

Abstract
Background
Allopatric divergence across lineages can lead to post-zygotic reproductive isolation upon secondary contact and disrupt coevolution between mitochondrial and nuclear genomes, promoting emergence of genetic incompatibilities. A previous FST scan on the transcriptome of the Baltic clam Macoma balthica highlighted several genes potentially involved in mito-nuclear incompatibilities (MNIs). As proteins involved in the mitochondrial oxidative phosphorylation (OXPHO) chain are prone to MNIs and can contribute to the maintenance of genetic barriers, the mitochondrial genomes of six Ma. balthica individuals spanning two secondary contact zones were sequenced using the Illumina MiSeq plateform.
Results
The mitogenome has an approximate length of 16,806 bp and encodes 13 protein-coding genes, 2 rRNAs and 22 tRNAs, all located on the same strand. atp8, a gene long reported as rare in bivalves, was detected. It encodes 42 amino acids and is putatively expressed and functional. A large unassigned region was identified between rrnS and tRNAMet and could likely correspond to the Control Region. Replacement and synonymous mutations were mapped on the inferred secondary structure of all protein-coding genes of the OXPHO chain. The atp6 and atp8 genes were characterized by background levels of replacement mutations, relative to synonymous mutations. However, most nad genes (notably nad2 and nad5) were characterized by an elevated proportion of replacement mutations.
Conclusions
Six nearly complete mitochondrial genomes were successfully assembled and annotated, providing the necessary roadmap to study MNIs at OXPHO loci. Few replacement mutations were mapped on mitochondrial-encoded ATP synthase subunits, which is in contrast with previous data on nuclear-encoded subunits. Conversely, the high population divergence and the prevalence of non-synonymous mutations at nad genes are congruent with previous observations from the nuclear transcriptome. This further suggest that MNIs between subunits of Complex I of the OXPHO chain, coding for NADH dehydrogenase, may play a role in maintaining barriers to gene flow in Ma. balthica.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-014-0259-z) contains supplementary material, which is available to authorized users.
Keywords: NADH dehydrogenase, ATP synthase, Next-generation sequencing, OXPHO chain, Positive selection, Inter-specific hybridization, Hybrid zone
Background
The mitochondrial electron transport chain, a central component of cellular energy production, relies on the coevolution of mitochondrial- and nuclear-encoded genes to function [1,2]. Mito-nuclear coevolution is however easily disrupted because genomes have different mutation rates, modes of inheritance, number and speed of recombination events, effective population size and selection pressures [3-5]. Such disruption can cause the emergence and maintenance of mito-nuclear incompatibilities (MNIs, among other types of genomic incompatibilities [6-9]) that are described by the Dobzhansky-Muller model [10,11].
Dobzhansky-Muller incompatibilities have been implicated in intrinsic post-zygotic barriers [8] and involved in mito-nuclear gene interactions in the fruit fly Drosophila [5], the parasitoid wasp Nasonia [12-14], the Atlantic eel Anguilla [15,16] and the marine copepod Trigriopus californicus [4,17,18]. Studying reproductive isolation is especially relevant in the marine environment, where many marine organisms have a high fecundity, large population sizes and a high dispersive potential [19], and where physical barriers to dispersal are not readily apparent. Endogenous barriers to gene flow may therefore be an important factor structuring marine populations in this environment. In addition, marine taxa can be sensitive to glaciation cycles that can promote the establishment of secondary contact zones and therefore lead to admixture of genetically divergent backgrounds [20].
In this context, the Baltic clam Macoma balthica (Mollusca: Bivalvia: Tellinidae) is a noteworthy model for studying the role of MNIs in structuring marine populations. This infaunal tellinid bivalve occurs on sandy-mud flats from the upper intertidal to the subtidal, in parts of the northern hemisphere. Its natural European range extends from eastern Russia (Pechora Sea) to southern France (Gironde Estuary). Ma. balthica is characterized by a complex colonization history in Europe. With the opening of the Bering Strait, several trans-arctic invasion events of Pacific populations into the Atlantic have occurred, leading to secondary contact of divergent lineages in the Bay of Biscay [21] and in the Baltic and White seas [22], allowing gene flow between previously-isolated populations. The long periods of reproductive isolation between these populations (~0.1-1.8 My between Bay of Biscay populations [23]; ~ 2–3.5 My between Ma. balthica balthica and Ma. balthica rubra subspecies [22]) represent strong potential for the accumulation of genetic incompatibilities.
A recent FST scan based on transcriptome data highlighted outlier loci coding for nuclear subunits of the FOF1-ATP synthase complex (subunits alpha, gamma and O) and a putative isoform of the NADH deshydrogenase [24], which are involved in the oxidative phosphorylation (OXPHO) mitochondrial chain [1,2]. These protein complexes are composed of subunits encoded by both nuclear and mitochondrial genes [2] and require tight mito-nuclear coevolution to be fully operational. They are therefore prime candidates for the establishment of MNIs following secondary contact between previously allopatric populations [6,17], which may constitute endogenous barriers to gene flow.
Sequences from only two mitochondrial genes have been published to date for this species (cox3 [22]; cox1 [21,23]). To work toward the diagnosis of MNIs in Ma. balthica and further test the hypothesis of mito-nuclear coevolution breakdown among Ma. balthica lineages, additional mitochondrial data are therefore required for comparison with the nuclear transcriptomic data at hand. Although several mitogenomes have been sequenced for marine bivalves (e.g. [25,26]) only one is currently available for the Tellinidae (Moerella iridescens [26]). However, mitochondrial sequences of Mo. iridescens are highly divergent from those of Ma. balthica (raw p-distance at cox1 of 17%). We have therefore set to sequence the mitogenomes of six Ma. balthica individuals spanning two secondary contact zones (i.e. across the Kattegat strait and the Brittany peninsula) to test whether mitochondrial genes interacting with previously-detected nuclear outlier loci show stronger divergence and selection pressures, compared to other mitochondrial genes. Our intents were (i) to detect and map protein-coding genes (PCGs) involved in the OXPHO chain, (ii) to estimate the degree of divergence and selection pressures across lineages for all PCGs, and (iii) to map mutations onto predicted secondary structure of mitochondrial membrane-embedded protein to help detect potential incompatibilities among mitochondrial and nuclear OXPHO subunits. In particular, we characterize a putative atp8 gene and discuss its functionality, as (i) it is absent in about half of the published bivalve mitogenomes, and (ii) its role is potentially relevant to the study of MNIs in Ma. balthica.
Methods
Sample collection and DNA extraction
Previous work on Ma. balthica suggested the presence of at least three divergent mitochondrial clades in Europe [21-23], which were targeted in our sampling: a Baltic lineage, and two Atlantic lineages that are separated by the French Finistère peninsula. Ma. balthica specimens were collected from 2003 to 2006 (A10, M12, W4, W20 and F17) and 2013 (A6) along European coasts, ranging from Aytré, France to Tvärminne, Finland (Table 1). Total genomic DNA was extracted from foot muscle tissue using the Dneasy™ Tissue Kit (Qiagen, Germany) following the manufacturer's protocol and stored at −20°C until further analyses.
Table 1.
Sampling sites and mitochondrial cox1 haplotypes of the sequenced specimens of Ma. balthica . cox1 haplotypes are described in Becquet et al. [21] and are noted H1 to H5. H1 and H2 are common of the Bay of Biscay, H3 of the English Channel, H4 and H5 of North and Baltic seas, respectively
| Sampling site | Country | Code | Latitude | Longitude | GB Accession no. | cox1 haplotype |
|---|---|---|---|---|---|---|
| Aytré | France | A | 46.126 | −1.1306 | A10: KM373200 | H1 |
| Aytré | France | A | 46.126 | −1.1306 | A6: KM373201 | H2 |
| Mont-Saint-Michel | France | M | 48.438 | −1.5153 | M12: KM373202 | H3 |
| Balgzand | Netherlands | W | 52.9301 | +4.7953 | W4: KM373203 | H3 |
| Balgzand | Netherlands | W | 52.9301 | +4.7953 | W20: KM373204 | H4 |
| Tvärminne | Finland | F | 59.4883 | +21.2051 | F17: KM373205 | H5 |
Initially eight mitogenomes were sequenced, corresponding to two individuals per mitochondrial lineage, as inferred from 313 bp of cox1 by Becquet et al. [21] (and hereon referred to as “cox1 haplotypes”). Six nearly-complete mitogenomes were eventually obtained, as Long-Range PCR (detailed below) failed for two individuals. They correspond to cox1 haplotypes H1 to H5 (Table 1). H1-H4 were originally described in [21] and H5 is a newly described haplotype.
