.), individually. The luciferase activity was determined and normalized with Renilla luciferase activity of plasmid pRL-TK. Quantification of the data is shown as mean ± SEM (n = 5). (B) Hep 3B cells were transfected with N.C. or miR-224. The cells with miR-224 were cotransfected with pRK5-Smad4 or control vector plasmids. (C) Similarly, Hep 3B cells were transfected with anti-N.C. or anti-miR-224. The cells with anti-miR-224 were cotransfected with si-Smad4 or si-RNA negative control (si-N.C.). The protein expression of Smad4 and β-actin was determined by western blotting. The transfected cells were injected subcutaneously into NOD/SCID mice (n = 5) for 14 days and the quantification of the tumors is shown in (B,C). (D) Two representative IHC staining of Smad4 expression from 46 paired HBV-associated HCC specimens. The analysis and quantification of protein expression are the same as in Fig. 1A. Scale bar = 20 μm. P values were obtained using a paired Student t test. (E) Correlation of Smad4 expression with miR-224 was analyzed using 46 paired HBV-associated HCC specimens and linear regression coefficient and statistical significance are indicated. N, adjacent nontumor tissue; T, tumor tissue. (F) The survival rates were determined by Kaplan-Meier analysis of HCC patients 4 years after surgery. P values were obtained by log rank test. High-risk group: low-Atg5, high-miR-224, and low-Smad4 expression in the HCC tumor parts compared to the adjacent nontumor parts. The remaining specimens are defined as the low-risk group. *P < 0.05 and **P < 0.01.
When Smad4 was silenced by small interfering RNA (siRNA) in Hep 3B cells and the cells were injected subcutaneously into NOD/SCID mice, tumor formation was increased, indicating that Smad4 plays a tumor suppressor role in HCC tumorigenesis (Supporting Fig. 13). To clarify the effect of miR-224 and the target gene Smad4 on tumor formation, the cells expressing different levels of miR-224 and Smad4 were injected subcutaneously into NOD/SCID mice. MiR-224 induced tumor formation was suppressed by overexpression of Smad4 (Fig. 6B). In contrast, anti-miR-224 suppressed tumor formation was further reversed by silencing Smad4 (Fig. 6C). A similar experimental design was used to reveal that miR-224 enhanced migration activity is through silencing Smad4 expression (Supporting Fig. 14). In conclusion, we demonstrated that miR-224 promotes tumor formation and migration through suppressing Smad4 expression.
In HBV-associated HCC specimens and HBx-transgenic mice, Smad4 expression was lower in the tumor portions as compared to the adjacent nontumor parts (Fig. 6D; Supporting Fig. 15A). Furthermore, Smad4 expression was inversely correlated with miR-224 expression (Fig. 6E) and positively correlated with Atg5 expression (Supporting Fig. 15B). Differently, the expression of Smad4 was not changed in the HCV-associated HCC (Supporting Fig. 15C). Finally, HCC patients are classified as the high-risk group (low-Atg5, high-miR-224, and low-Smad4 expression in the tumor parts compared to the adjacent nontumor parts), and the remaining HCC patients belong to the low-risk group. The former high-risk group shows significant correlation with poor overall survival rate (Fig. 6F). Altogether, our results suggest that low autophagic level accompanied with high miR-224 and low Smad4 expression promote HCC tumorigenesis.
