

Abstract
The preparation of a crystalline, peracetyl adamantanyl thiosialoside donor protected by an isothiocyanate group is described. On activation at -78 C in the presence of typical carbohydrate acceptors this donor gives high yields of the corresponding sialosides with exquisite α-selectivity. The high selectivity extends to the 4-O-benzyl-protected 3-OH acceptors that are typically less reactive and selective than galactose 3,4-diols. Treatment of the α-sialosides with tris(trimethylsilyl)silane or allyltris(trimethylsilyl)silane sialosides replaces the C5-N5 bond by a C-H or a C-C bond. Reaction of the isothiocyanate-protected sialosides with thioacids achieves conversion into amides. Reaction of the isothiocyanate with an amine gives a thiourea, which can be converted to a guanidine. The very high α-selectivities observed with the new donor and the rich chemistry of the isothiocyante function considerably extend the scope for optimization at the sialoside 5-position.
Keywords: glycosylation, isothiocyanate, guanidine, thiourea, deamination
In recent years major steps have been taken toward the establishment of high-yielding and highly α-selective chemical sialidation reactions.[1] For the most part advances have centered around modification of the N-5 protecting group,[2] culminating in the discovery of the 4-O-,5-N-oxazolidine systems[3] and their N-acetyl variants,[3c, 4] which afford excellent yields and selectivities. Nevertheless, the potential applications of sialic acid glycosides and their oligomers in medicine,[5] and the consequent need for larger scale synthesis, drive the continued search for improved methods.
The N-5 position also plays a prominent role in the development of sialic acid glycosides with improved properties for application as therapeutic agents and/or vaccines.[6] Such N-5 modified systems are either produced chemoenzymatically,[6b, 7] or chemically by removal of the N-5 protecting group post-glycosylation followed by derivatization.[6a, 6c, 8] We now reveal that protection of N-5 in the form of an isothiocyanate provides a sialyl donor that is not only exquisitely α-selective in its coupling reactions but which also, by taking advantage of the versatile chemistry[9] of the isothiocyanate group, affords facile access to an unprecedented range of functionality in the so-formed glycosides. The isothiocyanate 2, previously obtained in low yield as a by-product in the synthesis of an N-acetyl-4-O,5-N-oxazolidinthione-proected sialyl donor,[10] was procured in 59% yield by treatment of the β-S-adamantanyl thiosialoside 1[4b, 10] with HCl in ether, followed by phenyl chlorothionoformate and aqueous sodium bicarbonate, and finally acetic anhydride in pyridine (Scheme 1). Donor 2 is a stable, readily handled white crystalline solid.
Scheme 1.
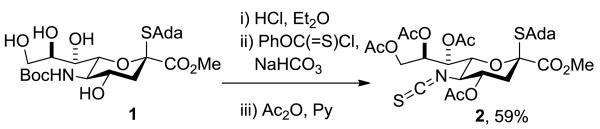
Synthesis of the Isothiocyanate 2. Abbreviations: Ada = 1-adamantanyl, Boc = tert-butyloxycarbonyl, Py = pyridine
Activation of donor 2 at -78 °C in 2/1 dichloromethane and acetonitrile in the presence of 1.2 equivs of various acceptors (Scheme 2) afforded the corresponding glycosides 3-7 (Table 1). The anomeric configuration of the products was assigned on the basis of the heteronuclear 3JC1,H3ax coupling constant, which followed the typical pattern.[11] An authentic sample of the β-anomer of 5 was obtained in 20% yield by coupling 2 with methyl 2,4,6-tri-O-benzyl-α-d-galactopyranoside 10 in pure dichloromethane at -30 °C (Figure 1), and enabled confirmation of its absence in the couplings conducted at -78 °C in the dichloromethane/acetonitriloe mixture. A by-product of this latter reaction conducted at -30 °C, obtained in 27% yield, was methyl 3-O-(1-adamantanyl)-2,4,6-tri-O-benzyl-β-d-galactopyranoside 13 (Figure 1) arising from formation of the 1-adamantanyl cation in the reaction mixture at the higher temperature.
Scheme 2.
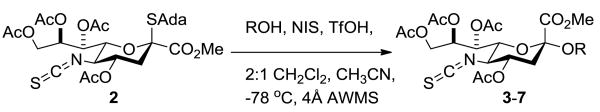
Glycosylation with Isothiocyanate 2. Abbreviations: NIS = N-iodosuccinimide, TfOH = trifluoromethanesulfonic acid, AWMS = acid-washed molecular sieves.
