

ABSTRACT
The human respiratory syncytial virus (HRSV) core viral RNA polymerase comprises the large polymerase protein (L) and its cofactor, the phosphoprotein (P), which associate with the viral ribonucleoprotein complex to replicate the genome and, together with the M2-1 protein, transcribe viral mRNAs. While cellular proteins have long been proposed to be involved in the synthesis of HRSV RNA by associating with the polymerase complex, their characterization has been hindered by the difficulty of purifying the viral polymerase from mammalian cell culture. In this study, enhanced green fluorescent protein (EGFP)-tagged L- and P-protein expression was coupled with high-affinity anti-GFP antibody-based immunoprecipitation and quantitative proteomics to identify cellular proteins that interacted with either the L- or the P-proteins when expressed as part of a biologically active viral RNP. Several core groups of cellular proteins were identified that interacted with each viral protein including, in both cases, protein chaperones. Ablation of chaperone activity by using small-molecule inhibitors confirmed previously reported studies which suggested that this class of proteins acted as positive viral factors. Inhibition of HSP90 chaperone function in the current study showed that HSP90 is critical for L-protein function and stability, whether in the presence or absence of the P-protein. Inhibition studies suggested that HSP70 also disrupts virus biology and might help the polymerase remodel the nucleocapsid to allow RNA synthesis to occur efficiently. This indicated a proviral role for protein chaperones in HRSV replication and demonstrates that the function of cellular proteins can be targeted as potential therapeutics to disrupt virus replication.
IMPORTANCE Human respiratory syncytial virus (HRSV) represents a major health care and economic burden, being the main cause of severe respiratory infections in infants worldwide. No vaccine or effective therapy is available. This study focused on identifying those cellular proteins that potentially interact specifically with the viral proteins that are central to virus replication and transcription, with a view to providing potential targets for the development of a specific, transient therapeutic which disrupts virus biology but prevents the emergence of resistance, while maintaining cell viability. In particular, protein chaperones (heat shock proteins 70 and 90), which aid protein folding and function, were identified. The mechanism by which these chaperones contribute to virus biology was tested, and this study demonstrates to the field that cellular protein chaperones may be required for maintaining the correct folding and therefore functionality of specific proteins within the virus replication complex.
INTRODUCTION
Human respiratory syncytial virus (HRSV) is the leading cause of severe respiratory tract infections in newborn children worldwide (1). It infects almost all infants within the first 2 years of life and is the main cause of infant bronchiolitis. Currently, only ribavirin is approved for therapeutic treatment, and there is a pressing need for additional therapies to be developed (2, 3). HRSV belongs to the genus Pneumovirus of the family Paramyxoviridae and the order Mononegavirales (1). The viral genome consists of a nonsegmented, approximately 15-kb RNA of negative polarity that encodes 11 proteins. As with all the members of the Mononegavirales, the genomic RNA of HRSV is associated with the viral nucleoprotein (N) and maintained as a helical N-RNA ribonucleoprotein (RNP) complex (4). The RNP is used as a template for transcription and replication by the RNA-dependent RNA polymerase (RdRp) complex, which consists of the large polymerase protein (L) and its cofactor, phosphoprotein (P). Whereas N-, P-, and L-proteins are sufficient to mediate viral genome replication, transcription activity also requires the M2-1 protein, which functions as a polymerase cofactor (5). The specific recognition of the viral N-RNA complex by the RdRp constitutes a prerequisite for viral transcription and replication. This recognition is mediated by the P-protein (6), which interacts with the L-, N-, and M2-1-proteins. A feature of the Mononegavirales is that the RdRp is present in the virus particle to initiate viral mRNA transcription upon release of the RNP in the cytoplasm.
Cellular proteins have been shown to play crucial roles in viral RNA synthesis for a number of other viruses that encode RdRps. These roles include involvement in the formation of virus factories (7, 8) and acting directly as cofactors to regulate RdRp activity (9, 10). Several cellular proteins have been shown to potentially associate with the HRSV L-, P-, N-, and M2-1-proteins, including heat shock proteins (HSPs). Expression and purification of the L-protein (with the P-protein) from insect cells previously suggested an association with HSP70 (11), and data from coimmunoprecipitation studies suggested that HSP70 associates with the P-protein alone (12). The association of the polymerase complex with HSP70 was also observed within lipid raft membranes in virus-infected cells (13). HSP90 has been identified within the virion proteome (14), and ablation of the HSP90 chaperone function in HRSV-infected cells resulted in a decrease in progeny virus (14, 15). Geller et al. postulated that HSP90 contributed to L-protein stability (15), an effect which had been shown previously for vesicular stomatitis virus (VSV) (16), another member of the Mononegavirales. Further, Geller et al. and Radhakrishnan et al. demonstrated potential antiviral activities of HSP90 inhibitors on HRSV biology (14, 15), and these were refractive to resistance (15).
A particular difficulty in studying the HRSV polymerase complex is the lack of immunological reagents to the L-protein to couple with immunoprecipitation (IP) strategies. L expression in eukaryotic cell culture systems also seems to require coexpression of the P-protein (11, 17). To overcome these difficulties, and to map which cellular proteins associate with the HRSV polymerase complex in mammalian cells, we made use of a plasmid-based HRSV rescue system in which enhanced green fluorescent protein (EGFP) is inserted either between domains V and VI of the L-protein (EGFP-L) (17) or within the disordered N-terminal region of the P-protein (EGFP-P). These fusion proteins maintain biological activity in the context of reconstituted RNPs (a “minireplicon” system) and are able to both replicate and transcribe viral RNA when the N- and M2-1-proteins are expressed from additional support plasmids (17). The activity of the replicon can be monitored via the expression of a reporter protein, such as luciferase. The EGFP moiety allows the use of a highly specific, single-chain antibody to EGFP to facilitate detection or purification of either the EGFP-L or the EGFP-P protein in the context of the minireplicon system. GFP-Trap technology was coupled with label-free quantitative proteomics for the identification and discrimination of specific interacting proteins. Previous studies investigating the interactomes of HRSV NS1 and NS2, porcine reproductive and respiratory syndrome (PRRSV) and infectious bronchitis virus (IBV) N-proteins and Zaire ebolavirus (EBOV) VP24, have also used this approach (18–21). Similar to the study investigating EBOV VP24, small-molecule inhibitors were subsequently applied to decipher the potential role(s) of identified binding partners in the context of wild-type (WT)_infection and other assays.
Our data indicated that the EGFP-L and EGFP-P proteins associate with a number of cellular proteins, including chaperones. Ablation of the function of these proteins had a detrimental effect on virus biology. Specifically, the protein chaperone function of HSP90 was required to maintain the stability of the L-protein, both in association with the P-protein and in isolation, and it was determined that HSP70 plays a positive role in HRSV RNA synthesis. This adds to the body of evidence that the functions of cellular proteins is critical for virus biology and suggests that these functions can be targeted for the development of antiviral therapies (15, 22, 23).
MATERIALS AND METHODS
Cells and virus.
HEp2 and A549 cells were obtained from the Public Health England Culture Collection and were grown at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS) and 1% (vol/vol) penicillin-streptomycin. For transfection experiments using EGFP-L (see below), BSRT7 cells (baby hamster kidney [BHK] cells expressing T7 RNA polymerase [12]; maintained by the addition of G418) were grown in DMEM supplemented with 1× Glutamax (Invitrogen) and 10% (vol/vol) FBS. HRSV strain A2 was propagated in HEp2 cells and concentrated by using a sucrose gradient-based method which also removes cellular contaminants (24). Virus was concentrated in the 30% phase of a discontinuous sucrose gradient and stored in this fraction for stability during long-term storage. Virus titers were calculated as previously described (25). HRSV strain A2 for experiments related to in vitro RNA synthesis was also propagated in HEp2 cells but was not sucrose purified.
pP-EGFP plasmid construction.
A unique BamHI site was introduced at nucleotide 96 of the P gene in the pP plasmid by site-directed mutagenesis using the QuikChange kit (Stratagene). The EGFP gene was then inserted in frame with the P open reading frame (ORF) at the BamHI site.
EGFP-L and EGFP-P IPs.
For IPs, BSRT7 cells were routinely seeded in two 10-cm2 dishes per condition and were 90% confluent prior to transfection. Cell monolayers were cotransfected with 12.5 μg of MiniG-Luc (which carries the genes for the HRSV minigenome, including the firefly luciferase reporter gene), 12.5 μg of pN, 12.5 μg of pP or pP-EGFP, 6.25 μg of pL or pL-EGFP, and 12.5 μg of pM2-1 plasmids and using 50 μl of Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cells were harvested 24 h posttransfection, and cell lysate was incubated with GFP-Trap beads (Chromotek) for 2 h. The bead pellet was washed and suspended in 50 μl of glycine elution buffer (0.2 M glycine, pH 2.5). The supernatant was neutralized with 5 μl of 1 M Tris base (pH 10.4) in preparation for liquid chromatography-tandem mass spectrometry (LC-MS/MS).
IP sample preparation for proteomics.