Liver Tumor Formation Is Suppressed by Manipulation of Autophagic Level or MiR-224 Expression
To clarify whether autophagy and miR-224 could be potential targets for HCC therapy, the effects of autophagy and miR-224 on tumor formation was evaluated in a syngeneic orthotopic rat model of liver tumor formation. Rat hepatoma N1-S1 cells were orthotopically transplanted into the livers of SD rats and liver tumors were formed in 1 week. The N1-S1 cell line was established from a liver tumor induced by a carcinogen without virus infection (Supporting Fig. 16). Our data showed that the autophagy related genes (Atg5 and Beclin 1) were down-regulated and miR-224 was up-regulated in N1-S1 derived tumors, indicating that low autophagy level together with high miR-224 expression are involved in rat liver tumorigenesis (Supporting Fig. 17). Rat liver transplantation of N1-S1 cells was followed the next day by intraperitoneal injection of amiodarone or double-distilled water (DDW) (Supporting Fig. 18A). One week after injection, the body weights of the treated rats showed no difference (Supporting Fig. 18B), but tumor weight was reduced in the group receiving amiodarone compared to the DDW group (Fig. 7A, upper panels). Similarly, tumor weight was reduced in the group transplanted with anti-miR-224-transfected N1-S1 cells as compared with anti-negative control (Fig. 7A, lower panels). Autophagy induction in the rats by amiodarone was confirmed by overexpression of LC3-type II (Fig. 7B) and formation of double membrane autophagosomes in the amiodarone treatment group (Fig. 7C,D). Similarly, only mature miR-224 expression was decreased (Fig. 7E; Supporting Fig. 18C) and Smad4 expression was increased after amiodarone treatment (Fig. 7B). Furthermore, the inhibition of miR-224 by amiodarone or anti-miR-224 was equivalent (Fig. 7E). Collectively, the results demonstrate that induction of autophagy or suppression of miR-224 effectively suppresses liver tumor formation.
Figure 7.

Liver tumors are suppressed by manipulation level of autophagy or miR-224 expression in an orthotopic rat model. In the former group, rat N1-S1 hepatoma cells were orthotopically injected into the rat livers, followed by intraperitoneal injection of DDW or amiodarone (30 mg/kg). In the latter group, rat N1-S1 cells were transfected with miR-224 antagonist (anti-miR-224) or negative control (anti-N.C), and injected into the rats’ livers. (A) Rats were sacrificed to collect the livers 1 week after orthotopic injection. The arrows point to tumors in the liver, and five mice were used in each group. Tumor formation was quantified by measuring the tumor weight. (B) Protein was extracted from the tumors of the former group, and the expression of LC3, Smad4 and β-actin was determined by western blotting. (C,D) Tumor sections of DDW (C) or amiodarone (D) treatment were further examined under TEM. Scale bar = 2 μm; White boxes in (C,D) are further enlarged. Scale bar = 500 nm. The arrows point the autophagosomes. N, nucleus. (E) The expression of miR-224 in the tumors after various treatments was evaluated by real-time PCR. Quantification of miR-224 expression is shown as mean ± SEM (n = 5). P values were obtained by Student t test. *P < 0.05 and **P < 0.01.
Discussion
Accumulating evidence suggests that autophagy is involved in tumor suppression.6 We reveal a new mechanism of autophagy, by which mature form miR-224 is preferentially recruited to the autophagosome followed by lysosomal degradation (Fig. 8A). Furthermore, reduction of autophagy correlated with high miR-224 expression was detected in HBV-related HCC specimens and tumors in HBx-transgenic mice. In the cell line and tumor models with a low level of autophagy, miR-224 expression was high, and accordingly tumorigenesis was induced through Smad4 silencing mediated by miR-224 (Fig. 8B). Interestingly, HCC patients with low-Atg5, high-miR-224, and low-Smad4 expression showed significant correlation with poor overall survival rate (Fig. 6F). However, the mechanism by which the level of autophagy is decreased in HBV-related HCC remains unclear.
Figure 8.
