

ABSTRACT
Adenoviruses encode a set of highly abundant microRNAs (mivaRNAs), which are generated by Dicer-mediated cleavage of the larger noncoding virus-associated RNAs (VA RNAs) I and II. We performed deep RNA sequencing to thoroughly investigate the relative abundance of individual single strands of mivaRNA isoforms in human A549 cells lytically infected with human adenovirus 5 (Ad5) at physiologically relevant multiplicities of infection (MOIs). In addition, we investigated their relative abundance in the endogenous RNA-induced silencing complexes (RISCs). The occupation of endogenous RISCs by mivaRNAs turned out to be pronounced but not as dominant as previously inferred from experiments with AGO2-overexpressing cells infected at high MOIs. In parallel, levels of RISC-incorporated mRNAs were investigated as well. Analysis of mRNAs enriched in RISCs in Ad5-infected cells revealed that only mRNAs with complementarity to the seed sequences of mivaRNAs derived from VA RNAI but not VA RNAII were overrepresented among them, indicating that only mivaRNAs derived from VA RNAI are likely to contribute substantially to the posttranscriptional downregulation of host gene expression. Furthermore, to generate a comprehensive picture of the entire transcriptome/targetome in lytically infected cells, we determined changes in cellular miRNA levels in both total RNA and RISC RNA as well, and bioinformatical analysis of mRNAs of total RNA/RISC fractions revealed a general, genome-wide trend toward detargeting of cellular mRNAs upon infection. Lastly, we identified the direct targets of both single strands of a VA RNAI-derived mivaRNA that constituted one of the two most abundant isoforms in RISCs of lytically infected A549 cells.
IMPORTANCE Viral and cellular miRNAs have been recognized as important players in virus-host interactions. This work provides the currently most comprehensive picture of the entire mRNA/miRNA transcriptome and of the complete RISC targetome during lytic adenovirus infection and thus represents the basis for a deeper understanding of the interplay between the virus and the cellular RNA interference machinery. Our data suggest that, at least in the model system that was employed, lytic infection by Ad5 is accompanied by a measurable global net detargeting effect on cellular mRNAs, and analysis of RISC-associated viral small RNAs revealed that the VA RNAs are the only source of virus-encoded miRNAs. Moreover, this work allows to assess the power of individual viral miRNAs to regulate cellular gene expression and provides a list of proven and putative direct targets of these miRNAs, which is of importance, given the fact that information about validated targets of adenovirus-encoded miRNAs is scarce.
INTRODUCTION
Over the past few years, a number of microRNAs (miRNAs) have been identified and characterized in their ability to posttranscriptionally regulate the expression of human and nonhuman genes (1, 2). The presence of miRNA-encoding genes is not restricted to eukaryotes; they are also present in the genomes of certain viruses (3–6). Depending on the serotype, human adenoviruses encode one or two so-called virus-associated RNAs (VA RNAs) that can be processed into functional miRNAs (7–10). Members of adenovirus species C encode 2 well-structured VA RNAs, VA RNAI and VA RNAII (11–14). These RNAs are approximately 160 nucleotides (nt) long and are generated by RNA polymerase III. They are expressed throughout the infection cycle but reach their highest concentration during the late phase of infection (15). Whereas VA RNAII is not vital for virus replication, VA RNAI, the VA RNA produced predominantly, is essential for efficient translation of mRNAs during the late phase of infection (16). A well-known function of VA RNAI is the inhibition of interferon-inducible protein kinase R (PKR), which is activated in infected cells by viral double-stranded RNA (dsRNA) (17, 18) and thus constitutes a component of the antiviral response. By binding to PKR, VA RNAI prevents PKR from phosphorylating the translation initiation factor eIF-2α, which would otherwise result in the shutdown of cellular protein synthesis (19, 20, 21). Furthermore, VA RNAs I and II have been been shown to be processed by Dicer into miRNA-like small RNAs (mivaRNAs) (7, 8, 10, 22). Because large amounts of VA RNAs are exported from the nucleus by Exportin-5, which functions as the exporter of cellular miRNAs (23), and are processed by Dicer, and because some of the processing products are incorporated into the RNA-induced silencing complex, VA RNAs and mivaRNAs have been suggested to inhibit the cellular RNA interference (RNAi) pathway (7, 8, 9, 10, 22).
MivaRNAs exist in several isoforms that possess slightly variable ends (24, 25, 26). In VA RNAI-derived mivaRNAs, one end is determined by the transcriptional start site of RNA polymerase III, which initiates transcription predominantly either at a specific “A” position [mivaRNAI(A)] or 3 nucleotides downstream at a specific “G” position [mivaRNAI(G)] (24, 27). The other end of the small RNA (sRNA) double strand is determined by Dicer, which cleaves within the stem region of VA RNAI predominantly at positions 137 and 138, based on the numbering of the shorter VA RNAI(G) form by Xu et al. (22), within the 3′-arm of the VA RNA and at the corresponding opposite site located on the 5′-arm. This generates mivaRNAI(A)-137/mivaRNAI(G)-137 and mivaRNAI(A)-138/mivaRNAI(G)-138, respectively. MivaRNAs derived from VA RNAII of lytically infected cells were reported to be less heterogeneous, with the most abundant mivaRNA generated by Dicer-mediated cleavage predominantly at position 138 (mivaRNAII-138) (25). RNAs derived from both VA RNAI and VA RNAII have been shown to be produced by both lytically and persistently infected cells (8, 22, 24, 26).
A previous study demonstrated that adenoviral mivaRNAs are able to target cellular mRNAs (28). This study focused on one of the most abundant mivaRNA single strands derived from the 3′-arm of VA RNAI [3′mivaRNAI(G)-138] encoded by adenovirus 5 (Ad5). A set of cellular genes that are downregulated in the presence of this mivaRNA was revealed, and for one mRNA (TIA1), direct targeting by the mivaRNA was demonstrated. However, because (even more abundant) VA RNAI-derived mivaRNA isoforms exist, of which both single strands may target cellular mRNAs [e.g., 5′mivaRNAI(A) and 3′mivaRNAI-137], and because TIA1 is the only direct target of a mivaRNA that has been reported so far, the spectrum of proven, direct, mivaRNA targets is basically still unknown. It is furthermore unknown which mivaRNAs are the likely key players in downregulating cellular gene expression, and no comprehensive picture of the targetome of lytically infected cells exists to date.
MATERIALS AND METHODS
Cell lines and viruses.
HEK293 (human embryonic kidney; ATCC CRL-1573) and A549 (human epithelial lung carcinoma; ATCC CCL-185) cells were cultivated in Dulbecco's modified Eagle's medium (DMEM) with stabilized glutamine (PAA Laboratories, Pasching, Austria) supplemented with 10% fetal bovine serum (FBS; PAA Laboratories) in a humidified 5% CO2 atmosphere at 37°C. Wild-type Ad5 (ATCC VR-5), the VA RNAI and II double mutant dl-sub720 (29), and the recombinant, replication-deficient adenoviruses Ad-Luc-as, Ad-mi-, and Ad-Fluc-mi1 (30) were amplified in HEK293 cells. Viruses were purified using standard CsCl density gradient ultracentrifugation. Titers of infectious virus particles were determined with A549 or HEK293 cells by 50% tissue culture infective dose (TCID50) assays.
MivaRNA mimics.
Single-stranded RNA (ssRNA) oligonucleotides mimicking mivaRNAI(A)-137 [mivaRNAI(A)-137-5′, AGCGGGCACUCUUCCGUGGUCUGG; mivaRNAI(A)-137-3′, AGACAACGGGGGAGUGCUCCUUUU] and mivaRNAI(G)-137 [mivaRNAI(G)-137-5′, GGGCACUCUUCCGUGGUCUGG; mivaRNAI(G)-137-3′, AGACAACGGGGGAGUGCUCCU] were purchased from RiboTask (Odense, Denmark). Oligonucleotides were annealed by combining equimolar amounts (20 μM) of the 2 single strands in a 75-μl reaction mixture containing 50 mM Tris (pH 7.5) and 100 nM NaCl in nuclease-free H2O. The solution was incubated for 2 min at 95°C and allowed to cool gradually to room temperature over 1 h and finally to 4°C over another hour. The mivaRNA mimics were analyzed by gel electrophoresis and stored at −80°C.
Plasmids and dual-luciferase-based reporter assays.
All plasmid reporter vectors were based on psiCHECK-2 (Promega, Mannheim, Germany). Vectors pmiVAI5 and pmiVAI3 carry sequences that are 100% complementary to both mivaRNAI(A)-137 and mivaRNAI(G)-137 single strands derived from the 5′-arm of VA RNAI (target sequence, 5′-ACCAGACCACGGAAGAGTGCCCGCT-3′) or 100% complementary to the respective mivaRNA single strands derived from the 3′-arm of VA RNAI (target sequence, 5′-AAAAGGAGCACTCCCCCGTTGTCTG-3′) inserted into the SgfI and NotI sites within the Renilla luciferase 3′-untranslated region (3′-UTR). Dual-luciferase reporter vectors for validating natural target sequences of mivaRNAs contain the respective target sequences inserted either into the SgfI and PmeI sites within the 3′-UTR or into the AatI and NheI sites within the 5′-UTR of the Renilla luciferase gene. The exact coordinates of the individual sequences are shown in Table S1 in the supplemental material. The vector pAdVAntage, containing the adenovirus 2 VAI and VAII genes under the control of their own promoters, was purchased from Promega.
For dual-luciferase assays, 1.2 × 105 HEK293 cells were seeded into the wells of 96-well plates and cotransfected with 50 ng of individual dual-luciferase reporter vectors or the empty parental psiCHECK-2 vector and either 100 nM mivaRNA mimics or a nontargeting negative-control siRNA (Ambion/Life Technologies Austria, Vienna, Austria). In some cases, the vector pAdVAntage was used instead of mivaRNA mimics at a concentration of 200 ng per reaction volume. Transfections were performed using Lipofectamine 2000 based on the recommendations of the manufacturer (Invitrogen/Life Technologies Austria, Vienna, Austria). Firefly and Renilla luciferase activities were determined at 48 h posttransfection on an EnSpire Multimode plate reader (PerkinElmer, Brunn am Gebirge, Austria) using the Dual-Glo luciferase assay according to the manufacturer's instructions (Promega).
RT-qPCR-based analysis of RNA.
To investigate the downregulation of the expression of candidate target genes, 1 × 105 A549 cells were seeded into the wells of 24-well plates and transfected with 100 nM mivaRNA mimics or a nontargeting control siRNA (Ambion/Life Technologies) by using Lipofectamine 2000. Total RNA was isolated at 48 h posttransfection using an RNeasy minikit (Qiagen, Hilden, Germany). Residual DNA was removed using RQ1 DNase (Promega), and reverse transcription (RT) was performed using the High Capacity cDNA reverse transcription kit (Applied Biosystems/Life Technologies Austria, Vienna, Austria). All quantitative PCRs (qPCRs) were performed in duplicate with the iQSYBRGreen Supermix (Bio-Rad Laboratories, Vienna, Austria) or, when employing TaqMan primer/probe sets, with the Light Cycler TaqMan master mix (Roche Diagnostics, Vienna, Austria) according to the instructions of the manufacturers. Thermocycling conditions consisted of a predenaturation step at 95°C followed by 50 cycles of amplification: denaturation for 10 s at 95°C, annealing for 10 s at 63°C, and elongation for 15 s at 72°C. RNA levels of candidate targets in the input and immunoprecipitation (IP) fractions obtained in the RNA-IP (RIP) experiments were determined similarly. Levels of target RNAs were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) RNA levels. Sequences of all primers and probes specific for individual cellular mRNAs and for the firefly (FLuc) and Renilla (RLuc) luciferase mRNAs are listed in Table S1 in the supplemental material. Hsa-miR-16 levels in the input and IP fractions of RIP experiments were determined using a predesigned miRNA quantification assay (Invitrogen/Life Technologies). Briefly, 10 ng of RNA was reverse transcribed using the TaqMan MicroRNA reverse transcription kit (catalog no. 4366596; Applied Biosystems/Life Technologies) according to the manufacturer's instructions. Hsa-miR-16 levels were determined in a subsequent RT-qPCR performed on a LightCycler 2.0 instrument (Roche Diagnostics). Briefly, 1.4 μl of the reverse transcription product was combined with 10 μl of Light Cycler TaqMan master mix (Roche Diagnostics) and 1 μl of TaqMan microRNA assay mix (20×; forward primer, reverse primer, and probe; catalog no. 4427975; Applied Biosystems/Life Technologies) in a 20-μl reaction mixture. Thermocycling conditions consisted of a predenaturation step at 95°C, followed by 50 cycles of amplification: denaturation for 10 s at 95°C, annealing and elongation for 15 s at 60°C.
