

ABSTRACT
During DNA encapsidation, herpes simplex virus 1 (HSV-1) procapsids are converted to DNA-containing capsids by a process involving activation of the viral protease, expulsion of the scaffold proteins, and the uptake of viral DNA. Encapsidation requires six minor capsid proteins (UL6, UL15, UL17, UL25, UL28, and UL33) and one viral protein, UL32, not found to be associated with capsids. Although functions have been assigned to each of the minor capsid proteins, the role of UL32 in encapsidation has remained a mystery. Using an HSV-1 variant containing a functional hemagglutinin-tagged UL32, we demonstrated that UL32 was synthesized with true late kinetics and that it exhibited a previously unrecognized localization pattern. At 6 to 9 h postinfection (hpi), UL32 accumulated in viral replication compartments in the nucleus of the host cell, while at 24 hpi, it was additionally found in the cytoplasm. A newly generated UL32-null mutant was used to confirm that although B capsids containing wild-type levels of capsid proteins were synthesized, these procapsids were unable to initiate the encapsidation process. Furthermore, we showed that UL32 is redox sensitive and identified two highly conserved oxidoreductase-like C-X-X-C motifs that are essential for protein function. In addition, the disulfide bond profiles of the viral proteins UL6, UL25, and VP19C and the viral protease, VP24, were altered in the absence of UL32, suggesting that UL32 may act to modulate disulfide bond formation during procapsid assembly and maturation.
IMPORTANCE Although functions have been assigned to six of the seven required packaging proteins of HSV, the role of UL32 in encapsidation has remained a mystery. UL32 is a cysteine-rich viral protein that contains C-X-X-C motifs reminiscent of those in proteins that participate in the regulation of disulfide bond formation. We have previously demonstrated that disulfide bonds are required for the formation and stability of the viral capsids and are also important for the formation and stability of the UL6 portal ring. In this report, we demonstrate that the disulfide bond profiles of the viral proteins UL6, UL25, and VP19C and the viral protease, VP24, are altered in cells infected with a newly isolated UL32-null mutant virus, suggesting that UL32 acts as a chaperone capable of modulating disulfide bond formation. Furthermore, these results suggest that proper regulation of disulfide bonds is essential for initiating encapsidation.
INTRODUCTION
The products of herpes simplex virus 1 (HSV-1) DNA replication are head-to-tail concatemers that are resolved into monomeric genomic units and packaged into a procapsid shell in the nucleus of the infected cell (reviewed in references 1 to 3). The procapsid is comprised of the major capsid protein (VP5), triplex proteins (VP19C and VP23), and a dodecameric UL6 portal ring. The precursor of the viral protease (UL26) and the scaffolding protein (UL26.5) form a scaffold around which the capsid shell assembles (3, 4). During DNA encapsidation, the viral protease (VP24) is activated, scaffold proteins are released, and viral DNA is taken up (1, 4–7). HSV procapsid maturation involves dramatic structural remodeling, resulting in the conversion of a relatively fragile and porous procapsid into a more stable angular icosahedron filled with DNA (6–8). Three types of capsids, designated type A, B, or C, can be separated by sucrose gradient centrifugation. A capsids have released the scaffold but contain no DNA as a result of an abortive packaging event. B capsids resemble procapsids, in that they retain the scaffold and contain no DNA. C capsids contain viral genomes and are destined to mature into infectious virions. We have recently demonstrated disulfide linkages between capsid proteins VP5, VP23, and VP19C and other components of the mature virion, including UL17, UL25, and tegument proteins; furthermore, we showed that disulfide bond formation is required for capsid stability (9). Intersubunit disulfide bonds also exist between monomeric subunits of the portal ring and contribute to ring formation and/or stability (10). The disulfide bonds in the portal ring can be detected in the nucleus. This result was somewhat surprising since this compartment is normally thought to be a reducing environment.
The cleavage and packaging machinery consists of several required viral gene products, six of which are transiently or stably associated with capsids. The UL6 portal ring is found at a unique vertex on the procapsid and acts as a channel through which DNA is taken up (11–15). UL15, UL28, and UL33 make up a heterotrimeric complex believed to act as a molecular motor facilitating DNA uptake and cleavage known as the terminase (15–18). Terminase subunits are transiently associated with procapsids (19–21). UL17 and UL25 form a heterodimeric structure referred to as the capsid vertex-specific component (CVSC) that appears to stabilize DNA-containing capsids (19, 22–27). A seventh viral protein, UL32, is also essential for encapsidation; however, it is apparently not associated with procapsids or mature virions, and its precise role in infection remains elusive (28). UL32 also differs from the other cleavage and packaging proteins with respect to its intracellular localization. While other cleavage and packaging proteins are found in the nucleus in globular domains known as replication compartments, UL32 has been shown to localize primarily in the cytoplasm, with a small fraction being observed in replication compartments (28, 29). In the present study, an HSV-1 isolate expressing hemagglutinin (HA)-tagged UL32 was generated and used to demonstrate that UL32 localization is more dynamic than previously reported. At early times during infection, UL32 localized predominantly within replication compartments; however, as infection progressed, UL32 was observed primarily in the cytoplasm. This dynamic staining pattern is unique among encapsidation proteins and may provide clues into the function of UL32.
UL32 is a cysteine-rich protein, and 15 of its 22 cysteines are conserved among the members of the herpesvirus family. Cysteines contain a reactive thiol group that can be modified in response to changes in oxidation state. Interestingly, cysteines are involved in many cellular processes, including redox signaling and dithiol-disulfide exchange reactions; furthermore, they play important roles at the catalytic site of many enzymes (reviewed in reference 30). UL32 contains three C-X-X-C motifs which are reminiscent of the highly conserved motifs found in thiol-disulfide oxidoreductases known to catalyze redox reactions essential for maintaining the redox balance within cells through the formation and disruption of disulfide bonds in client proteins (31–34). In this study, we tested the hypothesis that UL32 is a redox-sensitive protein involved in the modulation of disulfide bonds in HSV-1 proteins. We show that UL32 is redox sensitive and that two of these three conserved C-X-X-C motifs are essential for protein function. Furthermore, the viral proteins UL6, UL25, and VP19C and the viral protease, VP24, exhibit an altered disulfide bond pattern in cells infected with a UL32-null mutant, supporting the hypothesis that UL32 functions either directly or indirectly to regulate disulfide bond formation. These results also suggest that appropriate disulfide bond formation is required for the formation of procapsids that are competent for encapsidation.
MATERIALS AND METHODS
Cell lines and viruses.
African green monkey kidney (Vero CC181) cells (American Type Culture Collection, Manassas, VA) and UL32 mutant virus-complementing cell line 158 were maintained and propagated in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 5% fetal bovine serum and 1% penicillin-streptomycin. The KOS strain of HSV-1 was used as the wild-type virus. The HSV-1 UL32-null mutant virus hr64 with an ICP6::LacZ insertion was previously described by Lamberti and Weller (28). A replication-competent virus, HA32, expressing an HA-tagged UL32 protein, and a newly isolated truly null UL32 mutant, hr64FS, were generated in this study (see below). Cell maintenance and infections were carried out at 37°C.
Construction of HA32.
A plasmid (pUL32) capable of expressing the full-length UL32 open reading frame (pUL32) was kindly provided by A. Patel (University of Glasgow Centre for Virus Research). PCR was used to insert a sequence encoding the influenza virus HA tag (YPYDVPDY) between codons 2 and 3 of the UL32 gene, generating pHA-UL32. Plasmid pHA-UL32 and plasmid pUL33 (35), containing a fragment encoding the adjacent UL33 gene, were then used to generate p33-HA-32, in which a complete UL32-UL33 locus identical to that in wild-type virus, except for the sequence encoding the tag, was reconstructed. A fragment spanning the UL32-UL33 region was excised from p33-HA-32 and gel purified. Monolayers of BHK cells (seeded at 1 × 106 cells per 35-mm petri dish) were transfected with 0.5 μg hr64 DNA and 0.5 μg linearized plasmid using the calcium phosphate method as previously described (36). Cells were incubated in Glasgow minimum essential medium supplemented with 5% serum at 37°C for 3 days. Serial 2-fold dilutions were used to infect BHK cells in a 96-well plate, wells containing single plaques were identified, and the resultant virus was analyzed for the ability to express HA32 by Western blotting. One clone was selected, and working stocks of the resulting virus, HA32, were prepared.
