

Abstract
DNA damage and repair are linked to cancer. DNA damage that is induced endogenously or from exogenous sources has the potential to result in mutations and genomic instability if not properly repaired, eventually leading to cancer. Inflammation is also linked to cancer. Reactive oxygen and nitrogen species (RONs) produced by inflammatory cells at sites of infection can induce DNA damage. RONs can also amplify inflammatory responses, leading to increased DNA damage. Here, we focus on the links between DNA damage, repair, and inflammation, as they relate to cancer. We examine the interplay between chronic inflammation, DNA damage and repair and review recent findings in this rapidly emerging field, including the links between DNA damage and the innate immune system, and the roles of inflammation in altering the microbiome, which subsequently leads to the induction of DNA damage in the colon. Mouse models of defective DNA repair and inflammatory control are extensively reviewed, including treatment of mouse models with pathogens, which leads to DNA damage. The roles of microRNAs in regulating inflammation and DNA repair are discussed. Importantly, DNA repair and inflammation are linked in many important ways, and in some cases balance each other to maintain homeostasis. The failure to repair DNA damage or to control inflammatory responses has the potential to lead to cancer.
Keywords: Colon cancer, DNA damage, infection, microbiota, pathogens, reactive oxygen, nitrogen species
Introduction
In 1863, Rudolf Virchow hypothesized that the origin of cancer was at sites of chronic inflammation (Virchow, 1863), based on observations of the presence of a “lymphoreticular infiltrate” in human tumors. Inflammation is part of a complex biological response of vascular tissues to harmful stimuli, such as pathogens, damaged cells or irritants. In recent years, inflammation has taken its stage to be listed as the seventh hallmark of cancer (Hanahan & Weinberg, 2000). Epidemiological and clinical studies estimate that approximately 25% of all cancers are linked to chronic inflammation, inflammation induced by environmental exposures, or infection of pathogens (Balkwill & Mantovani, 2010, 2012; Saigo et al., 2008). Many studies have characterized the roles that inflammation plays in the induction of a carcinogenic phenotype.
A mutator phenotype resulting from aberrant DNA repair and replication is also an important driver of cancer (Loeb, 2011). Aberrant mismatch repair (Fishel & Kolodner, 1995; Fishel et al., 1993, 1994) deficient MutYH DNA glycosylase activity (for recent reviews, see Raetz et al., 2012; Wallace et al., 2012) and DNA polymerase misincorporation (Zhao et al., 2013) are all examples of how aberrant functions of DNA repair and replication contribute to a mutator phenotype. A common link between inflammation and DNA repair is DNA damage. DNA damage is present during inflammation and accumulates during chronic inflammation. DNA repair plays an important role in counteracting this damage but is not always successful, often leading to cancer promotion. The focus of this review is on the interplay between inflammation and DNA repair and how an imbalance in these processes could lead to human cancer.
Endogenous RONs-induced DNA damage and repair
Reactive oxygen and nitrogen species (RONs) damage many cellular molecules including DNA (Nathan & Cunningham-Bussel, 2013; West & Marnett, 2006). Reactive oxygen species (ROS) arise endogenously as a result of mitochondrial respiration, cellular signaling and during the breakdown of fatty acids by the peroxisome. Endogenous reactive nitrogen species also arise from the production of nitric oxide from L-arginine and during cellular signaling processes. Redox homeostasis plays an important role in mitigating the effects of RONs through the actions of enzymes including superoxide dismutase, glutathione-S-transferase, and glutathione peroxidase. RONs that escape these defense systems damage DNA at a rate of at least 20 000 DNA base lesions per cell per day (Barnes & Lindahl, 2004). ROS can damage all four canonical bases in DNA along with the methylated form of deoxycytidine to generate a large number of oxidatively damaged bases (Svilar et al., 2011). Chronic ROS can generate significant levels of lipid peroxidation products, which react with DNA to generate etheno-base DNA adducts. Nitric oxide (NO) can also generate etheno-base adducts (Nair et al., 1998). In addition, peroxynitrite reacts with guanine to create 8-nitroguanine, which can spontaneously depurinate to generate abasic sites (Dedon & Tannenbaum, 2004;Mutamba et al., 2011; Yermilov et al., 1995). Peroxynitrite also induces single-strand breaks in DNA (Yermilov et al., 1996).
DNA damage induced by RONs is generally repaired by the base excision repair (BER) pathway (for a review see Wallace et al., 2012), which is a critically important pathway for the maintenance of genome integrity. Repair by BER involves removal of the damaged base and resulting abasic site by a DNA glycosylase with associated lyase activity or the combined actions of a glycosylase and AP endonuclease, followed by filling of the small DNA gap by a DNA polymerase, and concluding with ligation. Mutations in BER genes are associated with chronic inflammation. DNA damage induced by alkylating agents and reactive nitrogen species, including 1-methyladenine and 3-methylcytosine and 1,N6 – ethenoadenine (εA) and are removed by BER, but can also be eliminated by direct reversal. The ALKBH2 and ALKBH3 proteins catalyze direct reversal of the damage in the presence of α-ketoglutarate, oxygen and iron (Falnes et al., 2002; Trewick et al., 2002). Mismatch repair (MMR) is also affected by chronic inflammation. Replication errors, including base mismatches and small insertion/deletion loops (IDL), are repaired by the MMR system (Jiricny, 2006; Pena-Diaz & Jiricny, 2012). Following recognition of the mismatch or IDL on the newly synthesized DNA strand, a large fragment of DNA including the mismatch is excised. DNA synthesis then takes place to fill the large gap, followed by ligation of the nick. Overwhelming the system with RONs-induced DNA damage, or the presence of compromised DNA repair due to genetic abnormalities, can result in incomplete repair of DNA damage, leading to mutagenesis and/or chromosomal instability.
Inflammation-associated DNA damage and repair
Acute inflammation is a protective response against pathogens, but also important for the regeneration of damaged tissues after injury and infection (Ben-Neriah & Karin, 2011). RONs are generated by inflammatory cells that are recruited to the sites of infection and tissue regeneration.
Chronic inflammation results from persistent pathogen or viral infection, autoimmune reactions or persistent foreign bodies. One of the hallmarks of chronic inflammation is the infiltration of inflammatory cells including macrophages, neutrophils, and various types of lymphocytes. The presence of these cells leads to sustained production of pro-inflammatory mediators, such as cytokines, chemokines, interferon-γ and lipid mediators that coordinately constitute a cancer-prone microenvironment. Chronic inflammation often results in tissue damage, an increased mutation rate and genomic instability. If accumulated in excess the chronic inflammatory environment leads to the initiation of carcinogenesis (Coussens & Werb, 2002). Therefore, the balance between the pro and anti-inflammatory immune responses of the host cells during inflammation is critical to determine the fate of the cell. DNA repair is also the counter response of the host cells to chronic inflammation (Ferguson, 2010).
Interplay between chronic inflammation, DNA damage and repair
A growing body of evidence indicates that RONs can cause damage to cellular macromolecules. DNA is the most sensitive biological target for damage from oxidative stress (for a recent review see Federico et al., 2007), as it is not well protected and has limited chemical stability (Lindahl, 1993; Taha et al., 2010). Genomic damage that is either left unrepaired or is repaired with errors may result in mutations of critical genes which ultimately leads to an elevated cancer risk (De Bont & van Larebeke, 2004; Trabulus et al., 2012). Inflammation of the tissue surrounding a tumor can hasten the oncogenic process by directly promoting genetic instability and favoring or inducing gene mutations. RONs, which are abundant during inflammation, can induce DNA mutations, epigenetic alterations, and post-translational modifications of proteins that control the cell cycle or survival (Colotta et al., 2009). In recent years new data suggest that the DNA damage response can initiate an immune response. The DNA damage response (DDR) directly activates a variety of transcription factors such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and interferon regulatory factors (IRFs) (for a review see McCool & Miyamoto, 2012). These transcription factors induce the expression of various immune genes, including inflammatory cytokines and chemokines. In addition, the DDR and oxidative stress induce the expression of a number of ligands for activating immune receptors such as natural killer group 2, member D (NKG2D) and DNAX accessory molecule 1 (DNAM-1), which are mainly expressed by cytotoxic immune cells such as T cells and natural killer (NK) cells.
The presence of inflammatory cells at sites of chronic inflammation results in the local release of RONs, which results in DNA damage, as discussed earlier. Damage to DNA by RONs leads to mutations. Early studies, in which naked DNA was incubated with a variety of reactive oxygen generating systems followed by repair and replication in E. coli, demonstrated that oxidative DNA damage could induce mutations and produce mutagenic signatures (for a review see Feig et al., 1994). Although repair pathways have evolved to process this DNA damage, they are imperfect leading to the induction of mutations and genomic instability. For example, exposure of mammalian cells to nitric oxide induces homologous recombination between direct repeat sequences (Kiziltepe et al., 2005), much of which result from strand breaks and oxidized bases. A second example is that co-culture of mammalian cells with activated human promyelocytic cells or macrophages induces an increased frequency of mutations over background that were mediated by RONs (Kim et al., 2003). Importantly, the increase in mutagenesis was reversed by incubation of the cells with oxygen radical scavengers.
Chronic inflammation and NF-κB
Nuclear factor of kappa light gene polypeptide gene enhancer in B cells (NF-κB) is a master regulator of the immune response by inducing transcription of proinflammatory genes (for a review see Tak & Firestein, 2001). NF-κB is also activated during chronic inflammation and during the cellular response to DNA damage. Ataxia telangiectasia mutated (ATM) and NF-κB essential modulator (NEMO) cooperate to activate NF-κB in response to DNA damage, which involves transport of NF-κB from the cytoplasm to the nucleus (for a review see McCool & Miyamoto, 2012). Once in the nucleus NF-κB is a transcription factor that upregulates the expression of both pro- and anti-apoptotic factors (Roos & Kaina, 2013), ultimately protecting the cell from apoptosis. Interestingly, DNA intercalators, which alter the structure of DNA, also induce activation of NF-κB, leading to a significant increase in DNA damage and ultimately, apoptosis (Karl et al., 2009). Therefore the status of NF-κB and its associated factors during chronic inflammation has great potential to influence the outcomes of inflammation, including accumulation of mutations, cell survival and cancer.
The DNA damage sensor PARP1 is a scaffold that coordinates assembly of inhibitor of kappa B kinase gamma (IKKγ), Ataxia Telangiectasia mutated (ATM) and the protein inhibitor of activated STAT gamma 1 (PIASγ1), small ubiquitin-like modifier (SUMO) ligase (Stilmann et al., 2009). Sumoylation of IKKγ results in activation of NF-κB. Damage in the form of single-strand breaks (SSBs) and stalled replication forks is sensed by poly-ADP ribose polymerase 1 (PARP1) and can therefore result in activation of NF-κB. NF-κB itself stimulates DNA DSB repair (Volcic et al., 2012). Specifically, inhibitor of kappa B alpha (IκBα) releases NF-κB dimers that associate with and stimulate the activity of the BRCA1-CtIP nuclease complex, resulting in end resection and initiation of homology dependent repair (HDR). This is followed by NF-κB-dependent upregulation of BRCA2 and ATM. The presence of the BRCA2 recombination mediator and stimulation of end-resection channel DSB repair into HDR. Therefore if bases damaged by ROS during inflammation lead to an accumulation of BER intermediates and eventual DSB formation, much of the repair will be channeled into HDR.
DNA damage and upregulation of IRFs
DNA damage induced in the chronic inflammatory environment has the capacity to upregulate interferon responsive factors (IRFs) including IRF1, 3 and 7. IRF1 is induced in response to DNA damage in an ATM dependent manner (Pamment et al., 2002). Activation of IRF1 leads to expression of genes that function in cell cycle regulation, apoptosis and activation of T cells (Kirchhoff & Hauser, 1999; Matsuyama et al., 1993; Stevens & Yu-Lee, 1992, 1994; Tamura et al., 1995). Mouse cells deficient in IRF-1 are compromised in their abilities to repair damaged DNA (Prost et al., 1998). DNA damage induces phosphorylation of IRF-3, resulting in its transport from the cytoplasm to the nucleus and activation of transcription (Kim et al., 1999), leading to downregulation of proliferation. IRF-7 is also upregulated in response to DNA damage (Kim et al., 2000).
DNA damaging agents and NKG2D
The natural killer group 2D (NKG2D) is an activating receptor in natural killer cells (NK) and in T cells. Binding of ligands to the NKG2D receptor induces lysis of cells expressing the receptor. Interestingly, NKG2D ligands (NKG2DL) are upregulated in response to oxidative stress, such as RONs (Yamamoto et al., 2001). DNA damaging agents and replication inhibitors also upregulate the expression of NKG2D ligands in an ATM dependent manner (Gasser et al., 2005). Thus, DNA damage sustained in a chronic inflammatory environment could result in the activation of the immune response, leading to the recruitment of natural killer cells which lyse severely damaged cells, resulting in cell death (Gasser & Raulet, 2006a,b).
DNA damage and the inflammasome
Following cellular damage the NOD-like receptor family pryin domain-containing 3 (NLRP3) protein forms a multi-protein complex called the inflammasome that culminates with the activation of caspase-1 and the maturation and secretion of proinflammatory cytokines interleukin 1B (IL1B) and 18 (IL18) which can then drive inflammatory responses. Other activators of the inflammasome include bacteria and other pathogens and danger-associated molecular patterns (DAMPS) that work via a common downstream ligand, which is ROS. Recently it was shown that treatment of cells with MSU salt crystals or IR, both of which induce ROS and DNA damage activates the inflammasome (Licandro et al., 2013). This stabilizes p53 and p21 and leads to cell death. Activation of the inflammasome also downregulates DSB repair and BER. Downregulation of DNA repair in conjunction with caspase 1 activation leads to cell death, which could be another mechanism used by the immune system to eliminate cells with severe DNA damage, such as that sustained under conditions of chronic inflammation.
DNA damage, repair and STING
One of the major oxidized bases found in DNA exposed to RONs is 8-hydroxyguanine (8-oxoG). Nucleic acids in the cytoplasm are sensed by the innate immune system through the presence of molecular recognition patterns. It has recently been shown that the presence of 8-oxoG in DNA from pathogens decreases its susceptibility to degradation by the TREX1 nuclease. This leads to the potentiation of cytosolic immune recognition through the endoplasmic reticulum-associated protein STING, and the release of type I interferon (Gehrke et al., 2013). Thus, oxidized bases in DNA have the potential to activate immune signaling. In the case of chronic inflammation, RONs-induced DNA damage could potentiate immune responses, with the goal of leading to cell death. The meiotic recombination 11 homolog A (MRE11) and its associated protein, RAD50, function in DSB repair, and were recently shown to be necessary for activation of STING and release of interferon-β (IFN-β) through their ability to recognize and bind to double-stranded DNA, but not pathogens, in the cytoplasm (Kondo et al., 2013). The origin of this DNA is not known at present. Because secretion of IFN-β could potentially be harmful, it is suggested that once MRE11-RAD50 binds to DNA and initiates signaling through STING, it processes this DNA using the nuclease activity of MRE11 to terminate the downstream signaling.
Chronic inflammation and SASP
Cellular senescence is a state of irreversible growth arrest induced by telomere shortening (replicative senescence), oncogene activation and persistent DNA damage (Collado et al., 2007; Kuilman et al., 2010). Cellular senescence significantly contributes as an anti-tumorigenic mechanism via induction of growth arrest (Kuilman et al., 2010) and triggering of immune-mediated clearance of pre-malignant cells (Kang et al., 2011). However, unlike apoptotic cells, senescent cells remain metabolically active and undergo widespread gene expression changes (Campisi & d’Adda di Fagagna, 2007). The signature of senescent cells is the acquisition of a senescent-associated secretory phenotype (SASP) (Coppe et al., 2008). SASP proteins are generally induced at the level of mRNA (Coppe et al., 2008) and include a wide range of growth factors, proteases, chemokines and cytokines. Proteins that are known to stimulate inflammation include interleukin 6, 8 and 1 (IL6, IL8, IL1), granulocyte macrophage colony stimulating factor (GM-CSF) and growth regulated oncogene (GRO) factors are secreted by senescent cells. Recent evidence suggests that monocyte chemotactic proteins 2 and 3 (MCP-2, MCP-3), matrixme-talloproteinase 1 and 3 (MMP-1, MMP-3) and many of the insulin-like growth factor (IGF)-binding proteins (Kumar et al., 1992) are among the most robustly induced and secreted of these factors. Although the immune system plays a major role in modulating the levels of pro- and anti-inflammatory factors, it is not the only source of these factors. However, SASP is also considered to be a mechanism responsible for chronic inflammation observed during aging (Coppe et al., 2010; Freund et al., 2010). On the other hand, SASP causes abundant secretion of various bioactive proteins from senescent cells and thereby activates neighboring non-senescent cancer cells, leading to promotion of tumor growth (Bavik et al., 2006; Coppe et al., 2008). Therefore, the SASP has the ability to induce and amplify local and chronic inflammation that can eventually lead to DNA damage in surrounding cells.
Mouse models of chronic inflammation: interaction with microbiota
It has been reported that 80% of immune cells reside and form a symbiotic relationship with commensal microbiota in the healthy gut. The colonic microbiota represents the largest microbe population in humans. Importantly, the commensal bacteria of patients with inflammatory bowel disease (IBD) and ulcerative colitis (UC) is altered compared to that of healthy individuals, suggesting that the microbiome represents a source of chronic inflammation (Frank et al., 2011).
Due to the constant influx of foreign antigens and the presence of myriads of commensal bacteria, the control of inflammation in the intestinal tract has been extensively studied in very recent years. These studies suggest that microbe-induced chronic inflammation results in DNA damage in the gut. A possible scheme involves the emergence of certain members of the colonic microbial community as a result of proinflammatory immune responses leading to the remodeling of the microbiome. This results in the emergence of bacteria that can induce DNA damage and the DDR and promote colon cancer (Sears & Pardoll, 2011). For example, it has been suggested that various bacterial products such as polysaccharides have immunomodulatory effects (Hall et al., 2008) (for a review see Chow et al., 2010).