Western blot analysis.
Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on NuPage 10% Bis-Tris gels (Invitrogen/Life Technologies) and transferred to nitrocellulose membranes (Whatman, Dassel, Germany). Argonaute (AGO) proteins were detected using an anti-AGO antibody that recognizes all 4 human AGO proteins, AGO1 to AGO4 (Clone 2A8, MABE56; Merck Millipore, Vienna, Austria). A control antibody (sc-32233; Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used to detect GAPDH, which served as a reference. Bands were visualized using a secondary fluorescent goat anti-mouse antibody (catalog no. 35521; Pierce Biotechnology, Rockford, IL, USA). Fluorescence was measured using a Li-Cor Odyssey detection system (Li-Cor Biosciences, Lincoln, NE, USA).
Immunoprecipitation of Argonaute-containing RNP complexes.
To establish the AGO-IP protocol, an artificial test system consisting of an artificial miRNA and a corresponding artificial target mRNA was generated. Briefly, 3 × 106 A549 cells were cotransduced at a multiplicity of infection (MOI) of 30 TCID50/cell with the adenoviral, replication-incompetent, dual-luciferase reporter vector pAd-Luc-as (30) bearing an artificial target site within the 3′-UTR of the Renilla luciferase gene and the adenoviral vector Ad-FLuc-mi1 (30) encoding a miRNA that targets the corresponding site in Ad-Luc-as. Vector Ad-mi− (30), encoding a negative-control miRNA, served as a control. AGO-containing complexes were isolated at 24 h postransduction. For IPs using mivaRNA mimic-containing cells, 3e6 A549 cells were transfected with 10 nM mivaRNAI(A)-137, mivaRNAI(G)-137, or a negative-control siRNA (Ambion/Life Technologies) by using Lipofectamine 2000, and IPs were conducted at 20 to 24 h posttransfection. AGO-containing complexes from mock- or Ad5-infected cells (MOI = 10 TCID50/cell) were isolated at 30 h postinfection.
To prepare cell lysates, cells were washed 5 times with phosphate-buffered saline (PBS) and lysed on ice in a buffer containing 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM MgCl2, 0.5% Igepal CA-630 (Sigma-Aldrich, St. Louis, MO, USA), 5 mM dithiothreitol (DTT), protease inhibitors (10 μg/ml aprotinin, 5 μg/ml leupeptin, 100 μg/ml phenylmethylsulfonyl fluoride, and 1 μg/ml pepstatin), and 250 U/ml RiboLock RNase inhibitor (Fermentas, St. Leon-Rot, Germany). Nuclei and debris were removed by centrifugation, and the lysates were precleared. For IPs, we used the human pan-AGO antibody (clone 2A8; Merck Millipore) and a mouse IgG1 isotype control antibody (Clone 1E2.2; Merck Millipore); the antibodies (15 μg) were coupled to protein G Sepharose beads (Invitrogen/Life Technologies). For a single IP, 25 μl of beads was washed three times with 1 ml of nuclease-free ice-cold PBS, and antibodies were allowed to attach to the beads at 4°C for 6 h in 1 ml of nuclease-free PBS containing protease inhibitors. Beads were subsequently blocked overnight at 4°C with 1.2 mg/ml bovine serum albumin and 0.6 mg/ml yeast tRNA. Immediately before immunoprecipitation, beads were washed with PBS containing the blocking reagents and with cell lysis buffer. Cell lysates were precleared by adding 60 μl of unconjugated, blocked beads and incubated under rotation for 1 h at 4°C; 5% of the precleared lysate was used to isolate total RNA (referred to as input) using TRIzol (Invitrogen/Life Technologies), and another 5% was saved for detecting AGO proteins by Western blotting. The remaining precleared lysate was combined with the antibody-immobilized protein G Sepharose beads and incubated under rotation for 3 h at 4°C. Beads were washed twice with lysis buffer containing the blocking solution, three times with high-salt buffer (25 mM Tris-HCl [pH 8.0], 900 mM NaCl, 2 mM MgCl2, 1% Igepal CA-630, 5 mM DTT, blocking reagents, protease and RNase inhibitors), and twice with low-salt buffer (25 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM MgCl2, 0.05% Igepal CA-630, 5 mM DTT, blocking reagents, protease and RNase inhibitors). For quality control, 5% of the beads were saved to probe for AGO by Western blotting, and the remaining beads were subjected to RNA extraction using TRIzol. Contaminating DNA present in input and IP samples was removed using Turbo DNase (Invitrogen/Life Technologies), followed by TRIzol extraction and a final cleanup step using RNeasy MinElute cleanup columns (Qiagen). RNA to be used for microarray analysis was amplified using the Whole Transcriptome Expression kit (Ambion/Life Technologies) according to the instructions of the manufacturer.
Microarray analysis.
Microarray analyses of RNAs isolated from the input and IP fractions of cells transfected with mivaRNA mimics or infected with Ad5 were performed at the Competence Center for Fluorescent Bioanalysis (Regensburg, Germany; www.kfb-regensburg.de). Purified ssDNA, containing dUTP residues, was fragmented using a combination of uracil DNA glycosylase (UDG) and apurinic/apyrimidinic endonuclease 1 (APE 1) followed by terminal labeling with biotin (WT Terminal Labeling kit; Affymetrix, Inc., Santa Clara, CA, USA). Between 0.6 and 3 μg of fragmented and labeled DNA was hybridized to Affymetrix Human Gene 1.1 ST arrays. For hybridization, washing, staining, and scanning, an Affymetrix GeneTitan system was used. Summarized log2 probe set signals were calculated by using the RMA algorithm (31) with the Affymetrix GeneChip Expression Console software. RMA was run separately on samples containing total RNA, RNA-induced silencing complex (RISC) RNA, and adenovirus-infected samples. Subsequent analyses were performed in the R statistical environment using Bioconductor packages (31). To compare distinct groups, quantile normalization (32) was used to achieve identical empirical distributions. When the overall decrease in cellular miRNA levels in RISCs after Ad5 infection had to be taken into account, miRNA reads were additionally normalized to copurifying small RNA (i.e., rRNA) read counts in IP libraries.
Cross-hybridizing probe sets were excluded. Replicate samples were averaged, and log2 ratios were used to assess enrichment in the IP samples. The data sets were deposited in the Gene Expression Omnibus (GEO) database (accession number GSE50541).
RNA sequencing.
Sequencing of sRNA libraries derived from RNA that had been isolated from the input and IP fractions of Ad5-infected cells was carried out at GATC Biotech (Constance, Germany); 4 sRNA libraries (RNA of 22 to 30 nt in length) covering the content of the 2 IPs and the associated total RNA input fractions were sequenced. Sequencing (50-bp single reads) was performed on an Illumina HiSeq2000 platform. Processing and basic analysis of raw data were performed at the Beijing Genomics Institute (China) using an sRNA analysis pipeline. Reads were mapped to the human genome (hg19), human sequences stored in NCBI GenBank (http://www.ncbi.nlm.nih.gov/GenBank/), Rfam 11.0 (http://rfam.sanger.ac.uk/), and miRBAse 19.0 (http://www.mirbase.org/) and were assigned to individual classes of small RNAs. Reads were additionally mapped to the Ad5 genome (AC_000008). For counting the read tags in the human and adenovirus genome, no mismatches were allowed, and the tags that aligned to exactly the same coordinates were not collapsed into one unique read. Sequencing data as well as quality control and alignment settings can be retrieved from the Gene Expression Omnibus (GEO) database (accession number GSE50576).
Prediction of target sites and overrepresentation of seed-matching sequences.
Targets of mivaRNAs in the human genome (33) were predicted using TargetScan 6.0 for human transcripts. Perl scripts were downloaded from http://www.targetscan.org/. Targets were identified using default parameters in all Refseq transcripts downloaded from ftp://ftp.ncbi.nlm.nih.gov/refseq/, where rna.fa was converted to the TargetScan input format using a custom Perl script. Further analysis was conducted in the R statistical environment. To test for overrepresentation of mivaRNA targets in distinct sets of genes defined by the microarray analysis, hypergeometric tests were performed using the R function phyper (q, m, N-m, k, lower.tail = TRUE). The parameters for these tests were the following: population size (N) = number of genes that were present (19,484 after filtering) on the Affymetrix Human Gene 1.1 ST Arrays; m = the number of enriched genes (at a specified cutoff) from array analysis; k = number of target genes for a given mivaRNA; q = intersection of k and m. In Fig. 5 and 10, the P values from these tests are presented as −log10 (P value). Alternatively, the Sylamer algorithm (34) was used to test for the overrepresentation of motifs complementary to the seed sequences of miRNAs. Hypergeometric testing was carried out for 6-mer, 7-mer, and 8-mer sequences, with 6-mer sequences consistently giving the best results. Proven targets of cellular miRNAs were retrieved from miRTarBase (35).
FIG 5.

Predicted mivaRNAI targets are overrepresented in AGO IP fractions of mivaRNA-transfected cells. TargetScan was used to predict the cellular targets of the individual single strands of mivaRNAI(G)-137 and mivaRNAI(A)-137, respectively. We used hypergeometric testing to investigate the overrepresentation of RNAs with matches to mivaRNA-137 seed sequences within the top 25 and top 50 RNAs most highly enriched in RISCs (after normalizing to input RNA levels) of cells transfected with mivaRNAI(G)-137 and mivaRNAI(A) compared to RISCs of cells transfected with the negative-control siRNA. As a control, we also tested RNAs with seed sequence matches to VA RNAI 3′-arm-derived single strands of mivaRNAI-134 to -141 and mivaRNAII-136 to -140 derived from the 3′-arm of VA RNAII. y axis, P values (−log10) representing the grade of overrepresentation of RNAs with seed sequence matches to the individual mivaRNAs within the sets of RNAs enriched in RISCs compared to the entire set of RNAs. The yellow line represents a P value of 0.05; the red line, a P value of 0.001. Sequences of mivaRNAs derived from the 5′-and 3′-arms of the respective VA RNAs are indicated as 5′ and 3′, respectively. (A) Overrepresentation of RNAs with seed matches to the individual mivaRNA single strands in RISCs of cells transfected with mivaRNAI(G)-137. (B) Overrepresentation of RNAs with seed matches to the individual mivaRNA single strands in RISCs of cells transfected with mivaRNAI(A)-137 in a purged list of RNAs lacking the top 1,000 genes that were enriched by both mivaRNAI(G)-137 and mivaRNAI(A)-137. (C) Analysis as in panel B with the difference that a purged list lacking RNAs that were exclusively enriched by mivaRNAI(A)-137 was used.
FIG 10.
