

ABSTRACT
Interleukin-1 beta (IL-1β) is an inflammatory cytokine that is secreted in response to inflammasome activation by innate microbe-sensing pathways. Although some retroviruses can trigger IL-1β secretion through the DNA-sensing molecule IFI16, the effect of IL-1β on the course of infection is unknown. To test whether IL-1β secretion affects retroviral replication in vivo, I constructed a novel murine leukemia virus strain (FMLV-IL-1β) that encodes the mature form of IL-1β. This virus replicated with kinetics similar to that of wild-type virus in tissue culture but caused a dramatically more aggressive infection of both C57BL/6 and BALB/c mice. By 7 days postinfection (PI), mice infected with FMLV-IL-1β exhibited splenomegaly and viral loads 300-fold higher than those in mice infected with wild-type FMLV. Furthermore, the enlarged spleens of FMLV-IL-1β-infected mice correlated with a large expansion of Gr-1+ CD11b+ myeloid-derived suppressor cells, as well as elevated levels of immune activation. Although FMLV-IL-1β infection was controlled by C57BL/6 mice by 14 days p.i., FMLV-IL-1β was able to establish a significant persistent infection and immune activation in BALB/c mice. These results demonstrate that IL-1β secretion is a powerful positive regulator of retroviral infection and that FMLV-IL-1β represents a new model of proinflammatory retroviral infection.
IMPORTANCE Interleukin-1 beta (IL-1β) is an inflammatory cytokine released in response to activation of innate pathogen-sensing pathways during microbial infection. To examine the potential impact of IL-1β on retroviral replication in vivo, I constructed a novel mouse retrovirus strain (FMLV-IL-1β) that encodes IL-1β and promotes abundant IL-1β secretion from infected cells. This virus replicates with normal kinetics in cultured cells but displays a dramatically enhanced ability to replicate in mice and caused persistent infection and immune activation in the BALB/c strain of mice. These results establish IL-1β as a positive regulator of retroviral replication and suggest that targeting this pathway may have therapeutic benefits in infections with proinflammatory retroviruses. This virus can also be used to further study the impact of inflammatory pathways on retroviral infection.
INTRODUCTION
Retroviruses are a large and diverse family of viruses that all share a replication strategy, involving reverse transcription of viral genomic RNA into double-stranded DNA, followed by integration into host cell DNA. Some retroviruses, such as human immunodeficiency virus (HIV) and human T cell leukemia virus (HTLV), are significant human pathogens for which no effective vaccine exists (1). As such, there is still a major need to characterize and understand immunological parameters that regulate the outcome of retroviral infection.
Acute retroviral infection in immunocompetent hosts typically involves a transient wave of host cytokine secretion, resulting from the detection of infection by the innate immune system (2). However, the precise mechanistic roles played by most individual cytokines in retroviral infection are still unclear. Inflammatory cytokines could potentially facilitate retroviral replication by promoting the recruitment of susceptible cells to sites of infection, by triggering cellular activation and proliferation, or by activating transcription factors that positively regulate viral gene expression. Conversely, innate cytokine responses could help to activate potent innate and adaptive immune responses at sites of infection and thus lead to improved viral clearance. As such, the innate cytokine response likely has complex effects in vivo that collectively influence the outcome of infection.
Innate host pathways that respond to retroviruses and promote cytokine secretion are therefore promising clinical targets for modulating the course of retroviral infection. Unfortunately, the identities of the retrovirus-sensing pathways that contribute to the cytokine response are not clear, and until recently, little was known about how the innate immune system detects retroviruses (3). Nevertheless, some recent reports have begun to identify and characterize retrovirus-sensing pathways. Work from several laboratories has shown that Toll-like receptor 7 (TLR7), a single-stranded RNA sensor expressed in endosomes, can act as a sensing pathway for retroviruses (4–8). The HIV-1 genome contains potential TLR7 ligands (9), and HIV-1 triggers alpha interferon (IFN-α) and tumor necrosis factor alpha (TNF-α) secretion by plasmacytoid dendritic cells (pDCs) in a TLR7-dependent manner (10). However, this response requires very high doses of virus, and TLR7-expressing conventional dendritic cells (cDCs) are curiously inert to HIV-1 (11), suggesting that HIV-1 is a relatively weak stimulator of TLR7. Furthermore, in vivo evidence from humans and mice has shown that TLR7 is a protective factor against retroviral infection—TLR7-deficient mice exhibit greatly elevated virus levels after infection with murine gammaretroviruses (5, 8), and a loss-of-function TLR7 allele in humans correlates with higher viral loads and accelerated HIV-1 disease progression (12, 13). Furthermore, TLR7-deficient mice mount an elevated inflammatory cytokine response to retroviral infection, indicating the existence of an alternative pathway that drives inflammation and cytokine secretion (8).
The inflammasome has recently been described as a retrovirus-sensing pathway that triggers caspase-1 activation and secretion of the inflammatory cytokine interleukin-1 beta (IL-1β) in response to viral cDNA generated during abortive infection (14, 15). Secreted IL-1β binds to, and activates, its cognate receptor (IL-1β receptor 1 [IL-1R1]) and is a potent stimulator of the transcription factor NF-κB. Since IL-1R1 is broadly expressed in the immune system, IL-1β can affect many different immune lineages (16). IL-1β is also known to have important roles in promoting tumorigenesis and autoimmune diseases (17, 18). It has been proposed that the release of cytokines such as IL-1β in response to infection initiates an inflammatory positive feedback loop that promotes retroviral replication, immune activation, and pathogenesis (14, 19). However, the in vivo effects of IL-1β on retroviral infection and the host response are unknown and have not previously been investigated.
Mouse models of retroviral infection offer a system where the role of specific molecules such as IL-1β can be easily tested, but it is unclear if murine retroviruses trigger a similar inflammatory process. Murine retrovirus infection induces a modest IL-1β response (8, 20, 21), but IL-1R1-deficient mice exhibit little overt phenotype with respect to disease or viral loads following infection with the retroviral pathogen Friend virus (FV) (my unpublished data). To circumvent this limitation, and to test the role of virus-induced IL-1β in retroviral infection, I constructed a novel strain of murine leukemia virus (FMLV-IL-1β) that encodes the active mature form of murine IL-1β. FMLV-IL-1β thus permits direct examination of the virological and immunological consequences of virus-dependent IL-1β secretion and inflammation. FMLV-IL-1β was found to exhibit greatly elevated replication in vivo, demonstrating that coupling IL-1β secretion to infection potently enhances retroviral replication in mammalian hosts.
MATERIALS AND METHODS
Plasmids and FMLV-IL-1β construction.
A full-length clone of NB-tropic FMLV (generous gift from Mario Santiago, University of Colorado Denver) was modified by inserting an in-frame coding sequence for a 2A peptide from porcine teschovirus (P2A) at the 3′ end of the envelope gene, as well as a NotI restriction enzyme site. The parental FMLV plasmid was first digested with the restriction enzymes ClaI and BplI, which cut upstream and downstream of the envelope stop codon, respectively, and a commercially synthesized double-stranded DNA sequence (IDT-DNA) that overlaps this region and contains the P2A and NotI sequences was ligated into the digested plasmid. Coding sequence for murine IL-1β from valine 118 to serine 269 (corresponding to the mature cleaved form of IL-1β) was then cloned in-frame into the NotI site downstream of the P2A sequence. The integrity of the construct was verified by DNA sequencing.
Virus stocks and focus-forming assays.