Primer design and Long-Range PCR amplification of mtDNA
Three pairs of primers were designed using Primer-BLAST [27] to amplify the mitochondrial genome of Ma. balthica (Additional files 1 and 2). The first two pairs were designed based on cox1 and cox3 sequences available on GenBank for Ma. balthica [22,23]. Primer length was maximized (28–31 bp) to enhance stability during Long-Range PCR (LR-PCR). When the two-step mitogenome amplification did not succeed, one additional primer set (Palumbi et al. 1996 in [28]) was used to prime within rrnL. These three primer pairs allowed the amplification of 3 to 16.5 kbp. LR-PCR products were visualised by electrophoresis on a 0.7% agarose gel stained with GelRed™ (Biotium, Hayward, CA, USA) to assess quality.
Library preparation and DNA sequencing
The quantity of LR-PCR DNA templates (prior and during library preparation) was assessed by spectrophotometry (Nanodrop ND-800, Thermo Scientific, Waltham, MA, USA), fluorometry (Qubit, Invitrogen, Carlsbad, NM, USA) and quantitative PCR (PicoGreen dosage with Quant-iT™ PicoGreen® dsDNA Assay Kit, Invitrogen on ABI 7900HT, Applied Biosystems). After quality controls, six libraries (one per individual) were prepared using the TruSeq DNA Sample Prep Kits v2 and TruSeq Universal Adapters (Illumina, San Diego, CA, USA). A TruSeq Universal Adapter was used for each DNA library in order to separate reads from different individuals after DNA sequencing. Library sizes were checked on BioAnalyzer chips (Agilent Technologies, Santa Clara, CA, USA). Paired-end library sequencing was carried out on the Illumina MiSeq plateform (2 × 250 bp chemistry) at the GeT-PlaGe lab (GenoToul, Toulouse, France).
Sequence cleaning, trimming, assembly and gene annotation
Genomes were assembled in two rounds, using different read quality filters and assembly parameters, to optimize contig length while controlling for quality. In the first round, reads were de-multiplexed and quality-filtered (length > 10 nucleotides; Q ≥ 28 on 50% of read length) using the FastX toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Duplicated sequences were removed and adapters were clipped. Sequences (Illumina 1.9 quality scores) were checked for quality before and after FastX filtering using FastQC v.0.10.1 [29]. Assembly was performed in Velvet v.1.2.09 [30], and parameters (coverage cut-off and k-mer size) were optimized using a custom R script to minimize the number of contigs and maximize contig lengths (total length and N50 parameter). Optimized k-mer sizes were 89 bp (W20), 97 bp (F17), 103 bp (A10, M12), 107 bp (A6) and 119 bp (W4). Optimized k-mer coverages were 250 (A6, A10, F17, M12, W4) and 300 (W20). Assemblies were checked in Tablet v.1.13.05.17 [31].
In the second round of assemblies, we used more stringent quality filters (length > 250 nucleotides; Q ≥ 29 on 90% of read length), and fixed k-mer length and k-mer coverage to 247 bp and 100, respectively. In the second assembly, contigs from the first round were used as a reference; Sanger sequences for parts of the cox1 and cox3 genes were included to fill the gaps flanking the LR-PCR primer sequences. Sanger sequencing was performed by GATC Biotech (Konstanz, Germany) using an ABI 3730xl automated DNA Analyzer (Perkin-Elmer Applied Biosystems, Foster City, CA, USA).
All contigs from the first and second assemblies and parts of cox1, cox3 sequences were aligned using Sequencher™ v.5.0.1 (Gene Codes Corporation, Ann Arbor, MI, USA). Resulting contigs (1 for A6, A10, W4, F17 and 2 for M12, W20) were searched on GenBank (Tellinidae non-redundant protein sequence database) in order to control for contaminants. We also searched for the presence of these contigs among the 454 transcriptome sequences available for Ma. balthica [24] using a local BLAST database and tblastx (BLAST toolkit v. 2.2.25; [32]). Mitochondrial genome annotations were then performed on the MITOS WebServer, first using default parameters [33] and second using advanced parameters (BLAST E-value = 1×10−4, Start/Stop Range = 40 and Final Maximum Overlap = 10) in order to improve annotation quality and start/stop codon delimitation.
Quality control and gene boundary delimitation
Mitochondrial genomes were aligned using Sequencher™ and each ambiguity and indel was individually checked by eye in Tablet. The gene boundaries automatically detected by MITOS were checked individually in Sequencher™ to correct for misalignments. The location and size of the 13 PCGs were evaluated by comparing the location of start and stop codons with that of published mitochondrial genomes (the tellinid Mo. iridescens: GenBank accession number JN398362.1 [26] and the venerid Meretrix lusoria: GQ903339.1 [25]). Congruence among the PCG hydrophobic profiles of these species was assessed using Unipro UGENE v1.12.0 [34]. The map of mitogenome of Ma. balthica was produced using GenomeVx [35].
tRNA and ATP8 protein structure characterization
tRNA secondary structures were inferred using MITOS in default search mode [33]. GC content was calculated with UGENE. Predictions of transmembrane alpha-helices and hydrophilic helices for the ATP8 protein were inferred with TMHMM v.2.0 [36] and were compared with previously characterized ATP8 proteins from other bivalves. ATP8 amino-acid sequences were aligned with MUSCLE v.3.8.31 [37] with manual adjustments and a graphical representation was prepared using Texshade [38].
Genetic divergence and phylogenetic analysis
Genetic divergence among four pairs of haplotypes of Ma. balthica individuals was analyzed using the ape v.3.0-8 [39] and seqinr v.3.0-7 [40] R packages [41]. The TN93 [42] model of nucleotide substitution was determined as the most likely for our dataset based on the BIC scores calculated in jModelTest 2 [43]. TN93 genetic distances within and between specimens with different cox1 haplotypes were calculated within a 200 bp window sliding every 10 bp.
Nucleotide diversity for synonymous (πS) and non-synonymous (πA) substitutions was calculated across all PCGs for all mitogenomes (n = 6) and Ma. balthica rubra mitogenomes (n = 5), using a 200 bp window sliding every 10 bp (DnaSP v.5.10.01 [44]). Haplotype networks for each of the 13 PCGs were built using the ape and pegas v.0.4-2 [45] R packages. Finally, the secondary structure of PCGs was inferred with Protter v.1.0 [46]. Fisher’s exact tests (as implemented in R) were computed on each PCG to test for the random distribution of (i) all mutations and (ii) non-synonymous mutations across mitochondrial compartments (extra-, inter- and intra-membrane).
Results
Mitochondrial genome assembly and annotation
One Illumina MiSeq run produced 22.4 M raw reads, representing 3.02 Gbp after quality filtering (i.e. 4.78% reads deleted). GC content varied between 36 and 40% (average: 37%). The second round of genome assemblies yielded a median contig size (N50) of 6,893 bp and a maximum contig size averaging 7,449 bp.
Mitochondrial genome organization, PCGs & codon usage
Six nearly complete mitochondrial genomes from five mitochondrial lineages (as represented by the cox1 haplotypes of Becquet et al. [21]) were obtained. The mitogenome of individual A10 from Aytré is considered as our reference genome hereon. The size of the mitogenome is estimated at 16,806 bp (A6, M12, W4: 16,805; W20: 16,807 and F17: 16,794 bp). It is composed of 37 genes, including 13 PCGs among which an atp8 gene, two ribosomal RNA genes and 22 transfer RNA genes (Table 2 and Figure 1). All genes are encoded on the same strand, and gene arrangement is identical among the six mitogenomes sequenced.
Table 2.