Construction of hr64FS.
A UL32 mutant containing a frameshift deletion at nucleotide 166 was generated by two-step sequential PCR mutagenesis using the following primers: forward primer 5′-CAGTCACTGATAGGTGTGCCGCCCGTCGGTAC-3′ and reverse primer 5′-CGGCACACxCTATCAGTGACTGGCCGTCAGG-3′. This led to a site-specific deletion of a single nucleotide (between the underlined nucleotides) and the presence of two consecutive in-frame stop codons (in bold). The following primers were used to amplify the full-length product containing the frameshift mutation: forward primer 5′-CACCATGGCAACTTCGCCCCCCGGGGTCC-3′ and reverse primer 5′-TCATACATAGGTACACAGGGTGTGCTC-3′. The forward primer introduced an NcoI restriction site at the 5′ end of the UL32 gene. This PCR product was ligated into pUL32lacZ (28), after digestion with NcoI and NheI, to create p32STOPlacZ. p32STOPlacZ was digested with NheI and XbaI, and the fragment was inserted in place of the NheI-XbaI fragment of p205-Pstl (28), which contains genes UL31 to UL33, resulting in p205-32STOP. Linearized p205-32STOP was cotransfected with infectious hr64 DNA into 158 cells at a molar ratio of 10:1. Infectious particles were harvested and used to infect 158 cells in a plaque assay, and blue-white screening with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) was utilized to select white plaques.
Western blot analysis.
Cells were harvested and proteins were separated by reducing SDS-PAGE as previously described and transferred to a polyvinylidene difluoride membrane (10). Membranes were blocked with 5% milk and incubated overnight with the following antibodies: mouse monoclonal anti-HA (1:1,000; catalog number Sc-7392; Santa Cruz), rabbit polyclonal anti-HA (1:1,000; catalog number 631207; Clontech), rabbit polyclonal anti-UL32 (1:1,000; antibody to synthetic antigenic peptide was generated by Open Biosystems), mouse monoclonal anti-ICP8 (1:2,000; catalog number ab20194; Abcam), mouse monoclonal anti-VP5 (1:1,000; catalog number HA018; EastCoast Bio), mouse monoclonal anti-VP24 9-2 (1:1,000) (37), and mouse anti-actin (1:10,000; catalog number A5441; Sigma).
Growth curves.
Confluent Vero or 158 cells were infected with the indicated viruses at a multiplicity of infection (MOI) of 0.1. Cells and supernatant were harvested at 0, 6, 12, and 24 h postinfection. After three freeze-thaw cycles, serial dilutions were plated on Vero cells (see Fig. 1) or 158 cells (see Fig. 6), overlaid with methylcellulose, incubated for 72 h, fixed in 4% formaldehyde, and stained with 1% crystal violet solution. Viral plaques were counted by eye.
FIG 1.
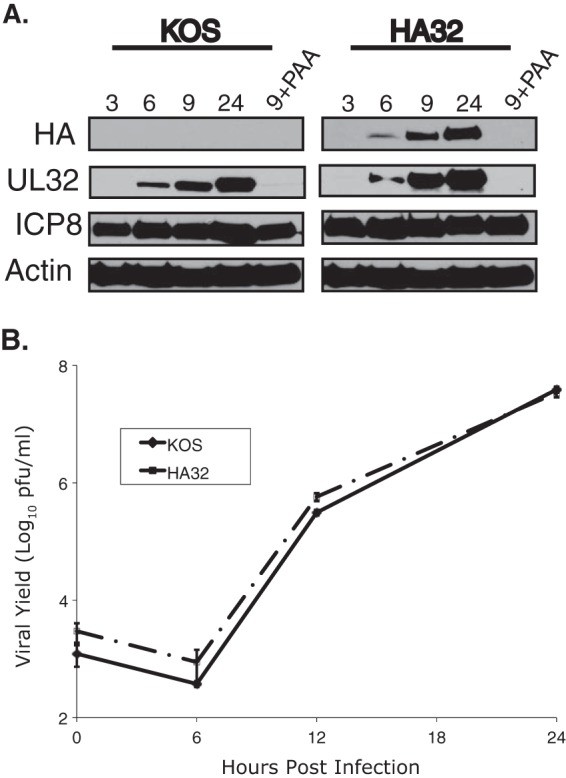
HA32 resembles wild-type virus in protein expression and viral growth properties. (A) Vero cells were infected with KOS or HA32 at an MOI of 3 and collected at the times postinfection (in hours) indicated above the gels. Samples were harvested in reducing SDS sample buffer and resolved by SDS-PAGE. Proteins were detected by immunoblotting with the antibodies indicated on the left. (B) Growth curves of KOS and HA32 propagated on Vero cells at an MOI of 0.1. Mean values from three independent experiments are plotted, and deviations are represented by error bars.
FIG 6.

UL32 mutants, defective in growth, synthesize wild-type levels of viral DNA but are unable to cleave and package it. (A) Growth curves of KOS, hr64FS, or hr64 propagated on Vero or 158 cells. Mean values from three independent experiments are plotted, and deviations are represented by error bars. (B) Vero or 158 cells were infected with KOS, hr64, or hr64FS, and viral DNA was analyzed by Southern blotting, as described in Materials and Methods. The blotted DNA was hybridized with a 32P-labeled probe containing the BamHI SQ fragment. SQ, viral DNA junctions; S and Q, viral DNA termini.
Immunofluorescent confocal microscopy.
Monolayers of Vero cells were grown on glass coverslips prior to infection. Infected cells were washed with 2× phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde, and permeabilized in ice-cold acetone for 2 min, followed by blocking in 3% normal goat serum (NGS) diluted in PBS. Cells were incubated in primary antibodies diluted in 3% NGS for 1 to 2 h at room temperature by inverting coverslips onto a drop of antibody dilution. The dilutions of primary antibodies in 3% NGS were as follows: monoclonal anti-ICP8, 1:200; polyclonal anti-ICP8 (367), 1:500; monoclonal anti-VP5 5C and monoclonal anti-VP23 1D2 (both generously provided by Jay Brown, University of Virginia), 1:200; monoclonal anti-VP21/VP22a MCA406 (Serotec Inc.), 1:500; and rat monoclonal anti-HA (catalog number 11867423001; Roche), 1:200. Cells were washed extensively before incubation in Alexa Fluor secondary antibodies (1:200; Molecular Probes), diluted in 3% NGS, and mounted onto slides with glycerol gelatin containing 2.5% diazobicyclo-[2.2.2] octane (DABCO; Sigma Chemical Co.). Images were captured using a Zeiss LSM 510 confocal NLO microscope equipped with argon and HeNe lasers and a Zeiss ×63 objective lens (numerical aperture, 1.4). Images were processed and arranged using Adobe Photoshop CS3 and Illustrator CS3 software.
Sucrose gradient sedimentation.
Capsids were isolated and separated by sucrose gradient sedimentation as previously described (10). Fractions were collected and prepared for Western blot analysis under reducing conditions as described above.
Analysis of conserved residues in UL32.
UL32 homologs were identified through a sequence similarity search against the KOS UL32 protein with the NCBI BLAST program. Over 100 UL32 homologs were identified using NCBI BLAST. The 12 homologs shown in Fig. 3 represent each of the following subfamilies of herpesviruses (GenBank accession numbers are given in parentheses): human herpesvirus 1 (HHV-1) KOS (AFE62860.1), HHV-2 (AHG54696.1), HHV-3 (AGL50983.1), HHV-4 (AFY97834.1), HHV-7 (YP_073776.1), HHV-8 (AAC57153.1), bovine herpesvirus 1 (BOHV-1; NP_045326.1), BOHV-5 (YP_003662492.1), equid herpesvirus 4 (EHV-4; NP_045245.1), suid herpesvirus 1 (PRV-1; AGW01108.1), gallid herpesvirus 2 (GaHV-2; YP_001033961.1), and GaHV-3 (AEI00238.1).
FIG 3.