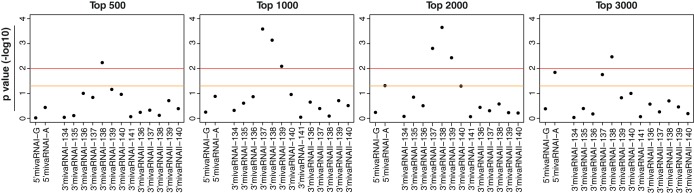
Overrepresentation of RNAs with seed sequence matches to VA RNAI- and VA RNAII-derived mivaRNAs among RNAs enriched in RISCs of Ad5-infected cells. Hypergeometric testing was performed to calculate significant overrepresentation of RNAs that were predicted by TargetScan to be targeted by a set of mivaRNA isoforms among the top 500, 1,000, 2,000, and 3,000 RNAs most highly enriched in RISCs of Ad5-infected cells compared to mock-infected cells. We tested RNAs with seed sequence matches to VA RNAI 5′-arm-derived single strands of mivaRNAI(G)-137 and mivaRNAI(A)-137, to VA RNAI 3′-arm-derived single strands of mivaRNAI-134, -135, -136, -137, -138, -139, -140, and -141, and to VA RNAII 3′-arm-derived mivaRNAII-136, -137, -138, -139, and -140. y axis, P values (−log10) representing the grade of overrepresentation of RNAs with seed sequence matches to the individual mivaRNAs within the sets of RNAs enriched in RISCs compared to the entire set of RNAs. The yellow line represents a P value of 0.05; the red line, a P value of 0.001.
Microarray and RNA sequencing data accession numbers.
Microarray and RNA sequencing data sets were deposited in the Gene Expression Omnibus (GEO) database and are accessible through accession numbers GSE50541 and GSE50576, respectively.
RESULTS
Abundance of VA RNAI- and VA RNAII-derived RNAs in RISCs of Ad5-infected cells.
To isolate RISCs and associated RNAs, including those derived from VA RNAs I and II (Fig. 1A), we performed IPs with an antibody that binds all 4 human AGO proteins (AGO1 to AGO4) and thus allows to isolate the entire pool of human RISCs (36). In addition, this approach was supposed to account for the possibility that mivaRNAs are differentially sorted into the different RISC subtypes. Subsequent deep sequencing of sRNA libraries was used to identify individual RISC-associated miRNAs as has been described previously (37). The performance of our RIP protocol was evaluated using an artificial test system (Fig. 1B): we cotransduced A549 cells with an adenoviral, replication-deficient vector bearing the humanized Renilla luciferase (hRLuc) and firefly luciferase (hFLuc) genes (Ad-Luc-as) and a second adenoviral vector carrying the sequence for an artificial miRNA (amiRNA) targeting a site within the 3′-UTR of the hRLuc mRNA (Ad-FLuc-mi1). A distinct vector carrying the sequence for a nontargeting amiRNA (Ad-mi-) was used as a control. RT-qPCR demonstrated that cotransduction of the target RNA-expressing and the amiRNA-expressing vector decreased hRLuc mRNA levels (Fig. 1C). Testing the input and IP fractions by anti-AGO Western blotting revealed that AGO proteins were enriched efficiently relative to the input fractions in anti-AGO IP fractions but not in isotype-control IgG IP fractions (Fig. 1D). Moreover, hRLuc target mRNA was enriched in immunopurified AGO complexes relative to control IPs from cells expressing the hRLuc-targeting amiRNA (Fig. 1E, left panel), but no significant hRLuc mRNA enrichment was detected in IP samples from cells expressing the negative-control amiRNA (Fig. 1E, right panel). Furthermore, no enrichment in the IP fraction was detected for the housekeeping gene GAPDH. In addition, AGO IP resulted in a pronounced enrichment of endogenous hsa-miR-16 in the IP fraction compared to the IP fraction obtained with the isotype control antibody (Fig. 1F) and in a concomitant enrichment of cyclin E1 (CCNE1), a proven target of hsa-miR-16; no such enrichment was detected in IPs with the isotype control antibody (Fig. 1G).
FIG 1.

Validation of the RIP-Chip protocol used for identifying mivaRNA targets. (A) Drawing of Ad5 VA RNAs I and II. (B) Schematic illustration of the test system: A549 cells were cotransduced with an adenoviral vector bearing humanized Renilla luciferase (hRLuc) and firefly luciferase (hFLuc) genes (Ad-Luc-as) and a second adenoviral vector carrying the sequence for an artificial miRNA (amiRNA) known to directly target the 3′-UTR of the hRLuc mRNA (Ad-FLuc-mi1). A distinct vector carrying the sequence for a nontargeting amiRNA (Ad-mi−) was used as a control. At 24 h postransduction, cells were lysed and RISC complexes were isolated by IP using the antibody that recognizes human AGO1 to -4. A distinct IgG isotype control antibody was used as a reference. Total RNA was isolated from 5% of the input fraction and from the IP fraction. (C) hRLuc mRNA levels are lower in the input fraction of cells expressing the targeting amiRNA (mi+) than in the input fractions of cells expressing the negative-control amiRNA (mi−). hRLuc mRNA levels at 24 h posttransduction were determined using RT-qPCR and normalized to hFLuc mRNA levels. Relative hRLuc mRNA levels (means ± standard deviations [SD], n = 3) of a representative experiment are shown. ***, P < 0.01. (D) Immunoblot analysis of the input and AGO IP fractions demonstrating the specific enrichment of AGO proteins in the IP fraction. An anti-GAPDH antibody served as a control. (E) Left panel, hRLuc mRNA is enriched in the AGO IP fractions (white bars) compared to the IgG control IP fractions (black bars) of cells expressing the hRLuc-targeting amiRNA. hRLuc mRNA levels were determined using RT-qPCR and normalized to hRLuc levels of the input fractions. GAPDH mRNA levels from the same fractions were determined as a control. Right panel, same setup as for the left panel, except that cells expressing the negative-control amiRNA were used. The data represent the mean results for 2 IP replicates of a representative experiment. (F) Endogenous hsa-miR-16 is enriched in the AGO IP fractions. Relative hsa-miR-16 levels of the AGO IP fractions, the IgG control IP fractions, and the input fractions were determined using RT-qPCR. The values for the input fraction were set as 1. Data represent the mean results for 2 IP replicates of a representative experiment. (G) Endogenous CCNE1 mRNA is enriched in the AGO IP fractions. Relative CCNE1 mRNA levels of the AGO IP fractions, the IgG control IP fractions, and the input fractions were determined using RT-qPCR. Relative CCNE1 mRNA levels of the IP fractions were calculated by normalizing against both the respective input levels and GAPDH levels. Data represent the mean results for 2 IP replicates of a representative experiment.
To isolate mivaRNAs from RISCs, we infected A549 cells with wild-type Ad5 at an MOI of 10 TCID50/cell or mock infected the cells. We immunoprecipitated RISCs and associated RNAs at 30 h postinfection and isolated RNAs from the IP and input fractions to generate a total of 8 sRNA libraries (2 separate IPs for each condition), which were subjected to deep sequencing. Reads were assigned to individual annotation classes and were additionally mapped to the Ad5 genome. Absolute read counts for mivaRNAs, cellular miRNAs, and other major classes of small RNAs with significant read counts that could unambiguously be mapped to the human or adenoviral genome are listed in Tables S4 and S5 in the supplemental material.
Essentially all reads of RISC-incorporated small RNAs that mapped to the adenoviral genome were derived from VA RNAs. There was no indication of miRNAs being generated from other parts of the genome to similar extents (Fig. 2A). Analysis of the group of cellular miRNAs and VA RNA-derived RNAs revealed that roughly one half of the reads represented cellular miRNAs and the other half represented VA RNA-derived sRNAs. Analysis of cellular miRNA/mivaRNA read counts of RISC-associated RNA revealed 85% cellular miRNA and 15% mivaRNA (Fig. 2B, lower left chart). Among RISC-associated sRNAs, those derived from VA RNAII were more abundant than those derived from VA RNAI (Fig. 2B, lower left chart).
FIG 2.

Relative abundance of cellular miRNAs and adenoviral sRNAs derived from VA RNAI and -II in RISCs of Ad5-infected cells. A549 cells were infected with Ad5 at an MOI of 10 TCID50/cell or were mock infected. At 30 h postinfection, AGO-containing complexes and associated RNAs were isolated by IP. RNAs from input and IP fractions were purified, 4 corresponding sRNA libraries were generated from 2 IPs, and the content of the libraries was subjected to RNA sequencing. Reads were assigned to individual sRNA annotation classes and mapped to the Ad5 genome. (A) Reads along the Ad5 genome. Left panel, input fraction. Right panel, IP fraction. The results of a single IP experiment are shown. VA RNA-derived sRNAs are indicated with arrows. (B) Relative abundance of sRNAs derived from VA RNAs I and II. Upper charts, relative abundance in the input fractions. Lower charts, relative abundance in the IP fractions. Left, abundance of all VA RNAI- and VA RNAII-derived sRNAs relative to cellular miRNAs. Middle, relative abundance of individual mivaRNA isoforms among the most prevalent isoforms derived from VA RNAI. Right, relative abundance of individual mivaRNA isoforms among the most prevalent isoforms derived from VA RNAII. Mean values from 2 IPs are shown. (C) Reads along the VA RNAI and -II regions. For consistency, coordinates based on the previous numbering of VA RNAs in which the 5′ end of 5′mivaRNAI-G represents the position +1 and the 5′ end of 5′mivaRNAI-A represents the position −3 of VA RNAI are shown. Parts representing the stem regions that give rise to RISC-incorporated mivaRNAs are indicated. Mean values from 2 IPs are shown. (D) Detailed read counts for major mivaRNA isoforms derived from the VA RNA I and II terminal stem regions that were obtained for the input (left panel) and IP (right panel) fractions. Read counts for the entire pool of cellular miRNAs, rRNA, Y RNA, 7SK RNA, and snRNA are given as a reference. Mean values from 2 IPs are shown.
Among the sRNAs derived from the 5′ arm of VA RNAI, those starting at the “A” position [position −3 relative to the start of VA RNAI(G)] were present in the libraries to an extent approximately 5 times higher than sRNAs starting at the “G” position. These 2 mivaRNA isoforms together accounted for approximately one-half of all VA RNAI reads in the input fraction (Fig. 2B, upper middle chart). The most abundant single strands derived from the 3′-arm of VA RNAI were those of mivaRNAI-137 and mivaRNAI-138. These 2 RNAs accounted for 23% and 14% of the VA RNAI-assigned reads, respectively. Moreover, these 2 RNAs were the most abundant VA RNAI-derived RNAs in the RISC fraction (36% and 32%, respectively; Fig. 2B, lower middle chart), confirming previous findings for lytically infected human cells (22). Whereas the 5′-arm-derived strand of VA RNAI starting at the “A” position, 5′mivaRNAI(A), was also highly incorporated into RISCs (27%), the incorporation of 5′mivaRNAI(G) was poor. The most abundant mivaRNA in the libraries derived from IP samples was 3′mivaRNAII-138 of the 3′-arm of VA RNAII, which accounted for 69% of the abundant VA RNAII-derived mivaRNA isoforms (Fig. 2B, lower right chart). 3′mivaRNAII-136 and 3′mivaRNAII-137 were also present in the same libraries in significant amounts (16% and 15%, respectively). The absolute numbers of reads mapping to the entire VA RNAI and II regions are presented in Fig. 2C; see also Table S2 in the supplemental material. Absolute read counts of selected mivaRNAs derived from the terminal stem regions of VA RNAI and II are shown in Fig. 2D, and read counts for cellular miRNAs are listed in Table S3 in the supplemental material.
Individual mivaRNAs have distinct targeting capacities.