IL10 deficient mice and microbiota
IL10 suppresses macrophage activation and inhibits production of inflammatory cytokines. IL10-deficient mice develop anemia associated with chronic enterocolitis (Kuhn et al., 1993). However, when the IL10-deficient mice are raised under specific pathogen-free (SPF) conditions, they develop only localized inflammation. This suggests that in the absence of IL10 there is little control over normal immune responses to enterobacteria, leading to overproduction of cytokines and resulting in chronic inflammation. The chronic inflammation associated with enterocolitis in the IL10-deficient mice was documented to involve uncontrolled cytokine production by activated macrophages (Berg et al., 1996). It was subsequently shown that IL10-deficient mice raised in germ free conditions did not develop colitis, showing that resident enteric bacteria are necessary for the development of colitis in the IL10-deficient mouse model (Sellon et al., 1998). Manipulation of the microbiota in the IL10-deficient mice influences the development of chronic inflammation and cancer in mice treated with azoxymethane (AOM) (Uronis et al., 2009). Treatment of IL10-deficient mice with AOM leads to the development of colorectal tumors in most mice, whereas few tumors are observed in wild-type mice also treated with AOM. In these studies tumor multiplicity correlated with the presence of chronic inflammation. IL10-deficient germ free and mice harboring mildly colitogenic bacterium and treated with AOM did not develop colitis or colon cancer. Importantly, enteric bacteria have the capacity to induce the DDR. For example, co-culture of eukaryotic cells with E. coli harboring the genomic island that encodes giant modular nonribosomal peptide and polyketide synthases (pks) induces DSBs and activation of the DDR (Nougayrede et al., 2006). Recent sequencing shows that AOM-induced inflammation of the gut of IL10-deficient mice alters the composition of the microbiota including significantly increased abundance of Enterobacteriaceae (Arthur et al., 2012), as shown in Figure 1. Specifically, the guts of IL10-deficient mice harbored a 100-fold increase in E. coli. Treatment of IL10-deficient mice with AOM either mono-associated with the human commensal Enterococcus faecalis or the adherent invasive E. coli NC101 resulted in severe colitis and similar induction of proinflammatory cytokines. However, only mice monoassociated with E. coli NC101 developed adenocarcinoma. Importantly, the NC101 E. coli harbor the polyketide synthetase (pks) island that was detected in 40% of patients with IBD, suggesting that it is related to disease pathology. Infection of non-transformed rat epithelial cells with E. coli harboring the pks island resulted increased levels of phosphorylated H2AX (γH2AX) but this was not observed in cells infected with E. coli deleted of the pks island. Interestingly, infection of the IL10-deficient mice with E. coli NC101 and with NC101 deleted of pks resulted in chronic inflammation, but a significantly greater tumor burden was observed in mice with bacteria harboring the pks island. The abundance of γH2AX foci in the crypts of mice infected with the pks-deleted E. coli was significantly less than that observed in mice infected with pks-proficient E. coli. These studies suggest that chronic inflammation alters the populations of microbiota in the gut, resulting in the expansion of microbes like E. coli NC101. These types of microbes induce DNA damage and activate the DDR, which likely leads to genotoxic damage and cancer, as shown in Figure 1. An elegant study demonstrated that the ablation of the TLR (Toll-like receptor)-mediated signaling pathway adaptor MyD88 impairs intestinal tumorigenesis (Rakoff-Nahoum et al., 2006) in IL10 deficient mice.
Figure 1.

NF-κB and the interplay between DNA repair and inflammation. NF-κB is a master regulator of the inflammatory response. After activation through TLR4, several proinflammatory genes are activated, including IL6, STAT3 and TNFα, which has the potential to lead to a cytokine storm. NF-κB is also induced by DNA damage, ROS and hypoxia, leading to the expression of proinflammtory genes. Induction of NF-κB by DNA damage is regulated by ATM, a master regulation of the DNA damage response, and this occurs with assistance from NEMO. Induction of NF-κB also leads to the induction of miR-21 and miR-210, resulting in induction of IL10, and anti-inflammatory cytokine, but also downregulation of PTEN, CDC25A and RAD52, proteins that are involved in the DNA damage response. Not only does hypoxia induce activation NF-κB, but it also induces expression of miR-155, which downregulates mismatch repair, with the potential to lead to genomic instability. However, IL10, which is indirectly induced by NF-κB, has the capacity to downregulate miR-155. Of special interest for this review is the finding that NF-κB stimulates the CtIP-BRCA1 complex, thereby promoting homology-directed repair (HDR). (see colour version of this figure at www.informahealthcare.com/bmg).
In contrast, recent identification of commensal colonization factors (CCF) in microbial genomes and the identification of the toll-like receptor 2 (TLR2)-mediated pathway in the host demonstrated the important factors of bacterial colonization and stabilization for gut homeostasis (Lee et al., 2013; Round & Mazmanian, 2009).
T-bet, chronic inflammation and DNA damage
Although several reports suggest that innate immunity via the TLR-MyD88 pathway is important, recent advances also indicate that there are MyD88-independent pathways that might be involved in colorectal cancer (CRC) development. T-bet is a T box transcription factor that is expressed only in immune cells, regulates host-commensal homeostasis, and chemokine and cytokine expression (Glimcher, 2007; Ma, 2007). T-bet plays an important role in the induction of inflammatory programs in both innate and adaptive immunity. Loss of T-bet in innate immune cells results in spontaneous colitis in the absence of adaptive immunity and influences the host microbiota to become coligenic, which could lead to induction of the DDR. Deletion of T-bet in Rag2-deficient mice results in the development of ulcerative colitis (TRUC mice) (Garrett et al., 2007). A further study demonstrates that TRUC mice spontaneously develop colitis-associated adenocarcinoma (Garrett et al., 2009). The lack of T-bet in dendritic cells results in increased RONs production in colonic epithelial cells (CECs) from TRUC mice, which induced the formation of DNA adducts such as 8-oxoG. Importantly, such events were independent of the conventional TLR-MyD88 pathway while the presence of commensal bacteria was still critical. A follow-up study showed that metabolites such as short fatty chain acids from commensal bacteria may play a key role in colonic inflammation (Smith et al., 2013).
Commensal bacteria protect tissue from inflammation
Commensal bacteria are also critical players in protection from inflammation in the gut. For example, dextran sodium sulfate (DSS) treatment induces inflammation in the colon, but oral administration of lactic acid bacteria (LAB) alleviated this inflammation (Kawashima et al., 2013). Reduced levels of inflammatory cells including neutrophils, eosinophils and macrophages were observed in the lamina propria of mice fed LAB compared to controls. Interferon β (IFN-β), an important anti-inflammatory mediator, is secreted in response to the presence of LAB as a result of TLR3 acting as a sensor to the dsRNA of the commensal LAB. Expression of inflammatory mediators including IL6 and interleukin 17 (IL17) decreases in the inflamed colons of mice treated with LAB. Therefore, the microbiota exerts an influence over the inflammatory responses of the gut, which could ultimately control the levels of DNA damage and mutation.
Chronic inflammation, DNA repair and inflammatory bowel disease
Inflammation is an accepted factor contributing to colorectal carcinoma (CRC) progression (Feagins et al., 2009; Thorsteinsdottir et al., 2011; Ullman & Itzkowitz, 2011), and several inflammatory pre-neoplastic states result in an increased risk of cancer progression in affected patients (Colotta et al., 2009; Grivennikov et al., 2010; Mantovani et al., 2008). In IBD patients, RONs generated during the inflammatory response result in oxidative damage within the colonic epithelium (Kruidenier et al., 2003a; Singer et al., 1996; Sohn et al., 2012) and chronic inflammation results in an increased mutational load throughout the diseased colon (Hussain et al., 2000; Risques et al., 2011; Salk et al., 2009; Willenbucher et al., 1999). Consistent with increased mutagenesis, MMR gene expression is epigenetically down-regulated in mouse models of IBD (Edwards et al., 2009), and oxidative stress can decrease expression of mut S homolog 6 (MSH6) and postmeiotic segregation 2 (PMS2) repair proteins by causing their denaturation (Chang et al., 2002). In contrast, alkyladenine glycosylase (AAG) and AP endonuclease can be overexpressed within inflamed tissues of ulcerative colitis patients (Hofseth et al., 2003). Aberrant expression of activation-induced cytidine deaminse (AID) is induced in ulcerative colitis and H. pylori inflamed patient epithelium (Gushima et al., 2011). This AID expression is induced by TNF-α through the NK-κB signaling pathway and by interleukin 4 and 13 (IL4 and IL13) cytokines (Endo et al., 2008). These studies suggest that chronic inflammation may promote mutagenesis by altering the expression of repair proteins in patient tissues, in addition to causing direct oxidative DNA damage.
Macrophages associated with IBD induce inflammation, cellular senescence and γH2AX foci, a marker of DNA damage (Sohn et al., 2012). The induction of cellular senescence is associated with secretion of iNOS from macrophages both in vivo and in vitro. Thus, the nitrosative stress induced by macrophages has a direct effect upon the induction of cellular senescence. Whether macrophage-induced senescent cells are part of the SASP remains to be determined.
Innate immunity and chronic inflammation
The TLR of the innate immune system was identified by Janeway (1989) and is considered as a receptor family that recognizes pathogen-associated molecular patterns (PAMPs). TLR signaling contributes to the growth of tumors in numerous organs and thus may represent an important component of tumorigenesis. PAMPs are present in all microorganisms regardless of their pathogenicity. Initial studies have shown that TLRs recognize microbial ligands as well as endogenous host-derived ligands. Stimulation of TLRs leads to the activation of NF-κB, c-jun, mitogen-activated protein kinase (MAPK) and IRF family-mediated gene transcription (for a review see Lee & Kim, 2007). Such a diverse set of signaling activation is also involved in tissue repair and regeneration. Initial studies regarding the role of TLRs in cancer has been focused on their functions as a receptor family to boost anti-tumor immunity upon testing of exogenous agonists of TLRs. Recent studies suggest that microbe-derived mutagens or microbe-induced chronic inflammation may be an oncogenic mechanism. Thus, it is reasonable to surmise that the protective role of TLRs in cancer may be associated with the prevention of pathogen-derived (e.g. EBV, HBV, H. pylori) cancers (for a review see Rakoff-Nahoum & Medzhitov, 2009). Recent studies also suggest that TLR signaling contributes to tumor growth in various organs as well. For example, MyD88 is an important stimulator of chemically induced tumors of both skin and connective tissue. MyD88-deficiency inhibited tumor formation upon administration of DMBA and TPA or 3′-methylcholanthrene (MCA), which led to the development of skin papillomas and sarcomas, respectively (Swann et al., 2008). It has been demonstrated that chronic and unregulated TLR signaling leads to DNA damage and mutation as well as aberrant chromosomal translocation by inducing free radicals or activation-induced cytidine deaminase (AID) (Ferrero, 2005; Uno et al., 2007; Xu et al., 2007). A previous study demonstrated that the ablation of TLR-mediated signaling pathway adaptor MyD88 impairs intestinal tumorigenesis (Rakoff-Nahoum & Medzhitov, 2007). However, these results cannot be directly translated since the ablation of TLRs in the Apc/Min+ mice model did not show a reduction of intestinal tumor formation. Thus, whether TLRs are involved in tumor initiation is yet to be determined. In contrast to the MyD88-deficient Apc/Min+ mice, the IL10−/− mouse model of colitis-associated carcinoma shows that colitis is dependent upon TLR recognition of the intestinal microflora (Rakoff-Nahoum et al., 2006). Therefore, the role of TLRs is yet to be determined in this context. This suggests that important yet unknown mechanisms may regulate TLRs and hence tumor immunity. One can envision several possible common pathways to govern the role of TLRs such as the Wnt and mTOR pathways that sense the metabolic status of bacteria and host cells.
MicroRNAs, inflammation and DNA repair
MicroRNAs and DNA repair
MicroRNAs (miRNAs) are small, non-coding RNA molecules that regulate post-transcriptional gene expression by targeting an mRNA transcript for translational repression or degradation. miRNA regulation is becoming increasingly more important in understanding disease initiation and progression due to its nearly ubiquitous role in cellular pathway regulation and function. In fact, it has been estimated that miRNAs target at least 60% of the protein coding genome (Friedman et al., 2009). Over the past decade, microRNAs have emerged as critical regulators of the DNA damage response, reviewed recently by Wang & Taniguchi (2013). Here, we discuss a few select microRNAs that have been shown to be involved in both DNA repair and inflammation, namely miR-155, miR-210 and miR-21, all of which are frequently overexpressed in cancer.
miR-155 is a widely studied microRNA that targets several genes essential for the DNA damage response. Overexpression of miR-155 has been observed in a variety of solid tumors and hematologic malignancies (Eis et al., 2005; Iorio et al., 2005; Shibuya et al., 2010) and has been correlated with poor prognosis in lung and pancreatic cancer (Papaconstantinou et al., 2013; Yanaihara et al., 2006). Overexpression of miR-155 has been demonstrated via multiple pathways. Most relevant, miR-155 expression is driven by inflammation, leading to an increase in mutation as a result of WEE1 repression, a miR-155 target (Tili et al., 2011). It has also been shown that miR-155 overexpression can be a consequence of hypoxia, driven by the transcription factor, HIF-1α (Babar et al., 2011; Bruning et al., 2011). Furthermore, it was shown that the induction of miR-155 in hypoxia promotes radiation resistance in hypoxic lung cancer cells, potentially through the repression of FOXO3a, a pro-apoptotic factor (Babar et al., 2011). Additional transcriptional regulation of miR-155 exists, including the recent finding that BRCA1 epigenetically regulates miR-155 expression (Chang et al., 2002). Importantly, BRCA1, a key player in the DNA damage response, is often repressed in cancer, which could result in the overexpression of miR-155. It has also been demonstrated that miR-155 targets mismatch repair proteins, MLH1, MSH2 and MSH6 promoting microsatellite instability (Valeri et al., 2010). Taken together, these data suggest that miR-155 overexpression in cancer might lead to mismatch repair inhibition and increased mutagenesis as well as therapeutic radiation resistance, driving tumor progression.
Another microRNA of interest, miR-210, is also involved in both the DNA damage response and inflammation. Similar to miR-155, miR-210 overexpression has been observed in several types of cancer (Camps et al., 2008; Foekens et al., 2008; Huang et al., 2009; Malzkorn et al., 2010; Papaconstantinou et al., 2013). miR-210 expression is also induced by hypoxia, again driven by HIF-1α (Crosby et al., 2009) and has been shown to promote radiation resistance in hypoxic lung cancer cells (Grosso et al., 2013). Interestingly, miR-210 was recently shown to stabilize HIF-1α under normal oxygen conditions and promote a hypoxic-like environment, rendering normoxic cells more resistant to radiation (Grosso et al., 2013). Although targets of miR-210 have been difficult to identify, a few DNA damage response members have emerged as confirmed targets. miR-210 has been shown to target RAD52, a key player in the homology dependent repair (HDR) pathway (Crosby et al., 2009). Inhibition of RAD52 by miR-210 overexpression could lead to a decrease in HDR and promote mutagenesis. In addition to DNA repair targets, miR-210 has been shown to target SHIP1, an important negative regulator of cell proliferation involved in inflammation response (Lee et al., 2013). Interestingly, miR-155 has also been shown to target SHIP1 (O’Connell et al., 2009), suggesting a role for hypoxia-driven miRNAs in regulating the inflammation response. It is important to note that although the mechanism behind miR-210 pathogenesis remains largely unclear, it is well-established that miR-210 and its inhibition could become an important prognostic and therapeutic tool in cancer treatment (reviewed recently Hong et al., 2013).
Finally, another miRNA involved in both DNA repair and inflammation is miR-21. Similar to both miR-155 and miR-210, miR-21 is induced by hypoxia and deregulated in a number of different cancer types leading to a poorer prognosis in patients with miR-21 overexpressing tumors (de Oliveira et al., 2009; Nair et al., 2012). Overexpression of miR-21 could significantly impair the DNA damage response due to the involvement of multiple miR-21 targets in DNA damage recognition and repair. One of the first targets identified for miR-21 was phosphatase and tensin homolog (PTEN), a key regulator of the PI3K/AKT signaling pathway (Meng et al., 2006). In the presence of DNA damage, the PI3K/AKT pathway is activated, inhibiting apoptosis and enabling DNA repair. PTEN, a negative regulator of AKT, inhibits the PI3K/AKT pathway and promotes apoptosis. Overexpression of miR-21 in cancer likely results in PTEN inhibition and activation of the PI3K/AKT pathway and DNA repair, resulting in tumor cell survival. Another target of miR-21 is CDC25A, which is also involved in the DNA damage response during cell cycle checkpoint activation. Overexpression of miR-21 has been demonstrated to inhibit CDC25A, which halts the cell cycle, and facilitates DNA repair (Anastasov et al., 2012). The targeting of both PTEN and CDC25A by miR-21 overexpression results in radiation resistance, suggesting a mechanism by which miR-21 functions in tumor progression (Anastasov et al., 2012; Liu et al., 2013).
MicroRNAs and inflammation
It is commonly accepted that a proinflammatory tumor microenvironment may hamper anti-tumor immunity and favor cancer initiation and progression. The discovery of small non-coding regulatory RNAs, namely microRNAs (miRNAs) shed a new light on regulatory mechanisms linking inflammation and cancer. miRNA expression changes are observed in autoimmune diseases and cancers.
miR-21 is frequently up-regulated in human cancers such as breast cancer and glioma (Krichevsky & Gabriely, 2009). Functional roles for miR-21 have been reported in B cells, T (Th) cells, dendritic cells (DCs) and in various types of autoimmune diseases (Garchow et al., 2011; Lu et al., 2009, 2011). Dysregulation of miR-21 has been also found in some autoimmune conditions and mouse models of cancer (Fenoglio et al., 2011; Kiyohara & Hirohata, 1994; Ruan et al., 2011). DNA methylation is regulated by miRNAs that target the DNA methylation machinery. Pan et al. showed that miR-21 was overexpressed in CD4+ T cells from SLE patients and MRL/lpr mice, where miR-21 promoted cell hypomethylation by inhibiting DNA methyltransferase 1 (DNMT1) expression (Pan et al., 2010). Detailed analysis indicated that miR-21 indirectly targeted RASGRP1 and down-regulated DNMT1 expression. This process is mediated by the Ras-MAPK pathway upstream of DNMT1. In contrast, repressing miR-21 expression in CD4+ T cells from SLE patients up-regulated DNMT1 expression and attenuated DNA hypomethylation.
miR-155 plays a role in innate immunity in macrophages. Lipopolysaccharide (LPS)-mediated activation of TLR signaling induces miR-155 in human macrophages. miR-155 is involved in the activation of tumor necrosis factor-α (TNF-α) and interleukin 6 (IL6), suggesting its critical role in tumor microenvironment formation. miR-155-deficient dendritic cells showed impaired antigen presentation and are subsequently unable to activate T cells to promote inflammation (Rodriguez et al., 2007).
Macrophage inflammatory responses to infection involve the upregulation of several miRNAs including miR-21 (Bazzoni et al., 2009; Liu et al., 2009; Sheedy et al., 2010). Regulation of the tumor suppressor protein programmed cell death 4 (PDCD4) is critical for the control of inflammation induced by LPS. LPS induces activation of TLR 4, which leads to upregulation of PDCD4, and proinflammatory mediators NF-κB and IL6. miR-21 is also induced in response to LPS in many cell types, and this is dependent on MyD88 and NF-κB. Once induced, miR-21 inhibits expression of PDCD4, thus promoting expression of IL10, an anti-inflammatory cytokine. LPS also induces the expression of miR-155, which results in a decrease in SHIP1 expression and promotion of the inflammatory response. However, in the presence of IL10, miR-155 expression is inhibited, which switches off the pro-inflammatory response (McCoy et al., 2010).