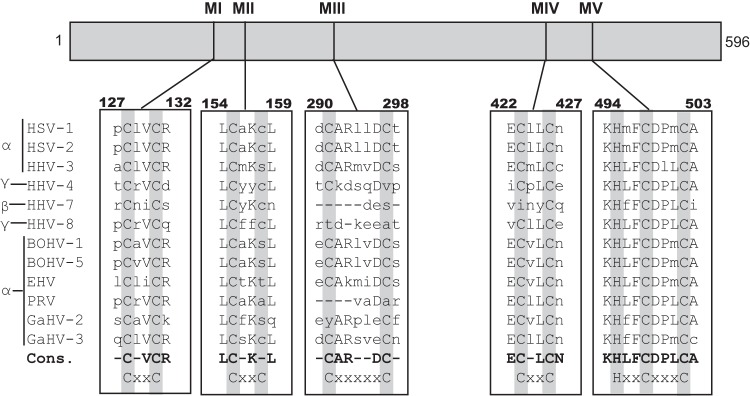
UL32 homologs are found in all three subfamilies of herpesviruses. Sequence alignments were performed on UL32 homologs from herpesviruses from the alpha-, beta-, and gammaherpesvirus subfamilies. Residues of interest are highlighted in gray, and the corresponding motifs are indicated in the last row. The consensus sequence (Cons.) is represented by the bold lettering. Lowercase letters represent residues with little to no conservation. The viruses analyzed are indicated in Materials and Methods. MI to MV, motifs I to V, respectively.
Generation of substitution mutations.
Point mutations were generated in pAPVUL32, where UL32 is driven off the ICP6 promoter (previously described in reference 28), by site-directed mutagenesis using a QuikChange II site-directed mutagenesis kit (Agilent Technologies) per the manufacturer's protocol. Primers are available upon request. The resulting plasmids were sequenced to confirm the mutations. Protein expression was confirmed by Western blotting.
Transient complementation assay.
Vero cells grown to ∼75% confluence in 35-mm dishes were transfected with 1 μg of empty, wild-type, or mutant plasmid using the Lipofectamine 2000 reagent according to the manufacturer's protocol. At 4 h posttransfection, cells were superinfected with hr64FS at an MOI of 3 PFU per cell. At 24 h postinfection, progeny viruses were harvested and assayed on the 158 cell line for total viral yield. The percent complementation was calculated by dividing the titer obtained for the mutant plasmid by the titer obtained for the wild-type plasmid and multiplying by 100. The background titers from the empty plasmid samples were subtracted.
DNA isolation and Southern blot analysis.
Vero and 158 cells were infected with KOS, hr64, or hr64FS at an MOI of 5 and harvested at 16 to 20 h postinfection (hpi). Total viral and cellular DNA was isolated, digested with BamHI, and resolved by agarose gel electrophoresis. DNA was transferred to a GeneScreen Plus nylon membrane (New England Nuclear Corp.) and hybridized with a 32P-labeled probe. The pKBK plasmid, which contains the 5.9-kb BamHI (SQ) junction fragment to detect viral termini from KOS (kindly provided by Neal Deluca, University of Pittsburgh), was radiolabeled using a Random Primers DNA labeling system (Invitrogen).
H2O2 treatment.
Vero cells were infected with KOS or HA32. At the indicated times before harvest, cells were treated with 5 mM H2O2 in 2 ml serum-free medium and harvested in 200 μl radioimmunoprecipitation assay (RIPA) buffer (reduced) or 10% trichloroacetic acid (TCA; nonreduced) at 9 hpi. TCA-precipitated samples were resuspended in denaturing N-ethylmaleimide (NEM) buffer (100 mM Tris, pH 6.8, 1% SDS, 1 mM EDTA, 50 mM NEM). Samples were subjected to SDS-PAGE in reducing or nonreducing SDS sample buffer.
Silver staining.
After SDS-PAGE, gels were fixed for 1 h in a solution containing 30% ethanol and 10% acetic acid and washed twice in 20% ethanol (10 min for each wash) and once for 20 min with water. The gel was sensitized in 0.1% sodium thiosulfate for 1 min, briefly rinsed with water, and incubated in cold 0.1% silver nitrate for 30 min at room temperature. The silver nitrate solution was removed, and the gel was washed twice with water (1 min each wash) and developed with 3% potassium bicarbonate and 0.05% formaldehyde. Once the protein bands were detected, staining was terminated by replacing the developing solution with 5% acetic acid solution for 10 min.
Cryo-electron microscopy and image reconstruction.
Purified B capsids (3 μl) were pipetted onto slightly glow-discharged Quantifoil R2/1 grids (Quantifoil, Jena, Germany), blotted, and vitrified in an FEI Vitrobot Mk III instrument (FEI, Hillsboro, OR). Grids were transferred onto a Gatan 626 cryoholder (Gatan, Pleasanton, CA) and imaged in an FEI Tecnai TF20 microscope operating at 200 kV and a ×50,000 nominal magnification. Images were collected under low-dose conditions on a Gatan UltraScan charge-coupled-device camera with an effective pixel size of 2.14 Å at the sample. Image reconstructions were carried out with the Auto3DEM package (38) using randomly oriented models as the starting point in each case (39). Resolutions were estimated where the Fourier shell correlation dropped below 0.5. For the control KOS sample of empty capsids, 412 capsid images were collected from 57 micrographs, and 370 were used to calculate the final map to a 33-Å resolution. The hr64FS sample yielded 1,001 capsid images from 104 micrographs, and 900 were included in the final map to a resolution of 21.4 Å.
Thiol modifications.
Vero cells were infected with KOS, hr64FS, or hr81-1 virus and harvested in 10% TCA at 10 hpi. Samples were centrifuged at 13,000 rpm for 30 min in a benchtop centrifuge. The TCA was aspirated, and the pellet was washed with 5% TCA, followed by another centrifugation. After aspiration, the pellets were allowed to dry and resuspended in denaturing buffer (6 M urea, 200 mM Tris, pH 8.8, 10 mM EDTA, 0.5% SDS) containing 20 mM NEM to block free thiols. Equal volumes of 20% TCA were added to the samples to precipitate the proteins in a total concentration of 10% TCA. After the wash, the samples were resuspended in denaturing buffer containing 50 mM dithiothreitol (DTT). DTT was quenched, and newly exposed thiols were modified with the addition of 100 mM 4-acetamido-4′-maleimidylstilbene (AMS). Reducing loading buffer was added to the samples, and proteins were separated by SDS-PAGE.
RESULTS
UL32 is a late protein with an unusually dynamic intracellular localization pattern.
In this study, we took advantage of an HSV isolate expressing HA-tagged UL32 (HA32) to monitor the kinetics of UL32 expression under natural infection conditions. Figure 1A shows that in cells infected with the wild-type virus (KOS strain), UL32 was maximally expressed at 24 hpi, consistent with late expression kinetics. In cells infected with HA32, a similar pattern was observed (Fig. 1A). In order to confirm that this expression pattern reflected true late kinetics, infected cells were treated with the viral DNA polymerase inhibitor phosphonoacetic acid (PAA) to prevent viral DNA synthesis. In PAA-treated cells infected with KOS or HA32, UL32 expression was prevented, demonstrating that UL32 is a true late protein (Fig. 1A). This is the first demonstration that the UL32 protein is expressed with true late kinetics, although a previous report suggested that UL32 mRNA was expressed with late kinetics (40). In order to demonstrate that the HA-tagged version of UL32 (HA-UL32) is functional, growth curves of virus produced from Vero cells infected with KOS or HA32 were performed. Figure 1B shows that these viruses exhibited identical growth patterns.