Table 1. Glycosylation reactions with isothiocyanate 2[a][b].
| Entry | Acceptor | Product, Yield, selectivity |
|---|---|---|
| 1 |
|
|
| 2 |
|
|
| 3 |
|
|
| 4[c] |
|
|
| 5 |
|
|
Bn = benzyl, Bz = benzoyl, isothiocyanyl sialyl = [methyl (4,7,8,9-tetra-O-acetyl-3-deoxy-5-isothiocyanyl d-glycero-d-galacto-α,β-nonulopyranosid)onate].
unless otherwise stated all reactions were conducted at -78 °C with 1.2 equiv of acceptor in 2:1 dichloromethane:acetonitrile.
After glycosylation the crude reaction mixture was acetylated to facilitate purification.
Figure 1. Structures of the Adamantanyl Ether 13, of Azido-Protected Sialyl Donors 14, and of the 5H4 Conformer the Hypothetical Oxocarbenium Ion 15.

The formation of a single anomer of 5 at -78 °C (Table 1, entry 3) is especially noteworthy. Typically, 4-O-protected galactopyranosyl 3-OH acceptors give only poor selectivity in sialidation reactions, even with the oxazolidinone-proected donors,[4] hence the common use of the more reactive and selective 3,4-galactosyl diols. The excellent selectivities obtained with the isothiocyanate-protected donor are all the more remarkable when contrasted with the coupling reactions of related azide-protected sialyl donors 14 (Figure 1),[12] which are reported to be competent donors for coupling to primary alcohols,[13] but to be much less selective with secondary alcohols.[2, 13c]
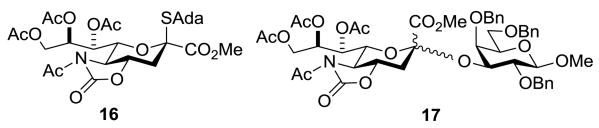
Two possibilities were envisaged for the greater selectivity of the isothiocyanate 2 over the structurally-related azides 14. First, as the isothiocyanate group is considerably more polar than the azido and isocyanate groups (dipole moments of C6H5N3, C6H5N=C=O, and PhN=C=S in Debye units, respectively: 1.82, 2.43, 2.69[14]), it is possible that the isothiocyanate simply serves as a strongly electron-withdrawing group and promotes SN2 glycosylation as has been proposed[15] for the oxazolidinone system. Alternatively, consistent with current models for the through-space stabilization of glycosyl oxocarbenium ions,[16] it is possible that a transient intermediate sialyl oxocarbenium ion preferentially adopts the 5H4 conformation 15 benefitting from stabilization by the pseudoaxial 4-O-acetate and the isothiocyanate groups, with the latter providing significant steric shielding to the β-face (Figure 1). In a competition experiment designed to begin to probe this question a 1:1:1 mixture of the isothiocyanate 2, the N-acetyloxazolidinone 16 (Figure 2), and acceptor 10 was activated at -78 °C by the addition of 0.2 equiv of triflic acid. After standard work up the disaccharides 5 and 17 (Figure 2) were isolated in 3 and 51% yield, respectively, the latter as a 4:1 α:β-mixture. Consistent with this result, donors 2 and 16 were recovered from this experiment in 73 and 17% yield, respectively. While this experiment does not exclude the involvement of oxocarbenium ions such as 15, it establishes the isothiocyanate-protected donor 2 is less reactive than the N-acetyloxazolidinone-protected donor 16 under the usual conditions consistent with the highly electron-withdrawing nature of the isothiocyanate moiety.
Figure 2. Structures of the N-acetyloxazolidionone-protected donor 16 and glycoside 17 from the competition experiment.
Turning to the post-glycosylation derivatization of the isothiocyanate group, in a modification of the Saegusa-Barton[17] radical deamination protocol heating of disaccharide 5 with tris(trimethylsilyl)silane[18] and AIBN in toluene at reflux afforded the 5-deamino-α-sialoside 18 (Scheme 3). Acetylation of the residual alcohol in 4 followed by AIBN-initiated reaction with allyltris(trimethylsilyl)silane[19] in toluene at reflux gave the 5-deamino-5-allyl-α-sialoside 19 as a single equatorial diastereoisomer consistent with earlier reports[20] on radical C-C-bond formation at the 4-position of glucopyranosides (Scheme 3). Reaction of N-Boc-l-methionine thioacid and benzyloxy thioacetic acid, both derived by deprotection of the corresponding 9-fluorenylmethyl thioesters,[21] with disaccharide 5 gave the modified sialosides 20 and 21 in good yield (Scheme 3). The greater ease of reaction of thioacids with isothiocyanates[22] than with unactivated azides[23] is noteworthy. In a further example of the isothiocyanate to amide transformation, the residual alcohol in disacccharide 7 was acetylated and the product 7-Ac allowed to react with benzyloxy thioacetic acid in the presence of piperidine in DMF at 40 °C to give the disialoside 22 containing a protected glycolyl amide and an acetamide (Scheme 3).