In adaptive immunity, the role of miRNAs in distinct effector T helper cell subsets has been recently reported. An important role of miR-155 is implicated in regulatory T (Treg) cell formation and function. Forkhead box P3 (FOXP3), a lineage specific transcription factor for Treg cells, may directly regulate the expression of miR-155 (Zheng & Rudensky, 2007). Likewise, miRNA-155-deficient mice are immunodeficient, indicating that miR-155 is critical for homeostasis and the immune system. The fact that miR-21 and miR-155 overexpression impair Treg suppressor activity suggests additional inflammatory effects of these two miRNAs (Rodriguez et al., 2007). Often miRNAs increase the buffering force of gene regulatory networks by targeting factors implicated in the same proinflammatory pathways. For example, miR-155 targets different DNA repair enzymes or DNA damage-induced checkpoints.
miR-210 is a typical hypoxia-induced miRNA, thus its expression is increased by low oxygen in a HIF-dependent manner (Biswas et al., 2010). Like miR-21, miR-210 expression was upregulated in mouse peritoneal macrophages treated with LPS, suggesting a regulatory role of miR-210 in LPS/TLR4 signaling (Qi et al., 2012). Likewise, over-expression of miR-210 inhibits TLR4-induced secretion of IL6 and tumor necrosis factor (TNF) via its targeting of NF-κB1 (p105) transcripts. This suggests that miR-210 is a negative feedback regulator in LPS/TLR4 signaling (Qi et al., 2012). miR-210 may represent a miRNA that links inflammatory signals in the hypoxic microenvironment. It is notable that the expression of miR-21, miR-155 or miR-210 is controlled by the same immune/inflammatory and oncogenic signals. The regulatory mechanism of the expression of these miRNAs may explain converging points between immune and tumorigenic signaling pathways. miR-21 and miR-210 are important for transformation, tumorigenicity, DNA repair and chemotactic cell invasion and are under the control of signal transducer and activator of transcription (STAT3)-mediated transcription (Iliopoulos et al., 2009). Since STAT3 is a target of IL6 signaling, the ability of these two miRNAs to transform cells places them in a positive regulatory circuit with NF-κB, IL6 and STAT3. Interestingly, a similar feedback loop was found in other cancer cell lines and in patient cancer tissues (Iliopoulos et al., 2009).
It has been suggested that the expansion of Tregs occurs in an inflammatory hypoxic environment. It has been shown that hypoxia is an intrinsic molecular cue that promotes FoxP3 expression, resulting in immunosuppressive mechanisms to prevent possible tissue damage in conditions of reduced oxygen availability (Clambey et al., 2012). It has been also suggested that Treg cells provide a complementary immunological mechanism to protect tissue injury that is driven by low oxygen tension (i.e. hypoxia) in inflamed or cancerous tissues mainly via the cAMP-elevating A2A and A2B adenosine receptors (Sitkovsky, 2009). Collectively, this suggests that the dysregulation of miRNA can both promote tumor microenvironment formation and cellular transformation.
miRNAs clearly play an important role in regulating both the DNA damage response and inflammation pathways. Although only a select few miRNAs were covered here, many other members of the DNA damage response are also subject to miRNA regulation. The three miRNAs discussed here were chosen for both their overlap with inflammation as well as their similarity in expression, such that overexpression is observed for each of these miRNAs in multiple cancer types and that this induction of expression can be driven, at least in part, by hypoxia. Further investigation will be necessary to determine the importance of the multifaceted role of these and other oncogenic miRNAs. Enhancing our understanding of aberrantly expressed miRNAs and their function in different oncogenic pathways will likely improve current cancer therapy.
Interplay between infection-induced inflammation and DNA repair
Introduction
Since the discovery that Helicobacter pylori infection leads to gastric cancer (Warren, 1983), other chronic bacterial infections have been shown to cause cancer (Houghton & Wang, 2005). Streptococcus bovis (S. bovis)/infantarius was traditionally considered a lower grade pathogen frequently involved in bacteremia and endocarditis (Biarc et al., 2004). This bacterium became important in human health as it was shown that 25–80% of patients who presented with S. bovis bacteremia also had a colorectal tumor (Biarc et al., 2004). It has been estimated that over 25% of human malignancies are attributable to chronic inflammatory status, which arises as a consequence of infection and immunologic abnormality (Bouvard et al., 2009). In developing countries, up to 23% of malignancies are caused by infectious agents, including hepatitis B and C viruses (liver cancer), human papilloma-viruses (cervical cancers) and H. pylori (stomach cancer) (de Martel et al., 2012). In contrast, in developed countries, cancers caused by chronic infections are estimated to amount to approximately 8% of all malignancies (de Martel et al., 2012). There are pathogenic factors or conditions that facilitate chronic inflammation subsequently increase the risk of cancer development. In many cases, one can observe a direct connection between inflammation caused by infectious pathogens and cancer (Darveau, 1999). The inflammatory response triggered by infection precedes tumor development and is a part of the normal host defense, the goal of which is pathogen elimination (Coussens & Werb, 2002). Some pathogens can directly induce cell transformation but most, if not all, also induce a state of chronic inflammation that favors initiation, promotion and progression of tumors (Balkwill & Coussens, 2004; Fitzpatrick, 2001). Many bacteria that cause persistent infections produce toxins that specifically disrupt cellular signaling to perturb the regulation of cell growth (Wei et al., 2012) or to induce inflammation. Other bacterial toxins directly damage DNA (Cortes-Bratti et al., 2001; Frisan et al., 2002). Such toxins mimic carcinogens and tumor promoters, like TNF-α, and might represent a paradigm for bacterially induced carcinogenesis (Rogers & Fox, 2004). In this part of the review, we will cover infection-associated inflammation that is mediated via infiltration of phagocytes and generation of ROS that induces genomic instability that leads to cell dysfunction, mutation and cancer.
Pathogens impair DNA repair function
Helicobacter pylori infection results in the development of gastric cancer in 5% of infected people (Suerbaum & Michetti, 2002; Uemura et al., 2001; Wroblewski et al., 2010). The presence of Helicobacter in the stomach results in a host immune response aiming to swiftly eradicate the invading organisms by phagocytosis or secreted anti-microbial substances (Karin & Greten, 2005). In response to H. pylori infection, mucosal leukocytes up-regulate enzymes such as iNOS, myeloperoxidases, NADPH oxidases and peroxidases which generate a great amount of reactive oxygen and nitrogen species (RONs). To inhibit the bactericidal effects of nitric oxide (NO), H. pylori itself produces a great amount of superoxide anions (Nagata et al., 1998) and releases anti-oxidant enzymes (e.g. catalase, superoxide dismutase) to escape the host immune response. A growing body of evidence indicates that RONs produced by neutrophils and macrophages can directly induce DNA base oxidation, single strand breaks, generate excessive base excision repair intermediates and deamination, and can indirectly lead to base alkylation (Coussens & Werb, 2002; Kumagai et al., 2003; Reuter et al., 2010) as discussed above. In the case of infection, tissues are injured resulting in the local release of cytokines and other chemotactic factors that lead to infiltration and activation of macrophages and neutrophils to produce large quantities of proinflammatory mediators as well as reactive chemical species that cause mutagenic and cytotoxic damage (Dale et al., 2008; Terzic et al., 2010). For example, RONs-induced DNA damage contributes to tumor development in the Aag glycosylase deficient mouse model suggesting that DNA repair is an important suppressor of colon cancer in response to chronic inflammation, thus providing robust evidence that repair of RONs-associated DNA lesions is important for the suppression of inflammation-induced tumorigenesis (Meira et al., 2008). The mechanism of how H. pylori infection leads to a disequilibrium in genomic integrity of the host cells is likely a multifaceted one. It is possible to identify three mechanisms by which H. pylori infection leads to loss of genomic integrity and promotes carcinogenesis.
Increased levels of oxidized base lesions such as 7,8-dihydro-8-oxoguanine (8oxoG) are found in the inflamed gastric mucosae of H. pylori-infected people (Baik et al., 1996; Tardieu et al., 2000) along with reduced DNA damage repair (MMR, BER and DSBR) (Colotta et al., 2009; Kim et al., 2002; Maddocks et al., 2009; Mangerich et al., 2012; Mirzaee et al., 2008). Defects in human DNA MMR result in microsatellite instability (MSI) and predisposition to several types of epithelial cancer (Ou et al., 2007; Seruca et al., 1995). In addition, lack of MMR enhances the mutation frequency in tumor suppressor genes such as p53 resulting in disregulation of the cell cycle (Wei et al., 2012). The resulting increased mutation frequency likely correlates with the increase of aberrant expression of activation-induced cytidine deaminase (AID) in inflamed epithelial cells infected with H. pylori (Endo et al., 2008). A second alternative pathway for the promotion of genomic instability by H. pylori is increased mutations in mitochondrial DNA that alter the normal respiratory functions including the oxidative phosphorylation pathway (Carew & Huang, 2002; Machado et al., 2010). Even though the random mutation frequency of tumor mtDNA is significantly lower than surrounding normal tissue (Ericson et al., 2012; Kidane & Sweasy, 2012), mutation frequencies in both the mitochondrial D-loop region and in several genes encoding respiratory subunits increase in human cells infected with H. pylori (Touati, 2010).
The third mechanism stems from pathogen-mediated epigenetic changes in DNA repair genes (Ando et al., 2009; Bierne et al., 2012). Diverse cellular functions including the expression of inflammatory genes, DNA repair and cell proliferation are regulated by infection-mediated epigenetic changes (Adcock et al., 2007). There is considerable evidence suggesting that epigenetic mechanisms may mediate development of chronic inflammation by modulating the expression of pro-inflammatory cytokines such as TNF-α, IL1B, tumor suppressor genes, oncogenes and activation of transcription factors such as NF-κB (Gou et al., 2012; Gupta et al., 2010; Pikarsky et al., 2004; Taylor & Cummins, 2009). Epigenetic inactivation of DNA repair genes in cancer has been reported for several DNA repair pathways including BER, NER, MMR and several other DNA damage processing mechanisms (Benachenhou et al., 1998; Dobrovic & Simpfendorfer, 1997; Guan et al., 2008; Lawes et al., 2005). Aberrant DNA methylation is one of the major inactivating mechanisms of tumor suppressor genes and is deeply associated with H. pylori infection (Ding et al., 2010; Gupta et al., 2010; Niwa et al., 2010). H. pylori infection frequently methylates the CpG island in the promoter region of the E-cadherin gene (Chan et al., 2003). It has been reported that IL1 can modulate CpG island methylation through activation of DNA methyltransferase (Hmadcha et al., 1999). Interleukin 1B (IL1B) modulates methylation-dependent gene silencing via nitric oxide (NO) production (Hmadcha et al., 1999), DNA hypermethylation of tumor suppressor genes, extensive genomic DNA hypomethylation and alteration of histone modification patterns (Lopez-Serra & Esteller, 2012). This suggests that IL1B is an inducing factor in gastric inflammation and carcinogenesis (El-Omar et al., 2000; El-Omar, 2001). Epigenetic chromatin marks are unstable and they rapidly change in response to any external stimulus including infection (Berger, 2007). Permanent changes to the DNA methylation can lead to development of cancer.
Chronic inflammation is deeply involved in the induction of aberrant DNA methylation (Niwa et al., 2010). It was suggested that severity of inflammation characterized by expression of specific types of inflammatory related genes such as IL1B and iNOS are necessary for the initiation and maintenance of methylation induction (Hur et al., 2011). The less severe gastric inflammatory response induced by H. pylori infection in interleukin 1 receptor, type 1 (IL1R1) deficient mice with less promoter methylation of E-cadherin (Huang et al., 2013) suggested that epigenetic changes on chromatin structure likely play a crucial role in H. pylori-induced gastric inflammation (Huang et al., 2013). The crosstalk between DNA repair and pro-inflammatory cytokines during chronic inflammation exacerbates the methylation of CpG islands of tumor suppressor genes including in mismatch and base excision repair genes (Kane et al., 1997; Kuniyasu et al., 2004). In addition, IL6 has been reported to control DNA methylation (Isomoto et al., 2007) and modulated iNOS expression during cancer development (Ma et al., 2008). In contrast, deletion of DNA repair genes such as OGG1 (Mabley et al., 2005) or expression of a mutated variant of BRCA1 (Zielinski et al., 2003) resulted in low or defective pro-inflammatory cytokines (TNF-α, interleukin 12A (IL12A) and IL1B).
Inflammatory immune response and infection
The immune response is characterized by mucosal infiltration of various cells of the innate immune system leading to an acute gastric inflammation. If the bacteria can be eliminated successfully, acute inflammation is readily resolved and the normal tissue architecture is restored. However, incomplete bacterial eradication leads to a persistent inflammatory process resulting in chronic inflammation for longer time (Han & Ulevitch, 2005). The majority of infected individuals develop a continuous and progressive chronic inflammatory process initiated by non-atrophic gastritis and followed by multifocal atrophic gastritis, intestinal metaplasia, and dysplasia, symptoms considered to be hallmarks of the truly precancerous stage of the disease cascade (Zabaleta et al., 2006, Zabaleta, 2012). The predisposing inflammation is most often caused by colonization of the gastric epithelium by H. pylori, and chronically infected individuals have an increased risk of developing gastric cancer (El-Omar, 2001). Cells of the innate immune system recognize the bacteria through receptors of so called PAMPs. PAMPs include microbial components such as LPS, peptidoglycan, flagellin and double-stranded RNA that are recognized by TLRs (Takeda & Akira, 2003). The initial response is characterized by neutrophil and macrophage infiltration (Peek, 2001) and the release of high levels of pro-inflammatory cytokines including interleukin 8 (IL8), IL1B, TNF-α, IL6 and iNOS (Bogdan, 2001; Crabtree et al., 1991; Rad et al., 2004) and chemokines such as RANTES and MIP-1α and β (Gasperini et al., 1999). For example, IL8 is a neutrophil- and lymphocyte-activating chemotactic cytokine and its secretion by gastrointestinal epithelial cells is enhanced by H. pylori colonization (Crabtree et al., 1995; Peek et al., 1995). In contrast, if H. pylori infection induces infiltration of T lymphocytes and expression of the CCR6 ligand, CCL20 chemokine, this potentially triggers inflammation to augment apoptosis in inflamed gastric tissues. While it is clear that inflammatory cytokines contribute to inflammation-associated cancer susceptibility, the possible, and likely, contribution of the DNA damage that accompanies chronic inflammation has not been fully tested during H. pylori infection. Furthermore, DNA repair gene polymorphisms could influence the degree of inflammation in response to H. pylori infection that eventually impact the outcome of the tumor latency after infection. However, the connection between genetic polymorphisms in DNA repair genes in relation to infection will shed light in the future on the mechanisms of how DNA repair gene polymorphisms shape the outcome of the infection-induced inflammatory response. In addition, the infection mediated DNA damage response can also propagate inflammatory signaling and lead to more DNA damage to release more pro-inflammatory cytokines. Therefore, it may be possible to use DNA repair inhibitors to not only treat familial cancers, but also to treat and even prevent cancers related to infections, particularly cancers associated with chronic inflammation as discussed below.
Minimizing inflammatory response by DNA repair inhibitors
From the standpoint of infection, the functional plasticity of inflammatory and pro-inflammatory cytokines and innate immune cells, macrophages in particular, may potentially offer new targets for the therapeutic use if they are connected with the role and function of DNA repair genes. Several studies with genetically modified mice, along with the analysis of human tumors, have highlighted that altered expression of selected pro-inflammatory (e.g. TNF-α, IL1B, IL6) and/or anti-inflammatory cytokines (e.g. IL10, TGF-β) have a crucial role in the promotion of different types of tumors, including gastric, colorectal, liver, breast and skin tumors (Lin & Karin, 2007). The molecular pathways underlying pathogen-induced inflammation are expected to provide novel anticancer strategies to develop drug targets that reverse inflammation-mediated genomic instability. Even though PARP-1 inhibitors have emerged as promising and effective therapeutics for breast, ovarian, melanoma and lymphoid cancers, the use of PARP-1 inhibitors for the treatment of pathogen-associated (e.g. H. pylori) cancers remains to be evaluated clinically. Few studies have demonstrated suppression of the inflammatory response by PARP inhibitors in mice infected with H. pylori (Nossa et al., 2009) and prevention of the formation of Helicobacter-induced precancerous conditions in mice deficient of anti-inflammatory cytokine. PARP-1 sits at the center of the game not only to assist in repair of RONS-induced DNA lesions, but it also can contribute to further damage by promoting inflammation. Together, this points to PARP inhibition as an attractive target to suppress the DNA damage response that may block excessive inflammation acting potentially as a preventative agent for genomic instability and carcinogenesis.
Mouse models of inflammation, DNA repair and cancer
Several mouse models of inflammation and DNA repair are discussed below. The findings are summarized in Table 1.
Table 1.
Phenotypes of mouse models of DNA repair and inflammation.
| Gene | Role of gene product | Phenotype of mouse deletion | References |
|---|---|---|---|
| AAG/MPH | Alkyladenine DNA glycosylase | Inflammation and colon cancer upon treatment with DSS/AOM | Meira et al., 2008 |
| ALKBH2 ALKBH3 | Direct reversal of alkyation; repair of ethenobases | Inflammation and colon cancer upon treatment with AOM plus DSS; lethal in combination with AAG-deletion in the presence of AOM plus DSS | Calvo et al., 2012 |
| OGG1 | DNA glycosylase that removes 8-oxoG and Fapy G | Inflammation resistant; moderate tumorigenesis | Klungland et al., 1999; Minowa et al., 2000; Mabley et al., 2005; Bacsi et al., 2013 |
| MUTYH | DNA glycosylase that removes adenine opposite 8-oxoG | Intestinal adenomas with G:C to T:A mutations in the APC gene; mice doubly deficient in OGG1 and MUTYH are predisposed to lung and ovarian tumors | Sakamoto et al., 2007; Russo et al., 2004 |
| NEIL1 NTH1 | DNA glycosylases that removed oxidized bases | Mice doubly deleted of these glycosylases develop lung and liver tumors | Chan et al., 2009 |
| FANC C | Member of Fanconi Anemia crosslink repair pathway | Hypersensitive to LPS-induced inflammation intrinsic to hematopoietic system. | Sejas et al., 2007; Du et al., 2014 |
| SMAD4 | Regulator of TGF-β signaling | Inflammation and spontaneous head and neck cancer | Bornstein et al., 2009 |
| RAG2 | Adaptive immune response; V(D)J recombination | Infection with Helicobacter hepaticus leads to inflammation and colon cancer | Mangerich et al., 2012 |
| IL 10 | Suppressor of macrophage induction; inhibitor of proinflammatory cytokines | Spontaneous chronic inflammation and colon cancer that depends on commensal bacteria | Kuhn et al., 1993 |
| T-bet | T box transcription factor | Spontaneous colitis | Garrett et al., 2007 |
AAG DNA glycosylase
Alkyladenine DNA glycosylase (AAG) functions in BER and has a broad substrate specificity. AAG recognizes a variety of base damage that is induced by RONs including 3-methyladenine, 7-methylguanine, hypoxanthine, ethenoadenine and ethenoguanine. Treatment of AAG-deficient mice with dextran sulfate sodium (DSS) or DSS plus azoxymethane (AOM), which mimics the episodic inflammation in individuals with chronic inflammatory disease of the colon, induces significant inflammation compared to treatment of wild-type control mice (Meira et al., 2008). These treatments also result in an increase in etheno-base lesions and in 8-oxoG in the DNA. Importantly, AAG deficient mice exhibit significantly elevated colon tumor multiplicity, suggesting a link between chronic inflammation, DNA damage, and cancer. These results also suggest that AAG protects against inflammation and cancer.