Protein content and volume were normalized with 25 mM ammonium bicarbonate. Rapigest (Waters) was added to the samples prior to heating at 80°C for 10 min. Samples were then reduced with 3 mM dithiothreitol (DTT; Sigma) at 60°C for 10 min and alkylated with 9 mM iodoacetimde (Sigma) at room temperature for 30 min in the dark. Proteomic-grade trypsin (Sigma) was added at a protein:trypsin ratio of 50:1, and samples were incubated at 37°C overnight. Rapigest was removed by adding trifluoroacetic acid (TFA) to a final concentration of 1% (vol/vol) and incubating at 37°C for 2 h. Peptide samples were centrifuged at 12,000 × g for 60 min (4°C) to remove precipitated Rapigest. Peptides were concentrated and desalted using C18 Stage tips (Thermo Scientific), then dried using a centrifugal vacuum concentrator (Jouan) and resuspended in a 0.1% (vol/vol) TFA, 3% (vol/vol) acetonitrile solution.
Nano-LC ESI-MS/MS analysis.
Peptide mixtures (2 μl) were analyzed by online nanoflow liquid chromatography (nano-LC) by using the nanoACQUITY-nLC system (Waters MS Technologies, Manchester, United Kingdom) coupled to an LTQ-Orbitrap Velos mass spectrometer (ThermoFisher Scientific, Bremen, Germany) equipped with the manufacturer's nano-electrospray ionization (ESI) ion source. The analytical column (nanoACQUITY ultraperformace LC BEH130 C18, 15-cm by 75-μm by 1.7-μm capillary column) was maintained at 35°C with a flow rate of 300 nl/min. The gradient consisted of 3 to 40% acetonitrile in 0.1% formic acid for 90 min and then a ramp of 40 to 85% acetonitrile in 0.1% formic acid for 3 min. Full-scan MS spectra (m/z range, 300 to 2,000) were acquired by the Orbitrap at a resolution of 30,000. Analysis was performed in data-dependent mode. The top 20 most intense ions from the MS1 scan (full MS) were selected for tandem MS by collision-induced dissociation (CID), and all product spectra were acquired in the LTQ ion trap.
Thermo RAW files were imported into Progenesis LC-MS (version 4.1; Nonlinear Dynamics). Runs were time aligned using default settings and an autoselected run as the reference. Peaks were picked by the software and filtered to include only a charge state of between +2 and +6. Peptide intensities were normalized against the reference run, and these intensities were used to highlight differences in protein expression between control and treated samples with supporting statistical analysis (analysis of variance P values) calculated by the Progenesis LC-MS software. Spectral data were transformed to .mgf files with the Progenesis LC-MS program and exported for peptide identification using the Mascot search engine (version 2.3.02; Matrix Science). Tandem MS data were searched against the Mus musculus, Cricetulus griseus, and human respiratory syncytial virus A (strain A2) predicted proteomes (Uniprot release 2013_04). Mascot search parameters were as described above. Results were imported into the Progenesis LC-MS program as .xml files.
HSP90 inhibition studies.
For A549 and BSRT7 cells, the chaperone activity of HSP90 was initially inhibited by applying 0.0625 to 5 μM concentrations of the geldanamycin derivative 17-AAG (Calbiochem) diluted in dimethyl sulfoxide (DMSO; vehicle control). Cell viability/cytotoxicity were assessed using a colorimetric MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. Cells were seeded at a density of 3.8 × 103 cells/well (for 40 to 50% confluence), 1 × 104 cells/well (for 60 to 70% confluence), or 1.5 × 104 cells/well (for 80 to 90% confluence) in clear 96-well microplates 24 h prior to 17-AAG treatment. Following 17-AAG treatment, MTT powder (Sigma) was dissolved in DMEM (plus 10% [vol/vol] FBS) at 37°C to make a 10 mM solution and filtered through a 0.2-μm filter. Medium was aspirated, and the wells were washed with phosphate-buffered saline (PBS). Filtered MTT (100 μl/well) was applied, and the plates were incubated for 45 min at 37°C. MTT was removed and replaced with 100 μl/well DMSO, and cultures were thoroughly mixed by pipetting until MTT (a yellow tetrazole) was reduced to purple formazan in living cells. Absorbance was then measured at wavelengths between 425 and 570 nm on a Dynex plate reader. A dose-response curve was then produced to assess cell viability (data not shown). A more precise 50% effective concentration (EC50) could then be established alongside immunoblot analysis results with 17-AAG-treated cells to establish a suitable time course (18, 24, and 40 h posttreatment) for both cell lines.
HRSV-infected cells (multiplicity of infetion [MOI], 3) and mock-infected cells were routinely treated with 0.5 μM 17-AAG 4 h postinfection (p.i.) in comparison to nontreated and vehicle control-treated cells. For minireplicon experiments, BSRT7 cells were transfected 4 h prior to treatment with 0.5 μM 17-AAG. RNA (for quantitative PCR [qPCR]) and cell pellets (for whole-cell lysis) were processed 22 h post-virus application or minireplicon transfection and 18 h post-17-AAG treatment. To assess 17-AAG activity, cell pellets were lysed in RIPA buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1% [vol/vol] NP-40 alternative, 0.5% [wt/vol] sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) supplemented with 1 EDTA-free Complete protease inhibitor mixture (Roche Applied Science) per 50 ml buffer and immunoblotted with an antibody to Cdc2 (A17; Abcam), a known HSP90 client protein, which was used as a marker of proteasome-mediated Cdc2 degradation.
HSP70 inhibition studies.
MTT assays (data not shown) were again used to establish an EC50 prior to the application of small-molecule inhibitors VER-155008 (VER; SML0271), Pifithrin-μ (PIF; P0122), MKT-077 (M5449; Sigma-Aldrich), and YM-1 (500615; Merck Millipore) in the context of HRSV infection (A549 cells) or the minireplicon (BSRT7 cells) (data not shown). HSP70 inhibitors were applied to infected A549 cells or transfected BSRT7 cells as described above.
Cell extract in vitro RNA synthesis assay for HSP70 function.
Analysis of HRSV transcription in crude cell extracts was based on a protocol published previously for vesicular stomatitis virus (26). HEp2 cells in six-well plates were infected or mock infected with RSV at an MOI of 5 for 17 h, following which the medium was replaced with medium containing 2 μg/ml actinomycin D. After 1 h, cells were washed with PBS and treated with 250 μg/ml lysolecithin in PBS for 1 min on ice. The lysolecithin/PBS mix was aspirated, and cells from each well were scraped into 62 μl of transcription buffer (2 μg/ml actinomycin D, 50 mM Tris-acetate [pH 8], 8 mM Mg-acetate, 300 mM K-acetate, 2 mM DTT, 1 mM spermidine, 10 mM creatine phosphate, 1 μg/ml aprotinin, 16 U creatine phosphokinase, 50 μM ATP, 1 mM CTP, and either 50 μM or 1 mM GTP or UTP, depending on the radiolabeled NTP used). Samples were centrifuged at 800 × g for 5 min, and the supernatant was collected. Aliquots (25 μl) of the cell extract were combined with additional transcription buffer containing 10 μCi [α-32P]GTP or [α-32P]UTP. RNase inhibitor and either DMSO or VER diluted in DMSO were added to a total reaction mixture volume of 50 μl. Reaction mixtures were incubated for 4 h at 30°C, following which total RNA was extracted using a Qiagen RNeasy kit, according to the manufacturer's instructions. Purified total RNA samples were further processed by RNase H digestion, to remove poly(A) tails from mRNA transcripts, thus allowing resolution of individual mRNA bands. Total RNA was incubated for 1 h at 30°C in 50 mM Tris-Cl (pH 7.5), 10 mM MgCl2, 1 mM EDTA, 10 mM DTT, 10% glycerol, 200 ng oligo(dT)15 and 2.5 U RNase H (NEB) in a total volume of 30 μl. Total and RNase H-treated RNA samples were subjected to denaturing gel electrophoresis on a 4% acrylamide gel containing 7 M urea and analyzed by autoradiography. The RNA signal was quantified by analyzing TIF images of the autoradiograms by using Quantity One software. To ensure that the values obtained were in the linear range, a standard curve was generated by preparing a 2-fold dilution series of a molecular weight marker. This standard was included on all scans.
Minimalist in vitro RNA synthesis assay using purified HRSV polymerase.
The HRSV L/P/HSP70 complex was purified from insect cells as described previously (11). An RNA oligonucleotide (Dharmacon) representing nucleotides 1 to 25 of the trc promoter sequence (2 μM) was combined with transcription buffer containing 50 mM Tris-HCl (pH 7.4), 8 mM MgCl2, 5 mM DTT, 10% glycerol, and 250 μM GTP, 50 μM ATP, 50 μM CTP, and 50 μM UTP, with 10 μCi of [α-32P]GTP and either DMSO or various concentrations of VER (diluted in DMSO) as indicated. Reaction mixtures were incubated at 30°C for 10 min, and then HRSV L/P/HSP70 (containing ∼200 ng of L-protein) was added to the transcription mix, such that the final reaction volume was 50 μl. Reaction mixtures were incubated for 3 h at 30°C, then heated to 90°C for 3 min to inactivate the RdRp and cooled briefly on ice. The RNA was analyzed by electrophoresis on a 20% polyacrylamide gel containing 7 M urea in Tris-borate-EDTA buffer. The molecular weight ladder was generated by alkali hydrolysis of a 32P-end-labeled RNA oligonucleotide representing the anticipated RNA products. RNA products were visualized and quantified by phosphorimage scanning and Quantity One software.
