

Abstract
Polymorphisms attenuating IL-10 signaling confer genetic risk for inflammatory bowel disease. Yet how IL-10 prevents mucosal autoinflammation is incompletely understood. We demonstrate using lineage-specific deletions of IL-10Rα that IL-10 acts primarily through macrophages to limit colitis. Colitis depends on IL-6 to support pathologic Th17 cell generation in wild type mice. However, specific ablation of macrophage IL-10Rα provokes excessive IL-1β production that overrides Th17 IL-6 dependence, amplifying the colonic Th17 response and disease severity. IL-10 not only inhibits pro-IL-1β production transcriptionally in macrophages, but suppresses caspase-1 activation and caspase-1 dependent maturation of pro-IL-1β to IL-1β. Therefore lineage-specific effects of IL-10 skew the cytokine dependency of Th17 development required for colitis pathogenesis. Coordinated interventions may be needed to fully suppress Th17-mediated immunopathology.
Keywords: IL-10, IL-1β, macrophage, Th17, Inflammasome
Introduction
Inflammatory bowel diseases (IBD), including Crohn’s disease and ulcerative colitis, are characterized by chronic relapsing intestinal inflammation and disorganized immune responses in the gastrointestinal tract1. Although IBD pathogenesis is incompletely understood, mucosal T-helper type 17 (Th17) cells play a key pathologic role2–4. Th17 induction is supported by IL-1β, IL-6, IL-23 and TGF-β5–7, cytokines that upregulate and stabilize retinoic acid receptor-related organ receptor (RORγT) and pro-inflammatory function8.
The cytokine IL-10 preserves gastrointestinal homeostasis9. Mice deficient in IL-10 or IL-10Rβ develop spontaneous enterocolitis10, 11. Bi-allelic mutations in IL-10 lead to infantile enterocolitis, polymorphisms in IL-10 are associated with ulcerative colitis and Crohn’s disease, and an association has been further reported between IL-10Rα SNPs and early onset UC12–15.
Dynamic interactions between IL-10 and different IL-10 responsive immune cell lineages participate in IBD pathogenesis16. Mice with a deletion of IL-10 or its receptor solely in Foxp3+ T cells can develop spontaneous colitis17, 18. The IL-10R-deficient Foxp3+ T cells display decreased IL-10 secretion itself, potentially linking these phenotypes. Further, two recent studies have implicated the IL-10 response by myeloid populations to mucosal homeostasis and colitis susceptibility. Zigmond, et. al. used a CX3CR1 promoter-directed Cre to selectively delete IL-10Rα. These mice developed spontaneous colitis, and implicated IL-10 in the generation of anti-inflammatory CX3CR1+ myeloid cells necessary for colonic homeostasis19. Shouval, et. al. demonstrated that IL-10Rβ−/− Rag2−/− mice are susceptible to colitis mediated by total CD4 T cell transfer. The IL-10Rβ deficiency impedes the generation and activity of anti-inflammatory macrophages, and impairs iTreg generation and Treg function20. IL-10-producing myeloid cells have also been shown to prevent the downregulation of Foxp3 in T cell transfer colitis, indicating a central role for IL-10 in the crosstalk between regulatory T cell and myeloid populations21. Implicating IL-10 in T cell effector function as well, T cell-specific blockade of IL-10 signaling using a dominant negative IL-10 receptor (IL-10RDN) leads to increased Th17 cells in an anti-CD3 Ab induced model of small intestinal inflammation22.
Despite the clear role of IL-10 in intestinal homeostasis, pharmacologic administration of IL-10 has not proven effective23. Further resolution of the core interactions, responsible cell types, and pertinent mechanisms underlying IL-10’s activity will be necessary to develop effective interventions targeting this pathway. Here we use the transfer of naive CD4+CD45RBhi T cells into lymphocyte-deficient strains to analyze how the lineage specific activities of IL-10 impact the pathologic T cell response.
Transferred naïve T cells are activated by microbial flora, provoking Th17-dependent colonic inflammation. Studies using IL-10RDN T cells have indicated that the T cell IL-10 response does not significantly restrain colitis development after CD45RBhi T cell transfer24. We verify this here using T cells conditionally deficient in the specific receptor for IL-10, IL-10Rα. However, we further show using additional targeted deletion models that IL-10 acts dominantly on macrophages (Mϕs) to mediate its inhibitory effects, and define how this occurs through a shift in the cytokine dependency of pathologic effector T cells. We show that IL-10 acts on Mϕs to potently suppress IL-1β production through several routes, including the inhibition pro-IL-1β production, caspase-1 activation, and IL-1β maturation. This not only modulates colitis, but also transforms the pathologic Th17 response from one that is IL-6-independent and IL-1-dependent to one with an obligate requirement for IL-6. Our findings demonstrate a redundancy in operative Th17 induction pathways during colitis, the critical role of the Mϕ response to IL-10 in controlling these pathways, and imply that coordinated therapies targeting redundant cytokines may be required to fully suppress disease.
Results
T cell IL-10 response does not impact colitis development
To evaluate lineage-specific effects of IL-10 in T cell transfer-mediated colitis, we first produced mice with a selective deletion of the specific IL-10 receptor, IL-10Rα, restricted to T cells (IL-10RαTdel)25. Colitis was then induced by transferring naive CD4+CD25−CD45RBhi T cells from wild-type (WT) or IL-10RαTdel mice into Rag1−/− recipients. Disease magnitude and quality were monitored through changes in body weight, colon histopathology, and colonic T cell and T cell subset (Th1, Th17, Foxp3+) infiltration (Supplementary Fig. 1). No differences were identified between recipients of WT and IL-10RαTdel T cells, indicating that IL-10 signaling into the transferred T cells does not modulate colitis severity in this T cell-dependent model.
Mϕ response to IL-10 restrains T cell-mediated colitis
We previously demonstrated that macrophage but not DC or T cell IL-10 signaling prominently alleviated inflammation in a distinct T cell-independent, toxin-induced model of acute colitis, DSS colitis26. Recent data have also implicated myeloid cells as regulators of disease severity in T cell transfer colitis19–21. To assess the impact of DC and Mϕ-selective deletion of IL-10Rα (IL-10RαDCdel, IL-10RαMdel) in T cell transfer colitis, we generated lineage-specific knock-outs on a Rag1−/− background and induced disease. Histologic and clinical disease did not differ between Rag1−/− and IL-10RαDCdelRag1−/− recipients (Supplementary Fig. 2a, b). In contrast, disease was intensified in IL-10RαMdelRag1−/− mice (Fig. 1a and Supplementary Fig. 2a, b). Weight loss was elevated and accelerated, with a mean±s.e.m. decrement at 8 wk of 19.1±1.1% vs 9.3±1.1% for Rag1−/− controls (Fig. 1a). IL-10RαMdelRag1−/− colons were shorter than Rag1−/− controls (8.2±0.3 vs 7.1±0.2 cm, Fig. 1b). Histopathology in IL-10RαMdelRag1−/− colons demonstrated increased inflammation, with more extensive cellular infiltrates, submucosal edema, and epithelial erosion (Fig. 1c). Total histologic score was 8.3±0.6 and 3.6±0.7 (scale 0–12) for IL-10RαMdelRag1−/− and Rag1−/− mice respectively. Moreover, fewer IL-10RαMdelRag1−/− mice survived with extended disease times (Fig. 1d), and these developed an elevated incidence of rectal prolapse (75% IL-10RαMdelRag1−/− mice vs 0% Rag1−/− controls at 12 wk).
Figure 1. Mϕ IL-10Rα expression attenuates T cell-induced colitis.