ALKBH2 and ALKBH3 proteins
The E. coli AlkB protein repairs etheno-bases and also repairs 1-methyladenine and 3-methylcytosine by direct reversal that depends on iron, oxygen and α-ketoglutarate. The human homologs of E. coli AlkB include the ALKBH2 and ALKBH3 proteins, which are known to repair etheno bases (Mishina et al., 2005; Ringvoll et al., 2008). Treatment of wild-type, ALKBH2-deficient, ALKBH3-deficient, and ALKBH2/3-deficient mice with AOM plus DSS resulted in colon tumorigenesis (Calvo et al., 2012). ALKBH2-deficient mice exhibited greater tumor multiplicities than wild-type mice. ALKBH3-deficient mice did not develop significantly more tumors than wild-type mice, but ALKBH2/3-deficient mice exhibited elevated tumorigenesis compared to ALKBH2-deficient mice alone. Under these conditions ALKBH3-deficient mice displayed significantly higher levels of inflammation than either ALKBH2-or ALKBH2/3-deficient mice. Similar levels of tumor development were observed in AAG−/− ALKBH2−/− and AAG−/−ALKBH3−/− mice. However, a single cycle of AOM/DSS or DSS alone resulted in lethality in the AAG−/−ALKBH2−/−ALKBH3−/− mice. This was accompanied by increased levels of etheno-bases but less colonic inflammation. AAG−/−ALKBH2−/−ALKBH3−/− mice recover from inflammation induced by cerulein but not by LPS. Importantly, DSS and LPS induce inflammation that is mediated by TLR signaling, whereas this is not the case with cerulean (Kim, 2008; Poltorak et al., 1998). This suggests that DNA repair by AAG, ALKBH2 and ALKBH3 is necessary for survival of inflammation that is mediated by TLR-signaling.
OGG1 and MUTYH DNA glycosylases
One of the most common mutagenic adducts that results from oxidative DNA damage is 7,8-dihyro-8-oxoguanine (8-oxoG) (Barnes & Lindahl, 2004). Adenine pairs with 8-oxoG during DNA synthesis, ultimately resulting in G to T transversions. The OGG1 DNA glycosylase, a member of the BER pathway, excises both 8-oxoG and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) (Bjoras et al., 1997; Girard et al., 1997; Ohtsubo et al., 2000; Thomas et al., 1997). The presence of 8-oxoG in DNA is considered to be a signature of DNA damage induced by ROS. The MUTYH DNA glycosylase, which also functions in BER, removes adenine opposite 8-oxoG and 2-hydroxyadenine opposite guanine, and a deficiency in this process leads to increased levels of mutagenesis. Importantly, biallelic mutations in MUTYH are associated with an inherited colorectal cancer syndrome resembling adenomatous polyposis (Al-Tassan et al., 2002; Jones et al., 2002; Sampson et al., 2003; Sieber et al., 2003).
Mice deleted of OGG1 exhibit increased levels of 8-oxoG in their tissues, but show only moderate increases in tumorigenesis (Klungland et al., 1999; Minowa et al., 2000). MUTYH deficient mice develop greater numbers of intestinal tumors than wild-type mice, and these adenomas harbor G:C to T:A mutations in the APC gene (Sakamoto et al., 2007). Mice doubly deleted of OGG1 and the MUTYH DNA glycosylase are predisposed to development of lung and ovarian tumors tumors (Russo et al., 2004). Treatment of these mice with KBrO3, an agent that induces oxidative base damage, significantly increases tumorigenesis. The lung tumors of MUTYH-deficient mice have G:C to T:A transversions in the K-RAS oncogene. Therefore, tumorigenesis results from the accumulation of G to T mutations as a result of increased levels of 8-oxoG and deficiency in the removal of adenine opposite 8-oxoG by the MUTYH DNA glycosylase, but the roles of inflammation in tumorigenesis were not characterized in these studies.
A recent report shows that OGG1-deficient mice are resistant to systemic inflammation induced by endotoxin (Mabley et al., 2005), as assessed by decreased levels of chemokines and cytokines in serum, compared to OGG1-proficient mice. The use of an airway inflammation model (Bacsi et al., 2013) yielded similar results in that down-regulation of OGG1 in the airways of mice challenged with ragweed pollen grain extract resulted in significantly less inflammation than in mice with normal levels of OGG1 in their airways. Lower levels of inflammation correlated with fewer strand breaks resulting from incision of the DNA by OGG1, suggesting that strand breaks augment the inflammatory process. Use of the OGG1-deleted mouse yielded similar results upon challenge with OVA or house dust mites (Li et al., 2012). The OGG1-deficient animals exhibited less interleukin 4, 6,10, 17 (IL4, 6, 10, 17) in challenged lung tissues and decreased expression of STAT6 and NF-κB. Interestingly, it has recently been shown that OGG1 glycosylase binds to the excised 8-oxoG base, and interacts with RAS family of GTPases to serve as a nucleotide exchange factor (Boldogh et al., 2012). Whether this novel activity of OGG1 is related to inflammation awaits further study. Treatment of MUTYH-deficient mice with dextran sulfate sodium (DSS) resulted in increased levels of 8-oxoG but significantly lower levels of inflammation compared to what was observed in wild-type mice (Casorelli et al., 2010). There was little change in the expression of pro- and anti-inflammatory cytokines in these mice, which correlated with the observed low levels of inflammation.
NTH1 and NEIL DNA glycosylases
Human NTH1, like all the EcoNth orthologs, possesses DNA-glycosylase/lyase activity on oxidized pyrimidines, formami-dopyrimidines, 5-formyluracil and incises AP sites (Asagoshi et al., 2000a,b; Eide et al., 2001; Ikeda et al, 1998; Jiang et al., 1997b; Marenstein et al., 2001, 2003; Melamede et al., 1994; Miyabe et al., 2002; Purmal et al., 1998; Sarker et al., 1998). In E.coli a backup activity to EcoNth for oxidized pyrimidines, EcoNei, was identified and characterized (Dizdaroglu et al., 2001; Jiang et al., 1997a; Melamede et al., 1994; Purmal et al., 1998; Zhang et al., 2000) and its first eukaryotic homologs were found in humans and designated NEIL1, NEIL2 and NEIL 3 (NEI-like). Like hNTH1, NEIL1 recognizes oxidized pyrimidines, formado-pyrimidines and thymine residues oxidized in the methyl group (Bandaru et al., 2002; Hazra et al., 2002; Hu et al., 2005; Wallace et al., 2003; Zhang et al., 2005). Unlike hNTH1, NEIL1 recognizes both stereoisomers of thymine glycol (McTigue et al., 2004, Miller et al., 2004). Thus far, the best substrates for hNEIL1 appear to be the hydantoin lesions, guanidinohydantoin (Gh) and spiroiminodihydantoin (Sp) (Krishnamurthy et al., 2008). NEIL1 is also capable of removing lesions from single-stranded DNA as well as from bubble and forked DNA structures (Dou et al., 2003, 2008). NEIL2 also prefers oxidized pyrimidines but shows a greater preference than NEIL1 for lesions in single-stranded and bubble structures (Dou et al., 2008). Because the expression of NEIL1 is cell-cycle dependent (Hazra et al., 2002), it acts on forked DNA structures (Hegde et al., 2008), and it interacts with PCNA (Dou et al., 2008) and FEN-1 (Hegde et al., 2008), it has been proposed that NEIL1 functions in replication associated repair. Although mice deleted of the NTH1 DNA glycosylase have no reported phenotype (Ocampo et al., 2002) and NEIL1 DNA glycosylase deletion in mice develop metabolic syndrome (Vartanian et al., 2006), mice deleted of both NEIL1 and NTH1 exhibit an increase in both pulmonary and hepatic carcinogenesis (Chan et al., 2009). The lung tumors contained GC to AT transitions at codon 12 of the KRAS gene, suggesting they arose as a result of oxidative damage at guanine residues in the DNA. Interestingly, 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) was enriched in the tissues of the NTH1 NEIL1-deficient mice, and 8-oxo-G was not. This suggests that oxidative damage to guanine occurred in the mice, leading to the induction of transition mutations that arose from deficient repair of FapyG due to lack of initiation of BER and perhaps bypass of the oxidative lesion. The oxidative DNA damage could have arisen from either endogenous ROS or ROS as a result of inflammation, but no information regarding lyphocytic infiltration of tissues or tumors was reported.
FANC C-deficient mice and inflammation
The FANC C protein is a member of the Fanconi anemia pathway, which is responsible for the repair of crosslinks and also functions in homology-directed repair (for a review see Kim & D’Andrea, 2012). Bone marrow failure is a leading cause of death in patients with Fanconi anemia. FANC C-deficient mice are hypersensitive to LPS-induced inflammation and septic shock that is intrinsic to the hematopoietic system and results in its suppression, which suggests that bone marrow failure may result in part from an inflammatory process (Sejas et al., 2007; Du et al., 2014). The hematopoietic suppression observed in the FANC C deficient mice appears to be dependent upon increased expression of tumor necrosis factor alpha (TNF-α) and ROS. Interestingly, inactivation of FANC C results in increased activation of toll like receptor 8 (TLR 8) (Vanderwerf et al., 2009) leading to an increase in the expression of TNF-α. Although specific mutants of FANC C were able to rescue the crosslink repair defects of FANC C deficient cells, they were not able to suppress TNF-α overexpression, suggesting a role for FANC C in suppression of TLR 8 that is independent of its role in crosslink repair.
SMAD 4 deletion, inflammation and cancer
SMAD 4 is a central regulator of TGF-β signaling and is found to be downregulated in human head and neck squamous cell carcinoma (HNSCC) and adjacent buccal mucosa (Bornstein et al., 2009). Loss of SMAD4 leads to increased expression of TGF-β. Homozygous deletion of SMAD 4 in mice results in spontaneous HNSCC, downregulation of BRCA1, FANC A and FANC D2, which are members of the Fanconi anemia pathway. SMAD 4 deficient mice also exhibit increased levels of inflammation in head and neck epithelia, similar to what is observed in human tumors, and this is correlated with increased expression of TGF-β. Increased levels of genomic instability were observed in SMAD 4 deficient mice, likely as a result of downregulation of BRCA1 and genes of the Fanconi anemia pathway. These results suggest that SMAD4 is a “guardian gene”, at least within the context of HNSCC.
Colon cancer in Rag2−/− mice infected with Helicobacter hepaticus
Rag2−/− mice have no B or T lymphocytes to fight infection, but do have cells of monocyte origin including macrophages and neutrophils. Infection of these mice with H. hepaticus results in the development of severe colitis by 10 weeks and colon carcinoma by 20 weeks post infection (Mangerich et al., 2012). Elevated levels of infiltration of neutrophils and macrophages accompanying the colitis are observed in H. hepaticus-infected Rag2−/− mice compared to controls, suggesting the presence of an inflammatory response. Importantly, significant DNA damage, including high levels of halogenated bases were identified in these mice. Transcriptional profiling demonstrated that many of the genes encoding proteins involved with the generation of reactive chemical species were upregulated in infected mice, whereas DNA repair genes were downregulated in colon tissue (Mangerich et al., 2012). Therefore, the lack of regulatory T cells disturbs the balance of immune cells in the lamina propria, leading to high levels of inflammation and cancer. Use of a iNOS inhibitor was shown to block NO production and a reduction in colitis-associated pathology (Erdman et al., 2009) as was depletion of GR-1 cells. Given that much of the damage observed was in the form of halogenated lesions, it is likely the neutrophils, which are the source of hypochlorous acid via activation of myeloperoxidase (MPO), induce the colitis-associated DNA damage. This correlates with the observation that MPO and neutrophils are associated with the intestinal lesions of IBD patients (Knutson et al., 2013; Kruidenier et al., 2003a,b; Robinson et al., 1997).
APC (Adenomatous polyposis coli)
APC has been characterized in multiple studies, but its main role as a tumor suppressor was recognized in colorectal cancer. Approximately 81% of hypermutated tumors and 53% of non-hypermutated tumors in the colon harbor mutations in the APC gene. The function(s) of the Apc protein are diverse and include cellular proliferation, differentiation, apoptosis, migration, and cytoskeletal regulation (Zeineldin & Neufeld, 2013). APC has been known as a tumor suppressor particularly in human colorectal cancer while acting as an inhibitor for hyperactivation of canonical Wnt signaling (Fearon, 2011). However, the critical role of APC is not limited to only colon cancer but includes other types of cancer with the importance of the Wnt signaling pathway in cell proliferation and differentiation in general (Karim et al., 2004). Germline mutations in the APC gene predispose individuals to an inherited form of colon cancer termed familial adenomatous polyposis (FAP) (Groden et al., 1991; Joslyn et al., 1991; Kinzler et al., 1991). Colorectal carcinogenesis results from one inherited germline mutation in the APC gene, followed by loss of heterozygosity (LOH) in the second copy of the APC gene, resulting in lack of functional APC protein in the cells.
Karin et al. demonstrated that chronic epithelial NF-κB activation that occurs in intestinal epithelia cells (IEC) constitutively active for Iκκ-β expression accelerates the loss of APC through iNOS (inducible nitric oxide synthase) upregulation (Shaked et al., 2012). Here the constitutive expression of active Iκκ-β induced chronic activation of the NF-κB pathway and did not interfere with the Wnt pathway-mediated β-catenin accumulation, suggesting that the decoupling of the Wnt pathway and chronic inflammation does occur with the loss APC protein in intestinal epithelial cells (IECs). Also such activation induced iNOS, and resulted in excessive DNA damage, particularly elevated amounts of 8,5′-cyclo-2′-deoxyadenosine (cdA) and 8,5′-cyclo-2′-deoxyguanosine (cdG), which are oxidatively induced DNA lesions in Iκκ-β in the IEC of mice. Of note, cdA and cdG are two of many endogenous reactive oxygen species (ROS)-induced DNA lesions. This study also elegantly demonstrated that IECs are undergoing constant DNA damage and repair. Consistent with this, very recent study (Pribluda et al., 2013), which suggested that inhibition of the chronic NF-κB pathway by NSAIDs is beneficial in preventing DNA damage in IECs. Inhibition of NOS by pharmacological treatment of APCmin/+ mice with guanidinoethyldisulfide (GED) reduced the intestinal tumor load and oxidative stress in these mice as does gene knockout of iNOS (Ahn & Ohshima, 2001; Mabley et al., 2004).
IL17 and the linkage between DNA repair and inflammation
In recent years, interleukin 17 (IL17) has been mostly described as a proinflammatory cytokine for tumorigenesis and autoimmune diseases. IL17 was initially identified as an important CD4 T cell-derived proinflammatory cytokine in autoimmune diseases models such as EAE (Baeten et al., 2013). It has been suggested that IL6 and TGF-β1 are two critical cytokines that influence the development of Th17 cells, while other proinflammatory cytokines including IL1β and IL23 further develop pathogenic Th17 cells’ pathogenicity. IL17 has six family members (IL17A-F). Two isoforms, IL17A and IL17F, have similar structures whereas the others are distant family members (Iwakura et al., 2011). The differential role of IL17A and IL17F has been documented previously (Ishigame et al., 2009). Of note, the particular presence of IL17 in the intestinal lamina propria raised questions regarding its role in gut inflammation. Several groups have reported IL17 as a critical stimulator of intestinal tumorigenesis in spontaneous and a bacterial toxin-mediated pathway (Blatner et al., 2012; Chae & Bothwell, 2011; Chae et al., 2010; Wu et al., 2009). The molecular mechanism of IL17-mediated tumorigenesis is still unclear. However, there are several studies that indicate the importance of a constant level of IL17 and its recognition of commensal microbiota in intestinal tumorigenesis. It has been reported that IL17 is constantly expressed by CD4 T cells and this was maintained by commensal microbiota (Atarashi et al., 2008). Also the recognition of microbiota by the adaptor protein MyD88 has been found to be important to develop intestinal tumorigenesis (Rakoff-Nahoum & Medzhitov, 2007). Hence, it is very likely that the recognition of commensal bacteria and subsequent maintenance of a basal level of inflammation by IL17 is protective under normal circumstances. However, when a critical component of a tumor suppressor/DNA repair related component such as APC is affected, this protective process could turn into a “feeding” role for transformed cells. For example, a very recent study suggested that Tpl2 (tumor progression locus 2) activity is modulated by IL17 via AP-1 to induce neoplastic transformation in epidermal cells and human breast cancer cells MCF7 (Kim et al., 2013).
In a broader sense the involvement of IL17 in different types of cancers, previous and recent studies reported that polymorphisms in IL17 gene are associated with various types of cancer such as gastric, cervical, colorectal and breast cancer (Wu et al., 2010). It is known that IL17 can markedly activate the NF-κB pathway (Chang et al., 2011).
Although very much speculative in its current stage, it could be possible that IL17 may lead to hyperactivation of the NF-κB pathway to induce APC loss. Such an LOH event driven by a constantly activated NF-κB pathway has been suggested previously (Shaked et al., 2012). Loss of APC protein eventually leads to the accumulation of mutations, which may in part result from its ability to interfere with BER. Future studies need to address whether IL17 is directly or indirectly regulating DNA damage control in the above cancer types.
Seminal studies showed that Th17 cells are an important cell subset to induce both spontaneous and Enterotoxigenic Bacteroides fragilis (EBTF)-driven intestinal tumorigenesis (Chae et al., 2010; Wu et al., 2009). Notably the maintenance of such Th17 cell populations was dependent on the bacterial ATP production in the gut (Atarashi et al., 2008).
Tumor microenvironment, inflammation and DNA repair
Introduction
Chronic inflammation has been considered as the “fuel” for tumorigenesis is to promote the generation of a tumor microenvironment that enables angiogenesis and recruitment of inflammatory mediators and cell types. The complex interaction between intrinsic mutations and tumor microenvironment eventually leads to cancer. Thus, the formation of a permissive tumor environment could be considered as a sum of collective counter responses as well as manipulation of host immune system by tumors that accumulated mutations to evade intrinsic and extrinsic checkpoints in the host.