Immunofluorescence confocal microscopy.
Coverslip-adhered cell monolayers were fixed with paraformaldehyde, made permeable with PBS containing 0.1% (vol/vol) Triton, and probed with anti-HSP90 antibody (4F10; sc-69703; Santa Cruz Biotechnology) diluted 1:50 in PBS containing 2% FBS. The primary antibody was detected using an Alexa Fluor-conjugated secondary antibody (A4116) diluted 1:200 in PBS containing 2% FBS, prior to mounting with Prolong Gold antifade reagent (Invitrogen). Confocal microscopy images were captured on an LSM510 META microscope (Carl Zeiss Ltd.) equipped with 40× and 63×, 1.4-numerical aperture, oil immersion lenses. Pinholes were set to allow optical sections of 1 to 2 μm to be acquired. Where possible, all fluorescence was measured in the linear range.
Immunoblot analysis.
Soluble cellular proteins were obtained as detailed above, and total protein concentrations were determined via the bicinchoninic acid assay (Pierce). Proteins (5 to 7.5 μg) were resolved by 7.5 or 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore), using a Bio-Rad semidry transfer apparatus, and the following antibodies were applied: CDC2 (A17; Ab18), HSP90 (S88; Ab1429), HRSV (polyclonal to detect structural proteins; ab20745) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 6C5; Ab8245) (all antibodies were obtained from Abcam). HSP90 (4F10; sc-69703), Luciferase (251-150; sc-32896), and STI1 (28; sc-136082) were obtained from Santa Cruz Biotechnology. The following horseradish peroxidase-conjugated secondary antibodies were detected with enhanced chemiluminescence: anti-rabbit (Ab6154) and anti-mouse (A4116) antibodies were obtained from Sigma, and anti-goat antibody (Ab6741) was obtained from Abcam.
Quantitative real-time PCR.
To measure mRNA levels, cDNA was synthesized from 1 μg of total RNA using the ThermoScript reverse transcription-PCR (RT-PCR) system (Invitrogen) according to the manufacturer's instructions. Twenty microliters of cDNA product was then diluted with 80 μl of nuclease-free water. For each qPCR mixture, 1 μl of diluted cDNA and 1 μl of a primer pair (0.5 μl of each of the forward primer and reverse primer) were mixed with 10 μl of SYBR green mastermix (Bio-Rad) to make up a final reaction volume of 20 μl with nuclease-free water. qPCR was performed on a DNA Engine Opticon 2 system (Bio-Rad), following the recommended protocol. The results of three independent experiments were averaged and normalized by using either a murine or a human GAPDH gene control, depending on the experiment.
Luciferase assay.
BSRT7 cells were routinely transfected with the complete (+L) luciferase-based or incomplete (−L) luciferase-based HRSV minireplicon reporter plasmid system 4 h prior to drug treatment and lysed in an appropriate amount of passive lysis buffer (Promega) for 15 min at room temperature 22 h posttransfection. Lysate (20 μl) was pipetted into a 96-well white flat-bottom plate and assayed using the dual luciferase system (Promega) according to the manufacturer's instructions. The luminescent signal was recorded using a luminescence reader (BMG Labtech). The results of three independent experiments were averaged, with normalization to the Renilla control. Luciferase expression was expressed in relative light units (RLU), relative to a no-drug control (0 μl).
Pulse-chase labeling.
Cells were prepared for pulse-chase 22 h posttransfection with EGFP plasmids; all steps of the pulse-chase were performed in the presence or absence of 1 μM 17-AAG. Cells were depleted of methionine by incubation in methionine-free medium (Gibco) for 2 h. Cells were then pulse-labeled with [35S]methionine (PerkinElmer) for 30 min and either harvested immediately or chased with medium containing an excess of cold methionine (50 mM) for 2 h at 37°C. GFP-tagged proteins were isolated from the cell lysates by using GFP-Trap beads (Chromotek), and proteins were analyzed by SDS-PAGE and autoradiography. A plasmid carrying the EGFP-ADAR1 genes was the kind gift of M. Carmo-Fonseca (27) and was used as a control.
Flow cytometry.
Flow cytometric analysis of GFP-positive cells was performed on a FACSCalibur apparatus and using CellQuest software (BD Biosciences). Data analysis was performed using FlowJo (version X.0.7; Tree Star Inc.). Live cells were defined based on forward and side scatter. Cells in the live gate were then plotted against side scatter and FL1 for GFP expression, and the mean percentage of GFP-positive cells was obtained for each condition.
RESULTS
Identification of potential interacting partners of the HRSV polymerase complex.
To obtain a more complete picture of the potential interaction partners of the HRSV polymerase complex, IP coupled with quantitative proteomics was used to identify cellular proteins that associated with the L- and P-proteins. This system utilized the expression of either a biologically active L-EGFP or P-EGFP in the context of an HRSV minireplicon harboring the luciferase gene, together with the appropriate wild-type proteins (N, M2-1, P [if EGFP-L] and L [if EGFP-P]) expressed from support plasmids in BSRT7 cells (17). Replication of the minireplicon RNA was dependent on the presence of functional L- (or EGFP-L), P- (or EGFP-P), and N-proteins. Transcription of luciferase mRNA from the minigenome was enhanced in the presence of M2-1 (17, 28), allowing replicon activity to be monitored by measuring the amount of the reporter gene mRNA, protein, or activity.
A highly specific GFP-Trap was used to selectively precipitate the EGFP-L or EGFP-P fusion protein and its interacting partners, as described previously (18–20). To identify bound proteins, label-free quantitative proteomics was used to compare the abundance of immunoprecipitated peptides from this system with the abundance of those precipitated from a control sample, in which cells were transfected with the same minireplicon system except that a wild-type, untagged L- or P-protein was used. This control was used to identify cellular proteins that bound to the monovalent agarose GFP-Trap beads only. LC-MS/MS analysis of the IP proteins was conducted on material eluted in 0.2 M glycine, pH 2.5.
To provide a statistically robust data set, both EGFP-L and -P and the untagged control were purified independently in triplicate, and each eluted sample was further analyzed in triplicate by LC-MS/MS. Progenesis software was used to analyze the LC-MS/MS data against mouse and hamster databases to derive a list of cellular proteins that potentially bound to EGFP-L or EGFP-P. The results from the mouse database were used, as they provided higher coverage and subsequent protein identification than the hamster database. Viral proteins were identified directly from peptide assignment to a FASTA file listing the amino acid sequences of the L-, P-, N-, and M2-1-proteins.
LC-MS/MS analysis identified 337 cellular proteins that copurified with EGFP-L. To identify likely specific interactions over background/random interactions, these proteins were triaged based on several different criteria. First, proteins identified and quantified by a single peptide were removed, as single-peptide identification in many cases does not provide robust identification. The identification of a protein was based on use of unique peptides (e.g., some proteins can share the same peptide sequence, and in such cases the sequence was not used). Second, the false-discovery rate (FDR) was set at 1% or below, implying that theoretically 99 out of 100 proteins would be identified correctly. Third, using the quantitative aspect to compare replicate control and experimental samples, both the binding ratio and statistical significance could be assigned to potential interacting partners. The binding ratio refers to the relative quantification when comparing the tagged replicon system to the untagged control and is expressed as the fold change.
Proteins interacting specifically with the bait would be expected to be enriched to a greater extent than in the control and show a correspondingly high binding ratio. Proteins binding to the beads equally in both control and test samples (i.e., nonspecific binders) will have a binding ratio near 1.0. These can include common, low-order “hits,” such as vimentin, nucleolin, cytoskeletal, and ribosomal proteins, certain classes of helicases, and heat shock proteins, which associate with the binding matrix (29). Low-order hits with a binding ratio of 1.9 or less were therefore removed. Fourth, the data sets were further processed by comparison to previously determined EGFP and matrix interactomes to filter out likely false positives (18–20, 29, 30).
Using these criteria, 19 cellular proteins were found to interact with the EGFP-L protein (see Table S1 in the supplemental material). LC-MS/MS analysis also identified the EGFP-L, -P, and -N proteins from the EGFP-L and EGFP-P IPs, indicating that confidence could be placed in the approach, as these proteins form known interactions (31). The M2-1 protein was detected (data not shown) but was only identified by a single peptide and was therefore excluded from the data presented in Table S1. The PANTHER bioinformatic algorithm was applied to the data set in Table S1 (excluding viral proteins) to identify groups of polypeptides that had potentially common functionality. The output indicated that a number of the EGFP-L-interacting proteins with ratios greater than 2.0 clustered on proteins with folding/chaperone activities and membrane trafficking, nucleic acid binding, and transferase activities. When the data were analyzed as a volcano (scatter) plot of significance versus binding ratio, it was clear that proteins involved in chaperone function formed a major part of the EGFP-L interactome (Fig. 1).
FIG 1.

Analysis via label-free LC-MS/MS of cellular proteins that associated with the EGFP-L protein in the context of the HRSV minireplicon. Control and experimental samples (performed in triplicate) were each processed in triplicate by LC-MS/MS. Statistical analysis using a volcano plot identified proteins of interest, which are displayed both as large-magnitude ratio changes (x axis) as well as high statistical significance (y axis; −log10 of the P value). Dashed red lines show chosen cutoff ratios of change: >2 (log 2 = 1) and P < 0.05 [−log(100.05) = 1.3]. Points highlighted in black or green had a ratio change of more than 2, in comparison with those shown in gray; green highlights proteins with a chaperone or chaperone-associated function.