Based on the results reported above, we designed mimics of the major mivaRNA isoforms present in the libraries: 5′mivaRNAI(A)-137, 3′mivaRNAI(A)-137, and 3′mivaRNAII-138. To differentiate between the targeting by the 5′-arm- and 3′-arm-derived single strands of mivaRNAI(A) (see below), we also included a mimic of 5′mivaRNAI(G)-137. The capacity of these mimics to silence genes was evaluated using dual-luciferase assays. We generated reporter vectors carrying a firefly luciferase gene (hFLuc) for normalization and a Renilla luciferase gene (hRLuc) with a modified 3′-UTR harboring sequences complementary to individual mivaRNA single strands. Cotransfecting cells with these reporters and individual mivaRNA mimics or a nontargeting negative-control siRNA revealed that both single strands derived from the 5′-arm of VA RNAI could silence a complementary target, albeit to distinct extents (Fig. 3A, left panel). Whereas mivaRNAI(G)-137 reduced Renilla luciferase activity only moderately, mivaRNAI(A)-137 decreased Renilla luciferase activity by approximately 75%. However, both mivaRNAs silenced the target complementary to the 3′-arm of VA RNAI equally effectively (Fig. 3A, middle panel); this was as expected, because the respective seed sequences are identical in both mivaRNA mimics. The mivaRNAs derived from the 3′-arm of VA RNAI silenced genes more potently than mivaRNAs derived from the 5′-arm, agreeing with previous data demonstrating that mivaRNA strands derived from the 3′-arm were generally incorporated into RISCs more efficiently (24). In the same study, it was also reported that incorporation into RISC was nevertheless efficient for both strands of mivaRNAI(A) while a clear bias existed against the incorporation of the mivaRNAI(G) strand derived from the 5′-arm. In accordance with this finding, silencing by the mivaRNAI(G) strand derived from the 5′-arm was the weakest in our test system (Fig. 3A, left panel). We also observed targeting by the VA RNAII 3′-arm-derived single strand of mivaRNAII-138, albeit to a lesser degree than targeting by the respective VA RNAI 3′arm single strand (Fig. 3A, right panel). The silencing rates measured for the mivaRNA mimics were similar to those measured when cells were instead cotransfected with vector pAdVAntage containing the VA RNAI and VA RNAII genes of adenovirus 2 under the control of their own promoters (Fig. 3B). The reporter genes became further silenced when cells were infected with wild-type Ad5: the presence of Ad5 resulted in a substantial reduction in Renilla luciferase expression from the reporter for VA RNAI 3′-arm-derived mivaRNAs (Fig. 3C, second panel) and in a lesser but still pronounced reduction in luciferase expression from the reporter for the 5′-arm-derived mivaRNAs (Fig. 3C, first panel). Although we found that mivaRNA single strands of the 3′-arm of VA RNAII were highly abundant in RISCs, the knockdown rate of gene expression for this particular reporter was only moderate, especially compared to that of the reporter for the highly potent VA RNAI 3′-arm-derived mivaRNAs.
FIG 3.

Functional characterization of mivaRNA mimics. (A) HEK293 cells were cotransfected with the dual-luciferase reporter vectors pmiVAI5, pmiVAI3, and pmiVAII3 carrying sequences complementary to the mivaRNAI strands derived from the VA RNAI 5′- and 3′-arms and the 3′-arm of VA RNAII, respectively, and mimics of mivaRNAI(A)-137 or mivaRNAI(G)-137 or a nontargeting, negative-control siRNA. Luciferase activities were determined at 24 h posttransfection. Renilla luciferase activities normalized to firefly luciferase activities (means ± SD, n = 3) from a representative experiment are shown. **, P < 0.01; ***, P < 0.001; n.s., not significant. (B) The experimental setup was as described for panel A, except that cells were cotransfected with vector pAdVAntage instead of mivaRNA mimics. The parental reporter vector psiCHECK-2 lacking the mivaRNA target sites in the 3′-UTR of the Renilla luciferase mRNA was used as a control. Renilla luciferase activities normalized to firefly luciferase activities (means ± SD, n = 3) from a representative experiment are shown. ***, P < 0.001. (C) The experimental setup was as described for panel A, except that cells were not cotransfected with mivaRNA mimics or pAdVAntage but were instead infected with wild-type Ad5 at an MOI of 10 or were mock infected. Luciferase activities were determined at 24 h postinfection. Renilla luciferase activities normalized to firefly luciferase activities (means ± SD, n = 3) from a representative experiment are shown. ***, P < 0.001; n.s., not significant.
RNAs specifically enriched in RISCs in the presence of mivaRNAI(A)-137 and mivaRNAI(G)-137.
A previous study (28) focused on genes whose expression is downregulated in the presence of the single strand of mivaRNAI-138 derived from the 3′-arm of VA RNAI. However, because our RNA sequencing (RNA-Seq) data (Fig. 2) indicated that 3′mivaRNAI-137 was actually slightly more abundant and that, concomitantly, the opposite strand derived from the 5′-arm of mivaRNAI starting at the “A” position [5′mivaRNAI(A)] was also efficiently incorporated into RISCs, we sought to identify the direct targets of these particular mivaRNA single strands (Fig. 4A). We performed RNA immunoprecipitation followed by microarray analysis (RIP-Chip), a technique that has previously successfully been used to identify targets of viral miRNAs (38). Briefly, we transfected A549 cells with mimics of mivaRNAI(A)-137 or mivaRNAI(G)-137 or with a negative-control siRNA and isolated AGO-containing complexes as before. RISC-associated RNAs and the RNAs present in the input fraction were subjected to microarray analysis to identify the mRNAs enriched specifically in RISCs of mivaRNA-containing cells compared to RISCs of cells transfected with the control siRNA. All RIP experiments were performed in triplicate.
FIG 4.
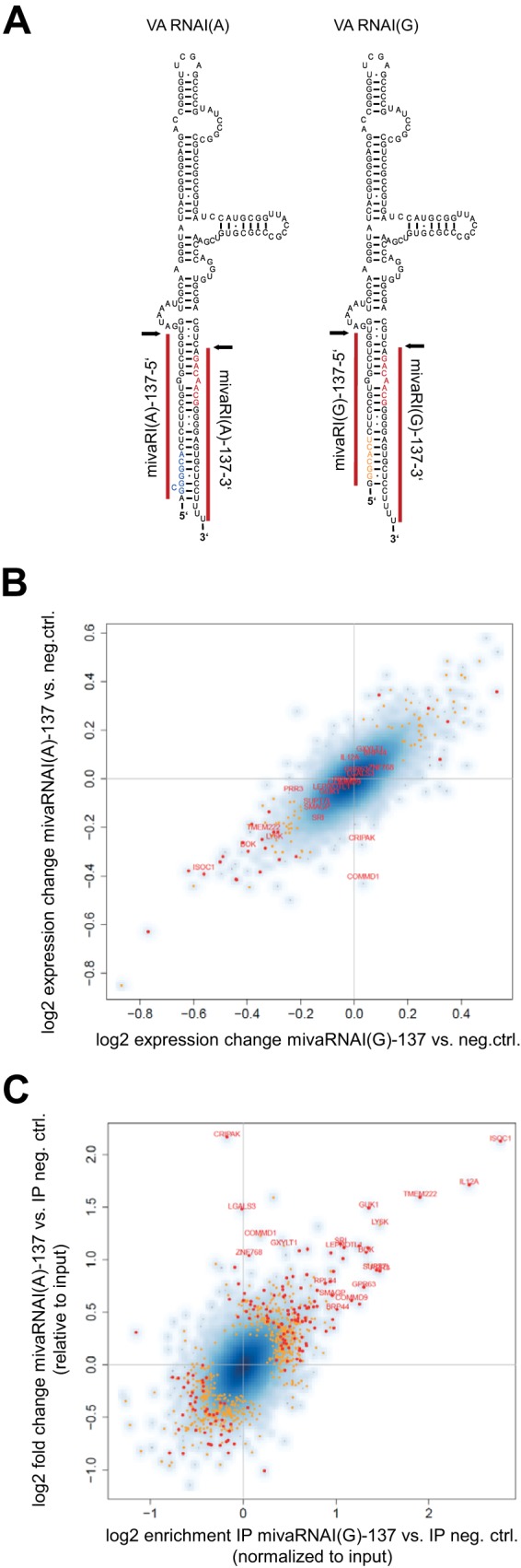
Sequences of VA RNAI variants and consequences of mivaRNA transfection on RNAs in the total RNA and RISC-associated RNA fractions. (A) VA RNAI variants VA RNAI(A) and VA RNAI(G). The mivaRNAs whose functions were analyzed in this study, mivaRNAI(A)-137 and mivaRNAI(G)-137, are indicated as red bars. The respective seed sequences of the 4 potential guide strands are indicated in color. Note that the single strands derived from the 3′-arm of the VA RNA contain identical seed sequences. The Dicer processing sites are indicated with arrows. (B and C) A549 cells were transfected with mimics of mivaRNAI(A)-137 or mivaRNAI(G)-137 or with a negative-control siRNA. AGO-containing complexes were isolated by IP at 24 h posttransfection. RNA was purified from total RNA fractions (input) and IP fractions and subjected to microarray analysis. Statistically significantly differing mRNAs are indicated as small orange dots (P < 0.05) or as larger red dots (P < 0.01). The names of genes that were eventually selected for further analysis are given in red. (B) Differences in total RNA levels (input) of A549 cells transfected with mivaRNAI(A) versus negative-control siRNA {y axis; shown as log2 [input mivaRNAI(A) − input neg.ctrl. siRNA]} and of cells transfected with mivaRNAI(G) versus negative-control siRNA {x axis; shown as log2 [input mivaRNAI(G) − input neg.ctrl. siRNA]}. RNA was isolated at 24 h posttransfection. The data represent the mean log2 changes in RNA levels in the input fractions of 3 independent IPs. (C) Differences in RNA levels in the AGO IP fractions of the same cells as in panel A that were transfected with mivaRNAI(A) versus negative-control siRNA {y axis; shown as log2 [IP mivaRNAI(A) − IP neg.ctrl. siRNA]} and of cells transfected with mivaRNAI(G) versus negative-control siRNA {x axis; shown as log2 [IP mivaRNAI(G) − IP neg.ctrl. siRNA]}. All mRNA levels in the IP fractions were corrected for their respective levels in the total RNA fraction (input). Mean log2 enrichments from 3 independent IPs are shown.
We included both mivaRNAI(G)-137 and mivaRNAI(A)-137 to differentiate between the targeting by the single strands derived from the 5′- and 3′-arms of VA RNAI, respectively: the 2 mivaRNAs contain 4 potential guide strands, of which the 2 strands derived from the 3′-arm contain identical seed sequences (nt 2 to 7 or 2 to 8 depending on the definition) (Fig. 4A). Therefore, these 2 particular strands would be expected to target the same set of mRNAs. The RNAs enriched in RISCs by both mivaRNAs would represent the targets of the single strands derived from the 3′-arm of VA RNAI (neglecting the potential existence of—probably very rare—transcripts containing functional target sites for both mivaRNAs). Conversely, RNAs enriched exclusively by either mivaRNAI(A)-137 or mivaRNAI(G)-137 would constitute targets of the single strands derived from the left arm of VA RNAI(A) and VA RNAI(G), respectively.
Isolation of RNA was performed at 20 to 24 h posttransfection. Accordingly, gene expression differences between cells transfected with the mivaRNA mimics or the negative-control siRNA were generally low. A picture of the global changes in gene expression after transduction with the 2 mivaRNA mimics is presented in Fig. 4B, and a detailed list of up- or downregulated genes is shown in Table S4 in the supplemental material.
Comparing microarray data for AGO IP samples revealed that several RNAs were enriched in RISCs upon mivaRNAI(A)-137 or mivaRNAI(G)-137 transfection in comparison to the negative-control siRNA. Figure 4C presents a scatter plot showing the log2-fold change of RNAs in RISCs of mivaRNAI(A)-137- and mivaRNAI(G)-137-transfected cells compared to RISCs of cells transfected with the negative-control siRNA after correction of RISC RNA levels for their respective levels in the total RNA fraction (input). This plot clearly indicates the enrichment of an overlapping set of RNAs in RISCs after transfection with either of the 2 mivaRNA mimics (dots arranged along the diagonal axis). The enrichment by both mivaRNA mimics identified these RNAs as putative targets of the VA RNAI 3′-arm-derived single strands that contain the same seed sequence. These RNAs represented the majority of enriched RNAs, which agrees with our results from the dual-luciferase assays indicating that both 3′mivaRNAI(A)-137 and 3′mivaRNAI(G)-137 exhibited the highest silencing potency (Fig. 3A).