To generate stocks of infectious FMLV or FMLV-IL-1β virus, 10 μg plasmid was transfected into subconfluent 293FT cells, a fast-growing subclone of 293T cells (Invitrogen), using LT1 transfection reagent (Mirus Bio, Madison, WI, USA). At 4 h posttransfection, medium was replaced. Two days after transfection, supernatant was clarified by low-speed centrifugation and filtered through a 0.45-μm filter to remove cell debris. To generate reliable high-titer stocks for infection of mice, this supernatant was used to infect Mus dunni cells, which were then passaged for 1 to 2 weeks, until the majority of cells showed evidence of infection. Supernatants from these cells were collected, filtered, and frozen in aliquots at −80°C.
To generate stocks of Friend virus complex (FV), a 10% spleen homogenate from BALB/c mice at 8 days after infection with B-tropic polycythemia-inducing FV was prepared in phosphate-buffered saline (PBS) and freeze-thawed once to release virus particles. This mixture was then centrifuged, filtered through a 0.45-μm filter, and frozen at −80°C in aliquots. The original FV stock was obtained from K. Hasenkrug (NIAID) and was confirmed to be free of lactate dehydrogenase-elevating virus by PCR.
To assay the titers of FMLV or FV in stocks, a focus-forming assay was performed. Serial 10-fold dilutions of virus samples were plated on subconfluent Mus dunni cells. At 3 days postinfection (dpi), the cells were washed with PBS, fixed with methanol for 10 min, and then stained with monoclonal antibody (MAb) 720, which recognizes the FMLV envelope protein (22). To visualize infected foci, the cells were then stained with anti-IgG1–horseradish peroxidase followed by treatment with aminoethyl carbazole substrate (Sigma). The number of foci was then used to calculate the number of infectious focus-forming units (FFU) in the original sample.
To measure expression of glycosylated Gag (glyco-Gag) or envelope protein on the surfaces of infected cells, MAb 34 (23) or MAb 720, respectively, was used to stain infected cells, followed by a fluorescein isothiocyanate (FITC)-labeled anti-mouse IgG antibody (eBioscience). Stained cells were then washed in PBS before flow cytometry to determine the mean fluorescence intensity (MFI) of infected cells. MAb 34 and MAb 720 hybridomas were obtained from K. Hasenkrug (NIAID).
Mouse strains and infections.
All mouse strains were purchased from The Jackson Laboratory. Specific strains and stock numbers are C57BL/6J (000664), BALB/c (000651), and IL-1 receptor 1 (IL-1R1) knockout mice (003245). For FMLV, FMLV-IL-1β, and FV infections, mice at 6 to 8 weeks of age were infected with 30,000 focus-forming units (FFU) of infectious virus by intraperitoneal injection. To measure the number of infected cells per spleen in infected mice, serial dilutions of splenocyte suspensions were plated on subconfluent Mus dunni cells. Three days later, splenocytes were washed away, and the number of infected foci determined by a focus-forming assay as described for assaying virus stocks. The number of foci was then used to calculate the number of infected cells per spleen in the original sample.
Ethics statement.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (24). These studies were approved by the Committee on Animal Care of the Massachusetts Institute of Technology (protocol 0712-075-15).
Organ processing and analysis by flow cytometry.
For analysis of mouse spleens, dissected organs were first mashed through a 70-μm mesh, pelleted, and then subjected to erythrocyte hypotonic lysis. Splenocytes were then resuspended in fluorescence-activated cell sorting (FACS) staining buffer (PBS with 2% fetal calf serum and 2 mM EDTA). For flow cytometry, splenocyte samples were stained with 1 to 2 million cells in 100 μl of FACS staining buffer. An Fc-blocking antibody (eBioscience) was used to prevent cross-reactivity of labeled antibodies with Fc receptors. Specific antibodies/fluorophores used to detect antigens were TCRβ-APC, CD8-FITC, B220-PerCP-Cy5.5, CD4-AF700, CD69-BV510, CD44-BV421, CD11b-APC-Cy7, CD11c-APC, Gr-1-FITC, CD86-PE, and PD1-PECy7. All antibodies were obtained from eBioscience (San Diego, CA) or BioLegend (San Diego, CA). The H2Db-GagL-PE tetramer was synthesized by Beckman-Coulter (Brea, CA). Samples were stained for 20 min on ice, followed by washing with PBS and resuspension in FACS buffer. Lineage-defining markers and activation markers were stained simultaneously. Samples were analyzed on an Accuri C6 or a BD LSRII flow cytometer.
Antibody assay.
To measure virus-specific antibodies in plasma samples, a rat kidney cell line that is chronically infected with FV and abundantly expresses the FV envelope protein on its surface (obtained from Leonard Evans, NIAID) was stained with samples of mouse plasma at 1:10 dilution in PBS with 2% fetal calf serum (FCS) for 30 min on ice. The cells were then washed in PBS and stained with a FITC-labeled anti-mouse IgG antibody. The mean fluorescent intensity (MFI) of staining was then assayed by flow cytometry.
IL-1β assay.
Murine IL-1β was assayed using a commercial IL-1β enzyme-linked immunosorbent assay (ELISA) kit (eBioscience).
Statistical analyses.
To determine significant statistical difference between experimental groups, Student's t test was used, except for virus-level data from infected mice, for which a Mann-Whitney analysis was used. Unless otherwise indicated, error bars on figures represent the standard deviations of the data set.
Nucleotide sequence accession numbers.
Accession numbers for genes/viruses discussed in this study are as follows: for IL-1β, NM_008361; for IL-1R1, NM_008362; for FMLV, NC_001362.
RESULTS
Construction and characterization of FMLV-lL-1β.
To test the effect of virus-driven IL-1β on retroviral replication and pathogenesis, I constructed a novel strain of murine leukemia virus (FMLV-IL-1β) that encodes the mature caspase 1-processed form of murine IL-1β. Inserting a cytokine-coding sequence into the viral genome allows direct manipulation of the infected cell's response to infection and mimics a spatially localized innate immune response more accurately than repeated dosing of mice with recombinant cytokines. FMLV-IL-1β was derived from a full-length replication competent clone of NB-tropic F-MLV (25). To permit viral expression of IL-1β, the coding sequence for the mature caspase-1-processed form of IL-1β (Val118-Ser269) was inserted immediately downstream of, and in frame with, the viral envelope gene but separated by a 2A peptide sequence from porcine teschovirus (P2A) (Fig. 1A). The 2A peptide sequence mediates “cleavage” between envelope and IL-1β by preventing peptide bond formation (26). In cells infected with FMLV-IL-1β, IL-1β is thus expressed and secreted independently of inflammasome and caspase 1 activation. DNA sequencing confirmed the integrity of all constructs.
FIG 1.

Construction of FMLV-IL-1β. (A) A full-length replication-competent clone of ecotropic FMLV was modified by the addition of coding sequence for the porcine teschovirus 1 2A peptide (P2A) followed by the coding sequence for a region of murine IL-1β from Val118 to Ser269. (B) The ability of the new virus (FMLV-IL-1β) as well as its parent virus to replicate in cultured murine fibroblasts was analyzed. Subconfluent Mus dunni cells were infected with virus at a multiplicity of infection (MOI) of 0.01. At 24-h intervals, supernatant samples were removed, and the concentration of focus-forming units (FFU) was measured by focus-forming assays. Each infection was carried out in triplicate, and data points represent the averages of three measurements. (C) The concentration of IL-1β released into the supernatant of infected Mus dunni cells at 3 dpi was measured by ELISA. Each data point is the average for three replicates. The asterisk indicates a P value of <0.05 (t test). ND, not detected. (D) Mus dunni cells were infected at an MOI of 0.001, and at 3 dpi, the cells were stained with MAb 34 or MAb 720, which recognize the viral proteins glyco-Gag and envelope, respectively, followed by a FITC-labeled secondary antibody. The mean fluorescence intensity (MFI) of the infected-cell gate was determined by flow cytometry and is represented in arbitrary units. Each bar represents the average of triplicate measurements. Error bars indicate the standard deviations of the data set. NS, not significant (P > 0.05).
To test the ability of FMLV-IL-1β to replicate in tissue culture, I infected Mus dunni cells with FMLV or FMLV-IL-1β at a low multiplicity of infection (MOI, 0.01) and monitored the level of infectious virus in the supernatant at 24-h intervals postinfection (1, 2, and 3 dpi) by focus-forming assay. Infectious virus was detectable in the supernatant by 1 dpi for both viruses and increased progressively until 3 dpi. Notably, FMLV-IL-1β replicated with kinetics indistinguishable from wild-type FMLV (Fig. 1B), indicating that P2A peptide sequences or heterologous inserts in this location do not dramatically affect FMLV replication. To confirm the ability of this virus to express IL-1β, I measured IL-1β in the supernatant of infected cells at 3 dpi by enzyme-linked immunosorbent assay (ELISA). As expected, abundant IL-1β was detectable in the supernatant of FMLV-IL-1β-infected cells but not in that of cells infected with wild-type FMLV (Fig. 1C). To investigate whether modification of FMLV altered expression of viral gene products, I infected Mus dunni cells with FMLV or FMLV-IL-1β at a low MOI (0.001) and then, at 3 dpi, stained these cells with monoclonal antibodies specific for glycosylated Gag (MAb 34) or envelope (MAb 720). The expression level of these proteins was then quantified by staining with a fluorescently labeled secondary antibody and flow cytometry to determine the mean fluorescence intensity (MFI) of infected cells. Notably, FMLV-infected cells and FMLV-IL-1β-infected cells expressed glycosylated Gag and envelope at similar levels (Fig. 1D). These results verify that FMLV-IL-1β replicates normally in tissue culture and promotes secretion of IL-1β from infected cells.
FMLV-IL-1β exhibits enhanced replication in vivo.
To examine the effect of virus-induced IL-1β secretion on retroviral replication in vivo, I infected C57BL/6 (B6) mice with FMLV or FMLV-IL-1β and examined the level of infected cells in the spleen at 7 dpi by focus-forming assay. Adult B6 mice are relatively resistant to FMLV infection, and as expected, by 7 dpi, the spleens of mice infected with FMLV exhibited only limited numbers of infected cells (Fig. 2A). In contrast, FMLV-IL-1β infection resulted in a dramatically higher number of infected cells, approximately 300-fold higher than for FMLV. Also, FMLV-IL-1β-infected mice displayed significant splenomegaly, with an average 3-fold increase in spleen size (Fig. 2B).
FIG 2.
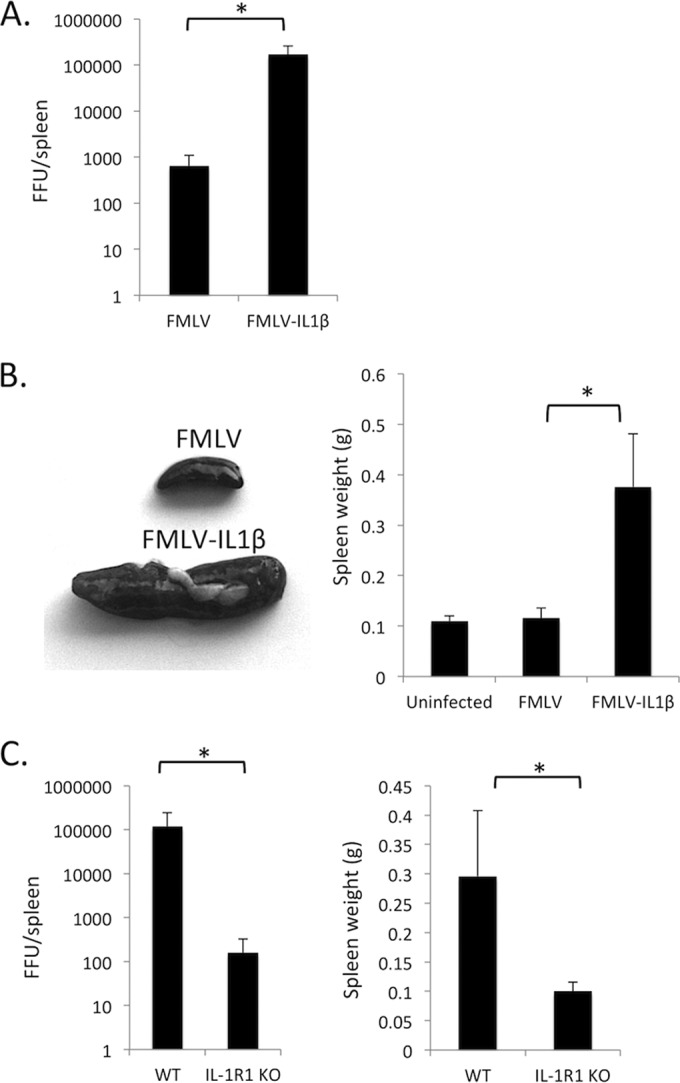
FMLV-IL-1β exhibits enhanced replication in vivo. (A) C57BL/6 (B6) mice were infected with FMLV or FMLV-IL-1β. At 7 dpi, mice were dissected, and the number of focus-forming units (FFU) per spleen was measured by plating splenocyte dilutions on Mus dunni cells followed by a focus-forming assay. The results of the focus-forming assay were used to calculate FFU/spleen in each infected mouse. (B) The weights of spleens from FMLV- and FMLV-IL-1β-infected mice were measured (right). Representative spleens are shown on the left. (C) Wild-type B6 (WT) or congenic IL-1 receptor 1-deficient (IL-1R1 KO) mice were infected with FMLV-IL-1β. At 7 dpi, FFU per spleen (left) and spleen weights (right) were measured by plating splenocyte dilutions on Mus dunni cells followed by a focus-forming assay. Each bar represents the average for 5 mice. The asterisks indicate P values of <0.05 (t test or Mann-Whitney test). Error bars indicate the standard deviations of the data set.
To rule out the possibility that the FMLV-ILβ virus had acquired an additional mutation that enhanced its ability to replicate in vivo or had become contaminated with another more aggressive MLV strain, I examined whether FMLV-IL-1β viral loads or splenomegaly was altered in IL-1 receptor 1 (IL-1R1)-deficient mice. Consistent with the hypothesis that the enhanced replication of FMLV-IL-1β in vivo is due to virus-driven IL-1β expression, IL-1R1 knockout mice had much lower viral loads and spleen sizes, similar to those observed for mice infected with wild-type virus (Fig. 1C). These data thus demonstrate that IL-1β-dependent signaling is a potent positive regulator of retroviral infection in vivo and that coupling retrovirus infection with secretion of IL-1β results in a significantly more aggressive acute infection.
FMLV-IL-1β infection causes an expansion of CD11b+ Gr-1+ myeloid-derived suppressor cells.
