

1. Introduction
Reactive oxygen, nitrogen, and sulfur species, referred to as ROS, RNS, and RSS, respectively, are produced during normal cell function and in response to various stimuli. An imbalance in the metabolism of these reactive intermediates results in the phenomenon known as oxidative stress. If left unchecked, oxidative molecules can inflict damage on all classes of biological macromolecules and eventually lead to cell death. Indeed, sustained elevated levels of reactive species have been implicated in the etiology (e.g., atherosclerosis, hypertension, diabetes) or the progression (e.g., stroke, cancer, and neurodegenerative disorders) of a number of human diseases.1 Over the past several decades, however, a new paradigm has emerged in which the aforementioned species have also been shown to function as targeted, intracellular second messengers with regulatory roles in an array of physiological processes.2 Against this backdrop, it is not surprising that considerable ongoing efforts are aimed at elucidating the role that these reactive intermediates play in health and disease.
Site-specific, covalent modification of proteins represents a prominent molecular mechanism for transforming an oxidant signal into a biological response. Amino acids that are candidates for reversible modification include cysteines whose thiol (i.e., sulfhydryl) side chain is deprotonated at physiological pH, which is an important attribute for enhancing reactivity. While reactive species can modify other amino acids (e.g., histidine, methionine, tryptophan, and tyrosine), this Review will focus exclusively on cysteine, whose identity as cellular target or “sensor” of reactive intermediates is most prevalent and established.3 Oxidation of thiols results in a range of sulfur-containing products, not just disulfide bridges, as typically presented in biochemistry textbooks. An overview of the most relevant forms of oxidized sulfur species found in vivo is presented in Chart 1.
Chart 1. Biologically Relevant Cysteine Chemotypesa.

a Red, irreversible modifications. Green, unique enzyme intermediates. Note: Additional modifications can form as enzyme intermediates including thiyl radicals, disulfides, and persulfides.
Sulfur occupies a unique position in biology because of its ability to adopt a wide range of oxidation states (−2 to +6) and chemically unique forms or “chemotypes”3a each with distinct pathways of formation, physical and reactivity properties. Redox reactions of cysteine residues can lead to an array of post-translational modifications that are an important mechanism for the regulation of proteins from all major functional categories (e.g., enzymes, contractile, structural, storage, and transport proteins). Among these modifications are reversible, regulatory disulfides, thiosulfinates, S-glutathionylation, sulfenic acids, sulfenamides, sulfinamides, S-nitrosylation, and persulfides in conjunction with largely irreversible species, such as sulfinic acids, sulfonic acids, and sulfonamides that are often viewed as hallmarks of oxidative stress and disease.4 In regards to terminology, we note that the “-yl-“ particle in the terms above has gained widespread use in recent years5 as an analogy to other post-translational modifications, such as phosphorylation or acetylation, and is not intended to indicate a specific mechanism of S-group attachment or a radical-associated process.
The reversibility of many oxidative post-translational modifications (oxPTMs) of cysteine thiols highlights their ability to function as a binary “switch”, regulating protein function, interactions and localization, akin to phosphorylation. Given this analogy, and the discovery of biological RO/N/S-generating systems, it not surprising that investigation of cell signaling pathways involving oxidation of cysteine residues has emerged as an extremely active area of research. However, elucidating the functional role of cysteine oxPTMs in normal physiology and disease has been hampered, in part, because of the difficultly in detecting these modifications in complex biological systems with chemical specificity. After a brief introduction reprising major RO/N/S species produced by cells and mechanisms of thiol oxidation, we focus this review on different oxPTMs of protein cysteine thiols, with particular emphasis on those chemical properties that differentiate one modification from another. In keeping with this general theme, we review recent progress in using chemical approaches to develop probes that enable selective and direct detection of individual modifications within their native cellular environment. Along the way, we complement this discussion with examples from the literature that highlight ways in which cysteine oxidation can be used to control protein function and cell signaling pathways.
2. Cysteine Reactivity and Oxidant Sensitivity
Ionization constants (pKa) for the low-molecular weight thiols, cysteine (Cys), and glutathione (GSH), are 8.3 and 8.8, respectively. However, pKa values for cysteine residues in proteins can be strongly influenced by the local environment. For example, the two active-site cysteines in the DsbA disulfide oxidoreductase have pKa values of 3.5 and 10.6 Low pKa protein thiols, particularly those ionized at physiological pH, are often referred to as “reactive cysteines”.7 Features of the protein environment that can facilitate thiol ionization include proximity to positively charged amino acids,8 hydrogen bonding,9 and location at the N-terminal end of an α-helix (Ncap).10 For example, Ncap effects on cysteine reactivity have recently been noted in the thiol peroxidase, peroxiredoxin 1 (Prx1),11 and the epidermal growth factor receptor (EGFR) kinase.11b,12
Although the molecular basis remains incompletely understood, empirical observations indicate that not all cysteine residues in an individual protein are equally sensitive to oxidation. Since thiolates are much stronger nucleophiles than thiol groups, one key factor in oxidization susceptibility is low pKa. This fact is highlighted by the observation that many biological oxidants, such as hydrogen peroxide (H2O2), react exclusively with the thiolate anion.13 On the other hand, as noted by Winterbourn and Hampton, low pKa is not the only determinant of oxidant reactivity.14 To illustrate this point, one need only to consider the 1 000 000-fold difference in reaction rate constants of H2O2 with the active site cysteine of peroxiredoxin 2 (pKa ≈ 5–6; 2 × 107 M–1 s–1)15 and protein tyrosine phosphatases (PTPs), such as PTP1B (pKa ≈ 5.4; 20 M–1 s–1).16 Structural and functional studies suggest that the superior reactivity of Prx2 is due to a protein environment that is preorganized to activate both the peroxidatic cysteine and the peroxide substrate, as well as to stabilize the transition state for the SN2 substitution reaction.11a,15 In short, low pKa protein thiols are prime candidates for oxidation, but it is also important to recognize that “reactive cysteine” and “oxidant-sensitive cysteine” are not always synonymous with one another. A more extensive discussion of this topic has been presented by Winterbourn and colleagues.2d,14
2.1. Methods to Identify Low-pKa Cysteine Residues
From first principles, we know that cysteine reactivity depends on features of the local protein microenvironment; however, there is still much to learn about sequence and structural motifs that are associated with lowering cysteine thiol pKa.9b One approach to understand these features is to generate a comprehensive list of proteins that harbor low pKa cysteines and collate this information with sequence and three-dimensional (3D) structural data. To this end, a number of methods have been developed to identify low pKa cysteine residues in proteins.
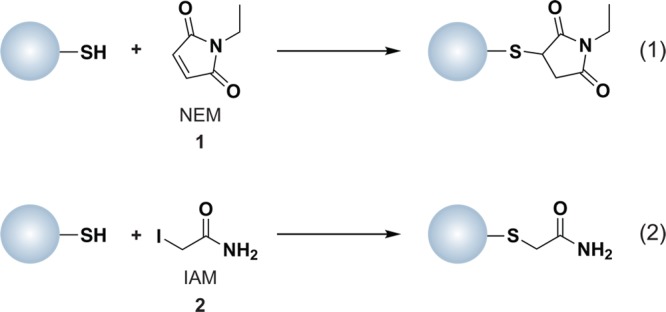
Computational methods to identify reactive cysteines in the proteome are often based on the conservation of redox-active cysteine residues, particularly those required for catalysis.17 Chemical methods typically employ reagents such as N-ethylmaleimide (NEM, 1) or iodoacetamide (IAM, 2), which form covalent adducts with sulfhydryl groups by Michael addition or nucleophilic substitution (SN2), respectively (Chart 2). The reaction of NEM with thiols is faster than IAM and less dependent on pH.18 However, IAM is more specific for thiols than NEM, which can modify side chain amines, such as histidine and lysine, when used in large excess or at basic pH.19 Since the thiol primarily reacts with IAM as the unprotonated thiolate anion, this reagent is most frequently used to identify low pKa cysteines, also referred to as the “reactive thiol proteome”.18,20 Both NEM and IAM can be conjugated to biotin or fluorophores to facilitate enrichment of labeled proteins, followed by one or two-dimensional (1 or 2D) gel electrophoresis with subsequent identification by liquid chromatography-tandem mass spectrometry (LC-MS/MS). In one recent example, N-(biotinoyl)-N′-(iodoacetyl)ethylenediamine, commonly referred to as biotinylated iodoacetamide (BIAM), was used to identify surface-exposed reactive cysteine residues in Saccharomyces cerevisiae.21 In yet earlier examples, BIAM and 5-iodoacetamido-fluorescein were used at low micromolar concentrations and mildly acidic pH to label reactive thiols.22 The majority of methods for profiling reactive cysteine residues use the alkylating reagent at a single concentration; however, a recent study by Weerapana et al. employed a range of IAM concentrations and differential isotopic labeling to identify reactive cysteines.23 Identifying low pKa cysteine thiols affords a list of proteins that are candidates for redox-mediated modification, but additional studies are required to evaluate oxidant sensitivity.
Chart 2. Protein Thiols React with N-Ethylmaleimide (NEM, 1, Equation 1) and Iodoacetamide (IAM, 2, Equation 2) by Michael Addition or SN2 Displacement, Respectively.
3. Reactive Oxygen Species (ROS) in Biological Systems
Among biologically relevant and abundant ROS (Chart 3), superoxide (O2•–) and H2O2 appear most important in receptor-mediated signaling. Although rates of cellular O2•– production can be high, in most mammalian cells the steady-state concentration is estimated to be in the low picomolar range (note that cellular concentrations and half-lives for ROS are approximate and can vary considerably depending on the cell type, nutritional and environmental conditions, as well as the stage of the cell-cycle).24 This is due to the rapid rate constant for spontaneous dismutation of O2•– to H2O2 and molecular oxygen (∼105 M–1 s–1) or as catalyzed by the superoxide dismutase (SOD) enzyme family, which is 104 times as fast (∼109 M–1 s–1).25 In turn, antioxidant enzymes, such as peroxiredoxin (Prx), catalase (CAT), and glutathione peroxidase (GPx), maintain steady-state intracellular H2O2 levels in the nanomolar to low micromolar range.24b,26 Compared to other ROS in Chart 3, H2O2 is a mild oxidant and has the longest cellular half-life (∼1 ms).2a,24b,26,27 Owing to its relative stability and selective reactivity, H2O2 appears well suited for a second messenger role.
Chart 3. Formation and Transformation of Biologically Relevant Reactive Oxygen Species (ROS)a.
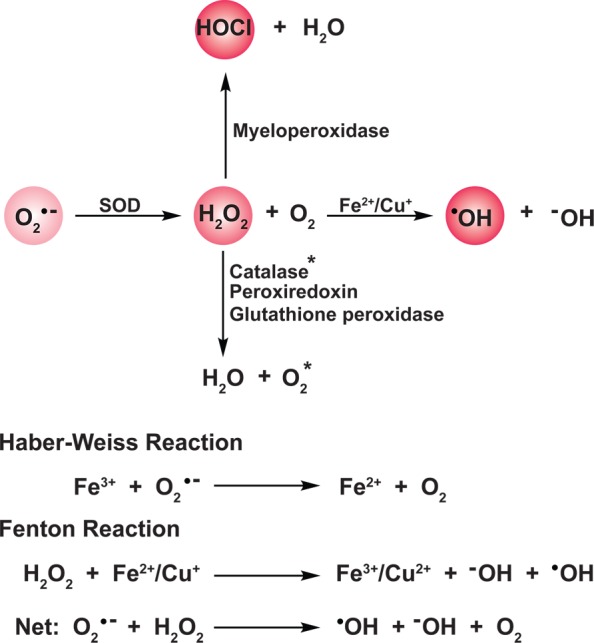
a Superoxide (O2•–), formed predominantly from the mitochondrial electron transport chain and NADPH oxidase enzyme complexes (not shown), is dismutated to hydrogen peroxide (H2O2) and oxygen by superoxide dismutases (SOD). H2O2 is in turn metabolized by catalases, peroxiredoxins, and glutathione peroxidases. Additionally, H2O2, alone or in concert with O2•—, can react with trace metal ions (Fe2+ or Cu+) to generate hydroxyl radical (•OH) via Fenton or Haber–Weiss chemistry, respectively. In phagosomes, H2O2 serves as a substrate for myeloid peroxidase to produce hypochlorous acid (HOCl) and water. Color intensity correlates to relative ROS reactivity.
The relative stability and uncharged nature of H2O2 may permit its diffusion through membranes, though this diffusion would be less rapid than that of gases, such as nitric oxide (•NO) and hydrogen sulfide (H2S). Recent studies indicate that aquaporins, a family of small (24–30 kDa) pore-forming integral membrane proteins, can also mediate H2O2 transport.28 Underscoring its diffusible nature and relative stability, H2O2 is known to function as a mobile paracrine signal to regulate plant cell differentiation29 as well as recruitment of immune cells for wound healing in eukaryotes.30 By contrast, the negatively charged O2•— does not freely diffuse across membranes (though evidence for its translocation via anion channels has been reported31). The protonated form of O2•– (HO2• pKa ≈ 4.9) is membrane permeable but is only present in low amounts at physiological pH (<0.2% at pH 7.4). Nonetheless, HO2• may be relevant in phagocytes where O2•– may reach a steady-state concentration of ∼25 μM.32
H2O2 alone, or in concert with O2•–, can also react with trace metal ions (Fe2+ or Cu+) to generate the hydroxyl radical (•OH) via Fenton or Haber–Weiss chemistry, respectively (Chart 3).33 Unlike O2•– and H2O2, whose production and metabolism are regulated processes, there are no known enzyme antioxidants for •OH neutralization. The •OH is a strong oxidant and reacts indiscriminately at diffusion-limited rates with protein, DNA, and lipid biomolecules,24b,34 which contributes to its short cellular half-life (∼1 ns).24b In healthy cells, •OH formation is low since H2O2 metabolism and metal ion concentrations are both tightly regulated to avoid toxicity. Conversely, pathologies that are associated with aberrant H2O2 metabolism or the presence of adventitious uncomplexed metal ions are often associated with increased •OH production and oxidative damage. For instance, mutations in Cu,Zn-SOD linked to familial amyotrophic lateral sclerosis (FALS) enhance •OH formation by Fenton and Haber–Weiss reactions and contribute to motor neuron degeneration.35
3.1. ROS Production and Metabolism
The subsections below outline important biological sources of ROS, which are formed as byproducts of respiration or by the action of enzymes. Although our discussion is focused primarily on the initial species generated by reduction of oxygen (O2•– and H2O2) important secondary products, such as hypohalous acids (HOX) are also briefly covered. The interested reader is also directed to these sources for more information about the regulation of ROS metabolism26,36 and methods for ROS detection.37
3.1.1. Mitochondrial Sources of ROS
The mitochondrial electron transport chain (ETC) funnels electrons from reduced metabolic components (NADH and FADH2) in the mitochondrial matrix through four protein complexes (I–IV) in which molecular oxygen serves as the terminal electron acceptor and is reduced to water (Figure 1a). The energy released during electron transfer is used to establish a proton gradient across the inner mitochondrial membrane that is harnessed to drive the production of the primary cellular energy source, adenosine-5′-triphosphate (ATP) via ATP synthase (complex V). This is an imperfect system, however, and electrons can leak prematurely from the ETC at complexes I and III resulting in the univalent reduction of molecular oxygen to O2•– in either the matrix (complex I and III) or the intermembrane space (complex III) (Figure 1a).26,38 It is estimated that 0.15–2% of molecular oxygen consumed is converted to O2•– by the mammalian ETC.38b,39 While this figure may seem low, mammals consume a large amount of oxygen resulting in the constitutive production of a significant amount of O2•– (and H2O2 through O2•– dismutation). For example, mutant mice lacking mitochondrial manganese-SOD (Mn-SOD) exhibit neonatal lethality resulting from neurodegeneration and cardiomyopathy, which may be rescued by small-molecule scavengers of O2•–.40 Deletion of individual SOD genes is also detrimental to bacteria41 and yeast42 survival further highlighting the impact of O2•– production in the ETC. Clearly, mitochondria are significant contributors to cellular H2O2 generation by dismutation of O2•– from the ETC.
Figure 1.

Biological sources of reactive oxygen species (ROS). (a) The mitochondrial electron transport chain (ETC). Four protein complexes (I–IV) funnel electrons (black arrows) from NADH and succinate in the matrix to ultimately reduce molecular oxygen to water and establish a proton gradient (gray arrows) that is harnessed by complex V to generate ATP. Electrons can leak prematurely from the ETC at complexes I and III (red arrows) to generate superoxide (O2•–) in either the matrix or intermembrane space. (b) p66 (Shc) facilitates pro-apoptotic O2•– or H2O2 production in the mitochondria. In response to UV irradiation or growth factor deprivation, p66 (Shc) localizes to the mitochondria where it interacts with complex III to divert electrons from cytochrome c directly to molecular oxygen to generate O2•– or H2O2. This H2O2 can translocate to the cytoplasm (not shown) where it can influence signaling, and can regulate opening of the mitochondrial permeability transition pore (mPTP), which initiates mitochondrial swelling and apoptosis. (c) NOX enzyme complexes assemble at distinct regions of the plasma membrane or intracellular membranes to regulate localized ROS production in response to diverse signals. Receptor stimulation initiates the recruitment of specific coactivating proteins or calcium to one of seven NOX catalytic cores. Once activated, NOX enzymes funnel electrons from NADPH in the cytoplasm through FAD and heme cofactors across the membrane to generate O2•– (NOX1-2) or H2O2 (Duox1-2) on the extracellular/lumenal face. O2•– is dismutated to H2O2 and oxygen either spontaneously or as enhanced by SOD, which can translocate across the membrane by diffusion or, more likely, through aquaporin channels to regulate protein activity and signaling in the cytoplasm.
The amount of mitochondrial-derived O2•– is variable43 and regulated by a number of factors, such as oxygen concentration, proton motive force,44 ETC efficiency,45 and the availability of electron donors. Pathologies that include neurodegenerative disorders, cancer, and diabetes are associated with mitochondrial dysfunction and enhanced ROS production.46 Mitochondrial stress and ROS-dependent AMP kinase activation have also been implicated in maternally inherited hearing loss.47 Recent studies in mice and yeast have revealed an evolutionarily conserved mechanism that cells use to control mitochondrial O2•– production.48 This is accomplished by adjusting the flux through metabolic pathways that regulate the flow of electrons into the ETC. Interestingly, these studies show that ROS-dependent inactivation of pyruvate kinase or a switch in isoform expression can redirect metabolic flow through the pentose phosphate pathway, which makes the reduced nicotinamide adenine dinucleotide phosphate (NADPH) required to maintain cellular redox homeostasis.
Extrinsic and intrinsic signals can also regulate mitochondrial O2•– production. This process is strictly dependent on the adaptor protein p66(Shc), which regulates the level of ROS, apoptosis induction, and lifespan in mammals.49 Cell signals including growth factor deprivation, oxidative stress, or UV irradiation induce translocation of p66(Shc) into the mitochondria where it promotes electron transfer from Complex III directly to oxygen, enhancing O2•— production (Figure 1b).50 After conversion to H2O2 through dismutation, this ROS diffuses into the cytoplasm where it decreases the activity of FoxO3, a transcription factor that regulates the expression of mitochondrial antioxidant enzymes, including Mn-SOD and catalase.51 The reduction in antioxidant capacity further increases mitochondrial oxidative stress and enhances the pro-apoptotic function of p66(Shc).52 Of note, mutant Mn-SOD heterozygous knockout mice exhibit marked sensitization of the mitochondrial permeability transition pore (mPTP) and premature induction of apoptosis.53 Mice lacking p66(Shc) live ∼30% longer and show increased resistance to oxidative stress and age-related pathologies, marking it as a potential therapeutic target for diseases that are associated with oxidative damage.26,49,50,54 Several studies suggest an additional role for mitochondrial ROS in immune system function.55 For instance, a recent report demonstrated recruitment of mitochondria to phagosomes in infected activated murine macrophages and that mitochondrial-derived ROS was required for microbial killing.56 Mice lacking p66(Shc) also exhibit decreased O2•– production in macrophages, highlighting another potential role for p66(Shc)-regulated mitochondrial ROS production.57
Although a thorough review of the plant literature in this area is beyond the scope of this review, we would be remiss if we did not note that in plant cells O2•– is also produced in the mitochondria by the ETC, as well as other subcellular compartments, such as chloroplasts and peroxisomes through photorespiration.58 The amount of ROS generated via photorespiration can increase in response to environmental constraints, including biotic and abiotic stresses. The interested reader is referred to the following extensive reviews for additional information on this topic.59
3.1.2. Enzymatic Generation of ROS
In addition to mitochondrial sources of O2•–, this reactive intermediate can be generated as a byproduct during the catalytic cycle of numerous enzymes, such as “nonspecific” peroxidases (i.e., haem-containing peroxidases capable of using H2O2 to oxidize a range of substrates), as well as xanthine and aldehyde oxidases.3a,60 Electron leakage from NADPH cytochrome P450 reductases present in the endoplasmic reticulum (ER) can also generate O2•– during hormone and drug metabolism.61 The autoxidation of glyceraldehydes, reduced flavin mononucleotide (FMNH2), and reduced flavin adenine dinucleotide (FADH2) can also produce O2•–, albeit with slow reaction kinetics.24b,24c As noted above, the dismutation of O2•– provides a major source of H2O2 in cells. In addition, there are numerous enzymes that produce H2O2 without the intermediacy of O2•–, including xanthine, glucose, lysyl, monoamine, and d-amino acid oxidases, as well as the peroxisomal pathway for beta-oxidation of fatty acids.62 The contribution of these sources of O2•– and H2O2 to redox signaling remains to be determined.
In activated phagocytes of the immune system, myeloperoxidase- and eosinophil peroxidase-catalyzed oxidation of halide (Cl–, Br–, I–) and pseudohalide (SCN–) ions converts H2O2 to the corresponding hypohalous acid (HOX), such as hypochlorous acid (HOCl) (Chart 3).2d,32e,63 HOXs react preferentially with thiols and methionine residues and these potent oxidants are generally believed to be responsible for much of the bactericidal activity of neutrophils. The reaction of HOCl with O2•– is also known to generate •OH and is proposed to serve as the primary source of •OH in neutrophils.64 The interested reader is referred to the following sources for additional information on this unique class of oxidants.2d,32e
A variety of extracellular signals including peptide growth factors, cytokines, and G-protein-coupled receptor (GPCR) agonists and, more recently, mechanical distortion in cardiomyocytes65 trigger deliberate production of ROS through activation of NADPH oxidase (NOX) complexes.66 NOX-derived ROS is required for propagation of many pathways12,65,67 and the maintenance of essential stem cell populations in the brain.68 NOX complexes produce ROS with one of seven enzymatic cores (NOX1-5, Duox1, and Duox2) that exhibit differential cell- and tissue-specific expression patterns. As illustrated in Figure 1c, activation of NOX requires association of a flavin adenine dinucleotide (FAD) cofactor, distinct membrane and cytoplasmic coactivator proteins (Nox1-4, Duox1, and Duox2) or binding of calcium to the intracellular domain (Nox5, Duox1 and Duox2).36a,36b,36d As follows, NOX activation can be tightly controlled by signal-mediated recruitment of these coactivating proteins69 or cofactors,69c,70 which are likely to be pathway- and isoform-specific.
The activated NOX transports an electron from cytoplasmic NADPH through FAD and heme cofactors across plasma and intracellular membranes to produce O2•– on the extracellular/lumenal face (Figure 1c).36a,36b,36d,71 O2•– is then dismutated to H2O2 and molecular oxygen, either spontaneously or via extracellular SOD,72 though some NOX isoforms (Duox1 and Duox2) are equipped with an extracellular peroxidase domain that is believed to directly mediate two-electron reduction of molecular oxygen to H2O2.73 Translocation of electrons from the cytoplasm across biological membranes with the concomitant release of protons from NADPH results in local acidification proportional to oxidant production. In neutrophil phagosomes, where NOX2 is estimated to produce O2•– at steady-state levels of 25 μM,32d sustained NOX2 activity is coupled to voltage-gated proton channels to mitigate local acidification.74 A similar dependence on a voltage-gated proton channel has been demonstrated for prolonged NOX activation in active B cells.75 The efflux of electrons also results in net positive charge accumulation on the ROS-producing face, which may promote electron transfer through NOX. Recently, a nonselective cation (Ca2+, Na+, K+) channel called, TRPM2, was shown to be activated by NOX-derived ROS.76 TRPM2 activation depolarized the plasma membrane, which dampened NOX-mediated ROS production in phagosomes. This finding presents a novel mechanism by which cells can regulate the amplitude and duration of NOX activity.
Within a given signaling pathway, identifying which NOX isoform is acting as the primary ROS source is usually accomplished by determining the relative expression level of each isoform using isoform-specific antibodies12 or by overexpressing the isoform of interest.77 However, inherent differences in antibody affinity and specificity issues can complicate these determinations, and protein overexpression does not reflect native conditions. Many cell types express multiple NOX isoforms, making it difficult to discern isoform-specific roles in a given signaling pathway, as knockout or siRNA knockdown studies are not always feasible. The participation of NOX in a given signaling pathway is commonly assessed using a number of small molecule inhibitors, including apocynin or the flavin analog, diphenyleneiodonium (DPI). These results should be interpreted with caution, as both compounds have been shown to have off-target effects in some cell types.78 Isoform-specific NOX inhibitors would greatly assist in dissecting the role of individual NOX family members in signaling pathways.79 For example, a peptide inhibitor that is highly specific for NOX2 has been used to study its role in vascular O2•– production in mice80 and during mechanical distortion in cardiomyocytes.65 High-throughput screens have also identified small-molecule inhibitors of NOX181 and NOX2.82
H2O2 that results from NOX activation can enter the cytoplasm through diffusion, or as recently shown, by transport through aquaporin channels where it can mediate distinct physiological responses, such as proliferation, differentiation, and apoptosis.26,83 Since H2O2 that is produced extracellularly or in the luminal space must enter the cytoplasm to modulate intracellular signaling pathways, one key question is how can its effects be localized? Much remains to be understood about this important aspect of redox signaling, however, one possible answer is that aquaporins are directed to lipid raft membrane microdomains84 that are also enriched for NOX. Indeed, NOX isoforms are both temporally and spatially localized to distinct membrane regions via lipid rafts,36b activated receptors,12,70 and focal adhesions.85 Depending on the stimuli and cell type, NOX family members also localize to distinct subcellular compartments, such as the ER86 and nucleus.87 As will be discussed in more detail below, the localized activities of NOX, as well as antioxidant enzymes that metabolize ROS may also help restrict H2O2 to regions where signaling proteins are similarly localized.
3.1.3. ROS-Metabolizing Enzymes
As stated above, dismutation of O2•– by SOD produces H2O2. The peroxiredoxin (Prx) and glutathione peroxidase (GPx) families are primarily responsible for the metabolism of H2O2 in cells. These enzymes decompose H2O2 to form water and molecular oxygen in a mechanism involving the oxidation of an active site cysteine (or selenocysteine in GPxs from higher eukaryotes).88 The enzymes are recycled back to their active, reduced form by thioredoxin/thioredoxin reductase (Trx/TrxR) or glutathione/glutathione reductase (GSH/GR) systems using reducing equivalents from NADPH. Another H2O2-metabolizing enzyme, known as catalase, is present mainly in peroxisomes. Plants synthesize high concentrations of ascorbate,59b which is used as a substrate by ascorbate peroxidases to regulate H2O2 bioavailability in these systems.89 Ascorbate peroxidases are subsequently reduced by a complex metabolic pathway, known as the glutathione-ascorbate cycle.90 A growing list of antioxidant enzymes, including Prxs, are themselves subject to redox regulation, which could permit localized accumulation of H2O2 for signaling while simultaneously limiting the range of H2O2 diffusion.11b,91
3.2. Modification of Protein Cysteine Thiols by ROS
The reaction of ROS with protein thiols provides a mechanism by which cells can “sense” changes in the redox balance. Though H2O2 is most often associated with a second messenger role, there is also evidence to suggest that O2•– functions in this capacity. For instance, a recent study demonstrated that disparate gradients of O2•– and H2O2 differentially regulated plant root proliferation and differentiation, respectively implicating distinct activities for these ROS.92 O2•– is a relatively unreactive radical and its primary cellular targets appear to be other radical species, such as nitric oxide (•NO) or metals. In proteins, O2•– can react with iron–sulfur clusters and heme centers leading to release and/or oxidation of iron.13 Numerous iron–sulfur cluster- and heme-containing proteins are sensitive to O2•–, including aconitase,93 the bacterial transcription factor SoxR,94 guanylate cyclase,95 and myeloperoxidase.96 Reactivity at protein metal centers is not unique to O2•–, however, as metal-dependent peroxide sensors like Bacillus subtilis PerR have also been reported.2a,83c,97 In contrast to redox switches based on peroxide-sensitive cysteine residues, PerR senses H2O2 by metal-catalyzed oxidation of histidine residues involved in coordinating Fe2+ (note that the mechanism involves reduction of H2O2 by Fe2+ to generate •OH, which then reacts rapidly with histidine). H2O2 may also modify tryptophan and tyrosine residues through a radical-based mechanism, but such reactions are much less favored and may not be physiologically relevant.98
H2O2 can directly oxidize the thioether group of methionine to yield two diastereomeric methionine sulfoxide products;99 however, a large body of evidence identifies cysteine as the most sensitive amino acid residue to H2O2-mediated oxidation. The two-electron oxidation of a thiolate by H2O2 yields sulfenic acid, which is increasingly implicated in a number of important biochemical transformations. Second-order rate constants for this reaction can vary dramatically in proteins (e.g., 20–107 M–1 s–1).14 Once formed, the sulfenic acid is subject to several alternative fates (Figure 2). Depending on the microenvironment, the sulfenic acid modification can be stabilized as observed in human serum albumin (HSA)100 and more than 40 protein crystal structures.9b,101 In this regard, there are several factors that appear to stabilize protein sulfenic acids, including the absence of thiols proximal to the site of formation or inaccessibility to low-molecular-weight thiols, such as GSH (γ-l-Glu-l-Cys-Gly).3b Reaction of sulfenic acid with a protein thiol or GSH yields an inter/intramolecular disulfide bridge or protein-S-GSH disulfide, respectively. Alternatively, in some proteins lacking a neighboring cysteine, a nitrogen atom of a backbone amide can react with sulfenic acid, forming a cyclic sulfenamide.102 The formation of disulfide and sulfenamide states protects against irreversible overoxidation, as S–S and S–N bonds can be reduced through the activity of Trx/TrxR or GSH/glutaredoxin (Grx)/GR systems.103 Sulfenic acid can also be reduced directly by the Trx system, through hydride transfer (H–) from FADH2 in a reaction catalyzed by NADH oxidase and NADH peroxidase enzymes from Streptococcus faecalis,104 or through the DsbD/DsbG system in the bacterial periplasm.105 In the presence of excess H2O2, sulfenic acid can be further oxidized to sulfinic (RSO2H) and sulfonic (RSO3H) oxyacids, though the observed rate constants for such reactions are generally slower (0.1–100 M–1 s–1) than the initial thiolate oxidation event (Figure 2).15,104b,106
Figure 2.
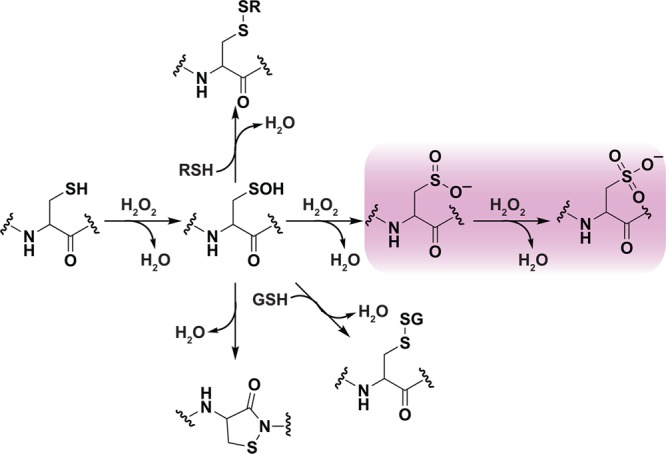
Oxidative modification of cysteine residues by hydrogen peroxide (H2O2). The initial reaction product of a low pKa protein thiolate with H2O2 yields a sulfenic acid, whose stability is determined, in part, by its accessibility to additional thiols. Reaction with a second cysteine in the same or neighboring protein yields a disulfide. Alternatively, reaction with the low molecular weight thiol, glutathione (GSH) affords a specialized mixed disulfide called a glutathione disulfide. In some proteins in which a neighboring cysteine is not present, nucleophilic attack of a backbone amide on the sulfenic acid yields a cyclic sulfenyl amide. Each of these oxoforms can be reduced by the GSH/glutaredoxin or thioredoxin/thioredoxin reductase systems to regenerate the reduced thiolate (not shown). In the presence of excess H2O2, such as under conditions of oxidative stress, the sulfenic acid can be hyperoxidized to the largely irreversible sulfinic and sulfonic acid forms (red box).
HOXs, such as HOCl, also mediate two-electron oxidation of cysteine. These reactions proceed through X+ transfer to give an unstable sulfenyl halide, which rapidly hydrolyzes to sulfenic acid (>107 M–1 s–1 for HOBr and HOCl).107 HOXs are aggressive oxidants and halogenating agents, which react with a wide range of cellular targets, including methionine, histidine, tryptophan, lysine, tyrosine, the protein backbone, nucleic acids and fatty acids. On the whole, the modifications of biomolecules that are mediated by HOX are numerous and highly damaging, which makes these oxidants highly effective toxic defense molecules that can be exploited by the human immune system to fight off microbial infection. As a final comment in this section, we note that the oxidation of cysteine thiols can also occur by one-electron redox pathways to give thiyl radicals, which undergo distinct sets of reactions. These transformations are briefly discussed in section 5 below (Reactive Sulfur Species (RSS) in Biological Systems) and we also refer the interested reader to the following sources for additional information.3a,14,108
3.3. Methods for Detecting ROS-Modified Cysteines
The reversible nature of cysteine sulfenic acid, disulfide and S-glutathionylation makes them well suited to control protein function during cell signaling. With the discovery of Sulfiredoxin (Srx) proteins,109 which can convert the sulfinic acid modification back to the thiol form, cysteine sulfinic acids have also emerged as a potential regulatory mechanism. Consequently, there has been considerable effort to develop methods to study changes in protein cysteine oxPTM. These techniques include indirect and direct methods for detection. The majority of indirect methods to detect cysteine oxidation rely upon the loss of reactivity with thiol-modifying reagents (Figure 3a) or restoration of labeling by reducing agents such as dithiothreitol (DTT) (Figure 3b). The latter method requires a complete blocking of free thiols with alkylating agents prior to the reduction step and is therefore limited to studies in cell lysates or with purified proteins.
Figure 3.

General overview of indirect and direct chemical methods to study protein oxidation. (a) Loss of labeling of oxidized thiols by an alkylating agent indirectly monitors protein oxidation. In response to oxidant treatment, susceptible cysteines are oxidized (purple) and thus are less reactive with alkylating agents such as NEM or IAM. Use of a biotinylated or fluorophore-conjugated alkylating agent permits detection by avidin blot or in-gel fluorescence, in which oxidized proteins exhibit a loss of signal. (b) Differential alkylation of reduced and oxidized thiols indirectly monitors protein oxidation. Free thiols (blue) are blocked with an alkylating agent such as NEM or IAM, reversibly oxidized thiols (purple) are reduced with a reducing agent such as DTT or TCEP, and nascent thiols are labeled with a second alkylating agent conjugated to biotin or a fluorophore. Oxidized proteins exhibit enhanced signal by avidin blot or in-gel fluorescence. (c) Direct chemical method to detect specific cysteine oxoforms. Samples are treated with a biotin or fluorophore-conjugated probe that selectively reacts with a distinct cysteine chemotype (purple) in which signal by avidin blot or in-gel fluorescence increases with increased protein oxidation.
More recently, chemical biology approaches have facilitated the development of small molecule- and protein-based methods for direct detection of distinct oxidative cysteine modifications (Figure 3c). In the event that these small molecules are cell permeable, specific cysteine modifications can be detected directly in their native environment without cell disruption (i.e., lysis). This is an attractive approach since it preserves labile cysteine modifications and maintains the integrity of subcellular organelles. The latter is especially important as organelles like the nucleus, mitochondria, and cytoplasm have more reduced redox potentials whereas the secretory system and the extracellular space are more oxidizing environments.110 Not surprisingly, cell lysis disrupts these individual redox environments and can result in substantial protein oxidation artifacts. The net result is to increase the challenges related to detecting low abundance modifications and in deciphering their biological significance. Likewise, cell disruption can hamper the detection of labile or transient cysteine modifications.
Methods to decrease oxidation artifacts in lysates have been reported, but these are often dependent upon the addition of trichloroacetic acid (which denatures proteins and can lead to acid-catalyzed overoxidation of labile modifications such as sulfenic acid) or on the addition of ROS-metabolizing enzymes to the lysis buffer.111 Even with these considerations, lysis buffers can never accurately mimic the intracellular redox potential, thereby exposing redox-sensitive proteins to oxygen and a different redox environment. Direct detection methods may also be associated with their own limitations as the addition of a small-molecule probe to cells could alter the biological function under investigation. This issue can be addressed, at least in part, by adding the probe to cells after signal pathway activation and/or by monitoring the effect of probe addition on relevant downstream biological markers.12 Another important consideration with direct detection methods is the rate at which probes react with the modified cysteine residue. If the reaction is slow, transient cysteine oxidation events may be missed. Conversely, if the reaction is too fast it could diminish the chemical selectivity of the probe or disrupt the biological process under study. In this way, moderately reactive probes for detecting individual oxidative cysteine modifications may be viewed as “spectators”, which sample the redox-signaling environment with minimal biological impact. Increasing the concentration of probe can also compensate for modest rates of reaction, but appropriate controls must be performed to ensure that the underlying biology is not disturbed.
Collectively, indirect and direct methods to monitor cysteine oxidation have enabled the discovery of many proteins that can undergo redox modification in a wide range of organisms and different cell types. To highlight the progress made over the past few years in the redox biology field, the following subsections will independently address the chemical properties of ROS-mediated cysteine modifications and methods for their detection. We also discuss selected examples from the recent literature that highlight the ways in which distinct cysteine modifications can mediate critical biological events.
3.3.1. Indirect Approaches for Detecting ROS-Sensitive Cysteines
Several methods have been developed to monitor global changes in cysteine oxidation, but do not reveal the chemical nature of the modification. One of the most commonly used reagents for this purpose is the BIAM alkylating reagent. In these experiments, the diminished nucleophilicity of the oxidized cysteine residue results in lower reactivity with BIAM and correlates with a loss of protein labeling (Figure 4a). An adaptation of this methodology that permits simultaneous identification and quantification of oxidant-sensitive cysteine thiols employs an acid-cleavable BIAM-based isotope-coded affinity tag (ICAT).112 In this method, free thiols are differentially alkylated with isotopic versions of the ICAT reagent and the extent of cysteine oxidation is determined by the ratio of light (12C) and heavy (13C) ICAT label by LC-MS/MS (Figure 4b).
Figure 4.

Indirect chemical methods to study general cysteine oxidation. (a) Loss of labeling of oxidized thiols by biotinylated-iodoacetamide (BIAM) indirectly monitors protein oxidation. Oxidized cysteines (purple) exhibit decreased reactivity with BIAM than reduced thiols, and are observed as a loss of signal by avidin blot. (b) Isotope-coded affinity tag (ICAT) reagents determine the ratio of oxidized thiols. Samples are untreated or subjected to oxidant. Free thiols are subsequently labeled with a light (12C) ICAT reagent in the untreated sample and with a heavy (13C) ICAT reagent in the oxidant-treated sample. As in panel a, reactive thiols (purple) exhibit decreased labeling upon oxidation. The samples are mixed, trypsinized, and enriched via the biotin affinity tag on the ICAT reagent. Eluted peptides are analyzed by LC-MS and heavy and light ICAT-labeled peptides are chemically identical, but differ in mass by 9 Da. The fraction of a thiol oxidized in the sample is determined by the ratio of heavy (13C) to light (12C) signal intensity, whereby thiols that are susceptible to oxidation (purple) will exhibit decreased signal intensity with the heavy ICAT reagent.
A subsequent alternative approach incorporates treatment with a reducing agent into the workflow (Figure 3b). Such protocols require free thiol alkylation, a reduction step with DTT or tris(2-carboxyethyl)phosphine (TCEP), and labeling of nascent thiols with a tagged alkylating agent, such as BIAM. In this approach, changes in cysteine oxidation are detected as differences in sample BIAM alkylation as assessed by avidin blot and oxidized proteins can be identified by enrichment and LC-MS/MS analysis (Figure 3b). In addition to BIAM, alternative biotinylated or fluorophore-modified alkylating reagents can be used to differentially alkylate thiols and these methodologies have been used to monitor protein oxidation in response to exogenous oxidants (e.g., H2O2 or diamide)21,113 or to ROS-promoting stimuli (e.g., peptide growth factors).114 A similar workflow has also been used to identify substrates of the Trx/TrxR and GSH/Grx/GR systems.113 Alternatively, protein substrates of the aforementioned reducing systems can be identified through their inclusion in the reduction step.21 For instance, BIAM-alkylated nascent thiols will represent oxidized proteins that were selectively reduced by the Trx/TrxR or GSH/Grx/GR systems. Together with the ICAT technology, this method has been used to identify protein disulfide targets of the Trx/TrxR system in plant extracts.115 In addition to studying the oxidized proteome, changes in total thiol content in protein and low molecular weight thiols, including GSH and homocysteine, can be indicative of fluctuations in biological redox balance and, in some cases, serves as a diagnostic function for disease. In this vein, an active area of research is the development of sensitive probes to monitor fluctuations in total thiol content.116
3.3.2. Direct and Selective Approaches for Detecting ROS-Sensitive Cysteines
3.3.2.1. Disulfides
Disulfide bond formation in proteins is a widely recognized cysteine modification that has important roles in protein folding and stability. Under normal cellular conditions, disulfide bond formation occurs largely in the extracellular space or the endoplasmic reticulum (ER). In this organelle, a class of enzymes called protein disulfide isomerases (PDI) inserts disulfides into nascent proteins that are destined for export to the extracellular milieu.117 By comparison, disulfide bonds are rare and generally transiently formed in the cytoplasm, mitochondria, or nucleus where thiol-dependent reductases maintain a reducing environment. Exceptions exist, however, as the sulfhydryl oxidase Erv1 and oxidoreductase Mia40 form a relay system that introduces disulfide bonds in substrate proteins in the mitochondrial inner membrane.118 Under oxidative stress conditions the intracellular redox balance can shift to support disulfide bond formation in reducing compartments until redox homeostasis is restored.
A major route of disulfide formation is by thiol condensation with sulfenic acid (Figure 2). These processes can occur either intra- or intermolecularly, and the rate of disulfide bond formation is dependent, in part, upon the distance between the two cysteine residues. Estimated rate constants for intra- and intermolecular disulfide bond formation are 10 s–1 and 105 M–1 s–1, respectively.119 Once formed, disulfides are relatively stable to most physiological nucleophiles and are generally cleaved by other thiols as in thiol-disulfide exchange (nucleophilic substitution) reactions (Figure 5).120 The thiol in a disulfide with the lower pKa will be the better leaving group and often dictates which cysteine is released in thiol-disulfide exchange. Indeed, this strategy is employed by the thiol-disulfide exchange catalysts in the cell, such as protein disulfide isomerases (PDI).121 Disulfides can also be oxidized to generate a thiosulfinate, which can subsequently react with a thiol to give disulfide and sulfenic acid products (Figure 5). The prevalence or biological significance of the thiosulfinate is unknown, however, it is interesting to note that this species forms as an intermediate during Srx-catalyzed sulfinic acid reduction of Prxs.122 Although the intermediate thiosulfinate is formed via a mechanism distinct from disulfide oxidation, its formation implies that the thiosulfinate may be a physiologically relevant, yet understudied modification. Further oxidation of a disulfide yields a thiosulfonate (Figure 5), which releases a disulfide and sulfinic acid subsequent to reaction with a thiol. Thiosulfonates have not been detected in cells, but could possibly be formed as an enzyme intermediate in sulfonic acid reduction akin to sulfinic acid reduction via sulfiredoxin, though an enzyme capable of catalyzing such a reaction is currently unknown.3a
Figure 5.

Possible fates of protein disulfides. Once formed, a protein disulfide (inter- or intramolecular) can undergo thiol-disulfide exchange with a third cysteine within the same or neighboring protein (eq 1). Herein, pKa of the disulfide thiols and thiol accessibility influence which cysteine is expelled. In the presence of high concentrations of H2O2, disulfides can additionally be oxidized to the thiosulfinate and thiosulfonate forms, though these reactions are very slow. Because of the potential for resonance stabilization or decreased pKa, subsequent reaction of these intermediates with a third cysteine affords a disulfide and a sulfenic acid (eq 2) or sulfinic acid (eq 3). The biological relevance of the thiosulfinate and thiosulfonate modifications is unknown due to a lack of means to study these oxoforms, however, a thiosulfinate forms as an intermediate during the sulfiredoxin catalytic cycle.
Global studies to identify proteins that undergo disulfide bond formation implicate this modification in the regulation of, among others, redox homeostasis, chaperone activity, metabolism, transcriptional regulation, and protein translation.111b,113 Once formed, disulfides can impact enzyme activity, subcellular localization, as well as protein–protein interactions.71 For example, the activity of certain PTPs is inhibited by disulfide bond formation involving the active site cysteine and the so-called backdoor cysteine.106b,123 This regulatory mechanism is also observed in certain members of the caspase family of cysteine proteases.124 Numerous studies have demonstrated an increase in protein phosphorylation in response to receptor activation that is dependent upon endogenous H2O2 production.12,65,67,68 Owing to this observation and their conserved catalytic cysteine residue, PTPs were initially proposed as the major cellular targets of signaling-derived H2O2.125 Kinases are now also believed to be redox regulated, though in many cases the molecular details are much less well characterized. Nonetheless, it has been established that serine/threonine kinases PKG1α126 and ATM127 are activated by intermolecular disulfide formation between homodimers that, in the case of PKG1α, enhances its affinity for target proteins. By contrast, intermolecular disulfide formation between Src tyrosine kinase monomers appears to inhibit kinase activity,128 though Src has also been shown to be activated by H2O2.129 Differential regulation by H2O2 may be explained, in part, by modification of multiple cysteine residues. For example, oxidative inhibition of Src involves Cys277, which is not conserved in all Src family kinases.128 The Src-family kinase Lyn, which encodes a glutamine at the site corresponding to Cys277, is activated by ROS in neutrophils suggesting that oxidative activation of this enzyme involves a different cysteine residue.30e Additional proteins whose activity have recently been shown to be modulated by disulfide bond formation include the bacterial chaperone Hsp33,130 the nonspecific cation channel TRPA1,131 and the glycolytic enzyme pyruvate kinase M2 (PKM2).48a
Disulfide bond formation can also influence the subcellular localization of a protein and/or protein–protein interactions. For example, intramolecular disulfide formation in the Saccharomyces cerevisiae transcription factor Yap1 induces a conformational change that masks the nuclear export signal (NES) and precludes interaction with the nuclear export receptor, Crm1. This results in nuclear accumulation of Yap1 and active transcription of genes involved in the oxidative stress response.132 Intramolecular disulfide formation in the small molecular chaperone, DnaJb5 and the class II histone deacetylase, HDAC4 results in sequential dissociation of the DnaJb5-HDAC4 complex, unmasking of the HDCA4 NES to mediate its cytoplasmic localization and derepression of target genes involved in hypertrophy (Figure 6a).71,133 A recent study by Shacter and colleagues indicates that oxidative stress-induced formation of two intramolecular disulfides in the actin-regulatory protein, cofilin leads to dissociation of the actin-cofilin complex. Additionally, oxidation of cofilin enables its mitochondrial accumulation (by an unresolved mechanism) where it can interact with the mPTP to promote mitochondrial swelling, cytochrome c release, and ultimately induction of apoptosis (Figure 6b).134
Figure 6.

Disulfide-mediated redox regulation of subcellular localization and protein–protein interactions. (a) Model for redox regulation of cardiac hypertrophy by HDAC4. The class II histone deacetylase HDAC4 normally deacetylates histones to suppress expression of genes involved in cardiac hypertrophy. Nuclear localization of HDAC4 is mediated by its association with importin α (Imp) via a multiprotein complex including the small molecular chaperone DnaJb5, the thioredoxin binding protein TBP-2, and thioredoxin (Trx1). In response to oxidant, HDAC4 and DnaJb5 undergo intramolecular disulfide bond formation, which causes dissociation and nuclear export of the complex permitting derepression of genes involved in hypertrophy. Upon removal of H2O2, Trx1 is believed to reduce the disulfides in HDAC4 and DnaJb5 to restore assembly and nuclear localization of the complex (not shown). (b) Model for redox regulation of apoptosis by cofilin. Cofilin associates with actin in the cytoplasm to disassemble actin filaments for cytoskeletal reorganization. In the presence of H2O2, two intramolecular disulfides form in cofilin permitting its relocation to the mitochondria by an unresolved mechanism. In the mitochondria, cofilin interacts with the mPTP to stimulate pore opening, mitochondrial swelling, cytochrome c release, and ultimately induction of apoptosis.
Methods to detect protein disulfide formation often use reducing and nonreducing SDS-PAGE gel electrophoresis (Figure 7a). Intermolecular disulfides are detected as reducing agent-sensitive protein complexes that migrate at a molecular mass equal to the that of the two oxidized proteins, as seen for PKG1α,126 Src,128 and ATM127 dimers (Figure 7a, right). Intramolecular disulfide bond formation can also lead to altered migration on gels, as observed for S. cerevisiae thiol peroxidase Gpx3,66c,135 PKM2,48a or PTEN (Figure 7a, left).123b Cysteine residues involved in disulfide bond formation can also be identified by the differential alkylation-type approach mentioned above. In this method, thiols are alkylated prior to sample separation by nonreducing SDS-PAGE; the protein band corresponding to the oxidized proteins of interest is then reduced in-gel with DTT or TCEP, and nascent thiols are labeled with a second alkylating agent. The protein is then digested in-gel and the differentially alkylated cysteine residues are identified by LC-MS/MS analysis.127,134
Figure 7.

Methods for detection and identification of protein disulfides. (a) Differential migration of proteins containing intra- and intermolecular disulfide bonds. Samples are resolved under nonreducing SDS-PAGE conditions. Intramolecular disulfides can facilitate enhanced protein migration in some proteins as compared to the reduced species (left). Intermolecular disulfide complexes migrate at the combined molecular weight of the individual proteins (right). (b) Redox 2D-PAGE. Protein samples are first separated by nonreducing gel electrophoresis to separate disulfide-bonded complexes by size (top). The proteins are subsequently reduced in-gel with DTT, alkylated with NEM or IAM, and separated in the second dimension under reducing conditions (down). Proteins that are not involved in intermolecular disulfide complexes run at the diagonal. Proteins involved in disulfide complexes migrate off the diagonal and can be identified by in-gel digestion and LC-MS/MS (not shown). (c) OxICAT method combines the ICAT technology with differential alkylation of reduced and oxidized thiols to permit quantification of oxidized residues. Cell lysates are generated in the presence of trichloroacetic acid and detergents to facilitate exposure of all protein cysteines while inhibiting thiol/disulfide exchange. Reduced thiols (blue) are subsequently blocked with the light (12C) ICAT reagent (blue), oxidized proteins (purple) are reduced with TCEP, and nascent thiols are alkylated with the heavy (13C) ICAT reagent (purple). Samples are trypsinized and labeled peptides are avidin enriched. Eluted peptides are analyzed by LC-MS and heavy and light ICAT-labeled peptides are chemically identical, but differ in mass by 9 Da. The percentage of a particular thiol that is oxidized in a sample is determined by the ratio of heavy (13C) to light (12C) signal intensity from the corresponding peptide. While TCEP can reduce all reversible oxoforms (e.g., disulfides, sulfenic acid, S-nitrosothiols), sulfenic acids and S-nitrosothiols are often acid-labile and likely lost during sample preparation. As such, OxICAT is likely most suitable to detect cysteines involved in disulfide bonds.
The differential migration of disulfide-containing proteins by nonreducing and reducing gel electrophoresis have also been exploited to develop the only direct and high-throughput method to identify oxidant induced, disulfide-bonded protein complexes. This approach, termed diagonal SDS-PAGE136 or redox 2D-PAGE137 involves sequential nonreducing/reducing two-dimensional SDS-PAGE (Figure 7b). The protein mixture is first resolved by nonreducing gel electrophoresis to separate complexes by size, followed by excision of a narrow gel strip in the sample lane over the entire molecular weight range. The proteins are then reduced and alkylated in-gel to prevent disulfide bond reformation, the gel strip laid at a 90° angle across a second gel, and the proteins are subsequently resolved under reducing conditions. Proteins that are not involved in disulfide bond formation will lie in a diagonal line on the 2D gel, whereas proteins that form disulfide bonds will appear as distinct spots above or below the diagonal line. Protein identity is subsequently determined by LC-MS/MS analysis. A major limitation of this method, as with all 2D SDS-PAGE based methods, is that it cannot reliably visualize or produce analytical quantities of low abundance proteins that are present in less than 1000 copies per cell.138 Nonetheless, this procedure has been used to detect disulfide-linked proteins in whole cell lysates derived from oxidant-treated rodent nerve cell cultures139 and cardiac myocytes.140 As outlined above, redox 2D-PAGE identifies proteins that form disulfides but does not provide information as to which proteins form which complexes. An alternative approach is to first isolate the protein of interest using a protein-specific antibody or affinity tag. This procedure permits identification of proteins that form disulfides with a protein of interest, and was recently used to identify of a novel reducing system in the bacterial periplasm.105
One limitation of the redox SDS-PAGE approach is that it does not provide quantitative information about the extent or fraction of cysteine oxidized under a given condition. To enable identification and quantification of reversibly oxidized protein cysteine residues, including disulfides, the Jakob group has reported an extension of the ICAT technology, known as OxICAT (Figure 7c).111b Lysates are first generated in the presence of TCA to precipitate proteins and prevent thiol/disulfide exchange. Free thiols are then alkylated with a light (12C) ICAT reagent, followed by reduction of with TCEP, which serves to reduce reversible modifications (Chart 1). Nascent thiols are subsequently labeled with a heavy (13C) ICAT reagent, protein samples are digested and ICAT-modified peptides are isolated by avidin affinity chromatography. The eluted peptides are then analyzed by LC-MS/MS and the extent of oxidation for a particular cysteine is determined by the ratio of the heavy to light MS signals. While this procedure is not specific for disulfide-bonded cysteines per se, sulfenic acids and S-nitrosothiols are exquisitely sensitive to changes in pH and may be lost during sample preparation.104a,141 Consequently, the OxICAT method seems best suited for disulfide detection, including both protein and low molecular weight (e.g., S-glutathionylation) disulfides.
3.3.2.2. S-Glutathionylation
The thiol-containing tripeptide, GSH is maintained at millimolar concentrations inside cells. Under normal conditions, 98% or more of GSH is maintained in its reduced state, however, in oxidative stress-associated disorders like cancer and neurodegenerative diseases, an appreciable amount of the GSH pool exists in the oxidized state, GSSG.142 The GSH/Grx/GR system maintains protein thiols in their reduced state through thiol-disulfide exchange and redox reactions. Additionally, GSH undergoes nucleophilic addition and displacement reactions to purge the cell of toxic electrophilic and oxidizing reagents as catalyzed by glutathione S-transferase (GST), glyoxalase, GR, and Grx.143
Protein S-glutathionylation can occur during reduction of disulfides by the GSH/Grx/GR system and is readily reversible. When the GSH/GSSG redox balance shifts toward a more oxidizing state, protein S-glutathionylation can function as a regulatory mechanism or protect against irreversible oxidation.120 If the GSH/Grx/GR system is compromised during oxidative stress, the accumulation of S-glutathionylated proteins can occur and has been associated with aging.144 Within the context of redox signaling, protein S-glutathionylation can take place through two possible mechanisms: (i) thiol–disulfide exchange of GSSG with a thiolate or (ii) condensation of GSH with a sulfenic acid (Figure 8) or an S-nitrosothiol. In a study of sulfenic acid-modified HSA, S-glutathionylation was estimated to occur with a rate constant of 2–100 M–1 s–1.106c Thiol–disulfide exchange between GSSG and a protein thiolate is very slow,145 but may be catalyzed by Grx, which appears to promote S-glutathionylation of the ETC complex I.146 In this case, Grx-mediated S-glutathionylation may occur through free radical formation.147 Specificity in S-glutathionylation may depend upon the steric properties, surrounding environment, and oxidation sensitivity of the cysteine. Like disulfides, S-glutathione protein adducts are stable to nonthiol nucleophiles. Deglutathionylation is catalyzed by members of the Grx family,148 but Srx,149 Trx,150 and PDI150a may also perform this function, albeit with decreased efficiency.151
Figure 8.

Mechanisms for glutathionylation. Protein glutathionylation products can be formed by (a) thiol/disulfide exchange of a protein thiolate with oxidized glutathione (GSSG) or (b) condensation of GSH with a protein sulfenic acid.
Enzymes such as trypsin,152 collagenase,153 and fructose-1,6-bisphosphatase154 are activated by S-glutathionylation, whereas glyceraldehyde 3-phosphate dehydrogenase (GAPDH),155 26S proteasome,156 cysteine protease caspase-1,157 and ETC complex I158 are inactivated by this modification. As previously mentioned, many PTPs are regulated by intramolecular disulfide bond formation at their catalytic cysteine.159 However, some PTPs do not contain a second cysteine proximal to their active site. In some of these cases, for example in PTP1B, the phosphatase undergoes S-glutathionylation to guard against hyperoxidation (defined as oxidation to irreversible sulfinic and sulfonic acid states).160 In addition to regulating enzyme activity, S-glutathionylation can also influence protein–DNA and protein–protein interactions. For instance, S-glutathionylation of cysteines in the DNA binding domain of transcriptional regulator, p53 weakens its association with DNA.161 Similarly, S-glutathionylation of the transcriptional regulator, interferon regulatory factor 3 (IRF3) inhibits its interaction with CBP/p300 coactivators and prevents activation of target genes involved in induction of an antiviral response.162
To date, several methods have been developed to detect protein S-glutathionylation based on immunological, metabolic labeling, and differential alkylation approaches.138 A common method to detect S-glutathionylation in proteins employs an antibody specific for the protein-S-GSH adduct.162,163 This antibody is amenable to immunoprecipitation, Western blot on nonreducing gels, and immunofluorescence analysis. The anti-GSH antibody has also been used in conjunction with 2D SDS-PAGE, where samples are separated by isoelectric focusing in the first dimension and by molecular weight in the second dimension, with ensuing MALDI-TOF MS to identify S-glutathionylated proteins in HeLa cells.163b Given the differences in the surrounding environment of the modified cysteine, a limitation of the antibody is that not all protein-S-GSH adducts are detected with the same affinity.164 An alternative immunological approach, called GST overlay, exploits the specificity and affinity of GST for GSH. In this method, Western blots from nonreducing SDS-PAGE gels are exposed to biotinylated-GST, which recognizes and binds selectively to protein-S-GSH disulfides; biotin-GST is subsequently detected by avidin blot (Figure 9a).165 Protein S-glutathionylation can also be monitored indirectly by differential alkylation. In this workflow, free thiols are alkylated, protein-S-GSH adducts are selectively reduced by Grx, and nascent thiols are tagged by a biotinylated or fluorescent alkylating reagent (Figure 9b).166 In theory, this approach could also be coupled to the OxICAT method to measure the extent of protein-S-GSH disulfides.
Figure 9.

Methods to detect protein S-glutathionylation. (a) GST overlay. Samples are separated by nonreducing SDS-PAGE to preserve protein-GSH disulfides. Blots are subsequently treated with biotinylated glutathione S-transferase (GST), which binds selectively to GSH and permits detection of S-glutathionylated proteins by avidin blot. (b) Indirect differential alkylation of S-glutathionylated proteins. Free thiols are blocked with NEM or IAM, protein-GSH disulfides are selectively reduced with glutaredoxin (Grx), and nascent thiols are labeled with an alkylating agent conjugated to biotin or a fluorophore. S-Glutathionylated proteins are detected by avidin blot or in-gel fluorescence. (c) Biotinylated glutathione ethyl ester (BioGEE, 3) enables in situ detection of glutathionylated proteins. N,N-Biotinyl glutathione disulfide (4) permits detection of proteins that become S-glutathionylated by thiol/disulfide exchange with GSSG.
Approaches have been developed to facilitate detection of S-glutathionylated proteins in cells. One such method involves inhibiting protein synthesis with cycloheximide, which does not affect GSH synthesis, with subsequent metabolic labeling of the GSH pool through35S-cysteine incorporation.167 Cells are subsequently lysed in the presence of a thiol alkylating agent to minimize thiol-disulfide exchange, samples are separated under nonreducing conditions, and analyzed by radiography. This technique has been used to identify proteins, such as enolase and 6-phosphogluconolactonase, that undergo S-glutathionylation in human T lymphocytes exposed to exogenous oxidants (e.g., H2O2 and diamide).5b Alternatively, Finkel, Eaton and colleagues have used biotinylated-GSH ethyl ester (BioGEE, 3)155,167 and N,N-biotinyl glutathione disulfide (4)164 (Figure 9c) to monitor protein S-glutathionylation in lysates, isolated cells, and tissues. While the biotin tag facilitates enrichment and identification of proteins that undergo S-glutathionylation, limitations of these methods include steric occlusion of biotinylated GSH analogues and poor cellular trafficking of biotinylated probes.168
3.3.2.3. Sulfenic Acids
Because of their reactive nature, sulfenic acids are often deemed unstable intermediates en route to additional cysteine modifications (Figure 2 and Chart 4). The formal oxidation state of the sulfur atom in a sulfenic acid is 0, enabling it to function as both a weak nucleophile and a soft electrophile (Chart 4 and 5, eq 1).3b The dual nature of its reactivity is clearly illustrated by the condensation of two sulfenic acids to generate a thiosulfinate (Chart 4). Thiosulfinate formation via sulfenic acid condensation may be most facile when sulfenate and sulfenic acid states are equally present.119b As previously discussed, the prevalence of thiosulfinates in cells is currently unknown; however, given the abundance of cellular thiols, interfacing of two sulfenic acids is likely to be a rare event.3a
Chart 4. Sulfenic Acids Exhibit Both Nucleophilic and Electrophilic Character, As Illustrated by Condensation of Two Sulfenic Acids to Afford a Thiosulfinate (Black Box)a.

a As a nucleophile (purple boxes), sulfenic acids can undergo SN2 displacement with halogenated compounds, such as 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl, 5), reaction with alkynes (6) and alkenes (7) to form the corresponding sulfoxides, and reaction with two equivalents of triphenylphosphines (8) to afford the free thiol and oxidized phosphine (not shown). Sulfenic acids can also function as an electrophile (green boxes) to react with thiols to yield a disulfide, and with 1,3-cyclohexadiones including dimedone (9), to yield a thioether adduct. As an electrophile, sulfenic acids can also react with hydrazines (10) to yield the thiol and azo compound (not shown), or with amines (11) to yield sulfenyl amides.
Chart 5. Reaction Schemes of Condensation of Two Sulfenic Acids to Yield a Thiosulfinate (Equation 1) and Electrophilic Reaction of Sulfenic Acid with Dimedone (9, Equation 2) and Hydrazines (10, Equation 3).

Analogous to the reactivity of sulfur in a cysteine thiol, the nucleophilic character of a sulfenic acid is likely to be influenced, in part, by pKa. Studies of sulfenic acids in small molecules have shown that electron-withdrawing substituents reduce the pKa to favor sulfenate formation and enhance the stability of this species.169 The pKa of sulfenic acids in proteins could be similarly modulated to regulate their stability and reduce its reactivity toward a thiol. Stabilization of the sulfenate anion through decreased pKa could also enhance the nucleophilic character of the sulfur atom, marking potential sites of cysteine hyperoxidation.
The pKa of sulfenic acids in small molecules has been estimated to be in the range of 4.5–12.5.104a,170 The pKa of protein sulfenic acids has not been as extensively studied, but two measurements have been made, both with bacterial Prxs. There are three classes of Prxs: typical 2-Cys, atypical 2-Cys, and 1-Cys Prxs. Both typical and atypical 2-Cys Prxs form sulfenic acid at their active site cysteine after reaction with H2O2, which then condenses with a second cysteine in the same (atypical) or neighboring (typical) Prx to generate a disulfide that is reduced by Trx/TrxR to complete the catalytic cycle.171 1-Cys Prxs do not contain a resolving cysteine and the sulfenic acid intermediate may be reduced by GSH or ascorbate.172 The first pKa measurement reported for the sulfenyl group of a protein sulfenic acid was obtained using a mutant form of 2-Cys Prx from Salmonella typhimurium, AhpC in which the resolving cysteine was changed to serine. Key to the success of these experiments, the sulfenic acid and sulfenate forms exhibit distinct spectral shifts in AhpC, allowing a pKa determination of 6.1.173 Consistent with this measurement, a tryptophan fluorescence study revealed a pKa of 6.6 for the sulfenic acid in a 1-Cys Prx from Mycobacterium tuberculosis.106a
Analogous to cysteine thiolate reactivity with H2O2, the propensity for sulfenic acid to undergo further oxidation to sulfinic acid can be strongly influenced by the local protein environment. Relative to their prokaryotic counterparts, 2-Cys Prxs from eukaryotic organisms appear uniquely sensitive to hyperoxidation and may be related, at least in part, to sulfenic acid pKa.91,174 For example, oxidation of bacterial peroxiredoxin AhpE sulfenic acid by H2O2 occurs at 40 M–1 s–1, whereas HSA sulfenic acid reacts at 0.4 M–1 s–1.106a,106c While the pKa of the protein sulfenic acids were not reported in these studies, it is interesting to note that initial formation of sulfenic acid was also significantly slower in HSA (2.7 M–1 s–1)106c compared to AhpE (8.2 × 104 M–1 s–1).106a To better understand how some protein environments facilitate sulfenic acid oxidation, additional physical organic and computational studies of both small-molecule and protein model systems will be required.
Sulfenic acids have been identified in the catalytic cycle of multiple enzymes, including Prx, NADH peroxidase, and methionine sulfoxide- and formylglycine-generating enzymes.66c,71,106a Formation of sulfenic acid has also been linked to oxidative stress-induced transcriptional changes in bacteria due to altered DNA binding of OxyR and OhrR and changes in the activity of the yeast Prx and Yap1 protein.66c,175 Less is known about the mechanisms that underlie sulfenic acid-mediated regulation of mammalian protein function and signaling pathways; however, cysteines from several transcription factors (i.e., NF-κB, Fos, and Jun), or proteins involved in cell signaling or metabolism (e.g., GAPDH, GR, PTPs, kinases, and proteases) can be converted to sulfenic acid in vitro. Sulfenic acid formation has also been implicated in the regulation of apoptosis, immune cell activation and proliferation, and growth factor (GF) signaling pathways.12,123c,176
Although sulfenic acids are often transient, an advantage to studying this modification is that it represents the initial product of two-electron oxidants with the thiolate anion and can therefore serve as a marker for oxidant-sensitive cysteine residues. A variety of indirect and direct chemical methods have been developed to detect protein sulfenic acid modifications (also termed sulfenylation5a,12). An early indirect chemical method that was reported involves thiol alkylation, reduction of sulfenic acids by arsenite, and labeling of nascent thiols with biotinylated NEM (Figure 10a).177 This methodology was subsequently used to profile sulfenic acid formation in rat kidney cell extracts;178 however, as with other indirect differential alkylation methods, a significant limitation is the debatable selectivity of the arsenite-mediated reduction step.179
Figure 10.

Methods to detect protein sulfenic acids. (a) Indirect differential alkylation of protein sulfenic acids. Free thiols (blue) are blocked with NEM or IAM, protein sulfenic acids (purple) are reduced with arsenite, and nascent thiols are labeled with biotinylated NEM (NEM-Biotin). Sulfenylated proteins are detected by avidin blot where increased protein oxidation is observed as an increase in signal intensity. (b) Direct in situ labeling of protein sulfenylation. Cells are treated with or without stimulant (e.g., oxidant, growth factor) and subsequently incubated with azido or alkyne dimedone analogues, such as 18 to chemically modify sulfenylated proteins. Afterward, excess probe is removed, cell lysates are generated, and probe-modified proteins are conjugated to biotin or a fluorophore by a coupling reaction (e.g., Staudinger ligation or Huisgen [3 + 2] cycloaddition). The samples can then be avidin enriched and subjected to proteomics analysis or analyzed by avidin blot or in-gel fluorescence where increased protein sulfenylation correlates to enhanced signal intensity. (c and d) High-throughput immunological detection of dimedone (9)-modified proteins using arrays. (c) Proteins immobilized on a microarray that are susceptible to sulfenylation are irreversibly modified by 9. The protein–dimedone adduct forms an epitope for selective detection by the antibody. (d) Cells are treated with or without stimulant (e.g., oxidant, growth factor) and are subsequently incubated with 9 to irreversibly modify sulfenylated proteins. Subsequent to cell lysis, proteins within a given signaling pathway are immobilized on an antibody array and dimedone-modified proteins are detected by addition of the antibody. (e) Isotope-coded dimedone 2-iododimedone (ICDID) permits quantification of protein sulfenylation. Sulfenic acids are labeled by d6-dimedone (21, purple), then excess reagent is removed and free thiols are labeled by d0-2-iododimedone (22, blue) generating chemically identical adducts that differ by 6 Da. The samples are trypsinized and analyzed by LC-MS where the extent of sulfenic acid occupancy is determined by the ratio of d6-dimedone to d0-dimedone peak intensities. (f) Quantification and site-identification of protein sulfenic acids with d6-DAz-2 or d6-DYn-2 and an acid-cleavable linker (ACL) coupling reagent. Sulfenic acids are labeled with d0-DAz-2 (16) in the untreated sample and with d6-DAz-2 (19) in the oxidant-treated sample. Excess probe is removed and the samples are combined and biotinylated by coupling with the alkyne-ACL (23) to generate chemically identical adducts that differ by 6 Da. The sample is then trypsinized, avidin enriched, and trifluoroacetic acid-eluted peptides are analyzed by LC-MS/MS where the increase in sulfenic acid modification in response to oxidant is determined by the ratio of d6 to d0 peak intensities. Biotin can complicate spectra and decreases peptide recovery, and the removal of biotin with the ACL permits increased sample elution and direct identification of modified peptides in the MS.
Direct methods for sulfenic acid detection have been developed that take advantage of the chemical reactivity of this oxyacid. Nucleophilic substitution of halonitroarenes, such as 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl, 5), and nucleophilic addition to electron-deficient alkynes (6), alkenes (7), and triphenyl phosphines (8) are reported to trap sulfenic acids (Chart 4).169 Of these, the most commonly used in the detection of protein sulfenic acids is NBD-Cl. This reagent reacts with thiols, sulfenic acids, and at higher pHs, amine-containing residues, but the resulting products are distinguished on the basis of their spectral properties and molecular weight.180 As NBD-Cl can react with a variety of protein functional groups, this reagent appears best suited for use with recombinant proteins, especially those with a single cysteine residue.16 Consequently, NBD-Cl does not have utility in global detection of protein sulfenic acids in complex protein mixtures, necessitating the development of methods for selective detection that exploit the electrophilic properties of the sulfur atom in sulfenic acid.
As first reported by Benitez and Allison in 1974, protein sulfenic acids react with cyclic 1,3-diketone carbon nucleophiles, like 5,5-dimethyl-1,3-cyclohexadione (dimedone, 9) and with hydrazines (10) or amines (11) (Chart 4 and 5)181 Dimedone has proven useful in revealing the requirement for protein sulfenic acid modifications in the S. cerevisiae Yap1-Gpx3 H2O2-sensing pathway,66c T cell activation,123c and EGFR signaling.12 Unlike sulfur, nitrogen, or phosphorus-based nucleophiles, under aqueous conditions cyclic 1,3-diketones do not cross react with cysteine thiols, sulfinic acid, or other functional groups commonly found in biomolecules, making this reaction an extremely attractive avenue for developing chemically selective detection methods. All chemoselective methods for detecting protein sulfenic acids reported to date depend upon this chemistry.138 Two recent reports expand the scope of reactive templates to 1,3-cyclopentadione182 and linear β-ketoester183 analogues (though caution should be exercised with linear derivatives since they have been reported to cross react with amines, such as lysine184). The lack of an enrichment or visualization “handle” for protein-S-dimedone adducts subsequently motivated the development of biotinylated (12,13)185 and fluorophore-conjugated (14) analogues185b,186 (Chart 6). These probes have been used in a proteomic study with isolated rat hearts185a and to identify AKT2 as a target of PDGF-induced H2O2.187 Depending upon the application, one potential drawback for direct conjugation of any probe to biotin or a fluorophore is that the bulky chemical tags can reduce cell-permeability.168b Naturally, not all conjugated probes are entirely impermeant (e.g., DCFH diacetate, DCP-Bio1) however, comparative studies show time and again that tagged derivatives often suffer from diminished cell uptake and trafficking properties.168,188 Alternative mechanisms of uptake are possible (e.g., active transport of BioGEE), but may limit probe distribution to specific cellular compartments. A further consideration when functionalizing probes with large chemical tags is that increased steric bulk can lead to a significant bias in protein target labeling.188a,189 The poor permeability of many biotin- and fluorophore-tagged probes typically necessitates labeling of proteins in lysates and is, therefore, subject to the aforementioned limitations. In this context, it is also important to bear in mind that labile or transient sulfenic acid modifications may be further oxidized or insufficiently trapped during the lysis procedure.
Chart 6. Biotin, Fluorophore, and Chemical Handle Derivatives of Dimedone.

A subsequent alternative approach that has emerged is the development of azido- and alkyne-functionalized dimedone analogous (Chart 6), termed DAz-1 (15),168b,180c DAz-2 (16),190 DYn-1 (17), and DYn-2 (18),12 which enable the trapping and tagging of protein sulfenic acid modifications directly in living cells. In later steps, proteins covalently modified by DAz or DYn probes can be coupled to biotin or fluorophores by Staudinger ligation191 or Huisgen [3 + 2] cycloaddition reactions (Chart 7, eqs 1 and 2, and Figure 10b).192 Application of DAz-2 to identify proteins that undergo sulfenic acid modifications in HeLa cells identified upward of 200 candidates, including the majority of known sulfenic acid-modified proteins.190 Cross-comparison of these data with those from disulfide and S-glutathionylation proteomes revealed modest overlap between these “redoxomes”, suggesting that a significant portion of sulfenic acid modifications may not be intermediates en route to S-thiolated forms and, instead, can be stabilized by the protein local environment.138 Alternatively, or in addition, it is also possible that (i) lysate-based approaches employed in the S-thiolation proteomic studies resulted in fewer identifications and, therefore, lower overlap with the “sulfenome”, and (ii) the modest rate constant for the reaction of many dimedone analogues with sulfenic acid (103 M–1 min–1)111a may not be sufficient to trap especially transient modifications. Azido dimedone analogues have also been used to show that sulfenic acid modification of the thiol peroxidase, Gpx3 is essential for yeast to sense oxidative stress66c and to identify a unique reducing system in the bacterial periplasm that protects single cysteine residues from oxidation.105
Chart 7. Bioorthogonal Coupling Reactions Staudinger Ligation and Huisgen [3 + 2] Cycloadditiona.
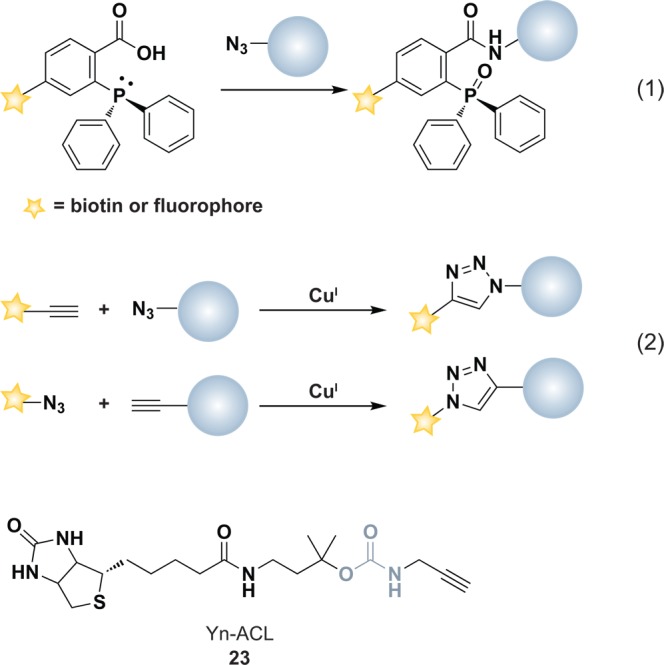
a Staudinger ligation (equation 1) functionalizes an azide-modified molecule, while Huisgen [3 + 2] cycloaddition (equation 2) couples azide-modified or alkyne-modified proteins to detection tags. Alkyne acid cleavable linker (Yn-ACL, 23) reagent for Huisgen [3 + 2] cycloaddition with azide-modified proteins. Blue, acid cleavable moiety.
More recently, DYn-2 was used in global profiling studies to reveal dynamic protein sulfenylation during EGF signaling in human epidermoid A431 cells and to identify the EGFR kinase as a prominent target of endogenous signaling H2O2 (Figure 11).12 Three PTPs involved in the regulation of EGFR signaling, PTP1B, PTEN, and SHP2 were also shown to undergo sulfenic acid modification in response to EGF stimulation of cells. Interestingly, PTPs and EGFR displayed differential sensitivity to oxidation by EGF-induced endogenous H2O2 that correlated with the relative proximity of each enzyme to the oxidant source itself, NOX2 (Figure 11). This study was the first of its kind to provide evidence for sulfenic acid modification of PTP in cells during growth factor signaling. Prior studies performed in lysates had led to speculation as to the likelihood of PTP oxidation due to their modest reactivity with H2O2 and their low abundance in comparison to the abundant and reactive Prxs.2d,123c,193 Interestingly, while this study found ER-localized PTP1B to be only moderately sensitive to H2O2 derived from plasma membrane-bound NOX2, a study by Keaney and colleagues indicates that this oxidation reaction becomes relevant during ER-localized NOX4 activation.194 As it is unlikely that the intrinsic reactivity of the active site cysteine in PTP1B differs in these two systems, these data suggest that proximity of PTP1B (and other proteins) to the NOX oxidant source may be an important determinant of target selectivity. Hence, the apparent sensitivity and physiological relevance of PTP1B oxidation, and protein thiols in general, is likely to be a signaling pathway and cell type-specific phenomena.
Figure 11.
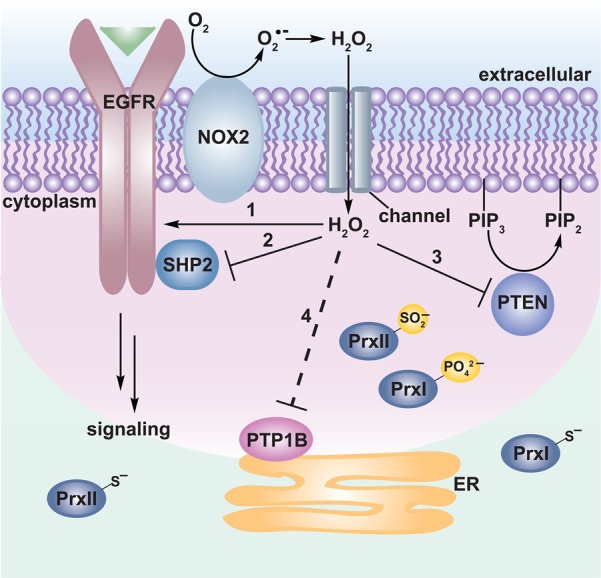
Redox regulation of epidermal growth factor (EGF) signaling by protein sulfenylation. Binding of EGF to the EGF receptor (EGFR) facilitates receptor dimerization, activation (not shown), and promotion of NOX2 complex assembly. NOX2-derived H2O2 translocates into the cytoplasm likely through channels such as aquaporins where it has been shown to regulate the activity of proteins involved in the EGFR signaling cascade. EGFR and the phosphatases SHP2, PTEN, and PTP1B were all found to be sulfenylated in response to EGF stimulation, albeit with differential sensitivities (ranked 1–4 in order of decreasing susceptibility). The sensitivity of each protein to oxidation correlates to their relative proximity to the oxidant source. In this way, EGFR, which forms a complex with NOX2, exhibited the highest sensitivity. Moreover, the EGFR-associated phosphatase SHP2 exhibited increased susceptibility to sulfenylation as compared to the cytoplasmic phosphatase PTEN, which regulates the levels of PIP3, and PTP1B, which is localized to the cytoplasmic face of the endoplasmic reticulum (ER). Co-localization of antioxidant enzymes such as peroxiredoxins (Prx) to the signaling regions is thought to limit the range of H2O2 diffusion (green area). Interestingly, NOX-derived reactive oxygen species (ROS) have also been shown to inactivate Prxs by hyperoxidation (PrxII) or phosphorylation (PrxI).11b These regulatory mechanisms have been proposed to permit localized accumulation of ROS for redox regulation of proteins located near the oxidant source (pink area).
In 2009, our group reported the first immunological method for detecting protein sulfenic acid modifications. Antibodies were elicited by a synthetic hapten mimicking dimedone-modified cysteine conjugated to KLH (Figure 10c) and are highly specific and sensitive for detecting protein-S-dimedone adducts by Western blot and immunofluorescence.195 Application of this immunochemical approach to protein arrays and breast cancer cell lines revealed considerable differences in the level of protein sulfenic acid modifications among tumor subtypes (Figure 10c). This method has also been used to demonstrate the cysteine sulfenylation and colocalization of oxidized proteins with NOX2 during EGF signaling.12 Subsequently, in 2011, Eaton and colleagues reported a similar antibody and used this reagent to study sulfenic acid modification of GAPDH in cardiac myocytes exposed to exogenous H2O2.196 A future application of these antibodies will be to combine their use with antibody arrays to facilitate unbiased investigation of protein sulfenic acid modifications in signaling pathways (Figure 10d).
Beyond detection, one approach to determine which protein sulfenic acid modifications are relevant to signaling in normal cells as well as in pathological processes is to quantify the extent of oxidation. To this end, our laboratory has recently developed two methods to facilitate relative quantification of sulfenic acid modifcations: (1) isotope-coded dimedone and 2-iododimedone (ICDID) (Figure 10e),197 and (2) isotopically light and heavy derivatives of DAz-2 (19) and DYn-2 (20) (Chart 6).198 The ICDID workflow uses deuterium-labeled dimedone (d6-dimedone, 21) to trap sulfenic acids, followed by alkylation of free thiols with 2-iododimedone (22). Importantly, the covalent adducts afforded by these two reagents are structurally/chemically identical, and have identical efficiencies of ionization. Nevertheless, the thiol and sulfenic acid-tagged species are differentiated from each other by 6 Da and the extent of sulfenic acid modification at a cysteine is determined from the ratio of heavy to light isotope-labeled peak intensities (Figure 10e). Alternatively, isotopically light and heavy forms of DAz-2 can be used to monitor relative changes in sulfenic acid modification. This strategy has been combined with an acid cleavable linker (ACL) that is suitable for Huisgen [3 + 2] cycloaddition coupling (Chart 7).198 With this method, samples are labeled by heavy or light DAz-2, combined and conjugated to the alkyne-biotin ACL reagent (Yn-ACL, 23) digested with trypsin, enriched on avidin cartridges, and tagged peptides are eluted by trifluoroacetic acid (TFA)-mediated cleavage. Peptides are then mapped by LC-MS/MS analysis to identify the sulfenic acid-modified protein and map the site of modification. The relative change in protein sulfenic acid modfication between two samples is determined by the ratio of heavy to light isotope-labeled peak intensities (Figure 10f).
Analogous to irreversible electrophilc inhibitors that modify semiconserved cysteines residues in protein tyrosine kinases (PTKs) currently in phase II and III cancer clinical trials,199 we envision the development of nucleophile-functionalized small molecules that target a sulfenic acid-modified cysteine in a specific protein. Our design strategy is to conjugate the nucleophile “warhead” to a high affinity ligand that binds proximal to the target cysteine sulfenic acid. As proof of principle, we have developed small molecules that target PTPs, termed redox-based probes (RBPs, Figure 12, 24–26), comprised of three parts: (i) a cyclohexanedione nucleophile, (ii) a chemical scaffold that binds to the conserved PTP active site, and (iii) an azide (or alkyne) chemical reporter to facilitate downstream detection and isolation of labeled PTPs (Figure 12a).189 The RBPs exhibited enhanced binding and sensitivity for detecting sulfenic acid modification of the catalytic cysteine in the YopH and PTP1B phosphatases, compared to the parent compound, DAz-1 (15), which lacks the additional binding element. The RBP approach should facilitate cellular investigations of PTP redox regulation. Methods to study PTP redox modulation are often thwarted by issues of low abundance and studies of this nature would greatly benefit from a targeted approach, as exemplified by RBPs. Neel and colleagues have also reported an indirect immunochemical method for global proteomic assessment of the PTP “redoxome” that relies on performic acid hyperoxidation of cysteine oxyacids (Figure 12b).200
Figure 12.

Targeted approaches to monitor cysteine oxPTM with in specific or family of proteins. (a) Redox-based probes (RBPs, 24–26) for protein tyrosine phosphatases (PTPs) are comprised of three parts: a warhead that permits chemoselective reaction with sulfenic acid, a PTP-directed scaffold that exhibits enhanced affinity for target binding, and an azide chemical reporter to facilitate downstream detection and isolation of labeled protein. (b) Indirect two-stage immunochemical approach to monitor PTP oxidation. In stage 1, free thiols in one aliquot of sample are alkylated with NEM, oxidized cysteines are reduced with DTT, and nascent thiols are hyperoxidized to sulfonic acid with pervanadate. The proteins are subsequently trypsinized, and sulfonic acid-modified peptides are isolated with a monoclonal antibody that recognizes hyperoxidized PTPs. In stage 2, a second aliquot of sample is reduced with DTT and all thiols are oxidized to sulfonic acid with pervanadate and processed as in stage 1. The enriched peptides are analyzed by LC-MS/MS and the extent of PTP oxidation is determined by the ratio of stage 1 to stage 2 signal intensities. (c) Conformation-sensing single-chain variable antibodies that selectively recognize the unique conformation of the sulfenyl amide oxoform of PTP1B.
In addition to pan-PTP recognition, RBPs can be refined to target a single member of the PTP family. Such a reagent would not only be useful to study redox-regulation of a specific PTP, but might possibly serve as lead compounds for the development of a new class of therapeutics to ameliorate diseases associated with aberrant PTP activity, as in diabetes.201 In support of this approach, Tonks and colleagues recently reported the development of antibodies as single-chain variable fragments that selectively recognize the unique conformation that PTP1B adopts when its activate site cysteine exists in the sulfenamide form (Figure 12c).202 These conformation-sensing antibodies were able to trap PTP1B in the inactive conformation permitting sustained insulin signaling in human embryonic kidney (HEK) cells. Lastly, the RBP or “nucleophilic inhibitor” approach can be extended to other classes of proteins that contain a redox-sensitive cysteine, such as EGFR.12
3.3.2.4. Sulfinic Acids
In the presence of excess oxidant, sulfenic acid can be oxidized to sulfinic acid (Figure 2). The formal oxidation number of the sulfur atom in sulfinic acid is +2. On this basis, this oxyacid might be expected to have enhanced electrophilicity compared to sulfenic acid. However, sulfinic acid does not undergo self-condensation or nucleophilic attack by thiols. This can be explained by the increase in partial positive charge on the sulfur in sulfinic acid, which converts the sulfur atom into a harder electrophile making it less prone to reaction with soft nucleophiles, such as thiols. With a pKa of 2, sulfinic acid is deprotonated at physiologic pH and can exist in two resonance forms (Chart 8).203 Sulfinic acids function as nucleophiles (Chart 9), reacting largely from sulfur to undergo alkylation (27), as well as nucleophilic addition to activated alkenes (28), aldehydes (29), lactones (30), α,β-unsaturated compounds (31), and diazonium salts (32) to give the corresponding sulfones.3b,204 The preceding reactions are established for sulfinic acids under synthetic organic conditions, but it is not established whether all of these reactions would take place with protein sulfinic acids. The reactions in Chart 9 exhibit a wide range of rates and some go to completion on the hour time scale (27,20528,20630,20731208), while others, such as 29 and 32,209 undergo rapid equilibrium-based transformations. Of note, the reaction of sulfinic acid with aldehydes serves as the basis for the Schiff’s test. As an ambidentate nucleophile, sulfinic acid can also react at oxygen as illustrated by nucleophilic attack of the sulfinate oxygen on the γ-phosphate of ATP (33) to form the sulfinic acid phosphoryl ester intermediate in the Srx catalytic cycle (Chart 9).122a,210
Chart 8. Resonance Structures of Sulfinic Acids.
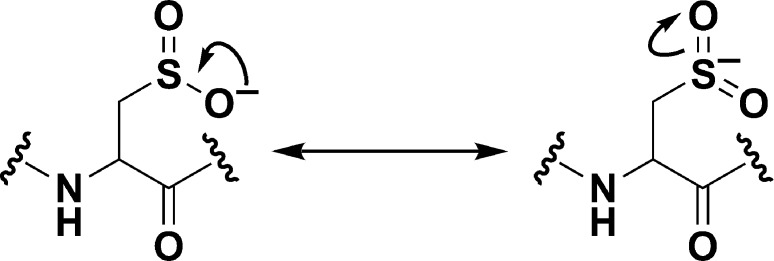
Chart 9. Sulfinic Acids Function as Soft Nucleophiles Reacting Primarily from the Sulfur to Undergo Alkylation (27) or Nucleophilic Addition to Activated Alkenes (28), Aldehydes (29), Lactones (30), α,β-Unsaturated Compounds (31), and Diazonium Salts (32)a.

a Sulfinic acids can also function as nucleophiles involving reactivity from the oxygen as exemplified by the sulfiredoxin (Srx)-catalyzed reaction with ATP (33) to yield a transient sulfinic acid phosphoryl ester. Though not shown, reactions 27 and 28 undergo acid-catalyzed SN1 reactions and thus require the protonated sulfinic acid species.
Cysteine sulfinylation can also modify protein metal binding properties. Oxidized sulfur ligands are weaker donors and can increase the Lewis acidity of the liganded metal center, which influences affinity and coordination. In matrix metalloproteases (MMPs), sulfinic acid oxidation of a zinc-coordinated active site cysteine thiolate activates protease function, in part by reducing the ability to coordinate the zinc cation.211 In contrast, nonheme FeIII coordination in nitrile hydratases (NHases) is accomplished by a unique CXXCXC binding sequence in which two cysteines are present as the sulfinic and sulfenic acid states.154,212 Cysteine oxidation is necessary for hydratase activity, and the increased Lewis acidity of the FeIII afforded by cysteine oxidation is believed to regulate the affinity of a catalytic water molecule for the metal center.3b,212a,213 A similar coordination motif has also been identified in the unique noncorrin cobalt center in a NHase from Pseudonacardia thermophila(214) and in thiocyanate hydrolase.215 It has also been suggested that cysteine oxidation alters the metal coordination from zinc (thiol) to iron or cobalt (sulfenic/sulfinic acid). This preference may not be as strictly defined as once thought, however, as a peptide mimetic inhibitor of neurotoxin F from Clostridium botulinum was recently shown to coordinate to an essential zinc by a cysteine sulfinate ligand.216
To date, the cysteine sulfinic acid modification has been most extensively characterized in Prxs and the Parkinson’s disease protein, DJ-1.217 Eukaryotic Prxs appear most susceptible to sulfinic acid modification,91,174 a feature that evolutionarily coincides with Srx expression, the only known sulfinic acid reductase.109a,218 Srx was recently identified in cyanobacteria,219 which also appear to have 2-Cys peroxiredoxins that are susceptible to hyperoxidation.220 Recent work shows that Srx-mediated reduction of Prx proceeds by a sulfinic acid phosphoryl ester that undergoes nucleophilic attack by Srx Cys84 to form a thiosulfinate intermediate that is subsequently resolved by Srx Cys48 to release Prx sulfenic acid and oxidized Srx, which are both recycled by the Trx/TrxR system.122a,210,221 The reaction of Srx with sulfinic acid is slow (kcat ≈ 0.2 min–1), and it is currently unknown whether any accessory proteins enhance this reaction in vivo.109b
The biological reversibility of sulfinic acid (at least in some proteins) hints at a regulatory function, analogous to a disulfide or sulfenic acid. In this vein, it has been proposed that reversible inactivation of Prx by sulfinic acid modification facilitates the accumulation of endogenous H2O2 to regulate signaling events in the so-called “floodgate hypothesis”.91 While Prx II appears to be particularly sensitive to hyperoxidation, it has recently been shown that phosphorylation inactivates Prx I,11b with both mechanisms of Prx inactivation serving to facilitate localized accumulation of H2O2 for signaling purposes. Reversible Prx oxidation has also been proposed to regulate eukaryotic circadian rhythms, though the molecular details remain largely unknown.222
Evidence of a regulatory role for reversible sulfinic acid Prx inactivation also stems from the observation that many signaling pathways, including neuronal N-methyl-d-aspartate (NDMA) receptor activity223 and macrophage activation by lipopolysaccharides224 induce Srx expression. In both cases, induction of Srx was dependent upon redox-regulated transcription factors, AP-1 and Nrf2.225 Srx can also translocate to the mitochondria, to reduce hyperoxidized Prx III and protect against oxidative damage and apoptosis.226 The molecular details are not entirely clear, however, it is possible that Srx-mediated reactivation of Prx III maintains low mitochondrial ROS levels to prevent opening of the mPTP. Srx overexpression also stabilizes PTEN and PTP1B,109c which is reminiscent of Prx I-mediated protection of PTEN tumor suppressor activity.227 The aforementioned studies suggest an important biological role for the reversibility of Prx hyperoxidation. Nonetheless, further studies are required, including the development of an Srx knockout mouse model to assess the physiological relevance of Prx reactivation. It should also be noted that Srx may also carry out other biological functions independent of sulfinic acid reduction.109c,224
Although sulfinic acid has gained recognition as a regulatory modification, the full scope of its biological formation remains poorly understood, due in part, to the lack of methods that are suited to general detection. Methods to detect protein sulfinic acids include the a molecular mass increase of 32 Da,228 acidic electrophoretic gel shifts,335,336,228 and antibodies that recognize a sulfinic/sulfonic acid peptide from a specific protein.200,229 Such approaches facilitate study of sulfinic acids in individual proteins, but have limited utility in global analysis. As thiols are good nucleophiles, a challenge to developing chemical methods for sulfinic acid detection lies in is its behavior as a weak nucleophile. An alternative approach is to design a reaction in which the product of the reaction with sulfinic acid is uniquely stabile. Along these lines, our lab has recently investigated the reaction sulfinic acids with aryl-nitroso compounds (Chart 10, eq 1). The initial sulfinic acid-derived N-sulfonyl hydroxylamine product is reversible, but can be trapped by ester-functionalized aryl-nitroso 34 to give an irreversible N-sulfonylbenzisoxazolone adduct (Chart 10, eq 1).203a The reaction of 34 with a thiol yields a sulfenamide species that can be cleaved with nucleophiles (Chart 10, eq 2) and, importantly, 34 does not cross react with other sulfur and nonsulfur containing biological functional groups.
Chart 10. Chemoselective Approach to Detect Sulfinic Acidsa.

a Reaction of a sulfinic acid with ester-protected aryl-nitroso compound 34 and subsequent intramolecular nucleophilic attack of the nitroso anion intermediate on the ester yields a stable N-sulfonylbenzisoxazolone adduct (equation 1). In contrast, analogous reaction with a thiol yields a sulfenyl amide adduct that is susceptible to subsequent reaction with a second thiol or reducing agent (equation 2).
3.3.2.5. Sulfonic Acids
While the sulfinic acid modification is relatively stable, it can undergo further oxidization to give sulfonic acid, the most highly oxidized thiol species (Figure 2). Like sulfinic acid, the sulfur atom in sulfonic acid (formal oxidation number of +4) functions as a hard electrophile and does not undergo self-condensation or nucleophilic attack by thiols. With a pKa <2, the sulfonic acid is a both strong acid and a weak base, which makes it a good leaving group in SN1, SN2, E1, and E2 reactions.3b Moreover, organic sulfonic acids can undergo nucleophilic attack on alkenes (35), alkynes (36), and allenes (37) to generate the corresponding sulfonic acid esters (Chart 11), where the reaction initiates exclusively at the oxygen.230 However, it is currently unknown whether any of the reactions presented in Chart 11 are amenable to protein studies.
Chart 11. Reactions of Sulfonic Acidsa.

a Sulfonic acids function as soft nucleophiles and react exclusively from the oxygen atom to undergo acid-catalyzed reaction with alkenes (35), alkynes (36), and allenes (37) to generate the corresponding sulfonic acid esters. These reactions occur only in organic solvent as the resulting products are largely unstable in water.
The sulfonic acid modification has been characterized in a small group of proteins, including mammalian Cu,Zn-SOD, where it has been speculated that damage resulting from hyperoxidation plays an important role in diseases like familial amyotrophic lateral sclerosis.229b Sulfonic acid is also present in mammalian cells as the naturally occurring low molecular weight compound, taurine. This biomolecule plays functions as a general osmolyte and modulator of neuronal activity.231 As with sulfinic acid, elucidation of the biological and pathological role of sulfonic acid modification has been hindered by a lack of means to selectively detect this oxyacid. Recently, a method has been developed that permits selective enrichment of sulfonic acid-modified peptides using poly arginine (PA)-coated nanodiamonds as high affinity probes.232 BSA, used as a model system in this study, was oxidized with performic acid, digested, and sulfonic acid-containing peptides were enriched and eluted from PA-coated nanodiamonds with phosphoric acid, with subsequent identification of oxidized peptides by MALDI-MS analysis (Figure 13a). This methodology might have an application in the characterization of protein sulfonic acids in cell lysates by first alkylating reduced and reversibly oxidized thiols with IAM or NEM. Sulfinic acid and sulfonic acid-modified peptides might also identified through a scheme involving performic acid oxidation (Figure 13b). A limitation of this method, however, is that sulfonic acid-modified peptides are in competition with phosphorylated peptides for binding to the PA-coated nanodiamonds,233 though this could potentially be reduced by phosphatase treatment of lysates.
Figure 13.
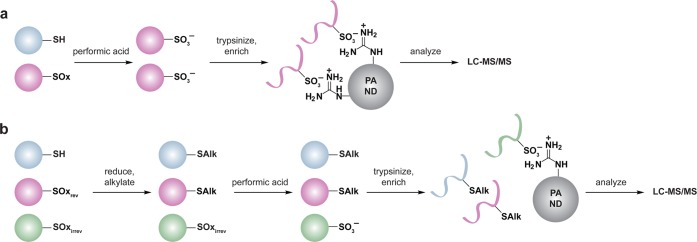
Method for selective enrichment of sulfonic acid-modified peptides. (a) All cysteine residues are oxidized to sulfonic acid with performic acid. Proteins are then trypsinized and sulfonic acid-modified peptides are enriched using polyarginine (PA)-coated nanodiamonds (ND). Eluted peptides are analyzed by LC-MS/MS. (b) A plausible extension of the PA-ND enrichment technology to identify sulfinylated and sulfonylated cysteines. Samples are first treated with a reducing agent to reduce all reversibly oxidized cysteines (purple), and alkylated with NEM or IAM. Irreversibly oxidized cysteines (green) are subsequently oxidized to sulfonic acid with performic acid. The sample is then trypsinized, sulfonic acid-modified peptides are enriched with PA-ND, and eluted peptides are analyzed by LC-MS/MS to identify sites of hyperoxidation.
4. Reactive Nitrogen Species (RNS) in Biological Systems
The prototypical RNS produced in biological systems is nitric oxide (•NO). In cells, the estimated steady state concentration and half-life of this species is 100 pM–5 nM and ∼0.1–2 s, respectively.234 Although •NO is more stable than H2O2 in cells, protein and small molecule NO-donors are believed to be a relevant source of •NO in biological systems. In general, •NO is a modestly reactive radical and does not inflict indiscriminate damage on biomolecules. Due to its gaseous and neutral nature, •NO is four times more soluble in membranes than in aqueous solution,235 which permits its diffusion across membranes and, in this context, •NO can function as an autocrine and a paracrine signal within a 100–200 μm radius of the production site.236 For example, •NO was recently shown to function as a paracrine signal to regulate active T cell expansion in lymph nodes.237 Initially deemed toxic, •NO was later identified as the first gas known to act as a biological second messenger in mammals where it regulates vasodilation/relaxation of underlying smooth muscle cells.238 Since these seminal discoveries, roles for •NO have been established in a range of biological processes including proliferation, apoptosis, angiogenesis, and host defense.239•NO appears to be metabolized by autoxidation to nitrite (NO2–) and nitrate (NO3–), which occurs about 30-fold faster within the interior of lipid bilayers than in aqueous solution (Chart 12).240•NO can also react rapidly (1.1 × 109 M–1 s–1) with nitrogen dioxide (•NO2) to generate additional nitrosating compounds such as dinitrogen trioxide (N2O3) (Chart 12).241 The production of N2O3 is a trimolecular reaction with oxygen and two molecules of •NO and is, therefore, not favorable at low •NO concentrations (Chart 12). In turn, these •NO oxidation products play important roles in physiological and pathological processes.241,242 In addition to autoxidation, •NO reacts rapidly (1010 M–1 s–1) with O2•— to generate peroxynitrite (ONOO–), which is reactive and damaging to biomolecules, analogous to •OH.242c,243 Though it will not be discussed further here, ONOO– is an important RNS in many biological settings; the interested reader is referred to the following source for additional information.242c
Chart 12. Formation and Transformation of Biologically Relevant Reactive Nitrogen Species (RNS)a.
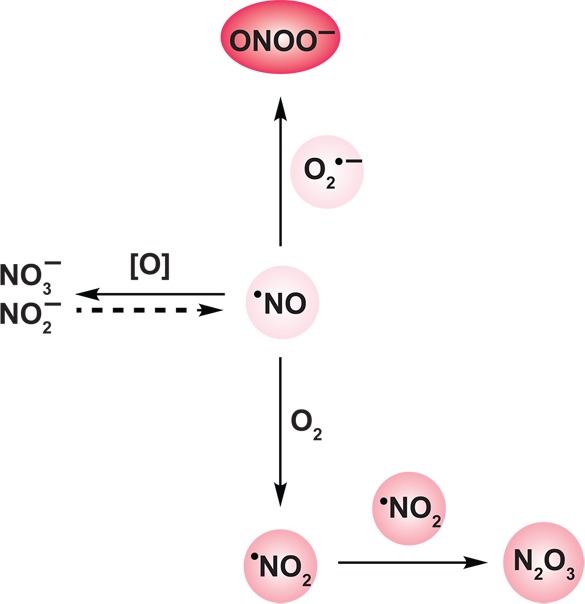
a Metabolism of nitric oxide (•NO) ultimately involves oxidation to nitrite (NO2–) and nitrate (NO3–) and there is evidence for this process being reversible (dashed arrow). Though a generally unreactive radical, •NO can react with molecular oxygen to generate nitrogen dioxide radical (•NO2). Radical-radical coupling of •NO2 with a second molecule of •NO affords dinitrogen trioxide (N2O3) via an ultimately trimolecular reaction. The trimolecular nature of this reaction makes N2O3 generation poorly favored at low •NO concentrations. In the presence of O2•–, •NO can react to yield peroxynitrite (ONOO–). Color intensity correlates to relative RNS reactivity.
4.1. •NO Production and Metabolism
4.1.1. •NO Synthases (NOS)
Enzymatic •NO production is predominantly mediated by the heme- and flavin-containing •NO synthases (NOSs), which catalyze the formation of •NO from NADPH, molecular oxygen, and l-arginine (Figure 14a).244 The linear arrangement of NOSs reveal three domains: the N-terminal oxygenase domain, C-terminal reductase domain, and the connecting calmodulin (CaM)-binding site. The oxygenase domain contains the heme and (6R)-5,6,7,8-tetrahydrobiopterin (BH4) cofactors, and the l-arginine binding site, while the reductase domain has a binding site for NADPH and houses the FAD and FMN flavin cofactors (Figure 14b).245•NO is produced by the flow of electrons derived from NADPH through the flavins in the reductase domain to the heme in the oxygenase domain, where oxygen and l-arginine are bound. NOS functions as a dimer in which the large (3000 Å) dimer interface in the oxygenase domain includes the BH4 binding site and is stabilized by a zinc ion that is coordinated by two cysteine residues in a conserved CXXXXC motif per monomer.246 Dimerization helps to structure the active-site pocket containing the heme cofactor and the l-arginine binding site, and there is evidence for electron flow occurring between monomers (Figure 14b).247
Figure 14.
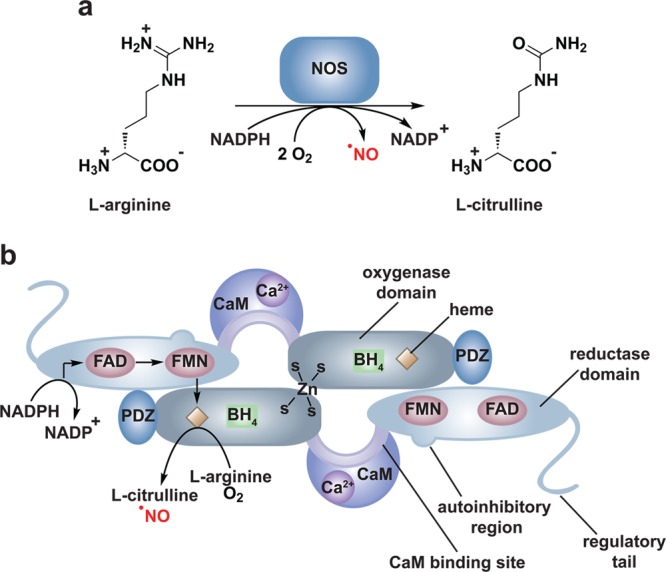
Nitric oxide (•NO) production by nitric oxide synthases (NOS). (a) Reaction catalyzed by NOS. (b) Linear arrangement of NOS. NOS contain three distinct domains: the N-terminal oxygenase domain (gray), the C-terminal reductase domain (light blue), and the connecting calmodulin (CaM) binding site (purple). All three NOS isoforms encode a C-terminal regulatory tail and endothelial NOS (eNOS) and neuronal NOS (nNOS) additionally contain an autoinhibitory region in the reductase domain. Activation of eNOS and nNOS requires binding of Ca2+/CaM whereas inducible NOS (iNOS) is expressed with Ca2+/CaM tightly bound. The production of •NO by NOS involves translocation of electrons from NADPH bound in the reductase domain through the FAD and FMN cofactors. The electrons are then transferred to the heme prosthetic group in the oxygenase domain where l-arginine and molecular oxygen bind. The tetrahydrobiopterin (BH4) cofactor in the oxygenase domain appears to regulate the nature of reactive intermediates generated by NOS (e.g., •NO versus O2•–). The CaM binding site, autoinhibitory region, and regulatory tail are believed to regulate enzyme activity by influencing the efficiency of electron transfer between the reductase and oxygenase domains. All NOS isoforms function as homodimers that are stabilized by a zinc ion coordinated by two cysteine residues from each monomer and there is evidence suggesting that electrons are transferred between monomers (as depicted). NOS enzymes additionally contain sequences such as PDZ domains (deep blue) that facilitate protein–protein interactions, which are involved in subcellular targeting of NOS and for mediating direct interactions with protein targets of •NO.
There are three known NOS isoforms that exhibit 51–57% sequence homology among the human enzymes: inducible NOS (iNOS), endothelial NOS (eNOS), and neuronal NOS (nNOS). iNOS is expressed in a wide range of cell types and tissues including phagocytic cells where it produces •NO for cytotoxic purposes. eNOS is expressed primarily in vascular endothelial cells where •NO functions in a paracrine manner to regulate vasodilation. Lastly, nNOS is expressed primarily in the brain where •NO is involved in neurotransmission.245d,245e,248 The NOS isoforms can be classified as those that exhibit constitutive (eNOS, nNOS) and inducible (iNOS) expression as well as those that are activated in a Ca2+-dependent (eNOS, nNOS) and independent (iNOS) manner. All NOS isoforms have a C-terminal tail that appears to regulate enzyme activity.249 Moreover, eNOS and nNOS additionally contain an autoinhibitory loop in the flavin-binding domain that is believed to hinder efficient electron transfer between the FAD and FMN cofactors.250 In response to receptor activation, a number of growth factors, cytokines, and G-protein coupled receptor (GPCR) agonists have been shown to induce an increase in intracellular calcium, which binds tightly to CaM.251 The Ca2+/CaM complex then binds to the CaM binding site in NOS and is believed to induce a conformational change involving the C-terminal tail and the autoinhibitory loop to optimally orient the reductase and oxygenase domains for efficient electron transfer, which is the rate limiting step in •NO production.249,250,252 In contrast to eNOS and nNOS, iNOS, whose expression is controlled by cytokines and interleukins, is expressed with tightly bound Ca2+/CaM and thus functions independent of the intracellular calcium concentration. Rate constants for •NO production range from 200 min–1 for iNOS, which produces high concentrations of •NO over the course of hours in immune cells, to 100 min–1 for nNOS and 20 min–1 for eNOS.249,253 The diverse •NO production rates suggest structural and regulatory differences between isoforms that influence inherent electron flux rates. Moreover, NOS isoforms appear to be regulated, in part, by the rate of product (•NO) release.254
In addition to intrinsic factors, extrinsic factors such as phosphorylation and protein–protein interactions also regulate NOS activity.245e,248 The serine/threonine kinase AKT has been shown to phosphorylate eNOS in the reductase domain and the C-terminal regulatory tail.255 Accessibility of these phosphorylation sites appears to be regulated by Ca2+/CaM binding, and both AKT-mediated eNOS phosphorylation and sustained eNOS activation were recently found to be necessary for the tumorigenic properties of oncogenic Ras.256
Additional extrinsic factors that regulate NOS activity are protein–protein interactions, as illustrated by Ca2+/CaM-mediated eNOS and nNOS activation. Interestingly, eNOS and nNOS have also been shown to interact with the ATP-dependent molecular chaperone, Hsp90, which may facilitate Ca2+/CaM-induced conformational changes.257 Protein–protein interactions have also been shown to mediate membrane localization of the cytoplasmic NOS enzymes for cell signaling. In contrast to certain NOX complexes, which have been shown to assemble at activated membrane receptors,12,70 NOS appears to preassociate with receptors and distinct membrane microdomains prior to ligand stimulation. For example, nNOS has a unique N-terminal PDZ domain, which mediates protein–protein interactions and directs intracellular proteins into multiprotein complexes.258 In neuronal cells, nNOS is targeted to postsynaptic sites through binding of its PDZ domain to corresponding domains of proteins including PSD-95 and PSD-93.259 PSD-95 binds to the NMDA receptor (NMDAR) thereby mediating a link between the receptor and nNOS, and this complex forms in the absence of NMDA.260 By an independent mechanism, eNOS is localized to the membrane through direct interaction with the bradykinin 2 receptor (B2R).261 All three NOS isoforms contain the conserved sequence FXXFXXXXW, which is a putative caveolin binding site, in their oxygenase and reductase domains.246b,262 In endothelial cells and cardiac myocytes, eNOS is localized to caveolae, a specialized form of lipid raft, by direct interaction with caveolin-1 and caveolin-3.263 Interestingly, eNOS is held in an inactive conformation by its interaction with caveolin and B2R, which is released upon Ca2+/CaM-binding or receptor activation, respectively.261,264
Membrane localization of eNOS regulates •NO production in endothelial cells by mediating l-arginine availability. In endothelial cells, eNOS forms a complex with the cationic amino acid transporter CAT-1 and arginosuccinate lyase (ASL).265 CAT-1 is responsible for arginine transport266 and ASL works in concert with arginosuccinate synthase (ASS1) to recycle l-citrulline, the amino acid product formed by NOS, back to l-arginine. Additionally, ASL funnels l-arginine imported by CAT-1 to eNOS.265 In this way, eNOS complex formation with CAT-1 and ASL regulates •NO production by modulating local substrate availability, somewhat like the regulation of flavin availability through NOX complex formation with riboflavin kinase.70
Under certain circumstances, NOS can form O2•– instead of •NO.267 Such conditions include the absence of the BH4 cofactor268 and uncoupling of electron transfer within NOS via conformational changes that permit direct oxygen interaction with the flavins in the reductase domain264b or S-glutathionylation of Cys689 and Cys908 in the reductase domain of eNOS.163a The more recent finding that S-glutathionylation influences reactive intermediate production by NOS is interesting given that coproduction of ROS and RNS can result in generation of the aggressive oxidant ONOO– and might be a mechanism to deter ONOO– production.163a Although further studies are required to determine whether ROS mediate NOS glutathionylation, the cross-regulation proposal is also supported by the observation that •NO can inhibit NOX-mediated oxidant production in plant immune cells.269
•NO has been shown to regulate a range of processes including proliferation, apoptosis, angiogenesis, host defense, and regulation of vasodilation.239 To elucidate the role of unique NOS isoforms in regulating these diverse biological processes, mouse models of NOS deficiency have been generated.270 Additionally, much effort has been aimed at developing selective small molecule inhibitors for individual NOS isoforms.245d Selective inhibitors have been developed for iNOS that act in competition with l-arginine in which selectivity is achieved through interactions with the novel substrate-binding site in this isoform, as compared to nNOS and eNOS.271 Two iNOS inhibitors have been used to probe the roles of this isoform in several animal models of diseases in which iNOS has been implicated. More recently, a therapeutic role for iNOS selective inhibitors has been shown for lung regeneration in a mouse model of full-established emphysema.272 Selective iNOS inhibitors have also been used in clinical studies for medical conditions involving lung damage including chronic obstructive pulmonary disease (COPD) and asthma.273 The continued development of NOS inhibitors will further our understanding of distinct roles for each of these isoforms in diverse biological processes and will certainly continue to uncover additional avenues for therapeutic intervention for diseases where NOS are implicated.
In plants, •NO has been shown to be involved in seed germination, root growth, respiration, stromal closure, and adaptive responses to biotic and abiotic stresses.274 Although the existence of bona fide NOS isoforms in plants remains controversial, the cytoplasmic enzyme nitrate reductase is a recognized source of •NO in these organisms, as reviewed elsewhere.275 Alternative pathways for NOS-independent •NO production have also been identified in different plant cell compartments, such as peroxisomes, mitochondria, and the apoplasm.275
The discovery that RNS are produced as second messengers to regulate a number of biological processes has spurred the development of methods to specifically detect these species in cells. Historically, •NO production has been detected indirectly by monitoring its oxidation products, namely N2O3, NO2–, and NO3– by colorimetric, spectroscopic and fluorescent means.276 The field has more recently seen the development of direct methods to specifically detect not only •NO, but also ONOO– and nitroxyl (HNO) by exploiting the unique reactivity of each of these species. These methods include nanotube-,277 cell-,278 protein-,279 small molecule-,280 and electrochemical-based281 assays. To date, no RNS probes are available that permit species detection in specific subcellular compartments or organelles. Improvements to the current technology including reversibility are required for regio- and spatiotemporal resolution of RNS production and the interested reader is referred to the following review for additional information.282
4.1.2. •NO-Metabolizing Enzymes
Unlike O2•– and H2O2, for which ROS metabolizing enzymes exist to regulate their levels, far less is known about enzymatic regulation of •NO availability. As previously mentioned, •NO autooxidizes to NO2– and NO3–, however, it was recently shown that •NO oxidation to NO2– can also be catalyzed by the abundant plasma multicopper oxidase, ceruloplasmin.283 NO2– and NO3– have traditionally been thought of as inert byproducts of •NO; however, there is increasing evidence for enzymatic reduction of NO2— to regenerate •NO by xanthine oxidase,284 by nitrate reductase in plants,275 or through reaction with deoxyhemoglobin in the vasculature.285 NO2– reduction could also facilitate •NO release at sites distant from NOS. Along these lines, fatty acids and proteins modified by •NO can similarly be reduced to release •NO or act to transfer •NO to sites distal from NOS. Through protein–protein interactions, NOS has been found to localize to the plasma membrane, endoplasmic reticulum, sarcoplasmic reticulum, and sarcolemmal caveolae where NOS regulates a distinct set of proteins in each location.286 This has spurred the hypothesis that NOS is placed where it is needed for local action of •NO, akin to NOX.287 However, it is possible that the aforementioned alternative mechanisms of •NO release and transport may extend •NO signaling to subcellular regions that are inaccessible by NOS, such as the nucleus,288 or may enhance the paracrine activity of •NO.289
4.2. Modification of Protein Cysteine Thiols by RNS
Similar to O2•–, •NO is a relatively unreactive radical and its primary targets in cells include other radical species such as O2•– and metals. Indeed, the propensity for •NO to coordinate to metals has been exploited in the development of •NO-specific small molecule fluorescent detectors.280a−280i The first identified cellular target of •NO was soluble guanylyl cyclase (sGC) in which •NO activates sGC through binding reversibly to the prosthetic heme.290 In endothelial cells, the •NO produced migrates through the vasculature to activate sGC in the underlying vascular smooth muscle cells to promote vasodilation.291•NO-mediated sGC activation also stimulates mitochondrial biogenesis in brown adipose tissue.292 In addition to sGC, •NO can regulate other heme-containing proteins including ETC Complex IV, where •NO binding inhibits cellular respiration and ROS production under hypoxic conditions.293•NO can also control protein function through iron–sulfur clusters, as documented for bacterial transcriptional regulators, such as NsrR, SoxR, and FNR.294 This form of regulation is thought to occur via •NO-mediated iron–sulfur cluster nitrosylation and degradation.295
It was recognized early on in the field that, in addition to regulating protein function by coordination to metal-based prosthetic groups, •NO could covalently modify protein cysteines, a modification subsequently termed S-nitrosylation.5c Analogous to other oxPTMs, specificity in modification appears to be imparted by cysteine reactivity, local protein environment, and proximity to the oxidant source.3b,12,194 In contrast to NOX signaling, (or, more likely, as is less well established for NOX signaling) the proximity of protein targets of •NO to the RNS source is frequently imparted by direct interaction with NOS. As discussed above, NOS enzymes contain structural features that facilitate protein–protein interactions, and a number of NOS-interacting proteins including caspase-3,296 cyclooxygenase-2,297 and the postsynaptic scaffolding protein PSD-95,260 have been shown to be S-nitrosylated after NOS activation.
Though still an active area of research, three prominent mechanisms have been proposed to account for de novo S-nitrosothiol formation, none of which involve direct reaction of •NO with thiols (Figure 15a–c). As mentioned above, •NO can be converted to the nitrosating compound N2O3 (Chart 12). The initial reaction of •NO with molecular oxygen to generate •NO2 and subsequent radical–radical combination of •NO with •NO2 permits N2O3 production with a rate constant of 109 M–1 s–1.241 N2O3 can subsequently react with a protein or low molecular weight thiolate to yield an S-nitrosothiol (Figure 15a). Given the requirement for two molecules of •NO in this reaction, the route is not favorable at low concentrations of this species. Alternatively, •NO2 or other radical species such as O2•–298 can promote the one-electron oxidation of a protein or low molecular weight thiolate to generate a thiyl radical that can undergo radical–radical combination with •NO to yield the S-nitrosothiol (Figure 15b). While evidence exists to support both of these mechanisms,299 a third route has been postulated to account for S-nitrosylation of some proteins. This mechanism, which has been demonstrated for both hemoglobin300 and nitrophorin,301 relies on the propensity of •NO to bind to heme prosthetic groups in which the heme-bound NO undergoes reductive nitrosylation of the heme prosthetic group and autotransfer to a thiol within the same protein to generate an S-nitrosothiol (Figure 15c). Though it will not be further discussed here, •NO and •NO-derived species can also modify other amino acids, including tyrosine.
Figure 15.
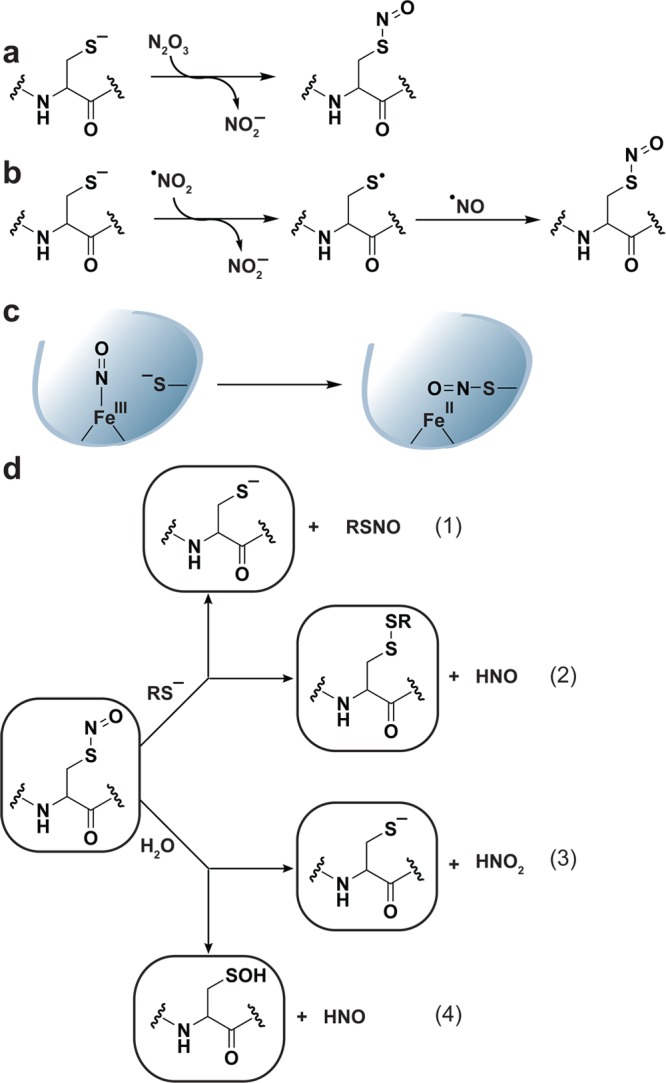
Formation and subsequent reactions of S-nitrosothiols. Three prominent mechanisms for S-nitrosothiol formation include (a) reaction of a protein or low molecular weight thiolate with N2O3, (b) formation of a thiyl radical upon initial reaction of a thiolate with •NO2 and other radical species and subsequent radical–radical combination with •NO, and (c) autotransfer of heme-bound +NO to a nearby cysteine thiolate as has been demonstrated for hemoglobin and nitrophorin. (d) Once formed, an S-nitrosothiol can react with a neighboring cysteine residue either within the same or an adjacent protein, or with GSH (not shown) to undergo transnitrosylation (eq 1) or disulfide bond formation (eq 2). Alternatively, an S-nitrosothiol can be hydrolyzed to release the free thiolate and nitrite (HNO2) (eq 3) or a sulfenic acid and HNO (eq 4). In each case, the pKa of the sulfur in the S-nitrosothiol, in part, influences which product is formed. In most cases, transnitrosylation and release of a free thiolate are favored upon reaction with a second cysteine or water, respectively due to the high pKa of the HNO leaving group.
Like sulfenic acid, the formal oxidation number of the sulfur atom in S-nitrosothiol is 0; despite this apparent similarity, there are many important differences between these modifications. The S-nitrosothiol group is not ionizable,302 can undergo hydrolysis to give sulfenic acid,303 or react with a thiol (Figure 15d). Interestingly, reaction of an S-nitrosothiol with a protein thiol or GSH does not always yield the mixed disulfide, but can instead (and perhaps more frequently) facilitate a process known as transnitrosylation (Figure 15d). The ability to undergo transnitrosylation is due to the different chemical properties of +NO compared to the hydroxyl in sulfenic acid, and will be discussed further in the following subsection. Transnitrosylation is increasingly viewed as another physiologically relevant mechanism for S-nitrosothiol formation5c,287,304 and studies of protein S-nitrosylation often use +NO donors such as GSNO, S-nitrosocysteine (SNOC), and S-nitroso-N-acetyl-d,l-penicillamine (SNAP).260,297,305 In vitro rate constants for de novo thiol S-nitrosylation in human and bovine serum albumin are on the order of 103 to 104 M–1 s–1.306 In contrast, in vitro S-nitrosylation rate constants for glutathione and other low molecular weight thiols are on the order of 105 to 107 M–1 s–1.306,307 Since de novo S-nitrosothiol formation depends on the combined reactivity of two •NO, molecular oxygen, and a thiol (Chart 12, Figure 15a and b), GSH and abundant proteins such as Trx, albumin, and hemoglobin could be primary targets of S-nitrosylation. Indeed, as previously mentioned, the aforementioned protein and low molecular weight thiols can function as +NO donors, and are proposed to extend •NO signaling to proteins distal to its site of production both within cells and as a paracrine signal.288,289
The tendency for a particular cysteine residue to undergo transnitrosylation appears to be regulated, in part, through steric (e.g., accessibility to +NO donors) and electrostatic factors.287,305h,308 Computational studies to identify a consensus sequence for S-nitrosylation have uncovered an acid–base motif, located distal to the modified cysteine in the protein tertiary structure among some S-nitrosothiols.304,309 This charged nature of the acid–base motif has been proposed to engage in protein–protein and protein-GSH interactions to facilitate transnitrosylation. A recent structural study, has revealed to two additional sequence motifs proximal to the S-nitrosothiol that facilitate reduction by Trx, though whether these particular cysteines also participate in transnitrosylation from nitrosylated Trx has not been established.304 Beyond these putative protein–protein interaction motifs, manual inspection of S-nitrosothiol sites incidate that S-nitrosylated cysteines may be directly flanked by an acid–base motif that enhance reactivity or decrease pKa.310 An additional feature of the environment surrounding S-nitrosylation sites is hydrophobicity.311 Hydrophobic protein surfaces could potentially concentrate nonpolar •NO and molecular oxygen, permitting the formation of N2O3 directly at the site of S-nitrosylation. Neither the local acid–base motif nor the hydrophobic environment are uniformly conserved, which is consistent with a similar lack of sequence bias for sulfenylation9b and may be reflective of the numerous mechanisms for de novo and transnitrosylation.304,312
Given the propensity for S-nitrosothiols to participate in transnitrosylation reactions, reducing systems are in place to reverse S-nitrosylated thiols and the interested reader is referred to the following reviews for a more thorough coverage of the topic.287,313 GSH can reduce a S-nitrosothiol to give the free thiol and GSNO. In turn, GSNO is reduced to regenerate GSH and release HNO by GSNO reductases (GSNOR).314 GSNOR acts exclusively on GSNO and deficiency of this enzyme increases the steady-state level of protein S-nitrosylation (which can be further enhanced by iNOS activation and may support a physiological role for GSNO as an +NO donor).314a,315 Protein S-nitrosothiols can also be reduced by the Trx/TrxR system.305e Enzymes with primary functions unrelated to protein S-nitrosylation may also act as denitrosylases, including PDI, xanthine oxidase, and SOD,313 though the physiological relevance of these activities remains unclear.
Since its discovery, S-nitrosylation has been implicated in the regulation of proteins involved in cellular trafficking,316 muscle contractility,317 apoptosis,305a,305e circulation,318 neural transmission,260,319 and host defense.320 However, it is important to keep in mind that most S-nitrosylated proteins that have been identified to date are derived from studies with exogenous +NO donors employed at unphysiological concentrations (though protein S-nitrosylation from endogenous •NO production has been observed in neurons and immune cells, the latter of which produce high concentrations of •NO for bactericidal purposes).245d,248
In plants, attempted microbial invasion triggers the hypersensitive response, a programmed execution of plant cells at the sites of infection. This process involves the generation of NOS-derived •NO with subsequent production of NOX-derived O2•– and the chemical messenger, salicylic acid.320 Interestingly, S-nitrosylation of NPR1, a master regulator of salicylic acid-mediated defense genes, promotes its oligomerization and cytoplasmic retention. S-nitrosylation is reversed by Trx in a salicylic acid-stimulated manner to facilitate NPR1 monomerization and nuclear translocation.305f Whether microbial invasion in plants induces cell death appears to be regulated, in part, by the extent of •NO and O2•– production, which together produce the more reactive ONOO–. Interestingly, both NOS321 and NOX269 have been found to be inhibited by S-nitrosylation, shedding light on a potential regulatory mechanism to control ROS and RNS coproduction in immune responses, which could be conserved across species. More recently, it was shown in a mouse model of Clostridium difficile infection that host-derived •NO S-nitrosylates and inhibits clostridial small molecule-activated glucosylating toxins, thereby preventing toxin cleavage and cell entry.322 This represents a unique mechanism for •NO-mediated pathogen detoxification.
In addition to being involved in the immune response, NOS may also play a role in synaptic plasticity (the strength of connection between two neurons), which is relevant to processes, such as spatial learning.323 nNOS is recruited to the membrane prior to synaptic signaling through its interaction with PSD-95, which physically links nNOS to NMDAR (Figure 16).259a,324 Stimulation of NMDAR triggers calcium entry and activates •NO production via the proximal nNOS. PSD-95 is localized to the membrane through a dynamic reversible cycling of S-palmitoylation, a posttranslational lipid modification, of two N-terminal cysteine residues.325 It was recently shown that nNOS activation mediates S-nitrosylation of these same cysteine residues in PSD-95 thereby preventing S-palmitoylation and reducing PSD-95 and hence nNOS membrane localization subsequent to neuron activation (Figure 16).260 This study highlights the intriguing possibility that differential modification of cysteines may represent a general paradigm in cell signaling and, in this context, S-nitrosylation of PSD-95 may function to regulate the duration of NMDA signaling. NMDAR activation also regulates the recruitment of AMPA receptors (AMPAR) to the synapse to propagate signaling. PSD-95 regulates AMPAR through its interaction with stargazin326 and was recently shown to be S-nitrosylated in response to NMDA signaling, thereby enhancing its binding to AMPAR (Figure 16).319a Lastly, nNOS-derived •NO can also regulate neural cells at the level of gene transcription. For example, S-nitrosylation of histone deacetylase 2 was found to induce its release from chromatin, permitting increased acetylation of histones surrounding genes involved in neural development and promoting transcription.305d
Figure 16.
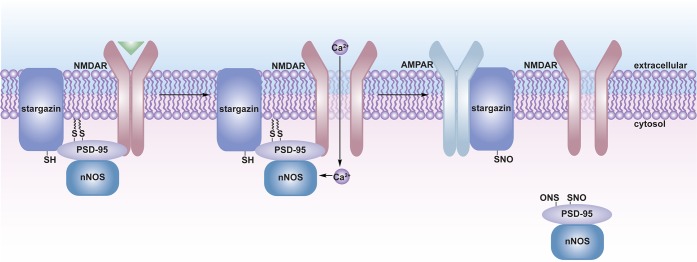
Regulation of neuronal signaling by S-nitrosylation. PSD-95 is a scaffolding protein that localizes to postsynaptic densities by reversible S-palmitoylation of two cysteine residues in the N-terminal region. In the absence of ligand, nNOS is physically linked to the NMDA receptor (NMDAR) at the neuronal plasma membrane via a mutual interaction with PSD-95. PSD-95 similarly localizes stargazin near NMDAR. NMDA binding to NMDAR facilitates calcium entry, which activates nNOS mediating •NO production and subsequent S-nitrosylation of PSD-95 on the same cysteine residues that undergo S-palmitoylation. S-nitrosylation thereby prevents PSD-95 lipidation and decreases membrane association of PSD-95 and, hence, nNOS. Stargazin is similarly S-nitrosylated, which enhances its interaction with the AMPA receptor facilitating its recruitment to the postsynaptic densities.
Expression of NOS isoforms is regulated by Ca2+/CaM binding. S-Nitrosylation of calcium transporters has been increasingly demonstrated, revealing a potential positive feedback loop. In one instance, S-nitrosylation of ryanodine receptors in skeletal muscle327 and neurons328 releases intracellular calcium stores to potentiate signaling that, in the latter case, are required for neural synaptic plasticity and can also contribute to neuronal cell death. S-Nitrosylation can also regulate entry of extracellular calcium as S-nitrosylation of transient receptor potential (TRP) cation channels mediates a conformational change in endothelial cells that opens the pore to permit calcium entry,329 which may similarly function as a positive feedback loop to potentiate NOS activity.
In addition to promoting cell signaling responses, dysregulation of S-nitrosylation has been implicated in disease, including neurodegenerative disorders.256,287,305a,305b,330 The E3 ubiquitin ligase parkin, which regulates the degradation of proteins important to survival of dopamine neurons, is S-nitrosylated in a mouse model of Parkinson’s disease (PD) and in brains of patients with PD.305b Parkin S-nitrosylation inhibits its ubiquitin ligase activity, which impairs ubiquitination of its substrate proteins and may contribute to the degenerative process. It has also been shown that PDI, an ER-resident enzyme that facilitates proper protein folding and protects neuronal cells against ER dysfunction, is S-nitrosylated in brain samples manifesting sporadic PD or Alzheimer’s disease.305c PDI S-nitrosylation inhibits its activity, resulting in activation of ER stress pathways (including the unfolded protein response) and abrogates PDI-mediated attenuation of neuronal cell death triggered by ER stress, which could contribute to neurodegenerative disorders. More recently, amyloid-β, a key mediator in Alzheimer’s disease was found to induce •NO production, which triggered mitochondrial fission, synaptic loss, and neuronal damage.305a This effect was attributed, in part, to S-nitrosylation of dynamin-related protein 1 (Drp1), a protein involved in regulation of mitochondrial fission. S-nitrosylated Drp1 is increased in brains of human Alzheimer’s disease patients where it is postulated to contribute to disease pathogenesis.
4.3. Methods for Detecting RNS-Modified Cysteines
The discovery of protein S-nitrosylation has spurred the development of methods for its detection.138,331 Initial indirect chemiluminescent, colorimetry and electrochemical approaches relied upon detection of NO liberated from S-nitrosothiols by mercury.332 However, these methods are artifact prone because of interference from other metabolites in the sample, such as NO2–. Moreover, indirect spectroscopic methods report only on the total amount of S-nitrosothiols and do not permit identification of the target proteins. While NO is released with metal treatment, the protein thiol remains coordinated to mercury and strategies to identify metal-coordinated proteins have been reported.304,333 A limitation to this method is that other metal-interacting modifications, including protein-S-GSH disulfides and, perhaps cysteine sulfenic acid and persulfide, can similarly be detected and complicate selective analysis. While alternative methods for S-nitrosothiol detection have since been developed, these indirect spectroscopic methods are still used to quantify the total amount of S-nitrosylated protein in purified samples.305g S-Nitrosylated proteins can also be identified using an anti-S-nitrosocysteine antibody or by MS, however, these methods are not well suited to identify S-nitrosothiols in complex protein mixtures and do not facilitate enrichment of oxidized proteins.
Chemical methods for direct and selective detection of protein S-nitrosylation have also been reported. In contrast to sulfenic acids, which have one electrophilic center, S-nitrosothiols contain two, allowing nucleophiles to attack the sulfur or nitrogen (the reaction site is influenced by the relative stability (i.e., pKa) of the leaving group). As previously discussed, the pKa of a free thiol group is ∼8.5 but can be significantly modulated in the protein environment.334 The pKa of HNO, the alternative leaving group, is approximated at 11.4.242a,335 On the basis of these relative pKa values, in the majority of cases, the thiol is predicted as the preferred leaving group. This leaving group preference provides a chemical rationale for transnitrosylation; however, S-nitrosothiols can also form en route to disulfide bonds (Figure 15d). In these cases, it is possible that features of the S-nitrosothiol environment favor disulfide bond formation by increasing the electrophilicity of the sulfur through pKa modulation (e.g., increasing the thiol pKa) or by promoting protonation of HNO in the S-nitrosothiol. The latter would be analogous to protonation of a hydroxyl group prior to nucleophilic attack to facilitate expulsion as water. Importantly, the disparate electrophilic centers in sulfenic acids and S-nitrosothiols can be exploited to permit chemical discrimination between these forms.
To date, the most popular procedure for S-nitrosothiol detection, is known as the biotin switch technique (BST).336 As shown in Figure 17a, the BST is an indirect method that involves blocking free thiols by S-methylthiolation with methylmethane thiosulfonate (MMTS, 38), selective reduction of S-nitrosothiols with ascorbate, and labeling nascent thiols with N-[6-(biotinamido)-hexyl]-3′-(2′-pyridyldithio)-propionamide (biotin-HPDP). The reaction of thiols with biotin-HPDP yields a mixed disulfide adduct that can be detected by avidin blot. Additionally, the biotin handle permits enrichment of labeled proteins for proteomics analysis. As with the two previously described indirect chemical methods, the success of the BST is dependent upon complete blocking of free thiols and the selectivity and efficiency of the reducing agent. Though the mechanism is not entirely clear, S-nitrosothiol reduction may involve nucleophilic attack of ascorbate (39) at the electrophilic nitrogen center to release the thiol (Chart 13). In accordance with the leaving group bias of transnitrosylation versus disulfide bond formation, some S-nitrosothiols cannot be reduced efficiently by ascorbate,331a,337 which might be due to competing reaction at the electrophilic sulfur center. Recently, the use of ascorbate as a selective reductant for S-nitrosothiols has been questioned, because of the observation that ascorbate can reduce some disulfides179,338 and sulfenic acid, as recently shown for some 1-Cys Prxs.172 In one instance, sinapinic acid was used in place of ascorbate as it does not appear to react with disulfides.339 Despite the limitations of the BST, this technique is routinely used in diverse protein systems and led to important advances in S-nitrosylation research.226,260,269,288,329
Figure 17.

Indirect and direct chemical methods for S-nitrosothiol detection. (a) The biotin switch technique (BST) is an indirect differential alkylation method that involves blocking free thiols (blue) with methylmethane thiosulfonate (MMTS, 38), reducing S-nitrosothiols (purple) with ascorbate, and labeling nascent thiols with Biotin-HPDP. The samples are then analyzed by nonreducing avidin blot in which S-nitrosylation correlates to increased signal intensity. (b) Quantification of protein S-nitrosylation with d-Switch. The d-Switch technique combines the BST with isotopically labeled NEM in which free thiols (blue) are blocked with d0-NEM, S-nitrosothiols (purple) are reduced with ascorbate and labeled with d5-NEM. The samples are subsequently separated by SDS-PAGE, digested in-gel with trypsin, and the resulting peptides are analyzed by LC-MS/MS. The extent of protein S-nitrosylation is determined by the ratio of d5-NEM to d0-NEM signal intensity. (c–e) Triarylphosphine-based methods to directly modify S-nitrosothiols. (c) Triarylphosphine reagent 40 reacts with S-nitrosothiols to yield a disulfide-bonded biotin adduct. (d) Compound 41 is oxidized upon reaction with S-nitrosothiols to yield a fluorescent compound that comments on the presence of S-nitrosothiols, but does not covalently modify oxidized proteins. (e) Water-soluble triarylphosphine 42 appears to form a stable S-alkylphosphonium adduct as monitored by 31P NMR and mass spectrometry.
Chart 13. Predicted Mechanism for the Reaction of Ascorbate with S-Nitrosothiol.
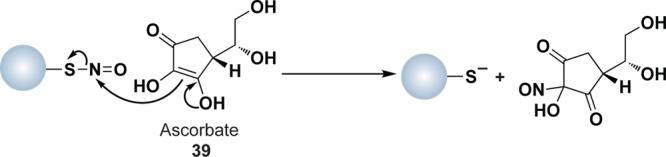
Improvements to the BST have been made that involve biotin enrichment of trypsin-digested peptides,310,340 resin-assisted capture,341 fluorescence labeling,342 and a microarray-based assay.305h The latter case shows a small percentage of false positives and does not cover the entire proteome; nonetheless, it permits rapid identification of candidate S-nitrosylated proteins and allows for the direct comparison and assessment of chemically distinct +NO donors. More recently, Thatcher and colleagues developed a quantitative approach termed d-Switch that combines the BST with isotopically labeled NEM (d5-NEM) (Figure 17b).343 Future adaptations of the d-Switch technique could incorporate a biotin affinity handle to permit sample enrichment analogous to ICAT.
Methods for direct chemical modification of S-nitrosothiols have also been reported. For example, triarylphosphines have shown promise as chemical probes for S-nitrosylation344 and the interested reader can find additional information about this chemistry from the following review.331b In the first demonstration of this approach, a small-molecule S-nitrosothiol model underwent reductive ligation with a triarylphosphine ester.344 A variation on this theme involves reductive ligation of an S-nitrosothiol with a biotinylated triarylphosphine thioester (40) in a THF-PBS system to generate a disulfide linkage with biotin (Figure 17c).345 The triarylphosphine reduction reaction has also been adapted to generate a new fluorescent probe (41) to monitor S-nitrosothiol content in recombinant proteins, but it is not currently amenable to identification of S-nitrosylated proteins in complex mixtures (Figure 17d).346 King and colleagues have reported the water-soluble triarylphosphine (42) that reacts with S-nitrosothiols to give a stable S-alkylphosphonium adduct detectable by 31P NMR and MS (Figure 17e).347 A future modification of this reagent could incorporate an affinity handle for protein enrichment (though the anionic nature of 42 likely precludes membrane permeability for cellular studies). Interestingly, while the S–P bond is usually labile, steric hindrance imparted by the substituted aryl ligands and aromatic stabilization of the phospho cation is believed to stabilize the S-alkylphosphonium adduct. Future work with triarylphosphine reagents will need to address cross reactivity with disulfides and sulfenic acids. From the perspective of selectivity, only the strategy presented in Figure 17c would be able to rigorously discriminate between S-nitrosothiols and disulfides or sulfenic acids, as biotin disulfide formation would be unique to this species (Chart 14).
Chart 14. Reaction Mechanism of Triarylphosphine 40 with a Protein S-Nitrosothiol to Yield a Disulfide-Bonded Biotin Adduct (Equation 1) and Potential Reaction of Sulfenic Acids or Disulfides with 40 (Dashed Arrows) Should Not Yield the Same Adduct (Equation 2).
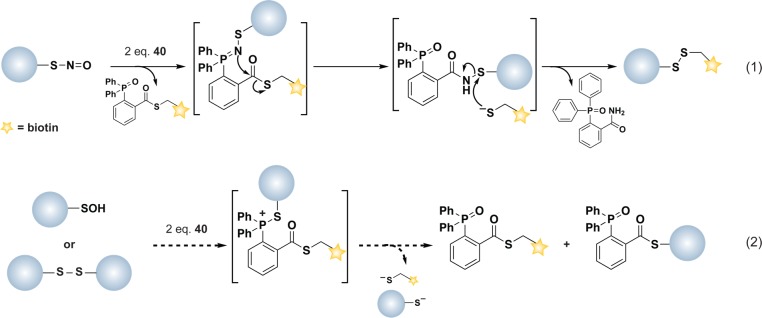
5. Reactive Sulfur Species (RSS) in Biological Systems
As we have seen, oxidation of protein and low molecular weight thiols generates a wide range of sulfur-containing products including disulfides, thiosulfinates, sulfenic acids, and S-nitrosothiols. Each modification is capable of propagating redox transformations that involve oxidation of other thiols analogous to ROS and RNS (Figures 2, 5, and 15d). As follows, these chemically reactive forms of cysteine can be classified as reactive sulfur species (RSS).213,348 Several “nonspecific” peroxidases, such as horseradish peroxidase, can also oxidize thiol substrates by one-electron oxidation to form thiyl radicals, which also represent an important class of RSS.349 Once formed, this radical species can participate in a variety of chemical reactions. Of particular note, thiyl radicals can react with a thiolate affording a disulfide radical anion intermediate, which culminates later in disulfide and O2•– formation.
In addition to reactive cysteine species in proteins, inorganic sulfur-containing species are also classified as RSS. The prototypical inorganic RSS is hydrogen sulfide (H2S), which is the most stable reactive intermediate considered in this review with a half-life on the minute time-scale.350 Along with •NO and carbon monoxide, H2S is produced in biological systems where it functions as a gasotransmitter to regulate diverse biological processes as an autocrine, paracrine, and endocrine signal.351 H2S is a weak acid with a pKa1 and pKa2 of 6.9 and >12 and, therefore, exists primarily in the dissociated thiolate form (HS—) at physiological pH (though H2S is commonly used to refer to all species: H2S, HS—, S2-).352 Similar to other reactive intermediates, H2S was first recognized as a toxic species when it was found to emanate from sewers, and is produced as a toxic byproduct of industrial processes. Research over the past two decades, however, has implicated H2S in a number of physiological and pathological systems. Roles for H2S in biology were initially suggested in vasodilation/relaxation, subsequently as a synaptic modulator and neuroprotectant, and as a regulator of inflammation.350,351,352b,353 The latter has motivated the development of H2S-releasing drugs, which are currently under investigation for their use as anti-inflammatory agents.354 More recently, H2S was also implicated in the control of cell proliferation and survival in cardiomyocytes.355
Given its role in similar physiological settings, H2S has been said to engender many of the same effects of •NO without the generation of hyperreactive (and possibly toxic) intermediates.356 H2S can scavenge reactive intermediates including •NO,357 ONOO–,358 O2•–,359 HOCl, or H2O2; however a general biological role for this activity remains largely speculative at this stage, owing to the low concentration of H2S in many systems (vide infra) and modest reactivity compared to GSH.301,350,352b,360 Nonetheless, Nudler and colleagues recently reported that microbial H2S production and subsequent oxidant scavenging can serve as defense against oxidant-generating antibiotics.361 On the other hand, H2S has also been shown to stimulate the production of ROS in prostate cancer cells362 in a pathway that depends on p66(Shc).363 Though a mechanism was not devulged, this study highlights the potential for complex interplay between RSS and ROS signaling, which is likely to be tissue and cell-type specific. Reaction of H2S with •NO and ONOO– can generate inert nitrosothiols,358 but in some cases, reaction with reactive intermediates facilitates production of additional RSS including polysulfides (H2Sn, n = 2–8) as has been recently described with HOCl (Chart 15).364 Polysulfide production proceeds through a sulfenyl chloride intermediate, which hydrolyzes to sulfenic acid, followed by condensation with a second H2S (Chart 15). Since H2S can participate in two nucleophilic reactions, subsequent oxidation of H2S2 and condensation with H2S facilitates higher order polysulfide formation.
Chart 15. Formation of H2S2 and Higher Order Polysulfides by Reaction of Hydrogen Sulfide (H2S) with ROS, Such as Hypochlorous Acid (HOCl) Initially Yields a Sulfenyl Chloride That Is Hydrolyzed by Water to Afford a Sulfenic Acid.

In addition to H2S, production of other inorganic RSS in cells, such as thiocyanate (−SCN), thiocyanogen [(SCN)2–], trithiocyanate [(SCN)3–] and hypothiocyanite [−OSCN] has been shown or postulated.119b,365 Nevertheless, the physiological significance of these RSS in redox signaling is not well established and, consequently, this section will focus exclusively on the role of H2S as an RSS in redox biology. For a more extensive overview of RSS, the interested reader is referred to the following sources.213,348,366
5.1. H2S Production and Metabolism
5.1.1. H2S-Generating Enzymes
H2S is primarily produced through an alternative metabolic pathway of the cytosolic pyridoxal 5′-phosphate (PLP)-dependent enzymes cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE). Together, these enzymes comprise the transsulfuration pathway that regulates cysteine biogenesis and can produce H2S in a variety of tissues (Figure 18a).350,351,352b,355,367 Little is known about the regulation of CBS and CSE activities, and it is thus largely unclear how H2S may be produced for signaling. To some extent, H2S production by CBS and CSE appears to be controlled by differences in tissue expression. CBS is expressed in many tissues including the liver, kidney, and brain367b,368 and CSE is expressed in, among others, the liver, kidney, cardiovascular system, and to some extent, the brain.367b,369 In the brain, H2S appears to function as an endogenous neuromodulator370 and CBS knockout mice show altered long-term potentiation.371 While the molecular mechanism(s) of H2S action in the context of neuronal signaling are not entirely clear, there is evidence that CBS is regulated at the transcriptional level by the second messenger cAMP.368a Moreover, CBS appears to be directly inhibited by binding of •NO to the heme cofactor,372 whereas CSE expression and activity have been shown to be enhanced by distinct NO donors,373 though these latter effects are controversial.374 Given that H2S can scavenge reactive intermediates, •NO-mediated regulation of CBS and CSE may influence •NO availability. Both CSE and CBS are believed to be activated by Ca2+/CaM binding similar to nNOS, suggesting that •NO and H2S production may be colocalized in some settings.371,375 Additionally, CBS has been shown to be abundant in actively proliferating cells where its activity is coupled to cellular metabolic demands through allosteric activation by AdoMET,370,376 which also appears to regulate CBS-mediated H2S production in neurons.377 More recently, H2S production in the liver of mice treated with the proinflammatory cytokine, tumor necrosis factor α (TNFα) was shown to be dependent upon CSE, which was regulated at the level of transcription.353
Figure 18.
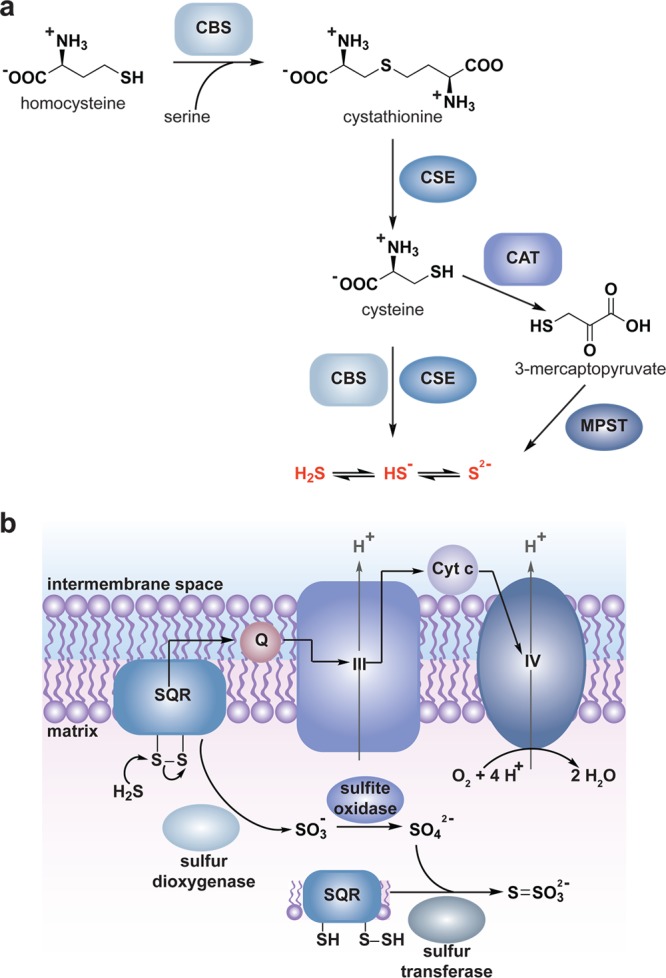
Generation and metabolism of H2S. (a) Pyridoxal 5′-phosphate (PLP)-dependent enzymes of the transsulfuration pathway, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) catalyze H2S production from homocysteine, cystathionine, and cysteine. Additionally, the combined activity of cysteine aminotransferase (CAT) and 3-mercaptopyruvate sulfurtransferase (3MST) in the cytoplasm and mitochondria can generate H2S. (b) Oxidative metabolism of H2S in the mitochondrial matrix is catalyzed by a series of enzymes to generate persulfide, sulfite (SO3–), thiosulfate (S2O32—), and sulfate (SO42—). The first step is catalyzed by SQR, which forms a protein-bound persulfide intermediate and funnels electrons from H2S oxidation directly into the ETC. The subsequent action of sulfur dioxygenase, sulfite oxidase and sulfur transferase are proposed to convert SQR persulfide into SO3–, SO42–, and S2O32–, respectively.
H2S is also produced by the combined activity of 3-mercaptopyruvate sulfurtransferase (3MST) and cysteine aminotransferase (CAT) in the cytosol and mitochondria (Figure 18a).352b,378 3MST is expressed in the liver, kidney, heart, lung, and brain,378d,379 and has been shown to produce H2S in brain homogenates from CBS knockout mice.379b Additionally, H2S is produced nonenzymatically from naturally occurring polysulfanes and other therapeutic compounds and the interested reader is referred to the following sources for additional information.351,380 H2S release from extracellular polysulfanes naturally occurring in garlic appears to occur via a GSH-coupled mechanism381 where extracellular H2S liberation leads to elevated levels of GSSG inside the cell.
To better understand the function of H2S in physiological and pathological settings, reliable assays are needed to accurately determine H2S concentrations in biological samples. A number of colorimetric, electrochemical, gas chromatrography, and metal-induced sulfide precipitation techniques have been developed.357,382 One such method for H2S detection involves the reaction of H2S with N,N-dimethyl-p-phenylenediamide (43) and ferric iron under acidic conditions to generate methylene blue (44), which is monitored by spectroscopy (Chart 16).356,383 One limitation to these methods is that they do not allow for rapid, accurate, and real-time determination of H2S concentrations. Additionally, many of these methods require the generation of cell lysates or tissue homogenates, and therefore afford variable estimates due to rapid H2S catabolism.352b Indeed, a significant challenge in this field is the disagreement as to what constitutes physiological concentrations of free H2S. Current estimates in biological samples and tissues range over a 105-fold concentration range (15 nM384–300 μM355) with 10–600 μM exogenous H2S frequently used to elicit cellular responses.370,375,385 At present, the most reliable estimate (∼100 pM in the blood and 15 nM in tissues) has been reported by Banerjee et al.384 using an innovative gas-chromatographic based chemiluminescent sulfur detection method, which avoids lengthy manipulation steps and the aggressive acidic or basic conditions utilized by early approaches that can leach sulfide from iron–sulfur proteins and lead to cysteine desulfuration through β-elimination.
Chart 16. Colorimetric and Fluorescent Methods for Detection and Quantification of H2Sa.

a Reaction of H2S with two equivalents of 43 in the presence of iron(III) chloride (FeCl3) yields methylene blue (44). Chemoselective reduction of azides in 45 and 46, two-step deprotection of 47, or two-step thioacetal formation from 48 upon reaction with H2S yields fluorescent compounds.
The need for detection methods with improved sensitivity and that measure H2S in cells has motivated the development of fluorescent small-molecule sensors. Recently, three approaches have been reported that rely on selective reaction of a caged fluorophore with H2S. Chang and colleagues have developed an azide-caged fluorophore (45) that becomes fluorescent after H2S-mediated reductive reaction to release the amine (Chart 16).386 The sensitivity of this probe for H2S was reported as 5 μM and was capable of detecting H2S in HEK293 cells treated with exogenous NaHS. Wang and colleagues similarly developed an azide-caged fluorophore (46) that underwent rapid and selective reductive reaction by H2S at concentrations ≥5 μM (Chart 16).387 Xian and colleagues have developed a two-step deprotection strategy (47) to liberate a fluorophore upon reaction with H2S (Chart 16).388 This two-step strategy precludes background signal arising from the reaction of their probe with cysteine or GSH. The probe was sensitive to ∼1 μM H2S in buffer and ∼50 μM in plasma, though lower concentrations were not tested. Invoking alterative chemistry, He and colleagues developed a selective sulfur trapping strategy involving H2S addition to an aldehyde (48) with subsequent Michael addition of the hemithioacetal intermediate with an adjacent unsaturated acrylate ester to give a thioacetal (Chart 16).389 In this latter case, however, important controls were not included to verify a lack of reactivity of 48 with GSH or cysteine. The resulting products exhibit increased fluorescence upon reaction with NaHS and H2S and can detect H2S biogenesis in cells. Implementation of these tools to study H2S signaling and further development of probes with lower limits of detection should expand our understanding of pathways where H2S is proposed to function as a second messenger.
5.1.2. H2S-Metabolizing Enzymes
Unchecked H2S accumulation is toxic and metabolic pathways are in place to regulate its levels. H2S is metabolized through oxidation, in a process that occurs efficiently in the mitochondria352b,390 as catalyzed by a series of enzymes to generate persulfide, sulfite, thiosulfate (S2O32–), and sulfate (Figure 18b).352b,391 The electrons from H2S oxidation funnel directly into ETC complex III by the sulfide:quinone oxidoreductase (which is intriguing given that H2S is also a potent inhibitor of cellular respiration as will be discussed in a following subsection). H2S can also be methylated by thiol-S-methyltransferase to give methanethiol and dimethylsulfide, and it serves as a substrate for rhodanese, leading to the formation of –SCN.351
5.2. H2S-Mediated Modification of Protein Cysteine Thiols
The first signaling role attributed to H2S was as a physiologic vasorelaxant.369b,373 Mice lacking CSE display pronounced hypertension375 and H2S was subsequently shown to be produced by CSE in endothelial cells where it suppresses leukocyte-endothelial cell interactions in the circulation, thereby regulating the immune response.392 Interestingly, both the vasorelaxation and immune response effects of H2S are due, in part, to activation of the ATP-sensitive potassium channel (KATP).373,393 Analogous to protein regulation by •NO, H2S-mediated KATP activation is imparted by chelation to the heme prosthetic group.394 Additional enzymes in which H2S regulates activity by metal chelation include cytochrome c oxidase (complex IV),395 carbonic anhydrase,396 and some NOS isoforms.397 H2S-mediated inhibition of cytochrome c oxidase decreases the cellular metabolic rate and O2•— production, and therefore regulates cell respiration analogous to •NO.293,398 Higher doses of H2S induce hypothermia and establishes a state of suspended animation in mice, which has become a topic of significant interest in the medical field.399 Other membrane channels including the cysteine/glutamate antiporter may also be regulated by H2S.400 In this case, activation of this antiporter stimulates cysteine uptake and GSH production to modulate cellular redox balance.
A second mechanism by which H2S regulates biological processes that has gained increasing attention is by S-sulfhydration/persulfide modification of cysteine residues in proteins. Though considered as “new players” in the field of redox signaling, persulfides were first identified as an intermediate that forms to facilitate sulfur delivery in multiple biosynthetic pathways.401 Within these biosynthetic pathways, the sulfur originates from cysteine, and persulfides form in a number of enzymes, including sulfurtransferases (e.g., rhodanese) and cysteine desulfurases (e.g., IscS) with rhodanese homology domains. Many of these enzymes are involved in delivering sulfide for the production of sulfur-containing vitamins and cofactors, including iron–sulfur clusters through shuttling of persulfide intermediates (e.g., transsulfhydration) between proteins, thus preventing release of free H2S.401,402 In addition to shuttling sulfide from cysteine for biosynthetic pathways, rhodanese proteins are also involved in H2S oxidation in the mitochondria and elimination of toxic compounds like cyanide through its thiosulfate:cyanide sulfurtransferase activity, involving sulfur transfer from a persulfide intermediate.352b,403 A common mechanism for detecting persulfide formation in recombinant protein utilizes a cyanolysis assay based on the reaction catalyzed by rhodanese (Figure 19a).367a,404
Figure 19.

Formation and reactions S-sulfhydryls. (a) The cyanolysis assay for persulfides relies upon reaction of persulfides with cyanide (CN–) to form thiocyanate (SCN–). SCN– is subsequently detected with iron nitrate (Fe(NO3)3•9H2O) to form [Fe(SCN)(H2O)5]2+, which absorbs at 460 nm. (b–d) Mechanisms of S-sulfhydryl formation. Protein S-sulfhydryls can form upon the reaction of H2S with (b) a protein sulfenic acid or (c) a disulfide, though the latter is very slow owing to the poor reductant activity of H2S. (d) Alternatively, S-sulfhydryls can form from nucleophilic attack of a protein thiolate on H2S2. (e) Once formed, protein S-sulfhydryls can be shuttled between proteins via transsulfhydration, as is well established for the sulfurtransferase IscS (eq 1). Reaction of a protein S-sulfhydryl with a second cysteine can alternatively yield a disulfide (eq 2). Akin to S-nitrosothiols, the relative pKa of the protein and H2S thiols will influence which reaction occurs.
Another mechanism through which H2S is proposed to regulate biological processes is by neutralization of reactive electrophiles. Though not mentioned previously, reactive electrophiles, such as the lipid oxidation product, 4-hydroxy-2-nonenal (4-HNE), can covalently modify protein cysteine, lysine, and histidine residues.143 To date, a number of proteins have been shown to be modified by specific reactive electrophiles. These include the transcription factor NF-kB,405 PTP1B,406 the MAPK kinases ERK and p38 MAPK,407 and transient receptor potential (TRP) channels.408 An additional system that is sensitive to reactive electrophiles is the mammalian Keap1-Nrf2 pathway that regulates expression of genes involved in oxidant and xenobiotic detoxification.409 A recent study also suggests that H2S can intercept reactive electrophiles, such as 4-HNE, to prevent protein modification.410 Though the 4-HNE modified H2S adduct was not confirmed in this work, the ability to neutralize reactive electrophiles is thought-provoking, as GSH is known to carry out an analogous function.411
S-sulfhydration (herein used to differentiate between a mechanism of sulfur transfer for the biogenesis of sulfur-containing cofactors and a redox regulated posttranslational modification) is now recognized as a mechanism by which H2S can modulate protein activity. Three primary mechanisms for protein S-sulfhydration have been postulated (Figure 19b–d). The first two mechanisms involve nucleophilic attack of H2S on sulfenic acid- or disulfide-modified proteins (Figure 19b and c). In regard to the latter reaction, we note that H2S is a poor reductant compared to GSH352b and reacts very slowly with protein disulfides in vitro.412 The final mechanism involves oxidation of H2S to generate H2S2 by reaction with ROS such as HOCl (Chart 15) and subsequent nucleophilic attack by a protein thiolate (Figure 19d). Given the low concentration of H2S that has been estimated in cells relative to the high concentration of GSH, the feasibility of direct reaction between H2S and a protein thiolate has been questioned.352b,413 In addition to H2S, the polysulfide diallyl trisulfide (DATS) and the H2S oxidation product, S2O32– have also been postulated as sulfur donors by transferring sulfane sulfur (S0), which could present an additional mechanism for S-sulfhydryl formation.413 Like S-nitrosothiols and disulfides, S-sulfhydryls contain two electrophilic centers and can undergo reaction with a second thiol. Reaction of S-sulfhydryls with a second protein thiol could conceivably yield a disulfide or facilitate transsulfhydration (Figure 19e, eq 2). The latter route is in line with persulfide transfer from IscC to associating proteins as a means for sulfur delivery in the biosynthesis of sulfur-containing cofactors and nucleotides (Figure 19e, eq 1).401,414 As previously mentioned, the pKa1 of H2S is 6.9,352a and therefore a preference for transsulfhydration is less certain than for transnitrosylation (pKa of 11.4 for HNO).242a,335 Transsulfhydration is thus likely to be highly protein specific, whereby cysteine pKa influences which thiol is expelled, analogous to thiol–disulfide exchange.
5.3. Methods for Detecting H2S-Modified Cysteine Thiols
Recently, three chemical procedures have been developed to study proteins susceptible to S-sulfhydration. The first method, developed by Snyder and colleagues, is a modified BST that sought to capitalize on the distinct reactivity of S-sulfhydryls in comparison to the other reversible modifications, including disulfides and protein-S-GSH adducts.415 In the first step, free thiols are labeled with MMTS. Next, excess MMTS is removed and S-sulfhydrated cysteines are alkylated with biotin-HPDP (the nature of the covalent adduct, disulfide or trisulfide, was not established in this work). The ability of S-sulfhydryls to react with biotin-HPDP implies that S-nitrosothiols detected by the original BST would be “contaminated” with persulfide-modified proteins. Indeed, the idea to modify the BST method for detection of protein S-sulfhydration originated from the observation that many proteins were still labeled by biotin-HPDP when ascorbate was omitted from the reaction sequence.336a Using the modified BST, Snyder and colleagues identified 39 potential targets of S-sulfhydration in liver homogenates treated with NaHS.415 GAPDH and actin, two targets of S-sulfhydration, exhibited a respective enhancement in activity and polymerization, after treatment. KATP was also found to be S-sulfhydrated by NaHS, though the effect of this modification on channel activity was not reported. Analogous to other differential alkylation strategies, specificity in this modified BST assay is dependent upon selectivity of the alkylating agents. Recent studies with small molecule and protein S-sulfhydryl models have shed light on the previously uncharacterized nucleophilic properties of the terminal sulfur (J. Pan and K. S. Carroll, unpublished results) as this functional group was found to react at the terminal sulfur atom with NEM, IAM, and MMTS (Chart 17). Thus, the underlying chemistry that mediates S-sulhydryl detection in the modified BST is yet unclear.
Chart 17. Protein S-Sulfhydryls React with NEM (Equation 1), IAM (Equation 2), and MMTS (Equation 3) to Yield the Corresponding Di- or Trisulfides.
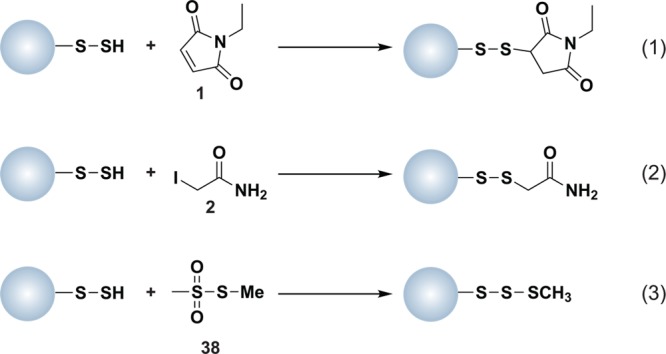
Snyder and colleagues subsequently demonstrated that S-sulfhydration of the p65 subunit of transcription factor NF-κB mediates its antiapoptotic activity using a second indirect chemical method to monitor S-sulfhydryl formation (Figure 20a).353 This two-step method involves modifying free thiols and S-sulfhydryls (at the terminal sulfur) with a fluorescently labeled maleimide compound. The reaction of the S-sulfhydryl with the maleimide reagent yields a disulfide linked adduct that can be reduced by DTT (Figure 20a). In this assay, an increase in S-sulfhydryl formation correlates with a decrease in signal, as monitored by in-gel fluorescence. This method was also extended to include the standard BST, such that S-sulfhydration and S-nitrosylation could be simultaneously monitored using two fluorescently labeled maleimide compounds (Figure 20b). This new method underscores the nucleophilic properties of S-sulfhydryl groups, however, its subtractive nature precludes its use for global identification of S-sulfhydrated proteins.
Figure 20.

Indirect chemical methods to detect S-sulfhydryls. (a) Free thiols (blue) and S-sulfhydryls (purple) are modified with a red fluorescent maleimide reagent. The disulfide linked maleimide adducts derived from S-sulfhydryls are reduced by DTT. Protein modification by S-sulfhydration is detected as a decrease in signal by in-gel fluorescence. (b) Indirect method to simultaneously monitor S-sulfhydryl and S-nitrosothiol formation. Free thiols (blue) and S-sulfhydryls (purple) are modified with a red fluorescent maleimide reagent. S-nitrosothiols (green) are reduced with ascorbate and nascent thiols modified with a green fluorescent maleimide reagent. The disulfide linked maleimide adducts derived from S-sulfhydryls are reduced by DTT. Protein modification by S-sulfhydration and S-nitrosylation are detected as a decrease in red signal and increase in green signal by in-gel fluorescence, respectively. (c) Free thiols (blue) and S-sulfhydryls (purple) are modified by IAM. The disulfide-linked alkylation adduct and all other reversible cysteine modifications (green) are reduced with DTT and nascent thiols are modified with IAP-biotin. Cysteine oxidation is monitored as an increase in signal by avidin blot.
An alternative indirect chemical method was recently reported by Tonks and co-workers to demonstrate S-sulfhydration of PTP1B in H2S-treated human embryonic kidney HEK293T cells.416 This three-step method involves IAM blocking of free thiols and persulfides, DTT reduction of reversibly oxidized thiols, and labeling of nascent thiols with IAP-biotin (Figure 20c). A limitation to this method is that the DTT reducing step is not selective for alkylated S-sulfhydryls, and thus, biotin signal will increase due to the presence of any reversible cysteine modification, including disulfides and sulfenic acids. This study additionally indicated that HeLa cells subjected to ER stress produce H2S in a CSE-dependent manner, leading to PTP1B modification. In these experiments, S-sulfhydration of PTP1B was confirmed in cell lysates by LC-MS/MS analysis. As the authors of this study point out, caution must be taken when assessing S-sulfhydration by MS as S-sulfhydryl and sulfinic acid modifications give a similar mass increase and necessitates high instrument resolution to differentiate between adducts.
6. Conclusions and Future Perspectives
Reactive intermediates, including ROS, RNS, and RSS, are increasingly emerging as major contributors to regulation of numerous physiological and pathological processes. Within this capacity, reactive intermediates function as second messengers to regulate the activity of an ever-expanding number of proteins through covalent modification of cysteine residues. Nonetheless, our understanding of the contribution that each class of reactive intermediate makes is at strikingly different stages. Research over the past five years has provided insight into mechanisms to regulate ROS and RNS production in response to diverse stimuli and the continued development of inhibitors for specific NOX and NOS isoforms will further our understanding of the individualized role of each of these enzymes. In this way, selective inhibitors could prove helpful in further dissecting signaling pathways susceptible to redox modulation and could shed light on novel therapeutic targets. Moreover, the continued improvement of ROS and RNS detection methods will facilitate regiotemporal resolution of their production. In stark contrast, our understanding of regulation of H2S production is still in its infancy. Continued studies to elucidate how and when H2S production is regulated by diverse signals and the development of probes with enhanced sensitivity for H2S will further our understanding of how this RSS participates in redox signaling as a physiologically relevant second messenger.
Likewise, work over the past decade has led to the development of methods that permit selective detection of specific cysteine modifications and, more recently, targeted detection within a subclass of signaling proteins, like PTPs. These collective techniques have greatly expanded the known inventory of proteins susceptible to cysteine oxidation and have shed light on diverse ways in which redox regulation can influence signaling. Nonetheless, the repertoire of reactive cysteine residues and related oxPTMs (particularly those identified by chemically selective, biocompatible approaches) is not complete. Future work is also needed to develop more selective methods for detection of sulfonic acid and S-sulfhydryl modifications. Techniques for cell-based discovery and identification of S-nitrosothiols and S-sulfhydryls are vital, as both modifications can be transferred among cysteines, and sample processing is important to minimize these side reactions as a means to accurately identify relevant sites of modification. Knowledge regarding the extent of site-specific cysteine oxidation is also important for understanding the function and regulation of oxPTMs. In addition, the ability to quantify changes in cysteine oxidation should help overcome a major hurdle in the field-namely, the prioritization of proteins within the “redoxome” selected for further characterization and functional analysis. Indeed, the transformative and paradigmatic discoveries in this exciting field lie in elucidating the functional consequences of these oxidative cysteine modifications in physiological and pathological processes.
Acknowledgments
The authors acknowledge funding from the Camille Henry Dreyfus Teacher Scholar Award (to K.S.C.) and the National Institutes of Health (Grant No. GM102187). Additionally, we would like to acknowledge Mauro LoConte, Vinayak Gupta, and Jia Pan for many helpful discussions.
Biographies

Candice Paulsen, originally from Portland, Oregon, received her B.S. in genetic biology from Purdue University in 2006. Paulsen earned her Ph.D. in chemical biology from the University of Michigan in 2011, where she worked under the advisement of Prof. Kate Carroll to study the role of protein sulfenylation in eukaryotic signal transduction. After continuing as a postdoctoral fellow with Prof. Carroll at Scripps Florida, Paulsen began as a postdoctoral fellow at the University of California, San Francisco in the department of physiology with Prof. David Julius in July of 2012. Her current research interests are focused on elucidating the gating mechanism of transient receptor potential (TRP) ion channels by noxious chemical and thermal stimuli as it pertains to pain signaling.

Kate S. Carroll is an Associate Professor in the Department of Chemistry at The Scripps Research Institute in Jupiter, Florida. She received her B.A. degree in Biochemistry from Mills College in 1996 and Ph.D. in Biochemistry from Stanford University in 2003. Her postdoctoral work was completed at the University of California, Berkeley, where she was a Damon Runyon-Walter Winchell Chancer Fund Fellow with Prof. Carolyn Bertozzi. She was an Assistant Professor at the University of Michigan until 2010, when she joined the Chemistry faculty at Scripps. Her research interests span the disciplines of chemistry and biology with an emphasis on studies of sulfur biochemistry pertinent to disease states. Her lab focuses on the development of novel tools to study redox modifications of cysteine thiols, profiling changes in protein oxidation associated with disease, and exploiting this information for development of diagnostic and therapeutic approaches. In addition, her group investigates sulfur pathways that are essential for infection and long-term survival of human pathogens such as Mycobacterium tuberculosis. She has received the Pfizer Award in Enzyme Chemistry (2013), the Camille Dreyfus Teacher-Scholar Award (2010), the Scientist Development Award from the American Heart Association (2008), and the Special Fellow Award from the Leukemia and Lymphoma Society (2006).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- a Hirooka Y.; Sagara Y.; Kishi T.; Sunagawa K. Circ. J. 2010, 74, 827. [DOI] [PubMed] [Google Scholar]; b Koh C. H.; Whiteman M.; Li Q. X.; Halliwell B.; Jenner A. M.; Wong B. S.; Laughton K. M.; Wenk M.; Masters C. L.; Beart P. M.; Bernard O.; Cheung N. S. J. Neurochem. 2006, 98, 1278. [DOI] [PubMed] [Google Scholar]; c Henriksen E. J.; Diamond-Stanic M. K.; Marchionne E. M. Free Radical Biol. Med. 2011, 51, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Suh Y. A.; Arnold R. S.; Lassegue B.; Shi J.; Xu X.; Sorescu D.; Chung A. B.; Griendling K. K.; Lambeth J. D. Nature 1999, 401, 79. [DOI] [PubMed] [Google Scholar]; e Grek C. L.; Tew K. D. Curr. Opin. Pharmacol. 2010, 10, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a D’Autreaux B.; Toledano M. B. Nat. Rev. Mol. Cell Biol. 2007, 8, 813. [DOI] [PubMed] [Google Scholar]; b Finkel T. IUBMB Life 2001, 52, 3. [DOI] [PubMed] [Google Scholar]; c Rhee S. G. Science 2006, 312, 1882. [DOI] [PubMed] [Google Scholar]; d Winterbourn C. C. Nat. Chem. Biol. 2008, 4, 278. [DOI] [PubMed] [Google Scholar]
- a Jacob C.; Winyard P. G.. Introduction in Redox Signaling and Regulation in Biology and Medicine; Jacob C., Winyard P. G., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp 1–12. [Google Scholar]; b Reddie K. G.; Carroll K. S. Curr. Opin. Chem. Biol. 2008, 12, 746. [DOI] [PubMed] [Google Scholar]
- a Andersen J. K. Nat. Med. 2004, 10SupplS18. [DOI] [PubMed] [Google Scholar]; b Klaunig J. E.; Kamendulis L. M. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 239. [DOI] [PubMed] [Google Scholar]
- a Finkel T. Sci. Signalinging 2012, 5, pe10. [DOI] [PubMed] [Google Scholar]; b Fratelli M.; Demol H.; Puype M.; Casagrande S.; Eberini I.; Salmona M.; Bonetto V.; Mengozzi M.; Duffieux F.; Miclet E.; Bachi A.; Vandekerckhove J.; Gianazza E.; Ghezzi P. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 3505. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hess D. T.; Matsumoto A.; Kim S. O.; Marshall H. E.; Stamler J. S. Nat. Rev. Mol. Cell Biol. 2005, 6, 150. [DOI] [PubMed] [Google Scholar]
- Nelson J. W.; Creighton T. E. Biochemistry 1994, 33, 5974. [DOI] [PubMed] [Google Scholar]
- Marino S. M.; Gladyshev V. N. J. Biol. Chem. 2012, 287, 4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutolf M. P.; Tirelli N.; Cerritelli S.; Cavalli L.; Hubbell J. A. Bioconjugate Chem. 2001, 12, 1051. [DOI] [PubMed] [Google Scholar]
- a Roos G.; Foloppe N.; Messens J. Antioxid. Redox Signaling 2013, 18, 94. [DOI] [PubMed] [Google Scholar]; b Salsbury F. R. Jr.; Knutson S. T.; Poole L. B.; Fetrow J. S. Protein Sci. 2008, 17, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kortemme T.; Creighton T. E. J. Mol. Biol. 1995, 253, 799. [DOI] [PubMed] [Google Scholar]; b Iqbalsyah T. M.; Moutevelis E.; Warwicker J.; Errington N.; Doig A. J. Protein Sci. 2006, 15, 1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hall A.; Parsonage D.; Poole L. B.; Karplus P. A. J. Mol. Biol. 2010, 402, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Woo H. A.; Yim S. H.; Shin D. H.; Kang D.; Yu D. Y.; Rhee S. G. Cell 2010, 140, 517. [DOI] [PubMed] [Google Scholar]
- Paulsen C. E.; Truong T. H.; Garcia F. J.; Homann A.; Gupta V.; Leonard S. E.; Carroll K. S. Nat. Chem. Biol. 2012, 8, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn C. C.; Metodiewa D. Free Radical Biol. Med. 1999, 27, 322. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C.; Hampton M. B. Free Radical Biol. Med. 2008, 45, 549. [DOI] [PubMed] [Google Scholar]
- Peskin A. V.; Low F. M.; Paton L. N.; Maghzal G. J.; Hampton M. B.; Winterbourn C. C. J. Biol. Chem. 2007, 282, 11885. [DOI] [PubMed] [Google Scholar]
- Denu J. M.; Tanner K. G. Biochemistry 1998, 37, 5633. [DOI] [PubMed] [Google Scholar]
- a Fomenko D. E.; Xing W.; Adair B. M.; Thomas D. J.; Gladyshev V. N. Science 2007, 315, 387. [DOI] [PubMed] [Google Scholar]; b Marino S. M.; Gladyshev V. N. Antioxid. Redox Signaling 2011, 15, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying J.; Clavreul N.; Sethuraman M.; Adachi T.; Cohen R. A. Free Radical Biol. Med. 2007, 43, 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill B. G.; Reily C.; Oh J. Y.; Johnson M. S.; Landar A. Free Radical Biol. Med. 2009, 47, 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lees W. J. In Folding of Disulfide Proteins; Chang R. J. Y., Ventura S., Eds.; Springer: New York City, 2011. [Google Scholar]; b Shaked Z.; Szajewski R. P.; Whitesides G. M. Biochemistry 1980, 19, 4156. [DOI] [PubMed] [Google Scholar]
- Marino S. M.; Li Y.; Fomenko D. E.; Agisheva N.; Cerny R. L.; Gladyshev V. N. Biochemistry 2010, 49, 7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kim J. R.; Yoon H. W.; Kwon K. S.; Lee S. R.; Rhee S. G. Anal. Biochem. 2000, 283, 214. [DOI] [PubMed] [Google Scholar]; b Wu Y.; Kwon K. S.; Rhee S. G. FEBS Lett. 1998, 440, 111. [DOI] [PubMed] [Google Scholar]
- Weerapana E.; Wang C.; Simon G. M.; Richter F.; Khare S.; Dillon M. B.; Bachovchin D. A.; Mowen K.; Baker D.; Cravatt B. F. Nature 2010, 468, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gardner P. R.; Raineri I.; Epstein L. B.; White C. W. J. Biol. Chem. 1995, 270, 13399. [DOI] [PubMed] [Google Scholar]; b Halliwell B.; Gutteridge J.. Free Radicals in Biology and Medicine; Halliwell B., Gutteridge J., Eds.; Oxford University Press: New York, 1999. [Google Scholar]; c Tyler D. D. Biochem. J. 1975, 147, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hsu J. L.; Hsieh Y.; Tu C.; O’Connor D.; Nick H. S.; Silverman D. N. J. Biol. Chem. 1996, 271, 17687. [DOI] [PubMed] [Google Scholar]; b McCord J. M.; Fridovich I. J. Biol. Chem. 1969, 244, 6049. [PubMed] [Google Scholar]
- Giorgio M.; Trinei M.; Migliaccio E.; Pelicci P. G. Nat. Rev. Mol. Cell Biol. 2007, 8, 722. [DOI] [PubMed] [Google Scholar]
- Reth M. Nat. Immunol. 2002, 3, 1129. [DOI] [PubMed] [Google Scholar]
- a Bienert G. P.; Moller A. L.; Kristiansen K. A.; Schulz A.; Moller I. M.; Schjoerring J. K.; Jahn T. P. J. Biol. Chem. 2007, 282, 1183. [DOI] [PubMed] [Google Scholar]; b Miller E. W.; Dickinson B. C.; Chang C. J. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 15681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienert G. P.; Schjoerring J. K.; Jahn T. P. Biochim. Biophys. Acta 2006, 1758, 994. [DOI] [PubMed] [Google Scholar]
- a Feng Y.; Santoriello C.; Mione M.; Hurlstone A.; Martin P. PLoS Biol. 2010, 8, e1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Klyubin I. V.; Kirpichnikova K. M.; Gamaley I. A. Eur. J. Cell Biol. 1996, 70, 347. [PubMed] [Google Scholar]; c Moreira S.; Stramer B.; Evans I.; Wood W.; Martin P. Curr. Biol. 2010, 20, 464. [DOI] [PubMed] [Google Scholar]; d Niethammer P.; Grabher C.; Look A. T.; Mitchison T. J. Nature 2009, 459, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yoo S. K.; Starnes T. W.; Deng Q.; Huttenlocher A. Nature 2011, 480, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbengegwi D. R.; Li Q.; Li C.; Bear C. E.; Engelhardt J. F. Mol. Cell. Biol. 2008, 28, 3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hackam D. J.; Rotstein O. D.; Zhang W. J.; Demaurex N.; Woodside M.; Tsai O.; Grinstein S. J. Biol. Chem. 1997, 272, 29810. [DOI] [PubMed] [Google Scholar]; b Lukacs G. L.; Rotstein O. D.; Grinstein S. J. Biol. Chem. 1990, 265, 21099. [PubMed] [Google Scholar]; c Pitt A.; Mayorga L. S.; Stahl P. D.; Schwartz A. L. J. Clin. Invest. 1992, 90, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Winterbourn C. C.; Hampton M. B.; Livesey J. H.; Kettle A. J. J. Biol. Chem. 2006, 281, 39860. [DOI] [PubMed] [Google Scholar]; e Winterbourn C. C.; Kettle A. J. Antioxid. Redox. Signal. 2013, 642. [DOI] [PubMed] [Google Scholar]
- Hurd T. R.; Murphy M. P.. Biological systems relevant for redox signalign and control. In Redox Signaling and Regulation in Biology and Medicine; Jacob C., Winyard P. G., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp 13–34. [Google Scholar]
- Halliwell B.; Gutteridge J. M.; Aruoma O. I. Anal. Biochem. 1987, 165, 215. [DOI] [PubMed] [Google Scholar]
- Bush A. I. Curr. Opin. Chem. Biol. 2000, 4, 184. [DOI] [PubMed] [Google Scholar]
- a Bedard K.; Krause K. H. Physiol. Rev. 2007, 87, 245. [DOI] [PubMed] [Google Scholar]; b Chen K.; Craige S. E.; Keaney J. F. Jr. Antioxid. Redox Signaling 2009, 11, 2467. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Klutz L. O.; Sies H.. Cellular generation of oxidants: Relation to oxidative stress. In Redox Signaling and Regulation in Biology and Medicine; Jacob C., Winyard P. G., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp 45–61. [Google Scholar]; d Petry A.; Weitnauer M.; Gorlach A. Antioxid. Redox Signaling 2010, 13, 467. [DOI] [PubMed] [Google Scholar]
- a Dickinson B. C.; Chang C. J. Nat. Chem. Biol. 2011, 7, 504. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Miller E. W.; Chang C. J. Curr. Opin. Chem. Biol. 2007, 11, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rhee S. G.; Chang T. S.; Jeong W.; Kang D. Mol. Cells 2010, 29, 539. [DOI] [PubMed] [Google Scholar]; d Soh N. Anal. Bioanal. Chem. 2006, 386, 532. [DOI] [PubMed] [Google Scholar]
- a Raha S.; Robinson B. H. Trends Biochem. Sci. 2000, 25, 502. [DOI] [PubMed] [Google Scholar]; b St-Pierre J.; Buckingham J. A.; Roebuck S. J.; Brand M. D. J. Biol. Chem. 2002, 277, 44784. [DOI] [PubMed] [Google Scholar]
- Chance B.; Sies H.; Boveris A. Physiol. Rev. 1979, 59, 527. [DOI] [PubMed] [Google Scholar]
- Lebovitz R. M.; Zhang H.; Vogel H.; Cartwright J. Jr.; Dionne L.; Lu N.; Huang S.; Matzuk M. M. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr S. B.; D’Ari R.; Touati D. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loon A. P.; Pesold-Hurt B.; Schatz G. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Green K.; Brand M. D.; Murphy M. P. Diabetes 2004, 53Suppl 1S110. [DOI] [PubMed] [Google Scholar]; b Korshunov S. S.; Skulachev V. P.; Starkov A. A. FEBS Lett. 1997, 416, 15. [DOI] [PubMed] [Google Scholar]; c Nicholls D. G. Aging Cell 2004, 3, 35. [DOI] [PubMed] [Google Scholar]
- Rottenberg H.; Covian R.; Trumpower B. L. J. Biol. Chem. 2009, 284, 19203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussmaul L.; Hirst J. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kirkinezos I. G.; Moraes C. T. Semin. Cell Dev. Biol. 2001, 12, 449. [DOI] [PubMed] [Google Scholar]; b de Moura M. B.; dos Santos L. S.; Van Houten B. Environ. Mol. Mutagen. 2010, 51, 391. [DOI] [PubMed] [Google Scholar]; c Victor V. M.; Rocha M.; Banuls C.; Bellod L.; Hernandez-Mijares A. Curr. Pharm. Des. 2011, 17, 1986. [DOI] [PubMed] [Google Scholar]
- Raimundo N.; Song L.; Shutt T. E.; McKay S. E.; Cotney J.; Guan M. X.; Gilliland T. C.; Hohuan D.; Santos-Sacchi J.; Shadel G. S. Cell 2012, 148, 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Anastasiou D.; Poulogiannis G.; Asara J. M.; Boxer M. B.; Jiang J. K.; Shen M.; Bellinger G.; Sasaki A. T.; Locasale J. W.; Auld D. S.; Thomas C. J.; Vander Heiden M. G.; Cantley L. C. Science 2011, 334, 1278. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gruning N. M.; Rinnerthaler M.; Bluemlein K.; Mulleder M.; Wamelink M. M.; Lehrach H.; Jakobs C.; Breitenbach M.; Ralser M. Cell Metab. 2011, 14, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E.; Giorgio M.; Mele S.; Pelicci G.; Reboldi P.; Pandolfi P. P.; Lanfrancone L.; Pelicci P. G. Nature 1999, 402, 309. [DOI] [PubMed] [Google Scholar]
- a Giorgio M.; Migliaccio E.; Orsini F.; Paolucci D.; Moroni M.; Contursi C.; Pelliccia G.; Luzi L.; Minucci S.; Marcaccio M.; Pinton P.; Rizzuto R.; Bernardi P.; Paolucci F.; Pelicci P. G. Cell 2005, 122, 221. [DOI] [PubMed] [Google Scholar]; b Orsini F.; Migliaccio E.; Moroni M.; Contursi C.; Raker V. A.; Piccini D.; Martin-Padura I.; Pelliccia G.; Trinei M.; Bono M.; Puri C.; Tacchetti C.; Ferrini M.; Mannucci R.; Nicoletti I.; Lanfrancone L.; Giorgio M.; Pelicci P. G. J. Biol. Chem. 2004, 279, 25689. [DOI] [PubMed] [Google Scholar]
- a Guo J.; Gertsberg Z.; Ozgen N.; Steinberg S. F. Circ. Res. 2009, 104, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nemoto S.; Finkel T. Science 2002, 295, 2450. [DOI] [PubMed] [Google Scholar]
- Bernardi P.; Petronilli V.; Di Lisa F.; Forte M. Trends Biochem. Sci. 2001, 26, 112. [DOI] [PubMed] [Google Scholar]
- Kokoszka J. E.; Coskun P.; Esposito L. A.; Wallace D. C. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Francia P.; delli Gatti C.; Bachschmid M.; Martin-Padura I.; Savoia C.; Migliaccio E.; Pelicci P. G.; Schiavoni M.; Luscher T. F.; Volpe M.; Cosentino F. Circulation 2004, 110, 2889. [DOI] [PubMed] [Google Scholar]; b Pani G.; Koch O. R.; Galeotti T. Int. J. Biochem. Cell Biol. 2009, 41, 1002. [DOI] [PubMed] [Google Scholar]; c Pinton P.; Rizzuto R. Cell Cycle 2008, 7, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Rota M.; LeCapitaine N.; Hosoda T.; Boni A.; De Angelis A.; Padin-Iruegas M. E.; Esposito G.; Vitale S.; Urbanek K.; Casarsa C.; Giorgio M.; Luscher T. F.; Pelicci P. G.; Anversa P.; Leri A.; Kajstura J. Circ. Res. 2006, 99, 42. [DOI] [PubMed] [Google Scholar]
- a Arsenijevic D.; Onuma H.; Pecqueur C.; Raimbault S.; Manning B. S.; Miroux B.; Couplan E.; Alves-Guerra M. C.; Goubern M.; Surwit R.; Bouillaud F.; Richard D.; Collins S.; Ricquier D. Nat. Genet. 2000, 26, 435. [DOI] [PubMed] [Google Scholar]; b Rousset S.; Emre Y.; Join-Lambert O.; Hurtaud C.; Ricquier D.; Cassard-Doulcier A. M. Cytokine 2006, 35, 135. [DOI] [PubMed] [Google Scholar]; c Sonoda J.; Laganiere J.; Mehl I. R.; Barish G. D.; Chong L. W.; Li X.; Scheffler I. E.; Mock D. C.; Bataille A. R.; Robert F.; Lee C. H.; Giguere V.; Evans R. M. Genes Dev. 2007, 21, 1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A. P.; Brodsky I. E.; Rahner C.; Woo D. K.; Erdjument-Bromage H.; Tempst P.; Walsh M. C.; Choi Y.; Shadel G. S.; Ghosh S. Nature 2011, 472, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomilov A. A.; Bicocca V.; Schoenfeld R. A.; Giorgio M.; Migliaccio E.; Ramsey J. J.; Hagopian K.; Pelicci P. G.; Cortopassi G. A. J. Biol. Chem. 2010, 285, 1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor G.; Veljovic-Jovanovic S.; Driscoll S.; Novitskaya L.; Foyer C. H. Ann. Bot. 2002, 89Spec No841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dietz K. J.; Pfannschmidt T. Plant Physiol. 2011, 155, 1477. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Foyer C. H.; Noctor G. Plant Cell 2005, 17, 1866. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Joo J. H.; Wang S.; Chen J. G.; Jones A. M.; Fedoroff N. V. Plant Cell 2005, 17, 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot H.; Littauer A. Free Radical Biol. Med. 1989, 6, 541. [DOI] [PubMed] [Google Scholar]
- Cross A. R.; Jones O. T. Biochim. Biophys. Acta 1991, 1057, 281. [DOI] [PubMed] [Google Scholar]
- a Finkel T. Curr. Opin. Cell Biol. 1998, 10, 248. [DOI] [PubMed] [Google Scholar]; b Kamata H.; Hirata H. Cell. Signal. 1999, 11, 1. [DOI] [PubMed] [Google Scholar]; c Niimura Y.; Poole L. B.; Massey V. J. Biol. Chem. 1995, 270, 25645. [DOI] [PubMed] [Google Scholar]
- Klebanoff S. J. J. Leukocyte Biol. 2005, 77, 598. [DOI] [PubMed] [Google Scholar]
- Miller R. A.; Britigan B. E. Clin. Microbiol. Rev. 1997, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser B. L.; Ward C. W.; Lederer W. J. Science 2011, 333, 1440. [DOI] [PubMed] [Google Scholar]
- a Dickinson B. C.; Tang Y.; Chang Z.; Chang C. J. Chem. Biol. 2011, 18, 943. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Finkel T. J. Cell Biol. 2011, 194, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Paulsen C. E.; Carroll K. S. Chem. Biol. 2009, 16, 217. [DOI] [PubMed] [Google Scholar]
- a Bae Y. S.; Kang S. W.; Seo M. S.; Baines I. C.; Tekle E.; Chock P. B.; Rhee S. G. J. Biol. Chem. 1997, 272, 217. [PubMed] [Google Scholar]; b Sundaresan M.; Yu Z. X.; Ferrans V. J.; Irani K.; Finkel T. Science 1995, 270, 296. [DOI] [PubMed] [Google Scholar]
- Dickinson B. C.; Peltier J.; Stone D.; Schaffer D. V.; Chang C. J. Nat. Chem. Biol. 2011, 7, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Choi H.; Leto T. L.; Hunyady L.; Catt K. J.; Bae Y. S.; Rhee S. G. J. Biol. Chem. 2008, 283, 255. [DOI] [PubMed] [Google Scholar]; b Kim Y. S.; Morgan M. J.; Choksi S.; Liu Z. G. Mol. Cell 2007, 26, 675. [DOI] [PubMed] [Google Scholar]; c Sharma P.; Chakraborty R.; Wang L.; Min B.; Tremblay M. L.; Kawahara T.; Lambeth J. D.; Haque S. J. Immunity 2008, 29, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Honda F.; Kano H.; Kanegane H.; Nonoyama S.; Kim E. S.; Lee S. K.; Takagi M.; Mizutani S.; Morio T. Nat. Immunol. 2012, 13, 369. [DOI] [PubMed] [Google Scholar]
- Yazdanpanah B.; Wiegmann K.; Tchikov V.; Krut O.; Pongratz C.; Schramm M.; Kleinridders A.; Wunderlich T.; Kashkar H.; Utermohlen O.; Bruning J. C.; Schutze S.; Kronke M. Nature 2009, 460, 1159. [DOI] [PubMed] [Google Scholar]
- Paulsen C. E.; Carroll K. S. ACS Chem. Biol. 2010, 5, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshikawa J.; Urao N.; Kim H. W.; Kaplan N.; Razvi M.; McKinney R.; Poole L. B.; Fukai T.; Ushio-Fukai M. PLoS One 2010, 5, e10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameziane-El-Hassani R.; Morand S.; Boucher J. L.; Frapart Y. M.; Apostolou D.; Agnandji D.; Gnidehou S.; Ohayon R.; Noel-Hudson M. S.; Francon J.; Lalaoui K.; Virion A.; Dupuy C. J. Biol. Chem. 2005, 280, 30046. [DOI] [PubMed] [Google Scholar]
- a DeCoursey T. E.; Morgan D.; Cherny V. V. Nature 2003, 422, 531. [DOI] [PubMed] [Google Scholar]; b Korshunov S. S.; Imlay J. A. Mol. Microbiol. 2002, 43, 95. [DOI] [PubMed] [Google Scholar]; c Petheo G. L.; Demaurex N. Biochem. J. 2005, 388, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ramsey I. S.; Ruchti E.; Kaczmarek J. S.; Clapham D. E. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso M.; Bhamrah M. K.; Henley T.; Boyd R. S.; Langlais C.; Cain K.; Dinsdale D.; Pulford K.; Khan M.; Musset B.; Cherny V. V.; Morgan D.; Gascoyne R. D.; Vigorito E.; DeCoursey T. E.; MacLennan I. C.; Dyer M. J. Nat. Immunol. 2010, 11, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di A.; Gao X. P.; Qian F.; Kawamura T.; Han J.; Hecquet C.; Ye R. D.; Vogel S. M.; Malik A. B. Nat. Immunol. 2011, 13, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anilkumar N.; Weber R.; Zhang M.; Brewer A.; Shah A. M. Arterioscler., Thromb., Vasc. Biol. 2008, 28, 1347. [DOI] [PubMed] [Google Scholar]
- a Heumuller S.; Wind S.; Barbosa-Sicard E.; Schmidt H. H.; Busse R.; Schroder K.; Brandes R. P. Hypertension 2008, 51, 211. [DOI] [PubMed] [Google Scholar]; b Li Y.; Trush M. A. Biochem. Biophys. Res. Commun. 1998, 253, 295. [DOI] [PubMed] [Google Scholar]; c Majander A.; Finel M.; Wikstrom M. J. Biol. Chem. 1994, 269, 21037. [PubMed] [Google Scholar]; d Touyz R. M. Hypertension 2008, 51, 172. [DOI] [PubMed] [Google Scholar]
- Jaquet V.; Scapozza L.; Clark R. A.; Krause K. H.; Lambeth J. D. Antioxid. Redox Signaling 2009, 11, 2535. [DOI] [PubMed] [Google Scholar]
- Rey F. E.; Cifuentes M. E.; Kiarash A.; Quinn M. T.; Pagano P. J. Circ. Res. 2001, 89, 408. [DOI] [PubMed] [Google Scholar]
- Gianni D.; Nicolas N.; Zhang H.; Der Mardirossian C.; Kister J.; Martinez L.; Ferguson J.; Roush W. R.; Brown S. J.; Bokoch G. M.; Hodder P.; Rosen H.. Probe Reports from the NIH Molecular Libraries Program; NIH: Bethesda, MD, 2010. [Google Scholar]
- Smith S. M.; Min J.; Ganesh T.; Diebold B.; Kawahara T.; Zhu Y.; McCoy J.; Sun A.; Snyder J. P.; Fu H.; Du Y.; Lewis I.; Lambeth J. D. Chem. Biol. 2012, 19, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kwon S. H.; Pimentel D. R.; Remondino A.; Sawyer D. B.; Colucci W. S. J. Mol. Cell Cardiol. 2003, 35, 615. [DOI] [PubMed] [Google Scholar]; b Owusu-Ansah E.; Banerjee U. Nature 2009, 461, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Veal E. A.; Day A. M.; Morgan B. A. Mol. Cell 2007, 26, 1. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y.; Cho G.; Yuan Z.; Inoue N.; Nakae Y. Biochim. Biophys. Acta 2006, 1758, 1053. [DOI] [PubMed] [Google Scholar]
- Wu R. F.; Xu Y. C.; Ma Z.; Nwariaku F. E.; Sarosi G. A. Jr.; Terada L. S. J. Cell Biol. 2005, 171, 893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Basuroy S.; Bhattacharya S.; Leffler C. W.; Parfenova H. Am. J. Physiol.: Cell Physiol. 2009, 296, C422. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Serrander L.; Cartier L.; Bedard K.; Banfi B.; Lardy B.; Plastre O.; Sienkiewicz A.; Forro L.; Schlegel W.; Krause K. H. Biochem. J. 2007, 406, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Weyemi U.; Lagente-Chevallier O.; Boufraqech M.; Prenois F.; Courtin F.; Caillou B.; Talbot M.; Dardalhon M.; Al Ghuzlan A.; Bidart J. M.; Schlumberger M.; Dupuy C. Oncogene 2011, 31, 1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hahn N. E.; Meischl C.; Wijnker P. J.; Musters R. J.; Fornerod M.; Janssen H. W.; Paulus W. J.; van Rossum A. C.; Niessen H. W.; Krijnen P. A. Cell. Physiol. Biochem. 2011, 27, 471. [DOI] [PubMed] [Google Scholar]; b Meischl C.; Krijnen P. A.; Sipkens J. A.; Cillessen S. A.; Munoz I. G.; Okroj M.; Ramska M.; Muller A.; Visser C. A.; Musters R. J.; Simonides W. S.; Hack C. E.; Roos D.; Niessen H. W. Apoptosis 2006, 11, 913. [DOI] [PubMed] [Google Scholar]
- a Klomsiri C.; Karplus P. A.; Poole L. B. Antioxid. Redox Signaling 2011, 14, 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hall A.; Karplus P. A.; Poole L. B. FEBS J. 2009, 276, 2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raven E. L. Subcell. Biochem. 2000, 35, 317. [DOI] [PubMed] [Google Scholar]
- Noctor G.; Foyer C. H. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 249. [DOI] [PubMed] [Google Scholar]
- Wood Z. A.; Poole L. B.; Karplus P. A. Science 2003, 300, 650. [DOI] [PubMed] [Google Scholar]
- Tsukagoshi H.; Busch W.; Benfey P. N. Cell 2010, 143, 606. [DOI] [PubMed] [Google Scholar]
- Gardner P. R.; Fridovich I. J. Biol. Chem. 1991, 266, 19328. [PubMed] [Google Scholar]
- Hidalgo E.; Demple B. EMBO J. 1994, 13, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune B.; Schmidt K. U.; Ullrich V. Eur. J. Biochem. 1990, 192, 683. [DOI] [PubMed] [Google Scholar]
- Kettle A. J.; Anderson R. F.; Hampton M. B.; Winterbourn C. C. Biochemistry 2007, 46, 4888. [DOI] [PubMed] [Google Scholar]
- Lee J. W.; Helmann J. D. J. Biol. Chem. 2006, 281, 23567. [DOI] [PubMed] [Google Scholar]
- a Ali F. E.; Barnham K. J.; Barrow C. J.; Separovic F. J. Inorg. Biochem. 2004, 98, 173. [DOI] [PubMed] [Google Scholar]; b Ji J. A.; Zhang B.; Cheng W.; Wang Y. J. J. Pharm. Sci. 2009, 98, 4485. [DOI] [PubMed] [Google Scholar]
- a Hoshi T.; Heinemann S. J. Physiol. 2001, 531, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schoneich C. Biochim. Biophys. Acta 2005, 1703, 111. [DOI] [PubMed] [Google Scholar]
- Carballal S.; Alvarez B.; Turell L.; Botti H.; Freeman B. A.; Radi R. Amino Acids 2007, 32, 543. [DOI] [PubMed] [Google Scholar]
- Poor C. B.; Chen P. R.; Duguid E.; Rice P. A.; He C. J. Biol. Chem. 2009, 284, 23517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lee J. W.; Soonsanga S.; Helmann J. D. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 8743. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Salmeen A.; Andersen J. N.; Myers M. P.; Meng T. C.; Hinks J. A.; Tonks N. K.; Barford D. Nature 2003, 423, 769. [DOI] [PubMed] [Google Scholar]; c Sarma B. K.; Mugesh G. J. Am. Chem. Soc. 2007, 129, 8872. [DOI] [PubMed] [Google Scholar]; d Sivaramakrishnan S.; Keerthi K.; Gates K. S. J. Am. Chem. Soc. 2005, 127, 10830. [DOI] [PubMed] [Google Scholar]; e Tanner J. J.; Parsons Z. D.; Cummings A. H.; Zhou H.; Gates K. S. Antioxid. Redox Signaling 2011, 15, 77. [DOI] [PubMed] [Google Scholar]; f van Montfort R. L.; Congreve M.; Tisi D.; Carr R.; Jhoti H. Nature 2003, 423, 773. [DOI] [PubMed] [Google Scholar]; g Yang J.; Groen A.; Lemeer S.; Jans A.; Slijper M.; Roe S. M.; den Hertog J.; Barford D. Biochemistry 2007, 46, 709. [DOI] [PubMed] [Google Scholar]
- Berndt C.; Lillig C. H.; Holmgren A. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1227. [DOI] [PubMed] [Google Scholar]
- a Claiborne A.; Miller H.; Parsonage D.; Ross R. P. FASEB J 1993, 7, 1483. [DOI] [PubMed] [Google Scholar]; b Crane E. J. 3rd; Parsonage D.; Poole L. B.; Claiborne A. Biochemistry 1995, 34, 14114. [DOI] [PubMed] [Google Scholar]
- Depuydt M.; Leonard S. E.; Vertommen D.; Denoncin K.; Morsomme P.; Wahni K.; Messens J.; Carroll K. S.; Collet J. F. Science 2009, 326, 1109. [DOI] [PubMed] [Google Scholar]
- a Hugo M.; Turell L.; Manta B.; Botti H.; Monteiro G.; Netto L. E.; Alvarez B.; Radi R.; Trujillo M. Biochemistry 2009, 48, 9416. [DOI] [PubMed] [Google Scholar]; b Sohn J.; Rudolph J. Biochemistry 2003, 42, 10060. [DOI] [PubMed] [Google Scholar]; c Turell L.; Botti H.; Carballal S.; Ferrer-Sueta G.; Souza J. M.; Duran R.; Freeman B. A.; Radi R.; Alvarez B. Biochemistry 2008, 47, 358. [DOI] [PubMed] [Google Scholar]
- a Davies M. J. J. Clin. Biochem. Nutr. 2011, 48, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nagy P. Antioxid. Redox Signaling 2013, 10.1089/ars.2012.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rio L. A.; Sandalio L. M.; Palma J. M.; Bueno P.; Corpas F. J. Free Radical Biol. Med. 1992, 13, 557. [DOI] [PubMed] [Google Scholar]
- a Biteau B.; Labarre J.; Toledano M. B. Nature 2003, 425, 980. [DOI] [PubMed] [Google Scholar]; b Chang T. S.; Jeong W.; Woo H. A.; Lee S. M.; Park S.; Rhee S. G. J. Biol. Chem. 2004, 279, 50994. [DOI] [PubMed] [Google Scholar]; c Lei K.; Townsend D. M.; Tew K. D. Oncogene 2008, 27, 4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukosz M.; Jakob S.; Buchner N.; Zschauer T. C.; Altschmied J.; Haendeler J. Antioxid. Redox Signaling 2010, 12, 713. [DOI] [PubMed] [Google Scholar]
- a Klomsiri C.; Nelson K. J.; Bechtold E.; Soito L.; Johnson L. C.; Lowther W. T.; Ryu S. E.; King S. B.; Furdui C. M.; Poole L. B. Methods Enzymol. 2010, 473, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Leichert L. I.; Gehrke F.; Gudiseva H. V.; Blackwell T.; Ilbert M.; Walker A. K.; Strahler J. R.; Andrews P. C.; Jakob U. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sethuraman M.; Clavreul N.; Huang H.; McComb M. E.; Costello C. E.; Cohen R. A. Free Radical Biol. Med. 2007, 42, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sethuraman M.; McComb M. E.; Huang H.; Huang S.; Heibeck T.; Costello C. E.; Cohen R. A. J. Proteome Res. 2004, 3, 1228. [DOI] [PubMed] [Google Scholar]
- Le Moan N.; Clement G.; Le Maout S.; Tacnet F.; Toledano M. B. J. Biol. Chem. 2006, 281, 10420. [DOI] [PubMed] [Google Scholar]
- a Boivin B.; Tonks N. K. Methods Enzymol. 2010, 474, 35. [DOI] [PubMed] [Google Scholar]; b Cuddihy S. L.; Winterbourn C. C.; Hampton M. B. Antioxid. Redox Signaling 2011, 15, 167. [DOI] [PubMed] [Google Scholar]
- Hagglund P.; Bunkenborg J.; Maeda K.; Svensson B. J. Proteome Res. 2008, 7, 5270. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zhou Y.; Peng X.; Yoon J. Chem. Soc. Rev. 2010, 39, 2120. [DOI] [PubMed] [Google Scholar]
- Nakamura T.; Yamamoto T.; Abe M.; Matsumura H.; Hagihara Y.; Goto T.; Yamaguchi T.; Inoue T. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fass D. Biochim. Biophys. Acta 2008, 1783, 557. [DOI] [PubMed] [Google Scholar]; b Hell K. Biochim. Biophys. Acta 2008, 1783, 601. [DOI] [PubMed] [Google Scholar]
- a Lee C.; Lee S. M.; Mukhopadhyay P.; Kim S. J.; Lee S. C.; Ahn W. S.; Yu M. H.; Storz G.; Ryu S. E. Nat. Struct. Mol. Biol. 2004, 11, 1179. [DOI] [PubMed] [Google Scholar]; b Nagy P.; Ashby M. T. J. Am. Chem. Soc. 2007, 129, 14082. [DOI] [PubMed] [Google Scholar]
- Ghezzi P.; Di Simplicio P.. Protein glutathiolation. In Redox Signaling and Regulation in Biology and Medicine; Jacob C., Winyard P. G., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp 123–140. [Google Scholar]
- Kadokura H.; Beckwith J.; Gilbert H. F.. Oxidative folding. In Redox Biochemistry; Banergee R., Ed.; John Wiley & Sons: Hoboken, NJ, 2008; pp 113–120. [Google Scholar]
- a Jonsson T. J.; Tsang A. W.; Lowther W. T.; Furdui C. M. J. Biol. Chem. 2008, 283, 22890. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Roussel X.; Bechade G.; Kriznik A.; Van Dorsselaer A.; Sanglier-Cianferani S.; Branlant G.; Rahuel-Clermont S. J. Biol. Chem. 2008, 283, 22371. [DOI] [PubMed] [Google Scholar]
- a Chiarugi P. IUBMB Life 2001, 52, 55. [DOI] [PubMed] [Google Scholar]; b Kwon J.; Lee S. R.; Yang K. S.; Ahn Y.; Kim Y. J.; Stadtman E. R.; Rhee S. G. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16419. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Michalek R. D.; Nelson K. J.; Holbrook B. C.; Yi J. S.; Stridiron D.; Daniel L. W.; Fetrow J. S.; King S. B.; Poole L. B.; Grayson J. M. J. Immunol. 2007, 179, 6456. [DOI] [PubMed] [Google Scholar]; d Savitsky P. A.; Finkel T. J. Biol. Chem. 2002, 277, 20535. [DOI] [PubMed] [Google Scholar]
- Choi K.; Ryu S. W.; Song S.; Choi H.; Kang S. W.; Choi C. Cell Death Differ. 2010, 17, 833. [DOI] [PubMed] [Google Scholar]
- Ostman A.; Hellberg C.; Bohmer F. D. Nat. Rev. Cancer 2006, 6, 307. [DOI] [PubMed] [Google Scholar]
- Burgoyne J. R.; Madhani M.; Cuello F.; Charles R. L.; Brennan J. P.; Schroder E.; Browning D. D.; Eaton P. Science 2007, 317, 1393. [DOI] [PubMed] [Google Scholar]
- Guo Z.; Kozlov S.; Lavin M. F.; Person M. D.; Paull T. T. Science 2010, 330, 517. [DOI] [PubMed] [Google Scholar]
- Kemble D. J.; Sun G. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoni E.; Buricchi F.; Raugei G.; Ramponi G.; Chiarugi P. Mol. Cell. Biol. 2005, 25, 6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J.; Ilbert M.; Graf P. C.; Ozcelik D.; Jakob U. Cell 2008, 135, 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N.; Kuwaki T.; Kiyonaka S.; Numata T.; Kozai D.; Mizuno Y.; Yamamoto S.; Naito S.; Knevels E.; Carmeliet P.; Oga T.; Kaneko S.; Suga S.; Nokami T.; Yoshida J.; Mori Y. Nat. Chem. Biol. 2011, 7, 701. [DOI] [PubMed] [Google Scholar]
- a Kuge S.; Toda T.; Iizuka N.; Nomoto A. Genes Cells 1998, 3, 521. [DOI] [PubMed] [Google Scholar]; b Wood M. J.; Storz G.; Tjandra N. Nature 2004, 430, 917. [DOI] [PubMed] [Google Scholar]
- Ago T.; Liu T.; Zhai P.; Chen W.; Li H.; Molkentin J. D.; Vatner S. F.; Sadoshima J. Cell 2008, 133, 978. [DOI] [PubMed] [Google Scholar]
- Klamt F.; Zdanov S.; Levine R. L.; Pariser A.; Zhang Y.; Zhang B.; Yu L. R.; Veenstra T. D.; Shacter E. Nat. Chem. Biol. 2009, 11, 1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay A.; Pflieger D.; Barrault M. B.; Vinh J.; Toledano M. B. Cell 2002, 111, 471. [DOI] [PubMed] [Google Scholar]
- Sommer A.; Traut R. R. Proc. Natl. Acad. Sci. U. S. A. 1974, 71, 3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming R. C. Methods Mol. Biol. 2008, 476, 165. [DOI] [PubMed] [Google Scholar]
- Leonard S. E.; Carroll K. S. Curr. Opin. Chem. Biol. 2011, 15, 88. [DOI] [PubMed] [Google Scholar]
- Cumming R. C.; Andon N. L.; Haynes P. A.; Park M.; Fischer W. H.; Schubert D. J. Biol. Chem. 2004, 279, 21749. [DOI] [PubMed] [Google Scholar]
- Brennan J. P.; Wait R.; Begum S.; Bell J. R.; Dunn M. J.; Eaton P. J. Biol. Chem. 2004, 279, 41352. [DOI] [PubMed] [Google Scholar]
- Gu J.; Lewis R. S. Ann. Biomed. Eng. 2007, 35, 1554. [DOI] [PubMed] [Google Scholar]
- a Owen J. B.; Butterfield D. A. Methods Mol. Biol. 2010, 648, 269. [DOI] [PubMed] [Google Scholar]; b Townsend D. M.; Tew K. D.; Tapiero H. Biomed. Pharmacother. 2003, 57, 145.12818476 [Google Scholar]
- Rudolph T. K.; Freeman B. A. Sci. Signaling 2009, 2, re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi R.; Cardaioli E.; Scaloni A.; Amiconi G.; Di Simplicio P. Biochim. Biophys. Acta 1995, 1243, 230. [DOI] [PubMed] [Google Scholar]
- a Di Simplicio P.; Cacace M. G.; Lusini L.; Giannerini F.; Giustarini D.; Rossi R. Arch. Biochem. Biophys. 1998, 355, 145. [DOI] [PubMed] [Google Scholar]; b Gilbert H. F. Methods Enzymol. 1984, 107, 330. [DOI] [PubMed] [Google Scholar]; c Ziegler D. M. Annu. Rev. Biochem. 1985, 54, 305. [DOI] [PubMed] [Google Scholar]
- Beer S. M.; Taylor E. R.; Brown S. E.; Dahm C. C.; Costa N. J.; Runswick M. J.; Murphy M. P. J. Biol. Chem. 2004, 279, 47939. [DOI] [PubMed] [Google Scholar]
- Starke D. W.; Chock P. B.; Mieyal J. J. J. Biol. Chem. 2003, 278, 14607. [DOI] [PubMed] [Google Scholar]
- a Gravina S. A.; Mieyal J. J. Biochemistry 1993, 32, 3368. [DOI] [PubMed] [Google Scholar]; b Ruoppolo M.; Lundstrom-Ljung J.; Talamo F.; Pucci P.; Marino G. Biochemistry 1997, 36, 12259. [DOI] [PubMed] [Google Scholar]
- Park J. W.; Mieyal J. J.; Rhee S. G.; Chock P. B. J. Biol. Chem. 2009, 284, 23364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jung C. H.; Thomas J. A. Arch. Biochem. Biophys. 1996, 335, 61. [DOI] [PubMed] [Google Scholar]; b Mannervik B.; Axelsson K.; Sundewall A. C.; Holmgren A. Biochem. J. 1983, 213, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chrestensen C. A.; Starke D. W.; Mieyal J. J. J. Biol. Chem. 2000, 275, 26556. [DOI] [PubMed] [Google Scholar]; b Holmgren A. Proc. Natl. Acad. Sci. U. S. A. 1976, 73, 2275. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Holmgren A.; Ohlsson I.; Grankvist M. L. J. Biol. Chem. 1978, 253, 430. [PubMed] [Google Scholar]
- Steven F. S.; Griffin M. M.; Smith R. H. Eur. J. Biochem. 1981, 119, 75. [DOI] [PubMed] [Google Scholar]
- Macartney H. W.; Tschesche H. Eur. J. Biochem. 1983, 130, 85. [DOI] [PubMed] [Google Scholar]
- Nakashima K.; Horecker B. L.; Pontremoli S. Arch. Biochem. Biophys. 1970, 141, 579. [DOI] [PubMed] [Google Scholar]
- Eaton P.; Wright N.; Hearse D. J.; Shattock M. J. J. Mol. Cell Cardiol. 2002, 34, 1549. [DOI] [PubMed] [Google Scholar]
- Zmijewski J. W.; Banerjee S.; Abraham E. J. Biol. Chem. 2009, 284, 22213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner F.; Molawi K.; Zychlinsky A. Nat. Immunol. 2008, 9, 866. [DOI] [PubMed] [Google Scholar]
- Hurd T. R.; Requejo R.; Filipovska A.; Brown S.; Prime T. A.; Robinson A. J.; Fearnley I. M.; Murphy M. P. J. Biol. Chem. 2008, 283, 24801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonks N. K. Cell 2005, 121, 667. [DOI] [PubMed] [Google Scholar]
- Barrett W. C.; DeGnore J. P.; Konig S.; Fales H. M.; Keng Y. F.; Zhang Z. Y.; Yim M. B.; Chock P. B. Biochemistry 1999, 38, 6699. [DOI] [PubMed] [Google Scholar]
- Velu C. S.; Niture S. K.; Doneanu C. E.; Pattabiraman N.; Srivenugopal K. S. Biochemistry 2007, 46, 7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinarakis E.; Chantzoura E.; Thanos D.; Spyrou G. EMBO J. 2008, 27, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chen C. A.; Wang T. Y.; Varadharaj S.; Reyes L. A.; Hemann C.; Talukder M. A.; Chen Y. R.; Druhan L. J.; Zweier J. L. Nature 2010, 468, 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Townsend D. M.; Findlay V. J.; Fazilev F.; Ogle M.; Fraser J.; Saavedra J. E.; Ji X.; Keefer L. K.; Tew K. D. Mol. Pharmacol. 2006, 69, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan J. P.; Miller J. I.; Fuller W.; Wait R.; Begum S.; Dunn M. J.; Eaton P. Mol. Cell Proteomics 2006, 5, 215. [DOI] [PubMed] [Google Scholar]
- Cheng G.; Ikeda Y.; Iuchi Y.; Fujii J. Arch. Biochem. Biophys. 2005, 435, 42. [DOI] [PubMed] [Google Scholar]
- a Aesif S. W.; Janssen-Heininger Y. M.; Reynaert N. L. Methods Enzymol. 2010, 474, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lind C.; Gerdes R.; Hamnell Y.; Schuppe-Koistinen I.; von Lowenhielm H. B.; Holmgren A.; Cotgreave I. A. Arch. Biochem. Biophys. 2002, 406, 229. [DOI] [PubMed] [Google Scholar]
- Sullivan D. M.; Wehr N. B.; Fergusson M. M.; Levine R. L.; Finkel T. Biochemistry 2000, 39, 11121. [DOI] [PubMed] [Google Scholar]
- a Cohen M. S.; Hadjivassiliou H.; Taunton J. Nat. Chem. Biol. 2007, 3, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Seo Y. H.; Carroll K. S. Bioorg. Med. Chem. Lett. 2009, 19, 356. [DOI] [PubMed] [Google Scholar]
- Sulfenic Acids and Derivatives, Patai’s Chemistry of Functional Groups; Patai S., Ed.; John Wiley & Sons: Hoboken, NJ, 1990. [Google Scholar]
- McGrath A. J.; Garrett G. E.; Valgimigli L.; Pratt D. A. J. Am. Chem. Soc. 2010, 132, 16759. [DOI] [PubMed] [Google Scholar]
- Poole L. B.; Hall A.; Nelson K. J. Curr. Protoc. Toxicol. 2011, 49, 7.9.1. 10.1002/0471140856.tx0709s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro G.; Horta B. B.; Pimenta D. C.; Augusto O.; Netto L. E. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole L. B.; Ellis H. R. Methods Enzymol. 2002, 348, 122. [DOI] [PubMed] [Google Scholar]
- Karplus P. A.; Hall A. Subcell. Biochem 2007, 44, 41. [DOI] [PubMed] [Google Scholar]
- a Antelmann H.; Helmann J. D. Antioxid. Redox Signaling 2011, 14, 1049. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chen H.; Xu G.; Zhao Y.; Tian B.; Lu H.; Yu X.; Xu Z.; Ying N.; Hu S.; Hua Y. PLoS One 2008, 3, e1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantano C.; Reynaert N. L.; van der Vliet A.; Janssen-Heininger Y. M. Antioxid. Redox Signaling 2006, 8, 1791. [DOI] [PubMed] [Google Scholar]
- Saurin A. T.; Neubert H.; Brennan J. P.; Eaton P. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 17982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyther R.; Ahmeda A.; Johns E.; McDonagh B.; Sheehan D. J. Proteome Res. 2010, 9, 2678. [DOI] [PubMed] [Google Scholar]
- Giustarini D.; Dalle-Donne I.; Colombo R.; Milzani A.; Rossi R. Nitric Oxide 2008, 19, 252. [DOI] [PubMed] [Google Scholar]
- a Ellis H. R.; Poole L. B. Biochemistry 1997, 36, 15013. [DOI] [PubMed] [Google Scholar]; b Ma L. H.; Takanishi C. L.; Wood M. J. J. Biol. Chem. 2007, 282, 31429. [DOI] [PubMed] [Google Scholar]; c Reddie K. G.; Seo Y. H.; Muse Iii W. B.; Leonard S. E.; Carroll K. S. Mol. Biosyst. 2008, 4, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hogg D. R. In The Chemistry of Sulphenic Acids and Their Derivatives; Patai S., Ed.; John Wiley & Sons: Hoboken, NJ, 1990. [Google Scholar]; b Benitez L. V.; Allison W. S. J. Biol. Chem. 1974, 249, 6234. [PubMed] [Google Scholar]
- Qian J.; Klomsiri C.; Wright M. W.; King S. B.; Tsang A. W.; Poole L. B.; Furdui C. M. Chem. Commun. 2011, 47, 9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J.; Wani R.; Klomsiri C.; Poole L. B.; Tsang A. W.; Furdui C. M. Chem. Commun. 2012, 48, 4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Tanaka F.; Lerner R. A.; Barbas C. F. 3rd; Wilson I. A. J. Am. Chem. Soc. 2009, 131, 18206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Charles R. L.; Schroder E.; May G.; Free P.; Gaffney P. R.; Wait R.; Begum S.; Heads R. J.; Eaton P. Mol. Cell Proteomics 2007, 6, 1473. [DOI] [PubMed] [Google Scholar]; b Poole L. B.; Klomsiri C.; Knaggs S. A.; Furdui C. M.; Nelson K. J.; Thomas M. J.; Fetrow J. S.; Daniel L. W.; King S. B. Bioconjug. Chem. 2007, 18, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole L. B.; Zeng B. B.; Knaggs S. A.; Yakubu M.; King S. B. Bioconjug. Chem. 2005, 16, 1624. [DOI] [PubMed] [Google Scholar]
- Wani R.; Qian J.; Yin L.; Bechtold E.; King S. B.; Poole L. B.; Paek E.; Tsang A. W.; Furdui C. M. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hang H. C.; Loureiro J.; Spooner E.; van der Velden A. W.; Kim Y. M.; Pollington A. M.; Maehr R.; Starnbach M. N.; Ploegh H. L. ACS Chem. Biol. 2006, 1, 713. [DOI] [PubMed] [Google Scholar]; b Speers A. E.; Cravatt B. F. Chem. Biol. 2004, 11, 535. [DOI] [PubMed] [Google Scholar]
- Leonard S. E.; Garcia F. J.; Goodsell D. S.; Carroll K. S. Angew. Chem., Int. Ed. Engl. 2011, 50, 4423. [DOI] [PubMed] [Google Scholar]
- Leonard S. E.; Reddie K. G.; Carroll K. S. ACS Chem. Biol. 2009, 4, 783. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Baskin J. M.; Prescher J. A.; Lo A.; Bertozzi C. R. ACS Chem. Biol. 2006, 1, 644. [DOI] [PubMed] [Google Scholar]
- Truong T. H.; Carroll K. S. Curr. Protoc. Chem. Biol. 2012, 4, 101. [Google Scholar]
- a Goldkorn T.; Balaban N.; Matsukuma K.; Chea V.; Gould R.; Last J.; Chan C.; Chavez C. Am. J. Respir. Cell Mol. Biol. 1998, 19, 786. [DOI] [PubMed] [Google Scholar]; b Lee S. R.; Kwon K. S.; Kim S. R.; Rhee S. G. J. Biol. Chem. 1998, 273, 15366. [DOI] [PubMed] [Google Scholar]; c Lee S. R.; Yang K. S.; Kwon J.; Lee C.; Jeong W.; Rhee S. G. J. Biol. Chem. 2002, 277, 20336. [DOI] [PubMed] [Google Scholar]; d Meng T. C.; Fukada T.; Tonks N. K. Mol. Cell 2002, 9, 387. [DOI] [PubMed] [Google Scholar]; e Roos G.; Messens J. Free Radical Biol. Med. 2011, 51, 314. [DOI] [PubMed] [Google Scholar]; f Stone J. R.; Yang S. Antioxid. Redox Signaling 2006, 8, 243. [DOI] [PubMed] [Google Scholar]
- Chen K.; Kirber M. T.; Xiao H.; Yang Y.; Keaney J. F. Jr. J. Cell Biol. 2008, 181, 1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y. H.; Carroll K. S. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maller C.; Schroder E.; Eaton P. Antioxid. Redox Signaling 2011, 14, 49. [DOI] [PubMed] [Google Scholar]
- Seo Y. H.; Carroll K. S. Angew. Chem., Int. Ed. 2011, 50, 1342. [DOI] [PubMed] [Google Scholar]
- Truong T. H.; Garcia F. J.; Seo Y. H.; Carroll K. S. Bioorg. Med. Chem. Lett. 2011, 21, 5015. [DOI] [PubMed] [Google Scholar]
- a Singh J.; Petter R. C.; Kluge A. F. Curr. Opin. Chem. Biol. 2010, 14, 475. [DOI] [PubMed] [Google Scholar]; b Zhou W.; Ercan D.; Chen L.; Yun C. H.; Li D.; Capelletti M.; Cortot A. B.; Chirieac L.; Iacob R. E.; Padera R.; Engen J. R.; Wong K. K.; Eck M. J.; Gray N. S.; Janne P. A. Nature 2009, 462, 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karisch R.; Fernandez M.; Taylor P.; Virtanen C.; St-Germain J. R.; Jin L. L.; Harris I. S.; Mori J.; Mak T. W.; Senis Y. A.; Ostman A.; Moran M. F.; Neel B. G. Cell 2011, 146, 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elchebly M.; Payette P.; Michaliszyn E.; Cromlish W.; Collins S.; Loy A. L.; Normandin D.; Cheng A.; Himms-Hagen J.; Chan C. C.; Ramachandran C.; Gresser M. J.; Tremblay M. L.; Kennedy B. P. Science 1999, 283, 1544. [DOI] [PubMed] [Google Scholar]
- Haque A.; Andersen J. N.; Salmeen A.; Barford D.; Tonks N. K. Cell 2011, 147, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lo Conte M.; Carroll K. S. Angew. Chem., Int. Ed. 2012, 51, 6502. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schubart D. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- a Cremlyn J. W. In An Introduction to Organosulfur Chemistry; Cremlyn J. W., Ed.; John Wiley & Sons: Hoboken, NJ, 1996. [Google Scholar]; b Babbs C. F.; Gale M. J. Anal. Biochem. 1987, 163, 67. [DOI] [PubMed] [Google Scholar]; c Babbs C. F.; Steiner M. G. Methods Enzymol. 1990, 186, 137. [DOI] [PubMed] [Google Scholar]
- Liu C. R.; Li M. B.; Cheng D. J.; Yang C. F.; Tian S. K. Org. Lett. 2009, 11, 2543. [DOI] [PubMed] [Google Scholar]
- Aleksiev D. I.; Ivanova S. Phosphorus, Sulfur, Silicon 1994, 90, 41. [Google Scholar]
- Gresham T. L.; Jansen J. E.; Shaver F. W.; Frederick M. R.; Fiedorek F. T.; Bankert R. A.; Gregory J. T.; Beears W. L. J. Am. Chem. Soc. 1952, 74, 1323. [Google Scholar]
- a Ogata Y.; Sawaki Y.; Isono M. Tetrahedron 1969, 25, 2715. [Google Scholar]; b Zvezdova D.; Stoeva S.; Aleksiev D. J. Chin. Chem. Soc. 2007, 54, 447. [Google Scholar]
- Ritchie C. D.; Saltiel J. D.; Lewis E. S. J. Am. Chem. Soc. 1961, 82, 4601. [Google Scholar]
- a Jonsson T. J.; Johnson L. C.; Lowther W. T. J. Biol. Chem. 2009, 284, 33305. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lowther W. T.; Haynes A. C. Antioxid. Redox Signaling 2011, 15, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Roussel X.; Kriznik A.; Richard C.; Rahuel-Clermont S.; Branlant G. J. Biol. Chem. 2009, 284, 33048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thomas D. D.; Ridnour L.; Donzelli S.; Espey M. G.; Mancardi D.; Isenberg J. S.; Feelisch M.; Roberts D. D.; Wink D. A. In Redox Proteomics: From Protein Modifications to Cellular Dysfunction and Diseases; Dalle-Donne I., Scaloni A., Butterfield D. A., Desiderio D. M., Eds.; John Wiley & Sons: Hoboken, NJ, 2006. [Google Scholar]; b Fu X.; Kassim S. Y.; Parks W. C.; Heinecke J. W. J. Biol. Chem. 2001, 276, 41279. [DOI] [PubMed] [Google Scholar]
- a Dey A.; Jeffrey S. P.; Darensbourg M.; Hodgson K. O.; Hedman B.; Solomon E. I. Inorg. Chem. 2007, 46, 4989. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Murakami T.; Nojiri M.; Nakayama H.; Odaka M.; Yohda M.; Dohmae N.; Takio K.; Nagamune T.; Endo I. Protein Sci. 2000, 9, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Noguchi T.; Nojiri M.; Takei K.; Odaka M.; Kamiya N. Biochemistry 2003, 42, 11642. [DOI] [PubMed] [Google Scholar]
- Jacob C.; Giles G. I.; Giles N. M.; Sies H. Angew. Chem., Int. Ed. 2003, 42, 4742. [DOI] [PubMed] [Google Scholar]
- Miyanaga A.; Fushinobu S.; Ito K.; Wakagi T. Biochem. Biophys. Res. Commun. 2001, 288, 1169. [DOI] [PubMed] [Google Scholar]
- Arakawa T.; Kawano Y.; Katayama Y.; Nakayama H.; Dohmae N.; Yohda M.; Odaka M. J. Am. Chem. Soc. 2009, 131, 14838. [DOI] [PubMed] [Google Scholar]
- Agarwal R.; Schmidt J. J.; Stafford R. G.; Swaminathan S. Nat. Struct. Mol. Biol. 2009, 16, 789. [DOI] [PubMed] [Google Scholar]
- a Andres-Mateos E.; Perier C.; Zhang L.; Blanchard-Fillion B.; Greco T. M.; Thomas B.; Ko H. S.; Sasaki M.; Ischiropoulos H.; Przedborski S.; Dawson T. M.; Dawson V. L. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14807. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Blackinton J.; Lakshminarasimhan M.; Thomas K. J.; Ahmad R.; Greggio E.; Raza A. S.; Cookson M. R.; Wilson M. A. J. Biol. Chem. 2009, 284, 6476. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Canet-Aviles R. M.; Wilson M. A.; Miller D. W.; Ahmad R.; McLendon C.; Bandyopadhyay S.; Baptista M. J.; Ringe D.; Petsko G. A.; Cookson M. R. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9103. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Junn E.; Jang W. H.; Zhao X.; Jeong B. S.; Mouradian M. M. J. Neurosci. Res. 2009, 87, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Witt A. C.; Lakshminarasimhan M.; Remington B. C.; Hasim S.; Pozharski E.; Wilson M. A. Biochemistry 2008, 47, 7430. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zhou W.; Zhu M.; Wilson M. A.; Petsko G. A.; Fink A. L. J. Mol. Biol. 2006, 356, 1036. [DOI] [PubMed] [Google Scholar]
- Woo H. A.; Chae H. Z.; Hwang S. C.; Yang K. S.; Kang S. W.; Kim K.; Rhee S. G. Science 2003, 300, 653. [DOI] [PubMed] [Google Scholar]
- Boileau C.; Eme L.; Brochier-Armanet C.; Janicki A.; Zhang C. C.; Latifi A. New Phytol. 2011, 191, 1108. [DOI] [PubMed] [Google Scholar]
- Pascual M. B.; Mata-Cabana A.; Florencio F. J.; Lindahl M.; Cejudo F. J. J. Biol. Chem. 2010, 285, 34485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jonsson T. J.; Johnson L. C.; Lowther W. T. Nature 2008, 451, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jonsson T. J.; Murray M. S.; Johnson L. C.; Lowther W. T. J. Biol. Chem. 2008, 283, 23846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a O’Neill J. S.; Reddy A. B. Nature 2011, 469, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]; b O’Neill J. S.; van Ooijen G.; Dixon L. E.; Troein C.; Corellou F.; Bouget F. Y.; Reddy A. B.; Millar A. J. Nature 2011, 469, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S.; Soriano F. X.; Leveille F.; Martel M. A.; Dakin K. A.; Hansen H. H.; Kaindl A.; Sifringer M.; Fowler J.; Stefovska V.; McKenzie G.; Craigon M.; Corriveau R.; Ghazal P.; Horsburgh K.; Yankner B. A.; Wyllie D. J.; Ikonomidou C.; Hardingham G. E. Nat. Neurosci. 2008, 11, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Jung Y.; Shin B. S.; Song H.; Bae S. H.; Rhee S. G.; Jeong W. J. Biol. Chem. 2010, 285, 34419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Abate C.; Patel L.; Rauscher F. J. 3rd; Curran T. Science 1990, 249, 1157. [DOI] [PubMed] [Google Scholar]; b Chen W.; Sun Z.; Wang X. J.; Jiang T.; Huang Z.; Fang D.; Zhang D. D. Mol. Cell 2009, 34, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh Y. H.; Baek J. Y.; Jeong W.; Rhee S. G.; Chang T. S. J. Biol. Chem. 2009, 284, 8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J.; Schulte J.; Knight A.; Leslie N. R.; Zagozdzon A.; Bronson R.; Manevich Y.; Beeson C.; Neumann C. A. EMBO J. 2009, 28, 1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann M.; Zhang T.; Hendrich S.; Thomas J. A. Methods Enzymol. 2002, 348, 146. [DOI] [PubMed] [Google Scholar]
- a Woo H. A.; Kang S. W.; Kim H. K.; Yang K. S.; Chae H. Z.; Rhee S. G. J. Biol. Chem. 2003, 278, 47361. [DOI] [PubMed] [Google Scholar]; b Fujiwara N.; Nakano M.; Kato S.; Yoshihara D.; Ookawara T.; Eguchi H.; Taniguchi N.; Suzuki K. J. Biol. Chem. 2007, 282, 35933. [DOI] [PubMed] [Google Scholar]
- a Cobb R. L.; Mahan J. E.; Fahey D. R. J. Org. Chem. 1977, 42, 2601. [Google Scholar]; b Tully P. S. In Kirk-Othmer Encyclopedia of Chemical Technology; Kirk-Othmer, Ed.; John Wiley & Sons: Hoboken, NJ, 2011. [Google Scholar]; c Vasilyev A. V.; Walspurger S.; Chassaing S.; Pale P.; Sommer J. Eur. J. Org. Chem. 2007, 2007, 5740. [Google Scholar]
- Stadtman E. R.; Moskovitz J.; Levine R. L. Antioxid. Redox Signaling 2003, 5, 577. [DOI] [PubMed] [Google Scholar]
- Chang Y. C.; Huang C. N.; Lin C. H.; Chang H. C.; Wu C. C. Proteomics 2010, 10, 2961. [DOI] [PubMed] [Google Scholar]
- Chang C. K.; Wu C. C.; Wang Y. S.; Chang H. C. Anal. Chem. 2008, 80, 3791. [DOI] [PubMed] [Google Scholar]
- a Jacob C.; Doering M.; Burkholz T.. The chemical basis of biological redox control. In Redox Signaling and Regulation in Biology and Medicine; Jacob C., Winyard P. G., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp 63–85. [Google Scholar]; b Hall C. N.; Garthwaite J. Nitric Oxide 2009, 21, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller M.; Botti H.; Batthyany C.; Rubbo H.; Radi R.; Denicola A. J. Biol. Chem. 2005, 280, 8850. [DOI] [PubMed] [Google Scholar]
- Malinski T.; Taha Z.; Grunfeld S.; Patton S.; Kapturczak M.; Tomboulian P. Biochem. Biophys. Res. Commun. 1993, 193, 1076. [DOI] [PubMed] [Google Scholar]
- Lukacs-Kornek V.; Malhotra D.; Fletcher A. L.; Acton S. E.; Elpek K. G.; Tayalia P.; Collier A. R.; Turley S. J. Nat. Immunol. 2011, 12, 1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ignarro L. J.; Buga G. M.; Wood K. S.; Byrns R. E.; Chaudhuri G. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 9265. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Palmer R. M.; Ferrige A. G.; Moncada S. Nature 1987, 327, 524. [DOI] [PubMed] [Google Scholar]
- Bogdan C. Nat. Immunol. 2001, 2, 907. [DOI] [PubMed] [Google Scholar]
- Moller M. N.; Li Q.; Vitturi D. A.; Robinson J. M.; Lancaster J. R. Jr.; Denicola A. Chem. Res. Toxicol. 2007, 20, 709. [DOI] [PubMed] [Google Scholar]
- Czapski G.; Goldstein S. Free Radical Biol. Med. 1995, 19, 785. [DOI] [PubMed] [Google Scholar]
- a Fukuto J. M.; Dutton A. S.; Houk K. N. ChemBioChem 2005, 6, 612. [DOI] [PubMed] [Google Scholar]; b Gladwin M. T.; Schechter A. N.; Kim-Shapiro D. B.; Patel R. P.; Hogg N.; Shiva S.; Cannon R. O. 3rd; Kelm M.; Wink D. A.; Espey M. G.; Oldfield E. H.; Pluta R. M.; Freeman B. A.; Lancaster J. R. Jr.; Feelisch M.; Lundberg J. O. Nat. Chem. Biol. 2005, 1, 308. [DOI] [PubMed] [Google Scholar]; c Szabo C.; Ischiropoulos H.; Radi R. Nat. Rev. Drug Discovery 2007, 6, 662. [DOI] [PubMed] [Google Scholar]
- a Ferrer-Sueta G.; Radi R. ACS Chem. Biol. 2009, 4, 161. [DOI] [PubMed] [Google Scholar]; b Kissner R.; Nauser T.; Bugnon P.; Lye P. G.; Koppenol W. H. Chem. Res. Toxicol. 1997, 10, 1285. [DOI] [PubMed] [Google Scholar]
- a Knowles R. G.; Moncada S. Biochem. J. 1994, 298Pt 2249. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Marletta M. A. Cell 1994, 78, 927. [DOI] [PubMed] [Google Scholar]; c Nathan C.; Xie Q. W. Cell 1994, 78, 915. [DOI] [PubMed] [Google Scholar]
- a McMillan K.; Masters B. S. Biochemistry 1995, 34, 3686. [DOI] [PubMed] [Google Scholar]; b Richards M. K.; Marletta M. A. Biochemistry 1994, 33, 14723. [DOI] [PubMed] [Google Scholar]; c Ghosh D. K.; Stuehr D. J. Biochemistry 1995, 34, 801. [DOI] [PubMed] [Google Scholar]; d Alderton W. K.; Cooper C. E.; Knowles R. G. Biochem. J. 2001, 357, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Masters B. S.Role of nitric oxide synthases in redox signaling. In Redox Biochemistry; Banergee R., Ed.; John Wiley & Sons: Hoboken, NJ, 2008; pp 148–152. [Google Scholar]
- a Li H.; Raman C. S.; Glaser C. B.; Blasko E.; Young T. A.; Parkinson J. F.; Whitlow M.; Poulos T. L. J. Biol. Chem. 1999, 274, 21276. [DOI] [PubMed] [Google Scholar]; b Raman C. S.; Li H.; Martasek P.; Kral V.; Masters B. S.; Poulos T. L. Cell 1998, 95, 939. [DOI] [PubMed] [Google Scholar]
- Siddhanta U.; Wu C.; Abu-Soud H. M.; Zhang J.; Ghosh D. K.; Stuehr D. J. J. Biol. Chem. 1996, 271, 7309. [DOI] [PubMed] [Google Scholar]
- Roman L. J.; Martasek P.; Masters B. S. Chem. Rev. 2002, 102, 1179. [DOI] [PubMed] [Google Scholar]
- Roman L. J.; Martasek P.; Miller R. T.; Harris D. E.; de La Garza M. A.; Shea T. M.; Kim J. J.; Masters B. S. J. Biol. Chem. 2000, 275, 29225. [DOI] [PubMed] [Google Scholar]
- Salerno J. C.; Harris D. E.; Irizarry K.; Patel B.; Morales A. J.; Smith S. M.; Martasek P.; Roman L. J.; Masters B. S.; Jones C. L.; Weissman B. A.; Lane P.; Liu Q.; Gross S. S. J. Biol. Chem. 1997, 272, 29769. [DOI] [PubMed] [Google Scholar]
- Clapham D. E. Cell 2007, 131, 1047. [DOI] [PubMed] [Google Scholar]
- a Gachhui R.; Presta A.; Bentley D. F.; Abu-Soud H. M.; McArthur R.; Brudvig G.; Ghosh D. K.; Stuehr D. J. J. Biol. Chem. 1996, 271, 20594. [DOI] [PubMed] [Google Scholar]; b Matsuda H.; Iyanagi T. Biochim. Biophys. Acta 1999, 1473, 345. [DOI] [PubMed] [Google Scholar]; c Nishida C. R.; Ortiz de Montellano P. R. J. Biol. Chem. 1998, 273, 5566. [DOI] [PubMed] [Google Scholar]; d Su Z.; Blazing M. A.; Fan D.; George S. E. J. Biol. Chem. 1995, 270, 29117. [DOI] [PubMed] [Google Scholar]
- Roman L. J.; Miller R. T.; de La Garza M. A.; Kim J. J.; Siler Masters B. S. J. Biol. Chem. 2000, 275, 21914. [DOI] [PubMed] [Google Scholar]
- a Whited C. A.; Warren J. J.; Lavoie K. D.; Weinert E. E.; Agapie T.; Winkler J. R.; Gray H. B. J. Am. Chem. Soc. 2012, 134, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hurshman A. R.; Marletta M. A. Biochemistry 1995, 34, 5627. [DOI] [PubMed] [Google Scholar]; c Abu-Soud H. M.; Wang J.; Rousseau D. L.; Fukuto J. M.; Ignarro L. J.; Stuehr D. J. J. Biol. Chem. 1995, 270, 22997. [DOI] [PubMed] [Google Scholar]; d Santolini J.; Adak S.; Curran C. M.; Stuehr D. J. J. Biol. Chem. 2001, 276, 1233. [DOI] [PubMed] [Google Scholar]
- a Fulton D.; Gratton J. P.; McCabe T. J.; Fontana J.; Fujio Y.; Walsh K.; Franke T. F.; Papapetropoulos A.; Sessa W. C. Nature 1999, 399, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lane P.; Gross S. S. J. Biol. Chem. 2002, 277, 19087. [DOI] [PubMed] [Google Scholar]
- Lim K. H.; Ancrile B. B.; Kashatus D. F.; Counter C. M. Nature 2008, 452, 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cardena G.; Fan R.; Shah V.; Sorrentino R.; Cirino G.; Papapetropoulos A.; Sessa W. C. Nature 1998, 392, 821. [DOI] [PubMed] [Google Scholar]
- a Kim S. K. Curr. Opin. Cell Biol. 1995, 7, 641. [DOI] [PubMed] [Google Scholar]; b Cho K. O.; Hunt C. A.; Kennedy M. B. Neuron 1992, 9, 929. [DOI] [PubMed] [Google Scholar]
- a Brenman J. E.; Chao D. S.; Gee S. H.; McGee A. W.; Craven S. E.; Santillano D. R.; Wu Z.; Huang F.; Xia H.; Peters M. F.; Froehner S. C.; Bredt D. S. Cell 1996, 84, 757. [DOI] [PubMed] [Google Scholar]; b Christopherson K. S.; Bredt D. S. J. Clin. Invest. 1997, 100, 2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho G. P.; Selvakumar B.; Mukai J.; Hester L. D.; Wang Y.; Gogos J. A.; Snyder S. H. Neuron 2011, 71, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju H.; Venema V. J.; Marrero M. B.; Venema R. C. J. Biol. Chem. 1998, 273, 24025. [DOI] [PubMed] [Google Scholar]
- Couet J.; Li S.; Okamoto T.; Ikezu T.; Lisanti M. P. J. Biol. Chem. 1997, 272, 6525. [DOI] [PubMed] [Google Scholar]
- a Kurzchalia T. V.; Parton R. G. Curr. Opin. Cell Biol. 1999, 11, 424. [DOI] [PubMed] [Google Scholar]; b Garcia-Cardena G.; Fan R.; Stern D. F.; Liu J.; Sessa W. C. J. Biol. Chem. 1996, 271, 27237. [DOI] [PubMed] [Google Scholar]; c Ju H.; Zou R.; Venema V. J.; Venema R. C. J. Biol. Chem. 1997, 272, 18522. [DOI] [PubMed] [Google Scholar]
- a Harris M. B.; Ju H.; Venema V. J.; Liang H.; Zou R.; Michell B. J.; Chen Z. P.; Kemp B. E.; Venema R. C. J. Biol. Chem. 2001, 276, 16587. [DOI] [PubMed] [Google Scholar]; b Michel J. B.; Feron O.; Sacks D.; Michel T. J. Biol. Chem. 1997, 272, 15583. [DOI] [PubMed] [Google Scholar]
- Erez A.; Nagamani S. C.; Shchelochkov O. A.; Premkumar M. H.; Campeau P. M.; Chen Y.; Garg H. K.; Li L.; Mian A.; Bertin T. K.; Black J. O.; Zeng H.; Tang Y.; Reddy A. K.; Summar M.; O’Brien W. E.; Harrison D. G.; Mitch W. E.; Marini J. C.; Aschner J. L.; Bryan N. S.; Lee B. Nat. Med. 2011, 17, 1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Huang W.; Harris M. B.; Goolsby J. M.; Venema R. C. Biochem. J. 2005, 386, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Xia Y.; Tsai A. L.; Berka V.; Zweier J. L. J. Biol. Chem. 1998, 273, 25804. [DOI] [PubMed] [Google Scholar]; b Stuehr D. J.; Santolini J.; Wang Z. Q.; Wei C. C.; Adak S. J. Biol. Chem. 2004, 279, 36167. [DOI] [PubMed] [Google Scholar]
- a Vasquez-Vivar J.; Hogg N.; Martasek P.; Karoui H.; Pritchard K. A. Jr.; Kalyanaraman B. J. Biol. Chem. 1999, 274, 26736. [DOI] [PubMed] [Google Scholar]; b Pou S.; Pou W. S.; Bredt D. S.; Snyder S. H.; Rosen G. M. J. Biol. Chem. 1992, 267, 24173. [PubMed] [Google Scholar]
- Yun B. W.; Feechan A.; Yin M.; Saidi N. B.; Le Bihan T.; Yu M.; Moore J. W.; Kang J. G.; Kwon E.; Spoel S. H.; Pallas J. A.; Loake G. J. Nature 2011, 478, 264. [DOI] [PubMed] [Google Scholar]
- Huang P. L. J. Am. Soc. Nephrol. 2000, 11Suppl 16S120. [PubMed] [Google Scholar]
- a Garvey E. P.; Oplinger J. A.; Tanoury G. J.; Sherman P. A.; Fowler M.; Marshall S.; Harmon M. F.; Paith J. E.; Furfine E. S. J. Biol. Chem. 1994, 269, 26669. [PubMed] [Google Scholar]; b Garvey E. P.; Oplinger J. A.; Furfine E. S.; Kiff R. J.; Laszlo F.; Whittle B. J.; Knowles R. G. J. Biol. Chem. 1997, 272, 4959. [DOI] [PubMed] [Google Scholar]; c Young R. J.; Beams R. M.; Carter K.; Clark H. A.; Coe D. M.; Chambers C. L.; Davies P. I.; Dawson J.; Drysdale M. J.; Franzman K. W.; French C.; Hodgson S. T.; Hodson H. F.; Kleanthous S.; Rider P.; Sanders D.; Sawyer D. A.; Scott K. J.; Shearer B. G.; Stocker R.; Smith S.; Tackley M. C.; Knowles R. G. Bioorg. Med. Chem. Lett. 2000, 10, 597. [DOI] [PubMed] [Google Scholar]
- Seimetz M.; Parajuli N.; Pichl A.; Veit F.; Kwapiszewska G.; Weisel F. C.; Milger K.; Egemnazarov B.; Turowska A.; Fuchs B.; Nikam S.; Roth M.; Sydykov A.; Medebach T.; Klepetko W.; Jaksch P.; Dumitrascu R.; Garn H.; Voswinckel R.; Kostin S.; Seeger W.; Schermuly R. T.; Grimminger F.; Ghofrani H. A.; Weissmann N. Cell 2011, 147, 293. [DOI] [PubMed] [Google Scholar]
- a Brindicci C.; Ito K.; Torre O.; Barnes P. J.; Kharitonov S. A. Chest 2009, 135, 353. [DOI] [PubMed] [Google Scholar]; b Singh D.; Richards D.; Knowles R. G.; Schwartz S.; Woodcock A.; Langley S.; O’Connor B. J. Am. J. Respir. Crit. Care Med. 2007, 176, 988. [DOI] [PubMed] [Google Scholar]
- a Besson-Bard A.; Pugin A.; Wendehenne D. Annu. Rev. Plant Biol. 2008, 59, 21. [DOI] [PubMed] [Google Scholar]; b Borisjuk L.; Macherel D.; Benamar A.; Wobus U.; Rolletschek H. New Phytol. 2007, 176, 813. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Moreau M.; Lindermayr C.; Durner J.; Klessig D. F. Physiol. Plant. 2010, 138, 372. [DOI] [PubMed] [Google Scholar]
- Gupta K. J.; Fernie A. R.; Kaiser W. M.; van Dongen J. T. Trends Plant Sci. 2011, 16, 160. [DOI] [PubMed] [Google Scholar]
- a Gomes A.; Fernandes E.; Lima J. L. J. Fluoresc. 2006, 16, 119. [DOI] [PubMed] [Google Scholar]; b Lim M. H.; Lippard S. J. Acc. Chem. Res. 2007, 40, 41. [DOI] [PubMed] [Google Scholar]; c Nagano T.; Yoshimura T. Chem. Rev. 2002, 102, 1235. [DOI] [PubMed] [Google Scholar]; d Hetrick E. M.; Schoenfisch M. H. Annu. Rev. Anal. Chem. 2009, 2, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H.; Heller D. A.; Jin H.; Barone P. W.; Song C.; Zhang J.; Trudel L. J.; Wogan G. N.; Tannenbaum S. R.; Strano M. S. Nat. Chem. 2009, 1, 473. [DOI] [PubMed] [Google Scholar]
- a Sato M.; Hida N.; Umezawa Y. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 14515. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sato M.; Nakajima T.; Goto M.; Umezawa Y. Anal. Chem. 2006, 78, 8175. [DOI] [PubMed] [Google Scholar]
- Boon E. M.; Marletta M. A. J. Am. Chem. Soc. 2006, 128, 10022. [DOI] [PubMed] [Google Scholar]
- a Chen O.; Uzlaner N.; Priel Z.; Likhtenshtein G. I. J. Biochem. Biophys. Methods 2008, 70, 1006. [DOI] [PubMed] [Google Scholar]; b Do L.; Smith R. C.; Tennyson A. G.; Lippard S. J. Inorg. Chem. 2006, 45, 8998. [DOI] [PubMed] [Google Scholar]; c Huang J. C.; Li D. J.; Diao J. C.; Hou J.; Yuan J. L.; Zou G. L. Talanta 2007, 72, 1283. [DOI] [PubMed] [Google Scholar]; d Lim M. H.; Wong B. A.; Pitcock W. H. Jr.; Mokshagundam D.; Baik M. H.; Lippard S. J. J. Am. Chem. Soc. 2006, 128, 14364. [DOI] [PubMed] [Google Scholar]; e Lim M. H.; Xu D.; Lippard S. J. Nat. Chem. Biol. 2006, 2, 375. [DOI] [PubMed] [Google Scholar]; f McQuade L. E.; Lippard S. J. Inorg. Chem. 2010, 49, 7464. [DOI] [PubMed] [Google Scholar]; g Ortiz M.; Torrens M.; Mola J. L.; Ortiz P. J.; Fragoso A.; Diaz A.; Cao R.; Prados P.; de Mendoza J.; Otero A.; Antinolo A.; Lara A. Dalton Trans. 2008, 27, 3559. [DOI] [PubMed] [Google Scholar]; h Ouyang J.; Hong H.; Shen C.; Zhao Y.; Ouyang C.; Dong L.; Zhu J.; Guo Z.; Zeng K.; Chen J.; Zhang C.; Zhang J. Free Radical Biol. Med. 2008, 45, 1426. [DOI] [PubMed] [Google Scholar]; i Smith R. C.; Tennyson A. G.; Won A. C.; Lippard S. J. Inorg. Chem. 2006, 45, 9367. [DOI] [PubMed] [Google Scholar]; j Sun Z. N.; Wang H. L.; Liu F. Q.; Chen Y.; Tam P. K.; Yang D. Org. Lett. 2009, 11, 1887. [DOI] [PubMed] [Google Scholar]; k Yang D.; Wang H. L.; Sun Z. N.; Chung N. W.; Shen J. G. J. Am. Chem. Soc. 2006, 128, 6004. [DOI] [PubMed] [Google Scholar]; l Yuan L.; Lin W.; Xie Y.; Chen B.; Zhu S. J. Am. Chem. Soc. 2012, 134, 1305. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Arbault S.; Chen Y.; Crozatier C.; Tapsoba I. Lab Chip 2007, 7, 233. [DOI] [PubMed] [Google Scholar]
- McQuade L. E.; Lippard S. J. Curr. Opin. Chem. Biol. 2010, 14, 43. [DOI] [PubMed] [Google Scholar]
- Shiva S.; Wang X.; Ringwood L. A.; Xu X.; Yuditskaya S.; Annavajjhala V.; Miyajima H.; Hogg N.; Harris Z. L.; Gladwin M. T. Nat. Chem. Biol. 2006, 2, 486. [DOI] [PubMed] [Google Scholar]
- a Zweier J. L.; Wang P.; Samouilov A.; Kuppusamy P. Nat. Med. 1995, 1, 804. [DOI] [PubMed] [Google Scholar]; b Li H.; Samouilov A.; Liu X.; Zweier J. L. J. Biol. Chem. 2001, 276, 24482. [DOI] [PubMed] [Google Scholar]; c Modin A.; Bjorne H.; Herulf M.; Alving K.; Weitzberg E.; Lundberg J. O. Acta Physiol. Scand. 2001, 171, 9. [DOI] [PubMed] [Google Scholar]
- a Cosby K.; Partovi K. S.; Crawford J. H.; Patel R. P.; Reiter C. D.; Martyr S.; Yang B. K.; Waclawiw M. A.; Zalos G.; Xu X.; Huang K. T.; Shields H.; Kim-Shapiro D. B.; Schechter A. N.; Cannon R. O., 3rd; Gladwin M. T. Nat. Med. 2003, 9, 1498. [DOI] [PubMed] [Google Scholar]; b Nagababu E.; Ramasamy S.; Abernethy D. R.; Rifkind J. M. J. Biol. Chem. 2003, 278, 46349. [DOI] [PubMed] [Google Scholar]
- a Barouch L. A.; Harrison R. W.; Skaf M. W.; Rosas G. O.; Cappola T. P.; Kobeissi Z. A.; Hobai I. A.; Lemmon C. A.; Burnett A. L.; O’Rourke B.; Rodriguez E. R.; Huang P. L.; Lima J. A.; Berkowitz D. E.; Hare J. M. Nature 2002, 416, 337. [DOI] [PubMed] [Google Scholar]; b Hare J. M.; Stamler J. S. J. Clin. Invest. 2005, 115, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Iwakiri Y.; Satoh A.; Chatterjee S.; Toomre D. K.; Chalouni C. M.; Fulton D.; Groszmann R. J.; Shah V. H.; Sessa W. C. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 19777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth D.; Stamler J. S. Curr. Opin. Chem. Biol. 2011, 15, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg M. D.; Sen N.; