

Introduction
NeuroAIDS is in essence a glial driven disease. Microglia and astrocytes harbor the infection and release cellular and viral toxins responsible for bystander injury of neurons [1–4]. Emerging evidence implies that, besides peripheral immune dysregulation, opiates can exacerbate the neuropathological effects of human immunodeficiency virus type 1 (HIV-1) infection in the central nervous system (CNS) though direct actions on μ-opioid receptor (MOR) expressing glia [5–10]. The concept that HIV-1 and opiates can enhance CNS neuropathology is supported both experimentally [1] and in patients with HIV-associated neurocognitive disorders (HAND) [11, 12]. The CNS is preferentially vulnerable to opiate and HIV-1 interactions most likely due to the complexity and interrelatedness of MOR actions across neurons, astroglia and microglia. In fact, glial cells are implicated as the primary mediators of opiate-driven HIV-1 exacerbation seen with acute morphine and Tat [5] and also perhaps gp120 [9] co-exposure.
Ibudilast (3-isobutyryl-2-isopropylpyrazolo[1,5-a]pyridine; AV411, MN-166) is a non-selective phosphodiesterase (PDE) inhibitor that has been marketed for over two decades in Japan for the treatment of chronic asthma [13]. Both AV411 and its amidated analogue, AV1013, have a low molecular weight (230 Da), with the ability to readily cross the blood brain barrier [13], and have pronounced anti-inflammatory and anti-nociceptive effects [14–17] that are reported to be principally mediated by glia [16, 18–20]. AV411 reduces neuroinflammation by elevating cAMP [21–23] and by decreasing tumor necrosis factor-α (TNF-α) release, which restricts the activity of glia, while promoting the production of the anti-inflammatory cytokine interleukin (IL)-10 and increasing the release of various neurotrophic factors by astrocytes and microglia [18–20]. In addition, AV411 has been shown to be neuroprotective against LPS-activated microglia and in neuronal injury from excitotoxic ischemia [24, 25]. AV411 reduces post-herpetic neuralgia [26], limits white matter damage following chronic hypoxia [27], is protective in an oligodendrocyte precursor cell line (CG-4) [21, 23], and is potentially neuroprotective in multiple sclerosis [28, 29]. Thus, a variety of evidence implicates AV411 in restricting glial inflammation. In addition, a link between epigenetic changes in IL-10 expression in microglia and reinstatement of opiate-induced conditioned place preference suggests a role for glia in mediating lasting changes in drug-seeking behavior [30]. In the context of opiate abuse, AV411 attenuates the effects of opioid withdrawal, limits morphine-induced dopamine release in the nucleus accumbens, and prevents morphine induction of pro-inflammatory cytokine expression [31].
The beneficial effects of AV411 on drug abuse extend beyond the opiates. We have previously found that both AV411 and AV1013 attenuate methamphetamine-induced locomotor activity and its sensitization [32], block prime- and cue-induced relapse of methamphetamine-maintained responding [33, 34], and reduce methamphetamine self-administration. In part, because of these and other preclinical data AV411 is in clinical trials for methamphetamine abuse [35] and for the treatment of opiate withdrawal [36]. Prompted by evidence that AV411 and AV1013 reduces CNS inflammation and addictive behavior, as well as emerging findings that these drugs can also attenuate inflammation in HIV-exposed glia, we evaluated the properties of ibudilast and AV1013 as potential therapies for the management of HIV-associated inflammation and neurodegenerative effects.
Methods
Experiments were conducted in accordance with procedures reviewed and approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee (IACUC).
Reagents and drug treatments
HIV-1 Tat 1–86 IIIB was purchased from ImmunoDX, LLC (Woburn, MA, USA) and was used at a concentration of 100 nM (~0.8 μg/mL). Soluble Tat levels in HIV-1 patient sera have been measured up to 40 ng/mL [37, 38]. Morphine sulfate was obtained from NIDA and used at a concentration of 500 nM. Ibudilast (AV411) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA) and AV1013 was obtained from MediciNova (San Diego, CA, USA); both were used at 100 nM, 1 μM and 10 μM concentrations. For time-lapse experiments treatments were done in vitro using murine mixed-glia and neurons in culture. Cells were left untreated or exposed to HIV-1 Tat1–86 (100 nM) ± morphine sulfate (500 nM). Additionally, the above treatments were challenged with increasing concentrations of AV411 or AV1013. For experiments examining intracellular signaling, murine mixed-glial cultures lacking neurons were treated in vitro with vehicle control (serum-free DMEM) or the same drug treatments as above.
Murine primary cell cultures
Striatal mixed glial cultures
Brains from P0–P1 ICR mouse pups were removed and striata dissected out. Cells were plated at 1.0 × 105 cells/cm2 on poly-L-lysine coated cell culture dishes and grown until confluent in DMEM supplemented with 10% FBS.
Striatal neuron cultures
E14–E15 ICR embryos were aseptically removed by cesarean section, decapitated, and the striata were dissected from the cerebrum. The neuron-enriched cell isolates were plated at 0.5 × 105 cells/cm2 onto a bedlayer of 9–10 day old mixed glia.
Transfection and HIV-1 infection of human microglia
Primary human microglial cells (ScienCell, Carlsbad, CA, USA) in culture, were transfected in vitro as described previously [39]. For detailed methods see Supplemental Digital Content 1.
ELISA
ELISA was performed as described previously [39]. For detailed methods see Supplemental Digital Content 1.
Quantitative real time PCR (qRT-PCR)
Tat RNA expression levels were examined 24 h post-infection by qRT-PCR and Nested PCR. For detailed methods see Supplemental Digital Content 1.
Cell viability
Cell viability in mixed glia cultures was assessed after 12, 24 and 48 h treatment with AV411 and AV1013 at concentrations of 100 nM, 1 and 10 μM using propidium iodide (Molecular Probes, Eugene, OR, USA), and viable cells were quantified by flow cytometry using a FACSCanto II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Subcellular localization of p65
Immunofluorescence labeling was performed as described previously [7]. For detailed methods see Supplemental Digital Content 1.
Statistical analyses
Mean data values and the standard error of the mean (SEM) were calculated for each variable. One-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons was used to analyze data involving multiple sample groups. For time-lapse microscopy studies, a two-way repeated measures ANOVA was performed followed by Duncan’s post hoc analysis. A value of p < 0.05 was designated as statistically significant.