In our experience, and that of others, there is good agreement between using quantitative proteomics to identify interacting partners from IP-based experiments and subsequent follow-on experiments using specific analysis techniques (18–20, 32, 33). Aspects of the EGFP-L interactome were validated by conducting repeat IPs of control and experimental systems. Specific virus and cellular proteins were subsequently probed by immunoblot analysis (Fig. 2). Biological activity of the HRSV RNPs was validated by probing for luciferase, which was expected to be expressed from the minireplicon in the presence of functional support proteins: L, N, P, and M2-1 for the control system and EGFP-L, N, P, and M2-1 for the experimental system. There was less luciferase observed in the experimental system (Fig. 2, top panel), in agreement with previously published work indicating that the insertion of EGFP into the HRSV L-protein attenuates but does not abolish activity (17). No luciferase was detected in the bound fraction (Fig. 2). The expression of N, P, and M2-1 from the support plasmids was confirmed in the input samples for both the control and experimental systems. However, no viral proteins were precipitated in the sample lacking an EGFP tag, and only the N- and P-proteins could be detected in the bound fraction of the EGFP-L sample (Fig. 2, middle panel). M2-1 was not detected by immunoblotting in the bound fraction from either the control or experimental system (Fig. 2); this is consistent with previous reports (34) in which M2-1 probably binds to RNA rather than a direct interaction with the P-protein. In the bound fraction for L-EGFP, the immunoblot analysis, though not quantitative, indicated that more N was being captured than P and M2-1. This was consistent with the quantitative LC-MS/MS analysis that indicated that N had a higher binding ratio than P (19.9 and 4.0, respectively) (see Table S1 in the supplemental material). This was indicative that RNP complexes may have been precipitated.
FIG 2.

Expression of luciferase from the control and experimental minireplicon system was confirmed via immunoblotting with a probe for an antiluciferase antibody. Luciferase was below the limit of detection in the immunoprecipitated (bound) fraction, as expected. Expression of the N-, P-, and M2-1-proteins was confirmed via immunoblotting with a probe for a polyclonal anti-RSV antibody, for both the control and experimental system. Only the N- and P-proteins could be detected in the bound fraction from the experimental system and were not found in the bound fraction from the control system. The presence of the cellular proteins HSP90 and STIP1 was also investigated in the IPs by immunoblotting.
To validate potential cellular interactors identified by LC-MS/MS, we probed the IP samples using immunoblot analysis: stress-induced phosphoprotein 1 (STIP1; ratio, 15.5) and HSP90 (ratio, 8.5) were precipitated in greater quantities from the experimental system than the control precipitation, again in agreement with the proteomics screen (Fig. 2, lower panels). HSP90 was also observed in the bound fraction from the control experiment to a lesser extent, and this was in line with previous observations that chaperone proteins associate with the binding matrix in an IP (29), but in the absence of a specific interaction, they generally have a binding ratio of 1.0 or less.
The mini-replicon system required the stable expression of the L-protein together with the N-, P-, and M2-1-proteins to generate a functional RNP that mimicked those found in infected cells (17). Thus, binding data from the EGFP-L IP could reflect a mixture of cellular proteins that bound to the L-protein and/or to the other viral components. To explore this, and also to investigate whether cellular proteins could interact directly with the polymerase cofactor P-protein, we engineered a biologically active P-EGFP protein. Sequence alignment between HRSV P and pneumovirus of mice (PVM) P-proteins revealed a region of low sequence conservation located in the N-terminal region of P (data not shown), which is a naturally disordered region. Insertion of EGFP at residue K32 of the HRSV P-protein resulted in an EGFP-P fusion protein that still localized to inclusion bodies (IBs) and retained 10% of activity compared to WT P, as assayed using a minireplicon system (data not shown). Accordingly, P-EGFP was used as bait in further IPs, in the context of reconstituted minigenome RNPs. LC-MS/MS analysis of the EGFP-P IP identified both N and L (see Table S2 in the supplemental material), thus confirming known interactions. M2-1 was also identified, but again, only with a single peptide and was therefore excluded from Table S2. Thirty-nine cellular proteins with a binding ratio of 2 or more and identified by two or more peptides had the potential to interact with P-EGFP (see Table S2). As with the EGFP-L IP, cellular polypeptides involved in chaperone function were prominent, including STIP1, DNAJA2, and HSP90.
HRSV macromolecular synthesis requires cellular chaperone activity.
Previous studies have indicated that HRSV titers decreases or that virion assembly and transmission are impaired in infected cells when they are treated with 17-AAG (14) or other HSP90 inhibitors, depending on the time point of inhibitor application (14, 15), suggesting a key role for HSP90 in the life cycle of the virus. In the current study, an assay point of 4 h p.i. for inhibitor application and 22 h p.i. were chosen to assess effects on virus biology. This was based on a one-step growth curve analysis of the A2 strain in A549 cells with an MOI of 3, allowing us to delineate a time point after the release of progeny virus from the first round of infection but before the onset of release of virus from reinfection (data not shown). The quantitative interactome analysis indicated that HSP90 had a higher binding ratio with the L- than the P-protein (8.5- versus 2.9-fold over the control, respectively) (see Tables S1 and S2 in the supplemental material) and thus may be involved directly in L-protein function. To investigate whether the decrease in viral titer observed in the presence of 17-AAG was due to a reduction in viral mRNA synthesis and/or viral protein abundance, we assessed the effect of titrating 17-AAG on HRSV-infected cells in comparison to untreated and vehicle control (DMSO)-treated cells. The inhibitor was applied 4 h p.i., and a concentration range was used that maintained cell viability (data not shown). The activity of the inhibitor was confirmed at higher doses by the depletion of Cdc2 (Fig. 3A), a protein whose abundance is dependent on HSP90 chaperone activity (13). Conversely, at the concentrations tested, there was no significant effect on the abundance of HSP90 (Fig. 3A). However, as the concentration of 17-AAG increased, there was a marked dose-dependent decrease in the abundance of viral proteins (Fig. 3A). This was mirrored by a corresponding decrease in all viral mRNA species in HRSV-infected cells treated with 17-AAG (at 0.5 μM, applied at 4 h p.i.), as determined by qRT-PCR (Fig. 3B). This was similar to the observations that treatment of infected cells with 17-AAG has a detrimental effect on HRSV biology (14, 15).
FIG 3.

(A) Immunoblot analysis of mock-infected and HRSV-infected A549 cells that were treated with 17-AAG from 4 h p.i. in comparison to a vehicle control (DMSO) and untreated cells. Viral proteins visually decreased in abundance with increased 17-AAG concentration. Cdc2, an HSP90 client protein, was used as a marker of inhibitor activity. The abundance of HSP90 was shown to be unchanged in nontreated, vehicle control-treated, and 17-AAG-treated cells and was also used as a loading control. (B) The mRNA level of each viral transcript was compared by qRT-PCR at 22 h p.i. in the presence or absence of 0.5 μM 17-AAG, which was added at 4 h p.i. The data were normalized to the control gene GAPDH and are shown as the fold change for each mRNA between 17-AAG-treated HRSV-infected cells and mock-treated HRSV-infected cells.
The ablation of viral gene expression in infected cells treated with 17-AAG may have been due to an overall decrease in stability of all viral proteins or, specifically, one or more of the proteins involved in RNA synthesis (e.g., the L-, P-, N-, and M2-1-proteins). The work of Geller et al. (15) suggested the latter was the most likely possibility, given that the L-protein could not be definitively identified. To investigate this further, we made use of the minireplicon system in which the wild-type L-, P-, N-, and M2-1-proteins were provided from support plasmids, and hence their synthesis was not dependent on any viral processes. In this system, HRSV-driven luciferase expression showed a dose-dependent inhibition by the addition of 17-AAG (Fig. 4A). The EC50 was approximately 0.2 μM, similar to that seen for authentic viral gene expression (Fig. 3). When polypeptide expression was detected by immunoblot analysis, luciferase was readily detected in the absence, but not the presence of 17-AAG (Fig. 4B), consistent with the activity assays (Fig. 4A). Likewise, qRT-PCR revealed an approximately 5-fold decrease in the amount of luciferase mRNA in replicon-expressing cells treated with 0.5 μM 17-AAG compared to the amount of luciferase mRNA in replicon-expressing cells in the absence of 17-AAG (Fig. 4C). However, immunoblot analysis indicated that the levels of P, N, and M2-1 expressed from the support plasmids were unchanged in the absence or presence of 17-AAG (Fig. 4B). The L-protein could not be detected due to the lack of a specific antibody. Thus, 17-AAG inhibits the process of viral gene expression but not by destabilizing all virus polypeptides.
FIG 4.