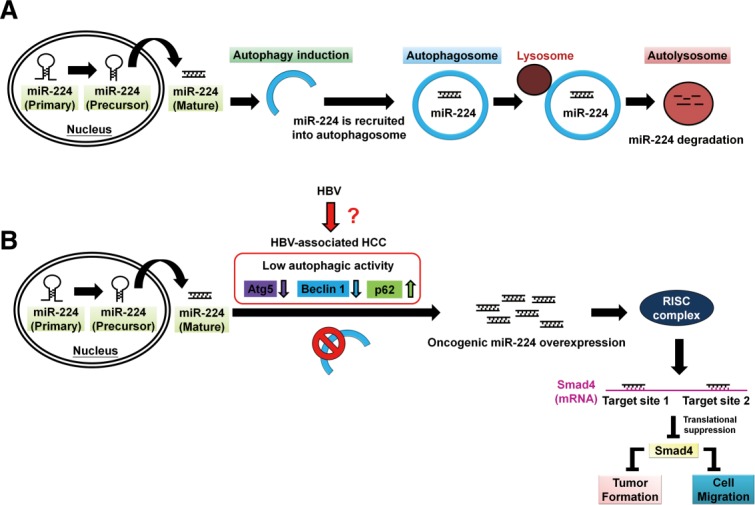
A schematic hypothetical model of autophagy regulation of miR-224 and reduction of autophagy is associated with high miR-224 expression in HBV-related HCC. (A) The mature form miR-224 is preferentially recruited into the autophagosome and proceeds with fusion with lysosome to form autolysosome, in which miR-224 was degraded. (B) Low level of autophagy occurs during HBV-associated HCC development demonstrated by low expression of autophagy-related genes (Atg5, Beclin 1) and accumulation of p62 protein through an undermined mechanism. Low autophagic activity may play a pivotal role in miR-224 overexpression, which promotes cell migration and tumor formation through silencing Smad4 protein expression.
In this study we demonstrated that various autophagy inducers suppress miR-224 expression in HBV-, HCV-, and nonvirus-infected cell lines, suggesting that it is a general event and is independent of viral status. Nevertheless, the reduction of autophagy was more frequently observed in HBV-associated HCC. The univariate and multivariate analyses of various clinical-pathological factors in 93 paired HCC specimens showed that only virus status is correlated with Atg5, miR-224, and Smad4 expression either alone or in combination (Supporting Table 2). Gao et al.21 reported that miR-224 is overexpressed in premalignant dysplastic nodules and persistent HBV-associated HCC. Together, these pieces of evidence indicate that HBV infection is a critical pathological factor in the autophagy-miR-224-Smad4 pathway-related tumorigenesis of HCC.
HBV and HCV cause severe liver diseases including hepatitis, cirrhosis, and HCC. HBV surface and X proteins as well as HCV nonstructural protein NSP5B induces autophagy.22,23 Autophagy induced by these viruses promotes viral replication. For virus-associated HCC, we demonstrated that reduction of autophagy was only associated with HBV-associated HCC (Fig. 1; Supporting Fig. 2), which was confirmed in the tumors of HBx-transgenic mice (Fig. 2B). Our results are consistent with the report that heterozygous deletion of Beclin 1 in the HBV transgenic mice increases spontaneous malignancies and accelerates HBV-induced HCC.24 Even though autophagy promotes the replication of both HBV and HCV, it only demonstrates a suppressive role in HBV-related tumorigenesis. It is possible that in contrast to HCV (RNA virus), HBV genomic DNA integrates into host chromosomes and causes insertional mutagenesis.25 Further study is needed to clarify whether such insertional mutagenesis affects Atg members in HBV-associated tumorigenesis. For nonvirus-infected HCC, the level of autophagy and miR-224 play similar roles in liver cancer formation, which was demonstrated in our orthotopic model of rat liver tumor formation (Fig. 7). Autophagy inducers such as amiodarone could be used as a potential therapy for various HCC with different etiology. However, our findings suggest that the stage of HBV infection as well as the tumorigenesis status of the HCC patient should be carefully evaluated before considering an autophagy-related therapy.
MicroRNA promoters are regulated either by transcriptional factors or by epigenetic modification. The expression of miR-224 is in concert with the expression of the adjacent GABRE gene regulated at the transcriptional level by HDACs and p300 in HCC.26 However, in this study miR-224 showed no correlation with GABRE expression in 75 paired HCC specimens analyzed (data not shown). Furthermore, we reveal that the mature form miR-224 is regulated at the posttranscriptional level by autophagic degradation. This discrepancy may be due to the variation in genetic background as well as the involvement of different etiologic agents during liver cancer formation.