Main features of the mitogenome of Ma. balthica
| Gene | Position | Size | Intergenic nucleotides 1 | Codon 2 | |||
|---|---|---|---|---|---|---|---|
| From | To | Nucleotides | Amino acids | Start | Stop | ||
| tRNA-Met | 1 | 65 | 65 | 9 | |||
| atp8 | 75 | 203 | 129 | 42 | 3 | ATT | TAA |
| tRNA-Ser1 | 207 | 275 | 69 | 10 | |||
| nad6 | 286 | 819 | 534 | 177 | −32 | ATT | TAA |
| rrnL | 788 | 2148 | 1361 | 25 | |||
| atp6 | 2174 | 2884 | 711 | 236 | 121 | ATG (GTG - F17) | TAA |
| cox3 | 3006 | 3818 | 813 | 270 | 60 | GTG | TAA |
| nad2 | 3879 | 4889 | 1011 | 336 | 0 | ATA | TAA |
| tRNA-Pro | 4890 | 4955 | 66 | 13 | |||
| tRNA-Gln | 4969 | 5034 | 66 | −1 | |||
| tRNA-Cys | 5034 | 5094 | 61 | 1 | |||
| tRNA-Ala | 5096 | 5161 | 66 | 0 | |||
| tRNA-Phe | 5162 | 5227 | 66 | 46 | |||
| cox1 | 5274 | 6944 | 1671 | 556 | 0 | ATA | TAA |
| nad4 | 6945 | 8285 | 1341 | 446 | 6 | ATA | TAG |
| tRNA-His | 8292 | 8356 | 65 | 1 | |||
| tRNA-Ser2 | 8358 | 8422 | 65 | 1 | |||
| tRNA-Glu | 8424 | 8490 | 67 | 0 | |||
| nad3 | 8491 | 8853 | 363 | 120 | 13 | TTG | TAG (TAA - W20) |
| tRNA-Ile | 8867 | 8935 | 69 | 2 | |||
| tRNA-Lys | 8938 | 9002 | 65 | 0 | |||
| nad4l | 9003 | 9293 | 291 | 96 | 5 | GTG | TAA |
| tRNA-Tyr | 9299 | 9360 | 62 | 0 | |||
| tRNA-Thr | 9361 | 9424 | 64 | 0 | |||
| tRNA-Leu1 | 9425 | 9489 | 65 | 0 | |||
| tRNA-Asp | 9490 | 9553 | 64 | 1 | |||
| tRNA-Leu2 | 9555 | 9620 | 66 | 1 | |||
| nad1 | 9622 | 10545 | 924 | 307 | 1 | ATG | TAG |
| tRNA-Asn | 10547 | 10611 | 65 | 0 | |||
| nad5 | 10612 | 12405 | 1794 | 597 | −50 | TTG | TAA |
| tRNA-Arg | 12356 | 12420 | 65 | 1 | |||
| cob | 12422 | 13660 | 1239 | 412 | 1 | ATT | TAA |
| cox2 | 13662 | 14516 | 855 | 284 | 3 | ATG | TAA |
| tRNA-Val | 14520 | 14586 | 67 | 0 | |||
| tRNA-Trp | 14587 | 14652 | 66 | 5 | |||
| tRNA-Gly | 14658 | 14723 | 66 | 3 | |||
| rrnS | 14727 | 15606 | 880 | 1200* | |||
Main features of the mitogenome of Ma. balthica, based on specimen A10.
1Nucleotide positions are indicated by the numbers separating the different mitochondrial genes. Overlapping nucleotides between adjacent genes are noticed by negative numbers.
2 atp6 and nad3 genes presented specific codon for one mitogenome. Codon is indicated in brackets following by mtDNA reference.
*See text.
Figure 1.
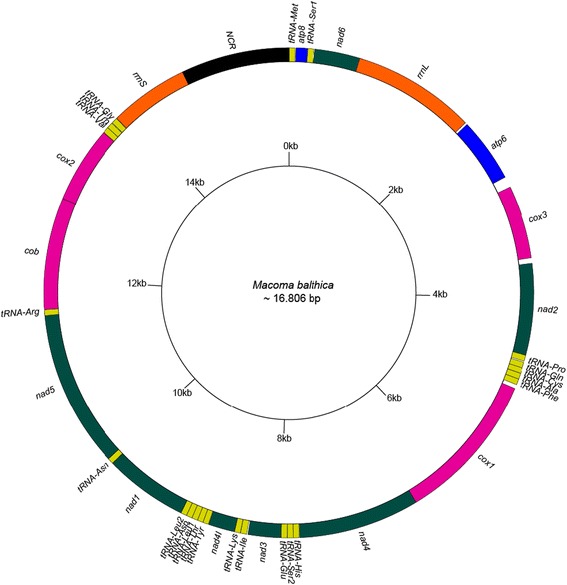
Map of the mitochondrial genome of Ma. balthica , based on specimen A10. NCR indicates the largest non-coding region.
In total, PCGs encode 3,879 amino acids (aa). With a size of 1,794 bp (597 aa), nad5 is the largest PCGs and cox1, nad4, cob and nad2 genes exceed 1,000 bp. Conversely, nad3 and nad4l genes are smaller than 400 bp lengths and atp8 gene is the smallest gene with 129 bp (42 aa).
Five different initiation codons were identified. Three PCGs begin with ATT (atp8, nad6, cob), three with ATG (atp6, nad1, cox2) and three with ATA (nad2, cox1, nad4). The last one is classically found in the invertebrate mitochondrial genetic code, particularly in bivalves. Four nad genes start with NTG codons, corresponding to ATG (nad1), TTG (nad3 and nad5) and GTG (nad4l). By contrast, PCGs are most often terminated by TAA (10 PCGs) rather than TAG (3 PCGs: nad4, nad3, nad1). These stop codons correspond to the only two possible combinations in the current invertebrate mitochondrial genetic code.
Transfer and ribosomal RNA genes
The mitogenome of Ma. balthica is composed of 22 tRNA genes, which range in size from 61 to 69 bp. All of them fold into cloverleaf secondary structures with four arms, some of them presenting folding differences (Additional file 3). Four tRNA (tRNAPro, tRNACys, tRNALeu2 and tRNAIle) have a small supplemental stem loop, tRNAAsn has not terminal TΨC loop, tRNASer1 has the dihydrouracil (DHU) stem replaced by a big DHU loop and three discriminator nucleotides were detected instead of the single nucleotide usually presents at the 5' end. Finally, tRNAAla, tRNALeu1 and tRNAVal do not possess a discriminator nucleotide.
rrnS and rrnL have respective lengths of 880 bp and 1,361 bp, and an AT content of 65.1% and 64.2%. Among specimens, average divergence is low (rrnS: 0 to 3%, rrnL: 0 to 5%). Overall, the same tRNA and rRNA features characterize all six mitogenomes of Ma. balthica.
Non-coding regions: intergenic spacers and putative CR
Intergenic nucleotides represent 1,532 bp in total (i.e. 9.12% of the whole mitogenome), and are divided into 24 non-coding regions (NCRs, Table 2). The largest NCR is about 1,200 bp long (i.e. 7.14% of the whole mitogenome), is located between rrnS and tRNAMet and is thought to contain the Control Region (CR). The putative CR could not be fully sequenced (either with MiSeq or Sanger sequencing, despite multiple attempts) due to its high AT content (estimated ~ 70%) and numerous tandem repeat motifs, but its size was estimated based on PCR products visualized by gel electrophoresis in strongly denaturing conditions. The other 23 NCRs ranged from 1 to 121 bp. Three overlapping gene regions were detected (nad6 – rrnL: −32 bp, tRNAGln – tRNACys: −1 bp and nad5 – tRNAArg: −50 bp).
Characterisation of a putative atp8 gene
Local tblastx searches detected the full set of 13 mitochondrial PCGs characterized here among the 454 transcriptome sequences available for Ma. balthica [24], including an atp8, a gene long reported as rare in bivalves. This is the first record of an atp8 gene in the Tellinidae. Up to now, this gene had not been highlighted in Mo. iridescens [26]. However, using the same data analysis approach as Ma. balthica, an atp8 gene was also detected between tRNAMet and tRNASer1 in publicly available Mo. iridescens sequences. Indeed, out of 84 mitogenomes of bivalves freely available on GenBank database, atp8 gene had been detected in only 42 species.