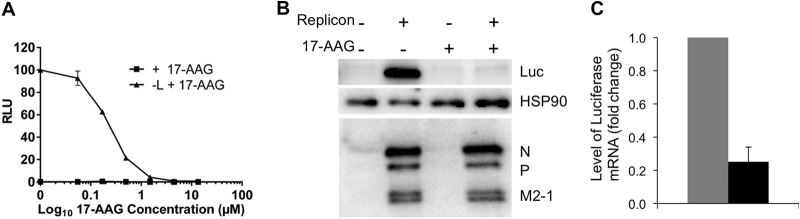
HSP90 inhibition reduces HRSV replication in the context of the minireplicon. (A) Luciferase activity from BSRT7 cells transfected with the complete (+ L) or incomplete (− L) minireplicon system incubated in the presence and absence of 17-AAG was analyzed. Results are expressed as the percentage of relative light units (RLU) produced in the absence of drug and are means ± standard deviations of triplicate samples. (B) Luciferase protein abundance was assessed and compared for BSRT7 cells transfected with the minireplicon system (+) or mock transfected (−) and either treated (+) or not treated (−) with 17-AAG. The abundance of the viral proteins N, P, and M2-1 were also compared and remained unaffected by the presence of 17-AAG. (C) The abundance of the luciferase mRNA from the minireplicon was compared by qRT-PCR at 24 h p.i. with (black bar) or without (gray bar) the treatment of 0.5 μM 17-AAG added at 4 h posttransfection. The data were normalized to the control gene GAPDH and are shown as the fold change for each mRNA between 17-AAG-treated transfected cells and mock-treated cells.
HSP90 chaperone activity supports HRSV replication by stabilizing the L-protein.
In the context of the minireplicon system, 17-AAG treatment 4 h posttransfection inhibited viral mRNA synthesis but did not affect the accumulation of the P-, N-, and M2-1-proteins expressed from support plasmids (Fig. 4B). However, the anti-HRSV polyclonal antibody used did not detect the L-protein in this system. To determine if 17-AAG directly affected L-protein abundance, we used the GFP moiety of the EGFP-L protein as a detection tag. Confocal microscopy was used to determine the intracellular localization of the L-protein in the context of the minireplicon system with and without 0.5 μM 17-AAG (Fig. 5A). In the absence of 17-AAG, the EGFP-L could be readily detected, showing a punctate cytoplasmic distribution reminiscent of the cytoplasmic IBs as previously observed either in infected cells or cells cotransfected with N- and P-proteins (17). Furthermore, the EGFP-L puncta strongly colocalized with HSP90. In contrast, no EGFP-L fluorescence could be detected in transfected cells that had been treated with 17-AAG, suggesting its destabilization (Fig. 5A). To quantify the percentage of cells that were positive for EGFP-L expression, in the presence and absence of 17-AAG, transfected cells were analyzed by flow cytometry. In the absence of drug, around 10% of the transfected cell population was positive for L-EGFP expression, either when expressed alone or with the P-protein (Fig. 5B). However, the numbers of positive cells were significantly reduced to almost undetectable levels (<1% positive cells; only slightly above the autofluorescent background attributable to the drug itself) in 17-AAG-treated cells, irrespective of P coexpression (Fig. 5B). In contrast, the percentage of EGFP-P-positive cells was not significantly affected by 17-AAG treatment, and only a modest effect was observed on the fluorescence of EGFP-ADAR1, which was used as an irrelevant control (Fig. 5B). Thus, 17-AAG treatment reduced the expression of the EGFP-L protein, but not that of other EGFP-tagged proteins, indicating the effect was specific to the L-protein domain.
FIG 5.

(A) Indirect immunofluorescence confocal microscopy, showing the localization of HSP90 with L-EGFP in cells in either the presence or absence of 0.5 μM 17-AAG, which was added at 4 h posttransfection. L-EGFP in untreated cells showed punctate staining. HSP90 was labeled in red, and L-EGFP was detected as green areas. Colocalization was determined by the yellow signal from the merged image. No L-EGFP protein was visualized in minireplicon-expressing cells treated with 17-AAG. (B) Flow cytometry analysis showing that HSP90 inhibition reduces the percentage of L-GFP-positive cells. BSRT7 cells were transfected with the following plasmids: L-EGFP (L), L-EGFP plus P (L+P), P-EGFP (P), ADAR1-EGFP (ADAR1), or L (−) and cultured in the presence or absence of 1 μM 17-AAG for 18 h prior to harvesting. The mean EGFP fluorescence from 20,000 cells was measured by flow cytometry, and the percentage of EGFP-positive cells was calculated following gating of EGFP-negative cells (−). Error bars represent the standard deviations from biological triplicate samples. ***, P < 0.0001 by unpaired two-tailed t test. (C). Pulse-chase analysis of the minireplicon system to test whether HSP90 inhibition destabilizes L-EGFP. BSRT7 cells were transfected with the indicated plasmids: L-EGFP, L-EGFP plus P-EGFP, or ADAR1-EGFP. Nontreated and 17-AAG-treated cells were pulse-labeled 24 h posttransfection with [35S]methionine for 30 min and either harvested immediately (t = 0) or chased with medium containing an excess of cold methionine for 2 h at 37°C (t = 2). EGFP-tagged proteins were isolated from the cell lysates by using EGFP-Trap beads, and proteins were analyzed by SDS-PAGE and autoradiography. HSP90 inhibition destabilizes L-EGFP, as shown by the absence of L-EGFP alone or L-EGFP and P in the presence of 1 μM 17-AAG. The species corresponding to L-EGFP, P-EGFP, and ADAR1-EGFP are indicated as L-GFP, P-GFP, and GFP-ADAR, respectively.
To determine whether reduced L expression in 17-AAG-treated cells was the result of protein destabilization caused by loss of HSP90 chaperone function, we pulse-labeled transfected cells with [35S]methionine. Subsequently, the accumulation of GFP-tagged polypeptides was analyzed by SDS-PAGE and autoradiography following GFP-Trap IP, either before or after a chase period with an excess of cold methionine (Fig. 5C). Inhibition of HSP90 chaperone function by 17-AAG had little effect on the quantity of EGFP-L, EGFP-P, or (as a further control) EGFP-ADAR1 synthesized during the pulse period (Fig. 5C; compare the + and – samples for t = 0). However, while both EGFP-P and ADAR1 proteins showed a slight increase in degradation at 2 h postchase in response to 17-AAG treatment, EGFP-L was undetectable in 17-AAG-treated cells (Fig. 5C, compare the + and – samples for t = 2). As expected, EGFP-L was also the least stable protein in the absence of 17-AAG. Although its stability was increased by coexpression of EGFP-P, the presence of EGFP-P was not sufficient to prevent the accelerated degradation of EGFP-L following treatment with 17-AAG. Thus, blocking HSP90 chaperone function directly destabilizes L protein, independently of P. Overall, we concluded that HSP90 is a positive viral factor in the biology of HRSV, being essential for viral gene expression by promoting the stability of the viral RNA polymerase.
Role of HSP70 in the replication of HRSV.
Several other chaperones were identified as potential interaction partners of L-EGFP and/or P-EGFP in the context of the replicon, including the chaperone HSP70 (with high significance but a lower binding ratio than HSP90) and cochaperones: STIP1 and DnaJ (Hsp40) homolog, subfamily A, member 2 (DNJA2). There are no inhibitors for the cochaperones STIP1 and DnaJ. Therefore, a small interfering RNA (siRNA) approach was used to ablate the mRNAs encoding these proteins to investigate whether they had a potential role in the virus life cycle. While both proteins were ablated/decreased in abundance by the siRNA approach, the data indicated that this has no apparent affect on virus biology (data not shown).
HSP70 was previously identified as interacting with the L- and P-proteins coexpressed and purified from insect cells (11) and also with the P-protein alone (12). Using the minireplicon system in BSRT7 cells, we investigated whether four inhibitors of HSP70 chaperone function (PIF, VER, MKT-077, and YM-1) had an effect on viral gene expression. The data indicated that luciferase activity decreased in a dose-dependent manner with increasing concentrations of PIF and VER (Fig. 6A). However, when cell viability was assessed by measuring intracellular ATP levels, both drugs were clearly toxic at 50 μM and above. For PIF, there was no evidence of selective inhibition of HRSV gene expression, while for VER, virus gene expression was around 10-fold more sensitive to the inhibitor than cellular ATP content (Fig. 6A). The actions of MKT-077 and YM-1 were also evaluated in this system. However, with both of these inhibitors there was no significant alteration in the amount of luciferase compared to the untreated controls (Fig. 6A). The data indicated that treated cells expressing the minireplicon VER had the greatest effect on its ability to produce luciferase.
FIG 6.

HSP70 inhibitors reduce HRSV gene expression. (A) Luciferase activity was analyzed from BSRT7 cells transfected with the complete (+ L) or incomplete (− L) minireplicon system and incubated in the presence and absence of PIF, VER, MKT-077, or YM-1. Results (black lines) are expressed as the percentage of RLU produced in the absence of drug. Cell viability was assessed by measuring intracellular ATP levels (gray lines). All data are the means ± standard deviations of triplicate samples. (B) Inhibition of HSP70 by either PIF, VER, MKT-077, or YM-01 resulted in a decrease in an abundance of HRSV proteins at higher concentrations. VER was shown to be the most effective inhibitor in the context of the replicon system and wild-type infection.