Various miRNAs have been reported to regulate autophagy pathway through targeting Atg family members.12 These data are in contrast to our result that miR-224 does not affect autophagic induction and degradation (Supporting Fig. 8). Gibbings et al.27 reported that autophagy selectively degrades Dicer and Ago2 (components of miRNA-binding complex), and the miRNAs (let-7a and miR-16) are not degraded by autophagy because they are not in the Dicer-Ago2 complex. They claimed that autophagy may maintain miRNAs stability through an undetermined mechanism. Similarly, let-7a expression was not affected by autophagy in our investigation. In contrast, we reveal that oncogenic miR-224 was accumulated in the autophagosomes (Figs. 4A, 5B,C) followed by autophagic degradation (Fig. 3B-E) as compared to let-7a. This is the first report to demonstrate that miR-224 is preferentially accumulated in the autophagosomes and diminished through autophagy-related degradation.
RNA-binding proteins bind with miRNA to regulate miRNA maturation. For example, the RNA-binding protein Lin28 specifically recruits TUT4 (a terminal uridylyl transferase) by recognizing the GGAG sequence of pre-let-7 to block the biogenesis of let-7.28 It is probable that miR-224 is recognized by an undetermined RNA-binding protein and then transported to the autophagosome for degradation with the help of the autophagy adapter proteins (such as p62). Further study is needed to clarify the putative binding protein(s) as well as the adapter protein of miR-224.
MiR-224 is oncogenic in HCC due to its involvement in cell proliferation, migration, invasion, and antiapoptosis, and the genes related to cell survival (BCL-2 and MAPK1) and migration (PAK4, MMP9) are up-regulated.11,29 These results only reveal miR-224-related genes. Although Yao et al.30 first reported that Smad4, a transcriptional factor of the transforming growth factor beta (TGF-β) signaling pathway, is the target gene of miR-224 and is responsible for the proliferation and function of granulosa cells, the role of Smad4 in HCC remains unclear. The TGF-β signaling pathway suppresses cell proliferation at an initial stage, but promotes metastasis at late stages. Loss of Smad4 switches the role of TGF-β from a tumor suppressor to a tumor promoter.31 Smad4 expression was lower in the tumors of HBx transgenic mice (Supporting Fig. 15A) and down-regulated in HBV- but not in HCV-associated HCC (Fig. 6D versus Supporting Fig. 15C). Our data demonstrated that miR-224 indeed suppresses Smad4 expression and overexpression of Smad4 reverses the biofunction of miR-224 in HCC. Together, our findings reveal that miR-224 promotes HBV-associated HCC tumorigenesis through inhibition of Smad4.
Amiodarone is a widely used antiarrhythmic drug but some patients under long-term treatment show side effects including pulmonary complications and thyroid disease.32 It is also a potential drug to treat glioma and prostate carcinoma.33 The dosage and time of amiodarone treatment may affect disease development. Further experimental trials are required to determine the optimal dosage of amiodarone for suppressing HCC. This study provides a new therapeutic strategy of HCC by manipulating the level of autophagy using amiodarone to decrease oncogenic miR-224 expression. Furthermore, autophagy-miR-224-Smad4 in combination could be used for prediction of HCC patient survival.
Acknowledgments
We thank Drs. Rudy Juliano, Rik Derynck, and Tamotsu Yahsimori for providing the plasmids pRK5-Smad4 and ptfLC3; Dr. Kung-Chia Young for providing the Con-1 cell line; Drs. Kuen-Jer Tsai and Ya-Chun Hsiao for conducting FACS-like tissue-cytometry, image acquisition, and data analysis; Sheng-Hsiang Lin and Shang-Chi Lee for providing statistical analysis of the data; and Drs. Robert Anderson and Chia-Jui Yen for critically editing the article. We also thank the Taiwan Liver Cancer Network, National Health Research Institutes, Zhunan, Taiwan for providing the HCC tissue samples and related anonymous clinical data.