To understand the cellular basis of the splenomegaly observed during FMLV-IL-1β infection, I compared the cellular composition of the spleens of FMLV- and FMLV-IL-1β-infected mice at 7 dpi by flow cytometry. Interestingly, forward-scatter (FSC) and side scatter (SSC) plots showed that, compared to FMLV-infected spleens, FMLV-IL-1β-infected spleens exhibited a significant increase in the proportion of cells within the myeloid compartment, defined as cells with a high degree of SSC (Fig. 3A). Specifically, the proportion of myeloid cells increased from approximately 10% to almost 40% (Fig. 3B). This suggests that IL-1β secretion by infected cells has had a substantial effect on the cellular composition of the spleen and promotes a dramatic expansion of high-SSC myeloid cells.
FIG 3.
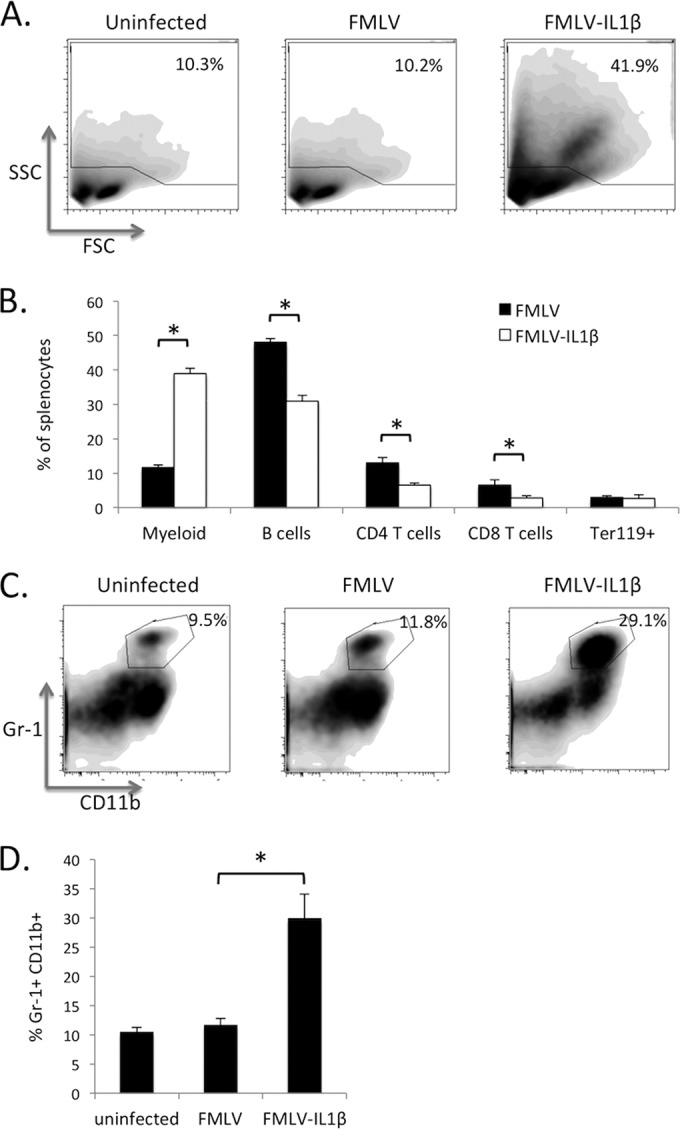
FMLV-IL-1β induces expansion of CD11b+ Gr-1+ myeloid tissue-derived suppressor cells. Splenocytes from mice infected with FMLV or FMLV-IL-1β at 7 dpi were stained with fluorescently labeled antibodies and analyzed by flow cytometry to measure the relative abundance of different cell lineages. (A) Data were first examined by forward scatter (FSC) and side scatter (SSC) to distinguish lymphocytes (low SSC) from myeloid cells (high SSC). (B) The proportion of splenocytes within specific immune lineages was measured; CD4 and CD8 T cells were identified by coexpression of TCRβ and CD4 or CD8, respectively, while B cells were defined as B220+ CD11c− lymphocytes. Erythroblasts were defined as Ter119+ cells within the low-SSC gate. (C) The myeloid gate was further subdivided by CD11b and Gr-1 expression, and the percentage of CD11b/Gr-1 double positive cells within this gate was calculated (D). Each flow cytometry plot is representative of 5 mice, while bars represent the averages for five mice. The asterisk indicates a P value of <0.05 (t test). Error bars indicate the standard deviations of the data set.
A more detailed analysis of individual lineages by flow cytometry showed that all lymphocyte subsets (B cells, CD4 T cells, and CD8 T cells) decreased as a proportion of the total splenocyte suspension (Fig. 3B), but within the lymphocyte gate, the relative proportions of these subsets did not change significantly. Significantly, FMLV-IL-1β infection had no effect on the proportion of Ter119+ erythroblasts, in contrast with published reports for infection with FV complex (27). Within the myeloid gate, however, there was a large increase in the proportion of CD11b+ Gr-1+ cells from roughly 10% to 30% (Fig. 3C and D). The CD11b+ Gr-1+ cell population has been characterized as myeloid-derived suppressor cells (MDSCs) that can inhibit pathogen-specific and tumor-specific immune responses. MDSCs have previously been shown to be induced by IL-1β secretion (28), likely as part of a negative feedback loop to limit immunopathogenesis. Notably, MDSC levels have been recently shown to be elevated in HIV-1 patients and may contribute to the suppression of HIV-1-specific immune responses (29, 30).
FMLV-IL-1β-infected mice exhibit elevated immune activation.
Chronically elevated immune activation and inflammation is a defining feature of HIV-1 infection, and immune activation is a strong predictor of disease progression, but the mechanisms behind this response are unclear (31). In addition to antigen-driven activation of lymphocytes, many immune cells can also be activated via pathogen-sensing pathways, such as Toll-like receptors (TLRs), and also by cytokines. Since the IL-1β receptor (IL-1R1) is broadly expressed on immune cells, I hypothesized that FMLV-IL-1β would induce elevated immune activation in infected mice. To measure the level of immune activation induced by FMLV and FMLV-IL-1β, splenocytes from infected mice at 7 dpi were examined by flow cytometry for the expression of known activation markers for each lineage. Strikingly, most immune lineages exhibited significantly elevated levels of activation in FMLV-IL-1β-infected mice (Fig. 4). CD4 T cells and CD8 T cells had elevated expression of the memory marker CD44, indicating antigen exposure, as well as the recent activation marker CD69, while CD69 and the costimulatory molecule CD86 were upregulated on B cells. CD86 was also upregulated on innate immune cell lineages such as plasmacytoid dendritic cells (pDCs), conventional dendritic cells (cDCs), and macrophages. These results demonstrate that increased expression of IL-1β in retrovirus-infected cells leads to a significantly more potent activation of both lymphoid and myeloid cell lineages during acute retroviral infection.
FIG 4.