Scheme 3.

Formation of Desamino and Amido Derivatives from Isothiocyanate 5. Abbreviation: TTMS = tris(trimethylsilyl)silyl
In a further demonstration of the possibilities afforded by the isothiocyanate group, disaccharide 5 was treated first with 2-phenylethylamine to give the thiourea 23 in 90% yield. Subsequent reaction with methyl iodide gave an isothiourea 24, which on heating with ammonia in DMF gave the guanidine 25 (Scheme 4).
Scheme 4.

Synthesis of Thiourea and Guanidine Derivatives. Abbreviations: DIPEA = diisopropylethylamine, DMAP = 4-dimethylaminopyridine, DMF = dimethylformamide
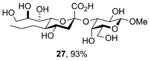
Finally, selected disaccharides were subjected to a two-step deprotection protocol involving the saponification of all esters followed by hydrogenolysis over palladium-charcoal in aqueous buffer (Scheme 5 and Table 2). In this manner novel sialosides either completely lacking subsitution at the 5-position (Table 2, entry 1) or in which the amido function has been replaced by an alkyl chain (Table 2, entry 2) become available for the first time. A variety of N5 amides can readily produced, as exemplified by the important N-glycoyl chain (Table 2, entry 3), and even guanidines (Table 2, entry 4) may be easily accessed in this manner.
Scheme 5. Deprotection of Selected Disaccharides.

Table 2. Deprotection Reactions.
| Entry | Substrate | Product, Yield, selectivity |
|---|---|---|
| 1 | 18 |
|
| 2[a] | 19 |
|
| 3 | 20 |
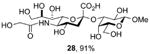
|
| 4 | 25 |
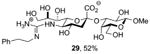
|
Concomitant hydrogenation of the allyl group took place in the course of the hydrogenolytic debenzylation.
Overall, the crystalline sialyl donor 2 affords very highly stereoselective access to a range of sialyl saccharides. Because of the richness of isothiocyanate chemistry, such isothiocyanate-protected saccharides offer direct introduction of a range of standard and novel functionality at the 5-position post-glycosylation, frequently in a single reaction step. In combination with the stereospecific oxidative deamination methods recently developed in our laboratory,[24] this chemistry opens up the 5-position of the sialic acid glycosides, beyond the modified amides accessible by current methods, as a promising locus for the optimization of their diverse biological properties.
Experimental Section
General coupling protocol
A solution of donor 2 (0.15 mmol), acceptor (0.18 mmol), and activated 4 Å acid-washed powdered molecular sieves (300 mg, 2.0 g/mmol) in anhydrous CH2Cl2:MeCN (2:1, 2 mL) was stirred for 5 h under Ar, and then cooled to -78 °C, followed by addition of NIS (42 mg, 0.18 mmol) and TfOH (2 μL, 0.02 mmol). The reaction mixture was stirred at -78 °C for 5 h and then quenched with DIPEA (7 μL). The mixture was diluted with CH2Cl2, filtered through Celite, washed with 20% aqueous Na2S2O3 solution, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with EtOAc:hexane mixtures to afford the desired coupled product.
General Protocol for Amide Formation from Isothiocyanates
To the required 9-fluorenylmethyl (thioester (0.03 mmol) at room temperature was added piperidine (0.21 mmol) in DMF (500 μL). The reaction mixture was stirred for 15 min, then diluted with CHCl3 (3 mL). The resulting solution was washed with 1N HCl aq. (3 mL) and brine (3 mL), dried over Na2SO4, and concentrated in vacuo. The residue was dried under high vacuum, and dissolved in dry CH2Cl2 (0.5 mL) before addition of the isothiocyanate (0.02 mmol). The reaction mixture was stirred for 36 h at room temperature before the volatiles were removed in vacuo. The residue was purified by column chromatography on silica gel eluting with EtOAc:hexane mixtures to afford the corresponding amide.