Glossary
- anti-miR-224
miR-224 antagonist
- AP
autophagosome fraction
- Atg
autophagy-related genes
- CQ
chloroquine
- DDW
double distilled water
- DIG
digoxigenin
- HBV
hepatitis B virus
- HBx
hepatitis B virus X protein
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- IHC
immunohistochemistry staining
- miRNA ISH
miRNA in situ hybridization
- miRNAs
microRNAs.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Fig.1
Supporting Fig.2
Supporting Fig.3
Supporting Fig.4
Supporting Fig.5
Supporting Fig.6
Supporting Fig.7
Supporting Fig.8
Supporting Fig.9
Supporting Fig.10
Supporting Fig.11
Supporting Fig.12
Supporting Fig.13
Supporting Fig.14
Supporting Fig.15
Supporting Fig.16
Supporting Fig.17
Supporting Fig.18
Supporting Figure Legends.
Supporting Table 1.
Supporting Table 2.
Supporting Table 3.
Supporting Data 1.
References
- 1.Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99–111. doi: 10.1016/j.ccr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 2.Aravalli RN, Steer CJ, Cressman EN. Molecular mechanisms of hepatocellular carcinoma. Hepatology. 2008;48:2047–2063. doi: 10.1002/hep.22580. [DOI] [PubMed] [Google Scholar]
- 3.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008;68:9167–9175. doi: 10.1158/0008-5472.CAN-08-1573. [DOI] [PubMed] [Google Scholar]
- 5.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lozy F, Karantza V. Autophagy and cancer cell metabolism. Semin Cell Dev Biol. 2012;23:395–401. doi: 10.1016/j.semcdb.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 9.Couzin J. MicroRNAs make big impression in disease after disease. Science. 2008;319:1782–1784. doi: 10.1126/science.319.5871.1782. [DOI] [PubMed] [Google Scholar]
- 10.Chen PJ, Yeh SH, Liu WH, Lin CC, Huang HC, Chen CL, et al. Androgen pathway stimulates microRNA-216a transcription to suppress the tumor suppressor in lung cancer-1 gene in early hepatocarcinogenesis. Hepatology. 2012;56:632–643. doi: 10.1002/hep.25695. [DOI] [PubMed] [Google Scholar]
- 11.Li Q, Wang G, Shan JL, Yang ZX, Wang HZ, Feng J, et al. MicroRNA-224 is upregulated in HepG2 cells and involved in cellular migration and invasion. J Gastroenterol Hepatol. 2010;25:164–171. doi: 10.1111/j.1440-1746.2009.05971.x. [DOI] [PubMed] [Google Scholar]
- 12.Fu LL, Wen X, Bao JK, Liu B. MicroRNA-modulated autophagic signaling networks in cancer. Int J Biochem Cell Biol. 2012;44:733–736. doi: 10.1016/j.biocel.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Hanna JA, Hahn L, Agarwal S, Rimm DL. In situ measurement of miR-205 in malignant melanoma tissue supports its role as a tumor suppressor microRNA. Lab Invest. 2012;92:1390–1397. doi: 10.1038/labinvest.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seglen PO, Brinchmann MF. Purification of autophagosomes from rat hepatocytes. Autophagy. 2010;6:542–547. doi: 10.4161/auto.6.4.11272. [DOI] [PubMed] [Google Scholar]
- 15.Lin X, Li X, Liang D, Lan K. MicroRNAs and unusual small RNAs discovered in Kaposi’s sarcoma-associated herpesvirus virions. J Virol. 2012;86:12717–12730. doi: 10.1128/JVI.01473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu BK, Li CC, Chen HJ, Chang JL, Jeng KS, Chou CK, et al. Blocking of G1/S transition and cell death in the regenerating liver of Hepatitis B virus X protein transgenic mice. Biochem Biophys Res Commun. 2006;340:916–928. doi: 10.1016/j.bbrc.2005.12.089. [DOI] [PubMed] [Google Scholar]