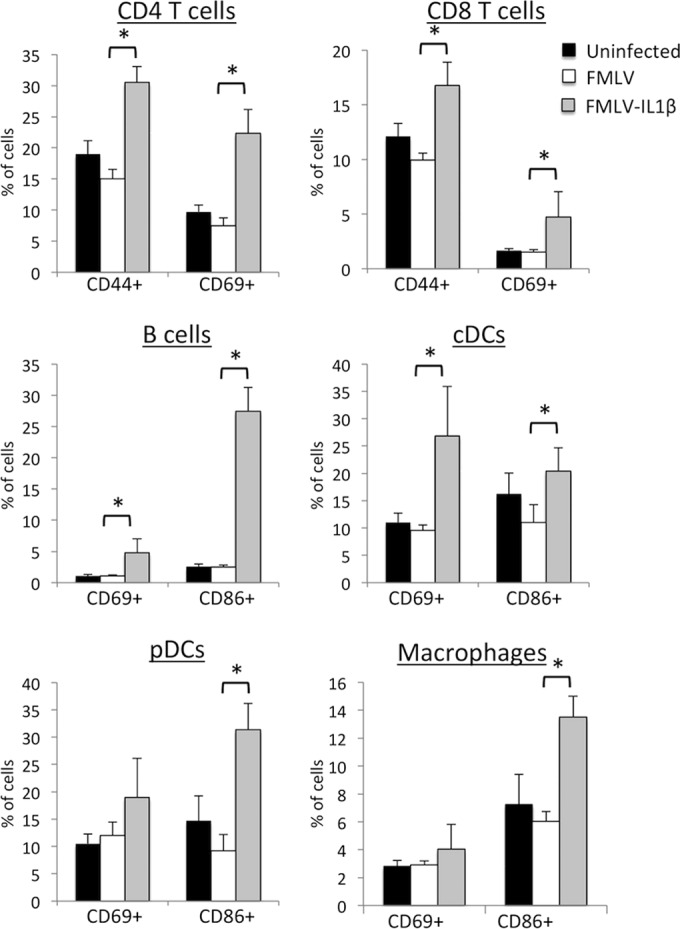
FMLV-IL-1β infection causes elevated immune activation. Splenocytes from mice infected with FMLV or FMLV-IL-1β at 7 dpi were stained for markers of immune lineages such as B cells (B220+ CD11c−), CD4 and CD8 T cells (TCRβ+ and CD4+ or CD8+), cDCs (CD11c+ B220−), pDCs (B220+ CD11cint), and macrophages (CD11b+ Gr-1−) as well as known markers of immune activation (CD69, CD86, and CD44), and analyzed by flow cytometry. Each bar represents the average for five mice. Asterisks indicate P values of <0.05 (t test). Error bars indicate the standard deviations of the data set.
FMLV-ILβ is controlled by B6 mice by 14 dpi.
FMLV-IL-1β is able to cause an elevated acute infection of B6 mice at 7 dpi, but the long-term outcome of FMLV-IL-1β infection was unknown. The inflammatory feedback loop induced by IL-1β could continue to promote infection, but B6 mice mount a potent adaptive immune response to gammaretroviral infection, with CD8 T cell responses and high titers of virus-specific antibodies appearing between 8 and 12 days postinfection (32). To investigate how IL-1β secretion affects retroviral replication in the context of a virus-specific adaptive immune response, I measured the level of infected cells in the spleens of mice at 14 dpi with FMLV or FMLV-IL-1β. Notably, at 14 dpi, infected cells were undetectable for both FMLV and FMLV-IL-1β, indicating that infected cells had been largely eliminated from the spleen by host immune responses, although significant splenomegaly remained (Fig. 5A). To examine the virus-specific adaptive immune responses, I measured FMLV-specific antibodies in the plasma (Fig. 5B) and CD8 T cells specific for the GagL peptide in the spleens (Fig. 5C) of infected mice at 14 dpi. The assay for FMLV-specific antibodies measures the total serum reactivity to FV surface antigens, which includes both the envelope glycoprotein and glyco-Gag. Mice infected with FMLV or FMLV-IL-1β both mounted a virus-specific antibody response, and this response was not significantly different between these two groups (P = 0.681). Similarly, the CD8 T cell response, which was assayed using the H2Db-GagL tetramer, was not significantly different between mice infected with FMLV and those infected with FMLV-IL-1β (P = 0.811). Thus, although IL-1β dramatically enhances retroviral infection in vivo, it does not counteract the B6 adaptive immune responses that limit and control infection by 14 dpi.
FIG 5.
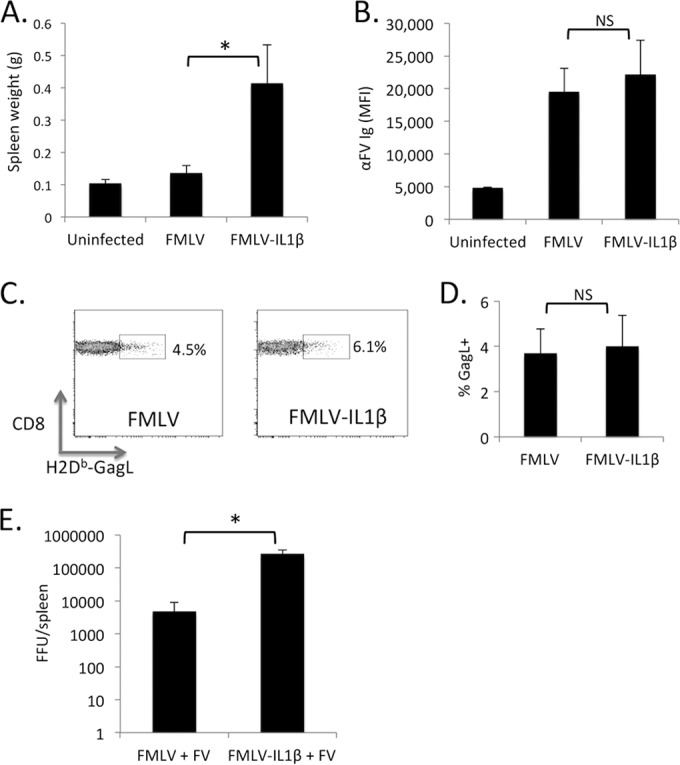
FMLV-ILβ is controlled by B6 mice by 14 dpi. (A) The weights of spleens from mice infected with FMLV or FMLV-IL-1β at 14 dpi were measured. (B) Serum from mice infected with FMLV or FMLV-IL-1β at 14 dpi was assayed for the presence of virus-specific antibodies via a flow-cytometry based assay. MFI values are in arbitrary units. (C) Splenocytes from mice at 14 dpi were assayed for virus-specific CD8 T cells by staining with an H2Db-GagL tetramer, and the percentage of tetramer-positive CD8 T cells was calculated (D). Error bars indicate standard errors. (E) Mice were coinfected with 30,000 FFU of FV (based on an assay for the infectivity of the FMLV component) and 30,000 FFU of FMLV or FMLV-IL-1β. At 14 dpi, the number of FFU per spleen was assayed by plating splenocyte dilutions on Mus dunni cells followed by a focus-forming assay. The focus-forming assay detects both FMLV and FMLV-IL-1β. Each bar represents the average for 5 mice. The asterisk indicates a P value of <0.05 (Mann-Whitney test). NS, not significant (P > 0.05). Error bars indicate the standard deviations of the data set.
FMLV-IL-1β enhances FV replication in B6 mice.
Friend virus (FV), a complex of FMLV and spleen focus-forming virus (SFFV), has been widely used as a model for retroviral infection and pathogenesis (33). SFFV encodes a glycoprotein (gp55) that activates the erythropoietin receptor (EpoR) and has a strong enhancing effect on replication of the complex (34). In B6 mice, FV is limited by potent virus-specific adaptive immune responses but is still able to establish a low-level persistent infection. B6 mice are also protected from FV disease due their expression of a truncated variant of the Stk kinase that mediates EpoR signaling (34). To investigate whether FMLV-ILβ could enhance FV replication, I coinfected B6 mice with FV and either FMLV or FMLV-IL-1β and assayed the number of infected cells in the spleen at 14 dpi. The focus-forming assay detects both FMLV and FMLV-IL-1β, and the viral load thus reflects the sum of these two viruses. Significantly, coinfection with FMLV-IL-1β resulted in viral loads that were approximately 100-fold elevated over those in mice coinfected with FMLV at 14 dpi (Fig. 5D). This demonstrates that although FMLV-IL-1β alone is cleared from B6 mice by 14 dpi, FMLV-IL-1β can still synergize with FV to result in much higher viral loads in B6 mice at 14 dpi.