A smaller set of RNAs were enriched exclusively in the presence of mivaRNAI(A)-137 (dots located around 0 [gray line] on the x axis in the upper part of the plot), identifying these RNAs as interacting partners of 5′mivaRNAI(A). We did not detect any RNAs enriched exclusively by 5′mivaRNAI(G) (no dots located around 0 on the y axis in the right half of the plot), reflecting the lower targeting and RISC incorporation capability of this RNA (Fig. 3A). A detailed list of enrichment values for all RNAs is presented in Table S4 in the supplemental material.
Predicted mivaRNAI targets are overrepresented in AGO IP fractions of mivaRNA-transfected cells.
To analyze if the enrichment of RNAs in the IP fractions was specific for the sequences of the mivaRNA mimics, we investigated whether RNAs matching the mivaRNA seed sequence were overrepresented within the group of enriched RNAs. We used TargetScan to retrieve RNAs with predicted seed sequence matches to the individual 5′-arm- and 3′-arm-derived single strands of mivaRNAI(A)-137 and mivaRNAI(G)-137 and then calculated the number of these RNAs present within the pools of the top 25 and top 50 RNAs enriched in the IP fractions. As a control, we also included existing and hypothetical 3′-arm-derived single strands of mivaRNAI-134 to -141 and mivaRNAII-136 to -140 derived from the 3′-arm of VA RNAII. Hypergeometric testing revealed that RNAs matching the seed sequence of 3′mivaRNAI(G)-137 were clearly overrepresented in RISCs upon transfection with mivaRNAI(G)-137 (Fig. 5A). No 5′mivaRNAI(G)-137 seed sequence-matching sequences were overrepresented. To be able to clearly see an overrepresentation of RNAs with seed sequence matches to either the 5′arm- or the 3′arm-derived mivaRNA(A) single strands after transfection with mivaRNA(A)-137, we cleared the list of RNAs from RNAs that were either (i) enriched by both mivaRNAI(A)-137 and mivaRNAI(G)-137, thus retaining only RNAs that were exclusively enriched by 5′mivaRNAI(A)-137) (Fig. 5B), or (ii) exclusively enriched by mivaRNAI(A)-137 [retaining only RNAs that were enriched by 3′mivaRNAI(A)-137] (Fig. 5C). Hypergeometric testing revealed that RNAs matching the seeds of both single strands of mivaRNAI(A)-137 were overrepresented in RISCs upon transfection with the mivaRNAI(A)-137. More-significant P values were obtained for the 3′-arm-derived single strand than for the 5′-arm-derived single strand, which confirmed the RIP-Chip data suggesting that the 5′-arm-derived mivaRNA single strand targeted a rather small set of RNAs. In general, no overrepresentation of sequences matching the seeds of mivaRNAs was revealed for nonenriched RNAs (bottom parts of the ranked lists of RISC-associated RNAs) (data not shown). In summary, these calculations suggested that RNAs were not enriched in the IP fractions because of an indirect effect of mivaRNA transfection and thus enabled the seed sequence matches to be used as an additional criterion for selecting candidate targets.
Enrichment of cellular transcripts in RISCs of adenovirus-infected cells.
We thought to further restrict the set of candidates to those that were also enriched in the RISCs of Ad5-infected cells. Thus, we conducted microarray analyses on the same RNAs isolated from the IP and input fractions of Ad5-infected cells that had been used for RNA sequencing. Generally, more RNAs were enriched in IP fractions of Ad5-infected cells than in the previous RIP-Chip experiments. This was expected because following infection with Ad5, a pool of distinct mivaRNAs recognizing a larger number of cellular RNAs is produced, as well as because mRNAs could also be enriched in RISCs due to increased targeting by a subset of cellular miRNAs whose expression increases upon infection with Ad5. A scatter plot depicting the overall pattern of statistically significant RNA enrichment in RISCs relative to gene expression changes after Ad5 infection is shown in Fig. 6A. Detailed enrichment values (means of 2 independent IPs) for RNAs are presented in Table S4 in the supplemental material. Comparing this set of RNAs with the set of RNAs enriched in cells transfected with the mivaRNA mimics revealed that the majority of the RNAs enriched most highly in RISCs by the mivaRNA mimics tended to be overrepresented in the group of RNAs enriched in RISCs of Ad5-infected cells as well (Fig. 6B and C).
FIG 6.
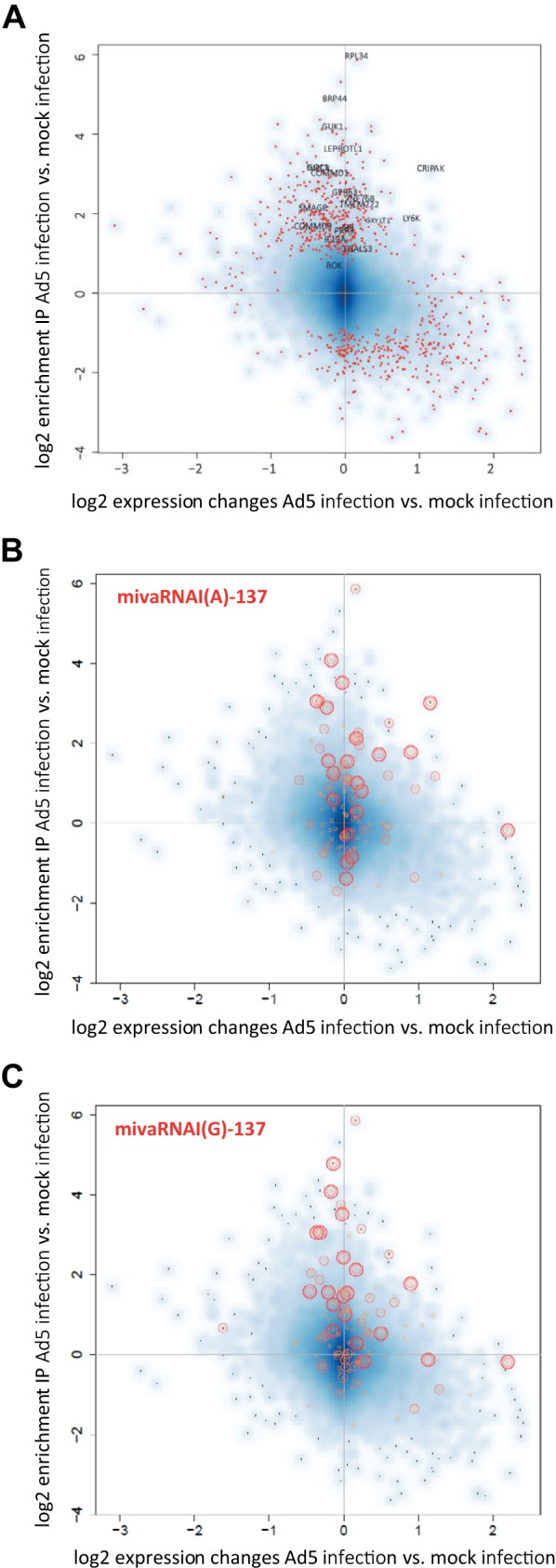
Changes in gene expression and incorporation of RNAs in RISCs upon infection of cells with Ad5. (A) A549 cells were infected with Ad5 at an MOI of 10. AGO-containing complexes were isolated using IP at 30 h postinfection. RNA was purified from total RNA fractions (input) and IP fractions and subjected to microarray analysis. Differences in RNA levels in the AGO IP fractions of cells infected with wild-type Ad5 versus mock-infected cells (y axis; shown as log2 [IP Ad5 − IP mock]), and the differences in total RNA levels (input fraction) of the same cells (x axis; shown as log2 [input Ad5 − input mock]) were calculated. The data represent the mean log2 changes in RNA levels of 2 independent IPs. Statistically significantly altered RNAs (P < 0.01; red dots) and the names of genes that were finally selected for further analysis are indicated. (B and C) Correlation of enrichment of RNAs in RISCs of mivaRNA-transfected cells with the enrichment of RNAs in RISCs of cells infected with Ad5. The scatter plots show the distribution of the top 20 (large red circles), top 50 (medium red circles), and top 100 (small orange circles) RNAs with the highest log2-fold changes in IP (normalized to input) of cells transfected with mivaRNA mimics versus cells transfected with a negative-control siRNA within the pool of RNAs that became up-/downregulated and enriched/depleted upon Ad5 infection, respectively. (B) RNAs enriched upon mivaRNAI(A)-137 transfection highlighted. (C) RNAs enriched upon mivaRNAI(G)-137 transfection highlighted.
To validate the targeting of RISC-enriched RNAs by mivaRNAs, we selected a set of most probable targets: we included all RNAs that were (i) enriched by at least 0.93 log2 (1.9-fold) in AGO complexes of cells transfected with mivaRNAI(A)-137 or mivaRNAI(G)-137 over AGO complexes of negative-control siRNA-transfected cells after normalization to the respective total RNA input levels; (ii) predicted by TargetScan to be targeted within their 5′-UTRs, coding sequences (CDSs), or 3′-UTRs by the VA RNAI 5′-arm-derived single strand of mivaRNAI(A)-137 or the 3′-arm-derived single strands of mivaRNAI(A)-137/mivaRNAI(G)-137; (iii) enriched in AGO complexes by at least 0.93 log2 (1.9-fold) in Ad5-infected cells compared to uninfected cells. This selection generated 14 candidates in total. We added to this list 6 more genes that did not fulfill these criteria entirely but appeared promising to us because they ranked very high in one or more of the lists of enriched RNAs or were not genuine TargetScan targets but nevertheless contained a match to nt 2 to 7 of the respective mivaRNA. The final set of 20 putative targets selected for validation is shown in Table 1. Most strikingly, all RNAs that were exclusively or predominantly enriched in RISCs by mivaRNAI(A)-137 (implying that they were targets of the single strand derived from the 5′-arm of VA RNAI) were also predicted by TargetScan to be exclusively recognized by this strand. For the remaining candidate targets, a clear bias was evident for a match to the mivaRNA single strand derived from the 3′-arm of VA RNAI (Table 1).
TABLE 1.
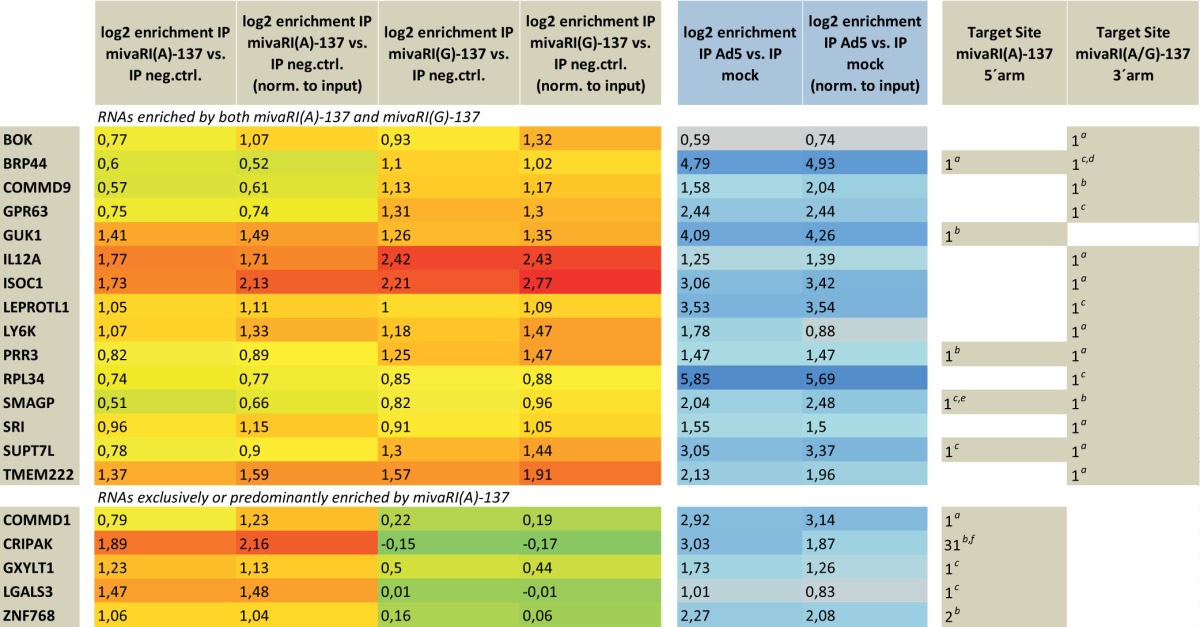
Predicted target site(s) located in 3′UTR.