Results
HIV-1 replication in human microglia is attenuated with AV411 and AV1013
The inhibitory potential of AV411 and the amino analog AV1013 on HIV-1 replication in primary human microglia was examined using transient transfection of a Tat-responsive long terminal repeat (LTR) construct with a luciferase reporter. Twenty-four hours post-infection, Tat expression was increased 2.2 and 2.5 fold in microglia after infection with HIV-1SF162 alone and with HIV-1 in combination with morphine, respectively, when compared to uninfected control cells (Fig 1A). Exposure to both AV411 and AV1013 caused a significant decrease in Tat expression, while concurrent exposure with morphine had a minimal effect on the inhibitory action of both AV411 and AV1013 (Fig. 1A).
Figure 1. AV411 and AV1013 inhibit HIV-1 SF162 replication in human microglia and displays non-specific interactions with morphine.
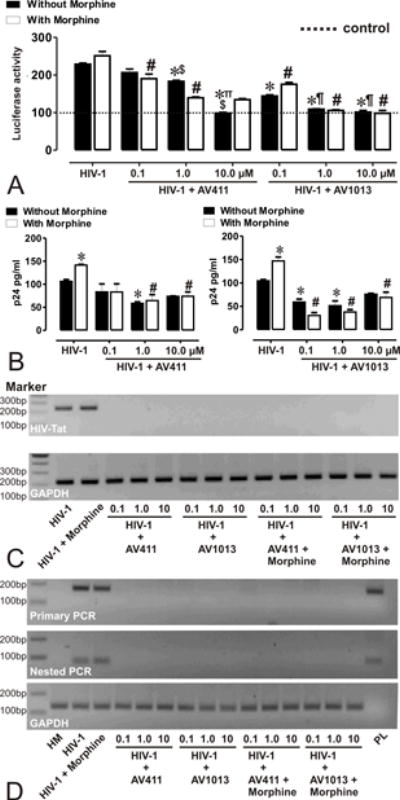
(A) HIV-1 Tat protein was monitored by transfecting human microglia with pBlue3’LTR-luc reporter followed by infection with R5 tropic HIV-1 ± morphine ± increasing concentrations of AV411 and AV1013. Relative Tat protein expression was examined 24h post-infection by measuring luciferase activity and the reported values represents the mean luciferase intensity ± SEM from 3 independent experiments (the dotted line represents un-infected control cells). *p<0.05 vs. HIV-1 R5; #p<0.05 vs. R5 + Morphine; ∏p<0.05 vs. 100 nM AV411; $p<0.05 vs. 1 μM AV411; ¶p<0.05 vs. 100 nM AV1013; §p<0.05 vs. 1 μM AV1013. (B) HIV-p24 Gag levels were monitored in human microglia infected with R5 tropic HIV-1 ± morphine ± increasing concentrations of AV411 and AV1013. Cells were infected for 3 days, and then incubated with control media ± morphine ± increasing concentrations of AV411 or AV1013 for another 2 days. Culture supernatants were used to measure p24 levels by ELISA. *p<0.05 vs. HIV-1 R5; #p<0.05 vs. R5 + Morphine. (C-D) HIV-1 Tat mRNA was monitored in human microglia infected with R5 tropic HIV-1 ± morphine ± increasing concentrations of AV411 and AV1013. Tat mRNA expression levels were examined 24 h post-infection by RT-PCR and Nested PCR, and the PCR products were detected using 2% agarose gels stained with ethidium bromide. Marker indicates the position of a 100 base pair (bp) ladder marker. GAPDH served as an input control HM = human microglia; PL= Tat expressing plasmid.
Twenty-four hours post-infection the PCR products shown in the top bands on the agarose gel correlate with the LTR luciferase data, while GAPDH, shown in the bottom bands, was used as a loading control. Lastly, HIV-1 p24 levels were examined in the supernatant from HIV-1SF162-infected microglia cells by ELISA (Fig. 1B). Forty-eight hours post-treatment with morphine caused a significant increase in viral replication when compared to HIV-1 alone. To confirm these initial studies, RNA expression levels of HIV-1 Tat in virally infected microglia were also examined by RT-PCR (Fig. 1C) and Nested PCR (Fig. 1D) to rule out lack of Tat expression. Exposure with both AV411 and AV1013 caused a significant decrease in viral replication, while concurrent exposure with morphine had no significant effect on the inhibitory actions of AV411.
AV411 and AV1013 block synergistic morphine and Tat-induced neurotoxicity in vitro
The therapeutic potential of AV411 and the amino analog AV1013 in attenuating the synergistic interactions seen with opiate abuse-HIV co-morbidity in neuronal cell death was examined in neuron-mixed-glia co-cultures from murine striatum exposed with Tat ± morphine. A within-subjects design was used, which assesses the same neurons throughout the experiment using a computer-aided stage encoder over 72 h, and that greatly reduces inter-subject variability and increases the sensitivity of the assay [1]. Neurons in control cultures showed some inherent toxicity in this assay, while treatment with Tat (100 nM) for 72 h significantly reduced the survival of isolated striatal neurons as compared with untreated neurons at each time point after 24 h (Fig. 2; *p < 0.05 vs. control). The Tat-induced decline in neuronal survival was significantly enhanced with collective morphine treatment at each time point after 28 h when compared to the Tat-only condition (Fig. 2B,D). At 100 nM AV411 had no effect on Tat and morphine mediated neurotoxicity, although at 10 μM concentration it did show some toxicity (Fig. 2B and data not shown). Co-administering 1.0 μM AV411 was not toxic and partially reversed Tat-mediated toxicity (Fig. 2A; dotted line), while significantly preventing the neurotoxic effects of combined Tat and morphine exposure (Fig. 2B). AV1013 at a concentration of 100 nM also was not toxic (Fig 2C; black line), and although it did not reverse Tat-mediated neuronal cell death (Fig. 2C; dotted line), it significantly prevented combined Tat and morphine-mediated neuronal cell death. At concentrations of 1μM and 10μM, AV1013 was intrinsically neurotoxic and displayed additive neurotoxicity when combined with Tat and morphine (Fig. 2D and data not shown). Thus, both AV411 and AV1013 can significantly reverse Tat-mediated neuronal cell death when co-exposed to morphine.