FMLV-IL-1β causes prolonged infection and immune activation in BALB/c mice.
Although FMLV-IL-1β is cleared from B6 mice by 14 dpi, I hypothesized that in a different mouse strain with a less potent FMLV-specific adaptive immune response, FMLV-IL-1β may persist for longer periods of time. BALB/c mice mount a suboptimal adaptive immune response to FMLV, since this strain lacks the H-2Db major histocompatibility complex class I (MHC-I) haplotype, which correlates with CD8 T cell-dependent control of FV in B6 mice (33). BALB/c mice also encode a variant of the retroviral restriction factor Apobec3 that fails to limit FV-induced B cell dysfunction (35). BALB/c mice are highly sensitive to FV complex, but FMLV alone replicates less well in this strain unless inoculated into neonates or after dosing with pristane, possibly due to insufficient cellular inflammation and proliferation (36–38).
To test this hypothesis, I infected BALB/c mice with FMLV or FMLV-IL-1β. At 5 weeks postinfection, FMLV was undetectable in the spleens of BALB/c mice, and splenic lymphocytes exhibited only minor levels of activation. In contrast, FMLV-IL-1β-infected mice exhibited large numbers of infected cells in their spleens at 5 weeks postinfection (Fig. 6A). Moreover, FMLV-IL-1β-infected mice exhibited splenomegaly (Fig. 6B) and greatly elevated levels of lymphocyte activation, as shown by expression of CD69, CD86, or CD44 (Fig. 6C). Interestingly, both CD4 and CD8 T cells also exhibited elevated expression of the exhaustion marker PD-1, a known marker of T cell immune dysfunction during HIV-1 infection (39, 40). These results demonstrate that inclusion of IL-1β in the viral genome is sufficient to induce an extended retroviral infection in BALB/c mice for at least 5 weeks, along with a state of chronically elevated immune activation.
FIG 6.

FMLV-IL-1β causes prolonged infection and immune activation in BALB/c mice. (A) BALB/c mice were infected with FMLV or FMLV-IL-1β. At 5 weeks postinfection, spleens were dissected, and assayed for focus-forming units (FFU) by plating splenocytes on Mus dunni cells, followed by a focus-forming assay. Assay results were used to calculate the FFU per spleen in each infected mouse. ND, not detected. (B) The weights of spleens from BALB/c mice at 5 weeks postinfection were measured (right). Representative spleens are shown on the left. (C) Splenocytes were analyzed for immune cell expression of activation markers by flow cytometry. Each bar represents the average for five mice. Asterisks indicate P values of <0.05 (t test). Error bars indicate the standard deviations of the data set.
DISCUSSION
Inflammatory cytokines have long been hypothesized to regulate the course of retroviral infection and pathogenesis, but the roles of most inflammatory pathways have not been tested. Validating the inflammatory feedback model and determining which innate sensing pathways and cytokines mediate it are critical for identifying new therapeutic targets for clinically significant retroviruses. These data identify IL-1β as a potent positive regulator of retroviral infection that promotes persistent infection and immune activation in mice, and they represent the first direct evidence for IL-1βs role in retroviral infection.
To demonstrate the role of IL-1β in retroviral infection, I constructed a novel IL-1β-expressing strain of FMLV (FMLV-IL-1β). This strain thus represents a new retroviral infection/disease model that may have relevance to human disease. Previous reports using foreign genes inserted into replication competent MLV strains have used internal ribosome entry site (IRES) sequences to drive insert expression (41). However, the 2A peptide approach offers an important advantage over IRESs due to its significantly smaller size. As such, inserts are more likely to be well tolerated, and larger inserts may be used. With respect to studying the role of virus-induced cytokine response in retroviral immunity, insertion of a cytokine-coding sequence into the viral genome offers advantages over repeated dosing with recombinant cytokines in terms of cost and in more physiological simulation of a localized innate immune response at the site of infection. Natural examples of virus-encoded cytokines have previously been described for herpesviruses—Kaposi's sarcoma-associated herpesvirus (KSHV) encodes an IL-6 homologue (42), and human cytomegalovirus (HCMV) expresses an IL-10 homologue (43).
Some previous work investigated IL-1β in the context of mouse retrovirus models. Modestly elevated IL-1β was detected in the plasma of mice acutely infected with Friend virus complex (8), and IL-1β transcription is upregulated in LP-BM5-infected mice (20). In these cases, the cellular source of the IL-1β was not identified, and it has not been demonstrated that the IL-1β response influences the course of infection or pathogenesis.
The mechanism by which virus-encoded IL-1β promotes retroviral replication in vivo will require further investigation. Significantly, elevated levels of immune cell activation were also observed in FMLV-IL-1β-infected mice, and activation of immune cells is known to increase permissiveness for retroviral infection (44). IL-1β secretion by infected cells may induce activation and/or proliferation of neighboring cells, thereby rendering them more susceptible to infection. Enhanced availability of susceptible cells will, in turn, amplify further rounds of infection and IL-1β secretion, establishing an inflammatory feedback loop (Fig. 7). Although it is also possible that IL-1β enhanced acute infection by transiently suppressing immune responses in B6 mice, FMLV-ILβ was cleared from this strain by 14 dpi, and these mice mounted apparently normal B and CD8 T cell responses. Alternatively, IL-1β may enhance susceptibility of cells to infection independently of activation. For example, exposure to a mixture of the cytokines IL-2, IL-4, IL-7, and IL-15 has been shown to be sufficient to promote infection of resting human T cells with HIV-1 in the absence of other stimuli (45).
FIG 7.

Possible roles for IL-1β in retroviral infection. IL-1β secreted by cells during, or in response to, retroviral infection engages IL-1R1 either on neighboring cells or on the infected cells themselves. This interaction activates transcriptional pathways that can enhance viral gene expression, promote cellular proliferation, and/or increase susceptibility to infection. The uninfected cells thus become infected more easily and express viral genes to a higher level. This then initiates further rounds of IL-1β secretion, resulting in an inflammatory positive feedback loop. IL-1β also promotes increased abundance of myeloid-derived suppressor cells (MDSCs) that may negatively regulate immune responses.
IL-1β may also play other roles in positively regulating retroviral replication; IL-1β triggers activation of the NF-κB transcription factor, which enhances HIV-1 transcription (46, 47). Secreted IL-1β may thus upregulate viral transcription in infected cells in an autocrine or paracrine fashion. IL-1β may also have a chemotactic role, enhancing virus dissemination by attracting susceptible cells closer to sites of active infection. For example, macrophage chemotaxis has been shown to be regulated by IL-1β (48). Also, IL-1β is a positive regulator of monocyte chemoattractant protein 1 (MCP-1) expression in endothelial cells, and MCP-1 is known to promote attraction of monocytes, T cells, and basophils (49).
The precise mechanisms behind elevated activation of lymphocytes and innate immune cells during FMLV-IL-1β infection are also still unclear. This response could be, in part, driven by the much higher loads of viral antigen and viral pathogen-associated molecular patterns (PAMPS) present in FMLV-IL-1β-infected mice, which will trigger pathogen-specific B cell/T cell receptors and innate pattern recognition receptors (PRRs), respectively. Alternatively, since the IL-1β receptor is widely expressed, the heightened immune activation could be a direct response of these cells to IL-1β-mediated signaling or due to IL-1β promoting the expression of other cytokines that stimulate immune cell activation.