Predicted target site(s) located in CDS.
Predicted target site(s) located in 5′UTR.
Target site only present in 1 of 3 protein-encoding transcript variants.
Predicted target site only present in 1 of 2 protein-encoding transcript variants.
High number of predicted target sites due to the highly repetitive nature of coding sequence.
RT-qPCR-based determination of relative abundances of candidate targets in input and IP fractions.
The enrichment of the 20 candidate targets in RISCs of mivaRNA-transfected cells was reanalyzed by RT-qPCR by using GAPDH levels for normalization (Fig. 7A). The overall enrichment pattern was the same as that obtained by microarray analysis, except for BRP44 and COMMD1, which appeared not to be enriched with the primer pairs used. However, because the data from RT-qPCR analysis largely reflected the data from the microarray analysis, we considered the microarray results to be sufficiently robust to continue with the whole set of 20 candidates. As with the microarray data set, we again observed a distinct pattern for RNAs that were enriched in RISCs exclusively by mivaRNAI(A)-137. When ignoring the general poor detection of COMMD1 in those fractions, all other RNAs of this subgroup (i.e., CRIPAK, GXYLT1, LGALS3, and ZNF768) showed the characteristic enrichment by mivaRNAI(A)-137 but not by mivaRNAI(G)-137. Enrichment values for the 20 candidate targets in RISCs of Ad5-infected cells as determined by RT-qPCR are shown in Fig. 7B.
FIG 7.

RT-qPCR-based calculation of the enrichment of selected candidate target RNAs in RISCs of cells transfected with mivaRNAI(A)-137 or mivaRNAI(G)-137 compared to RISCs of cells transfected with a negative-control siRNA (A) and enrichment of RNAs in RISCs of Ad5-infected cells compared to RISCs of mock-infected cells (B). Total RNA of AGO IP fractions were subjected to RT-qPCR using the respective gene-specific primer pairs, and individual RNA levels were normalized to GAPDH RNA levels. Mean enrichment levels ± SD in the RISC fractions are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Genes downregulated upon transfection with mivaRNAI(A)-137 or mivaRNAI(G)-137.
For the experiments described above, RNAs had been isolated early posttransfection and the gene expression changes had been accordingly low. Therefore, we repeated the transfection experiments and determined gene expression changes for the 20 candidate targets at 48 h posttransfection (Fig. 8). Most genes were generally downregulated after mivaRNA transfection, although the change was not statistically significant for all of them. Genes with target sites located exclusively in the 5′-UTR showed no or poor downregulation of expression. In contrast, 10 of the 15 genes with target sites located in their CDSs or 3′-UTRs showed statistically significant downregulation of expression, and the mRNA levels of all of these genes decreased to 60% or lower.
FIG 8.

Changes in the expression of selected candidate target genes upon transfection with mivaRNAs. A549 cells were transfected with mimics of mivaRNAI(A)-137 and mivaRNAI(G)-137 or with a negative-control siRNA. Putative target gene mRNA levels at 48 h posttransfection normalized to GAPDH mRNA levels were determined by RT-qPCR using the respective primer pairs. Relative mRNA levels (means ± SD from 2 independent experiments, each performed in triplicate) of cells transfected with mivaRNA mimics in comparison to cells transfected with a negative-control siRNA are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Predicted target regions confer downregulation of reporter gene expression in the presence of mivaRNA mimics.
Theoretically, downregulation of gene expression by mivaRNA mimics may represent a secondary effect, and conversely, targeting by mivaRNAs may not necessarily be accompanied by changes in transcript levels. Thus, to clearly demonstrate direct targeting by mivaRNAs, reporter constructs containing the suspected target regions were generated. We created 2 types of reporters: suspected target regions located in the CDSs or 3′-UTRs were inserted into the 3′-UTR of the Renilla luciferase gene, and target regions located in the 5′-UTR were inserted upstream of the Renilla luciferase gene in a way that replaced most of the original 5′-UTR. For genes containing multiple putative target sites at well-separated locations in the mRNA (i.e., genes PRR3, SMAGP, and SUPT7L), more than one construct was generated. The exact coordinates of the cloned sequences and the locations within their genes are listed in Table S1 in the supplemental material.
Suspected 5′-UTR target sequences did not mediate the downregulation of Renilla luciferase expression (data not shown). In contrast, 9 of 12 3′-UTR reporter constructs caused a downregulation of Renilla luciferase expression (Fig. 9). Agreeing with our IP experiments, most reporters were downregulated by both mivaRNAs, reflecting the fact that most of them were predicted targets of the mivaRNA single strands derived from the 3′-arm of VA RNAI. The reduction of transcripts was greatest for BOK, LY6K, and TMEM222. The PRR3, SMAGP, and SUPT7L transcripts contained predicted target sites for mivaRNA-137 single strands derived from both strands of VA RNAI. Targeting of these individual sites was analyzed using separate reporter vectors, and we observed efficient reduction of reporter activity for those reporters containing predicted target sites for the 3′-arm-derived mivaRNA mimics (reporters PRR3 [2], SMAGP [1], SUPT7L [1]).
FIG 9.

Effects of mivaRNA mimics on the expression of luciferase reporter genes. HEK293 cells were transfected with reporter constructs carrying the predicted mivaRNAI(A)-137 and mivaRNAI(G)-137 target regions as part of the 3′-UTR of a Renilla luciferase gene. The parental vector psiCHECK-2 lacking those sequences served as a negative control. Concomitantly, cells were transfected with mimics of mivaRNAI(A)-137 or mivaRNAI(G)-137 or with a negative-control siRNA. Renilla luciferase activities were normalized to firefly luciferase activities determined at 48 h posttransfection; the 2 luciferases were encoded by their respective reporter gene located on the same vectors. Relative light units (means ± SD; n = 3) in comparison to the negative-control siRNA are shown. ***, P < 0.001.
Furthermore, our data revealed that targets of the 5′-arm-derived single strand of mivaRNAI(A)-137 exist as well: cloning the respective predicted target region present in COMMD1 generated the expected pattern: Renilla luciferase activity was decreased by mivaRNAI(A)-137 but not by mivaRNAI(G)-137, thus verifying the RIP-Chip data that had revealed enrichment by only mivaRNAI(A)-137. However, the vast majority of validated targets represented targets of 3′mivaRNAI-137 (i.e., BOK, BRP44, ISOC1, LY6K, PRR3, SMAGP, SUPT7L, TMEM222). All identified target regions resided in the 3′-UTRs of their genes.
RNAs with seed matches to mivaRNAs of VA RNAI are overrepresented in RISCs of Ad5-infected cells.
Because RNAs with mivaRNA seed sequence matches were overrepresented among RNAs enriched in the RISCs of cells transfected with mivaRNA mimics, we investigated whether this overrepresentation also occurred for the set of RNAs enriched in RISCs of cells infected with Ad5. The goal was to identify the adenovirus-encoded mivaRNAs that are likely to contribute most to virus-mediated posttranscriptional downregulation of cellular gene expression. We conducted hypergeometrical testing for the mivaRNA isoforms that were shown by RNA-Seq data to be most prevalent in RISCs of Ad5-infected cells, as well as for the isoforms that would be produced by the cleavage of a few upstream or downstream nucleotides. Because the group of transcripts enriched in RISCs after Ad5 infection of cells was larger than the group enriched after mivaRNA transfection, we searched for overrepresentation of predicted mivaRNA targets within the top 500, 1,000, 2,000, and 3,000 most highly enriched transcripts (Fig. 10). The most significant P values (indicating the most marked overrepresentation of transcripts with respective seed sequence matches) were obtained for 3′mivaRNAI-137 and 3′mivaRNAI-138 for the groups of the top 1,000 and top 2,000 most highly enriched RNAs. This trend became weaker for 3′mivaRNAI-137 than for 3′mivaRNA-138 when the window was narrowed to the top 500 RNAs or extended to the top 3,000, which suggests that mivaRNAI-138 recognizes a slightly larger set of RNAs and/or interacts more strongly with its targets. As observed for the sets of RNAs enriched upon mivaRNA transfection, the RNAs with mivaRNA-139 seed sequence matches were also slightly overrepresented. Hypergeometrical tests with the top 2,000 and especially the top 3,000 most highly enriched RNAs revealed an additional overrepresentation of transcripts with seed sequence matches to 5′mivaRNAI(A). We did not observe an overrepresentation of RNAs with seed sequence matches to the single strand of mivaRNA(G) derived from the 5′-arm of VA RNAI, and we also did not obtain evidence for targeting by mivaRNAs derived from the 3′arm of VA RNAII.
Because the RNAs that are overrepresented in RISCs of Ad5-infected cells and predicted concomitantly to be targeted by the 3′mivaRNA-138 are enriched for true 3′mivaRNA-138 targets, we present a respective list of putative mivaRNAI-138 targets (in addition to the above-listed validated mivaRNAI-137 targets) in Table S5 in the supplemental material.
Impact of Ad5 infection on cellular miRNAs.
Ad5 infection changed the expression of several cellular miRNAs: 37 and 28 miRNAs were statistically significantly upregulated ≥1.5-fold and ≥2-fold, respectively, and 48 and 25 miRNAs were downregulated ≥1.5-fold and ≥2-fold, respectively (see Table S6 in the supplemental material). Among the most markedly upregulated miRNAs that were present in the cells at high levels were hsa-miR-125a-5p and hsa-miR-125b-5p (∼4- and ∼25-fold upregulated, respectively), hsa-miR-23a-3p (5-fold upregulated), and the mir-99 family members hsa-miR-99b-5p and hsa-miR-100-5p (∼2- and ∼8-fold upregulated, respectively).
The set of most strongly and statistically significantly downregulated miRNAs comprised the highly expressed let-7 family members that all share the same seed sequence. Hsa-let-7a was the most highly expressed miRNA in A549 cells, and together the hsa-let-7 family members accounted for 26% of all cellular miRNA reads in mock-infected cells. All of them were statistically significantly downregulated between 1.6- and 2.5-fold upon Ad5 infection. Other highly downregulated miRNAs with high read counts were hsa-miR-146a-5p and hsa-miR-146b-5p (∼2-fold decrease each), or hsa-miR-486-5p (∼10-fold decrease).
Up- or downregulated miRNAs or those that become enriched or depleted in RISCs of Ad5-infected cells may contribute to the change of cellular gene expression during infection. From the data in Fig. 6A, it is evident that a particular group of cellular mRNAs forms an asymmetrical extension into the lower right quadrant of the scatter plot, indicating that these RNAs were depleted in RISCs and concurrently upregulated in Ad5-infected cells. Thus, they represent mRNAs that may have become detargeted upon Ad5 infection. The upper left quadrant of the plot (containing mRNAs that became enriched in RISCs and concurrently downregulated upon infection) does not show a similarly pronounced asymmetrical extension. In fact, the number of RNAs that decreased statistically significantly in RISCs and whose steady-state levels concurrently increased in the cells upon infection (cutoff of 1 log2 for each condition) was more than 3-fold higher than those of RNAs that became enriched in RISCs and were concurrently downregulated upon infection. This pattern suggests a likely trend toward detargeting of cellular mRNAs that took place during infection. Moreover, in accordance with this observation, bioinformatic analysis revealed that proven targets of cellular miRNAs tended to be targeted by a higher number of miRNAs that were decreased in RISCs of Ad5-infected cells than for the rest of target mRNAs for which targeting miRNAs have been described. Importantly, this tendency increased with increasing mRNA depletion in RISCs/upregulation of gene expression (see the percentages in the lower right quadrants of the plots in Fig. 11A, increasing from left to right).
FIG 11.