UL32 was previously reported to localize predominantly to the cytoplasm of infected cells, although a small fraction was observed within replication compartments at 18 hpi (29). A similar pattern was observed in cells transiently expressing UL32 and superinfected with a UL32-null virus (28). The predominantly cytoplasmic localization pattern was somewhat surprising, considering reports that UL32 is essential for encapsidation and all of the other viral proteins essential for procapsid assembly and encapsidation have been reported to localize to replication compartments in the nucleus (28, 41–43). To reexamine the intracellular location of UL32, cells were infected with HA32 and processed for immunofluorescence at various times postinfection (Fig. 2). ICP8 staining served as an infection control and to mark replication compartments (44), and cells were counterstained with the DNA dye TOPRO3 to detect cell nuclei. Interestingly, at 6 hpi, UL32 was predominantly detected in replication compartments; however, at later times (24 hpi), a mostly cytoplasmic staining pattern was observed. To examine this phenomenon more quantitatively, we used ImageJ software to analyze the levels of fluorescent intensity in the cytoplasm and nucleus and within replication compartments (illustrated by the yellow boxes in Fig. 2B). At 6 hpi, the localization of UL32 strongly correlated with that of ICP8 within the nucleus, suggesting that it was present within replication compartments. At 9 hpi, UL32 was more diffusely localized within the nucleus, although most UL32 was observed in replication compartments. It should also be noted that some UL32 was observed in the cytoplasm at 9 hpi. At 24 hpi, UL32 localization no longer correlated with that of ICP8 and UL32 was detected in a diffuse pattern throughout the cell. These data reveal that the localization of UL32 progressed during infection from an initial localization within replication compartments to a diffuse localization in both the nucleus and the cytoplasm. Thus, although UL32 is initially observed in replication compartments, in contrast to other viral proteins that play roles in encapsidation, it is not retained there as the infection progresses. We also observed large perinuclear foci containing HA-tagged UL32 at 24 hpi (Fig. 2) or as early as 5 hpi at a higher MOI of 3 (data not shown). Although HA32 and KOS exhibited identical growth curves, we cannot rule out the possibility that these foci were caused by the HA tag on UL32; they were not observed in previous studies using a rabbit polyclonal antibody (29) or an EE-tagged UL32 (28). We ruled out the possibility that these foci represent aggresomes caused by the accumulation of misfolded proteins since they did not contain either Hsc70 or Hsp70 (data not shown), hallmarks of previously described aggresomes (45). We are intrigued by the possibility that they are related to a second function of UL32 in the cytoplasm, and additional experiments will be required to investigate this possibility.
FIG 2.

UL32 changes localization during infection. (A) Vero cells were infected with HA32 at an MOI of 0.1. At 6, 9, and 24 h postinfection, cells were fixed in 4% paraformaldehyde and permeabilized in acetone, followed by staining for ICP8 (red, mouse antibody) and HA (green, rat antibody). TOPRO3 staining was performed to visualize the nucleus. Bars = 5 μm. (B) The merged images in panel A were analyzed by ImageJ software. Yellow boxes, the area used to measure fluorescent intensity; blue arrows, the edges of the nucleus indicated by TOPRO3 staining. At least 100 cells were examined at each time point, and the staining patterns shown in all three panels represent those seen in nearly all cells on the coverslip.
Conserved C-X-X-C motifs are required for infection.
UL32 is cysteine rich and contains C-X-X-C motifs that are reminiscent of the cysteine-rich motifs present in thiol-disulfide oxidoreductases, such as thioredoxin. Oxidoreductases are conserved in species ranging from bacteria to humans and function as antioxidants involved in redox balance (31–34). For example, thioredoxin is sensitive to oxidation, and its antioxidant activity is coordinated through its C-X-X-C domain (31–34). Figure 3 shows that UL32 and homologs from all three subfamilies of herpesviruses contain five conserved cysteine-containing motifs. Sequence alignment using the Clustal W program revealed extensive conservation of cysteines, especially within motifs I, III, IV, and V (Fig. 3). Motifs I and IV fit the C-X-X-C pattern, while motif V contains a well-conserved H-X-X-C-X-X-X-C pattern reminiscent of a motif found in zinc-coordinating proteins responsive to disulfide stress (46, 47). This motif may be responsible for the observed zinc-binding properties of UL32 (29).
To test whether these conserved motifs are essential for UL32 function, each cysteine was mutated to alanine by site-directed mutagenesis to preserve the hydrophobic nature of cysteines, and the histidine in motif V was mutated to glutamine to preserve the hydrophilic nature at that position. Plasmids bearing these mutations were tested for their ability to complement a UL32-null virus (hr64FS) for production of infectious virus (Fig. 4). Consistent with the lack of conservation in motif II, mutants with mutations in cysteines 155 and 158 resemble wild-type UL32 in their ability to complement the null mutant. Most of the other cysteine mutants with mutations in motifs I, III, IV, and V failed to complement hr64FS, suggesting that these cysteines are essential for UL32 function, although mutants altered at cysteine 291 in motif III and cysteine 498 in motif V were capable of partial complementation. These data suggest that for two of the three most highly conserved motifs (motifs I and IV), both cysteine residues are essential, and for motifs III and V, at least one of the conserved residues is essential. The second cysteine in motif II is not conserved, and mutations in both cysteines are tolerated. Thus, the degree of conservation correlates well with the results of the mutational analysis.
FIG 4.
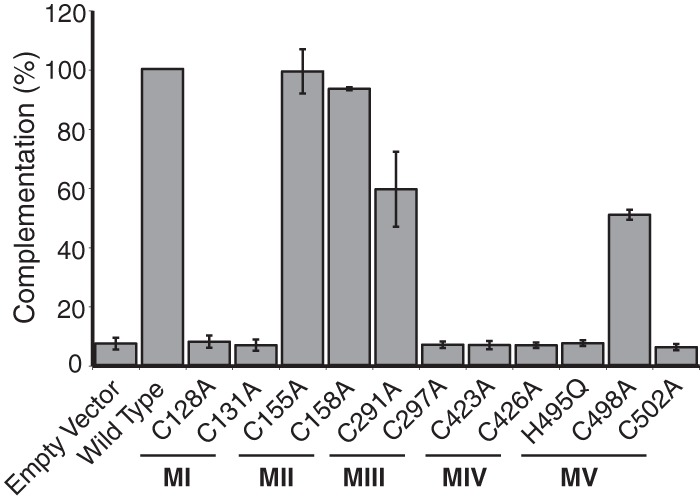
Mutagenesis of conserved residues in motifs I, III, IV, and V abolish complementation ability. Alanine substitution mutations were made at every cysteine in motifs I to V, as was a histidine-to-glutamine mutation at amino acid 498. A transient complementation assay was used to test the ability of the mutant proteins to rescue the UL32-null virus hr64FS. Plasmid DNAs containing each mutation were transfected into Vero cells, followed by superinfection with hr64FS, and the resulting titer was determined on 158 cells. Error bars represent the standard deviations from three separate experiments.
UL32 is sensitive to oxidation.
The sequence similarities between UL32 and redox-sensitive proteins led us to ask whether UL32 itself is sensitive to oxidative stress. Redox-sensitive proteins, such as Keap1, or thioredoxin-like proteins have exposed reactive thiols that serve as molecular switches that are modified under oxidizing conditions. These modified forms can be detected by changes in mobility observed by nonreducing SDS-PAGE (48–50). Vero cells were infected with HA32 at an MOI of 3 for 9 h, treated with 5 mM H2O2 for various times, and harvested in the presence of NEM buffer to prevent the spontaneous formation of disulfide bonds upon lysis (Fig. 5, lanes 4 to 8). Lanes 1 and 2 show control samples of cells infected with KOS or HA32 which were not treated with H2O2 and harvested in RIPA buffer. Lysates were subjected to SDS-PAGE under reducing (lanes 1 and 2) and nonreducing (lanes 4 to 8) conditions and analyzed by Western blotting, probing with anti-HA antibody. Lysates treated with NEM but not treated with H2O2 contained a prominent 64-kDa band representing NEM-modified monomeric UL32 (Fig. 5, lane 4). In cells treated with 5 mM H2O2, slower-migrating species, most notably, an ∼130-kDa band that became more prominent with time, were observed (Fig. 5, lanes 4 to 8) and may represent hetero- or homodimeric forms of UL32. The slower-migrating species were absent in samples treated with H2O2 for 30 min followed by treatment with 100 mM DTT (data not shown). These results suggest that exposed thiols in UL32 are redox sensitive and form disulfide-mediated complexes under oxidizing conditions.
FIG 5.