Figure 2. AV411 and AV1013 attenuate morphine and Tat-mediated interactive neurotoxicity in a concentration-dependent manner.
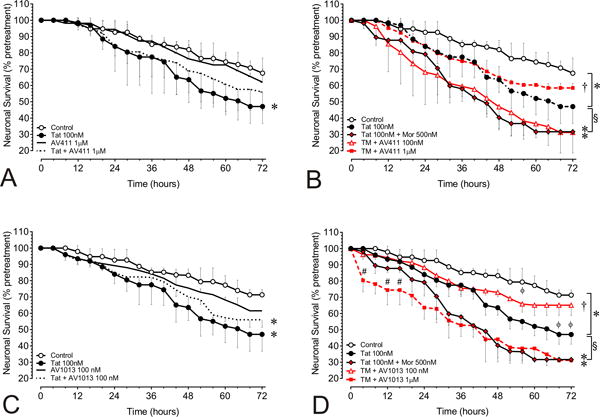
Tat ± morphine-induced neurotoxicity in neuron-mixed-glial co-cultures was monitored using a computer-assisted, time-lapse microscopy that permitted longitudinal analysis of individual neurons at 30 min intervals for 72 h. The mixed-glial cultures consisted of 90.2 ± 0.4% astrocytes; 8.8 ± 0.6% microglia and mimic proportions in the striatum [5]. Neuronal survival with (A) HIV-Tat ± 1 μM AV411 (B) with HIV-Tat ± morphine (M) ± 100 nM and 1.0 μM AV411 (C) with HIV-Tat ± 100 nM AV1013 (D) with HIV-Tat ± morphine (M) ± 100 nM and 1.0 μM AV1013. Data were analyzed by two-way repeated measures ANOVA followed by Duncan’s post hoc test. *p < 0.05, vs. controls; §p < 0.05, vs. Tat; †p < 0.05 vs. Tat + morphine group; **p vs. all other treatments; #p < 0.05 vs. 1 μM AV1013.
AV411 and AV1013 reduce the secretion of pro-inflammatory cytokines from Tat ± morphine treated cells
Activated microglia and astroglia are believed to be centrally involved in the neuronal loss associated with HIV-1 and opiate-induced cytotoxicity as they release pro-inflammatory factors, such as TNF-α, that eventually lead to cognitive deficits and other neuropathology linked to HAND [40, 41]. In an effort to determine a possible mechanism through which AV411 and the amino analog AV1013 were able to reverse Tat and morphine mediated neurotoxicity, the release of selective inflammatory cytokines were analyzed. The therapeutic potential of AV411 and AV1013 in attenuating MIF, a pro-inflammatory cytokine known for its effect on macrophage migration and activation, the pro-inflammatory mediator TNF-α, and the anti-inflammatory cytokine IL-10, were examined in glial co-cultures from murine striatum exposed with Tat ± morphine. The release of inflammatory molecules were measured in supernatants from mixed glial cultures after 1 h and 8 h treatment with 1 μM AV411 or 100 nM AV1013 (Fig. 3). MIF expression levels were relatively unchanged after 1 h treatment with AV411 or AV1013, when compared with vehicle-treated (control) cells (Fig. 3B). An 8 h exposure to Tat alone or in combination with morphine caused a significant increase in MIF secretion, and both AV411 and AV1013 significantly reduced this effect. Likewise, 8 h treatment, but not 1 h treatment, with Tat ± morphine elevated TNF-α secretion, and both AV411 and AV1013 returned TNF-α levels to vehicle-treated levels (Fig. 3C,D). On the contrary, both AV411 and AV1013 significantly increased IL-10 levels at both 1h and 8 h in the Tat and Tat + morphine groups, yet they had no effect on IL-10 in the vehicle-treated controls (Fig. 3E,F). Our data agrees with previously published data by others showing an inhibition of inflammatory responses and an increase in anti-inflammatory responses by AV411 [42–45].
Figure 3. Effects of AV411 and AV1013 on Tat ± morphine –induced inflammatory responses in mixed glial cultures.
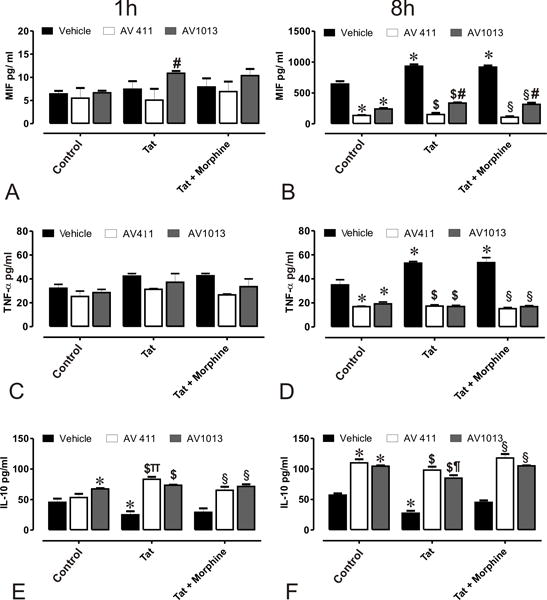
Supernatants from cells treated with control media, or medium with Tat ± Morphine with and without 1 μM AV411 and 100 nM AV1013 were used to detect MIF after 1 h (A) and 8 h (B); TNF-α after 1 h (C) and 8 h (D); and IL-10 after 1h (E) and 8h (F) by ELISA. One-way ANOVA followed by Bonferroni’s test for multiple comparisons was used to assess statistical differences. Values are determined from standard curves and given as the mean ± the SEM of 3 experiments. *p<0.05 vs. control; $p<0.05 vs. Tat; §p<0.05 vs. Tat + Morphine; πp<0.05 vs. AV411; ¶p<0.05 vs. AV1013.
AV411 and AV1013 modulate Tat + morphine-induced NF-ĸB nuclear translocation
Nuclear factor-kappa B (NF-κB) signaling plays an important role in Tat ± morphine -induced cytokine production in glial cultures [7]. It has been reported that AV411 can disrupt transcriptional activity of NF-ĸB and the stability of IκBα [42]. In an effort to explain how AV411 diminished MIF and TNF-α induced release by Tat ± morphine treatment, degradation of IκBα and nuclear translocation of NF-κB were determined by western blotting analysis (data not shown), and tracked by immunofluorescence labeling (Fig. 4A,B). Protein lysates from whole cell control showed detectable levels of IκBα by western blotting after 60 min, while incubation with Tat ± morphine led to the degradation of IκBα. Pretreatment with 1.0 and 10 μM AV411 in the presence or absence of Tat ± morphine led to the degradation of IκBα.