(A) CD4+CD25−CD45RBhi C57BL/6 T cells (5×105) were transferred into C57BL/6 Rag1−/− and IL-10RαMdelRag1−/− mice to induce colitis. Mean±1 s.e.m. percent of initial body weight is plotted. *, p<0.05; **, p<0.01; ***, p<0.001 (by t-test); (B) Colons were removed at week 8 and colon length measured from the indicated recipients or mice not receiving T cells (control). (C) Representative photomicrographs and tallied scores for disease parameters from H&E stained colon sections obtained 8 wk after naive T cells transfer. (D) Kaplan-Meier survival curves of Rag1−/− and IL-10RαMdelRag1−/− mice (n=10). (E) Colitis was induced in Rag1−/− and IL-10RαMdelRag1−/− mice with 5×105 CD4+CD25−CD45RBhi T cells from C57BL/6 mice. Anti-Ly6G or control Ab was administered i.p. 1 d pre-transfer and then weekly to deplete neutrophils. Mean±1 s.e.m. initial body weight is plotted (n=10/cohort). △, Rag1−/− vs Rag1−/−+ αLy6G; ○, Rag1−/− vs IL-10RαMdelRag1−/−; *, Rag1−/−+ αLy6G vs IL-10RαMdelRag1−/−+ αLy6G; p<0.05, p<0.01, and p<0.001 for 1, 2, and 3 symbols (by ANOVA). No significant difference was seen in IL-10RαMdelRag1−/− vs IL-10RαMdelRag1−/−+ αLy6G. (F) Histology scores for 8 week colon from mouse cohorts treated as in (e). Data are representative of three independent experiments, n=5–10 per cohort.
In a recent study, mice deleting IL-10R using a CX3CR1-driven Cre that is expressed by a large proportion of LPMϕs developed spontaneous colitis. We did not observe overt spontaneous disease in our IL-10RαMdelRag1−/− mice during the time frame of our assays. To further assess this, cohorts of these and IL-10Rαfl/flRag1−/− controls (n=10 for each) were aged for 6 months. There was no difference in weight gain, or presence of clinical signs of colitis or other illness in either population (Supplementary Fig. 2c). Histologic analysis of 5 aged IL-10RαMdelRag1−/− mice failed to identify evidence for colitis. Similarly, spontaneous colitis was not observed in 6 month aged IL-10RαMdelRag1+/+ mice.
IL-10’s protective role in colitis is well established. To determine whether the Mϕ-specific response to IL-10 can account for this, we also compared disease in IL-10RαMdelRag1−/− mice with Rag1−/− mice harboring a germline deletion in IL-10Rα (IL-10Rα−/−Rag1−/−; Supplementary Fig. 2a, b). No difference in clinical disease or histopathology was evident in these two recipient lines, implying that IL-10-mediated immunoprotection can largely be accounted for by its Mϕ-specific effects.
The Lys-M-Cre transgene used to ablate IL-10Rα in the IL-10RαMdelRag1−/− mice is expressed in granulocytes as well as Mϕs27. Neutrophils have a modest protective role in T cell transfer colitis28, and loss of IL-10Rα signaling there, rather than in Mϕs, may have impacted disease. To isolate the Mϕ-specific effect, we depleted neutrophils with specific Ab beginning prior to T cell transfer. Neutrophils were undetectable in the peripheral blood from αLy6G Ab but not control Ab treated animals throughout the experimental interval. In Rag1−/− recipients, weight loss was mildly increased on some disease days in neutrophil-depleted mice, though no significant difference in histopathology was identified (Fig. 1e, f). No significant change in disease severity was apparent with neutrophil depletion in IL-10RαMdelRag1−/− mice. Further, the prominent disease exacerbation in IL-10RαMdelRag1−/− compared with Rag1−/− recipients persisted after neutrophil depletion, indicating that the intensified immunopathology in IL-10RαMdelRag1−/− mice is primarily attributable to the Mϕ rather than granulocyte IL-10 response.
Increased numbers and activation of IL-10RαMdelRag1−/− LPMϕs
To identify causes of the enhanced IL-10RαMdelRag1−/− disease, we first analyzed lamina propria macrophages (LPMϕs). LPMϕs, typed as CD11bhiCD11c−/lo/modF4/80+Ly6G−/loSiglecF−, were significantly increased in IL-10RαMdelRag1−/− mice with colitis compared with Rag1−/− controls (Fig. 2a). This population was uniformly CD64+ and predominantly CX3CR1+, further delineating them as LPMϕs29, 30 (Supplementary Fig. 3a). The cells were further characterized by a uniformly elevated surface expression of activation markers, including CD40, CD80, and CD86 (Fig. 2b). Segregation of CD11b+CD64+CD103− and CD45+ cells based on Ly6C and class II MHC expression can distinguish pro-inflammatory (Ly6Chi) and anti-inflammatory (Ly6Clo) macrophage populations20, 30. Most LPMϕs analyzed using this alternative gating strategy were phenotypically pro-inflammatory, and the proportions of Ly6Chi and Ly6Clo macrophages did not differ between IL-10RαMdelRag1−/− mice and Rag1−/− controls (Supplementary Fig. 3b).
Figure 2. Analysis of lamina propria macrophages.

(A) Cells were isolated from large intestine lamina propria of Rag1−/− and IL-10RαMdelRag1−/− mice at wk 8 after colitis induction. Absolute numbers of LPMϕ (CD11bhiCD11c−/lo/modF4/80+Ly6G−/loSiglec-F−) were quantified. (B) Staining of LPMϕ gated as above, for CD40, CD80 and CD86. Gray line, isotype control; dashed black line, LPMϕs from Rag1−/− mice; Solid black line, LPMϕs from IL-10RαMdelRag1−/− mice. (C). Rag1−/− and IL-10RαMdelRag1−/− mice received 5×105 CD4+CD25−CD45RBhi C57BL/6 T cells. After 8 wk, BrdU was administered i.p. and the mice sacrificed 24 h later. Colon LP cells were isolated and BrdU positive Mϕs, DCs, neutrophils and CD4+ T cells measured by intracellular flow cytometry. Data are representative of three independent experiments, n=8–10 per cohort. **, p<0.01 (by t-test).
The increased IL-10RαMdelRag1−/− versus Rag1−/− LPMϕ numbers were further associated with an increased proliferation rate, as determined by incorporation of the nucleotide analog BrdU. Unlike for LPMϕs, no differences in BrdU incorporation were evident in colonic CD4 T cells, DCs, and neutrophils, or in Mϕs from other locations (Fig. 2c). Therefore, numbers, proliferation, and activation state of LPMϕs are increased in IL-10RαMdelRag1−/− mice.
Unimpaired IL-10RαMdelRag1−/− T regulatory response
A subset of T cells transferred into Rag1−/− mice upregulate Foxp3, and IL-10 signaling into Mϕs could contribute to the generation and maintenance of these regulatory T cells21, 22. Indeed, recent findings with IL-10Rβ−/−Rag2−/− mice have identified defective iTreg formation and Treg function. Treg co-transferred with naïve T cells even at a 1:1 ratio were incapable of preventing disease20. To assess Treg activity here, we quantified Foxp3+ iTreg forming in the LP, mesenteric lymph nodes (MLNs), and spleen of IL-10RαMdelRag1−/− and control Rag1−/− colitic mice. No differences were observed (Fig. 3a). We next analyzed whether sorted Treg transferred into IL-10RαMdelRag1−/− and Rag1−/− mice with the induction of colitis could prevent disease. The transferred cells were able to fully suppress disease development in both IL-10RαMdelRag1−/− and Rag1−/− recipients, indicating that a macrophage-specific defect in IL-10 response does not overtly impact Treg suppressive activity in this setting (Fig. 3b). Treg transfers led to a >2-fold increase in Treg in the spleens, MLNs, and colons of recipient mice and Treg percentages did not differ between IL-10RαMdelRag1−/− and Rag1−/− recipients in any of the locations (Fig. 3c). These results indicate that Treg are able to suppress disease in the absence of a macrophage-specific response to IL-10, and that Treg-specific mechanisms independent of IL-10 actions on macrophages are employed in this system.
Figure 3. T cell Foxp3 expression and IL-10 production.