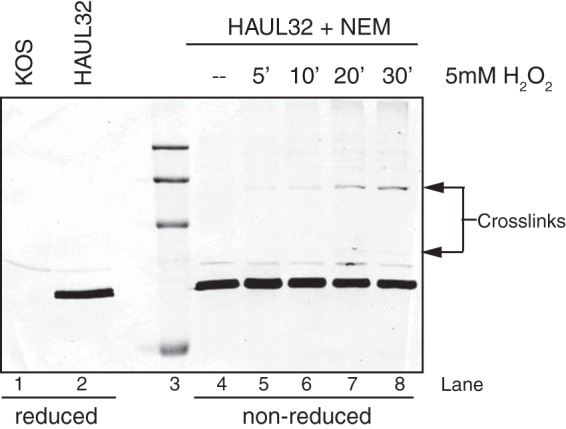
UL32 forms slower-migrating species in the presence of H2O2. Vero cells were infected with HA32 at an MOI of 3. At 9 hpi, cells were washed with PBS and overlaid with 2 ml of serum-free media either with or without 5 mM H2O2. Cells were washed with PBS and collected in RIPA buffer without NEM (lanes 1 and 2) or TCA precipitated, followed by resuspension in NEM buffer (lanes 4 to 8) at the times (in hours) indicated above lanes 5 to 8. Proteins were separated by reducing or nonreducing SDS-PAGE and analyzed by Western blotting using mouse anti-HA antibody. The molecular markers (lane 3) represent, from top to bottom, 250, 150, 100, and 50 kDa.
Generation and characterization of a new null UL32 mutant.
Our previously isolated UL32 mutant, hr64, expresses the N-terminal 273 residues of UL32, including the first two of the three C-X-X-C motifs (28), raising the possibility that this fragment of UL32 might mask the phenotype of a virus completely lacking UL32. Thus, a new UL32 mutant virus (hr64FS) containing a frameshift mutation was generated, resulting in the creation of two tandem premature stop codons at amino acids 56 and 57. This position was chosen to avoid altering the adjacent divergently transcribed UL31 gene. Figure 6A shows that hr64FS was unable to grow on Vero cells but replicated efficiently on the UL32-complementing cell line, named 158. Figure 6B shows that wild-type levels of viral DNA were observed in cells infected with hr64 or hr64FS; however, S and Q fragments representing cleavage products were not observed in hr64- or hr64FS-infected Vero cells, consistent with an encapsidation defect. The encapsidation defect was rescued when the virus was propagated on 158 cells. Thus, hr64FS exhibits the same growth phenotype and encapsidation defect as hr64.
Encapsidation of viral genomes requires the assembly of a competent procapsid containing major capsid protein VP5, triplex proteins VP19C and VP23, internal scaffold and protease, and a portal ring. In order to determine whether the defect in packaging was due to an aberrant capsid composition or morphology, we examined the ability of hr64FS to assemble structures that resemble procapsids. Procapsids cannot be isolated directly from infected cells because they are fragile and spontaneously mature into angular B capsids that have a protein composition similar to that of procapsids, except that the scaffold proteins have been proteolytically processed (51). B capsids from cells infected with KOS, hr64FS, or hr81-1 were purified by sucrose gradient sedimentation and analyzed by SDS-PAGE and silver staining to examine their protein composition. hr81-1 is a UL15-null virus, and since UL15 is a component of the HSV terminase, this mutant also exhibits a packaging defect. hr81-1 is capable of assembling B capsids and thus serves as a control to ensure that differences between KOS and hr64FS are due to the absence of UL32 and not to the encapsidation defect. Figure 7A demonstrates that B capsids isolated from hr64FS-infected cells have a protein composition indistinguishable from that of capsids isolated from KOS- or hr81-1-infected cells. Purified capsids were also analyzed by cryo-electron microscopy (cryo-EM), as described in Materials and Methods, and the cryo-EM reconstructions shown in Fig. 7B reveal no differences between the hr64FS and KOS capsids. We conclude that capsids assembled in the absence of UL32 resemble those assembled by wild-type virus, suggesting that there are no obvious defects in capsid composition or assembly.
FIG 7.
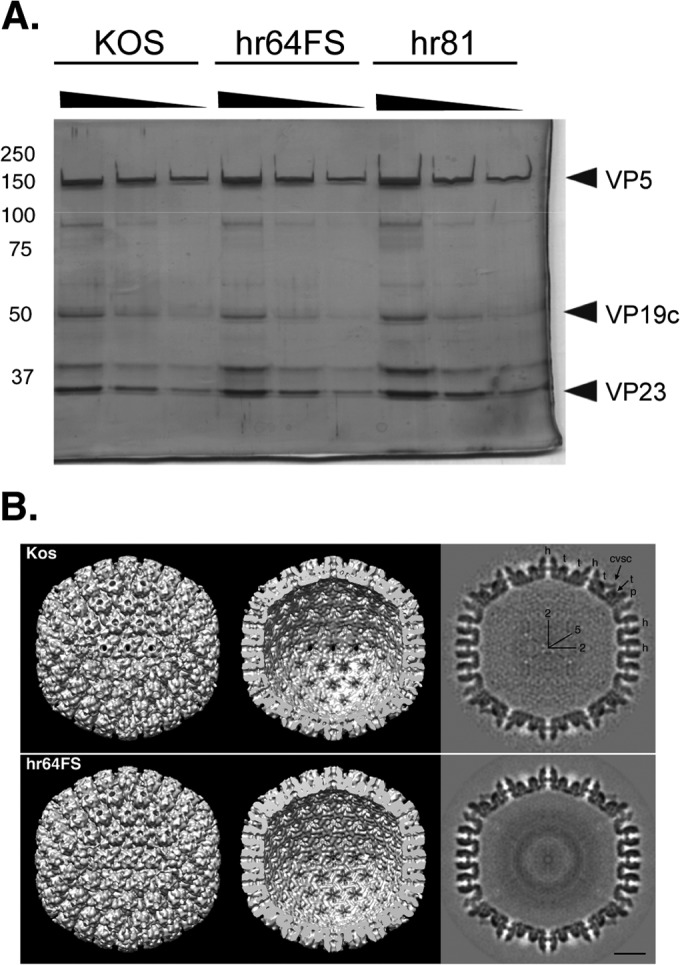
hr64FS capsids are indistinguishable from KOS capsids. (A) Viral capsids were isolated by sucrose gradient sedimentation from Vero cells infected with KOS, hr64FS, or hr81. Dilutions of B capsids were separated by reducing SDS-PGE and silver stained. Numbers on the left are molecular masses (in kilodaltons). (B) Cryo-EM analysis and reconstruction of B capsids isolated from KOS- or hr64FS-infected cells. Surface representations of the capsid exterior (left) and interior (middle) and a gray-scale central section (right) are shown. Capsid subunits are indicated: hexons (h), pentons (p), triplexes (t), and the capsid vertex specific component (CVSC). Icosahedral 2- and 5-fold symmetry axes are also marked. Bar = 200 Å.
We previously reported that hr64-infected cells exhibited inefficient localization of the major capsid protein (VP5) to replication compartments (28), leading to the hypothesis that UL32 may function as a virally encoded chaperone needed for efficient localization of one or more of the capsid proteins. We analyzed the localization patterns of VP5 and the rest of the viral capsid proteins in cells infected with the new UL32 mutant, hr64FS (Fig. 8). Surprisingly, despite our earlier report that VP5 was mislocalized in cells infected with hr64, in cells infected with hr64FS, VP5 localized to replication compartments, similar to the pattern seen in KOS-infected cells. This experiment was repeated with three different VP5 antisera, and in each case, VP5 was shown to localize to replication compartments in cells infected with either hr64 or hr64FS (data not shown). The reason for this apparent discrepancy is unclear but may be related to differences in culture conditions, such as the passage number of the cells or the levels of glutamine or oxygen in the medium. One or more of these differences could alter the folding environment in cells, leading to protein mislocalization. We also monitored the behavior of other capsid proteins, including VP22a and VP23, and both were found to localize to replication compartments in hr64FS-infected cells in a fashion similar to their localization in KOS-infected cells (Fig. 8). We conclude that UL32 does not influence the localization of capsid proteins, consistent with the observation that protein composition and capsid architecture appear to be normal in the absence of UL32 (Fig. 7A and B).
FIG 8.