Figure 4. AV411 and AV1013 inhibit Tat + Morphine-induced p65 NF-ĸB nuclear translocation and causes morphological changes in astrocytes at high concentrations.
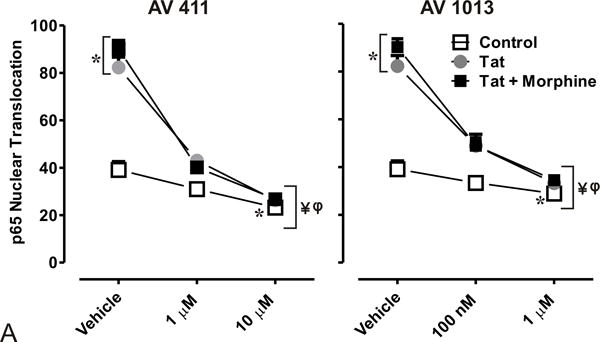
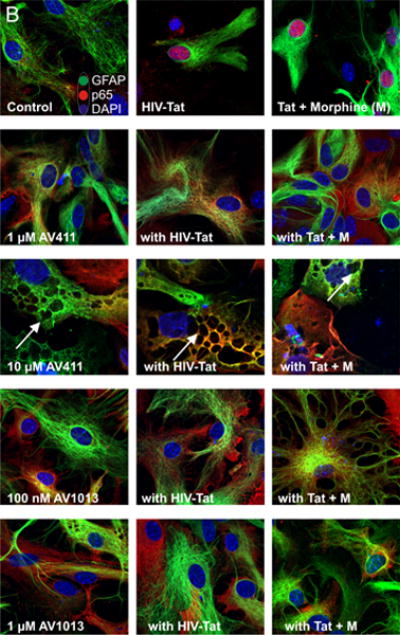
(A) Translocation of p65 in the nucleus was counted manually per 100 cells. DAPI was used as a reference for nuclear location. Total numbers were calculated using one-way ANOVA followed by Bonferroni’s test for multiple comparisons. Error bars show the mean ± the SEM from 3 independent experiments. *p<0.05 vs. vehicle; ¥p<0.05 vs. Tat; φp<0.05 vs. Tat + morphine. (B) Nuclear translocation of the p65 subunit (red) was readily detectable by immunofluorescence and visualized using confocal microscopy. Astrocytes were labeled with the cell type specific immunofluorescent markers, glial fibrillary acidic protein (GFAP; green) and their nuclei counterstained with DAPI (blue). Magnification = 63X.
We then determined p65 nuclear localization in mixed glial cell cultures labeled with the astrocyte specific marker glial fibrillary acidic protein (GFAP) by immunofluorescence (Fig. 4A). As expected, we observed p65 nuclear translocation in cells treated with Tat alone or in combination with morphine (Fig. 4A,B), while treatment with 1.0 μM and 10 μM AV411 in the presence or absence of Tat ± morphine attenuated p65 nuclear localization after 8 h treatment. In addition, astroglia displayed morphological changes including cytoplasmic retraction resembling stellation upon treatment (Fig. 4B; arrow), which can be caused by increases in cytoplasmic cAMP [46], as this phenomena was more noticeably with 10 μM concentrations of AV411. Interestingly, when cells were treated with AV1013, which lacks PDE activity, cytoplasmic retraction/stellation was not observed, suggesting that AV411 was responsible for the morphological changes, as cAMP-dependent pathways can influence important aspects of cellular growth, morphogenesis and differentiation. The data suggest that AV411 and AV1013 can disrupt Tat ± morphine- induced modulation of NF-κB signaling.
Effect of experimental treatments on cell viability
A viability assay was performed in mixed glia cells at 12 h, 24 h, and 48 h after treatment with increasing concentrations of AV411 and AV1013 (Fig. 5). Both AV411 and AV1013 showed minimal toxicity and caused negligible cell death after 12 h treatment. At the highest concentrations tested, 10 μM AV1013 caused a an ~23% decrease in glial viability after 24 h and a ~46% decrease in glial viability after 48 h, while 10 μM AV411 caused a loss in glial viability in about a quarter of the cells after 48 h. Importantly, however, beneficial reductions in HIV-1 replication, the release of inflammatory cytokines, and in neurodegeneration were evident at concentrations of AV411 or AV1013 that were an order of magnitude less than concentrations in which cytotoxicity was apparent.
Figure 5. Cell viability assay on mixed glial cultures treated with AV411 and AV1013.
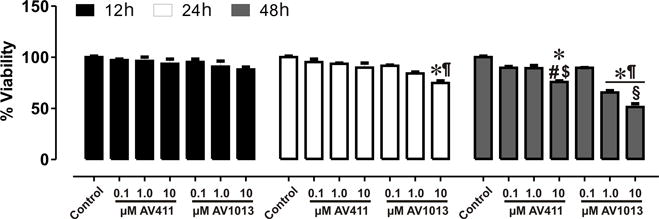
A cell viability assay was performed at 12 h, 24 h and 48 h after treatment with increasing concentrations of AV411 and AV1013. Data are the percentages of viable cells ± the SEM from 3 independent experiments. One-way ANOVA followed by Bonferroni’s test for multiple comparisons was used to assess statistical differences. *p<0.05 vs. control; #p<0.05 vs. 100 nM AV411; $p<0.05 vs. 1 μM AV411; ¶p<0.05 vs. 100 nM AV1013; §p<0.05 vs. 1 μM AV1013.
Discussion
Despite the advent of cART (combined antiretroviral therapy), the incidence of neurocognitive complications associated with HIV persists, which partially results from the limited efficiency by which many of these drugs cross the blood brain barrier [47]. Furthermore, cART does not directly target the inflammatory cascades thought to contribute to HIV encephalitis and HAND [42]. In fact, recent evidence indicates that the sustained production of Tat protein contributes to chronic immune activation seen in HIV-1-infected individuals maintained on cART [48], and provided partial justification for our use of Tat in the present study. Moreover, because microglia and astroglia are the principal cell types harboring HIV-1 infection in the brain parenchyma and are the main sources of CNS inflammation, glia are critical targets for therapeutic intervention. Therefore, suppression of neuroinflammation may provide new therapeutic approaches to neuroAIDS. HIV-1 and opiates selectively interact with microglia and astrocytes to enhance the release of proinflammatory cytokines [49–51] and to exacerbate bystander neurotoxicity [5]. The goal of this study was to evaluate the drug ibudilast (AV411), which has already been used clinically for almost two decades, for its ability to prevent viral replication and neuroinflammation, as well as its capacity to restrict the neurotoxic interactions between opiate drugs and HIV-1.