The actions of these inhibitors were also evaluated in HRSV-infected A549 cells by using immunoblotting to compare the level of viral proteins between untreated and treated, infected cells. No viral polypeptides were detected in mock-infected cells as expected, while cellular GAPDH levels remained constant across the drug concentrations tested, suggesting no major toxicity effects in this cell type when mock infected (Fig. 6B). For PIF, only the highest concentration of drug tested partially decreased the abundance of viral proteins, and GAPDH abundance remained unchanged. VER caused a dose-dependent decrease in viral protein accumulation compared to the untreated or vehicle control (DMSO) at 5 μM and above, but GAPDH abundance also decreased in infected cells treated with >50 μM VER (Fig. 6B). For MKT-077 and YM-1, the abundance of viral proteins began to decrease when the concentrations of these drugs were greater than 5 μM (Fig. 6B). Thus, inhibition of HSP70 had a deleterious consequence for virus protein expression, and treatment with VER appeared to be the most effective. However, because of the potential toxicity of the drugs in cells, we transferred our efforts to analyzing the more effective VER inhibitor in an in vitro system to avoid this potential complication.
HSP70 affects HRSV mRNA transcription but not HRSV RNA polymerization.
Whereas published data regarding HSP90 indicate that it has a role in polymerase stability, the data available regarding HSP70 indicate that it has a more direct role in HRSV polymerase activity. Previously, Brown et al. showed that following HRSV infection, a proportion of HSP70 relocalized to lipid raft-associated, cytoplasmic inclusions that are believed to be viral transcription/replication factories (13). Furthermore, it was shown that an antibody against HSP70 inhibited lipid raft-associated HRSV polymerase activity (13). More recent studies have shown that HSP70 copurifies with heterologously expressed HRSV L/P complexes (11). These findings suggest that HSP70 binds to the HRSV L/P complex and affects polymerase activity. This is consistent with the EGFP-Trap results (see Tables S1 and S2 in the supplemental material), although these may also reflect interaction with RNPs. To investigate this possibility, the effects of VER, the inhibitor that had the greatest effect on virus biology in the context of the minireplicon and wild-type-infected cells, were examined in two different HRSV polymerase assays. First, we examined HRSV polymerase activity using an in vitro cell extract assay (26). HRSV-infected HEp2 cells were gently disrupted and lysates were incubated in a buffer containing actinomycin D (to inhibit cellular polymerases) and [α-32P]NTP. The radiolabeled RNA produced was analyzed by denaturing polyacrylamide gel electrophoresis to measure the sum of multiple different polymerase functions on an intact nucleocapsid template, in a similar manner to that used by Brown and coworkers (13). Analysis of the RNA produced in this assay showed that in the absence of VER, a number of different RNA transcripts were generated in reaction mixtures containing extract from HRSV-infected cells but not mock-infected cells (Fig. 7A, compare lanes 1 and 2). This confirmed that the HRSV polymerase was active and that the signal was not contaminated with RNA generated from cellular polymerases. Addition of 0.5 μM VER led to a slight increase in the abundance of labeled RNA, but further increases led to a significant reduction (Fig. 7A, lanes 2 to 6, and C). These findings indicated that HSP70 is required for, or augments, HRSV polymerase activity on an intact nucleocapsid template.
FIG 7.

HSP70 affects HRSV transcription, but not RNA polymerization. (A) Addition of VER inhibits RSV mRNA synthesis in a cell extract in vitro. Lane 1, RNA from a reaction mixture containing mock-infected cell extract; lanes 2 to 6, RNA from a reaction mixture containing HRSV-infected cell extracts, with [α-32P]GTP as the labeled NTP and various concentrations of VER included in the reaction mixture, as indicated. The asterisk indicates the RNA band that was quantified to generate the graph in panel C. (B) Addition of VER did not inhibit HRSV RNA synthesis in an assay utilizing purified polymerase complexes. Lane 2, a negative control in which the L/P/HSP70 complex was omitted from the reaction mixture; lane 3, a negative control in which reaction mixtures were incubated with an L/P/HSP70 complex containing the N812A substitution at the catalytic site of the L-protein; lanes 3 to 8 show RNA generated from WT L/P/HSP70 complexes in the presence of various concentrations of VER. Lane 1 contains a size marker representing the expected products of de novo RNA synthesis from position 1 of the trc promoter. The bands representing RNA generated by de novo synthesis from the promoter, or 3′ extension of the input template, are indicated. (C) Bar chart showing levels of RNA synthesis in the HEp2 cell extract assay, based on quantification of the band marked with an asterisk in panel A. The data show the means and ranges of two independent experiments. (D) Bar chart showing levels of RNA synthesis in the minimalist RNA synthesis assay involving purified L/P/HSP70 complexes, based on quantification of the 21-nucleotide band marked with an asterisk in panel B. The data show the means and standard errors of the means from two or three independent experiments. In panels C and D, the data from each experiment were normalized to the level of labeled RNA produced in the absence of VER, which was set to a value of 1.
Next, we analyzed HRSV polymerase activity in a minimalist in vitro RNA synthesis assay, as described previously (11). This assay utilized recombinant HRSV L- and P-proteins, purified as a complex from insect cells. HSP70 consistently copurifies with L/P under the purification conditions used, in approximately an equimolar amount with L, and is not completely removed even under highly stringent washing conditions (11, 35; unpublished data). Thus, it is possible that HSP70 contributes to the RNA synthesis activity of the L/P complex. Purified L/P/HSP70 complexes were incubated with a naked RNA oligonucleotide, containing nucleotides 1 to 25 of the trailer-complement (trc) promoter sequence, in buffer containing NTPs, including [α-32P]GTP. The RNA products were analyzed by denaturing gel electrophoresis. In the absence of VER, WT L/P/HSP70 complexes yielded a series of bands representing products of de novo RNA synthesis from the promoter and RNA generated by back-priming and 3′ extension of the input template (Fig. 7B, lane 4) (for characterization of the different products, see reference 11). These bands could not be detected if WT L/P/HSP70 was omitted or replaced with a mutant version of the complex containing an amino acid substitution in the polymerization domain of L. In contrast to what was observed in the HEp2 cell extract assay, inclusion of VER in the reaction mixtures had no detectable effect on RNA synthesis (Fig. 7B, lanes 4 to 8, and D). This result showed that HSP70 activity does not affect the HRSV polymerization activity of the L/P complex per se. Together, these findings indicate that HSP70 affects HRSV polymerase activity in the context of a complete nucleocapsid but not on a short, naked RNA template.
DISCUSSION
In this study, the cellular interactomes of the HRSV L- and P-proteins were investigated. IP, coupled with quantitative proteomic analysis, indicated that HSP90 and several other chaperone proteins involved in the posttranslational folding of cellular proteins interacted with the L- and P-proteins. The ratio of N- and P-proteins is suggestive that RNPs were also isolated. Other potential cellular interacting proteins included the identification of the major vault protein (MVP), identified with the P-protein (see Table S2 in the supplemental material). MVP constitutes the main component of cytoplasmic ribonucleoprotein particles and is possibly a generic antiviral factor (36). Several other groups of proteins were identified, including translation factors, cytoskeletal components, hnRNPs, and splicing factors. However, extensive analyses of “bead proteomes” have suggested that these proteins may be common contaminants in IPs, unless they have a high binding ratio between the bait and control (29, 30). However, some of these interactions may be functionally significant, as demonstrated for the cytoskeleton proteins actin and profilin, which play important roles in HRSV RNA synthesis (37–39).
Interactors were identified by using GFP-Trap resin to selectively precipitate EGFP-tagged versions of the HRSV L- and P-proteins, which maintain biological activity in the context of a minireplicon system (17). Insertion of the EGFP open reading frame into either polypeptide reduced transcriptional activity in the minireplicon system compared to the native HRSV L- or P-proteins, as previously reported (17). This diminution of transcription activity or altered phenotype is commonly found for the insertion of additional ORFs in other RdRps and can depend on the location of the insertion site in the RdRp (40–42). Thus, one concern is that cellular proteins which form interactions with the L- and/or P-protein may not do so if the function of the viral protein is attenuated or the insertion of the EGFP disrupts a binding site; consequently, these proteins will not be identified in an interactome screen. Nevertheless, the use of tagged viral proteins that still maintained biological activity in the cell was advantageous in comparison to similar systems in which biological activity was not or could not be established. Furthermore, this approach permitted the isolation of L-protein-containing complexes, as no antibody to the HRSV L-protein (with which to perform IPs on the native protein) is publicly available.
Consistent with previously published data from investigations of the role of HSP90 in HRSV biology (14, 15), inhibiting the protein chaperone function of HSP90 by using the small-molecule inhibitor 17-AAG resulted in a decrease in viral mRNA and protein levels. In the context of the HRSV minireplicon system, 17-AAG treatment led to the ablation of reporter gene expression but not a decrease in the abundance of the proteins expressed from the support plasmids, indicating a striking effect on virus transcription and/or replication (Fig. 4B). Use of the replicon system (where synthesis of the viral proteins is dependent on T7 transcription of mRNAs rather than the HRSV RNA polymerase) allowed the mechanism of this global effect on viral gene expression to be dissected; 17-AAG treatment specifically reduced the accumulation of the viral L-protein. Expression of EGFP-L in isolation allowed us to perform kinetic analyses of protein expression and stability in the absence and presence of the P-protein cofactor (Fig. 5C). The simplest explanation consistent with our data is that HSP90 directly interacts with the L-protein to perform an essential chaperone function during its folding that is essential for its stability. This is also consistent with previous studies: Geller et al. postulated that the ablation of HSP90 activity had a detrimental effect on HRSV L-protein stability (15), and a similar hypothesis was proposed by Connor et al. based on their analysis of the effect of HSP90 inhibitors on the stability of the related VSV L-protein (16). Using metabolic labeling in conjunction with IP (based on a polyclonal RSV antibody), Geller et al. (15) observed specific degradation of a single ∼250-kDa viral protein, corresponding to the molecular mass of the HRSV L-protein in virus-infected cells. However, as discussed (15), this could not be positively identified due to the lack of specific sera. Using a tagged EGFP-L fusion protein which could be specifically identified through the EGFP moiety, we were able to confirm the effect of HSP90 activity on L-protein (in the presence or absence of P-protein) by using several different approaches.
Apart from HSP90, other chaperones that potentially interacted with the L- and P-proteins were HSP70, STIP1, and DNAJA2. In the absence of available inhibitors, siRNA was used to ablate STIP1 and DNAJA2 in HRSV infected cells, but with no apparent effect on virus biology (data not shown). Previous studies indicated that HSP70 might play a direct role in HRSV RNA synthesis and/or virus assembly (13). The data presented here potentially supported these findings. PIF and VER affected both replicon activity and protein synthesis in HRSV-infected cells, suggesting a positive role for HSP70 in replication of viral RNA, whereas MKT-077 and YM-1 appeared to negatively affect the abundance of viral proteins but not the synthesis of viral RNA. This illustrates the potentially difficulty of working with inhibitors and the necessity to use different experimental approaches to assess their impact on virus biology.
To investigate the effect of HSP70 in further detail, we employed an in vitro system. This assay, involving HRSV-infected HEp2 cell extract, showed that addition of the HSP70 inhibitor VER to the reaction mixture led to a 5-fold decrease in the level of HRSV RNA synthesis, indicating that HSP70 plays a positive role in HRSV RNA synthesis (Fig. 7A and C). In contrast to these findings, VER had no detectable effect in an experiment utilizing a minimalist assay, involving purified recombinant L- and P-proteins in complex with HSP70 (Fig. 7B and D).
A major difference between the two assays lies in the nature of the template. In the HEp2 cell-based extract assay, the template was an intact nucleocapsid, whereas in the minimalist assay (complex purified from insect cells), it was a naked RNA oligonucleotide. Thus, a possible explanation for why VER inhibited RNA synthesis in the HEp2 cell extract assay, but not in the minimalist assay, is that HSP70 might help the polymerase remodel the nucleocapsid to allow RNA synthesis to occur efficiently. This hypothesis is consistent with data obtained for other nonsegmented negative-strand RNA viruses. HSP70 has been shown to associate with the N-protein of the paramyxovirus measles virus and the rhabdovirus rabies virus and to positively affect RNA synthesis in both cases (43–45). Thus, HSP70 might be involved in nucleocapsid remodeling for a number of nonsegmented negative-strand RNA viruses, but it may accomplish this role by binding different proteins within the nucleocapsid complex, depending on the virus. Surprisingly, when a lower concentration of VER (0.5 μM) was included in the HEp2 cell extract assay mixture, there was a slight increase in RNA synthesis (Fig. 7A and C). This is also similar to results obtained with an HSP70 antibody, with which it was shown that a low concentration of antibody led to a slight increase in RNA synthesis, but higher concentrations inhibited synthesis (13). This intriguing finding suggests that the effect of HSP70 depends on its active concentration. It is possible that relatively lower HSP70 activity promotes nucleocapsid restructuring and RNA synthesis, whereas a relatively higher HSP70 activity renders the nucleocapsid inactive.
We postulate that HSP90 and other chaperone proteins, such as HSP70, may also be required for maintaining the correct folding, and therefore functionality, of viral proteins in the RNP complex. In this system, EGFP-L protein was ablated in the presence of 17-AAG, and heat shock proteins in general have been found to be essential for polymerase stability/activity and/or viral replication in other negative-strand RNA viruses, including VSV and other paramyxoviruses (16), La Crosse bunyavirus (16), rabies virus (44), Ebolavirus (46), and influenza virus (47–49).
Although HRSV replication is inhibited in cell culture by a range of HSP90 inhibitors (14, 15), continuous passage of the virus in the presence of the drugs did not select for resistant viruses (15), suggesting that inhibition of chaperone proteins could offer a generic antiviral therapeutic approach (23). The general approach of using quantitative proteomics to identify cellular proteins that are critical for virus biology may open up new avenues of therapeutic intervention. This illustrates the versatility of targeting cellular proteins, rather than viral proteins, as illustrated by work targeting EBOV biology (21, 46). Transiently targeting the function of host cell proteins which are crucial to virus biology offers an exciting new therapeutic avenue with the potential to solve the problem of resistance, as proviral cellular proteins are evolutionarily static on the time scale of lytic virus replication, as well as genetically separate from the genome, which would benefit from resistance. Many of these small-molecule inhibitors are preexisting clinically relevant therapeutics and could be potentially safely repurposed as part of an antiviral strategy.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Carmo-Fonseca for the gift of EGFP-ADAR1.
This research was supported by the award of a Medical Research Council (MRC) Project Grant (MR/K000276/1) to P.D., J.N.B., and J.A.H., an MRC studentship to J.N.B. and J.A.H., NIHR funding to J.A.H., and NIH grant R01AI074903 to R.F. W.A. is supported by the Ministry of Education, Kingdom of Saudi Arabia.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01783-14.
REFERENCES
- 1.Collins PL, Melero JA. 2011. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res 162:80–99. doi: 10.1016/j.virusres.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nokes JD, Cane PA. 2008. New strategies for control of respiratory syncytial virus infection. Curr Opin Infect Dis 21:639–643. doi: 10.1097/QCO.0b013e3283184245. [DOI] [PubMed] [Google Scholar]
- 3.Collins PL, Graham BS. 2008. Viral and host factors in human respiratory syncytial virus pathogenesis. J Virol 82:2040–2055. doi: 10.1128/JVI.01625-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagne N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eleouet JF, Rey FA. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283. doi: 10.1126/science.1177634. [DOI] [PubMed] [Google Scholar]
- 5.Fearns R, Collins PL. 1999. Role of the M2-1 transcription antitermination protein of respiratory syncytial virus in sequential transcription. J Virol 73:5852–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galloux M, Tarus B, Blazevic I, Fix J, Duquerroy S, Eleouet JF. 2012. Characterization of a viral phosphoprotein binding site on the surface of the respiratory syncytial nucleoprotein. J Virol 86:8375–8387. doi: 10.1128/JVI.00058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wileman T. 2006. Aggresomes and autophagy generate sites for virus replication. Science 312:875–878. doi: 10.1126/science.1126766. [DOI] [PubMed] [Google Scholar]
- 8.Kirkegaard K. 2009. Subversion of the cellular autophagy pathway by viruses. Curr Top Microbiol Immunol 335:323–333. doi: 10.1007/978-3-642-00302-8_16. [DOI] [PubMed] [Google Scholar]
- 9.Kidmose RT, Vasiliev NN, Chetverin AB, Andersen GR, Knudsen CR. 2010. Structure of the Qβ replicase, an RNA-dependent RNA polymerase consisting of viral and host proteins. Proc Natl Acad Sci U S A 107:10884–10889. doi: 10.1073/pnas.1003015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagata K, Kawaguchi A, Naito T. 2008. Host factors for replication and transcription of the influenza virus genome. Rev Med Virol 18:247–260. doi: 10.1002/rmv.575. [DOI] [PubMed] [Google Scholar]
- 11.Noton SL, Deflube LR, Tremaglio CZ, Fearns R. 2012. The respiratory syncytial virus polymerase has multiple RNA synthesis activities at the promoter. PLoS Pathog 8:e1002980. doi: 10.1371/journal.ppat.1002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliveira AP, Simabuco FM, Tamura RE, Guerrero MC, Ribeiro PG, Libermann TA, Zerbini LF, Ventura AM. 2013. Human respiratory syncytial virus N, P and M protein interactions in HEK-293T cells. Virus Res 177:108–112. doi: 10.1016/j.virusres.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Brown G, Rixon HW, Steel J, McDonald TP, Pitt AR, Graham S, Sugrue RJ. 2005. Evidence for an association between heat shock protein 70 and the respiratory syncytial virus polymerase complex within lipid-raft membranes during virus infection. Virology 338:69–80. doi: 10.1016/j.virol.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Radhakrishnan A, Yeo D, Brown G, Myaing MZ, Iyer LR, Fleck R, Tan BH, Aitken J, Sanmun D, Tang K, Yarwood A, Brink J, Sugrue RJ. 2010. Protein analysis of purified respiratory syncytial virus particles reveals an important role for heat shock protein 90 in virus particle assembly. Mol Cell Proteomics 9:1829–1848. doi: 10.1074/mcp.M110.001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geller R, Andino R, Frydman J. 2013. Hsp90 inhibitors exhibit resistance-free antiviral activity against respiratory syncytial virus. PLoS One 8:e56762. doi: 10.1371/journal.pone.0056762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connor JH, McKenzie MO, Parks GD, Lyles DS. 2007. Antiviral activity and RNA polymerase degradation following Hsp90 inhibition in a range of negative strand viruses. Virology 362:109–119. doi: 10.1016/j.virol.