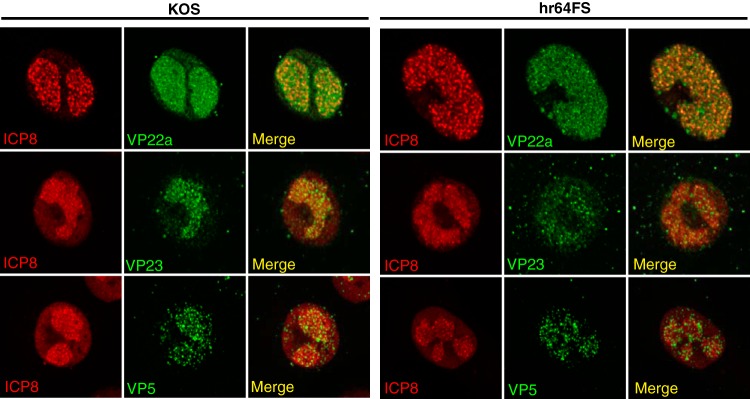
Capsid proteins localize to replication compartments in the absence of UL32. Vero cells were infected with KOS or hr64FS at an MOI of 3. Infected cells were fixed with 4% paraformaldehyde and permeabilized in acetone. Cells were stained with antibodies to ICP8 (red channel) plus the capsid proteins indicated (green channel), as described in Materials and Methods.
UL32 influences the disulfide bond status of viral proteins, including the viral protease (VP24).
We have previously reported that mature HSV capsids are extensively cross-linked by disulfide bonds (9) and that the UL6 portal ring contains intersubunit disulfide bonds (10). This raises the question of how disulfide bond formation in the portal is regulated, especially in a cellular environment that is generally thought to be reducing (10). We hypothesize that UL32 acts as a regulator of disulfide bond formation. To test this hypothesis, we took advantage of the thiol-modifying reagent AMS, a maleimide derivative that reacts with free cysteines, adding ∼500 Da to each cysteine that has a free sulfhydryl. If UL32 were responsible for disulfide bond regulation, we would expect a difference in the number of free cysteines available for modification with AMS in cells infected in the presence and absence of UL32. Vero cells were infected with KOS, hr64FS, or the packaging mutant hr81-1. At 11 hpi, infected cells were TCA precipitated, resuspended in denaturing buffer containing NEM to block free thiols, reduced with DTT to free thiols participating in disulfide bonds, and subsequently treated with AMS to modify the newly freed thiols, as described in Materials and Methods. Figure 9 shows that in the absence of UL32, both the viral protease (VP24) and UL6 migrate slower than they do in cells infected with KOS or hr81-1-infected cells. hr81-1 was included in this experiment to show that the disulfide bond alterations seen in hr64FS-infected cells were not the result of an encapsidation defect. These shifts were not observed in the absence of AMS and suggest that in hr64FS-infected cells, protease and UL6 contained disulfide bonds not observed in cells infected with KOS or hr81-1. Interestingly, similar shifts were observed in the minor capsid proteins UL25 and VP19C (data not shown). These results are consistent with the hypothesis that UL32 regulates disulfide bond formation and that the presence of UL32 may function to prevent premature disulfide bond formation in a number of viral capsid proteins.
FIG 9.
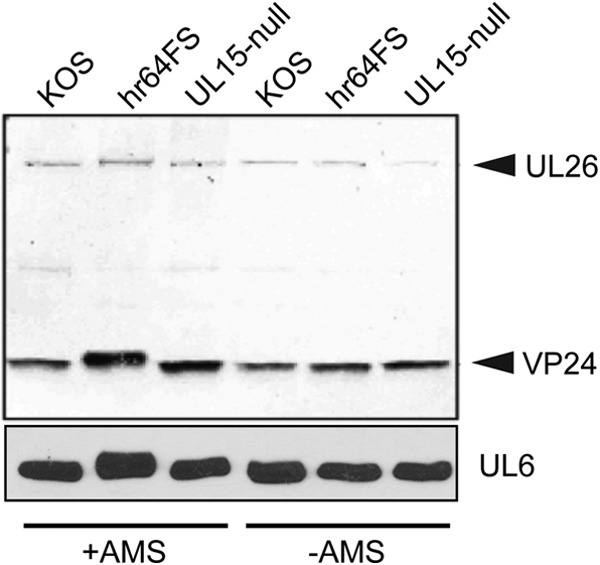
UL32 modulates disulfide bond formation in viral proteins. Vero cells were infected with KOS, hr64FS, or hr81 at an MOI of 3 and harvested in 10% TCA at 10 hpi. Free thiols were blocked with NEM, followed by reduction with DTT and then treatment with the thiol alkylating agent AMS. Proteins were resolved by reducing SDS-PAGE and detected by immunoblotting with anti-VP24 or anti-UL6 antibody.
DISCUSSION
In this study, two new viruses were used to examine the role of UL32 in the encapsidation of HSV-1 DNA: HA32, containing an HA-tagged UL32 protein (HA32), and a null mutant (hr64FS) containing a frameshift mutation. HA32 resembles wild-type virus in growth and encapsidation, while hr64FS is defective for both. Several observations were made. (i) hr64FS assembled B capsids that resemble wild-type B capsids in protein composition and appearance by cryo-EM (Fig. 7B); however, they were unable to encapsidate viral genomes. (ii) Although it was previously reported that UL32 is mostly cytoplasmic with some minor staining in the nucleus, a time course experiment (Fig. 2) revealed a dynamic localization pattern. UL32 was found predominantly in replication compartments early in infection, while later its localization became cytoplasmic/nuclear diffuse. (iii) We confirmed that UL32 is a true late protein. (iv) Two highly conserved C-X-X-C motifs within UL32 were shown to be essential for viral growth. (v) UL32 was shown to be redox sensitive. (vi) UL32 influenced the formation of disulfide bonds in the viral proteins UL25, VP19C, and UL6 and the viral protease, VP24.
The ability to assemble B capsids that are indistinguishable from those seen in cells infected with wild-type HSV suggests that UL32 is not required for the assembly of procapsids per se. On the other hand, we have confirmed that procapsids assembled in cells infected with a UL32 mutant fail to initiate encapsidation. Protease activation marks the initiation of the encapsidation process; however, the mechanism by which protease is regulated is not understood. The formation of inter- and intramolecular disulfide bonds is generally recognized to regulate a wide range of cellular processes. We have previously shown that disulfide bonds are essential for capsid stability and for the formation and/or stability of the UL6 portal ring (9, 10). It is possible that the HSV protease is regulated by the redox state. This suggestion may be consistent with previous reports suggesting that the human cytomegalovirus protease is regulated by disulfide bond formation. Alternatively, initiation of encapsidation may be triggered by redox-stimulated changes in the portal ring. Thus, encapsidation may be regulated through a thiol-disulfide exchange mechanism involving protease or portal, or both.
The suggestion that UL32 may perform a thiol-disulfide exchange reaction is consistent with the observations that UL32 itself is redox sensitive and that it contains C-X-X-C motifs reminiscent of those found in a number of other thiol-disulfide oxidoreductases. We now report that these conserved C-X-X-C motifs are essential for viral growth. UL32 may act directly on protease or UL6 by catalyzing the formation or breaking of disulfide bonds; alternatively, UL32 may exert indirect effects by altering the cellular milieu to provide an environment conducive for disulfide bond formation.
The localization of UL32 in infected cells may also provide some clues as to its function. Previous reports that UL32 is predominantly a cytoplasmic protein were hard to reconcile with the phenotype of UL32 mutants that suggested a role in encapsidation. The observation that UL32 was found to localize predominantly to replication compartments early in infection and in the cytoplasm and nucleus at later stages is consistent with additional functions for UL32 at a stage postencapsidation. Our finding that in the absence of UL32, capsid proteins UL6, UL25, and VP19C exhibited disulfide bonds not seen in wild-type infection is of interest. We and others have found that the cellular environment becomes oxidized during HSV-1 infection (52–56; B. S. Albright, unpublished data), and an oxidized environment may promote disulfide bond formation. Although some disulfide bonds may be beneficial for the virus, it is also possible that an oxidizing environment could lead to aberrant disulfide bond formation between exposed reactive thiols that would be deleterious to the virus. We suggest that UL32 may serve to protect sensitive thiols from aberrant or deleterious disulfide bond formation. Thus, in addition to a possible role in the nucleus regulating protease and/or UL6, UL32 may play additional roles required for capsid maturation in the cytoplasm, possibly by protecting reactive thiols. Alternatively, UL32 may catalyze the formation of disulfide bonds to regulate specific processes, such as protease activation.