In this study, AV411 and its analog AV1013: (i) inhibited replication of HIV-1 in microglia and additionally restricted Tat ± morphine-induced; (ii) inhibited inflammatory cytokine release; (iii) limited the activation of the p65 subunit of NF-κB; and (iv) reduced bystander neurotoxicity. The concentrations used to inhibit replication by HIV-1 were between 100 nM and 10 μM, while the most efficient concentration for achieving reduced neurotoxicity was 100 nM of AV1013 and 1 μM of AV411. Likewise, 100 nM of AV1013 and 1 μM of AV411 were sufficient to suppress MIF and TNF-α production and to inhibit p65 nuclear translocation. AV411 has a mean half-life of 19 h and inhibits PDE 3,4,10 and 11 at IC50s ranging from approximately 1 μM to 10 μM [13, 43]. AV441 displayed less neurotoxicity than AV1013 at equivalent concentrations, which might result from the proposed neuroprotective actions of inhibiting PDE [52]. Importantly, reductions in HIV-1 replication, the production of inflammatory cytokines, and in neuronal death, were achieved at concentrations of AV411 or AV1013 that were 10-fold lower than concentrations in which cytotoxicity was evident in mixed-glial cultures after prolonged exposure (Fig. 5).
In healthy adults, AV411 is generally well tolerated, and with the exception of hyperhidrosis, headache and nausea, no serious adverse side effects have been recorded at the clinically achievable concentration of 59.9 ng/ml AV411 in plasma [53]. Accordingly, the maximum serum concentration of AV411 after oral administration of 10 mg capsule is 30 ng/ml, or about 0.3 μM (www.kyorinpharm.co.jp/prodinfo/medicine/pdf/KETAS_Capsules.pdf). By administrating 20–30 mg/day, which is the prescribed treatment for asthma [54], the serum concentration of AV411 is ≤1 μM [18]. The steady-state elimination half-life of AV411 following repeated, daily 50 mg/kg b.i.d doses is 21–28 hours [54]. For the treatment of neurological conditions, higher target doses of 60–100 mg/day have been proposed based on evidence from preclinical and preliminary clinical studies [54]. Initial clinical trials have found ibudilast to be efficacious in the treatment of chronic pain, and disorders such as multiple sclerosis and drug withdrawal and relapse (including methamphetamine and opiates), which may be accompanied by marked neurodegeneration [54]. In a 2-week repeat-dose study comparing the tolerability and pharmacokinetics of ibudilast at 20 – 50 mg b.i.d. in 12 healthy subjects and 12 diabetics, peak ibudilast plasma concentrations approximating 120 ng/ml were obtained [13]. Obtaining this plasma level was well tolerated with gastrointestinal complaints being the most frequently reported, and then only by subjects who were also being co-medicated for their diabetes. This peak plasma concentration was within the range of our in vitro concentrations that inhibited replication of HIV-1 in microglia and reduced neurotoxicity. These observations, and considering that the maximum tolerated dose of ibudilast in humans has not yet been demonstrable [13], increase consideration of ibudilast’s potential clinical evaluation as an adjunct in the treatment of HIV and associated neurological complications.
MIF is a pivotal regulator of innate immunity [55, 56] and regulates immune sensitivity to glucocorticoids [57]. MIF expression can also affect adult neurogenesis, as well as learning and memory, and depression [58]. Within the central nervous system (CNS), astrocytes are the major site of MIF expression [59], while the major site of MIF action is at CXCR2 on macrophages and microglia [60]. Likewise, in the CNS, TNF-α is produced by astrocytes and microglia [61]. Both MIF and TNF-α are considered to play critical roles in the development of various pathological processes in the CNS, including neurodegeneration accompanying HIV-related pathology. MIF is involved in the pathogenesis of inflammatory and infectious diseases, and increased MIF levels are detected in HIV-infected individuals as compared to HIV-negative patients [62, 62, 63]. Moreover, the increases in TNF-α that are associated with HIV-1-infected or activated macrophages and microglia are thought to be a major contributor to HIV-1-induced neuroinflammation, which eventually leads to neuronal damage and cognitive impairment [64–66]. AV411 was shown by others to inhibit the catalytic and chemotactic functions of MIF [67], and to suppress TNF-α production by glia [45]; therefore, AV411 may play a strategic role in modifying direct astrocyte-to-microglial signaling via this pathway.
Transcriptional regulators of the NF-κB/IκB family promote the expression of a multitude of cytokines and chemokines [68]. In addition, HIV-1 can activate NF-κB to promote viral replication [69]. AV411 and AV1013 reduced Tat ± morphine-mediated p65 nuclear translocation and localization. Although the precise mechanism by which AV411 affects NF-κB transcriptional activity remains unknown, we conclude that blocking NF-κB activity by preventing nuclear translocation leads to the inhibition of MIF and TNF-α production. Alternatively, a reduction in p65 nuclear trafficking could result from an increase in IL-10, which is known to inhibit the NF-κB pathway [70].
Despite the significant ability of AV411 to inhibit HIV-1 replication and Tat-induced neurotoxicity, the mechanisms and cellular targets of AV411’s actions are not fully understood. We believe that opiate-HIV synergistic bystander neurotoxicity involves crosstalk between glia by blocking reciprocal positive feedback among glial cell types [1]. Although microglia are the main effectors of neuronal injury, their sustained activation, especially in the context of opiate and HIV co-exposure, appears to be driven by astroglial signals [1, 71]; the release of cytokines and chemokines, and protein oxidation are not self-sustaining in the absence of other cell inputs [10], which likely includes significant signals from astroglia [1, 7, 8]. Ongoing studies in our laboratory are examining the actions of AV411 and AV1013 independently in each glial type using murine and infective human models. Since AV411 appears to restrict neuroinflammatory processes in both astroglia and microglia, we speculate that by acting in both glial types AV411 is well-positioned to attenuate reverberating feedback cascades between astroglia and microglia, which otherwise sustains neuroinflammation and perhaps drives HIV-1 replication in microglia [1, 72]. Lastly, AV411 markedly attenuated HIV replication and prevented Tat-induced pro-inflammatory cytokine production both in the absence and presence of morphine. AV411 was also able to selectively attenuate morphine and Tat neurotoxic interactions, with less of an effect on neurotoxicity due to Tat alone. Further studies validating these outcomes in more therapeutically-relevant settings are needed to determine the utility of AV411 as an adjunctive therapeutic for neuroAIDS or for opiate-HIV comorbidity.