- 17.Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010;53:1123–1134. doi: 10.1016/j.jhep.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Aghajan M, Li N, Karin M. Obesity, autophagy and the pathogenesis of liver and pancreatic cancers. J Gastroenterol Hepatol. 2012;27(Suppl 2):10–14. doi: 10.1111/j.1440-1746.2011.07008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee YS, Pressman S, Andress AP, Kim K, White JL, Cassidy JJ, et al. Silencing by small RNAs is linked to endosomal trafficking. Nat Cell Biol. 2009;11:1150–1156. doi: 10.1038/ncb1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, et al. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One. 2009;4:e7124. doi: 10.1371/journal.pone.0007124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao P, Wong CC, Tung EK, Lee JM, Wong CM, Ng IO. Deregulation of microRNA expression occurs early and accumulates in early stages of HBV-associated multistep hepatocarcinogenesis. J Hepatol. 2011;54:1177–1184. doi: 10.1016/j.jhep.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 22.Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U S A. 2010;107:4383–4388. doi: 10.1073/pnas.0911373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, et al. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol. 2011;85:6319–6333. doi: 10.1128/JVI.02627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fallot G, Neuveut C, Buendia MA. Diverse roles of hepatitis B virus in liver cancer. Curr Opin Virol. 2012;2:467–473. doi: 10.1016/j.coviro.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Toh HC, Chow P, Chung AY, Meyers DJ, Cole PA, et al. MicroRNA-224 is up-regulated in hepatocellular carcinoma through epigenetic mechanisms. FASEB J. 2012;26:3032–3041. doi: 10.1096/fj.11-201855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibbings D, Mostowy S, Jay F, Schwab Y, Cossart P, Voinnet O. Selective autophagy degrades DICER and AGO2 and regulates miRNA activity. Nat Cell Biol. 2012;14:1314–1321. doi: 10.1038/ncb2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho J, et al. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Takahashi S, Tasaka A, Yoshima T, Ochi H, Chayama K. Involvement of microRNA-224 in cell proliferation, migration, invasion and anti-apoptosis in hepatocellular carcinoma. J Gastroenterol Hepatol. 2013;28:565–575. doi: 10.1111/j.1440-1746.2012.07271.x. [DOI] [PubMed] [Google Scholar]
- 30.Yao G, Yin M, Lian J, Tian H, Liu L, Li X, Sun F. MicroRNA-224 is involved in transforming growth factor-beta-mediated mouse granulosa cell proliferation and granulosa cell function by targeting Smad4. Mol Endocrinol. 2010;24:540–551. doi: 10.1210/me.2009-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang B, Halder SK, Kashikar ND, Cho YJ, Datta A, Gorden DL, et al. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology. 2010;138:969–980. doi: 10.1053/j.gastro.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maseeh UZ, Fatima N, Sajjad Z. Amiodarone therapy: don’t forget thyroid. J Pak Med Assoc. 2012;62:268–272. [PubMed] [Google Scholar]
- 33.Kim IY, Kang YJ, Yoon MJ, Kim EH, Kim SU, Kwon TK, et al. Amiodarone sensitizes human glioma cells but not astrocytes to TRAIL-induced apoptosis via CHOP-mediated DR5 upregulation. Neuro Oncol. 2011;13:267–279. doi: 10.1093/neuonc/noq195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Fig.1
Supporting Fig.2
Supporting Fig.3
Supporting Fig.4
Supporting Fig.5
Supporting Fig.6
Supporting Fig.7
Supporting Fig.8
Supporting Fig.9
Supporting Fig.10
Supporting Fig.11
Supporting Fig.12
Supporting Fig.13
Supporting Fig.14
Supporting Fig.15
Supporting Fig.16
Supporting Fig.17
Supporting Fig.18
Supporting Figure Legends.
Supporting Table 1.
Supporting Table 2.
Supporting Table 3.
Supporting Data 1.