This study provides the first evidence that HSV may encode a virally encoded oxidoreductase. The genomes of other large DNA viruses, which require disulfide bonds, have been shown to encode enzymes that function as oxidoreductases (57). For instance, the vaccinia virus genome encodes three oxidoreductases (E10R, A2.5L, and G4L) which participate in a thiol-disulfide relay responsible for modulating disulfide bonds in the cytoplasm (58). It is becoming increasingly clear that oxidative stress and cellular responses to it play critical roles in modulating a large number of biological processes, including pathogenesis by bacterial and viral infections. Many viruses, including HSV, have been shown to induce oxidative stress, and antioxidants have shown promise in the treatment of viral infection (54–56, 59–63). Viral proteins that play roles in sensing and responding to changes in redox status may thus provide targets for therapeutic intervention. The suggestion that UL32 encodes a redox-sensitive chaperone responsible for modulating disulfide bond formation in HSV-infected cells indicates that UL32 itself may represent a novel and promising drug target for controlling HSV infections.
ACKNOWLEDGMENTS
We thank Alexander M. Makhov (University of Pittsburgh) for his expert technical support for cryo-electron microscopy. We also thank the members of our laboratories for discussions and suggestions on the manuscript.
This project was supported by a National Institutes of Health grant awarded to S.K.W. (AI37549). N.D.S. was supported by the Medical Research Council of the United Kingdom, and E.A.C. was the recipient of a Medical Research Council studentship.
REFERENCES
- 1.Brown J, McVoy M, Homa F. 2002. Packaging DNA into herpesvirus capsids, p 111–153. In Holzenburg A, Bogner E (ed), Structure-function relationships of human pathogenic viruses. Springer, New York, NY. [Google Scholar]
- 2.Baines J, Weller S. 2005. Cleavage and packaging of herpes simplex virus 1 DNA, p 135–150. In Viral genome packaging machines: genetics, structure, and mechanism. Springer, New York, NY. [Google Scholar]
- 3.Conway JF, Homa FL. 2011. Nucleocapsid structure, assembly and DNA packaging of herpes simplex virus. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 4.Homa FL, Brown JC. 1997. Capsid assembly and DNA packaging in herpes simplex virus. Rev Med Virol 7:107–122. doi: 10.1002/(SICI)1099-1654(199707)7:2<107::AID-RMV191>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 5.Preston VG, Murray J, Preston CM, McDougall IM, Stow ND. 2008. The UL25 gene product of herpes simplex virus type 1 is involved in uncoating of the viral genome. J Virol 82:6654–6666. doi: 10.1128/JVI.00257-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steven A. 1997. Herpesvirus capsid assembly and envelopment. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 7.Steven AC, Heymann JB, Cheng N, Trus BL, Conway JF. 2005. Virus maturation: dynamics and mechanism of a stabilizing structural transition that leads to infectivity. Curr Opin Struct Biol 15:227–236. doi: 10.1016/j.sbi.2005.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roos WH, Radtke K, Kniesmeijer E, Geertsema H, Sodeik B, Wuite GJ. 2009. Scaffold expulsion and genome packaging trigger stabilization of herpes simplex virus capsids. Proc Natl Acad Sci U S A 106:9673–9678. doi: 10.1073/pnas.0901514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szczepaniak R, Nellissery J, Jadwin JA, Makhov AM, Kosinski A, Conway JF, Weller SK. 2011. Disulfide bond formation contributes to herpes simplex virus capsid stability and retention of pentons. J Virol 85:8625–8634. doi: 10.1128/JVI.00214-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albright BS, Nellissery J, Szczepaniak R, Weller SK. 2011. Disulfide bond formation in the herpes simplex virus 1 UL6 protein is required for portal ring formation and genome encapsidation. J Virol 85:8616–8624. doi: 10.1128/JVI.00123-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardone G, Winkler DC, Trus BL, Cheng N, Heuser JE, Newcomb WW, Brown JC, Steven AC. 2007. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology 361:426–434. doi: 10.1016/j.virol.2006.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang JT, Schmid MF, Rixon FJ, Chiu W. 2007. Electron cryotomography reveals the portal in the herpesvirus capsid. J Virol 81:2065–2068. doi: 10.1128/JVI.02053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newcomb WW, Juhas RM, Thomsen DR, Homa FL, Burch AD, Weller SK, Brown JC. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J Virol 7 5:10923–10932. doi: 10.1128/JVI.75.22.10923-10932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trus BL, Cheng N, Newcomb WW, Homa FL, Brown JC, Steven AC. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J Virol 78:12668–12671. doi: 10.1128/JVI.78.22.12668-12671.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White CA, Stow ND, Patel AH, Hughes M, Preston VG. 2003. Herpes simplex virus type 1 portal protein UL6 interacts with the putative terminase subunits UL15 and UL28. J Virol 77:6351–6358. doi: 10.1128/JVI.77.11.6351-6358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adelman K, Salmon B, Baines JD. 2001. Herpes simplex virus DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc Natl Acad Sci U S A 98:3086–3091. doi: 10.1073/pnas.061555698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beard PM, Taus NS, Baines JD. 2002. DNA cleavage and packaging proteins encoded by genes U(L)28, U(L)15, and U(L)33 of herpes simplex virus type 1 form a complex in infected cells. J Virol 76:4785–4791. doi: 10.1128/JVI.76.10.4785-4791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu D, Weller SK. 1998. Genetic analysis of the UL 15 gene locus for the putative terminase of herpes simplex virus type 1. Virology 243:32–44. doi: 10.1006/viro.1998.9041. [DOI] [PubMed] [Google Scholar]
- 19.Sheaffer AK, Newcomb WW, Gao M, Yu D, Weller SK, Brown JC, Tenney DJ. 2001. Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J Virol 75:687–698. doi: 10.1128/JVI.75.2.687-698.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu D, Weller SK. 1998. Herpes simplex virus type 1 cleavage and packaging proteins UL15 and UL28 are associated with B but not C capsids during packaging. J Virol 72:7428–7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beard PM, Duffy C, Baines JD. 2004. Quantification of the DNA cleavage and packaging proteins U(L)15 and U(L)28 in A and B capsids of herpes simplex virus type 1. J Virol 78:1367–1374. doi: 10.1128/JVI.78.3.1367-1374.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newcomb WW, Homa FL, Brown JC. 2006. Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J Virol 80:6286–6294. doi: 10.1128/JVI.02648-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogasawara M, Suzutani T, Yoshida I, Azuma M. 2001. Role of the UL25 gene product in packaging DNA into the herpes simplex virus capsid: location of UL25 product in the capsid and demonstration that it binds DNA. J Virol 75:1427–1436. doi: 10.1128/JVI.75.3.1427-1436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thurlow JK, Rixon FJ, Murphy M, Targett-Adams P, Hughes M, Preston VG. 2005. The herpes simplex virus type 1 DNA packaging protein UL17 is a virion protein that is present in both the capsid and the tegument compartments. J Virol 79:150–158. doi: 10.1128/JVI.79.1.150-158.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thurlow JK, Murphy M, Stow ND, Preston VG. 2006. Herpes simplex virus type 1 DNA-packaging protein UL17 is required for efficient binding of UL25 to capsids. J Virol 80:2118–2126. doi: 10.1128/JVI.80.5.2118-2126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trus BL, Newcomb WW, Cheng N, Cardone G, Marekov L, Homa FL, Brown JC, Steven AC. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol Cell 26:479–489. doi: 10.1016/j.molcel.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conway JF, Cockrell SK, Copeland AM, Newcomb WW, Brown JC, Homa FL. 2010. Labeling and localization of the herpes simplex virus capsid protein UL25 and its interaction with the two triplexes closest to the penton. J Mol Biol 397:575–586. doi: 10.1016/j.jmb.2010.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamberti C, Weller SK. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J Virol 72:2463–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang YE, Poon AP, Roizman B. 1996. Properties of the protein encoded by the UL32 open reading frame of herpes simplex virus 1. J Virol 70:3938–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Netto LE, de Oliveira MA, Monteiro G, Demasi AP, Cussiol JR, Discola KF, Demasi M, Silva GM, Alves SV, Faria VG, Horta BB. 2007. Reactive cysteine in proteins: protein folding, antioxidant defense, redox signaling and more. Comp Biochem Physiol C Toxicol Pharmacol 146:180–193. doi: 10.1016/j.cbpc.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 31.Chivers PT, Laboissiere MC, Raines RT. 1996. The CXXC motif: imperatives for the formation of native disulfide bonds in the cell. EMBO J 15:2659–2667. [PMC free article] [PubMed] [Google Scholar]