(A) Compared to the rest of the experimentally validated targets of cellular miRNAs, those targets whose levels increase upon Ad5 infection and that are concurrently depleted in RISCs are more frequently targeted by miRNAs whose levels decrease upon Ad5 infection. The scatter plots depict changes in mRNA levels in total RNA (x axis) and RISC IP fractions (y axis) upon infection with Ad5. Dots in the lower right quadrants represent mRNAs whose levels increase upon Ad5 infection and that are concurrently depleted in RISCs. Red dots indicate mRNAs with RISC enrichment levels relative to input that fall below a certain threshold (first panel, log2-fold change = 0; second panel, log2-fold change = −1; third panel, log2-fold change = −2; fourth panel, log2-fold change = −3). The numbers in the lower right corners indicate the percentages of these genes that are proven targets of miRNAs that were found to be depleted (log2 FC < −0.7) in RISCs of Ad5-infected cells (normalized to input) compared to uninfected cells. This percentage is increasing from left to right, indicating that mRNAs that are more highly depleted in RISCs represent to a higher extent targets of miRNAs that are downregulated upon Ad5 infection. For comparison, the markedly lower percentage of the rest of mRNAs that are validated targets of miRNAs depleted in RISCs of Ad5-infected cells (log2 FC < −0.7) is indicated in the upper left corner of the plots. (B) Sylamer enrichment landscape plot for 6-mer motifs. The x axis represents the list of RNAs (3′-UTRs) sorted from most enriched (left) to most depleted (right) in RISCs relative to input of Ad5-infected A549 cells versus mock-infected cells. The y axis shows the hypergeometric enrichment of 6-mer sequences along the sorted-gene list. Positive values indicate enrichment and negative values depletion. The sequences of the 10 most highly enriched and depleted 6-mer motifs in the data set are shown above and below the plot, respectively. The motif corresponding to the let-7 seed-complementary sequence and the corresponding enrichment P values along the sorted-gene list are shown in color.
Because hsa-let-7 family members were highly expressed in A549 cells (giving them the theoretical potency to significantly decrease target RNA levels), and because they were strongly downregulated upon Ad5 infection, their decrease may have contributed to the detargeting of a subset of those mRNAs.
Indeed, when we applied the Sylamer algorithm (34), which searches for significant overrepresentation of 6- to 8-nt motifs across entire ordered gene lists, to the set of mRNAs ranked according to their changes in RISC enrichment relative to input in Ad5-infected A549 cells versus mock-infected cells, the sequence complementary to the hsa-let-7 seed sequence emerged as one of the 10 most highly enriched motifs (Fig. 11B). This motif peaked at the right end of the plot, indicating a strong overrepresentation of sequences with complementarity to the hsa-let-7 seed sequence among those mRNAs that had become most highly depleted in RISCs/upregulated upon Ad5 infection. Thus, detargeting of a subset of RNAs during Ad5 infection is to some extent attributable to the downregulation of hsa-let-7 family members during Ad5 infection of A549 cells. Sylamer analysis did not reveal a comparably clear correlation for other differentially expressed miRNAs (neither upregulated nor downregulated miRNAs).
DISCUSSION
Adenovirus-encoded miRNAs are expressed at high levels and have been described to therefore saturate the cellular RNAi pathway (7, 9). Adenovirus may hypothetically benefit from unspecifically blocking RNAi. However, evolution should inevitably have forced these sRNAs to evolve into miRNAs with a targeting specificity that is in some way beneficial for the virus.
In this study, we determined direct targets of both single strands of mivaRNAI(A)-137. We focused on this particular mivaRNA because our RNA-Seq data indicated both single strands to be abundant in RISCs and because its single strand derived from the 3′arm of VA RNAI (3′mivaRNAI-137) was actually slightly more abundant in RISC than 3′mivaRNAI(A)-138, for which one direct target has previously been reported (28). The majority of targets identified in this study were targets of 3′mivaRNAI-137; we could identify only one direct target of 5′mivaRNAI(A)-137. This result was reflected by a lower number of predicted 5′mivaRNAI(A)-137 targets among RISC-associated RNAs than the number of predicted 3′mivaRNAI-137 targets (Fig. 5). It can be explained by an inherently lower silencing capacity of 5′mivaRNAI(A)-137 (Fig. 3), which in turn is likely due to the less efficient incorporation of this single strand into RISC, evident as an approximately 3-fold lower IP/input read count ratio than the ratio of its partner strand derived from the 3′-arm of VA RNAI. In agreement with previous data (22, 24), 5′mivaRNAI(G) was barely incorporated into RISCs (Fig. 2A). Accordingly, no target RNAs of 5′mivaRNAI(G) were enriched in RISCs (Fig. 4C), and no RNAs with a respective seed sequence match were overrepresented among the RISC-enriched RNAs of cells transfected with mivaRNA mimics (Fig. 5) or infected with Ad5 (Fig. 10). In contrast, 3′mivaRNAI-137 had the overall best gene-silencing capability, was highly abundant in RISCs, and had thus a higher number of interacting target mRNAs (Fig. 9).
In a previous study, targeting of primarily 5′-UTRs of mRNAs by a cytomegalovirus-encoded miRNA, followed by significant reduction in gene expression, was reported (38). To investigate a possible similar mechanism in adenovirus, we included a number of potential 5′-UTR target sites into our study. However, there was no indication for 5′-UTRs being targeted by the adenoviral mivaRNAs that were investigated by us: the target sites of all validated targets resided in the 3′-UTRs, which, however, is in accordance with the 3′-UTR being the main interaction site for miRNAs (1, 35).
A set of putative target genes whose expression is downregulated in the presence of 3′mivaRNAI-138, the second-most-abundant VA RNAI-derived mivaRNA in RISCs of Ad5-infected cells, has been reported previously (28). Although direct targeting by the mivaRNA was demonstrated for only one RNA (TIA1) in that study and thus the list may have also contained secondary targets, the set contained LY6K. Notably, this RNA also emerged as a target of 3′mivaRNAI-137 in our screen. TargetScan predicts that LY6K is a target of both mivaRNAs, which is due to an extended target site recognized by both mivaRNA isoforms. Thus, our reporter assays that unambiguously indicated direct targeting by 3′mivaRNAI-137 also suggest that direct interaction with 3′mivaRNAI-138 is highly likely, and the identification of LY6K as a mivaRNAI-137/138 target in screens using 2 distinct methods strengthens the validity of our data set. Notably, the set of RNAs that scored positive in our dual-luciferase assays contained 2 more RNAs (BRP44 and SMAGP) with a target site predicted to be shared by both 3′mivaRNAI-137 and 3′mivaRNAI-138. Whether targeting by both mivaRNA isoforms renders those RNAs more important targets remains to be seen.
Among other diverse biological processes, the identified targets are involved, for example, in apoptosis (BOK), mitochondrial processes (BRP44), membrane-associated processes (SMAGP, TMEM222), or cell growth (LY6K). Targeting of several of them is conceivable to support virus propagation. One of the most crucial tasks in the future will be to distinguish between “intentional” and “unintentional” targets. Some RNAs may be targeted as a side effect because they show complementarity to the mivaRNA seed sequence and share a target site with the “intentional” target. Targeting of such “unintentional” targets may neither positively nor negatively affect virus propagation substantially. Identifying intentional targets may be challenging, because an effect may be detected only when using distinct readout systems or in an in vivo setting. In preliminary experiments, we have assessed the effect of exogenously added mivaRNA mimics on the replication of an Ad5 VA RNAI and -II double mutant in cancer cell lines in vitro. Under the experimental conditions used, virus replication was unaffected (data not shown), an observation that is in accordance with that of Kamel et al. (39), who also did not detect a positive effect of mivaRNAs on lytic growth in tissue culture.
We determined that 3′mivaRNAII-138 was most abundant in Ad5-infected cells and that it was incorporated into RISCs as efficiently as 3′mivaRNAI-137 or 3′mivaRNAI-138 (see Table S2 in the supplemental material). Although 3′mivaRNAII-136 and 3′mivaRNAII-137 were present in RISCs at approximately 3-fold-smaller amounts than 3′mivaRNAII-138, they were present at levels similar to those of 3′mivaRNAI-137 and 3′mivaRNAI-138. Thus, in principle, 3′mivaRNAII-136, 3′mivaRNAII-137, and 3′mivaRNAII-138 could all target cellular RNAs, and their targets could be expected to be enriched in RISCs of Ad5-infected cells to the same extent as the RNAs targeted by 3′mivaRNAI-137 or 3′mivaRNAI-138. However, when analyzing mRNAs enriched in RISCs of Ad5-infected cells, we did not detect an overrepresentation of mRNAs with seed sequence matches to any 3′mivaRNAII isoforms (Fig. 10). Together with the observation that 3′mivaRNAII-138 exerted only a moderate silencing effect on reporter genes (Fig. 3), these data argue that this mivaRNA recognizes target mRNAs relatively poorly when taking its high RISC incorporation efficiency into account. Although the possibility that single cellular mivaRNAII targets exist cannot be ruled out completely, our data collectively argue that mivaRNAII isoforms do not seem to contribute substantially to posttranscriptional silencing of cellular genes; instead, our bioinformatic analyses suggest that broad modulation of host gene expression is carried out only by 3′mivaRNAI-137/138, and possibly, and in any case to a much lesser extent, by 5′mivaRNAI(A).
Zhao et al. (40) have recently reported the presence of sRNAs of the closely related adenovirus 2 that map to the central regions of VA RNAI and RNAII rather than to their respective stem regions, and they speculated that these sRNAs may also target cellular RNAs (i.e., function as miRNAs). We also observed these RNAs in our RNA-Seq data sets for total RNA of Ad5-infected cells (Fig. 2C; see also Table S2 in the supplemental material), and the most abundant RNA forms were obviously derived by the cleavage at the base of the stem-loop of the 3′-arm of VA RNAI (genome positions 10732 and 10733). However, we did not detect significant incorporation of any of these sRNAs into RISCs, which indicates that they do not function as miRNAs. Moreover, we did not find sRNAs derived from any other regions of the adenoviral genome to be significantly enriched in RISCs, suggesting that Ad5 does not generate further miRNAs encoded by other parts of the genome.
Xu et al. (22) reported, based on a small-scale Sanger sequencing study, that approximately 80% of miRNAs associated with RISCs of cells lytically infected at high MOIs were derived from VA RNAs. Our RNA deep-sequencing data revealed a significantly lower occupancy (15%) but a comparable relative abundance of individual mivaRNA isoforms in RISCs (Fig. 2A). This disparity in our results may be explained by the use of distinct cell lines in the 2 studies or, more likely, by our infecting of the cells by using a much lower number of viruses/cell. It should be noted that at the MOI that we used for infection (10 TCID50/cell), we still observed 100% infection of the cells. Moreover, while in the previous study sRNAs were isolated exclusively from RISCs containing FLAG-tagged AGO2, which was overexpressed in the cells, we isolated RNAs from the entire spectrum of endogenous RISCs composed of all 4 types of endogenous human AGO proteins (AGO1 to -4) to avoid any manipulation of the endogenous RNAi machinery. Unlike in the situation where AGO proteins are overexpressed, in the situation where AGO proteins are limiting and the RISCs are already occupied with cellular miRNAs, de novo-synthesized mivaRNAs are likely to become less readily incorporated into RISCs. In analogy, overexpression of AGO2 has been demonstrated to abrogate saturation of RISCs, consequently facilitating RISC incorporation of exogenous siRNAs or shRNAs by abolishing the competition with cellular miRNAs (41–44). Thus, our data indicate that RISC occupation by mivaRNAs appears to have been overestimated in the past, because under physiologically more relevant conditions, cellular miRNAs are still dominating over mivaRNAs in endogenous RISCs. This is in accordance with the presence of targets of cellular miRNAs in RISCs of infected cells even at late time points during infection. Importantly, these data indicate that posttranslational regulation of gene expression by cellular miRNAs does still take place in Ad5-infected cells and is not abolished by mivaRNAs even during the late phase of infection, a fact that needs to be taken into account in future transcriptomics-based studies aimed at the investigation of virus-host interactions. Moreover, these data keep the door open for possible therapeutic scenarios of blocking adenovirus replication by means of RNAi-based methods.