In Ma. balthica, this short gene was detected between tRNAMet and tRNASer1 and is separated from atp6 by 1,970 bp. It encodes a 42 aa protein (Table 2) terminated by a complete stop codon (TAA). Conversely to other bivalves (Additional file 4), but as in Mo. iridescens, ATP8 protein starts with an isoleucine rather than a methionine. Despite an important length polymorphism in bivalve ATP8 (range of 33 to 109 aa length), 5/42 aa in Ma. balthica are shared among all investigated bivalves with an atp8 (considering aa with > 75% conservation) and within the Tellinidae, Mo. iridescens and Ma. balthica have a 84% aa identity.
Secondary structure is highly conserved. Indeed, Ma. balthica ATP8 hydrophobicity patterns are congruent with the typical transmembraneous protein pattern for this gene in bivalves (Figure 2 and Additional file 4). It is composed of a central hydrophobic part corresponding to a transmembraneous helix (aa 7 to 29) and two hydrophilic N-terminus (inter-membraneous space, aa 1 to 6) and C-terminus (matrix space, aa 30 to 42) extremities. These two domains are positively charged whereas the central part is encoded by negatively charged aa. TMHMM transmembraneous helix prediction of the ATP8 protein from Ma. balthica are congruent with these observations.
Figure 2.
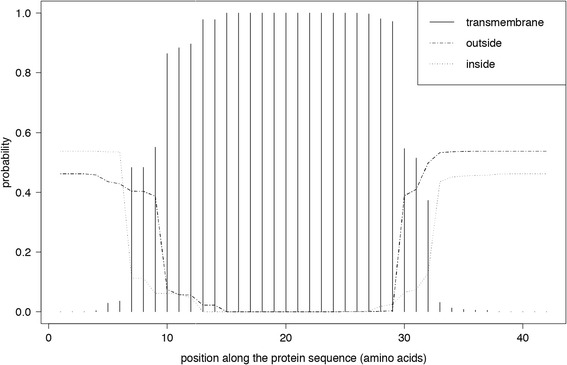
Prediction of transmembrane helices in the atp8 gene of Ma. balthica .
PCGs: sequence divergence and polymorphism
PCG-specific haplotype network topologies (Additional file 5) differ in resolution (built based on data from six specimens, number of haplotypes detected varied from three to six) but present nearly identical evolutionary relationships. The Ma. balthica balthica (F17) and Ma. balthica rubra (A6, A10, M12, W4 and W20) lineages are the most divergent; within the Ma. balthica rubra clade (denoted "intra-rubra" hereafter), W20 is separated from W4 and other, more southern individuals.
Some regions (rrnL, five tRNAs between nad2 and cox1, rrnS) are highly conserved and mutations are not evenly distributed along the 13 PCGs (Figure 3). The TN93 distance among the six mitogenomes ranged from 0.04 to 6.27% (median = 1.66, mean = 2.67) contrasting with 0.04 to 1.74% within intra-rubra (i.e. median = 0.7, mean = 0.92). The seven nad genes are highly mutated in comparison to the four cytochrome genes and two atp genes (Figure 3a). A10-F17 is the most divergent specimen pair (peak at 0.14 substitution/site at nad6 and nad5; and 0.125 substitution/site at nad2) compared to other specimen pairs (max. peak at 0.05 substitution/site in nad5 for the A10-W20 pair).
Figure 3.
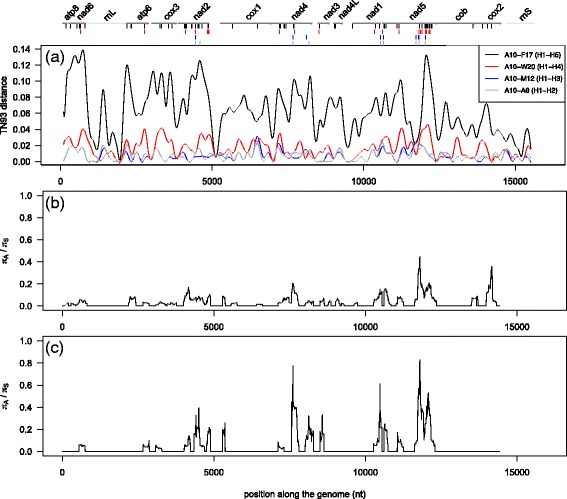
Nucleotide divergence among specimens along the mitogenome of Ma. balthica . Top: boundaries of protein-coding genes (black) and ribosomal genes (grey). Haplotypes H1-H5 represent cox1 haplotypes (see Methods). Vertical bars represent the position of non-synonymous mutations for the four cox1 haplotype comparisons, (a) divergence as measured by the Tamura and Nei [42] model of nucleotide substitution (TN93; substitution/site) and πA/πS ratio for (b) all six mtDNA and (c) the Ma. balthica rubra lineage (n = 5 mtDNA).
The ratio of intraspecific nucleotide diversity at non-synonymous (πA) and synonymous (πS) sites is lower when including both subspecies (Figure 3b) than within the intra-rubra group (Figure 3c). Overall, the nad gene group exhibits higher values of πA/πS than the atp and cytochrome genes (Figures 3b,c and Table 3). Among Ma. balthica lineages, πA/πS was most elevated in nad2, nad4, nad1, nad5 and unexpectedly in cox2 (Figure 3b; Table 3). The same pattern (except for cox2) was observed in the intra-rubra group (Figure 3c). Interestingly πA/πS ratios for these four nad genes are slightly higher in the intra-rubra group than among Ma. balthica lineages (Table 3). Minimal πA/πS ratio values were obtained in atp6, 8, cox1, 3 and cob genes (Table 3).
Table 3.
Distribution of amino-acid (aa) changes on the 13 protein coding genes (PCGs) of Macoma balthica
| Lineage comparison level | PCG | Gene length (aa) | Number of aa change (% of total length) | Extramembraneous compartment | Intermembraneous compartment | Intramembraneous compartment | Nucleotide divergence 1 | πA/πS ratio | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Length (aa) | Number of aa change | Length (aa) | Number of aa change | Length (aa) | Number of aa change | ||||||
| Among M. balthica (n = 6) | atp8 | 42 | 8 (19.1) | 5 | 0 | 26 | 5 | 11 | 3 | 0 - 8.74 (3.35 ± 1.64) | 0.03 |
| nad6 | 177 | 51 (28.8) | 37 | 10 | 109 | 32 | 31 | 9 | 0 - 13.76 (5.28 ± 2.25) | 0.027 | |
| atp6 | 236 | 38 (16.1) | 27 | 3 | 120 | 21 | 89 | 14 | 0 - 7.33 (2.98 ± 1.24) | 0.031 | |
| cox3 | 270 | 54 (20) | 87 | 23 | 141 | 24 | 42 | 7 | 0.12 - 9.21 (3.97 ± 1.58) | 0.017 | |
| nad2 | 336 | 66 (19.6) | 34 | 12 | 248 | 46 | 54 | 8 | 0.2 - 10.09 (3.96 ± 1.71) | 0.065 | |
| cox1 | 556 | 83 (14.9) | 108 | 14 | 286 | 43 | 162 | 26 | 0.06 - 6.71 (2.89 ± 1.13) | 0.004 | |
| nad4 | 446 | 75 (16.8) | 125 | 15 | 268 | 53 | 53 | 7 | 0 - 8.03 (3.21 ± 1.33) | 0.039 | |
| nad3 | 120 | 18 (15) | 8 | 2 | 72 | 9 | 40 | 7 | 0 - 7.26 (2.6 ± 1.15) | 0.027 | |
| nad4l | 96 | 18 (18.8) | 34 | 7 | 56 | 10 | 6 | 1 | 0 - 8.38 (3.29 ± 1.43) | 0.033 | |
| nad1 | 307 | 43 (14) | 46 | 8 | 203 | 29 | 58 | 6 | 0 - 6.24 (2.52 ± 1.09) | 0.035 | |
| nad5 | 597 | 78 (13.1) | 114 | 16 | 361 | 52 | 122 | 10 | 0.06 - 7.32 (3.31 ± 1.2) | 0.073 | |
| cob | 412 | 45 (10.9) | 72 | 8 | 182 | 20 | 158 | 17 | 0 - 4.83 (1.99 ± 0.81) | 0.008 | |
| cox2 | 284 | 32 (11.3) | 19 | 1 | 48 | 7 | 217 | 24 | 0 - 4.56 (1.86 ± 0.8) | 0.043 | |
| Among M. balthica rubra (n = 5) | atp8 | 42 | 2 (4.8) | 5 | 0 | 26 | 1 | 11 | 1 | 0 - 1.63 (0.65 ± 0.34) | 0 |
| nad6 | 177 | 17 (9.6) | 37 | 2 | 109 | 12 | 31 | 3 | 0 - 2.9 (1.58 ± 0.5) | 0.017 | |
| atp6 | 236 | 15 (6.4) | 27 | 1 | 120 | 13 | 89 | 1 | 0 - 2.04 (0.95 ± 0.29) | 0.02 | |
| cox3 | 270 | 24 (8.9) | 87 | 7 | 141 | 13 | 42 | 4 | 0.12 - 3.07 (1.42 ± 0.53) | 0.011 | |
| nad2 | 336 | 27 (8) | 34 | 2 | 248 | 21 | 54 | 4 | 0.2 - 2.37 (1.14 ± 0.4) | 0.084 | |
| cox1 | 556 | 38 (6.8) | 108 | 4 | 286 | 25 | 162 | 9 | 0.06 - 1.97 (1.05 ± 0.3) | 0 | |
| nad4 | 446 | 29 (6.5) | 125 | 7 | 268 | 18 | 53 | 4 | 0 - 1.75 (1.02 ± 0.25) | 0.064 | |
| nad3 | 120 | 6 (5) | 8 | 0 | 72 | 5 | 40 | 1 | 0 - 1.7 (0.73 ± 0.29) | 0.073 | |
| nad4l | 96 | 6 (6.3) | 34 | 3 | 56 | 3 | 6 | 0 | 0 - 1.4 (0.91 ± 0.2) | 0 | |
| nad1 | 307 | 15 (4.9) | 46 | 1 | 203 | 10 | 58 | 4 | 0 - 1.43 (0.72 ± 0.26) | 0.057 | |
| nad5 | 597 | 50 (8.4) | 114 | 11 | 361 | 30 | 122 | 9 | 0.06 - 2.64 (1.4 ± 0.45) | 0.118 | |
| cob | 412 | 17 (4.1) | 72 | 4 | 182 | 10 | 158 | 3 | 0 - 1.39 (0.67 ± 0.22) | 0 | |
| cox2 | 284 | 11 (3.9) | 19 | 4 | 48 | 1 | 217 | 6 | 0 - 1.07 (0.54 ± 0.17) | 0 | |
1global divergence: min - max range (mean ± standard-error) calculated using the TN93 model of nucleotide substitutions, as for Figure 1.
The number of aa changes (i.e. synonymous and non-synonymous mutations) is given for each gene overall and for the extra-, inter- and intra-membranous organellar compartments, as delimited in Protter (Additional file 6). The distribution of aa changes across different organellar compartments provides a preliminary roadmap for searching for MNIs. The positions of aa sites bearing synonymous and non-synonymous mutations are mapped on the inferred protein secondary structures presented in Additional file 6.
Synonymous and non-synonymous mutations were mapped onto the PCG secondary structured inferred by Protter in order to evaluate the potential for MNIs (Additional file 6). While the sliding window analysis was run using pairs of individuals, the Protter analyses were run jointly on all samples. Mutation mapping among Ma. balthica lineages reveals that nad6, cox3, nad2, atp8 and nad4l are the most mutated PCGs (number of aa changes ≥ 18.5% of total protein length), whereas only 11% of cob and cox2 are mutated (Table 3). Other PCGs possess intermediate divergence levels, which varied from 13 to 16%. Within intra-rubra group, the most mutated genes are nad6, cox3, nad5 and nad3 (number of aa changes ≥ 8% of total protein length), and atp8 was one of the most conserved genes, with cox2 and cob (number of aa changes ≤ 5% of total protein length). On average nad genes have more aa bearing bp mutations than any other genes (5/7 nad genes have ≥ 15% of mutated sites among Ma. balthica and ≥ 6% within intra-rubra group; Table 3). The number of non-synonymous mutations was twice higher among Ma. balthica lineages than within the intra-rubra group (57 vs 30 non-synonymous mutations), with a maximum occurrence observed within the nad2 and nad5 genes (total of 16 and 24 respectively, Table 3). By contrast to nad genes, atp6 and atp8 genes present few non-synonymous mutations (total of 1 and 4 respectively, Table 3). Finally, the type and number of mutations were not influenced by the location of the mutation relative to the mitochondrial membrane (extra, inter or intramembranous positions; Fisher exact test, P > 0.05).
Discussion
General mitogenome features
Studies of metazoan mtDNA have revealed a large degree of variation in gene order that can be explained by multiple rearrangement events as inversions, transpositions, inverse transpositions (for details see [47]). In bivalves, on average, the number of mtDNA PCGs is 37 ± 3 [48], and genome architecture varies depending on the subclass. Members of Paleoheterodonta present the same gene order whereas Pteriomorpha and most importantly Heterodonta (a sub-class including Macoma) are widely rearranged (for a review see [26,49-51]).
The mitogenome of Ma. balthica (16.8 kbp) is among the smallest within the heterodonts (mean of 19 kbp, NCBI Organelle Genome Resources) ranging between Acanthocardia tuberculata (the smallest heterodont mitogenome with 16,104 bp [52]) and Venerupis philippinarum (the largest heterodont mitogenome with 22,676 bp [53]). The mitogenomes of Ma. balthica and Mo. iridescens [26] are similar in PCG length but present some differences in their start and stop codons. Genomic architecture is generally highly conserved within mollusc species. For example, the mitogenomes of two individuals of the clam Meretrix lamarckii have the same number and order of genes, slightly differed in length, and have identical amino-acid sequences [54]. Similarly, total genome length varied by < 1% among the six mitogenomes sequenced here, and genome organisation was identical.
atp8, a dispensable gene in bivalves?
In metazoans, the number of genes is highly conserved (n = 37) and deviations from this number are rare. Among PCGs, the ATP synthase complex can be encoded by a maximum of three different genes corresponding to mitochondrial subunits 6, 8 and 9 (this latter subunit being present in most sponges [48]). However, in many bivalves, especially heterondonts, atp8 is lacking, as in the Mactridae (NC_023384 and [50]), Arcticidae (NC_022709), Solenidae [55,56], Semelidae, Solecurtidae and Psammobiidae [26,57]. The first bivalve atp8 was detected in [52] and was later found in the Cardiidae [49], Lucinidae (YP_003208152, YP_003208299) and Veneridae ([25,54,58,59] and YP_008854384). Here, the atp8 gene is reported for the first time in the Tellinidae. Strongly divergent features of atp8 among bivalves (lack of the MP** amino acid signature at the N-terminus, size length) could have previously hampered the annotation of this gene in the Tellinidae [60]. In other terms, this gene may evolve so fast in bivalves that it may be missed by homology analyses [60]. In mussels, Śmietanka et al. [61] highlighted that the number of nucleotide substitutions in PCGs is in average three times faster in the male mitochondrial lineage than in the female lineage, and varied differentially between respiratory complexes (from 2.6x for the first and fifth complexes to 8.5x for the third complex [61]). Strong divergence and fast evolution of this gene could also explain why it was not originally detected in Mo. iridescens.
Overall, gene length is highly variable among phyla (see [62]), raising the question of its role and dispensability in the functionality of the ATP synthase complex [48,63,64]. To date, the role of atp8 stays unclear and little documented in metazoans. The N- and C-terminal domains of ATP8 seem to be involved in ATP synthase activity, particularly assembly and function of the FO rotor [62] and was reported as an essential module in the stator stalk of yeast mtATPase [63,64]. Stephens et al. [64] have demonstrated that the N- and C-terminal parts of yeast ATP8 were located in the intermembranous space and the mitochondrial matrix, respectively, while the rest of the protein was a transmembraneous, central hydrophobic domain. Our results in Ma. balthica are congruent with these observations, supporting the potential functionality of the ATP8 protein.