FMLV-IL-1β also induced the dramatic accumulation of Gr-1+ CD11b+ MDSCs in infected mice. Interestingly, elevated MSDC levels in HIV-1 infected patients were recently described (29, 30). IL-1β has also previously been shown to promote MDSC accumulation in mice in the context of IL-1β expressing tumors (50). The origin of the IL-1β-induced MSDCs is unclear; IL-1β may promote proliferation of this lineage in situ or enhance recruitment from other sites. Alternatively, IL-1β may stimulate a different spleen-resident lineage to develop into MDSCs. MDSCs are characterized by their ability to inhibit pathogen-specific immune responses (51). Nevertheless, the clearance of FMLV-IL-1β from infected B6 mice as well as the apparently normal adaptive immune response in FMLV-IL-1β-infected mice suggests that these cells do not dramatically impact the course of infection. MDSCs could, however, still impact viral loads or host immunity in more subtle ways, such as modulating infection/immunity in specific tissues or compartments.
Reverse-transcribed HIV-1 DNA generated during abortive infection of resting CD4 T cells was recently shown to induce IL-1β secretion by binding to interferon gamma-inducible protein 16 (IFI16), which, in turn, forms an active inflammasome with apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC) (14, 15). This response is also associated with a highly inflammatory form of cell death known as pyroptosis (15, 52). As such, it is possible that the elevated viral loads and immune activation observed for FMLV-IL-1β are relevant to HIV-1 infection. Although, in the case of HIV-1, the IL-1β secretion occurs in cells that are not productively infected, this cytokine may still promote replication in neighboring cells by creating a localized inflammatory environment. Importantly, genetic studies in humans have identified variants of IL-1β and inflammasome subunits that affect susceptibility to HIV-1 infection (53, 54), suggesting that IL-1β does indeed play a functional role. Nevertheless, since many significant immunological differences exist between mice and humans, it will be important to test whether the findings with FMLV-IL-1β are directly relevant to HIV-1. Soluble recombinant IL-1R1 has previously been used in a phase I/II clinical trial and did not produce a consistent response (55). However, the data from the present study suggest that further investigation of the therapeutic potential of targeting of IL-1β in HIV-1 patients is warranted.
REFERENCES
- 1.Walker BD, Burton DR. 2008. Toward an AIDS vaccine. Science 320:760–764. doi: 10.1126/science.1152622. [DOI] [PubMed] [Google Scholar]
- 2.Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, Lebedeva M, DeCamp A, Li D, Grove D, Self SG, Borrow P. 2009. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol 83:3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medzhitov R, Littman D. 2008. HIV immunology needs a new direction. Nature 455:591. doi: 10.1038/455591a. [DOI] [PubMed] [Google Scholar]
- 4.Browne EP. 2011. Toll-like receptor 7 controls the anti-retroviral germinal center response. PLoS Pathog 7:e1002293. doi: 10.1371/journal.ppat.1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kane M, Case LK, Wang C, Yurkovetskiy L, Dikiy S, Golovkina TV. 2011. Innate immune sensing of retroviral infection via Toll-like receptor 7 occurs upon viral entry. Immunity 35:135–145. doi: 10.1016/j.immuni.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, Wen TF, Lindsay RJ, Orellana L, Mildvan D, Bazner S, Streeck H, Alter G, Lifson JD, Carrington M, Bosch RJ, Robbins GK, Altfeld M. 2009. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med 15:955–959. doi: 10.1038/nm.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rua R, Lepelley A, Gessain A, Schwartz O. 2012. Innate sensing of foamy viruses by human hematopoietic cells. J Virol 86:909–918. doi: 10.1128/JVI.06235-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Browne EP. 2013. Toll-like receptor 7 inhibits early acute retroviral infection through rapid lymphocyte responses. J Virol 87:7357–7366. doi: 10.1128/JVI.00788-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alter G, Suscovich TJ, Teigen N, Meier A, Streeck H, Brander C, Altfeld M. 2007. Single-stranded RNA derived from HIV-1 serves as a potent activator of NK cells. J Immunol 178:7658–7666. doi: 10.4049/jimmunol.178.12.7658. [DOI] [PubMed] [Google Scholar]
- 10.Beignon A-S, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, Larsson M, Gorelick RJ, Lifson JD, Bhardwaj N. 2005. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest 115:3265–3275. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, Littman DR. 2010. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 467:214–217. doi: 10.1038/nature09337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oh D-Y, Baumann K, Hamouda O, Eckert JK, Neumann K, Kücherer C, Bartmeyer B, Poggensee G, Oh N, Pruss A, Jessen H, Schumann RR. 2009. A frequent functional toll-like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. AIDS 23:297–307. doi: 10.1097/QAD.0b013e32831fb540. [DOI] [PubMed] [Google Scholar]
- 13.Said EA, Al-Yafei F, Zadjali F, Hasson SS, Al-Balushi MS, Al-Mahruqi S, Koh CY, Al-Naamani K, Al-Busaidi JZ, Idris MA, Balkhair A, Al-Jabri AA. 2014. Association of single-nucleotide polymorphisms in TLR7 (Gln11Leu) and TLR9 (1635A/G) with a higher CD4T cell count during HIV infection. Immunol Lett 160:58–64. doi: 10.1016/j.imlet.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. 2014. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343:428–432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doitsh G, Galloway NLK, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Muñoz-Arias I, Greene WC. 2014. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garlanda C, Dinarello CA, Mantovani A. 2013. The interleukin-1 family: back to the future. Immunity 39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y, Voronov E. 2006. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev 25:387–408. doi: 10.1007/s10555-006-9004-4. [DOI] [PubMed] [Google Scholar]
- 18.Lachmann HJ, Quartier P, So A, Hawkins PN. 2011. The emerging role of interleukin-1β in autoinflammatory diseases. Arthritis Rheum 63:314–324. doi: 10.1002/art.30105. [DOI] [PubMed] [Google Scholar]
- 19.Ipp H, Zemlin AE, Erasmus RT, Glashoff RH. 2014. Role of inflammation in HIV-1 disease progression and prognosis. Crit Rev Clin Lab Sci 51:98–111. doi: 10.3109/10408363.2013.865702. [DOI] [PubMed] [Google Scholar]
- 20.Cheung SC, Chattopadhyay SK, Morse HC, Pitha PM. 1991. Expression of defective virus and cytokine genes in murine AIDS. J Virol 65:823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allcock RJ, Peacock CD, Price P. 1996. The inflammatory macrophage response to MCMV in mice with a retroviral immunodeficiency syndrome (MAIDS). J Leukoc Biol 60:44–50. [DOI] [PubMed] [Google Scholar]
- 22.Robertson MN, Miyazawa M, Mori S, Caughey B, Evans LH, Hayes SF, Chesebro B. 1991. Production of monoclonal antibodies reactive with a denatured form of the Friend murine leukemia virus gp70 envelope protein: use in a focal infectivity assay, immunohistochemical studies, electron microscopy and western blotting. J Virol Methods 34:255–271. doi: 10.1016/0166-0934(91)90105-9. [DOI] [PubMed] [Google Scholar]
- 23.Chesebro B, Wehrly K, Cloyd M, Britt W, Portis J, Collins J, Nishio J. 1981. Characterization of mouse monoclonal antibodies specific for Friend murine leukemia virus-induced erythroleukemia cells: friend-specific and FMR-specific antigens. Virology 112:131–144. doi: 10.1016/0042-6822(81)90619-X. [DOI] [PubMed] [Google Scholar]