(A) Colitis was induced in Rag1−/− and IL-10RαMdelRag1−/− mice by the transfer of 5×105 CD4+CD25−CD45RBhi C57BL/6 T cells. The frequency of Foxp3+ Treg cells among CD4+ T cells in the spleen, MLN and colon 8 wk after initial T cell transfer is plotted. (B) Mice received sorted CD4+CD45RbloFoxp3-YFP+ Treg with naive T cells at a 1:1 ratio or naive (CD4+CD45RbhiFoxp3-YFP−)T cells alone, and were monitored for weight loss. **, p<0.01 for naïve T→Rag1−/− vs naïve T→IL-10RαMdelRag1−/− (by ANOVA). Differences were not significant between naïve T+Treg→Rag1−/− vs naïve T+Treg →IL-10RαMdelRag1−/− cohorts. (C) Splenic, MLN, and colonic Foxp3+ T cells were identified by flow cytometry in mice receiving or not receiving Treg as in (B).Upper-left and right plots depict results for colonic Treg. Lower-left plot depicts results for MLN and splenic Treg only in mice receiving supplemental Treg. ***, p<0.001 (by ANOVA). (D) Similar transfers were performed using CD4+CD25−CD45RBhi T cells from IL-10-GFP donors. Representative dot plots and summary analysis of the frequency of IL-10-GFP+ cells among CD4+ T cells in the spleen, MLN, and colon at 8 wk is shown. (E) Rag1−/− and IL-10RαMdelRag1−/− mice received 5×105 CD4+CD25−CD45RBhi T cells from C57BL/6 or IL-10−/− mice. Mean±1 s.e.m. percent initial body weight is plotted. *, WT→Rag1−/− vs WT→IL-10RαMdelRag1−/−; ●, IL-10−/−→Rag1−/− vs IL-10−/−→IL-10RαMdelRag1−/−; p<0.05, p<0.01, and p<0.001 for 1, 2, and 3 symbols (by ANOVA). Representative of 3 independent experiments, n=5 per cohort. (F). Histological scores for colons from the indicated mice at 8 wk.
Alternative regulatory T cell populations are demarcated by IL-10 production, and IL-10 itself may impact these directly or indirectly. To evaluate this, we induced colitis by transferring naïve T cells from IL-10-GFP knock-in reporter mice. Here too, no differences in population sizes were seen (Fig. 3d). To determine if T cell IL-10 production was itself functionally dispensable for the differential colitis in IL-10RαMdelRag1−/− mice, we performed transfers using IL-10−/− or WT T cells. Disease severity, measured clinically and histologically, was exclusively associated with recipient type. IL-10 production by transferred T cells did not influence disease magnitude clinically or histologically (Fig. 3e, f). Therefore, the Mϕ IL-10 response does not identifiably impact regulatory T cell presence. Further, while Mϕ response to IL-10 is critical in attenuating disease, T cells are not a significant source of this IL-10 here.
Pro-inflammatory cytokine production by IL-10RαMdel LPMϕs
To further evaluate the heightened disease severity in IL-10RαMdelRag1−/− mice, we measured in whole colons the levels of cytokines implicated in its pathogenesis, including IL-1β, IL-6, TNF-α, MCP-1, IL-10, IL-17, and IFN-γ31. With early disease (wk 4), IL-1β, IL-6, and MCP-1 were increased in IL-10RαMdelRag1−/− colon compared with Rag1−/− controls (Fig. 4a). As disease progressed (wk 8), the quantity of cytokine produced was altered. Additional elevations in IL-17 and TNF-α were identified at this time. Differences in IL-10 and IFN-γ were not seen.
Figure 4. Cytokine production by colonic macrophages.

(A) Colons from Rag1−/− and IL-10RαMdelRag1−/− mice, 8 wk after colitis induction, were homogenized and cytokines (IL-1β, IL-6, TNF-α, MCP-1, IL-10, IL-17 and IFN-γ) measured by ELISA or multiplex assay. Results from individual mice (circles) and cohort means (lines) are plotted. (B and C) Percent and absolute number of IL-17+ and IFN-γ+ cells among CD4+ T cells from colons of diseased mice (8 wk). (D) Relative expression of the indicated mRNAs (IL-1β, IL-6, IL-23p19 and iNOS) from LPMϕs, DCs, neutrophils and epithelial cells sorted from colon tissue and measured by qRT-PCR. (E) Relative expression of the indicated mRNAs (TNF-α, IL-17, IL-12p40, IL-10, and arginase) from LPMϕs sorted from colon tissue and measured by qRT-PCR. Data are representative of three independent experiments, n=10 per cohort. *, p<0.05; **, p<0.01, ***, p<0.001 (by t-test).
T cell transfer colitis is associated with colonic infiltration by both Th1 and Th17 cells, and the Th17 response is required for disease development. Our identification of increased colonic IL-6 and IL-1β in IL-10RαMdelRag1−/− mice, cytokines associated with Th17 induction, coupled with elevated IL-17 but not IFN-γ levels implied an intensification of the Th17 response. To test this, we enumerated IFN-γ and IL-17 producing T cells in the colonic infiltrate. Significantly increased percents and absolute numbers of CD4+IL-17A+ T cells were identified in IL-10RαMdel Rag1−/− compared with Rag1−/− colons (Fig. 4b). Likewise, after the induction of disease with CD4+CD25−CD45RBhi RORγT-GFP reporter T cells, increased GFP+ T cells were identified in the colon at 4 and 8 wk (Supplementary Fig. 4a). No differences in CD4+IFN-γ+ T cell quantities were evident (Fig. 4c).
To more specifically determine whether elevated production of Th17-promoting cytokines was specific to the Mϕ population, we sorted colonic CD11bhiCD11c−/lo/modF4/80+Ly6G−/loSiglecF− LPMϕs from colitic mice and assayed their cytokine expression profiles by qRT-PCR. A particularly prominent elevation in IL-1β production was apparent in IL-10RαMdelRag1−/− compared with Rag1−/− LPMϕs (mRNA expression ratio: 7.5±1.2). Lesser elevations in IL-6 (3.4±0.5), IL-23 (2.9±0.3) and other pro-inflammatory cytokines were also identified (Fig. 4d, e). IL-10 itself was unchanged and a modest decline in arginase with a corresponding increase in iNOS further indicated enhanced pro-inflammatory function of the IL-10RαMdelRag1−/− LPMϕs. For IL-1β, IL-6, IL-23p19, and iNOS, relative expression was compared in sorted colonic LPMϕs, DCs, neutrophils, and epithelial cells (Fig. 4d). Elevated expression was specific to the Mϕs, indicating that Mϕs are the primary source for the increased Th17-associated cytokines and implying that IL-10 acts directly on these cells to suppress their production.
As an alternative gating strategy, we also sorted CD11b+CD64+CD103−CD45+Ly6Chi LPMϕs and assessed similarly for IL-1β, IL-6, IL-23, IL-10, iNOS, and arginase. These were differentially expressed in a manner paralleling results above (Supplementary Fig. 4b). CD163 was additionally assessed as a marker for anti-inflammatory macrophages and found to be similar in the IL-10RαMdelRag1−/− and control Rag1−/− populations.
Colitis in IL-10RαMdelRag1−/− mice is Th17 dependent
Th17 cells have been shown to be essential to T cell transfer colitis in Rag1−/− mice32. The elevated colonic inflammation in IL-10RαMdelRag1−/− mice was correlated with an increased Th17 response, but may also have been mediated by alternative pathologic pathways. To verify a role for Th17 cells, we transferred RORγT−/− T cells into Rag1−/− and IL-10RαMdelRag1−/− recipients (Supplementary Fig. 5). RORγT, and hence Th17 cells, proved essential for colitis in both recipient types.