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fix J, Galloux M, Blondot ML, Eleouet JF. 2011. The insertion of fluorescent proteins in a variable region of respiratory syncytial virus L polymerase results in fluorescent and functional enzymes but with reduced activities. Open Virol J 5:103–108. doi: 10.2174/1874357901105010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu W, Tran KC, Teng MN, Heesom KJ, Matthews DA, Barr JN, Hiscox JA. 2012. The interactome of the human respiratory syncytial virus NS1 protein highlights multiple effects on host cell biology. J Virol 86:7777–7789. doi: 10.1128/JVI.00460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jourdan SS, Osorio F, Hiscox JA. 2012. An interactome map of the nucleocapsid protein from a highly pathogenic North American porcine reproductive and respiratory syndrome virus strain generated using SILAC-based quantitative proteomics. Proteomics 12:1015–1023. doi: 10.1002/pmic.201100469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emmott E, Munday D, Bickerton E, Britton P, Rodgers MA, Whitehouse A, Zhou EM, Hiscox JA. 2013. The cellular interactome of the coronavirus infectious bronchitis virus nucleocapsid protein and functional implications for virus biology. J Virol 87:9486–9500. doi: 10.1128/JVI.00321-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Dorival I, Wu W, Dowall S, Armstrong S, Touzelet O, Wastling J, Barr JN, Matthews D, Carroll M, Hewson R, Hiscox JA. 2014. Elucidation of the Ebola virus VP24 cellular interactome and disruption of virus biology through targeted inhibition of host cell protein function. J Proteome Res 13:5120–2135. doi: 10.1021/pr500556d. [DOI] [PubMed] [Google Scholar]
- 22.Neckers L, Tatu U. 2008. Molecular chaperones in pathogen virulence: emerging new targets for therapy. Cell Host Microbe 4:519–527. doi: 10.1016/j.chom.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geller R, Taguwa S, Frydman J. 2012. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim Biophys Acta 1823:698–706. doi: 10.1016/j.bbamcr.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ueba O. 1978. Respiratory syncytial virus. I. Concentration and purification of the infectious virus. Acta Med Okayama 32:265–272. [PubMed] [Google Scholar]
- 25.Munday DC, Emmott E, Surtees R, Lardeau CH, Wu W, Duprex WP, Dove BK, Barr JN, Hiscox JA. 2010. Quantitative proteomic analysis of A549 cells infected with human respiratory syncytial virus. Mol Cell Proteomics 9:2438–2459. doi: 10.1074/mcp.M110.001859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canter DM, Jackson RL, Perrault J. 1993. Faithful and efficient in vitro reconstitution of vesicular stomatitis virus transcription using plasmid-encoded L and P proteins. Virology 194:518–529. doi: 10.1006/viro.1993.1290. [DOI] [PubMed] [Google Scholar]
- 27.Desterro JM, Keegan LP, Lafarga M, Berciano MT, O'Connell M, Carmo-Fonseca M. 2003. Dynamic association of RNA-editing enzymes with the nucleolus. J Cell Sci 116:1805–1818. doi: 10.1242/jcs.00371. [DOI] [PubMed] [Google Scholar]
- 28.Tanner SJ, Ariza A, Richard CA, Kyle HF, Dods RL, Blondot ML, Wu W, Trincao J, Trinh CH, Hiscox JA, Carroll MW, Silman NJ, Eleouet JF, Edwards TA, Barr JN. 2014. Crystal structure of the essential transcription antiterminator M2-1 protein of human respiratory syncytial virus and implications of its phosphorylation. Proc Natl Acad Sci U S A 111:1580–1585. doi: 10.1073/pnas.1317262111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trinkle-Mulcahy L, Boulon S, Lam YW, Urcia R, Boisvert FM, Vandermoere F, Morrice NA, Swift S, Rothbauer U, Leonhardt H, Lamond A. 2008. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J Cell Biol 183:223–239. doi: 10.1083/jcb.200805092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boulon S, Ahmad Y, Trinkle-Mulcahy L, Verheggen C, Cobley A, Gregor P, Bertrand E, Whitehorn M, Lamond AI. 2010. Establishment of a protein frequency library and its application in the reliable identification of specific protein interaction partners. Mol Cell Proteomics 9:861–879. doi: 10.1074/mcp.M900517-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asenjo A, Mendieta J, Gomez-Puertas P, Villanueva N. 2008. Residues in human respiratory syncytial virus P protein that are essential for its activity on RNA viral synthesis. Virus Res 132:160–173. doi: 10.1016/j.virusres.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 32.Prevost M, Chamousset D, Nasa I, Freele E, Morrice N, Moorhead G, Trinkle-Mulcahy L. 2013. Quantitative fragmentome mapping reveals novel, domain-specific partners for the modular protein RepoMan (recruits PP1 onto mitotic chromatin at anaphase). Mol Cell Proteomics 12:1468–1486. doi: 10.1074/mcp.M112.023291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Wever V, Lloyd DC, Nasa I, Nimick M, Trinkle-Mulcahy L, Gourlay R, Morrice N, Moorhead GB. 2012. Isolation of human mitotic protein phosphatase complexes: identification of a complex between protein phosphatase 1 and the RNA helicase Ddx21. PLoS One 7:e39510. doi: 10.1371/journal.pone.0039510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia J, Garcia-Barreno B, Vivo A, Melero JA. 1993. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 195:243–247. doi: 10.1006/viro.1993.1366. [DOI] [PubMed] [Google Scholar]
- 35.Tremaglio CZ, Noton SL, Deflube LR, Fearns R. 2013. Respiratory syncytial virus polymerase can initiate transcription from position 3 of the leader promoter. J Virol 87:3196–3207. doi: 10.1128/JVI.02862-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S, Hao Q, Peng N, Yue X, Wang Y, Chen Y, Wu J, Zhu Y. 2012. Major vault protein: a virus-induced host factor against viral replication through the induction of type-I interferon. Hepatology 56:57–66. doi: 10.1002/hep.25642. [DOI] [PubMed] [Google Scholar]
- 37.Burke E, Dupuy L, Wall C, Barik S. 1998. Role of cellular actin in the gene expression and morphogenesis of human respiratory syncytial virus. Virology 252:137–148. doi: 10.1006/viro.1998.9471. [DOI] [PubMed] [Google Scholar]
- 38.Burke E, Mahoney NM, Almo SC, Barik S. 2000. Profilin is required for optimal actin-dependent transcription of respiratory syncytial virus genome RNA. J Virol 74:669–675. doi: 10.1128/JVI.74.2.669-675.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harpen M, Barik T, Musiyenko A, Barik S. 2009. Mutational analysis reveals a noncontractile but interactive role of actin and profilin in viral RNA-dependent RNA synthesis. J Virol 83:10869–10876. doi: 10.1128/JVI.01271-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duprex WP, Collins FM, Rima BK. 2002. Modulating the function of the measles virus RNA-dependent RNA polymerase by insertion of green fluorescent protein into the open reading frame. J Virol 76:7322–7328. doi: 10.1128/JVI.76.14.7322-7328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruedas JB, Perrault J. 2009. Insertion of enhanced green fluorescent protein in a hinge region of vesicular stomatitis virus L polymerase protein creates a temperature-sensitive virus that displays no virion-associated polymerase activity in vitro. J Virol 83:12241–12252. doi: 10.1128/JVI.01273-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finke S, Brzozka K, Conzelmann KK. 2004. Tracking fluorescence-labeled rabies virus: enhanced green fluorescent protein-tagged phosphoprotein P supports virus gene expression and formation of infectious particles. J Virol 78:12333–12343. doi: 10.1128/JVI.78.22.12333-12343.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lahaye X, Vidy A, Fouquet B, Blondel D. 2012. Hsp70 protein positively regulates rabies virus infection. J Virol 86:4743–4751. doi: 10.1128/JVI.06501-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X, Bourhis JM, Longhi S, Carsillo T, Buccellato M, Morin B, Canard B, Oglesbee M. 2005. Hsp72 recognizes a P binding motif in the measles virus N protein C-terminus. Virology 337:162–174. doi: 10.1016/j.virol.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, Glendening C, Linke H, Parks CL, Brooks C, Udem SA, Oglesbee M. 2002. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J Virol 76:8737–8746. doi: 10.1128/JVI.76.17.8737-8746.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith DR, McCarthy S, Chrovian A, Olinger G, Stossel A, Geisbert TW, Hensley LE, Connor JH. 2010. Inhibition of heat-shock protein 90 reduces Ebola virus replication. Antiviral Res 87:187–194. doi: 10.1016/j.antiviral.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chase G, Deng T, Fodor E, Leung BW, Mayer D, Schwemmle M, Brownlee G. 2008. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 377:431–439. doi: 10.1016/j.virol.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 48.Momose F, Naito T, Yano K, Sugimoto S, Morikawa Y, Nagata K. 2002. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J Biol Chem 277:45306–45314. doi: 10.1074/jbc.M206822200. [DOI] [PubMed] [Google Scholar]
- 49.Manzoor R, Kuroda K, Yoshida R, Tsuda Y, Fujikura D, Miyamoto H, Kajihara M, Kida H, Takada A. 2014. Heat shock protein 70 modulates influenza A virus polymerase activity. J Biol Chem 289:7599–7614. doi: 10.1074/jbc.M113.507798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.