Some reviews [25,52] have attempted to make the link between ATP8 functionality and its genomic position relative to ATP6. Indeed, as mentioned by Boore [47], in mammals these two PCGs are "translated from a bicistronic transcript, with translation initiating alternatively at the 5′ end of the mRNA for atp8 or at an internal start codon for atp6." Usually, these two genes are adjoined on the same strand, with overlapping reading frame [47]. Nonetheless, they can also be physically separated while both retaining functionality. Alternatively, atp8 may be disjoint from atp6 and non functional (for review see [62]). In Ma. balthica, atp6 and atp8 are separated by 1,970 bp but seemed to lead to functional proteins (at the least, both genes are expressed, as they were detected in our transcriptome database and presented the same PCG profile as other bivalves). This arrangement could be an "evolutionary stepping stone from the fully functional atp6-atp8 coupling, via decoupled but complete genes" [52].
Yeast ATP8 is thought to interact with ATP synthase subunits b, f, 6 and is also able to cross-link with stator subunit d [64]. Moreover, assembly of subunits 8 and 6 into ATP synthase requires direct interactions between them [64]. Yeast FO-F1-ATP synthase requires five indispensable subunits (nuclear: b, d, f and mitochondrial: 6, 8) for stator stalk stabilization and loss of any one of these subunits results in a deep functional uncoupling between the F1 stator and the FO rotor ([64] and references therein). This gene, if functional, may therefore coevolve with other ATP synthase subunits, and be involved in MNIs. Indeed, the sixth aa of the C-terminal domain of ATP8 is different in the Ma. balthica balthica and Ma. balthica rubra lineages, and multiple non-synonymous mutations were detected in the inter-membranous loops of ATP6. Furthermore, substitution of only one aa is sufficient to change protein stability [65]. This adaptive change could induce conformational modifications and impacted functionality role of PCGs.
PCGs: polymorphism, divergence and selective pressure
A previous FST scan based on nuclear transcriptome data, between populations of Ma. balthica spanning the same two secondary contact zones as considered here, detected strong population structure at several genes involved in the ATP synthase and NADH deshydrogenase complexes [24]. These complexes both rely on the coevolution of multiple nuclear- and mitochondrial-encoded protein subunits, and could therefore suffer from MNIs. Nuclear and mitochondrial data converge toward positive selection pressures on the NADH dehydrogenase (Complex I of the OXPHO chain). The nad gene group is characterized by high polymorphism levels and high relative prevalence of replacement mutations compared to other PCGs. In particular, the nad2 and nad5 genes were rich in replacement mutations. Comparing the mitogenomes of the European and American eels (Anguilla anguilla and A. rostrata), Jacobsen et al. [16] found some evidence for positive selection on atp6 (corroborating the results from [15]) and nad5. Inter-populational comparisons in the copepod Tigriopus californicus revealed that nad3 was also among the most divergent PCGs, and displayed dN/dS ratios that were twice as elevated as other PCGs [3]. Similarly, Gibson et al. [13] have recently shown that hybrid breakdown in Nasonia parasitoid wasps is likely partly due to incompatibilities between nuclear- and mitochondrial-encoded OXPHO genes of Complex I. They identified the nuclear gene Ndufa11, coding for a subunit of the NADH dehydrogenase complex as strongly affected by transmission ratio distortion and marked by a prevalence of replacement mutations far greater than what was measured for other nuclear OXPHO genes [13,14]. Complex I ensures essential functions in the OXPHO chain, as it initiates electron transport system (ETS) and interacts with the OXPHO complexes II and III. Differential evolution of the mitochondrial- and nuclear-encoded NADH dehydrogenase subunits may therefore lead to coadaptation breakdown and possibly to MNIs. Further, because of its key role in the OXPHO chain, dysfunction of Complex I may severely affect all processes downstream of the OXPHO chain, and, ultimately, ATP production.
The mitochondrial atp6 and atp8 genes were not detected as outlier mitochondrial genes among the six individuals sequenced based on the number of non-synonymous mutations and πA/πS ratios. This result contrasted with previous data, in which several nuclear-encoded ATP synthase subunits were highlighted as potentially being under positive selection [24]. Several factors could explain this result. First, the very small sample size used here (1–2 individuals sequenced per mitochondrial lineage) may have prevented us from detecting significant adaptive polymorphism. Indeed, the transcriptomic data suggested an asymmetric gene flow at the nuclear locus coding for the gamma subunit of ATP synthase, with a northern allele being apparently fixed in populations north of Brittany, and present in about 25% of reads in the population south of Brittany ([24] and unpublished data). It is therefore possible that the individuals selected here were not representative of the genetic diversity at mitochondrial atp genes. Second, a few adaptive mutations can have significant fitness impacts (e.g. Cytochrome c in Tigriopus californicus [66] and cMDH in Lottia [65]), and the relatively low prevalence of replacement mutations relative to neutral mutations may therefore not be indicative of the effect of protein evolution on fitness. Finally, genetic incompatibilities between ATP synthase subunits may arise solely between nuclear-encoded proteins.
We hope that future studies, focusing on experimental crosses and sequencing of additional ATP synthase subunits will help us determine whether these genes suffer from MNIs. Looking at the proportion of fixed and polymorphic mutations within and between Ma. balthica lineages using a larger sample size [67] will also help us investigate the adaptive value of protein divergence at OXPHO loci.
Conclusions
The participation of MNIs to barriers limiting gene flow in the marine environment remains poorly known. In this contribution we characterized the mitochondrial genome of Ma. balthica, a high gene flow marine bivalve, in an effort to test whether mitochondrial genes involved in epistatic interactions with previously-detected nuclear outlier loci show stronger nucleotide and protein divergence than average. Our results suggest that genes coding for subunits of the NADH dehydrogenase protein complex could be involved in genetic incompatibilities. Further investigations will focus on larger intra- and inter-lineage sampling for mitochondrial and nuclear genes of the OXPHO chain to more fully characterize MNIs. Also, experimental crossings Ma. balthica rubra individuals sampled on each side of the Brittany hybrid zone will help understand the functional effects of PCG divergence.
Availability of supporting data
Mitogenome DNA sequences of the six individuals have been submitted to GenBank (see Table 1 for Accession numbers) at www.ncbi.nlm.nih.gov. [68]. Raw Illumina reads were deposited on NCBI’s Sequence Read Archive (SRA; Project Accession Number SRP051152).
Acknowledgments
The authors thank the participants of the BIOCOMBE Project (contrat EVK3-2001-00146) for specimen collections, Olivier Bouchez and Emeline Lhuillier at the GeT-PlaGe lab, GenoToul (Toulouse) for their help with NGS data collection and analysis, the Molecular Core Facility at the University of La Rochelle, and Mikael Guichard, Marc-Henri Boisis-Delavaud and Frédéric Bret from the IT Center at the University of La Rochelle. The University of La Rochelle supercomputer “YMIR” was partly funded by the European Union (contract 31031–2008, European Regional Development Fund). We acknowledge the Axios Review editors Tim Vines and Michael Hellberg, and two anonymous reviewers for their constructive comments on an earlier version of the manuscript. This work was partly funded by the Agence Nationale de la Recherche (HySea project ANR-12-BSV7-0011) and by the European Union (contract 35722–2012, European Regional Development Fund). Salaries for AS and EP were covered by the Ministère de l'Education Nationale de la Recherche et de la Technologie and the European Regional Development Fund, respectively.
Additional files
Detail of the protocol used in LR-PCR.
List of primers used in LR-PCR.
Cloverleaf structures of the 22 tRNA genes in the reference mitogenome A10 of Ma. balthica. Nomenclature for portions of tRNA secondary structure is illustrated on tRNA Phe.
Complete alignment of all bivalve atp8 amino acid sequences available on GenBank (species – accession number of atp8 (aa length)). Accession numbers of atp8 sequences are not available for the two species of Tellinidae. For Ma. balthica and Mo. iridescens accession numbers of mitogenomes correspond to KM373200 and JN398362, respectively. Amino-acid hydrophobicity as described in [38] and references therein. Cytoplasmic side, transmembrane helix and mitochondrial matrix were defined following results for the subclass Heterodonta (transmembrane helix prediction in TMHMM).
Statistical parsimony haplotype networks for each PCG of Ma. balthica. The number of haplotypes present in each network is given in parentheses.
Mutation mapping for the 13 PCGs of Ma. balthica. A10 is taken as the reference and compared to other mitogenomes. Gray circles: synonymous changes in amino-acid (aa) among Ma. balthica lineages (i.e. all mitogenomes included); gray diamonds: synonymous changes in aa among Ma. balthica rubra lineages (i.e. excluding Ma. balthica balthica (F17)); black circles: non-synonymous changes in aa among Ma. balthica lineages; black diamonds: non-synonymous changes in aa among Ma. balthica rubra lineages.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AS, EP, VB and PG designed the study; AS, EP, VB, and NM conducted laboratory work; AS, EP and FE analyzed the data; AS and EP wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Alice Saunier, Email: saunier.alice@gmail.com.
Pascale Garcia, Email: pascale.garcia@univ-lr.fr.
Vanessa Becquet, Email: vanessa.becquet-buren@univ-lr.fr.
Nathalie Marsaud, Email: nmarsaud@insa-toulouse.fr.
Frédéric Escudié, Email: frederic.escudie@toulouse.inra.fr.
Eric Pante, Email: pante.eric@gmail.com.
References
- 1.Blier PU, Dufresne F, Burton RS. Natural selection and the evolution of mtDNA-encoded peptides: evidence for intergenomic co-adaptation. Trends Genet. 2001;17(7):400–406. doi: 10.1016/S0168-9525(01)02338-1. [DOI] [PubMed] [Google Scholar]
- 2.Rand DM, Haney RA, Fry AJ. Cytonuclear coevolution: the genomics of cooperation. Trends Ecol Evol. 2004;19(12):645–653. doi: 10.1016/j.tree.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Burton RS, Byrne RJ, Rawson PD. Three divergent mitochondrial genomes from California populations of the copepod Tigriopus californicus. Gene. 2007;403:53–59. doi: 10.1016/j.gene.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 4.Burton RS, Ellison CK, Harrison JS. The sorry state of F2 hybrids: Consequences of rapid mitochondrial DNA evolution in allopatric populations. Am Nat. 2006;168:S14–S24. doi: 10.1086/509046. [DOI] [PubMed] [Google Scholar]
- 5.Montooth KL, Meiklejohn CD, Abt DN, Rand DM. Mitochondrial-nuclear epistasis affects fitness within species but does not contribute to fixed incompatibilities between species of Drosophila. Evolution. 2010;64(12):3364–3379. doi: 10.1111/j.1558-5646.2010.01077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burton RS, Barreto FS. A disproportionate role for mtDNA in Dobzhansky–Muller incompatibilities? Mol Ecol. 2012;21(20):4942–4957. doi: 10.1111/mec.12006. [DOI] [PubMed] [Google Scholar]
- 7.Burton RS, Pereira RJ, Barreto FS. Cytonuclear genomic interactions and hybrid breakdown. Annu Rev Ecol Evol Syst. 2013;44(1):281–302. doi: 10.1146/annurev-ecolsys-110512-135758. [DOI] [Google Scholar]
- 8.Coyne J, Orr HA: Speciation. Inc. Sunderland, MA: Sinauer Associates; 2004.
- 9.Maheshwari S, Barbash DA. The genetics of hybrid incompatibilities. Annu Rev Genet. 2011;45(1):331–355. doi: 10.1146/annurev-genet-110410-132514. [DOI] [PubMed] [Google Scholar]
- 10.Dobzhanksy T. Studies on hybrid sterility II. Localization of sterility factors inDrosophila pseudoobscurahybrids. Genetics. 1936;21:113–135. doi: 10.1093/genetics/21.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller HJ. Isolating mechanisms, evolution, and temperature. Biol Symp. 1942;6:71–125. [Google Scholar]
- 12.Ellison CK, Niehuis O, Gadau J. Hybrid breakdown and mitochondrial dysfunction in hybrids of Nasonia parasitoid wasps. J Evol Biol. 2008;21(6):1844–1851. doi: 10.1111/j.1420-9101.2008.01608.x. [DOI] [PubMed] [Google Scholar]
- 13.Gibson JD, Niehuis O, Peirson BRE, Cash EI, Gadau J. Genetic and developmental basis of F2 hybrid breakdown in Nasonia parasitoid wasps. Evolution. 2013;67(7):2124–2132. doi: 10.1111/evo.12080. [DOI] [PubMed] [Google Scholar]
- 14.Gibson JD, Niehuis O, Verrelli BC, Gadau J. Contrasting patterns of selective constraints in nuclear-encoded genes of the oxidative phosphorylation pathway in holometabolous insects and their possible role in hybrid breakdown in Nasonia. Heredity. 2010;104:310–317. doi: 10.1038/hdy.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gagnaire PA, Normandeau E, Bernatchez L. Comparative genomics reveals adaptive protein evolution and a possible cytonuclear incompatibility between European and American eels. Mol Biol Evol. 2012;29(10):2909–2919. doi: 10.1093/molbev/mss076. [DOI] [PubMed] [Google Scholar]
- 16.Jacobsen MW, Pujolar JM, Gilbert MTP, Moreno-Mayar JV, Bernatchez L, Als TD, Lobon-Cervia J, Hansen MM. Speciation and demographic history of Atlantic eels (Anguilla anguilla and A. rostrata) revealed by mitogenome sequencing. Heredity. 2014;113(5):432–442. doi: 10.1038/hdy.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barreto FS, Moy GW, Burton RS. Interpopulation patterns of divergence and selection across the transcriptome of the copepod Tigriopus californicus. Mol Ecol. 2011;20(3):560–572. doi: 10.1111/j.1365-294X.2010.04963.x. [DOI] [PubMed] [Google Scholar]
- 18.Ellison CK, Burton RS. Interpopulation hybrid breakdown maps to the mitochondrial genome. Evolution. 2008;62(3):631–638. doi: 10.1111/j.1558-5646.2007.00305.x. [DOI] [PubMed] [Google Scholar]
- 19.Palumbi SR. Genetic divergence, reproductive isolation, and marine speciation. Annu Rev Ecol Syst. 1994;25:547–572. doi: 10.1146/annurev.es.25.110194.002555. [DOI] [Google Scholar]
- 20.Maggs CA, Castilho R, Foltz D, Henzler C, Jolly MT, Kelly J, Olsen J, Perez KE, Stam W, Väinölä R, Viard F, Wares J. Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology. 2008;89(11):S108–S122. doi: 10.1890/08-0257.1. [DOI] [PubMed] [Google Scholar]
- 21.Becquet V, Simon-Bouhet B, Pante E, Hummel H, Garcia P. Glacial refugium versus range limit: Conservation genetics of Macoma balthica, a key species in the Bay of Biscay (France) J Exp Mar Bio Ecol. 2012;432–433:73–82. doi: 10.1016/j.jembe.2012.07.008. [DOI] [Google Scholar]
- 22.Nikula R, Strelkov P, Väinölä R. Diversity and trans-artic invasion history of mitochondrial lineages in the north atlantic Macoma balthica complex (Bivalvia: Tellinidae) Evolution. 2007;61(4):928–941. doi: 10.1111/j.1558-5646.2007.00066.x. [DOI] [PubMed] [Google Scholar]
- 23.Luttikhuizen PC, Drent J, Baker AJ. Disjunct distribution of highly diverged mitochondrial lineage clade and population subdivision in a marine bivalve with pelagic larval dispersal. Mol Ecol. 2003;12:2215–2229. doi: 10.1046/j.1365-294X.2003.01872.x. [DOI] [PubMed] [Google Scholar]
- 24.Pante E, Rohfritsch A, Becquet V, Belkhir K, Bierne N, Garcia P. SNP detection from De Novo transcriptome sequencing in the bivalve Macoma balthica: marker development for evolutionary studies. PLoS One. 2012;7(12):1–10. doi: 10.1371/journal.pone.0052302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Zhang S, Li Y, Liu B. Complete mtDNA of Meretrix lusoria (Bivalvia: Veneridae) reveals the presence of an atp8 gene, length variation and heteroplasmy in the control region. Comp Biochem Physiol Part D. 2010;5:256–264. doi: 10.1016/j.cbd.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Yuan Y, Li Q, Yu H, Kong L. The complete mitochondrial genomes of six heterodont bivalves (Tellinoidea and Solenoidea): variable gene arrangements and phylogenetic implications. PLoS One. 2012;7(2):1–9. doi: 10.1371/journal.pone.0032353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden T. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13(1):134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burger G, Lavrov DV, Forget L, Lang BF. Sequencing complete mitochondrial and plastid genomes. Nat Protoc. 2007;2(3):603–614. doi: 10.1038/nprot.2007.59. [DOI] [PubMed] [Google Scholar]
- 29.Andrews S: FastQC: a quality control tool for high throughput sequence data. Available athttp://www.bioinformatics.babraham.ac.uk/projects/fastqc/ 2010.
- 30.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milne I, Stephen G, Bayer M, Cock PJA, Pritchard L, Cardle L, Shawand PD, Marshall D. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform. 2013;14(2):193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7(1–2):203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 33.Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69(2):313–319. doi: 10.1016/j.ympev.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 34.Okonechnikov K, Golosova O, Fursov M, Team U. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012;28(8):1166–1167. doi: 10.1093/bioinformatics/bts091. [DOI] [PubMed] [Google Scholar]
- 35.Conant GC, Wolfe KH. GenomeVx: Simple web-based creation of editable circular chromosome maps. Bioinformatics. 2007;24(6):861–862. doi: 10.1093/bioinformatics/btm598. [DOI] [PubMed] [Google Scholar]
- 36.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 37.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beitz E. TeXshade: shading and labeling of multiple sequence alignments using LaTeX2e. Bioinformatics. 2000;16(2):135–139. doi: 10.1093/bioinformatics/16.2.135. [DOI] [PubMed] [Google Scholar]
- 39.Paradis E, Claude J, Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20(2):289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 40.Charif D, Lobry J. SeqinR 1.0-2: A Contributed Package to the R Project for Statistical Computing Devoted to Biological Sequences Retrieval and Analysis. In: Bastolla U, Porto M, Roman HE, Vendruscolo M, editors. Structural Approaches to Sequence Evolution. Berlin Heidelberg: Springer; 2007. pp. 207–232. [Google Scholar]
- 41.R Core Team: R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [http://www.R-project.org/]; 2014.
- 42.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 43.Darriba D, Taboada G, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772–772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 45.Paradis E, Schliep K, Potts A, Winter D. pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics. 2010;26:419–420. doi: 10.1093/bioinformatics/btp696. [DOI] [PubMed] [Google Scholar]
- 46.Omasits U, Ahrens CH, Müller S, Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014;30(6):884–886. doi: 10.1093/bioinformatics/btt607. [DOI] [PubMed] [Google Scholar]
- 47.Boore JL. Survey and summary: animal mitochondrial genomes. Nucleic Acid Res. 1999;27(8):1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gissi C, Iannelli F, Pesole C. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:301–320. doi: 10.1038/hdy.2008.62. [DOI] [PubMed] [Google Scholar]
- 49.Imanishi Y, Tanaka M, Fujiwara M. Complete mitochondrial genome sequence of Japanese cockle Fulvia mutica (Cardiidae) Fisheries Science. 2013;79(6):949–957. doi: 10.1007/s12562-013-0662-1. [DOI] [Google Scholar]
- 50.Meng X, Zhao N, Shen X, Hao J, Liang M, Zhu X, Cheng H, Yan B, Liu Z. Complete mitochondrial genome of Coelomactra antiquata (Mollusca: Bivalvia): The first representative from the family Mactridae with novel gene order and unusual tandem repeats. Comp Biochem Physiol Part D Genomics Proteomics. 2012;7(2):175–179. doi: 10.1016/j.cbd.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 51.Ren J, Shen X, Sun M, Jiang F, Yu Y, Chi Z, Liu B. The complete mitochondrial genome of the clam Meretrix petechialis (Mollusca: Bivalvia: Veneridae) Mitochondrial DNA. 2009;20(4):78–87. doi: 10.1080/19401730902964425. [DOI] [PubMed] [Google Scholar]
- 52.Dreyer H, Steiner G. The complete sequences and gene organisation of the mitochondrial genomes of the heterodont bivalves Acanthocardia tuberculata and Hiatella arctica – and the first record for a putative Atpase subunit 8 gene in marine bivalves. Front Zool. 2006;3(13):1–14. doi: 10.1186/1742-9994-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Passamonti M, Scali V. Gender-associated mitochondrial DNA heteroplasmy in the venerid clam Tapes philippinarum (Mollusca Bivalvia) Curr Genet. 2001;39(2):117–124. doi: 10.1007/s002940100188. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Zhang S, Xiao G, Liu B. Complete mtDNA of the Meretrix lamarckii (Bivalvia: Veneridae) and molecular identification of suspected M. lamarckii based on the whole mitochondrial genome. Marine Genomics. 2011;4:263–271. doi: 10.1016/j.margen.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 55.Yuan Y, Li Q, Kong L, Yu H. The complete mitochondrial genome of the grand jackknife clam, Solen grandis (Bivalvia: Solenidae): a novel gene order and unusual non-coding region. Mol Biol Rep. 2012;39:1287–1292. doi: 10.1007/s11033-011-0861-8. [DOI] [PubMed] [Google Scholar]
- 56.Yuan Y, Li Q, Kong L, Yu H. The complete mitochondrial genome of Solen strictus (Bivalvia: Solenidae) Mitochondrial DNA. 2012;23(2):112–114. doi: 10.3109/19401736.2011.653802. [DOI] [PubMed] [Google Scholar]
- 57.Zheng R, Li J, Niu D. The complete DNA sequence of the mitochondrial genome of Sinonovacula constricta (Bivalvia: Solecurtidae) Acta Oceanologica Sinica. 2010;29(2):88–92. doi: 10.1007/s13131-010-0026-y. [DOI] [Google Scholar]
- 58.Xu X, Wu X, Yu Z. Comparative studies of the complete mitochondrial genomes of four Paphia clams and reconsideration of subgenus Neotapes (Bivalvia: Veneridae) Gene. 2012;494:17–23. doi: 10.1016/j.gene.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 59.He C, Wang J, Gao X, Song W, Li H, Li Y, Liu W, Su H. The complete mitochondrial genome of the hard clam Meretrix meretrix. Mol Biol Rep. 2011;38:3401–3409. doi: 10.1007/s11033-010-0449-8. [DOI] [PubMed] [Google Scholar]
- 60.Breton S, Stewart DT, Hoeh WR. Characterization of a mitochondrial ORF from the gender-associated mtDNAs of Mytilus spp. (Bivalvia: Mytilidae): Identification of the "missing" ATPase 8 gene. Marine Genomics. 2010;3(1):11–18. doi: 10.1016/j.margen.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 61.Śmietanka B, Burzynski A, Wenne R. Comparative genomics of marine mussels (Mytilus spp.) gender associated mtDNA: rapidly evolving atp8. J Mol Evol. 2010;71(5–6):385–400. doi: 10.1007/s00239-010-9393-4. [DOI] [PubMed] [Google Scholar]
- 62.Gissi C, Iannelli F, Pesole G. Complete mtDNA of Ciona intestinalis reveals extensive gene rearrangement and the presence of an atp8 and an extra trnM gene in Ascidians. J Mol Evol. 2004;58(4):376–389. doi: 10.1007/s00239-003-2559-6. [DOI] [PubMed] [Google Scholar]
- 63.Papakonstantinou T, Law RHP, Nagley P, Devenish RJ. Non-functional variants of yeast mitochondrial ATP synthase subunit 8 that assemble into the complex. Biochem Mol Biol Int. 1996;39(2):253–260. doi: 10.1080/15216549600201261. [DOI] [PubMed] [Google Scholar]
- 64.Stephens AN, Khan MA, Roucou X, Nagley P, Devenish RJ. The molecular neighborhood of subunit 8 of yeast mitochondrial F1F0-ATP synthase probed by cysteine scanning mutagenesis and chemical modification. J Biol Chem. 2003;278(20):17867–17875. doi: 10.1074/jbc.M300967200. [DOI] [PubMed] [Google Scholar]
- 65.Dong Y, Somero GN. Temperature adaptation of cytosolic malate dehydrogenases of limpets (genus Lottia): differences in stability and function due to minor changes in sequence correlate with biogeographic and vertical distributions. J Exp Biol. 2009;212(2):169–177. doi: 10.1242/jeb.024505. [DOI] [PubMed] [Google Scholar]
- 66.Rawson PD, Burton RS. Functional coadaptation between cytochrome c and cytochrome c oxidase within allopatric populations of a marine copepod. Proc Natl Acad Sci. 2002;99(20):12955–12958. doi: 10.1073/pnas.202335899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 68.Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2013;41(D1):D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]