- 32.Chivers PT, Prehoda KE, Raines RT. 1997. The CXXC motif: a rheostat in the active site. Biochemistry 36:4061–4066. doi: 10.1021/bi9628580. [DOI] [PubMed] [Google Scholar]
- 33.Fomenko DE, Gladyshev VN. 2003. Identity and functions of CxxC-derived motifs. Biochemistry 42:11214–11225. doi: 10.1021/bi034459s. [DOI] [PubMed] [Google Scholar]
- 34.Becerra A, Delaye L, Lazcano A, Orgel LE. 2007. Protein disulfide oxidoreductases and the evolution of thermophily: was the last common ancestor a heat-loving microbe? J Mol Evol 65:296–303. doi: 10.1007/s00239-007-9005-0. [DOI] [PubMed] [Google Scholar]
- 35.Beilstein F, Higgs MR, Stow ND. 2009. Mutational analysis of the herpes simplex virus type 1 DNA packaging protein UL33. J Virol 83:8938–8945. doi: 10.1128/JVI.01048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stow ND, Wilkie NM. 1976. An improved technique for obtaining enhanced infectivity with herpes simplex virus type 1 DNA. J Gen Virol 33:447–458. doi: 10.1099/0022-1317-33-3-447. [DOI] [PubMed] [Google Scholar]
- 37.Sheaffer AK, Newcomb WW, Brown JC, Gao M, Weller SK, Tenney DJ. 2000. Evidence for controlled incorporation of herpes simplex virus type 1 UL26 protease into capsids. J Virol 74:6838–6848. doi: 10.1128/JVI.74.15.6838-6848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan X, Sinkovits RS, Baker TS. 2007. AUTO3DEM—an automated and high throughput program for image reconstruction of icosahedral particles. J Struct Biol 157:73–82. doi: 10.1016/j.jsb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan X, Dryden KA, Tang J, Baker TS. 2007. Ab initio random model method facilitates 3D reconstruction of icosahedral particles. J Struct Biol 157:211–225. doi: 10.1016/j.jsb.2006.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holland LE, Sandri-Goldin RM, Goldin AL, Glorioso JC, Levine M. 1984. Transcriptional and genetic analyses of the herpes simplex virus type 1 genome: coordinates 0.29 to 0.45. J Virol 49:947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Higgs MR, Preston VG, Stow ND. 2008. The UL15 protein of herpes simplex virus type 1 is necessary for the localization of the UL28 and UL33 proteins to viral DNA replication centres. J Gen Virol 89:1709–1715. doi: 10.1099/vir.0.2008/000448-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scholtes L, Baines JD. 2009. Effects of major capsid proteins, capsid assembly, and DNA cleavage/packaging on the pUL17/pUL25 complex of herpes simplex virus 1. J Virol 83:12725–12737. doi: 10.1128/JVI.01658-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taus NS, Salmon B, Baines JD. 1998. The herpes simplex virus 1 UL 17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology 252:115–125. doi: 10.1006/viro.1998.9439. [DOI] [PubMed] [Google Scholar]
- 44.Quinlan MP, Chen LB, Knipe DM. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857–868. doi: 10.1016/0092-8674(84)90035-7. [DOI] [PubMed] [Google Scholar]
- 45.Muchowski PJ, Wacker JL. 2005. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 46.Kang JG, Paget MS, Seok YJ, Hahn MY, Bae JB, Hahn JS, Kleanthous C, Buttner MJ, Roe JH. 1999. RsrA, an anti-sigma factor regulated by redox change. EMBO J 18:4292–4298. doi: 10.1093/emboj/18.15.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song T, Dove SL, Lee KH, Husson RN. 2003. RshA, an anti-sigma factor that regulates the activity of the mycobacterial stress response sigma factor SigH. Mol Microbiol 50:949–959. doi: 10.1046/j.1365-2958.2003.03739.x. [DOI] [PubMed] [Google Scholar]
- 48.Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. 2002. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 111:471–481. doi: 10.1016/S0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 49.Zhang DD, Hannink M. 2003. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown JD, Day AM, Taylor SR, Tomalin LE, Morgan BA, Veal EA. 2013. A peroxiredoxin promotes H2O2 signaling and oxidative stress resistance by oxidizing a thioredoxin family protein. Cell Rep 5:1425–1435. doi: 10.1016/j.celrep.2013.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newcomb WW, Trus BL, Cheng N, Steven AC, Sheaffer AK, Tenney DJ, Weller SK, Brown JC. 2000. Isolation of herpes simplex virus procapsids from cells infected with a protease-deficient mutant virus. J Virol 74:1663–1673. doi: 10.1128/JVI.74.4.1663-1673.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalez-Dosal R, Horan KA, Rahbek SH, Ichijo H, Chen ZJ, Mieyal JJ, Hartmann R, Paludan SR. 2011. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: role for S-glutathionylation of TRAF3 and 6. PLoS Pathog 7:e1002250. doi: 10.1371/journal.ppat.1002250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mathew SS, Bryant PW, Burch AD. 2010. Accumulation of oxidized proteins in herpesvirus infected cells. Free Radic Biol Med 49:383–391. doi: 10.1016/j.freeradbiomed.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nucci C, Palamara AT, Ciriolo MR, Nencioni L, Savini P, D'Agostini C, Rotilio G, Cerulli L, Garaci E. 2000. Imbalance in corneal redox state during herpes simplex virus 1-induced keratitis in rabbits. Effectiveness of exogenous glutathione supply. Exp Eye Res 70:215–220. doi: 10.1006/exer.1999.0782. [DOI] [PubMed] [Google Scholar]
- 55.Palamara AT, Perno CF, Ciriolo MR, Dini L, Balestra E, D'Agostini C, Di Francesco P, Favalli C, Rotilio G, Garaci E. 1995. Evidence for antiviral activity of glutathione: in vitro inhibition of herpes simplex virus type 1 replication. Antiviral Res 27:237–253. doi: 10.1016/0166-3542(95)00008-A. [DOI] [PubMed] [Google Scholar]
- 56.Vogel JU, Cinatl J, Dauletbaev N, Buxbaum S, Treusch G, Cinatl J Jr, Gerein V, Doerr HW. 2005. Effects of S-acetylglutathione in cell and animal model of herpes simplex virus type 1 infection. Med Microbiol Immunol 194:55–59. doi: 10.1007/s00430-003-0212-z. [DOI] [PubMed] [Google Scholar]
- 57.Iyer LM, Aravind L, Koonin EV. 2001. Common origin of four diverse families of large eukaryotic DNA viruses. J Virol 75:11720–11734. doi: 10.1128/JVI.75.23.11720-11734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Senkevich TG, White CL, Koonin EV, Moss B. 2002. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc Natl Acad Sci U S A 99:6667–6672. doi: 10.1073/pnas.062163799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beck MA, Handy J, Levander OA. 2000. The role of oxidative stress in viral infections. Ann N Y Acad Sci 917:906–912. doi: 10.1111/j.1749-6632.2000.tb05456.x. [DOI] [PubMed] [Google Scholar]
- 60.Cai JY, Chen Y, Seth S, Furukawa S, Compans RW, Jones DP. 2003. Inhibition of influenza infection by glutathione. Free Radic Biol Med 34:928–936. doi: 10.1016/S0891-5849(03)00023-6. [DOI] [PubMed] [Google Scholar]
- 61.Garland M, Fawzi WW. 1999. Antioxidants and progression of human immunodeficiency virus (HIV) disease. Nutr Res 19:1259–1276. doi: 10.1016/S0271-5317(99)00072-X. [DOI] [Google Scholar]
- 62.Abdalla MY, Ahmad IM, Spitz DR, Schmidt WN, Britigan BE. 2005. Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. J Med Virol 76:489–497. doi: 10.1002/jmv.20388. [DOI] [PubMed] [Google Scholar]
- 63.Tian Y, Jiang W, Gao N, Zhang J, Chen W, Fan D, Zhou D, An J. 2010. Inhibitory effects of glutathione on dengue virus production. Biochem Biophys Res Commun 397:420–424. doi: 10.1016/j.bbrc.2010.05.108. [DOI] [PubMed] [Google Scholar]