Eradication of cancerous cells by immune system
Over the past 20 years, the availability of mouse genetic models facilitated study of the challenging question of how immunity can protect the host from cancer. One hypothesis is that the immune system plays three distinct roles in cancer prevention. First, the immune system protects the host from viral/bacterial infection that can induce tumors by eliminating such pathogens. Second, it abolishes the establishment of a proinflammatory microenvironment that facilitates tumorigenesis by eliminating pathogens and by prompt resolution of inflammation, so that it does not enter the chronic inflammation stage. Third, the immune system eliminates tumor cells in certain tissues by recognizing ligands for activating receptors on innate immune cells and tumor antigens that are co-expressed in nascent transformed cells. Such events lead to the activation of the adaptive immune system and eventually result in tumor clearance (for a review see Schreiber et al., 2011). A more sophisticated view of the control of tumor “quantity” as well as “quality” was established a decade ago (Dunn et al., 2002; Schreiber et al., 2011; Shankaran et al., 2001). These findings led to investigation of three stages of immune surveillance or so called cancer immunoediting – Elimination Equilibrium and Escape (Schreiber et al., 2011).
The concept of the Elimination phase is the step at which the innate and adaptive immune systems detect the presence of a developing tumor and destroy it. Several alerting mechanisms for the immune system are likely to include Type I IFNs, endogenous danger signal ligands from tumor cells such as HMGB1 or hyaluronan fragments, and expression of stress ligands such as RAE-1 and H60 (mouse) or MICA/B (human) (Steinle et al., 2001) (for reviews see Matzinger, 1994; Sims et al., 2010). Such mechanisms alert the innate immune cells to release proinflammatory and immunomodulatory cytokines to facilitate the development of tumor-specific adaptive immunity.
After undergoing the rigorous and robust Elimination step, only a few tumor cell variants may survive. At this point the Equilibrium stage is initiated. At this stage, the adaptive immune system prevents tumor cell outgrowth and also edits the antigenicity of the tumor cells. This can be considered as a functional state of dormancy. The evidence for the existence of an equilibrium phase came from a study showing that immunocompetent mice treated with low-dose carcinogen 3′-methylcholanthrene (MCA) harbored occult cancer cells for an extended time period, and then the depletion of IFN-γ and T cells resulted in rapid reappearance of the tumor at the original MCA injection site in half of the mice (Sims et al., 2010). Once tumor cells acquire the ability to outmaneuver immune recognition and/or destruction, they can emerge as visible tumors. By modulating their own response against the host immune system progressive growth of tumors is possible.
Tumor cell Escape can occur through several mechanisms (for a review see Dunn et al., 2004).At the tumor cell level, loss of antigens would abrogate immune recognition for the cytotoxic effects of immunity and subsequently promote tumor outgrowth. Such loss can be achieved by emergence of tumor cells that lack strong rejection antigens (DuPage et al., 2012; Monjazeb et al., 2013). Alternatively, loss of the major histocompatibility complex (MHC) class I proteins that load tumor antigenic peptide or loss of antigen processing functions within the tumor cell that are needed to produce the antigenic peptide epitope can achieve escape from immune surveillance (Siddle et al., 2013). Interestingly, these alterations are probably driven by a combination of genetic instability, inherent in tumor cells, and the process of immunoselection during the elimination and equilibrium stages (Khong & Restifo, 2002). The establishment of the tumor microenvironment is also an important mechanism of escape. Tumor cells can promote the development of an immunosuppressive microenvironment by producing immunosuppressive cytokines (VEGF, TGF-β) and recruitment of immunosuppressive lymphocytes (e.g. Tregs). The accumulation of Tregs in the tumor microenvironment often leads to a poor prognosis (Darrasse-Jèze & Podsypanina, 2013).
Control of DNA repair, inflammation and cancer by the immunosuppressive cytokine IL10
In addition to the proinflammatory role of IL17 in the intestine, recent studies identify regulatory T cells (Tregs) as an important player in colon cancer as well as other types of cancer such as ovarian cancer and renal cell carcinoma (Adeegbe & Nishikawa, 2013; Menetrier-Caux et al., 2012). Regulatory T cells are generated from both the thymus and periphery, and the transcription factor Foxp3 is a lineage specification marker (Josefowicz et al., 2012). Their main role is to suppress hyperactivation of the immune system, thus preventing autoimmunity and allergic reactions as well as excessive inflammation by infectious pathogens. Tregs are equipped with multiple mechanisms to perform negative regulatory functions by highly expressing negative co-stimulatory molecules for T cell activation such as programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4), and also secretion of immunosuppressive cytokines (IL10 and TGF-β). Growing evidence suggests that the accumulation of such immunosuppressive Tregs in the tumor microenvironment often results in a poor outcome for cancer patients. The important role of Tregs in the tumor microenvironment is often to inhibit effective anti-tumor immunity. Thus, targeting Tregs in the tumor microenvironment has been considered an effective approach to circumvent immunosuppression of multiple types of cancers such as non-small lung cancer (e.g. NSLCC and melanoma). Some of these approaches have now reached clinical trials in cancer patients (Hamid et al., 2013; Holmgaard et al., 2013). While targeting of immunosuppressive Tregs in the tumor microenvironment is beneficial, this systematic depletion of Tregs may have a possibility of causing autoimmunity. As cancer develops it may have accumulated myriads of mutations, which escape the host immune system, so it may be worth considering targeting of the immune system where inflammation and DNA repair are interacting at an early stage.
Interestingly, the inflammation event at an early stage would have more of a “feeding” role for tumorigenesis, and therefore the immunosuppressive nature of Treg cells could be beneficial. Such a hypothesis was supported by Erdman et al. demonstrating that the adoptive transfer of regulatory T cells could regress tumors within 1–3 weeks in the APC/Min+ mouse model (Erdman et al., 2005). They also elegantly showed that the transfer of IL-10 deficient regulatory T cells could not regress tumors, further suggesting that the immunosuppressive cytokine IL10 is critical to inhibit intestinal tumorigenesis (Erdman et al., 2003). In addition, other groups have shown that IL10 from regulatory T cells and CD4 T cells is a critical source of suppression for tumorigenesis (Dennis et al., 2013; Gounaris et al., 2009). Since IL10 is highly expressed in CD4+ T cells in the colonic lamina propria (Maynard & Weaver, 2008), it would be reasonable to think that IL10 may regulate intestinal tumorigenesis fed by IL17-mediated inflammation.
We have recently shown that the endogenous Tregs in APC/Min+ mice are not capable of regressing tumors while the depletion of endogenous Tregs regresses tumorigenesis (unpublished data). Further, we showed that the heterozygous mutation of APC in Tregs from otherwise wild-type mice restored their tumor regression capacity (unpublished data). These results demonstrate the effect of the tumor microenvironment on the development of Tregs function. They also suggest that IL10 from Tregs is critical at an early stage of intestinal tumorigenesis, and hereditary FAP colon cancer patients such as FAP patients may have further complications in their immune systems as a consequence of the germline mutation in the APC gene.
The contribution of the immunosuppressive cytokine IL10 to the DNA damage response needs to be addressed in future studies. A previous study showed that DNA damage triggers IL10 production after UV irradiation in mouse keratinocytes (Nishigori et al., 1996). This suggests that unrepaired DNA damage could induce systemic induction of the immunosuppressive cytokine. Additional evidence suggests that IL10 single nucleotide polymorphism (SNP) variants are associated with basal cell carcinoma alongside with DNA repair gene variants and also with genes such as iNOS that is associated with prostate cancer recurrence (Dluzniewski et al., 2012; Festa et al., 2005). Since the relationship between IL10 and the DNA damage response is unclear, future studies will require molecular and biochemical characterization of this pathway.
Antigenicity and tumor mutagenesis
Microsatellite DNA sequences comprise short tandem repeat (STR) motifs of one to six base pairs, and are ubiquitous in eukaryotic genomes, making up ~3% of total DNA in the human genome (Consortium, 2004). Microsatellites often display allele length alterations in tumor tissues, relative to normal tissue, or microsatellite instability (MSI) (Hile et al., 2013). Mismatch repair-deficient tumors display an MSI-High (MSI-H) phenotype due to widespread genome instability at microsatellites and other sequences. Loss of MMR protein expression and MSI-H is observed in Lynch syndrome tumors, and to varying degrees in sporadic colorectal cancer (CRC) and colitis-associated cancerous lesions (de la Chapelle & Hampel, 2010; Fleisher et al., 2000; Svrcek et al., 2007; Tahara et al., 2005). Tumor-specific loss of MMR destabilizes STRs genome-wide, including those present within protein-coding sequences. Consequently, translation of the resulting frameshift mutations generates truncated peptides. In another familial CRC predisposition syndrome, MUTYH-associated polyposis (MAP), loss of the base excision repair MUTYH glycosylase protein also results in a mutator phenotype, but one restricted to elevated transversion base substitution mutations (Al-Tassan et al., 2002; Nielsen et al., 2009).
Both MSI-H and MAP tumors carry a high mutational load due to loss of DNA repair, and are characterized by a strong immune response, relative to repair competent CRCs (de Miranda et al., 2012; Kloor et al., 2010). MSI-H tumors contain higher numbers of infiltrating, activated cytotoxic lymphocytes than do repair-proficient CRCs (Dolcetti et al., 1999; Guidoboni et al., 2001; Phillips et al., 2004). One hypothesis to explain these observations is that the high mutation rate within repair-deficient tumors produces aberrant tumor-specific antigens, thus triggering a strong, anti-tumor immune response (Linnebacher et al., 2001). Indeed, tumor infiltrating lymphocytes and peripheral blood lymphocytes from patients with MSI-H tumors can be reactive against epitopes representing tumor-specific frameshift antigens, and display cytotoxic activity towards MSI-H tumor cells (Ishikawa et al., 2003; Saeterdal et al., 2001). MSI-H tumors have a significantly better clinical prognosis than do repair-proficient CRCs (Popat et al., 2005), and the high density of infiltrating T cells in MSI-H tumors is correlated with favorable prognosis (Buckowitz et al., 2005; Guidoboni et al., 2001).
Paradoxically, the same increased mutation rate that enhances the immunogenicity of tumors also contributes to immune escape during tumor progression. One mechanism involved in immune escape is mutational inactivation of genes involved in antigen processing and recognition within repair-deficient tumors (de Miranda et al., 2012; Kloor et al., 2010). Defective HLA Class I expression is observed in both MMR-and BER repair-deficient tumors, and can be caused by somatic mutations at STRs within the β2-microglobin, TAP1 and TAP2 genes (Bicknell et al., 1996; de Miranda et al., 2009; Kloor et al., 2005). Despite their strong immunogenicity, 28% of MSI-H tumors do not express HLA Class II antigens (Lovig et al., 2002). Recently, somatic STR mutations within the cIITA and RFX5 genes have been reported in MSI-H CRCs, consistent with defective HLA Class II antigen presentation (Michel et al., 2010).
The potential relevance of STR targets within genes to tumorigenesis and cancer progression was recognized in early studies describing MSI-H tumors (Duval & Hamelin, 2002). Approximately 17% of human genes contain microsatellites within coding regions (Gemayel et al., 2010), and one report estimates that STRs are present in the exons of 92% of human genes (Madsen et al., 2008). Furthermore, STRs are over-represented in genes related to cancer and immune system diseases (Madsen et al., 2008). The current selective targets database (www.seltarbase.org) documents gene associated STRs that are mutated in cancers, and the large number of entries emphasizes the impact of this class of mutation to tumor development and immunological responses (Woerner et al., 2010). Intriguingly, intragenic microsatellite variation of S. cerevisiae genes can generate functional diversity of cell surface antigens (Verstrepen et al., 2005), and microsatellite length mutations underlie some forms of phase variation in bacterial species (Gemayel et al., 2010). Thus, mutations within repeated sequences also may be a process by which pathogens can elude the host immune system.
Summary
We have focused this review on links between DNA repair and inflammation as they relate to cancer. Although at this point in time, much remains to be elucidated, there are many links between DNA repair and inflammation. NF-κB, a master regulator of the immune response, and IRFs are upregulated in an ATM-dependent manner as part of the DNA damage response, as shown in Figure 3. DNA damage also upregulates NKG2D ligands, which can lead to death of severely damaged cells that reside in a chronic inflammatory environment. Activation of the inflammasome by DNA damage leads to downregulation of DNA repair and cell death, which could also be a protective mechanism in a chronic inflammatory environment. In some cases, as with the OGG1-deficient mouse model, the lack of DNA repair of 8-oxo-G results in tissues that are actually resistant to inflammation. Oxidized bases themselves have the capacity to potentiate immune responses. Specific miRNAs are upregulated under conditions of hypoxia and inflammation and some of these miRNAs downregulate DNA repair. Tumors with MSI and a high mutational load, as a result of deficient DNA repair, produce aberrant tumor-specific antigens and as a result are heavily infiltrated with lymphocytes, leading to a better prognosis.
Figure 3.

A model of the links between DNA damage and inflammation. Activation of the immune response occurs as a result of infection with pathogens, exposure to the microbiota of the gut and the secretory associated senescence phenotype (SASP). Acute inflammation can lead to chronic inflammation as described in this review, resulting in the generation of RONs and leading to DNA damage. Pathogens, genetic mutations in DNA repair genes and environment-associated expression of miRNAs can inactivate DNA repair, which results in a mutator phenotype, genomic instability and cancer. (see colour version of this figure at www.informahealthcare.com/bmg).
Activation of the immune inflammatory response to pathogens or microbiota and induction of the SASP in senescent cells leads to chronic inflammation, as depicted in Figure 2. It is known that inflammatory cells release RONs that result in DNA damage (Figure 1). An inability to regulate the inflammatory response or to repair the DNA, as a result of mutations in DNA repair or immune genes leads to cancer development. Overwhelming the cells with DNA damage, which might occur under conditions of chronic inflammation and deficient repair, as during infection with pathogens, can also lead to carcinogenesis. Thus, DNA repair and inflammation act together to maintain homeostasis.
Figure 2.
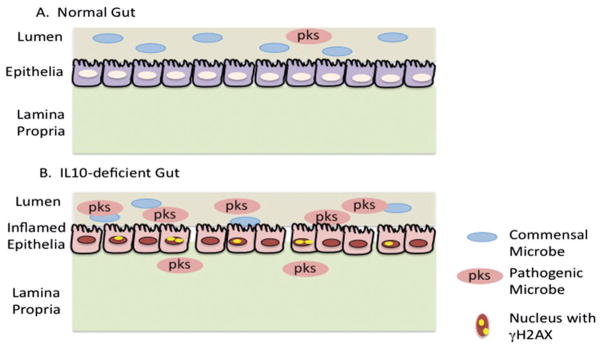
Interplay between microbiota, chronic inflammation and DNA damage in the gut. A. Healthy epithelia. Commensal microorganisms reside in the lumen, with very few pathogenic bacteria harboring the polyketide synthase (pks) pathogenicity island. B. Gut epithelia of an IL10-deficient mouse. The chronic inflammatory environment in the IL10-deficient mouse encourages enrichment of pathogenic E. coli that harbor the pks pathogenicity island. These pathogenic E. coli induce DNA damage in nearby epithelial cells that has the capacity to result in genomic instability and tumorigenesis. (see colour version of this figure at www.informahealthcare.com/bmg).
Acknowledgments
We thank Amrit Dhawan and Mary Jane Chisocki for their technical support.