Colitis is IL-6 independent in IL-10RαMdelRag1−/− mice
Though IL-6 is well established as a key inducer of Th17 cells, its isolated role in fostering the Th17 response fundamental to T cell transfer colitis has not been addressed. The increased IL-6 in IL-10RαMdelRag1−/− mice might have driven the increased immunopathology there. To assess this, we induced colitis by transfers of IL-6Rα−/− or WT naive T cells into IL-10RαMdelRag1−/− and Rag1−/− recipients. IL-6Rα−/− T cells were ineffective in inducing disease in Rag1−/− mice (Fig. 5a, b). Mice gained weight after T cell transfer and histologic lesions were mild. In contrast, IL-6Rα−/− and WT T cells proved equipotent in mediating severe clinical disease in IL-10RαMdelRag1−/− recipients. Although there was a trend toward modestly diminished histologic severity after IL-6Rα−/− transfer, this was not significant. Thus, the Mϕ response to IL-10 creates a dependency for IL-6 in disease pathogenesis.
Figure 5. Role of T cell IL-6 response in colitis development in Rag1−/− and IL-10RαMdelRag1−/− mice.

Colitis was induced in Rag1−/− and IL-10RαMdelRag1−/− mice by the transfer of 5×105 C57BL/6 or IL-6Rα−/− CD4+CD25−CD45RBhi T cells. Mice were sacrificed after 8 wk. (A) Representative weight curves, mean ±1 s.e.m., are plotted. *, WT→Rag1−/− vs IL-6Rα−/− →Rag1−/−; △, WT→Rag1−/− vs WT→IL-10RαMdelRag1−/−; □, IL-6Rα−/− →Rag1−/− vs IL-6Rα−/− →IL-10RαMdelRag1−/−; p<0.05, p<0.01, and p<0.001 for 1, 2, and 3 symbols (by ANOVA). No significant differences were observed for WT→IL-10RαMdelRag1−/− vs IL-6Rα−/− →IL-10RαMdelRag1−/−. (B) Histologic scores for colons analyzed at 8 wk. (C) Percent and absolute number of IL-17+ cells among CD4+ T cells in the spleen, MLN and colon. (D) Absolute numbers of CD4+ T cells. (E) Percent and absolute number of IFN-γ+ cells among CD4+ T cells. (F) Percent and absolute number of Foxp3+ Treg cells among CD4+ T cells. *, p<0.05; **, p<0.01; ***, p<0.001 (by t-test). Data are representative of three independent experiments, n=8 per cohort.
To better understand this, we analyzed the T cell responses leading to colonic injury. Th17 cell percentages among CD4+TCR+ T cells were reduced in the MLNs of mice receiving IL-6Rα−/− T cells (Fig. 5c). This was true for both IL-10RαMdelRag1−/− and Rag1−/− mice. Thus IL-6Rα signaling supports but is not essential for Th17 formation in the MLNs of both of these recipient lines. In the colon, the representation of Th17 cells was significantly decreased in Rag1−/− but not IL-10RαMdelRag1−/− recipients. This indicates a relative expansion of Th17 cells at the site of autoimmune inflammation specifically in IL-10RαMdelRag1−/− mice.
Absolute numbers of Th17 cells in all organs sampled dramatically differed between Rag1−/− and IL-10RαMdelRag1−/− recipients of IL-6Rα−/− T cells. Few Th17 cells were present in the spleens, MLNs, and colons of Rag1−/− recipients, whereas large numbers were present in IL-10RαMdelRag1−/− recipients (Fig. 5c). This difference reflected a pervasive decrease in total T cell number with the IL-6Rα−/−→Rag1−/− transfers (Fig. 5d). Therefore, IL-6 is broadly necessary for T cell expansion and colitis in Rag1−/− but dispensable in IL-10RαMdelRag1−/− mice. IL-10’s actions on Mϕs generate an IL-6 requirement for autoinflammatory disease and robust T cell expansion.
In contrast to Th17 development, no differences were identified in the proportions of Th1 or Foxp3+ CD4+ T cells in any of the transfer combinations tested, indicating that T cell IL-6 and Mϕ IL-10 responses do not similarly skew these maturation pathways (Fig. 5e, f). However, absolute numbers of Th1 and Foxp3+ cells were diminished in the IL-6Rα−/− →Rag1−/− combination, again reflecting the impaired T cell expansion in the absence of disease development.
IL-1 response is required for IL-10RαMdelRag1−/− colitis
IL-1β plays an essential role in the steady-state development of intestinal Th17 cells in healthy mice33 and further promotes the Th17 response during colitis34. Considering this, the lack of a requirement for IL-6 in IL-10RαMdelRag1−/− mice (Fig. 5a, b), and the dramatically elevated IL-1β expression in IL-10RαMdelRag1−/− LPMϕs (Fig. 4), we hypothesized that IL-1β plays a pathologic role in IL-10RαMdelRag1−/− mice that is able to supersede the IL-6 requirement for colitis. To test IL-1’s impact, we induced colitis by transferring IL-1R−/− T cells into IL-10RαMdelRag1−/− and Rag1−/− recipients.
IL-1R−/− T cell transfers into Rag1−/− mice did not lead to the virtually complete disease protection seen after IL-6Rα−/− T cell transfers. However IL-1R−/− →Rag1−/− transfers did show diminished clinical and histologic measures of colitis compared with control WT T cell transfers (Fig. 6a, b). IL-1R−/− T cells also provoked significantly milder disease than WT T cells In IL-10RαMdelRag1−/− recipients (Fig. 6a, b). This contrasts with IL-6Rα−/− T cells, which did not detectably alter disease severity. Therefore, while T cell IL-6 responsiveness is clinically important only in Rag1−/− colitis, T cell IL-1 response modulates both Rag1−/− and IL-10RαMdelRag1−/− disease.
Figure 6. Role of T cell IL-1 response in colitis development in Rag1−/− and IL-10RαMdelRag1−/− mice.

Colitis was induced in Rag1−/− and IL-10RαMdelRag1−/− mice by the transfer of 5×105 C57BL/6 or IL-1R−/− CD4+CD25−CD45RBhi T cells. Mice were sacrificed after 8 wk. (A) Representative weight curves, mean ±1 s.e.m., are plotted. *, WT→Rag1−/− vs IL-1R−/− →Rag1−/−; ○, WT→IL-10RαMdelRag1−/− vs IL-1R−/− → IL-10RαMdelRag1−/− △, WT→Rag1−/− vs WT→IL-10RαMdelRag1−/−; □, IL-1R−/− →Rag1−/− vs IL-1R−/− →IL-10RαMdelRag1−/−; p<0.01, and p<0.001 for 2 and 3 symbols (by ANOVA). (B) Histologic scores for colons analyzed at 8 wk. (C) Percent and absolute number of IL-17+ cells among CD4+ T cells in the spleen, MLN and colon. (D) Percent and absolute number of IFN-γ+ cells among CD4+ T cells. (E) Percent and absolute number of Foxp3+ Treg cells among CD4+ T cells. *, p<0.05; **, p<0.01; ***, p<0.001 (by t-test). Data are representative of three independent experiments, n=8 per cohort.
We performed similar analyses of the impact on T cell responses after IL-1R−/− transfers as for the IL-6Rα−/− transfers above. Whereas a decreased percent and absolute number of Th17 cells was seen in the MLNs, a site of T cell priming, after IL-6Rα−/− transfers (Fig. 5c), no differences in the MLNs were seen for IL-10RαMdelRag1−/− or Rag1−/− recipients receiving IL-1R−/− compared with WT T cells (Fig. 6c). However, substantially diminished proportions and absolute numbers of Th17 cells were seen in the colons of mice receiving IL-1R−/− T cells. The actions of IL-1 were specific to Th17 effectors; no differences were identified in Th1 and Foxp3+ Treg populations (Fig. 6d, e). This indicates that there is a selective defect in the Th17 response in the colon. T cell IL-1 but not IL-6 response in IL-10RαMdelRag1−/− mice promotes colonic inflammation by supporting Th17 cells at the site of autoinflammatory disease. In Rag1−/− mice, where IL-10 suppresses LPMϕ IL-1β production, IL-6 plays a more critical role.