Supplementary Material
Footnotes
Support of this work by the NIH (GM62160) is gratefully acknowledged. The 600 MHz NMR spectrometer in the Lumigen Instrument Center at Wayne State University was acquired with NSF support (MRI-084043). DC thanks Dr Peter Fügedi, Debrecen, for a helpful discussion.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- 1.a) Hanashima S. Trends in Glycosci Glycobiol. 2011;23:111–121. [Google Scholar]; b) Adak AK, Yu CC, Liang CF, Lin CC. Curr Op Chem Biol. 2013;17:1030–1038. doi: 10.1016/j.cbpa.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 2.De Meo C, Priyadarshani U. Carbohydr Res. 2008;343:1540–1552. doi: 10.1016/j.carres.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 3.a) Tanaka H, Nishiura Y, Takahashi T. J Am Chem Soc. 2006;128:7124–7125. doi: 10.1021/ja0613613. [DOI] [PubMed] [Google Scholar]; b) Farris MD, De Meo C. Tetrahedron Lett. 2007;48:1225–1227. [Google Scholar]; c) Hsu CH, Chu KC, Lin YS, Han JL, Peng YS, Ren CT, Wu CY, Wong CH. Chem Eur J. 2010;16:1754–1760. doi: 10.1002/chem.200903035. [DOI] [PubMed] [Google Scholar]
- 4.a) Crich D, Li W. J Org Chem. 2007;72:2387–2391. doi: 10.1021/jo062431r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Crich D, Li W. J Org Chem. 2007;72:7794–7797. doi: 10.1021/jo7012912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Angata T, Varki A. Chem Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]; b) Chen X, Varki AP. ACS Chem Biol. 2010;5:163–176. doi: 10.1021/cb900266r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Varki A, Gagneux P. Ann N Y Acad Sci. 2012;1253:16–36. doi: 10.1111/j.1749-6632.2012.06517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Boons GJ, Demchenko AV. In: Carbohydrate-Based Drug Discovery. Wong CH, editor. Vol. 1. Wiley-VCH; Weinheim: 2003. pp. 55–102. [Google Scholar]
- 6.a) Wang Q, Guo Z. ACS Med Chem Lett. 2011;2:373–378. doi: 10.1021/ml100313d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rillahan CD, Schwartz E, McBride R, Fokin VV, Paulson JC. Angew Chem Int Ed. 2012;51:11014–11018. doi: 10.1002/anie.201205831. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rillahan CD, Macauley MS, Schwartz E, He Y, McBride R, Arlian BM, Rangarajan J, Fokin VV, Paulson JC. Chem Sci. 2014;5:2398–2406. doi: 10.1039/C4SC00451E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X. ACS Chem Biol. 2012;7:1232–1240. doi: 10.1021/cb300125k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Song X, Yu H, Chen X, Lasanajak Y, Tappert MM, Air GM, Tiwari VK, Cao H, Chokhawala HA, Zheng H, Cummings RD, Smith DF. J Biol Chem. 2011;286:31610–31622. doi: 10.1074/jbc.M111.274217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Pazynina G, Tyrtysh T, Nasonov V, Belyanchikov I, Paramonov A, Malysheva N, Zinin AI, Kononov LO, Bovin N. Synlett. 2013:226–230. [Google Scholar]; b) Hsu Y, Ma HH, Lico LS, Jan JT, Fukase K, Uchinashi Y, Zulueta MML, Hung SC. Angew Chem Int Ed. 2014;53:2413–2416. doi: 10.1002/anie.201309646. [DOI] [PubMed] [Google Scholar]; c) Boltje TJ, Heise T, Rutjes FPT, van Delft FL. Eur J Org Chem. 2013 [Google Scholar]; d) Rich JR, Withers SG. Angew Chem Int Ed. 2012;51:8640–8643. doi: 10.1002/anie.201204578. [DOI] [PubMed] [Google Scholar]
- 9.a) Witczak ZJ. Adv Carbohydr Chem Biochem. 1986;44:91–145. doi: 10.1016/s0065-2318(08)60078-5. [DOI] [PubMed] [Google Scholar]; b) Fernandez JMG, Mellet CO. Adv Carbohydr Chem Biochem. 2000;55:35–135. [Google Scholar]; c) Braverman S, Cherkinsky M, Birsa ML. Science of Synthesis. 2005;18:65–320. [Google Scholar]
- 10.Rajender S, Crich D. J Carbohydr Chem. 2013;32:324–335. doi: 10.1080/07328303.2013.804081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Czarniecki MF, Thornton ER. J Am Chem Soc. 1977;99:8273–8279. [Google Scholar]; b) Hori H, Nakajima T, Nishida Y, Ohrui H, Meguro H. Tetrahedron Lett. 1988;29:6317–6320. [Google Scholar]; c) Haverkamp J, Spoormaker T, Dorland L, Vliegenthart JFG, Schauer R. J Am Chem Soc. 1979;101 [Google Scholar]; d) Prytulla S, Lauterwein J, Klessinger M, Thiem J. Carbohydr Res. 1991;215:345–349. [Google Scholar]; e) Kancharla PK, Crich D. J Am Chem Soc. 2013;135:18999–19007. doi: 10.1021/ja410683y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider R, Freyhardt CC, Schmidt RR. Eur J Org Chem. 2001:1655–1661. [Google Scholar]
- 13.a) Yu CS, Niikura K, Lin CC, Wong CH. Angew Chem Int Ed. 2001;40:2900–2903. doi: 10.1002/1521-3773(20010803)40:15<2900::AID-ANIE2900>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; b) Mukaiyama T, Mandai H, Jona H. Chem Lett. 2002:1182–1183. [Google Scholar]; c) Lu KC, Tseng SY, Lin CC. Carbohydr Res. 2002;337:755–760. doi: 10.1016/s0008-6215(02)00057-5. [DOI] [PubMed] [Google Scholar]
- 14.a) Cheng CL, Le Fevre RJW, Ritchie GLD. J Chem Soc B. 1971:435–437. [Google Scholar]; b) Förner W, Badawi HM. J Theo Comp Chem. 2010;9:511–529. [Google Scholar]
- 15.a) Kancharla PK, Navuluri C, Crich D. Angew Chem Int Ed. 2012;51:11105–11109. doi: 10.1002/anie.201204400. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kancharla PK, Kato T, Crich D. J Am Chem Soc. 2014;136:5472–5480. doi: 10.1021/ja501276r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Smith DM, Woerpel KA. Org Biomol Chem. 2006;4:1195–1201. doi: 10.1039/b600056h. [DOI] [PubMed] [Google Scholar]; b) Jensen HH, Bols M. Acc Chem Res. 2006;39:259–265. doi: 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]
- 17.a) Saegusa T, Kobayashi S, Ito Y, Yasuda N. J Am Chem Soc. 1968;90:4182–4182. [Google Scholar]; b) Barton DHR, Bringmann G, Lamotte G, Motherwell WB, Hay Motherwell RS, Porter AEA. J Chem Soc Perkin Trans 1. 1980:2657–2664. [Google Scholar]
- 18.Ballestri M, Chatgilialoglu C, Clark KB, Griller D, Giese B, Kopping B. J Org Chem. 1991:678–683. [Google Scholar]
- 19.a) Kosugi M, Kurata H, Kawata Ki, Migita T. Chem Lett. 1991:1327–1328. [Google Scholar]; b) Chatgilialoglu C, Ferreri C, Ballestri M, Curran DP. Tetrahedron Lett. 1996;37:6387–6390. [Google Scholar]
- 20.a) Giese B, Witzel T. Angew Chem Int Ed. 1986;25:450–451. [Google Scholar]; b) Gupta V, Kahne D. Tetrahedron Lett. 1993;34:591–594. [Google Scholar]
- 21.Crich D, Sana K, Guo S. Org Lett. 2007;9:4423–4426. doi: 10.1021/ol701583t. [DOI] [PubMed] [Google Scholar]
- 22.a) Kricheldorf HR, Leppert E. Makromol Chem. 1973;167:47–68. [Google Scholar]; b) Gonda J, Bednárikova M. Tetrahedron Lett. 1997;38:5569–5572. [Google Scholar]; c) Gonda J, Zavacká E, Budĕšínský M, Císařova I, Podlaha J. Tetrahedron Lett. 2000;41:525–529. [Google Scholar]; d) Gonda J, Martinková M, Walko M, Zavacká E, Budĕšínský M, Císařova I. Tetrahedron Lett. 2001;42:4401–4404. [Google Scholar]; e) Schoepfer J, Marquis C, Pasquier C, Neier R. J Chem Soc Chem Commun. 1994:1001–1002. [Google Scholar]; f) Crich D, Sasaki K. Org Lett. 2009;11:3514–3517. doi: 10.1021/ol901370y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Hakimelahi GH, Just G. Tetrahedron Lett. 1980;21:2119–2122. [Google Scholar]; b) Rakotomanomana N, Lacombe JM, Pavia A. Carbohydr Res. 1990;197:318–323. [Google Scholar]; c) Shangguan N, Katukojvala S, Greenberg R, Williams LJ. J Am Chem Soc. 2003;125:7754–7755. doi: 10.1021/ja0294919. [DOI] [PubMed] [Google Scholar]
- 24.Navuluri C, Crich D. Angew Chem Int Ed. 2013;52:11549–11552. doi: 10.1002/anie.201303781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.