Footnotes
References
- Adcock IM, Tsaprouni L, Bhavsar P, Ito K. Epigenetic regulation of airway inflammation. Curr Opin Immunol. 2007;19:694–700. doi: 10.1016/j.coi.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Front Immunol. 2013;4:190. doi: 10.3389/fimmu.2013.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn B, Ohshima H. Suppression of intestinal polyposis in Apc(Min/+) mice by inhibiting nitric oxide production. Cancer Res. 2001;61:8357–60. [PubMed] [Google Scholar]
- Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–32. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- Anastasov N, Höfig I, Vasconcellos IG, et al. Radiation resistance due to high expression of miR-21 and G2/M checkpoint arrest in breast cancer cells. Radiat Oncol. 2012;7:206. doi: 10.1186/1748-717X-7-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando T, Yoshida T, Enomoto S, et al. DNA methylation of microRNA genes in gastric mucosae of gastric cancer patients: its possible involvement in the formation of epigenetic field defect. Int J Cancer. 2009;124:2367–74. doi: 10.1002/ijc.24219. [DOI] [PubMed] [Google Scholar]
- Arthur JC, Perez-Chanona E, Muhlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asagoshi K, Odawara H, Nakano H, et al. Comparison of substrate specificities of Escherichia coli endonuclease III and its mouse homologue (mNTH1) using defined oligonucleotide substrates. Biochemistry. 2000a;39:11389–98. doi: 10.1021/bi000422l. [DOI] [PubMed] [Google Scholar]
- Asagoshi K, Yamada T, Okada Y, et al. Recognition of formamidopyrimidine by Escherichia coli and mammalian thymine glycol glycosylases. Distinctive paired base effects and biological and mechanistic implications. J Biol Chem. 2000b;275:24781–6. doi: 10.1074/jbc.M000576200. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Nishimura J, Shima T, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455:808–12. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- Babar IA, Czochor J, Steinmetz A, et al. Inhibition of hypoxia-induced miR-155 radiosensitizes hypoxic lung cancer cells. Cancer Biol Ther. 2011;12:908–14. doi: 10.4161/cbt.12.10.17681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacsi A, Aguilera-Aguirre L, Szczesny B, et al. Down-regulation of 8-oxoguanine DNA glycosylase 1 expression in the airway epithelium ameliorates allergic lung inflammation. DNA Repair (Amst) 2013;12:18–26. doi: 10.1016/j.dnarep.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeten D, Baraliakos X, Braun J, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382:1705–13. doi: 10.1016/S0140-6736(13)61134-4. [DOI] [PubMed] [Google Scholar]
- Baik SC, Youn HS, Chung MH, et al. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res. 1996;56:1279–82. [PubMed] [Google Scholar]
- Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature. 2004;431:405–6. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Mantovani A. Cancer and inflammation: implications for pharmacology and therapeutics. Clin Pharmacol Ther. 2010;87:401–6. doi: 10.1038/clpt.2009.312. [DOI] [PubMed] [Google Scholar]
- Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 2002;1:517–29. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–76. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- Bavik C, Coleman I, Dean JP, et al. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006;66:794–802. doi: 10.1158/0008-5472.CAN-05-1716. [DOI] [PubMed] [Google Scholar]
- Bazzoni F, Rossato M, Fabbri M, et al. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci USA. 2009;106:5282–7. doi: 10.1073/pnas.0810909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nature Immunol. 2011;12:715–23. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- Benachenhou N, Guiral S, Gorska-Flipot I, et al. Allelic losses and DNA methylation at DNA mismatch repair loci in sporadic colorectal cancer. Carcinogenesis. 1998;19:1925–9. doi: 10.1093/carcin/19.11.1925. [DOI] [PubMed] [Google Scholar]
- Berg DJ, Davidson N, Kuhn R, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–20. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Biarc J, Nguyen IS, Pini A, et al. Carcinogenic properties of proteins with pro-inflammatory activity from Streptococcus infantarius (formerly S. bovis) Carcinogenesis. 2004;25:1477–84. doi: 10.1093/carcin/bgh091. [DOI] [PubMed] [Google Scholar]
- Bicknell DC, Kaklamanis L, Hampson R, et al. Selection for beta2-microglobulin mutation in mismatch repair-defective colorectal carcinomas. Curr Biol. 1996;6:1695–7. doi: 10.1016/s0960-9822(02)70795-1. [DOI] [PubMed] [Google Scholar]
- Bierne H, Hamon M, Cossart P. Epigenetics and bacterial infections. Cold Spring Harb Perspect Med. 2012;2:a010272. doi: 10.1101/cshperspect.a010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S, Roy S, Banerjee J, et al. Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds. Proc Natl Acad Sci USA. 2010;107:6976–81. doi: 10.1073/pnas.1001653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjoras M, Luna L, Johnsen B, et al. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. Embo J. 1997;16:6314–22. doi: 10.1093/emboj/16.20.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatner NR, Mulcahy MF, Dennis KL, et al. Expression of RORgammat marks a pathogenic regulatory T cell subset in human colon cancer. Sci Translat Med. 2012;4:164ra159. doi: 10.1126/scitranslmed.3004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–16. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- Boldogh I, Hajas G, Aguilera-Aguirre, et al. Activation of ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. J Biol Chem. 2012;287:20769–73. doi: 10.1074/jbc.C112.364620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein S, White R, Malkoski S, et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest. 2009;119:3408–19. doi: 10.1172/JCI38854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvard V, Baan R, Straif K, et al. A review of human carcinogens – Part B: biological agents. Lancet Oncol. 2009;10:321–2. doi: 10.1016/s1470-2045(09)70096-8. [DOI] [PubMed] [Google Scholar]
- Bruning U, Cerone L, Neufeld Z, et al. MicroRNA-155 promotes resolution of hypoxia-inducible factor 1alpha activity during prolonged hypoxia. Mol Cell Biol. 2011;31:4087–96. doi: 10.1128/MCB.01276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckowitz A, Knaebel HP, Benner A, et al. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br J Cancer. 2005;92:1746–53. doi: 10.1038/sj.bjc.6602534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo JA, Meira LB, Lee CY, et al. DNA repair is indispensable for survival after acute inflammation. J Clin Invest. 2012;122:2680–9. doi: 10.1172/JCI63338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, D’adda Di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Camps C, Buffa FM, Colella S, et al. hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res. 2008;14:1340–8. doi: 10.1158/1078-0432.CCR-07-1755. [DOI] [PubMed] [Google Scholar]
- Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002;1:9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casorelli I, Pannellini T, De Luca G, et al. The Mutyh base excision repair gene influences the inflammatory response in a mouse model of ulcerative colitis. PLoS One. 2010;5:e12070. doi: 10.1371/journal.pone.0012070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae WJ, Bothwell AL. IL-17F deficiency inhibits small intestinal tumorigenesis in ApcMin/+ mice. Biochem Biophys Res Commun. 2011;414:31–6. doi: 10.1016/j.bbrc.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Chae WJ, Gibson TF, Zelterman D, et al. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci USA. 2010;107:5540–4. doi: 10.1073/pnas.0912675107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AO, Lam SK, Wong BC, et al. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut. 2003;52:502–6. doi: 10.1136/gut.52.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MK, Ocampo-Hafalla MT, Vartanian V, et al. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair (Amst) 2009;8:786–94. doi: 10.1016/j.dnarep.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Marra G, Chauhan DP, et al. Oxidative stress inactivates the human DNA mismatch repair system. Am J Physiol. 2002;283:C148–54. doi: 10.1152/ajpcell.00422.2001. [DOI] [PubMed] [Google Scholar]
- Chang S, Wang RH, Akagi K, et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011;17:1275–82. doi: 10.1038/nm.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow J, Lee SM, Shen Y, et al. Host-bacterial symbiosis in health and disease. Adv Immunol. 2010;107:243–74. doi: 10.1016/B978-0-12-381300-8.00008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clambey ET, McNamee EN, Westrich JA, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA. 2012;109:E2784–93. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–33. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Colotta F, Allavena P, Sica A, et al. Cancer-related inflammation, the seventh hallmark of cancer:links to genetic instability. Carcinogenesis. 2009;30:1073–81. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Bratti X, Frisan T, Thelestam M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon. 2001;39:1729–36. doi: 10.1016/s0041-0101(01)00159-3. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JE, Taylor JD, Wyatt JI, et al. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration, and gastric pathology. Lancet. 1991;338:332–5. doi: 10.1016/0140-6736(91)90477-7. [DOI] [PubMed] [Google Scholar]
- Crabtree JE, Xiang Z, Lindley IJ, et al. Induction of interleukin-8 secretion from gastric epithelial cells by a cagA negative isogenic mutant of Helicobacter pylori. J Clin Pathol. 1995;48:967–9. doi: 10.1136/jcp.48.10.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby M, Kulshreshtha R, Ivan M, Glazer P. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res. 2009;69:1221–9. doi: 10.1158/0008-5472.CAN-08-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. 2008;112:935–45. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- Darrasse-Jèze G, Podsypanina K. How numbers, nature, and immune status of Foxp3+ regulatory T-cells shape the early immunological events in tumor development. Front Immunol. 2013;4:292. doi: 10.3389/fimmu.2013.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau R. Infection, inflammation, and cancer. Nat Biotechnol. 1999;17:19. doi: 10.1038/5188. [DOI] [PubMed] [Google Scholar]
- De Bont R, Van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169–85. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- De La Chapelle A, Hampel H. Clinical relevance of microsatellite instability in colorectal cancer. J Clin Oncol. 2010;28:3380–7. doi: 10.1200/JCO.2009.27.0652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martel C, Ferlay J, Franceschi S, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–15. doi: 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- De Miranda NF, Hes FJ, Van Wezel T, Morreau H. Role of the microenvironment in the tumourigenesis of microsatellite unstable and MUTYH-associated polyposis colorectal cancers. Mutagenesis. 2012;27:247–53. doi: 10.1093/mutage/ger077. [DOI] [PubMed] [Google Scholar]
- De Miranda NF, Nielsen M, Pereira D, et al. MUTYH-associated polyposis carcinomas frequently lose HLA class I expression – a common event amongst DNA-repair-deficient colorectal cancers. J Pathol. 2009;219:69–76. doi: 10.1002/path.2569. [DOI] [PubMed] [Google Scholar]
- De Oliveira PE, Zhang L, Wang Z, Lazo JS. Hypoxia-mediated regulation of Cdc25A phosphatase by p21 and miR-21. Cell Cycle. 2009;8:3157–64. doi: 10.4161/cc.8.19.9704. [DOI] [PubMed] [Google Scholar]
- Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- Dennis KL, Wang Y, Blatner NR, et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res. 2013;73:5905–13. doi: 10.1158/0008-5472.CAN-13-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding SZ, Goldberg JB, Hatakeyama M. Helicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis. Future Oncol. 2010;6:851–62. doi: 10.2217/fon.10.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizdaroglu M, Burgess SM, Jaruga P, et al. Substrate specificity and excision kinetics of Escherichia coli endonuclease VIII (Nei) for modified bases in DNA damaged by free radicals. Biochemistry. 2001;40:12150–6. doi: 10.1021/bi015552o. [DOI] [PubMed] [Google Scholar]
- Dluzniewski PJ, Wang MH, Zheng SL, et al. Variation in IL10 and other genes involved in the immune response and in oxidation and prostate cancer recurrence. Cancer Epidemiol Biomarkers Prevention. 2012;21:1774–82. doi: 10.1158/1055-9965.EPI-12-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrovic A, Simpfendorfer D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997;57:3347–50. [PubMed] [Google Scholar]
- Dolcetti R, Viel A, Doglioni C, et al. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am J Pathol. 1999;154:1805–13. doi: 10.1016/S0002-9440(10)65436-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–84. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- Dou H, Theriot CA, Das A, et al. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J Biol Chem. 2008;283:3130–40. doi: 10.1074/jbc.M709186200. [DOI] [PubMed] [Google Scholar]
- Du W, Erden O, Pang Q. TNF-α signaling in Fanconi anemia. Blood Cells Mol Dis. 2014;52:2–11. doi: 10.1016/j.bcmd.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- Dupage M, Mazumdar C, Schmidt LM, et al. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–9. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002;62:2447–54. [PubMed] [Google Scholar]
- Edwards RA, Witherspoon M, Wang K, et al. Epigenetic repression of DNA mismatch repair by inflammation and hypoxia in inflammatory bowel disease-associated colorectal cancer. Cancer Res. 2009;69:6423–9. doi: 10.1158/0008-5472.CAN-09-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide L, Luna L, Gustad EC, et al. Human endonuclease III acts preferentially on DNA damage opposite guanine residues in DNA. Biochemistry. 2001;40:6653–9. doi: 10.1021/bi0028901. [DOI] [PubMed] [Google Scholar]
- Eis PS, Tam W, Sun L, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005;102:3627–32. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Omar EM. The importance of interleukin 1beta in Helicobacter pylori associated disease. Gut. 2001;48:743–7. doi: 10.1136/gut.48.6.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- Endo Y, Marusawa H, Kou T, et al. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology. 2008;135:889–98. 898, e1–3. doi: 10.1053/j.gastro.2008.06.091. [DOI] [PubMed] [Google Scholar]
- Erdman SE, Rao VP, Poutahidis T, et al. CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 2003;63:6042–50. [PubMed] [Google Scholar]
- Erdman SE, Rao VP, Poutahidis T, et al. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc Natl Acad Sci USA. 2009;106:1027–32. doi: 10.1073/pnas.0812347106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdman SE, Sohn JJ, Rao VP, et al. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res. 2005;65:3998–4004. doi: 10.1158/0008-5472.CAN-04-3104. [DOI] [PubMed] [Google Scholar]
- Ericson NG, Kulawiec M, Vermulst M, et al. Decreased mitochondrial DNA mutagenesis in human colorectal cancer. PLoS Genet. 2012;8:e1002689. doi: 10.1371/journal.pgen.1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falnes PO, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–82. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol. 2009;6:297–305. doi: 10.1038/nrgastro.2009.44. [DOI] [PubMed] [Google Scholar]
- Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- Federico A, Morgillo F, Tuccillo C, et al. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer. 2007;121:2381–6. doi: 10.1002/ijc.23192. [DOI] [PubMed] [Google Scholar]
- Feig DI, Reid TM, Loeb LA. Reactive oxygen species in tumorigenesis. Cancer Res. 1994;54:1890s–4s. [PubMed] [Google Scholar]
- Fenoglio C, Cantoni C, De Riz M, et al. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neurosci Lett. 2011;504:9–12. doi: 10.1016/j.neulet.2011.08.021. [DOI] [PubMed] [Google Scholar]
- Ferguson LR. Chronic inflammation and mutagenesis. Mutation Res. 2010;690:3–11. doi: 10.1016/j.mrfmmm.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Ferrero RL. Innate immune recognition of the extracellular mucosal pathogen, Helicobacter pylori. Mol Immunol. 2005;42:879–85. doi: 10.1016/j.molimm.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Festa F, Kumar R, Sanyal S, et al. Basal cell carcinoma and variants in genes coding for immune response, DNA repair, folate and iron metabolism. Mutation Res. 2005;574:105–11. doi: 10.1016/j.mrfmmm.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Fishel R, Kolodner RD. Identification of mismatch repair genes and their role in the development of cancer. Curr Opin Genet Dev. 1995;5:382–95. doi: 10.1016/0959-437x(95)80055-7. [DOI] [PubMed] [Google Scholar]
- Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–38. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1994;77:1. p following 166. [PubMed] [Google Scholar]
- Fitzpatrick FA. Inflammation, carcinogenesis and cancer. Int Immunopharmacol. 2001;1:1651–67. doi: 10.1016/s1567-5769(01)00102-3. [DOI] [PubMed] [Google Scholar]
- Fleisher AS, Esteller M, Harpaz N, et al. Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Res. 2000;60:4864–8. [PubMed] [Google Scholar]
- Foekens JA, Sieuwerts AM, Smid M, et al. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci USA. 2008;105:13021–6. doi: 10.1073/pnas.0803304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, Robertson CE, Hamm CM, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:179–84. doi: 10.1002/ibd.21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16:238–46. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisan T, Cortes-Bratti X, Thelestam M. Cytolethal distending toxins and activation of DNA damage-dependent checkpoint responses. Int J Med Microbiol. 2002;291:495–9. doi: 10.1078/1438-4221-00158. [DOI] [PubMed] [Google Scholar]
- Garchow BG, Bartulos Encinas O, Leung YT, et al. Silencing of microRNA-21 in vivo ameliorates autoimmune splenomegaly in lupus mice. EMBO Mol Med. 2011;3:605–15. doi: 10.1002/emmm.201100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Lord GM, Punit S, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Punit S, Gallini CA, et al. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell. 2009;16:208–19. doi: 10.1016/j.ccr.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini S, Marchi M, Calzetti F, et al. Gene expression and production of the monokine induced by IFN-gamma (MIG), IFN-inducible T cell alpha chemoattractant (I-TAC), and IFN-gamma-inducible protein-10 (IP-10) chemokines by human neutrophils. J Immunol. 1999;162:4928–37. [PubMed] [Google Scholar]
- Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–90. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser S, Raulet D. The DNA damage response, immunity and cancer. Semin Cancer Biol. 2006a;16:344–7. doi: 10.1016/j.semcancer.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Gasser S, Raulet DH. The DNA damage response arouses the immune system. Cancer Res. 2006b;66:3959–62. doi: 10.1158/0008-5472.CAN-05-4603. [DOI] [PubMed] [Google Scholar]
- Gehrke N, Mertens C, Zillinger T, et al. Oxidative Damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. 2013;39:482–95. doi: 10.1016/j.immuni.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet. 2010;44:445–77. doi: 10.1146/annurev-genet-072610-155046. [DOI] [PubMed] [Google Scholar]
- Girard PM, Guibourt N, Boiteux S. The Ogg1 protein of Saccharomyces cerevisiae: a 7,8-dihydro-8-oxoguanine DNA glycosylase/AP lyase whose lysine 241 is a critical residue for catalytic activity. Nucleic Acids Res. 1997;25:3204–11. doi: 10.1093/nar/25.16.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimcher LH. Trawling for treasure: tales of T-bet. Nat Immunol. 2007;8:448–50. doi: 10.1038/ni0507-448. [DOI] [PubMed] [Google Scholar]
- Gou Z, Liu R, Zhao G, et al. Epigenetic modification of TLRs in leukocytes is associated with increased susceptibility to Salmonella enteritidis in chickens. PLoS One. 2012;7:e33627. doi: 10.1371/journal.pone.0033627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gounaris E, Blatner NR, Dennis K, et al. T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer Res. 2009;69:5490–7. doi: 10.1158/0008-5472.CAN-09-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- Grosso S, Doyen J, Parks SK, et al. MiR-210 promotes a hypoxic phenotype and increases radioresistance in human lung cancer cell lines. Cell Death Dis. 2013;4:e544. doi: 10.1038/cddis.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H, Ji M, Hou P, et al. Hypermethylation of the DNA mismatch repair gene hMLH1 and its association with lymph node metastasis and T1799A BRAF mutation in patients with papillary thyroid cancer. Cancer. 2008;113:247–55. doi: 10.1002/cncr.23548. [DOI] [PubMed] [Google Scholar]
- Guidoboni M, Gafa R, Viel A, et al. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am J Pathol. 2001;159:297–304. doi: 10.1016/S0002-9440(10)61695-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799:775–87. doi: 10.1016/j.bbagrm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gushima M, Hirahashi M, Matsumoto T, et al. Expression of activation-induced cytidine deaminase in ulcerative colitis-associated carcinogenesis. Histopathology. 2011;59:460–9. doi: 10.1111/j.1365-2559.2011.03965.x. [DOI] [PubMed] [Google Scholar]