Supplementary Material
Acknowledgments
The authors would like to thank Mr. Ruturaj Masvekar for technical assistance and Dr. Sylvia Fitting for assistance with statistical analysis. The authors also wish to thank Dr. Kirk Johnson, (MediciNova, San Diego, CA, USA) for providing the AV1013 used in these studies. This work was funded by NIH grants R01 DA036154 (NEH); T32 DA007027 and F32 DA033898 (SMD); R01 DA018633 and K02 DA027374 (KFH), and R01 DA034231 (PEK and KFH). Flow cytometry was supported, in part, by NIH National Cancer Institute Cancer Center Support Grant P30 CA016059. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH
Footnotes
The authors have no conflict of interest to report.
Authors’ contributions
NEH and KFH conceived and designed the experiments. NEH, MR, SES, and SZ performed the experiments. NEH, EMP, PEK and KFH analyzed and interpreted the data. NEH, SMD, PMB, PEK, and KFH wrote the paper and provided important intellectual contributions. All authors read and approved the final manuscript.
Reference List
- 1.Hauser KF, Fitting S, Dever SM, Podhaizer EM, Knapp PE. Opiate drug use and the pathophysiology of neuroAIDS. Curr HIV Res. 2012;10(5):435–452. doi: 10.2174/157016212802138779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410(6831):988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5(1):69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 4.Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8(1):33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- 5.Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, et al. Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at μ-opioid receptor-expressing glia. Brain. 2011;134(12):3613–3628. doi: 10.1093/brain/awr281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Hage N, Podhaizer EM, Sturgill J, Hauser KF. Toll-like receptor expression and activation in astroglia: differential regulation by HIV-1 Tat, gp120, and morphine. Immunol Invest. 2011;40(5):498–522. doi: 10.3109/08820139.2011.561904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El-Hage N, Bruce-Keller AJ, Yakovleva T, Bakalkin G, Knapp PE, Hauser KF. Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca2+]i, NF-κB trafficking and transcription. PLoS ONE. 2008;3(12):e4093. doi: 10.1371/journal.pone.0004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, et al. HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006;53(2):132–146. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Podhaizer EM, Zou S, Fitting S, Samano KL, El-Hage N, Knapp PE, et al. Morphine and gp120 toxic interactions in striatal neurons are dependent on HIV-1 strain. J Neuroimmune Pharmacol. 2012;7(4):877–891. doi: 10.1007/s11481-011-9326-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turchan-Cholewo J, Dimayuga FO, Gupta S, Keller JN, Knapp PE, Hauser KF, et al. Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: possible role in cytokine regulation. J Neurochem. 2009;108:202–215. doi: 10.1111/j.1471-4159.2008.05756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byrd DA, Fellows RP, Morgello S, Franklin D, Heaton RK, Deutsch R, et al. Neurocognitive impact of substance use in HIV infection. J Acquir Immune Defic Syndr. 2011;58(2):154–162. doi: 10.1097/QAI.0b013e318229ba41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer VJ, Rubin LH, Martin E, Weber KM, Cohen MH, Golub ET, et al. HIV and recent illicit drug use interact to affect verbal memory in women. J Acquir Immune Defic Syndr. 2013;63(1):67–76. doi: 10.1097/QAI.0b013e318289565c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rolan P, Hutchinson M, Johnson K. Ibudilast: a review of its pharmacology, efficacy and safety in respiratory and neurological disease. Expert Opin Pharmacother. 2009;10(17):2897–2904. doi: 10.1517/14656560903426189. [DOI] [PubMed] [Google Scholar]
- 14.Hama AT, Broadhead A, Lorrain DS, Sagen J. The antinociceptive effect of the asthma drug ibudilast in rat models of peripheral and central neuropathic pain. J Neurotrauma. 2012;29(3):600–610. doi: 10.1089/neu.2011.1863. [DOI] [PubMed] [Google Scholar]
- 15.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10(1):23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutchinson MR, Lewis SS, Coats BD, Skyba DA, Crysdale NY, Berkelhammer DL, et al. Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast) Brain Behav Immun. 2009;23(2):240–250. doi: 10.1016/j.bbi.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hameed H, Hameed M, Christo PJ. The effect of morphine on glial cells as a potential therapeutic target for pharmacological development of analgesic drugs. Curr Pain Headache Rep. 2010;14(2):96–104. doi: 10.1007/s11916-010-0093-y. [DOI] [PubMed] [Google Scholar]
- 18.Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Res. 1999;837(1–2):203–212. doi: 10.1016/s0006-8993(99)01666-2. [DOI] [PubMed] [Google Scholar]
- 19.Mizuno T, Kurotani T, Komatsu Y, Kawanokuchi J, Kato H, Mitsuma N, et al. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004;46(3):404–411. doi: 10.1016/j.neuropharm.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 20.Beardsley PM, Hauser KF. Glial Modulators as Potential Treatments of Psychostimulant Abuse. Advances in Pharmacology. 2014;69:1–69. doi: 10.1016/B978-0-12-420118-7.00001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshioka A, Yamaya Y, Saiki S, Kanemoto M, Hirose G, Pleasure D. Cyclic GMP/cyclic GMP-dependent protein kinase system prevents excitotoxicity in an immortalized oligodendroglial cell line. J Neurochem. 2000;74(2):633–640. doi: 10.1046/j.1471-4159.2000.740633.x. [DOI] [PubMed] [Google Scholar]
- 22.Takuma K, Phuagphong P, Lee E, Mori K, Baba A, Matsuda T. Anti-apoptotic effect of cGMP in cultured astrocytes: inhibition by cGMP-dependent protein kinase of mitochondrial permeable transition pore. J Biol Chem. 2001;276(51):48093–48099. doi: 10.1074/jbc.M108622200. [DOI] [PubMed] [Google Scholar]