However, although occupation of endogenous RISCs by mivaRNAs appears to be less dominant in the light of our current experiments, the partial displacement of cellular miRNAs from RISCs was still obvious. It has been postulated previously that such a displacement of cellular miRNAs should consequently be manifested in a genome-wide derepression of cellular gene expression. The inhibitory effect of mivaRNA generation on cellular RNAi has so far been demonstrated only for single targets. Coincidentally, regardless of whether this was caused by global inhibition of cellular RNAi or by other gene-regulatory effects, we observed that the number of RNAs that became statistically significantly depleted in RISCs and whose steady-state levels concurrently increased in the cells upon infection exceeded that of RNAs that became enriched in RISCs/downregulated upon infection more than 3-fold, pointing toward a global detargeting effect.
To assess causal connections between global miRNA expression changes and changes in global mRNA expression patterns, we employed the Sylamer algorithm (34). Sylamer performs best when a single or few miRNAs are experimentally overexpressed or blocked. However, the sensitivity in the identification of overrepresented seed-matching motifs decreases markedly if the expression of a higher number of miRNAs is altered and the network of primary and secondary miRNA-mediated effects (on both the transcriptional and posttranscriptional level) becomes more complex. This situation is clearly the case when comparing gene expression data for Ad5-infected and mock-infected cells. In fact, we were able to see neither an overrepresentation of mivaRNA seed-matching motifs in the set of genes downregulated upon Ad5 infection nor, conversely, an overrepresentation of sequences matching the seeds of the highly abundant and strongly downregulated let-7 family members. However, both connections were clearly revealed when including the microarray data sets for the RNAs that had been isolated from RISCs (Fig. 11B). Thus, our results exemplify that a combination of RIP-Chip or RIP-Seq with computational approaches such as Sylamer can, at least in certain instances, significantly improve the analysis of miRNA-mRNA interaction networks.
Supplementary Material
ACKNOWLEDGMENTS
We thank Stijn van Dongen, European Bioinformatics Institute/European Molecular Biology Laboratory (EMBL-EBI), Hinxton, Cambridge, United Kingdom, for kindly providing support in Sylamer analysis.
This work was supported by the Austrian Science Fund (FWF) through grant L665-B13.
We declare that no competing interests exist.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02336-14.
REFERENCES
- 1.Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huntzinger E, Izaurralde E. 2011. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 3.Kincaid RP, Sullivan CS. 2012. Virus-encoded microRNAs: an overview and a look to the future. PLoS Pathog 8:e1003018. doi: 10.1371/journal.ppat.1003018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boss IW, Renne R. 2011. Viral miRNAs and immune evasion. Biochim Biophys Acta 1809:708–714. doi: 10.1016/j.bbagrm.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghosh Z, Mallick B, Chakrabarti J. 2009. Cellular versus viral microRNAs in host-virus interaction. Nucleic Acids Res 37:1035–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grundhoff A, Sullivan CS. 2011. Virus-encoded microRNAs. Virology 411:325–343. doi: 10.1016/j.virol.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersson MG, Haasnoot PC, Xu N, Berenjian S, Berkhout B, Akusjarvi G. 2005. Suppression of RNA interference by adenovirus virus-associated RNA. J Virol 79:9556–9565. doi: 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aparicio O, Razquin N, Zaratiegui M, Narvaiza I, Fortes P. 2006. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J Virol 80:1376–1384. doi: 10.1128/JVI.80.3.1376-1384.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu S, Cullen BR. 2004. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J Virol 78:12868–12876. doi: 10.1128/JVI.78.23.12868-12876.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sano M, Kato Y, Taira K. 2006. Sequence-specific interference by small RNAs derived from adenovirus VAI RNA. FEBS Lett 580:1553–1564. doi: 10.1016/j.febslet.2006.01.085. [DOI] [PubMed] [Google Scholar]
- 11.Coventry VK, Conn GL. 2008. Analysis of adenovirus VA RNAI structure and stability using compensatory base pair modifications. Nucleic Acids Res 36:1645–1653. doi: 10.1093/nar/gkn020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma Y, Mathews MB. 1996. Secondary and tertiary structure in the central domain of adenovirus type 2 VA RNA I. RNA 2:937–951. [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y, Mathews MB. 1996. Structure, function, and evolution of adenovirus-associated RNA: a phylogenetic approach. J Virol 70:5083–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma Y, Mathews MB. 1993. Comparative analysis of the structure and function of adenovirus virus-associated RNAs. J Virol 67:6605–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathews MB. 1995. Structure, function, and evolution of adenovirus virus-associated RNAs. Curr Topics Microbiol Immunol 199(Part 2):173–187. [DOI] [PubMed] [Google Scholar]
- 16.Thimmappaya B, Weinberger C, Schneider RJ, Shenk T. 1982. Adenovirus VAI RNA is required for efficient translation of viral mRNAs at late times after infection. Cell 31:543–551. doi: 10.1016/0092-8674(82)90310-5. [DOI] [PubMed] [Google Scholar]
- 17.Maran A, Mathews MB. 1988. Characterization of the double-stranded RNA implicated in the inhibition of protein synthesis in cells infected with a mutant adenovirus defective for VA RNA. Virology 164:106–113. doi: 10.1016/0042-6822(88)90625-3. [DOI] [PubMed] [Google Scholar]
- 18.Wahid AM, Coventry VK, Conn GL. 2009. The PKR-binding domain of adenovirus VA RNAI exists as a mixture of two functionally non-equivalent structures. Nucleic Acids Res 37:5830–5837. doi: 10.1093/nar/gkp595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitajewski J, Schneider RJ, Safer B, Munemitsu SM, Samuel CE, Thimmappaya B, Shenk T. 1986. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 45:195–200. doi: 10.1016/0092-8674(86)90383-1. [DOI] [PubMed] [Google Scholar]
- 20.Mathews MB, Shenk T. 1991. Adenovirus virus-associated RNA and translation control. J Virol 65:5657–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levin DH, Petryshyn R, London IM. 1980. Characterization of double-stranded-RNA-activated kinase that phosphorylates alpha subunit of eukaryotic initiation factor 2 (eIF-2 alpha) in reticulocyte lysates. Proc Natl Acad Sci U S A 77:832–836. doi: 10.1073/pnas.77.2.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu N, Segerman B, Zhou X, Akusjarvi G. 2007. Adenovirus virus-associated RNAII-derived small RNAs are efficiently incorporated into the RNA-induced silencing complex and associate with polyribosomes. J Virol 81:10540–10549. doi: 10.1128/JVI.00885-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yi R, Qin Y, Macara IG, Cullen BR. 2003. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu N, Gkountela S, Saeed K, Akusjarvi G. 2009. The 5′-end heterogeneity of adenovirus virus-associated RNAI contributes to the asymmetric guide strand incorporation into the RNA-induced silencing complex. Nucleic Acids Res 37:6950–6959. doi: 10.1093/nar/gkp764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersson G, Xu N, Akusjarvi G. 2007. In vitro methods to study RNA interference during an adenovirus infection. Methods Mol Med 131:47–61. doi: 10.1007/978-1-59745-277-9_4. [DOI] [PubMed] [Google Scholar]
- 26.Furuse Y, Ornelles DA, Cullen BR. 2013. Persistently adenovirus-infected lymphoid cells express microRNAs derived from the viral VAI and especially VAII RNA. Virology 447:140–145. doi: 10.1016/j.virol.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thimmappaya B, Jones N, Shenk T. 1979. A mutation which alters initiation of transcription by RNA polymerase III on the Ad5 chromosome. Cell 18:947–954. doi: 10.1016/0092-8674(79)90207-1. [DOI] [PubMed] [Google Scholar]
- 28.Aparicio O, Carnero E, Abad X, Razquin N, Guruceaga E, Segura V, Fortes P. 2010. Adenovirus VA RNA-derived miRNAs target cellular genes involved in cell growth, gene expression and DNA repair. Nucleic Acids Res 38:750–763. doi: 10.1093/nar/gkp1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhat RA, Thimmappaya B. 1984. Adenovirus mutants with DNA sequence perturbations in the intragenic promoter of VAI RNA gene allow the enhanced transcription of VAII RNA gene in HeLa cells. Nucleic Acids Res 12:7377–7388. doi: 10.1093/nar/12.19.7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ibrisimovic M, Kneidinger D, Lion T, Klein R. 2013. An adenoviral vector-based expression and delivery system for the inhibition of wild-type adenovirus replication by artificial microRNAs. Antivir Res 97:10–23. doi: 10.1016/j.antiviral.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 33.Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 34.van Dongen S, Abreu-Goodger C, Enright AJ. 2008. Detecting microRNA binding and siRNA off-target effects from expression data. Nat Methods 5:1023–1025. doi: 10.1038/nmeth.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu S, Jin L, Zhang F, Sarnow P, Kay MA. 2009. Biological basis for restriction of microRNA targets to the 3′ untranslated region in mammalian mRNAs. Nat Struct Mol Biol 16:144–150. doi: 10.1038/nsmb.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson PT, Hatzigeorgiou AG, Mourelatos Z. 2004. miRNP:mRNA association in polyribosomes in a human neuronal cell line. RNA 10:387–394. doi: 10.1261/rna.5181104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goff LA, Davila J, Swerdel MR, Moore JC, Cohen RI, Wu H, Sun YE, Hart RP. 2009. Ago2 immunoprecipitation identifies predicted microRNAs in human embryonic stem cells and neural precursors. PLoS One 4:e7192. doi: 10.1371/journal.pone.0007192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grey F, Tirabassi R, Meyers H, Wu G, McWeeney S, Hook L, Nelson JA. 2010. A viral microRNA down-regulates multiple cell cycle genes through mRNA 5′UTRs. PLoS Pathog 6:e1000967. doi: 10.1371/journal.ppat.1000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamel W, Segerman B, Oberg D, Punga T, Akusjarvi G. 2013. The adenovirus VA RNA-derived miRNAs are not essential for lytic virus growth in tissue culture cells. Nucleic Acids Res 41:4802–4812. doi: 10.1093/nar/gkt172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao H, Chen M, Pettersson U. 2013. Identification of adenovirus-encoded small RNAs by deep RNA sequencing. Virology 442:148–155. doi: 10.1016/j.virol.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 41.Diederichs S, Jung S, Rothenberg SM, Smolen GA, Mlody BG, Haber DA. 2008. Coexpression of Argonaute-2 enhances RNA interference toward perfect match binding sites. Proc Natl Acad Sci U S A 105:9284–9289. doi: 10.1073/pnas.0800803105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen CM, Chiu SL, Shen W, Cline HT. 2009. Co-expression of Argonaute2 enhances short hairpin RNA-induced RNA interference in Xenopus CNS neurons in vivo. Front Neurosci 3:63. doi: 10.3389/neuro.17.001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shao PL, Lu MY, Liau YJ, Chao MF, Chang LY, Lu CY, Kao CL, Chang SY, Chi YH, Huang LM. 2011. Argonaute-2 enhances suppression of human cytomegalovirus replication by polycistronic short hairpin RNAs targeting UL46, UL70 and UL122. Antivir Ther 16:741–749. doi: 10.3851/IMP1808. [DOI] [PubMed] [Google Scholar]
- 44.Borner K, Niopek D, Cotugno G, Kaldenbach M, Pankert T, Willemsen J, Zhang X, Schurmann N, Mockenhaupt S, Serva A, Hiet MS, Wiedtke E, Castoldi M, Starkuviene V, Erfle H, Gilbert DF, Bartenschlager R, Boutros M, Binder M, Streetz K, Krausslich HG, Grimm D. 2013. Robust RNAi enhancement via human Argonaute-2 overexpression from plasmids, viral vectors and cell lines. Nucleic Acids Res 41:e199. doi: 10.1093/nar/gkt836. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.