- Hall JA, Bouladoux N, Sun CM, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29:637–49. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. New Engl J Med. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Ulevitch RJ. Limiting inflammatory responses during activation of innate immunity. Nat Immunol. 2005;6:1198–205. doi: 10.1038/ni1274. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hazra TK, Izumi T, Boldogh I, et al. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci USA. 2002;99:3523–8. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde ML, Theriot CA, Das A, et al. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J Biol Chem. 2008;283:27028–37. doi: 10.1074/jbc.M802712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hile SE, Shabashev S, Eckert KA. Tumor-specific microsatellite instability: do distinct mechanisms underlie the MSI-L and EMAST phenotypes? Mutation Res. 2013;743–744:67–77. doi: 10.1016/j.mrfmmm.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hmadcha A, Bedoya FJ, Sobrino F, Pintado E. Methylation-dependent gene silencing induced by interleukin 1beta via nitric oxide production. J Exp Med. 1999;190:1595–604. doi: 10.1084/jem.190.11.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofseth LJ, Khan MA, Ambrose M, et al. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. J Clin Invest. 2003;112:1887–94. doi: 10.1172/JCI19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgaard RB, Zamarin D, Munn DH, et al. Indoleamine 23-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210:1389–402. doi: 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong L, Han Y, Zhang H, et al. miR-210: a therapeutic target in cancer. Expert Opin Ther Targets. 2013;17:21–8. doi: 10.1517/14728222.2012.732066. [DOI] [PubMed] [Google Scholar]
- Houghton J, Wang TC. Helicobacter pylori and gastric cancer: a new paradigm for inflammation-associated epithelial cancers. Gastroenterology. 2005;128:1567–78. doi: 10.1053/j.gastro.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Hu J, De Souza-Pinto NC, Haraguchi K, et al. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. J Biol Chem. 2005;280:40544–51. doi: 10.1074/jbc.M508772200. [DOI] [PubMed] [Google Scholar]
- Huang X, Ding L, Bennewith KL, et al. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell. 2009;35:856–67. doi: 10.1016/j.molcel.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FY, Chan AO, Lo RC, et al. Characterization of interleukin-1β in Helicobacter pylori-induced gastric inflammation and DNA methylation in interleukin-1 receptor type 1 knockout (IL-1R1(−/−)) mice. Eur J Cancer. 2013;49:2760–70. doi: 10.1016/j.ejca.2013.03.031. [DOI] [PubMed] [Google Scholar]
- Hur K, Niwa T, Toyoda T, et al. Insufficient role of cell proliferation in aberrant DNA methylation induction and involvement of specific types of inflammation. Carcinogenesis. 2011;32:35–41. doi: 10.1093/carcin/bgq219. [DOI] [PubMed] [Google Scholar]
- Hussain SP, Amstad P, Raja K, et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res. 2000;60:3333–7. [PubMed] [Google Scholar]
- Ikeda SBT, Roy R, Izumi T, et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J Biol Chem. 1998;273:1585–93. doi: 10.1074/jbc.273.34.21585. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–70. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- Ishigame H, Kakuta S, Nagai T, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–19. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Fujita T, Suzuki Y, et al. Tumor-specific immunological recognition of frameshift-mutated peptides in colon cancer with microsatellite instability. Cancer Res. 2003;63:5564–5572. [PubMed] [Google Scholar]
- Isomoto H, Mott JL, Kobayashi S, et al. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology. 2007;132:384–96. doi: 10.1053/j.gastro.2006.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–62. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- Jiang D, Hatahet Z, Blaisdell J, et al. Escherichia coli endonuclease VIII: cloning, sequencing, and overexpression of the nei structural gene and characterization of nei and nei nth mutants. J Bacteriol. 1997a;179:3773–82. doi: 10.1128/jb.179.11.3773-3782.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Hatahet Z, Melamede RJ, et al. Characterization of Escherichia coli endonuclease VIII. J Biol Chem. 1997b;272:32230–9. doi: 10.1074/jbc.272.51.32230. [DOI] [PubMed] [Google Scholar]
- Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–46. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- Jones S, Emmerson P, Maynard J, et al. Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C→T:A mutations. Hum Mol Genet. 2002;11:2961–7. doi: 10.1093/hmg/11.23.2961. [DOI] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joslyn G, Carlson M, Thliveris A, et al. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. 1991;66:601–13. doi: 10.1016/0092-8674(81)90022-2. [DOI] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–11. [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- Karim R, Tse G, Putti T, et al. The significance of the Wnt pathway in the pathology of human cancers. Pathology. 2004;36:120–8. doi: 10.1080/00313020410001671957. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Karl S, Pritschow Y, Volcic M, et al. Identification of a novel pro-apopotic function of NF–kappaB in the DNA damage response. J Cell Mol Med. 2009;13:4239–56. doi: 10.1111/j.1582-4934.2009.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Kosaka A, Yan H, et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-beta. Immunity. 2013;38:1187–97. doi: 10.1016/j.immuni.2013.02.024. [DOI] [PubMed] [Google Scholar]
- Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidane D, Sweasy JB. Tipping the balance in the powerhouse of the cell to “protect” colorectal cancer. PLoS Genet. 2012;8:e1002758. doi: 10.1371/journal.pgen.1002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G, Khanal P, Lim SC, et al. Interleukin-17 induces AP-1 activity and cellular transformation via upregulation of tumor progression locus 2 activity. Carcinogenesis. 2013;34:341–50. doi: 10.1093/carcin/bgs342. [DOI] [PubMed] [Google Scholar]
- Kim H. Cerulein pancreatitis: oxidative stress, inflammation, and apoptosis. Gut Liver. 2008;2:74–80. doi: 10.5009/gnl.2008.2.2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HW, Murakami A, Williams MV, Ohigashi H. Mutagenicity of reactive oxygen and nitrogen species as detected by co-culture of activated inflammatory leukocytes and AS52 cells. Carcinogenesis. 2003;24:235–41. doi: 10.1093/carcin/24.2.235. [DOI] [PubMed] [Google Scholar]
- Kim T, Kim TY, Song YH, et al. Activation of interferon regulatory factor 3 in response to DNA-damaging agents. J Biol Chem. 1999;274:30686–9. doi: 10.1074/jbc.274.43.30686. [DOI] [PubMed] [Google Scholar]
- Kim TK, Kim T, Kim TY, et al. Chemotherapeutic DNA-damaging drugs activate interferon regulatory factor-7 by the mitogen-activated protein kinase kinase-4-cJun NH2-terminal kinase pathway. Cancer Res. 2000;60:1153–6. [PubMed] [Google Scholar]
- Kim WJ, Vo QN, Shrivastav M, et al. Aberrant methylation of the ATM promoter correlates with increased radiosensitivity in a human colorectal tumor cell line. Oncogene. 2002;21:3864–71. doi: 10.1038/sj.onc.1205485. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–5. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- Kirchhoff S, Hauser H. Cooperative activity between HER oncogenes and the tumor suppressor IRF-1 results in apoptosis. Oncogene. 1999;18:3725–36. doi: 10.1038/sj.onc.1202704. [DOI] [PubMed] [Google Scholar]
- Kiyohara C, Hirohata T. Effects of vitamin C and PCB metabolites on the inhibition of aryl hydrocarbon hydroxylase and suppression of mutagenicity of benzo[a]pyrene. Toxicol In Vitro. 1994;8:1185–9. doi: 10.1016/0887-2333(94)90108-2. [DOI] [PubMed] [Google Scholar]
- Kiziltepe T, Yan A, Dong M, et al. Delineation of the chemical pathways underlying nitric oxide-induced homologous recombination in mammalian cells. Chem Biol. 2005;12:357–69. doi: 10.1016/j.chembiol.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Kloor M, Becker C, Benner A, et al. Immunoselective pressure and human leukocyte antigen class I antigen machinery defects in microsatellite unstable colorectal cancers. Cancer Res. 2005;65:6418–24. doi: 10.1158/0008-5472.CAN-05-0044. [DOI] [PubMed] [Google Scholar]
- Kloor M, Michel S, Von Knebel Doeberitz M. Immune evasion of microsatellite unstable colorectal cancers. Int J Cancer Journal International du Cancer. 2010;127:1001–10. doi: 10.1002/ijc.25283. [DOI] [PubMed] [Google Scholar]
- Klungland A, Rosewell I, Hollenbach S, et al. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA. 1999;96:13300–5. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson CG, Mangerich A, Zeng Y, et al. Chemical and cytokine features of innate immunity characterize serum and tissue profiles in inflammatory bowel disease. Proc Natl Acad Sci USA. 2013;110:E2332–41. doi: 10.1073/pnas.1222669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Kobayashi J, Saitoh T, et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci USA. 2013;110:2969–74. doi: 10.1073/pnas.1222694110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krichevsky AM, Gabriely G. miR-21: a small multi-faceted RNA. J Cell Mol Med. 2009;13:39–53. doi: 10.1111/j.1582-4934.2008.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy N, Zhao X, Burrows CJ, David SS. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 2008;47:7137–46. doi: 10.1021/bi800160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J Pathol. 2003a;201:28–36. doi: 10.1002/path.1409. [DOI] [PubMed] [Google Scholar]
- Kruidenier L, Kuiper I, Van Duijn W, et al. Imbalanced secondary mucosal antioxidant response in inflammatory bowel disease. J Pathol. 2003b;201:17–27. doi: 10.1002/path.1408. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai S, Jikimoto T, Saegusa J. Pathological roles of oxidative stress in autoimmune diseases. Rinsho Byori. 2003;51:126–32. [PubMed] [Google Scholar]
- Kumar S, Millis AJ, Baglioni C. Expression of interleukin 1-inducible genes and production of interleukin 1 by aging human fibroblasts. Proc Natl Acad Sci USA. 1992;89:4683–7. doi: 10.1073/pnas.89.10.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuniyasu H, Sasaki T, Sasahira T, et al. Repression of MLH1 and MGMT genes in colon mucosa adjacent to implanted cancer in athymic mouse. J Exp Clin Cancer Res. 2004;23:317–23. [PubMed] [Google Scholar]
- Lawes DA, Pearson T, Sengupta S, Boulos PB. The role of MLH1, MSH2 and MSH6 in the development of multiple colorectal cancers. Br J Cancer. 2005;93:472–7. doi: 10.1038/sj.bjc.6602708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem. 2007;76:447–80. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- Lee SM, Donaldson GP, Mikulski Z, et al. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature. 2013;501:426–9. doi: 10.1038/nature12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Yuan K, Yan C, et al. 8-Oxoguanine-DNA glycosylase 1 deficiency modifies allergic airway inflammation by regulating STAT6 and IL-4 in cells and in mice. Free Radic Biol Med. 2012;52:392–401. doi: 10.1016/j.freeradbiomed.2011.10.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licandro G, Ling Khor H, Beretta O, et al. The NLRP3 inflammasome affects DNA damage responses after oxidative and genotoxic stress in dendritic cells. Eur J Immunol. 2013;43:2126–37. doi: 10.1002/eji.201242918. [DOI] [PubMed] [Google Scholar]
- Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–83. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Linnebacher M, Gebert J, Rudy W, et al. Frameshift peptide-derived T-cell epitopes: a source of novel tumor-specific antigens. Int J Cancer. 2001;93:6–11. doi: 10.1002/ijc.1298. [DOI] [PubMed] [Google Scholar]
- Liu G, Friggeri A, Yang Y, et al. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci USA. 2009;106:15819–24. doi: 10.1073/pnas.0901216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZL, Wang H, Liu J, Wang ZX. MicroRNA-21 (miR-21) expression promotes growth, metastasis, and chemo- or radio-resistance in non-small cell lung cancer cells by targeting PTEN. Mol Cell Biochem. 2013;372:35–45. doi: 10.1007/s11010-012-1443-3. [DOI] [PubMed] [Google Scholar]
- Loeb LA. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat Rev Cancer. 2011;11:450–7. doi: 10.1038/nrc3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Serra P, Esteller M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene. 2012;31:1609–22. doi: 10.1038/onc.2011.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovig T, Andersen SN, Thorstensen L, et al. Strong HLA-DR expression in microsatellite stable carcinomas of the large bowel is associated with good prognosis. Br J Cancer. 2002;87:756–62. doi: 10.1038/sj.bjc.6600507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TX, Hartner J, Lim EJ, et al. MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-gamma pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J Immunol. 2011;187:3362–73. doi: 10.4049/jimmunol.1101235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TX, Munitz A, Rothenberg ME. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J Immunol. 2009;182:4994–5002. doi: 10.4049/jimmunol.0803560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A. Loss of T-bet sends host-microbe mutualism awry. Cell. 2007;131:15–17. doi: 10.1016/j.cell.2007.09.030. [DOI] [PubMed] [Google Scholar]
- Ma N, Kawanishi M, Hiraku Y, et al. Reactive nitrogen species-dependent DNA damage in EBV-associated nasopharyngeal carcinoma: the relation to STAT3 activation and EGFR expression. Int J Cancer. 2008;122:2517–25. doi: 10.1002/ijc.23415. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Pacher P, Bai P, et al. Suppression of intestinal polyposis in Apcmin/+ mice by targeting the nitric oxide or poly(ADP-ribose) pathways. Mutat Res. 2004;548:107–16. doi: 10.1016/j.mrfmmm.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Pacher P, Deb A, et al. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005;19:290–2. doi: 10.1096/fj.04-2278fje. [DOI] [PubMed] [Google Scholar]
- Machado AM, Figueiredo C, Seruca R, Rasmussen LJ. Helicobacter pylori infection generates genetic instability in gastric cells. Biochim Biophys Acta. 2010;1806:58–65. doi: 10.1016/j.bbcan.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Maddocks OD, Short AJ, Donnenberg MS, et al. Attaching and effacing Escherichia coli downregulate DNA mismatch repair protein in vitro and are associated with colorectal adenocarcinomas in humans. PLoS One. 2009;4:e5517. doi: 10.1371/journal.pone.0005517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen BE, Villesen P, Wiuf C. Short tandem repeats in human exons: a target for disease mutations. BMC Genomics. 2008;9:410. doi: 10.1186/1471-2164-9-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malzkorn B, Wolter M, Liesenberg F, et al. Identification and functional characterization of microRNAs involved in the malignant progression of gliomas. Brain Pathol. 2010;20:539–50. doi: 10.1111/j.1750-3639.2009.00328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangerich A, Knutson CG, Parry NM, et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc Natl Acad Sci USA. 2012;109:E1820–9. doi: 10.1073/pnas.1207829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Marenstein DR, Chan MK, Altamirano A, et al. Substrate specificity of human endonuclease III (hNTH1). Effect of human APE1 on hNTH1 activity. J Biol Chem. 2003;278:9005–12. doi: 10.1074/jbc.M212168200. [DOI] [PubMed] [Google Scholar]
- Marenstein DR, Ocampo MT, Chan MK, et al. Stimulation of human endonuclease III by Y box-binding protein 1 (DNA-binding protein B). Interaction between a base excision repair enzyme and a transcription factor. J Biol Chem. 2001;276:21242–9. doi: 10.1074/jbc.M101594200. [DOI] [PubMed] [Google Scholar]
- Matsuyama T, Kimura T, Kitagawa M, et al. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell. 1993;75:83–97. [PubMed] [Google Scholar]
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- Maynard CL, Weaver CT. Diversity in the contribution of interleukin-10 to T-cell-mediated immune regulation. Immunol Rev. 2008;226:219–33. doi: 10.1111/j.1600-065X.2008.00711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool KW, Miyamoto S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol Rev. 2012;246:311–26. doi: 10.1111/j.1600-065X.2012.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy CE, Sheedy FJ, Qualls JE, et al. IL-10 inhibits miR-155 induction by toll-like receptors. J Biol Chem. 2010;285:20492–8. doi: 10.1074/jbc.M110.102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue MM, Rieger RA, Rosenquist TA, et al. Stereoselective excision of thymine glycol lesions by mammalian cell extracts. DNA Repair (Amst) 2004;3:313–22. doi: 10.1016/j.dnarep.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Meira LB, Bugni JM, Green SL, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–25. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamede RJ, Hatahet Z, Kow YW, et al. Isolation and characterization of endonuclease VIII from Escherichia coli. Biochemistry. 1994;33:1255–64. doi: 10.1021/bi00171a028. [DOI] [PubMed] [Google Scholar]
- Menetrier-Caux C, Curiel T, Faget J, et al. Targeting regulatory T cells. Target Oncol. 2012;7:15–28. doi: 10.1007/s11523-012-0208-y. [DOI] [PubMed] [Google Scholar]
- Meng F, Henson R, Lang M, et al. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–29. doi: 10.1053/j.gastro.2006.02.057. [DOI] [PubMed] [Google Scholar]
- Michel S, Linnebacher M, Alcaniz J, et al. Lack of HLA class II antigen expression in microsatellite unstable colorectal carcinomas is caused by mutations in HLA class II regulatory genes. Int J Cancer Journal International du Cancer. 2010;127:889–98. doi: 10.1002/ijc.25106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller H, Fernandes AS, Zaika E, et al. Stereoselective excision of thymine glycol from oxidatively damaged DNA. Nucl Acids Res. 2004;32:338–45. doi: 10.1093/nar/gkh190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa O, Arai T, Hirano M, et al. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc Natl Acad Sci USA. 2000;97:4156–61. doi: 10.1073/pnas.050404497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaee V, Molaei M, Shalmani HM, Zali MR. Helicobacter pylori infection and expression of DNA mismatch repair proteins. World J Gastroenterol. 2008;14:6717–21. doi: 10.3748/wjg.14.6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina Y, Yang CG, He C. Direct repair of the exocyclic DNA adduct 1,N6-ethenoadenine by the DNA repair AlkB proteins. J Am Chem Soc. 2005;127:14594–5. doi: 10.1021/ja055957m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabe I, Zhang QM, Kino K, et al. Identification of 5-formyluracil DNA glycosylase activity of human hNTH1 protein. Nucl Acids Res. 2002;30:3443–8. doi: 10.1093/nar/gkf460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monjazeb AM, Zamora AE, Grossenbacher SK, et al. Immunoediting and antigen loss: overcoming the achilles heel of immunotherapy with antigen non-specific therapies. Front Oncol. 2013;3:197. doi: 10.3389/fonc.2013.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutamba JT, Svilar D, Prasongtanakij, et al. XRCC1 and base excision repair balance in response to nitric oxide. DNA Repair (Amst) 2011;10:1282–93. doi: 10.1016/j.dnarep.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Yu H, Nishikawa M, et al. Helicobacter pylori generates superoxide radicals and modulates nitric oxide metabolism. J Biol Chem. 1998;273:14071–3. doi: 10.1074/jbc.273.23.14071. [DOI] [PubMed] [Google Scholar]
- Nair J, Gal A, Tamir S, et al. Etheno adducts in spleen DNA of SJL mice stimulated to overproduce nitric oxide. Carcinogenesis. 1998;19:2081–4. doi: 10.1093/carcin/19.12.2081. [DOI] [PubMed] [Google Scholar]
- Nair VS, Maeda LS, Ioannidis JP. Clinical outcome prediction by microRNAs in human cancer: a systematic review. J Natl Cancer Inst. 2012;104:528–40. doi: 10.1093/jnci/djs027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13:349–61. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, De Miranda NF, Van Puijenbroek M, et al. Colorectal carcinomas in MUTYH-associated polyposis display histopathological similarities to microsatellite unstable carcinomas. BMC Cancer. 2009;9:184. doi: 10.1186/1471-2407-9-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishigori C, Yarosh DB, Ullrich SE, et al. Evidence that DNA damage triggers interleukin 10 cytokine production in UV-irradiated murine keratinocytes. Proc Natl Acad Sci USA. 1996;93:10354–9. doi: 10.1073/pnas.93.19.10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa T, Tsukamoto T, Toyoda T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010;70:1430–40. doi: 10.1158/0008-5472.CAN-09-2755. [DOI] [PubMed] [Google Scholar]