- 23.Yoshioka A, Shimizu Y, Hirose G, Kitasato H, Pleasure D. Cyclic AMP-elevating agents prevent oligodendroglial excitotoxicity. J Neurochem. 1998;70(6):2416–2423. doi: 10.1046/j.1471-4159.1998.70062416.x. [DOI] [PubMed] [Google Scholar]
- 24.Yoshioka M, Suda N, Mori K, Ueno K, Itoh Y, Togashi H, et al. Effects of ibudilast on hippocampal long-term potentiation and passive avoidance responses in rats with transient cerebral ischemia. Pharmacol Res. 2002;45(4):305–311. doi: 10.1006/phrs.2002.0949. [DOI] [PubMed] [Google Scholar]
- 25.Lee JY, Cho E, Ko YE, Kim I, Lee KJ, Kwon SU, et al. Ibudilast, a phosphodiesterase inhibitor with anti-inflammatory activity, protects against ischemic brain injury in rats. Brain Res. 2012;1431:97–106. doi: 10.1016/j.brainres.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 26.Bartley J. Post herpetic neuralgia, schwann cell activation and vitamin D. Med Hypotheses. 2009;73(6):927–929. doi: 10.1016/j.mehy.2009.06.039. [DOI] [PubMed] [Google Scholar]
- 27.Wakita H, Tomimoto H, Akiguchi I, Lin JX, Ihara M, Ohtani R, et al. Ibudilast, a phosphodiesterase inhibitor, protects against white matter damage under chronic cerebral hypoperfusion in the rat. Brain Res. 2003;992(1):53–59. doi: 10.1016/j.brainres.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 28.Barkhof F, Hulst HE, Drulovic J, Uitdehaag BM, Matsuda K, Landin R, et al. Ibudilast in relapsing-remitting multiple sclerosis: a neuroprotectant? Neurology. 2010;74(13):1033–1040. doi: 10.1212/WNL.0b013e3181d7d651. [DOI] [PubMed] [Google Scholar]
- 29.Fox RJ. Primary neuroprotection: the Holy Grail of multiple sclerosis therapy. Neurology. 2010;74(13):1018–1019. doi: 10.1212/WNL.0b013e3181d6b165. [DOI] [PubMed] [Google Scholar]
- 30.Schwarz JM, Hutchinson MR, Bilbo SD. Early-life experience decreases drug-induced reinstatement of morphine CPP in adulthood via microglial-specific epigenetic programming of anti-inflammatory IL-10 expression. J Neurosci. 2011;31(49):17835–17847. doi: 10.1523/JNEUROSCI.3297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bland ST, Hutchinson MR, Maier SF, Watkins LR, Johnson KW. The glial activation inhibitor AV411 reduces morphine-induced nucleus accumbens dopamine release. Brain Behav Immun. 2009;23(4):492–497. doi: 10.1016/j.bbi.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Snider SE, Vunck SA, van den Oord EJ, Adkins DE, McClay JL, Beardsley PM. The glial cell modulators, ibudilast and its amino analog, AV1013, attenuate methamphetamine locomotor activity and its sensitization in mice. Eur J Pharmacol. 2012;679(1–3):75–80. doi: 10.1016/j.ejphar.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beardsley PM, Shelton KL, Hendrick E, Johnson KW. The glial cell modulator and phosphodiesterase inhibitor, AV411 (ibudilast), attenuates prime- and stress-induced methamphetamine relapse. Eur J Pharmacol. 2010;637(1–3):102–108. doi: 10.1016/j.ejphar.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snider SE, Hendrick ES, Beardsley PM. Glial cell modulators attenuate methamphetamine self-administration in the rat. Eur J Pharmacol. 2013;701(1–3):124–130. doi: 10.1016/j.ejphar.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoptaw S. Safety interaction trial ibudilast and methamphetamine. ClinicalTrials.gov. 2011 [Google Scholar]
- 36.Cooper ZD, Jones JD, Comer SD. Glial modulators: a novel pharmacological approach to altering the behavioral effects of abused substances. Expert Opin Investig Drugs. 2012;21(2):169–178. doi: 10.1517/13543784.2012.651123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, et al. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375(6531):497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 38.Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, et al. Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci U S A. 2000;97(21):11466–11471. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El-Hage N, Dever SM, Podhaizer EM, Arnatt CK, Zhang Y, Hauser KF. A novel bivalent HIV-1 entry inhibitor reveals fundamental differences in CCR5-mu-opioid receptor interactions between human astroglia and microglia. AIDS. 2013;27(14):2181–2190. doi: 10.1097/QAD.0b013e3283639804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sippy BD, Hofman FM, Wallach D, Hinton DR. Increased expression of tumor necrosis factor-alpha receptors in the brains of patients with AIDS. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;10(5):511–521. [PubMed] [Google Scholar]
- 41.Achim CL, Wiley CA. Inflammation in AIDS and the role of the macrophage in brain pathology. Curr Opin Neurol. 1996;9(3):221–225. doi: 10.1097/00019052-199606000-00013. [DOI] [PubMed] [Google Scholar]
- 42.Kiebala M, Maggirwar SB. Ibudilast, a pharmacologic phosphodiesterase inhibitor, prevents human immunodeficiency virus-1 Tat-mediated activation of microglial cells. PLoS One. 2011;6(4):e18633. doi: 10.1371/journal.pone.0018633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Z, Liu S, Zhang L, Salem M, Greig GM, Chan CC, et al. Preferential inhibition of human phosphodiesterase 4 by ibudilast. Life Sci. 2006;78(23):2663–2668. doi: 10.1016/j.lfs.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 44.Lee JY, Cho E, Ko YE, Kim I, Lee KJ, Kwon SU, et al. Ibudilast, a phosphodiesterase inhibitor with anti-inflammatory activity, protects against ischemic brain injury in rats. Brain Res. 2012;1431:97–106. doi: 10.1016/j.brainres.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 45.Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Res. 1999;837(1–2):203–212. doi: 10.1016/s0006-8993(99)01666-2. [DOI] [PubMed] [Google Scholar]
- 46.Won CL, Oh YS. cAMP-induced stellation in primary astrocyte cultures with regional heterogeneity. Brain Res. 2000;887(2):250–258. doi: 10.1016/s0006-8993(00)02922-x. [DOI] [PubMed] [Google Scholar]