- 24.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 25.Portis JL, McAtee FJ, Kayman SC. 1992. Infectivity of retroviral DNA in vivo. J Acquir Immune Defic Syndr 5:1272–1273. doi: 10.1097/00126334-199212000-00011. [DOI] [PubMed] [Google Scholar]
- 26.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DAA. 2004. Correction of multi-gene deficiency in vivo using a single “self-cleaving” 2A peptide-based retroviral vector. Nat Biotechnol 22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 27.Zelinskyy G, Balkow S, Schimmer S, Werner T, Simon MM, Dittmer U. 2007. The level of friend retrovirus replication determines the cytolytic pathway of CD8+ T-cell-mediated pathogen control. J Virol 81:11881–11890. doi: 10.1128/JVI.01554-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elkabets M, Ribeiro VSG, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, Vosshenrich CAJ. 2010. IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol 40:3347–3357. doi: 10.1002/eji.201041037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vollbrecht T, Stirner R, Tufman A, Roider J, Huber RM, Bogner JR, Lechner A, Bourquin C, Draenert R. 2012. Chronic progressive HIV-1 infection is associated with elevated levels of myeloid-derived suppressor cells. AIDS 26:F31–37. doi: 10.1097/QAD.0b013e328354b43f. [DOI] [PubMed] [Google Scholar]
- 30.Qin A, Cai W, Pan T, Wu K, Yang Q, Wang N, Liu Y, Yan D, Hu F, Guo P, Chen X, Chen L, Zhang H, Tang X, Zhou J. 2013. Expansion of monocytic myeloid-derived suppressor cells dampens T cell function in HIV-1-seropositive individuals. J Virol 87:1477–1490. doi: 10.1128/JVI.01759-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hazenberg MD, Otto SA, van Benthem BHB, Roos MTL, Coutinho RA, Lange JMA, Hamann D, Prins M, Miedema F. 2003. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS 17:1881–1888. doi: 10.1097/00002030-200309050-00006. [DOI] [PubMed] [Google Scholar]
- 32.Hasenkrug KJ, Dittmer U. 2007. Immune control and prevention of chronic Friend retrovirus infection. Front Biosci 12:1544–1551. doi: 10.2741/2167. [DOI] [PubMed] [Google Scholar]
- 33.Hasenkrug KJ, Chesebro B. 1997. Immunity to retroviral infection: the Friend virus model. Proc Natl Acad Sci U S A 94:7811–7816. doi: 10.1073/pnas.94.15.7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Persons DA, Paulson RF, Loyd MR, Herley MT, Bodner SM, Bernstein A, Correll PH, Ney PA. 1999. Fv2 encodes a truncated form of the Stk receptor tyrosine kinase. Nat Genet 23:159–165. doi: 10.1038/13787. [DOI] [PubMed] [Google Scholar]
- 35.Santiago ML, Montano M, Benitez R, Messer RJ, Yonemoto W, Chesebro B, Hasenkrug KJ, Greene WC. 2008. Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science 321:1343–1346. doi: 10.1126/science.1161121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacDonald ME, Mak TW, Bernstein A. 1980. Erythroleukemia induction by replication-competent type C viruses cloned from the anemia- and polycythemia-inducing isolates of Friend leukemia virus. J Exp Med 151:1493–1503. doi: 10.1084/jem.151.6.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niho Y, Shibuya T, Mak TW. 1982. Modulation of erythropoiesis by the helper-independent Friend leukemia virus F-MuLV. J Exp Med 156:146–158. doi: 10.1084/jem.156.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukhopadhyaya R, Richardson J, Nazarov V, Corbin A, Koller R, Sitbon M, Wolff L. 1994. Different abilities of Friend murine leukemia virus (MuLV) and Moloney MuLV to induce promonocytic leukemia are due to determinants in both psi-gag-PR and env regions. J Virol 68:5100–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel M-R, Delwart E, Sepulveda H, Balderas RS, Routy J-P, Haddad EK, Sekaly R-P. 2006. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med 12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 40.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 41.Jespersen T, Duch M, Carrasco ML, Warming S, Pedersen FS. 1999. Expression of heterologous genes from an IRES translational cassette in replication competent murine leukemia virus vectors. Gene 239:227–235. doi: 10.1016/S0378-1119(99)00402-3. [DOI] [PubMed] [Google Scholar]
- 42.Nicholas J, Ruvolo VR, Burns WH, Sandford G, Wan X, Ciufo D, Hendrickson SB, Guo HG, Hayward GS, Reitz MS. 1997. Kaposi's sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat Med 3:287–292. doi: 10.1038/nm0397-287. [DOI] [PubMed] [Google Scholar]
- 43.Chang WLW, Baumgarth N, Yu D, Barry PA. 2004. Human cytomegalovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. J Virol 78:8720–8731. doi: 10.1128/JVI.78.16.8720-8731.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Datta SK, Melief CJ, Schwartz RS. 1975. Lymphocytes and leukemia viruses: tropism and transtropism of murine leukemia virus. J Natl Cancer Inst 55:425–432. [PubMed] [Google Scholar]
- 45.Unutmaz D, KewalRamani VN, Marmon S, Littman DR. 1999. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J Exp Med 189:1735–1746. doi: 10.1084/jem.189.11.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nabel G, Baltimore D. 1987. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326:711–713. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- 47.Granowitz EV, Saget BM, Wang MZ, Dinarello CA, Skolnik PR. 1995. Interleukin 1 induces HIV-1 expression in chronically infected U1 cells: blockade by interleukin 1 receptor antagonist and tumor necrosis factor binding protein type 1. Mol Med 1:667–677. [PMC free article] [PubMed] [Google Scholar]
- 48.Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. 2011. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol 187:4835–4843. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- 49.van der Velden VH, Verheggen MM, Bernasconi S, Sozzani S, Naber BA, van der Linden-van Beurden CA, Hoogsteden HC, Mantovani A, Versnel M. 1998. Interleukin-1beta and interferon-gamma differentially regulate release of monocyte chemotactic protein-1 and interleukin-8 by human bronchial epithelial cells. Eur Cytokine Netw 9:269–277. [PubMed] [Google Scholar]
- 50.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. 2006. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol 176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 51.Haile LA, Greten TF, Korangy F. 2012. Immune suppression: the hallmark of myeloid derived suppressor cells. Immunol Invest 41:581–594. doi: 10.3109/08820139.2012.680635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miao EA, Rajan JV, Aderem A. 2011. Caspase-1-induced pyroptotic cell death. Immunol Rev 243:206–214. doi: 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahir S, Chaudhari D, Chavan V, Samant-Mavani P, Nanavati R, Mehta P, Mania-Pramanik J. 2013. Polymorphisms in IL-1 gene cluster and its association with the risk of perinatal HIV transmission, in an Indian cohort. Immunol Lett 153:1–8. doi: 10.1016/j.imlet.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 54.Pontillo A, Oshiro TM, Girardelli M, Kamada AJ, Crovella S, Duarte AJS. 2012. Polymorphisms in inflammasome genes and susceptibility to HIV-1 infection. J Acquir Immune Defic Syndr 59:121–125. doi: 10.1097/QAI.0b013e3182392ebe. [DOI] [PubMed] [Google Scholar]
- 55.Takebe N, Paredes J, Pino MC, Lownsbury WH, Agosti J, Krown SE. 1998. Phase I/II trial of the type I soluble recombinant human interleukin-1 receptor in HIV-1-infected patients. J Interferon Cytokine Res 18:321–326. doi: 10.1089/jir.1998.18.321. [DOI] [PubMed] [Google Scholar]