- Nossa CW, Jain P, Tamilselvam B, et al. Activation of the abundant nuclear factor poly(ADP-ribose) polymerase-1 by Helicobacter pylori. Proc Natl Acad Sci USA. 2009;106:19998–20003. doi: 10.1073/pnas.0906753106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nougayrede JP, Homburg S, Taieb F, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–51. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci USA. 2009;106:7113–18. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo MT, Chaung W, Marenstein DR, et al. Targeted deletion of mNth1 reveals a novel DNA repair enzyme activity. Mol Cell Biol. 2002;22:6111–21. doi: 10.1128/MCB.22.17.6111-6121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo T, Nishioka K, Imaiso Y, et al. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucl Acids Res. 2000;28:1355–64. doi: 10.1093/nar/28.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou J, Niessen RC, Lutzen A, et al. Functional analysis helps to clarify the clinical importance of unclassified variants in DNA mismatch repair genes. Hum Mutat. 2007;28:1047–54. doi: 10.1002/humu.20580. [DOI] [PubMed] [Google Scholar]
- Pamment J, Ramsay E, Kelleher M, et al. Regulation of the IRF-1 tumour modifier during the response to genotoxic stress involves an ATM-dependent signalling pathway. Oncogene. 2002;21:7776–85. doi: 10.1038/sj.onc.1205981. [DOI] [PubMed] [Google Scholar]
- Pan W, Zhu S, Yuan M, et al. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. 2010;184:6773–81. doi: 10.4049/jimmunol.0904060. [DOI] [PubMed] [Google Scholar]
- Papaconstantinou IG, Manta A, Gazouli M, et al. Expression of microRNAs in patients with pancreatic cancer and its prognostic significance. Pancreas. 2013;42:67–71. doi: 10.1097/MPA.0b013e3182592ba7. [DOI] [PubMed] [Google Scholar]
- Peek RM., Jr IV. Helicobacter pylori strain-specific activation of signal transduction cascades related to gastric inflammation. Am J Physiol Gastrointest Liver Physiol. 2001;280:G525–30. doi: 10.1152/ajpgi.2001.280.4.G525. [DOI] [PubMed] [Google Scholar]
- Peek RM, Jr, Miller GG, Tham KT, et al. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab Invest. 1995;73:760–70. [PubMed] [Google Scholar]
- Pena-Diaz J, Jiricny J. Mammalian mismatch repair: error-free or error-prone? Trends Biochem Sci. 2012;37:206–14. doi: 10.1016/j.tibs.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Phillips SM, Banerjea A, Feakins R, et al. Tumour-infiltrating lymphocytes in colorectal cancer with microsatellite instability are activated and cytotoxic. Br J Surg. 2004;91:469–75. doi: 10.1002/bjs.4472. [DOI] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–18. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- Pribluda A, Elyada E, Wiener, et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell. 2013;24:242–56. doi: 10.1016/j.ccr.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Prost S, Bellamy CO, Cunningham DS, Harrison DJ. Altered DNA repair and dysregulation of p53 in IRF-1 null hepatocytes. FASEB J. 1998;12:181–8. doi: 10.1096/fasebj.12.2.181. [DOI] [PubMed] [Google Scholar]
- Purmal AA, Lampman GW, Bond JP, et al. Enzymatic processing of uracil glycol, a major oxidative product of DNA cytosine. J Biol Chem. 1998;273:10026–35. doi: 10.1074/jbc.273.16.10026. [DOI] [PubMed] [Google Scholar]
- Qi J, Qiao Y, Wang P, et al. microRNA-210 negatively regulates LPS-induced production of proinflammatory cytokines by targeting NF-kappaB1 in murine macrophages. FEBS Lett. 2012;586:1201–7. doi: 10.1016/j.febslet.2012.03.011. [DOI] [PubMed] [Google Scholar]
- Rad R, Dossumbekova A, Neu B, et al. Cytokine gene polymorphisms influence mucosal cytokine expression, gastric inflammation, and host specific colonisation during Helicobacter pylori infection. Gut. 2004;53:1082–9. doi: 10.1136/gut.2003.029736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz AG, Xie Y, Kundu S, et al. Cancer-associated variants and a common polymorphism of MUTYH exhibit reduced repair of oxidative DNA damage using a GFP-based assay in mammalian cells. Carcinogenesis. 2012;33:2301–9. doi: 10.1093/carcin/bgs270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity. 2006;25:319–29. doi: 10.1016/j.immuni.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–7. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9:57–63. doi: 10.1038/nrc2541. [DOI] [PubMed] [Google Scholar]
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–16. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringvoll J, Moen MN, Nordstrand LM, et al. AlkB homologue 2-mediated repair of ethenoadenine lesions in mammalian DNA. Cancer Res. 2008;68:4142–9. doi: 10.1158/0008-5472.CAN-08-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risques RA, Lai LA, Himmetoglu C, et al. Ulcerative colitis-associated colorectal cancer arises in a field of short telomeres, senescence, and inflammation. Cancer Res. 2011;71:1669–79. doi: 10.1158/0008-5472.CAN-10-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CE, Kottapalli V, D’astice M, et al. Regulation of neutrophils in ulcerative colitis by colonic factors: a possible mechanism of neutrophil activation and tissue damage. J Lab Clin Med. 1997;130:590–602. doi: 10.1016/s0022-2143(97)90109-8. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers AB, Fox JG. Inflammation and Cancer. I. Rodent models of infectious gastrointestinal and liver cancer. Am J Physiol Gastrointest Liver Physiol. 2004;286:G361–6. doi: 10.1152/ajpgi.00499.2003. [DOI] [PubMed] [Google Scholar]
- Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332:237–48. doi: 10.1016/j.canlet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nature reviews Immunology. 2009;9:313–23. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Q, Wang T, Kameswaran V, et al. The microRNA-21-PDCD4 axis prevents type 1 diabetes by blocking pancreatic beta cell death. Proc Natl Acad Sci USA. 2011;108:12030–5. doi: 10.1073/pnas.1101450108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo MT, De Luca G, Degan P, et al. Accumulation of the oxidative base lesion 8-hydroxyguanine in DNA of tumor-prone mice defective in both the Myh and Ogg1 DNA glycosylases. Cancer Res. 2004;64:4411–14. doi: 10.1158/0008-5472.CAN-04-0355. [DOI] [PubMed] [Google Scholar]
- Saeterdal I, Bjorheim J, Lislerud K, et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc Natl Acad Sci USA. 2001;98:13255–60. doi: 10.1073/pnas.231326898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saigo K, Yoshida K, Ikeda R, et al. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma. Hum Mutat. 2008;29:703–8. doi: 10.1002/humu.20701. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Tominaga Y, Yamauchi K, et al. MUTYH-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. 2007;67:6599–604. doi: 10.1158/0008-5472.CAN-06-4802. [DOI] [PubMed] [Google Scholar]
- Salk JJ, Salipante SJ, Risques RA, et al. Clonal expansions in ulcerative colitis identify patients with neoplasia. Proc Natl Acad Sci USA. 2009;106:20871–6. doi: 10.1073/pnas.0909428106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39–41. doi: 10.1016/S0140-6736(03)13805-6. [DOI] [PubMed] [Google Scholar]
- Sarker AH, Ikeda S, Nakano H, et al. Cloning and characterization of a mouse homologue (mNthl1) of Escherichia coli endonuclease III. J Mol Biol. 1998;282:761–74. doi: 10.1006/jmbi.1998.2042. [DOI] [PubMed] [Google Scholar]
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Sears CL, Pardoll DM. Perspective: alpha-bugs, their microbial partners, and the link to colon cancer. J Infect Dis. 2011;203:306–11. doi: 10.1093/jinfdis/jiq061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sejas DP, Rani R, Qiu Y, et al. Inflammatory reactive oxygen species-mediated hemopoietic suppression in Fancc-deficient mice. J Immunol. 2007;178:5277–87. doi: 10.4049/jimmunol.178.8.5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellon RK, Tonkonogy S, Schultz M, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–31. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seruca R, Suijkerbuijk RF, Gartner F, et al. Increasing levels of MYC and MET co-amplification during tumor progression of a case of gastric cancer. Cancer Genet Cytogenet. 1995;82:140–5. doi: 10.1016/0165-4608(95)00033-l. [DOI] [PubMed] [Google Scholar]
- Shaked H, Hofseth LJ, Chumanevich A, et al. Chronic epithelial NF-kappaB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc Natl Acad Sci USA. 2012;109:14007–12. doi: 10.1073/pnas.1211509109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- Sheedy FJ, Palsson-Mcdermott E, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nature Immunol. 2010;11:141–7. doi: 10.1038/ni.1828. [DOI] [PubMed] [Google Scholar]
- Shibuya H, Iinuma H, Shimada R, et al. Clinicopathological and prognostic value of microRNA-21 and microRNA-155 in colorectal cancer. Oncology. 2010;79:313–20. doi: 10.1159/000323283. [DOI] [PubMed] [Google Scholar]
- Siddle HV, Kreiss A, Tovar C, et al. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc Natl Acad Sci USA. 2013;110:5103–8. doi: 10.1073/pnas.1219920110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791–9. doi: 10.1056/NEJMoa025283. [DOI] [PubMed] [Google Scholar]
- Sims GP, Rowe DC, Rietdijk ST, et al. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–88. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- Singer II, Kawka DW, Scott S, et al. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology. 1996;111:871–85. doi: 10.1016/s0016-5085(96)70055-0. [DOI] [PubMed] [Google Scholar]
- Sitkovsky MV. T regulatory cells: hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol. 2009;30:102–8. doi: 10.1016/j.it.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–73. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JJ, Schetter AJ, Yfantis HG, et al. Macrophages, nitric oxide and microRNAs are associated with DNA damage response pathway and senescence in inflammatory bowel disease. PLoS One. 2012;7:e44156. doi: 10.1371/journal.pone.0044156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinle A, Li P, Morris DL, et al. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics. 2001;53:279–87. doi: 10.1007/s002510100325. [DOI] [PubMed] [Google Scholar]
- Stevens AM, Yu-Lee LY. The transcription factor interferon regulatory factor-1 is expressed during both early G1 and the G1/S transition in the prolactin-induced lymphocyte cell cycle. Mol Endocrinol. 1992;6:2236–43. doi: 10.1210/mend.6.12.1491701. [DOI] [PubMed] [Google Scholar]
- Stevens AM, Yu-Lee LY. Multiple prolactin-responsive elements mediate G1 and S phase expression of the interferon regulatory factor-1 gene. Mol Endocrinol. 1994;8:345–55. doi: 10.1210/mend.8.3.8015552. [DOI] [PubMed] [Google Scholar]
- Stilmann M, Hinz M, Arslan SC, et al. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell. 2009;36:365–78. doi: 10.1016/j.molcel.2009.09.032. [DOI] [PubMed] [Google Scholar]
- Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–86. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- Svilar D, Goellner EM, Almeida KH, Sobol RW. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid Redox Signal. 2011;14:2491–507. doi: 10.1089/ars.2010.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svrcek M, Cosnes J, Beaugerie L, et al. Colorectal neoplasia in Crohn’s colitis: a retrospective comparative study with ulcerative colitis. Histopathology. 2007;50:574–83. doi: 10.1111/j.1365-2559.2007.02663.x. [DOI] [PubMed] [Google Scholar]
- Swann JB, Vesely MD, Silva A, et al. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci USA. 2008;105:652–6. doi: 10.1073/pnas.0708594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha R, Seidman E, Mailhot G, et al. Oxidative stress and mitochondrial functions in the intestinal Caco-2/15 cell line. PLoS One. 2010;5:e11817. doi: 10.1371/journal.pone.0011817. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Tahara T, Inoue N, Hisamatsu T, et al. Clinical significance of microsatellite instability in the inflamed mucosa for the prediction of colonic neoplasms in patients with ulcerative colitis. J Gastroenterol Hepatol. 2005;20:710–15. doi: 10.1111/j.1440-1746.2005.03803.x. [DOI] [PubMed] [Google Scholar]
- Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Akira S. Toll receptors and pathogen resistance. Cell Microbiol. 2003;5:143–53. doi: 10.1046/j.1462-5822.2003.00264.x. [DOI] [PubMed] [Google Scholar]
- Tamura T, Ishihara M, Lamphier MS, et al. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature. 1995;376:596–9. doi: 10.1038/376596a0. [DOI] [PubMed] [Google Scholar]
- Tardieu D, Jaeg JP, Deloly A, et al. The COX-2 inhibitor nimesulide suppresses superoxide and 8-hydroxy-deoxyguanosine formation, and stimulates apoptosis in mucosa during early colonic inflammation in rats. Carcinogenesis. 2000;21:973–6. doi: 10.1093/carcin/21.5.973. [DOI] [PubMed] [Google Scholar]
- Taylor CT, Cummins EP. The role of NF-kappaB in hypoxia-induced gene expression. Ann N Y Acad Sci. 2009;1177:178–84. doi: 10.1111/j.1749-6632.2009.05024.x. [DOI] [PubMed] [Google Scholar]
- Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–14. e5. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- Thomas D, Scot AD, Barbey R, et al. Inactivation of OGG1 increases the incidence of G. C→T. A transversions in Saccharomyces cerevisiae: evidence for endogenous oxidative damage to DNA in eukaryotic cells. Mol Gen Genet. 1997;254:171–8. doi: 10.1007/s004380050405. [DOI] [PubMed] [Google Scholar]
- Thorsteinsdottir S, Gudjonsson T, Nielsen OH, et al. Pathogenesis and biomarkers of carcinogenesis in ulcerative colitis. Nat Rev Gastroenterol Hepatol. 2011;8:395–404. doi: 10.1038/nrgastro.2011.96. [DOI] [PubMed] [Google Scholar]
- Tili E, Michaille JJ, Wernicke D, et al. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc Natl Acad Sci USA. 2011;108:4908–13. doi: 10.1073/pnas.1101795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touati E. When bacteria become mutagenic and carcinogenic: lessons from H. pylori. Mutat Res. 2010;703:66–70. doi: 10.1016/j.mrgentox.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Trabulus S, Guven GS, Altiparmak MR, et al. DNA repair XRCC1 Arg399Gln polymorphism is associated with the risk of development of end-stage renal disease. Mol Biol Rep. 2012;39:6995–7001. doi: 10.1007/s11033-012-1529-8. [DOI] [PubMed] [Google Scholar]
- Trewick SC, Henshaw TF, Hausinger RP, et al. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–8. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–9. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–16. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- Uno K, Kato K, Atsumi T, et al. Toll-like receptor (TLR) 2 induced through TLR4 signaling initiated by Helicobacter pylori cooperatively amplifies iNOS induction in gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1004–12. doi: 10.1152/ajpgi.00096.2007. [DOI] [PubMed] [Google Scholar]
- Uronis JM, Muhlbauer M, Herfarth HH, et al. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4:e6026. doi: 10.1371/journal.pone.0006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeri N, Gasparini P, Fabbri M, et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci USA. 2010;107:6982–7. doi: 10.1073/pnas.1002472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderwerf SM, Svahn J, Olson S, et al. TLR8-dependent TNF-(alpha) overexpression in Fanconi anemia group C cells. Blood. 2009;114:5290–8. doi: 10.1182/blood-2009-05-222414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian V, Lowell B, Minko IG, et al. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc Natl Acad Sci USA. 2006;103:1864–9. doi: 10.1073/pnas.0507444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstrepen KJ, Jansen A, Lewitter F, Fink GR. Intragenic tandem repeats generate functional variability. Nature Genet. 2005;37:986–90. doi: 10.1038/ng1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virchow R. Cellular pathology as based upon physiological and pathological history. Philadelphia: J.B. Lippincott; 1863. [DOI] [PubMed] [Google Scholar]
- Volcic M, Karl S, Baumann B, et al. NF-kappaB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucl Acids Res. 2012;40:181–95. doi: 10.1093/nar/gkr687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SS, Bandaru V, Kathe SD, Bond JP. The enigma of endonuclease VIII. DNA Repair (Amst) 2003;2:441–53. doi: 10.1016/s1568-7864(02)00182-9. [DOI] [PubMed] [Google Scholar]
- Wallace SS, Murphy DL, Sweasy JB. Base excision repair and cancer. Cancer Lett. 2012;327:73–89. doi: 10.1016/j.canlet.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Taniguchi T. MicroRNAs and DNA damage response: implications for cancer therapy. Cell Cycle. 2013;12:32–42. doi: 10.4161/cc.23051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren JR. Unidentifed cruved bacilli on gastric epithelium in active chronic gastritis. The Lancet. 1983;321:1273. [PubMed] [Google Scholar]
- Wei J, Noto J, Zaika E, et al. Pathogenic bacterium Helicobacter pylori alters the expression profile of p53 protein isoforms and p53 response to cellular stresses. Proc Natl Acad Sci USA. 2012;109:E2543–50. doi: 10.1073/pnas.1205664109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JD, Marnett LJ. Endogenous reactive intermediates as modulators of cell signaling and cell death. Chem Res Toxicol. 2006;19:173–94. doi: 10.1021/tx050321u. [DOI] [PubMed] [Google Scholar]
- Willenbucher RF, Aust DE, Chang CG, et al. Genomic instability is an early event during the progression pathway of ulcerative-colitis-related neoplasia. Am J Pathol. 1999;154:1825–30. doi: 10.1016/S0002-9440(10)65438-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerner SM, Yuan YP, Benner A, et al. SelTarbase, a database of human mononucleotide-microsatellite mutations and their potential impact to tumorigenesis and immunology. Nucl Acids Res. 2010;38:D682–9. doi: 10.1093/nar/gkp839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewski LE, Peek RM, Jr, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23:713–39. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Rhee KJ, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nature Med. 2009;15:1016–22. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zeng Z, Chen B, et al. Association between polymorphisms in interleukin-17A and interleukin-17F genes and risks of gastric cancer. Int Journal Cancer. 2010;127:86–92. doi: 10.1002/ijc.25027. [DOI] [PubMed] [Google Scholar]
- Xu Z, Pone EJ, Al-Qahtani A, et al. Regulation of aicda expression and AID activity: relevance to somatic hypermutation and class switch DNA recombination. Crit Rev Immunol. 2007;27:367–97. doi: 10.1615/critrevimmunol.v27.i4.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Fujiyama Y, Andoh A, et al. Oxidative stress increases MICA and MICB gene expression in the human colon carcinoma cell line (CaCo-2) Biochim Biophys Acta. 2001;1526:10–12. doi: 10.1016/s0304-4165(01)00099-x. [DOI] [PubMed] [Google Scholar]
- Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–98. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Yermilov V, Rubio J, Ohshima H. Formation of 8-nitroguanine in DNA treated with peroxynitrite in vitro and its rapid removal from DNA by depurination. FEBS Lett. 1995;376:207–10. doi: 10.1016/0014-5793(95)01281-6. [DOI] [PubMed] [Google Scholar]
- Yermilov V, Yoshie Y, Rubio J, Ohshima H. Effects of carbon dioxide/bicarbonate on induction of DNA single-strand breaks and formation of 8-nitroguanine, 8-oxoguanine and base-propenal mediated by peroxynitrite. FEBS Lett. 1996;399:67–70. doi: 10.1016/s0014-5793(96)01288-4. [DOI] [PubMed] [Google Scholar]
- Zabaleta J. Multifactorial etiology of gastric cancer. Methods Mol Biol. 2012;863:411–35. doi: 10.1007/978-1-61779-612-8_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabaleta J, Camargo MC, Piazuelo MB, et al. Association of interleukin-1beta gene polymorphisms with precancerous gastric lesions in African Americans and Caucasians. Am J Gastroenterol. 2006;101:163–71. doi: 10.1111/j.1572-0241.2006.00387.x. [DOI] [PubMed] [Google Scholar]
- Zeineldin M, Neufeld KL. Understanding phenotypic variation in rodent models with germline Apc mutations. Cancer Res. 2013;73:2389–99. doi: 10.1158/0008-5472.CAN-12-4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QM, Miyabe I, Matsumoto Y, et al. Identification of repair enzymes for 5-formyluracil in DNA. Nth, Nei, and MutM proteins of Escherichia coli. J Biol Chem. 2000;275:35471–7. doi: 10.1074/jbc.M006125200. [DOI] [PubMed] [Google Scholar]
- Zhang QM, Yonekura S, Takao M, et al. DNA glycosylase activities for thymine residues oxidized in the methyl group are functions of the hNEIL1 and hNTH1 enzymes in human cells. DNA Repair (Amst) 2005;4:71–9. doi: 10.1016/j.dnarep.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Zhao S, Choi M, Overton JD, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci USA. 2013;110:2916–21. doi: 10.1073/pnas.1222577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8:457–62. doi: 10.1038/ni1455. [DOI] [PubMed] [Google Scholar]
- Zielinski CC, Budinsky AC, Wagner TM, et al. Defect of tumour necrosis factor-alpha (TNF-alpha) production and TNF-alpha-induced ICAM-1-expression in BRCA1 mutations carriers. Breast Cancer Res Treat. 2003;81:99–105. doi: 10.1023/A:1025761716283. [DOI] [PubMed] [Google Scholar]