- 47.McArthur JC. HIV dementia: an evolving disease. J Neuroimmunol. 2004;157(1–2):3–10. doi: 10.1016/j.jneuroim.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 48.Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R, et al. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc Natl Acad Sci U S A. 2013;110(33):13588–13593. doi: 10.1073/pnas.1308673110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia. 2005;50(2):91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahajan SD, Schwartz SA, Shanahan TC, Chawda RP, Nair MP. Morphine regulates gene expression of alpha- and beta-chemokines and their receptors on astroglial cells via the opioid mu receptor. J Immunol. 2002;169(7):3589–3599. doi: 10.4049/jimmunol.169.7.3589. [DOI] [PubMed] [Google Scholar]
- 51.Reddy PV, Pilakka-Kanthikeel S, Saxena SK, Saiyed Z, Nair MP. Interactive Effects of Morphine on HIV Infection: Role in HIV-Associated Neurocognitive Disorder. AIDS Res Treat. 2012;2012:953678. doi: 10.1155/2012/953678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen RW, Williams AJ, Liao Z, Yao C, Tortella FC, Dave JR. Broad spectrum neuroprotection profile of phosphodiesterase inhibitors as related to modulation of cell-cycle elements and caspase-3 activation. Neurosci Lett. 2007;418(2):165–169. doi: 10.1016/j.neulet.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 53.Rolan P, Gibbons JA, He L, Chang E, Jones D, Gross MI, et al. Ibudilast in healthy volunteers: safety, tolerability and pharmacokinetics with single and multiple doses. Br J Clin Pharmacol. 2008;66(6):792–801. doi: 10.1111/j.1365-2125.2008.03270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson KW, Matsuda K, Iwaki Y. Ibudilast for the treatment of drug addiction and other neurological conditions. Clinical Investigation. 2014;4(3):269–279. [Google Scholar]
- 55.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3(10):791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roger T, David J, Glauser MP, Calandra T. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature. 2001;414(6866):920–924. doi: 10.1038/414920a. [DOI] [PubMed] [Google Scholar]
- 57.Flaster H, Bernhagen J, Calandra T, Bucala R. The macrophage migration inhibitory factor-glucocorticoid dyad: regulation of inflammation and immunity. Mol Endocrinol. 2007;21(6):1267–1280. doi: 10.1210/me.2007-0065. [DOI] [PubMed] [Google Scholar]
- 58.Conboy L, Varea E, Castro JE, Sakouhi-Ouertatani H, Calandra T, Lashuel HA, et al. Macrophage migration inhibitory factor is critically involved in basal and fluoxetine-stimulated adult hippocampal cell proliferation and in anxiety, depression, and memory-related behaviors. Mol Psychiatry. 2011;16(5):533–547. doi: 10.1038/mp.2010.15. [DOI] [PubMed] [Google Scholar]
- 59.Bacher M, Weihe E, Dietzschold B, Meinhardt A, Vedder H, Gemsa D, et al. Borna disease virus-induced accumulation of macrophage migration inhibitory factor in rat brain astrocytes is associated with inhibition of macrophage infiltration. Glia. 2002;37(4):291–306. [PubMed] [Google Scholar]
- 60.Savaskan NE, Fingerle-Rowson G, Buchfelder M, Eyupoglu IY. Brain miffed by macrophage migration inhibitory factor. Int J Cell Biol. 2012;2012:139573. doi: 10.1155/2012/139573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sawada M, Kondo N, Suzumura A, Marunouchi T. Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res. 1989;491(2):394–397. doi: 10.1016/0006-8993(89)90078-4. [DOI] [PubMed] [Google Scholar]
- 62.Regis EG, Barreto-de-Souza V, Morgado MG, Bozza MT, Leng L, Bucala R, et al. Elevated levels of macrophage migration inhibitory factor (MIF) in the plasma of HIV-1-infected patients and in HIV-1-infected cell cultures: a relevant role on viral replication. Virology. 2010;399(1):31–38. doi: 10.1016/j.virol.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Delaloye J, De Bruin IJ, Darling KE, Reymond MK, Sweep FC, Roger T, et al. Increased macrophage migration inhibitory factor (MIF) plasma levels in acute HIV-1 infection. Cytokine. 2012;60(2):338–340. doi: 10.1016/j.cyto.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 64.Sui Z, Sniderhan LF, Schifitto G, Phipps RP, Gelbard HA, Dewhurst S, et al. Functional synergy between CD40 ligand and HIV-1 Tat contributes to inflammation: implications in HIV type 1 dementia. J Immunol. 2007;178(5):3226–3236. doi: 10.4049/jimmunol.178.5.3226. [DOI] [PubMed] [Google Scholar]
- 65.Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, et al. Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J Biol Chem. 1996;271(26):15303–15306. doi: 10.1074/jbc.271.26.15303. [DOI] [PubMed] [Google Scholar]
- 66.Brabers NA, Nottet HS. Role of the pro-inflammatory cytokines TNF-alpha and IL-1beta in HIV-associated dementia. Eur J Clin Invest. 2006;36(7):447–458. doi: 10.1111/j.1365-2362.2006.01657.x. [DOI] [PubMed] [Google Scholar]
- 67.Cho Y, Crichlow GV, Vermeire JJ, Leng L, Du X, Hodsdon ME, et al. Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc Natl Acad Sci U S A. 2010;107(25):11313–11318. doi: 10.1073/pnas.1002716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 69.Hiscott J, Kwon H, Genin P. Hostile takeovers: viral appropriation of the NF-kappaB pathway. J Clin Invest. 2001;107(2):143–151. doi: 10.1172/JCI11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hauser KF, El-Hage N, Stiene-Martin A, Maragos WF, Nath A, Persidsky Y, et al. HIV-1 neuropathogenesis: Glial mechanisms revealed through substance abuse. J Neurochem. 2007;100(3):567–586. doi: 10.1111/j.1471-4159.2006.04227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.El-Hage N, Bruce-Keller AJ, Knapp PE, Hauser KF. CCL5/RANTES gene deletion attenuates opioid-induced increases in glial CCL2/MCP-1 immunoreactivity and activation in HIV-1 Tat exposed mice. J Neuroimmune Pharmacol. 2008;3(4):275–285. doi: 10.1007/s11481-008-9127-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.