Il-10 inhibits pro-IL-1β production and maturation
Our data indicated that IL-1β, increased in the colon of IL-10RαMdel mice, supports the pathologic Th17 response mediating colitis. IL-10 inhibits this IL-1β production thereby attenuating disease. To determine whether IL-1β protein in the colon is primarily produced by LPMϕs, we used immunohistochemistry to colocalize IL-1β and CD11b in colon sections from diseased IL-10RαMdelRag1−/− and Rag1−/− mice (Supplementary Fig. 6a). This demonstrated that IL-1β is largely associated with the CD11b+ population.
IL-1β is generated from an inactive cytosolic precursor (pro-IL-1β). Caspase-1 cleaves pro-IL-1β, converting it into its active form which is then released from the cell35. Caspase-1, in turn, is activated by inflammasome stimulation36. To better evaluate the impact of IL-10 signaling on IL-1β production by LPMϕs, we first quantified pro-IL-1β levels by flow cytometry in gated CD11bhiCD11c−/lo/modF4/80+Ly6G−/loSiglecF− Mϕs. This demonstrated a 1.7-fold elevation in the percent of LPMϕs expressing pro-IL-1β in the colons of diseased IL-10RαMdelRag1−/− compared with Rag1−/− mice (mean±s.d.: 84.6±6.0% vs 48.6±8.4%; Fig. 7a). Further, the MFI of positive cells from IL-10RαMdelRag1−/− mice was nearly 2-fold greater than that from control mice (131.4±9.3 vs 76.6±5.7). In contrast to the colons of diseased mice, pro-IL-1β levels were low to undetectable in MLN and bone marrow macrophages, and levels did not differ between IL-10RαMdelRag1−/− mice and Rag1−/− controls. Differences in pro-IL-1β were also not apparent in mice in which disease was not induced (Supplementary Fig. 6b).
Figure 7. IL-10 inhibition of Mϕ inflammasome activity and IL-1β production.
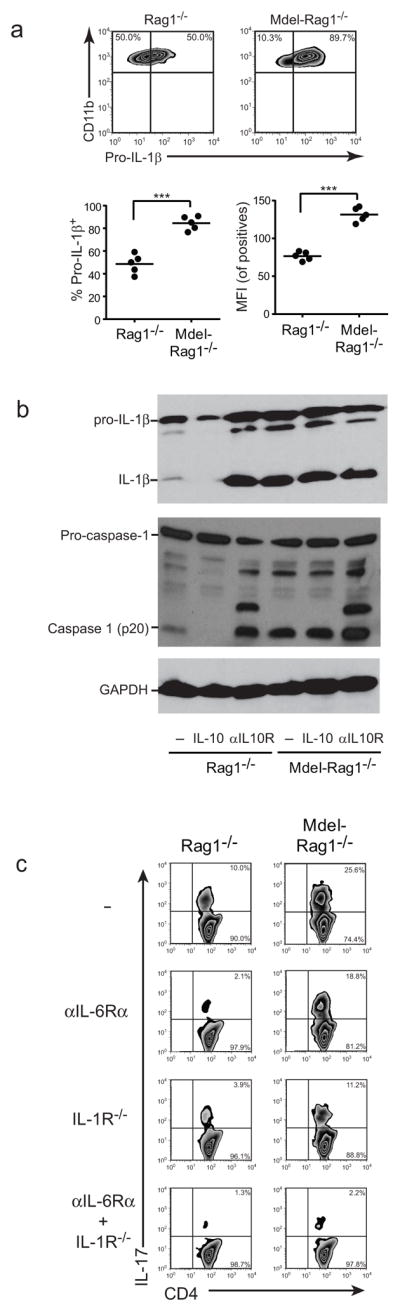
(A) Colitis was induced in Rag1−/− and IL-10RαMdelRag1−/− mice by transfer of 5×105 CD4+CD25−CD45RBhi T cells from WT mice. After 8 wks, LPMϕ pro-IL-1β expression was assessed by intracellular staining. Percent of Mϕs expressing pro-IL-1β and mean fluorescence intensity (MFI) of positive cells are plotted. Data are representative of three independent experiments, n=5 per cohort. ***, p<0.001 (by t-test). (B) Rag1−/− and IL-10RαMdelRag1−/− BMDMs were pretreated with recombinant murine IL-10 or blocking anti-IL-10Rα Ab for 4 h prior to the addition of LPS for 12 h and ATP for the final 30 minutes. Lysates were immunoblotted for IL-1β, caspase-1, and GAPDH. (C) Naïve CD4+ T cells from WT or IL-1R−/− mice and LPMϕs from colitic Rag1−/− and IL-10RαMdelRag1−/− mice were purified by flow cytometric sorting. The T cells were stimulated with anti-CD3 and anti-CD28 in the presence of anti-IL-4, anti-IFN-γ, and TGF-β. LPMϕs and T cell source, and the addition of blocking anti-IL-6R Ab are indicated. After 4 d, IL-17-producing cells were analyzed by intracellular staining.
We verified that pro-IL-1β was directly regulated in Mϕ by IL-10, analyzing its production in cultured and LPS and ATP-stimulated Mϕ by Western analysis, and simultaneously assessing for mature IL-1β formation (Fig. 7b and Supplemenatary Fig. 7). Substantially lower amounts of both pro-IL-1β and IL-1β were present in cultures of Rag1−/− than IL-10RαMdelRag1−/− Mϕ. After treatment with IL-10, pro-IL-β was diminished and mature IL-1β undetectable in Rag1−/− cultures, while this manipulation had no effect on IL-10RαMdelRag1−/−-derived Mϕs. In contrast, treatment with blocking anti-IL-10R antibody elevated Rag1−/− pro-IL-1β and IL-1β levels to those seen with IL-10RαMdelRag1−/− Mϕs.
The complete suppression of IL-1β maturation by IL-10 suggested that Mϕ IL-10R signaling also suppressed caspase-1-activation. To test this we assessed for caspase-1 cleavage to its activated form. LPS and ATP stimulation of Rag1−/− Mϕs led to a modest increase in activated caspase-1 (p20, Fig. 7b and Supplementary Fig. 7). Pre-treatment of Rag1−/− Mϕs with IL-10, however, abrogated this induction. In contrast, pre-treatment with anti-IL-10Rα Ab markedly increased activated caspase-1, indicating that autocrine IL-10 normally restrains caspase-1 activation. In the IL-10RαMdelRag1−/− Mϕs, p20 formation was similarly increased regardless of IL-10 or anti-IL-10Rα Ab treatment.
To determine whether increased IL-1 and IL-6 from LPMϕs can also directly impact Th17 cell maturation, we sorted LPMϕs from diseased IL-10RαMdelRag1−/− and control Rag1−/− mice, and co-cultured them with naïve T cells in the presence of added TGFβ but not IL-6. Some Th17 formed in the presence of Rag1−/− LPMϕs, however, this was markedly elevated with IL-10RαMdelRag1−/− LPMϕs (Fig. 7c). Addition of αIL-6Rα blocking antibody largely abrogated Th17 formation with Rag1−/− LPMϕs. However, consistent with our in vivo findings after IL-6R−/− T cell transfer, this effect was modest with IL-10RαMdelRag1−/−-derived LPMϕs. Blocking IL-1 signaling led to more substantial inhibition of Th17 development in IL-10RαMdel Rag1−/−-derived LPMϕs and inhibition of signaling by both cytokines essentially abrogated Th17 formation. Therefore, IL-10 can act on LPMϕs to directly impair their support of Th17 production. This occurs through the downregulation of IL-1 and to a lesser extent IL-6.
Discussion
IL-10’s anti-inflammatory signals maintain intestinal homeostasis. Yet the essential targets and mechanisms of IL-10 action are incompletely understood. Recent data has provided support for a myeloid response to IL-10 in restraining colonic inflammation, and indicated a critical role for IL-10 in the production of anti-inflammatory LPMϕs that are essential for maintaining immune homeostasis19, 20. We demonstrate here that Mϕs are the primary targets of IL-10 limiting colitis after naive T cell transfer into immunodeficient mice. We further identify how the macrophage-specific response to IL-10 skews the production and expansion of pathologic T cells, thereby promoting disease exacerbation. Colitis is Th17 dependent regardless of Mϕ IL-10 response. However, we show that IL-10 converts the disease from one that is independent of the Th17 inducing cytokine IL-6 to one that is highly dependent. In Rag1−/− recipients of CD4+CD45RBhi T cells, T cell responsiveness to IL-6 is necessary for Th17 formation and colitis development. However, in IL-10RαMdelRag1−/− recipients of IL-6Rα−/− T cells, a strong Th17 amplification occurs. Colitis develops that is clinically indistinguishable from that in recipients of IL-6RαWT T cells.
Our results further indicate that the Mϕ response to IL-10 downmodulates multiple pro-inflammatory cytokines in the colon, but its impact on IL-1β, recently documented to regulate Th17 formation during colitis34, appears paramount. IL-10 therefore shifts the cytokine requirements for the Th17 response. In IL-10’s absence, redundancy between IL-1β and IL-6 supports persistent colitis. Implicitly, though monotherapy with anti-IL-6 may be promising, tandem inhibition of the IL-1 pathway may be necessary for optimal suppression of the Th17 response in IBD, particularly where IL-10 signaling is attenuated.
Previous studies have indicated a role for IL-1β in promoting Th17 development both in humans and mice34, 37, 38. IL-1β levels in IL-10−/− mice with colitis are elevated39 and we extend this finding here to show that a Mϕ-selective deficit in IL-10 response is sufficient for this. Prior in vitro findings have also indicated that IL-1β synergizes with IL-23 to promote Th17 expansion7, and we did observe increased IL-23 along with IL-1β production by IL-10Rα-deficient macrophages.
Though we and others have identified an essential role for RORγT+ Th17 cells in colitis and elevated Th17 cells and/or cytokines have been observed in patients with IBD, the actual role of IL-17 itself is controversial and in a randomized control trial anti-IL-17A proved ineffective in Crohn’s disease40. Identifying effector mechanisms responsible for Th17 mediated immunopathology during colitis will be important as new therapeutic strategies are developed.
Zigmond, et. al., recently found that mice with a CX3CR1-restricted IL-10Rα deficiency develop spontaneous colitis19. This was hypothesized to be mediated by defective macrophage regulation by IL-10. Our findings are consistent with this and with a unique defining role for macrophages in colitis susceptibility. CX3CR1 is also expressed by DCs41 and monocytes42, and by using IL-10RαDCdelRag1−/−, IL-10RαMdelRag1−/−, IL-10Rα−/−Rag1−/− and neutrophil specific depleted mice, our findings support a dominant role for IL-10 action on macrophages in colitis development. This identification of Mϕ as a key target of IL-10 is consistent with a recently published report showing that ATP derived from commensal bacteria promotes Th17 differentiation through a subset of CD11c−/low LP cells43. Unlike Zigmond, et. al., we did not observe spontaneous autoimmunity in our animals, Differences in microflora, and particularly the presence of Helicobacter spp., may account for this. Our colony is maintained under helicobacter-free conditions, whereas Zigmond, et. al. report the presence of helicobacter. Consistently, helicobacter-free IL-10 deficient mice are protected from spontaneous disease44. Alternatively, differences in the subsets of Mϕs expressing the CX3CR1 versus Lys-M promoters, may distinguish spontaneous disease susceptibility in our two systems, and this needs to be further explored.
Shouval, et. al. recently identified a prominent role for IL-10Rβ signaling into myeloid cells in suppressing colitis development. A significant diminution of the Foxp3+ regulatory T cell response was seen. We did not identify a discernible effect of Mϕ IL-10Rα deficiency on iTreg development or transferred nTreg function. This will need resolution. IL-10Rβ is also utilized by IL-22, IL-26 and IFN-λ, which might explain the differences. Alternatively, cell types besides Lys-M+ macrophages may provide critical signals supporting Treg formation and maintenance, and IL-10 may be necessary for this. Regardless of these differences, Shouval, et. al. demonstrated a strongly pro-inflammatory phenotype of IL-10Rβ−/− BMDM, and extended this to Mϕs from IL-10R deficient patients, implying that these regulatory mechanics are translatable to human IBD. Cumulatively, these data provide strong evidence for macrophage as the critical target of IL-10, and assert several mechanisms through which this occurs.
After transfers of IL-6Rα-deficient T cells, we identified a decreased percent of Th17 cells in the MLN but not colon of IL-10RαMdelRag1−/− recipients. This may imply that Th17 cells primed in the MLN are amplified by IL-1β in the colon. However, the site(s) of priming of the Th17 response in colitis is not defined and it is possible that additional priming occurs within gut-associated lymphoid tissue itself. In this regards, we show that isolated LPMϕs from IL-10RαMdelRag1−/− mice have an enhanced ability to support Th17 development from naïve T cells. Hence, although without being able to track the initial site of pathologic T cell development we cannot distinguish whether IL-1β in our system is acting at priming or expansion of Th17 cells, increased colonic Mϕ IL-1β production can support either or both of these mechanisms.
Our results here are consistent with data demonstrating that IL-10 acts via STAT3 to suppress pro-IL-1β production45. Moreover, we extend these findings, showing that IL-10 further inhibits caspase-1 activity. Caspase-1 is activated through inflammasome induced oligomerization and autocatalytic cleavage and it will be important to identify the precise site at which IL-10 acts within the inflammasome cascade.
Though our study specifically interrogates the IL-10 response during colitis, its implications may extend beyond this. IL-1β also promotes the Th17 response in Helicobacter hepaticus colitis34. Administration of rIL-1-β selectively induced Th17 cells in the steady-state intestinal environment33. High levels of IL-1β are associated with an amplified Th17 response in autoimmune RA and EAE models46–48. These effects of IL-1β on Th17 cells may be similarly IL-10 and Mϕ dependent, and this can be assessed using the conditional knock-out mice we have developed.
Our results, as those using a Tg DN IL-10R24, do not support transferred naïve T cells as a significant target of IL-10 in this model. This does not indicate that IL-10 is not a significant T cell regulator. The effects of IL-10 on T cells is complex, and system and T cell subset dependent. Thus, the Treg response to IL-10 was found to sustain Treg in colitis, and mice with a Treg-selective deletion of IL-10R developed spontaneous colitis18, 21. Likewise, we have identified direct effects of IL-10 on T cells in regulating myelin-specific responses during experimental autoimmune encephalomyelitis25.
It will also be important to identify the source of IL-10 relevant to Mϕ targets. Our findings here and those of others indicate that neither effector nor the adaptive regulatory T cells that form after naïve T cell transfer are relevant sources49. In contrast, purified and adoptively transferred IL-10−/− Tregs were seen to be less potent than WT Tregs in preventing and treating established colitis, indicating that Treg IL-10 is significant in some circumstances. However, Treg only account for a portion of IL-10’s effects49–51. The absence of a physiologically relevant T cell IL-10 source here may suggest a myeloid source. Mϕ themselves are strong IL-10 producers and autocrine IL-10 signaling may well provide necessary signals that prevent overzealous Mϕ reactions. Indeed, we identified substantial IL-10 mediated autoregulation of pro-IL-1β production and caspase-1 activation in cultured Mϕ (Fig. 7). In contrast, Zigmond et. al. found no role for CX3CR1+ macrophage-produced IL-10 in the spontaneous colitis that they observed19, potentially implying that multiple sources may be relevant.
In summary, we show that the Mϕ response to IL-10 is critical for Th17 development during colitis. Further, IL-10’s suppression of pro-inflammatory cytokine production does not obviate the essential role for Th17 cells but does shift the cytokine requirements for that response from one primarily governed through IL-1β to one that is IL-6 dependent.
Methods
Mice
We previously produced and verified correct targeting of IL-10Rαfl/fl mice and lineage specific IL-10Rα deletions on a C57BL/6 background as described in prior publications25, 26. These were bred with B6.129S7-Rag1tm1Mom/J mice. B6.129P2-IL-10tm1Cgn/J, B6.129P2-Rorctm1Litt/J, B6.129S7-Il1r1tm1mx/J, B6.129(Cg)-Foxp3tm4(YFP/cre)Ayr/J, and B6;SJL-Il6Rαtm1.1Drew/J mice were obtained from The Jackson Laboratories. Colonies were maintained under spf, including detectable Helicobacter spp.-free, conditions. Mice of either sex were between 8 and 12 weeks of age at the time of study, and paired between experimental and control groups. Experimental protocols were approved by the St. Jude Animal Care and Use Committee.
Induction of colitis
Flow cytometrically sorted CD4+CD25−CD45RBhi T cells (5×105/mouse) derived from pooled splenocytes and LN cells of indicated mice were transferred i.v. into Rag1−/− or IL-10RαMdelRag1−/− mice. Body weight was monitored on a weekly basis. For neutrophil depletion studies, 1 mg anti-Ly6G mAb 1A8 (Bio-X-Cell) or control IgG26 was administered per mouse i.p. 1 d before cell transfer and weekly thereafter. Depletion was confirmed by flow cytometry. For analyses of Treg-mediated disease suppression, splenocytes from Foxp3-YFP reporter mice were collected and naïve T cells, defined as CD4+CD45RBhiYFP−, and Treg cells, defined as CD4+CD45RBlowYFP+, sorted. Purity after sorting was >99%. Age-matched Rag1−/− mice or IL-10RαMdelRag1−/− mice received 2×105 naive T cells with or without Treg cells at a 1:1 ratio i.v.
Histology
Colons were stained with hematoxylin and eosin. Three independent sections were assessed per mouse by a blinded reviewer. Inflammation scoring: 0, no or occasional inflammatory cells in the lamina propria (LP); 1, increased LP inflammatory cells; 2, confluence of inflammatory cells extending into the submucosa; 3, transmural infiltrate extension of the infiltrate. Ulceration scoring: 0, no ulceration; 1, mild (1–2 ulcers per 40 crypts analyzed); 2, moderate (3–4 ulcers); 3, severe (> 4 ulcers). Hyperplasia scoring: 0, normal; 1, crypts up to twice normal thickness with normal epithelium; 2, crypts >2 times normal thickness, hyperchromatic epithelium; reduced goblet cells, scattered arborization; 3, Crypts >4 times normal thickness, marked hyperchromasia, few to no goblet cells, high mitotic index, frequent arborization. Disease area scoring: 0, 0–5% involvement; 1, 5–30%; 2, 30–70%; 3, >70%. Total score is the sum of individual scores.
Cytokine levels
Frozen colon samples were homogenized in ice-cold PBS containing 1% NP-40 and complete protease inhibitor cocktail (Roche). Cytokines and chemokines were measured by Luminex (Bio-Rad) or ELISA (R&D Systems).
LP cell isolation
LP cells were isolated using a modification of a previously described protocol26. Large intestines were carefully excised, mesentery and fat removed, and intestines then opened longitudinally, rinsed in HBSS and cut into 1-cm pieces. Colon segments were vigorously shaken twice in medium with 1 mM EDTA (Sigma-Aldrich) for 20 min at 37°C, and suspended cells collected and filtered through a cell strainer. Tissue was further minced and incubated at 37°C for 1 h in medium with 1 mM collagenase type IV (Sigma-Aldrich) and 40 U ml−1 DNase I (Roche) with agitation. Cells were filtered, washed, and isolated over a percoll step gradient.
Cytokine PCR
Total RNA was isolated from sorted LPMϕs using the RNeasy mini kit (Qiagen), and cDNA synthesized using superscript III and oligo (dT) primers (Invitrogen). Expression levels of were normalized to HPRT (ΔCt) and compared with littermate controls using the ΔΔCt method52. Primer sequences are: TGF-β: F, CAC AGT ACA GCA AGG TCC TTG C; R, AGT AGA CGA TGG GCA GTG GCT; IL-12p35: F, ATG ACC CTG TGC CTT GGT AG; R, GAT TCT GAA GTG CTG CGT TG; IL-23p19: F, AGC GGG ACA TAT GAA TCT ACT AAG AGA; R, GTC CTA GTA GGG AGG TGT GAA GTT; IL-12p40: F, GAC CAT CAC TGT CAA AGA GTT TCT AGA T; R, AGG AAA GTC TTG TTT TTG AAA TTT TTT AA; IL-1β: F, GAT CCA CAC TCT CCA GCT GCA; R, CAA CCA ACA AGT GAT ATT CTC CATG; IL-10: F, GTG AAA ATA AGA GCA AGG CAG TG; R, ATT CAT GGC CTT GTA GAC ACC; TNF-α: F, AAT GGC CTC CCT CTC ATC AGT; R, CTA CAG GCT TGT CAC TCG AA; iNOS: F, TGA CGG CAA ACA TGA CTT CAG; R, GCC ATC GGG CAT CTG GTA; IL-6: F, TAT GAA GTT CCT CTC TGC AAG AGA; R, TAG GGA AGG CCG TGG TT; Arginase: F, TCA CTT TCC ACC ACC TCT TG AY; R, TCT CCA CCG CCT CAC GAC TC; IL-17A: F, GCT CCA GAA GGC CCT CAG, R, CTT TCC CTC CGC ATT GAC A; CD163: F, CCT TGG AAA CAG AGA CAG GC; R, TCC ACA CGT CCA GAA CAG TC; HPRT: F, GA CCG GTC CCG TCA TGC; R, TCA TAA CCT GGT TCAT CAT CGC. F, forward primer; R, reverse primer.
Flow cytometry
Cells were stained with Abs specific for mouse TCRβ, F4/80, CD11b, CD11c, CD40, CD64, CD80, CD86, Ly6G, Siglec-F, CD4, Foxp3, IL-17, IFN-γ, pro-IL-1β, or with isotype-matched controls (1:100 dilution for each antibody; BD Pharmingen or eBiosciences), and analyzed using a FACSCalibur or LSRII flow cytometer with Cell Quest (BD Biosciences) or FlowJo (TreeStar) software.
BRDU staining
Mice were injected i.p. with 150 μl BrdU (10 mg ml−1) in sterile 1× DPBS. After 16–20 h, lymphocytes were isolated, stained with Abs to cell surface markers, fixed and permeabilized with Cytofix/Cytoperm Buffer (BD Biosciences), treated with DNase (300 μg ml−1) at 37°C for 1 h, stained with anti–BrdU-APC (BD BRDU flow kit), and analyzed by flow cytometry.
BMDM culture and LPS stimulation
BMDMs were generated by culturing mouse bone marrow cells in L-cell-conditioned IMDM. The L-cell conditioned medium comprised supernant from cultures of L929 cells secreting M-CSF mixed at a 1:2 ratio with IMDM and then supplemented with 10% FBS, 1% non-essential amino acids and 1% penicillin–streptomycin. After 6 days of culture, cells were seeded in 12-well plates, and the next day treated with IL-10 (50ng ml−1) or anti-IL-10Rα Ab (1μg ml−1), and 4 h later stimulated with or without LPS (20 ng ml−1) for 12 h. For the final 30 min, 5mM ATP was added into the medium53.
Western blot
Culture samples were denatured in loading buffer containing SDS and 100 mM DTT, and boiled for 5 min. SDS-PAGE–separated proteins were transferred to polyvinylidene difluoride membranes and immunoblotted with primary Abs against caspase-1 (Adipogen; AG-20B-0042 or kind gift of Dr Peter Vandenabeele, Ghent University), IL-1β (R&D Systems), and GAPDH (Cell Signaling Technology; D16H11), followed by secondary anti-rabbit, anti-rat, anti-mouse, or anti-goat HRP Abs (Jackson ImmunoResearch Laboratories)54. Images have been cropped for presentation. Full size images are presented in Supplementary Fig. 7.
Th17 culture
Naive (CD4+CD45RBhighCD25−) T cells were purified by cell sorting to a purity >99%. These (5×105) were co-cultured at a ratio of 5:1 with or without sorted LPMϕs from 8 week diseased IL-10RαMdel Rag1−/− or Rag1−/− mice in 96-well plates pre-coated with 1 μg ml−1 anti-CD3 and 2 μg ml−1 anti-CD28. Cells were cultured in complete RPMI 1640 media containing 5 ng ml−1 TGF-β, 10 μg ml−1 anti-IL-4 and 10 μg ml−1 anti-IFN-γ Abs. 20 ng ml−1 IL-6 was added to a positive control Th17 culture condition only. After 4 days, cells were washed and restimulated with cytokine stimulation cocktail containing PMA, Ionomycin and Brefeldin A (Cell stimulation cocktail, eBioscience) for 4 hours at 37°C. Cells were washed and stained for the indicated cytokines.
Immunohistochemistry
Tissue cryosections were fixed in 4% PFA at 4°C overnight, embedded in optimal cutting temperature (OCT) compound, and sectioned in a cryostat (12 μm). For IL-1β immunostaining, sections were incubated with a polyclonal goat anti-mouse IL-1β primary antibody (1:200, R&D Systems) and monoclonal rat anti-mouse CD11b antibody (1:500, AbD Serotec). After washing 3 times with TBST, sections were incubated with Cy3-labeled donkey-anti-rat IgG antibody (1:200, Jackson Lab) and Alexa 488-labeled donkey anti-goat IgG antibody (1:200,Molecular Probes). Sections were mounted with mounting medium containing DAPI (Invitrogen), and confocal microscopy was performed.
Statistics
Statistics were calculated using Prism5 (GraphPad Software). Group comparisons were by two-sided Student’s t-test or, when multiple cohorts were present, ANOVA with Bonferroni correction. A p< 0.05 was considered significant.
Supplementary Material
Acknowledgments
Supported by the National Institutes of Health Grant AI056153 and AI106600 (to TLG) and the American Lebanese Syrian Associated Charities (ALSAC)/St. Jude Children’s Research Hospital (to all authors). We thank Jianmin Ye and Beatriz Sosa-Pineda for assistance with immunohistochemistry, and Richard Cross, Greig Lennon, and Parker Ingle for assistance with flow cytometric sorting.
Abbreviations
- IBD
inflammatory bowel disease
- IL-10Rα
interleukin 10 receptor α
- LPMϕ
lamina propria macrophage
- WT
wild type
- BMDM
Bone marrow derived macrophage
- RORγT
retinoic acid receptor-related organ receptor-γT
Footnotes
Disclosure/Competing Financial Interests
The authors declare no competing financial interests.
Author Contributions
B. L. designed and conducted research studies, analyzed data, prepared figures, and wrote the manuscript. P. G. and R. K. S. M. performed the Western analyses on IL-1b and caspase-1 with B. L. P. V. performed blinded histologic analyses and scoring of colon sections. T. D. K. provided guidance and advice on experimental design. T. L. G. coordinated the research efforts, designed the studies with B.L., assisted in data analysis, revised the manuscript and assisted in preparing figures.
References
- 1.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 3.Sarra M, Pallone F, Macdonald TT, Monteleone G. IL-23/IL-17 axis in IBD. Inflamm Bowel Dis. 2010;16:1808–1813. doi: 10.1002/ibd.21248. [DOI] [PubMed] [Google Scholar]
- 4.Maloy KJ, Kullberg MC. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol. 2008;1:339–349. doi: 10.1038/mi.2008.28. [DOI] [PubMed] [Google Scholar]
- 5.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 6.Ahern PP, Izcue A, Maloy KJ, Powrie F. The interleukin-23 axis in intestinal inflammation. Immunol Rev. 2008;226:147–159. doi: 10.1111/j.1600-065X.2008.00705.x. [DOI] [PubMed] [Google Scholar]
- 7.Chung Y, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivanov II, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 9.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 10.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 11.Spencer SD, et al. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med. 1998;187:571–578. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glocker EO, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moran CJ, et al. IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm Bowel Dis. 2013;19:115–123. doi: 10.1002/ibd.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pigneur B, et al. Phenotypic characterization of very early-onset IBD due to mutations in the IL10, IL10 receptor alpha or beta gene: a survey of the Genius Working Group. Inflamm Bowel Dis. 2013;19:2820–2828. doi: 10.1097/01.MIB.0000435439.22484.d3. [DOI] [PubMed] [Google Scholar]
- 15.Zhu H, Lei X, Liu Q, Wang Y. Interleukin-10-1082A/G polymorphism and inflammatory bowel disease susceptibility: a meta-analysis based on 17,585 subjects. Cytokine. 2013;61:146–153. doi: 10.1016/j.cyto.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 17.Rubtsov YP, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 18.Chaudhry A, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. 2011;34:566–578. doi: 10.1016/j.immuni.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zigmond E, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40:720–733. doi: 10.1016/j.immuni.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 20.Shouval DS, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–719. doi: 10.1016/j.immuni.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murai M, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huber S, et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(−) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 2011;34:554–565. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marlow GJ, van Gent D, Ferguson LR. Why interleukin-10 supplementation does not work in Crohn’s disease patients. World J Gastroenterol. 2013;19:3931–3941. doi: 10.3748/wjg.v19.i25.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamanaka M, et al. Memory/effector (CD45RB(lo)) CD4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J Exp Med. 2011;208:1027–1040. doi: 10.1084/jem.20102149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, et al. The T cell response to IL-10 alters cellular dynamics and paradoxically promotes central nervous system autoimmunity. J Immunol. 2012;189:669–678. doi: 10.4049/jimmunol.1200607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li B, Alli R, Vogel P, Geiger TL. IL-10 modulates DSS-induced colitis through a macrophage-ROS-NO axis. Mucosal Immunol. 2013 doi: 10.1038/mi.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 28.Kuhl AA, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133:1882–1892. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 29.Tamoutounour S, et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol. 2012;42:3150–3166. doi: 10.1002/eji.201242847. [DOI] [PubMed] [Google Scholar]
- 30.Bain CC, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6:498–510. doi: 10.1038/mi.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leppkes M, et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology. 2009;136:257–267. doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 33.Shaw MH, Kamada N, Kim YG, Nunez G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med. 2012;209:251–258. doi: 10.1084/jem.20111703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coccia M, et al. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med. 2012;209:1595–1609. doi: 10.1084/jem.20111453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 36.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gulen MF, et al. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity. 2010;32:54–66. doi: 10.1016/j.immuni.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aksentijevich I, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360:2426–2437. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Fu S, Sun S, Li Z, Guo B. Inflammasome activation has an important role in the development of spontaneous colitis. Mucosal Immunol. 2014;7:1139–1150. doi: 10.1038/mi.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hueber W, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niess JH, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 42.Landsman L, et al. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113:963–972. doi: 10.1182/blood-2008-07-170787. [DOI] [PubMed] [Google Scholar]
- 43.Atarashi K, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455:808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 44.Kullberg MC, et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect Immun. 1998;66:5157–5166. doi: 10.1128/iai.66.11.5157-5166.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guarda G, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 46.Horai R, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191:313–320. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El-Behi M, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sutton CE, et al. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 49.Uhlig HH, et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J Immunol. 2006;177:5852–5860. doi: 10.4049/jimmunol.177.9.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Asseman C, Read S, Powrie F. Colitogenic Th1 cells are present in the antigen-experienced T cell pool in normal mice: control by CD4+ regulatory T cells and IL-10. J Immunol. 2003;171:971–978. doi: 10.4049/jimmunol.171.2.971. [DOI] [PubMed] [Google Scholar]
- 52.Kullberg MC, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Opdenbosch N, et al. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun. 2014;5:3209. doi: 10.1038/ncomms4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gurung P, et al. FADD and Caspase-8 Mediate Priming and Activation of the Canonical and Noncanonical Nlrp3 Inflammasomes. J Immunol. 2014;192:1835–1846. doi: 10.4049/jimmunol.1302839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.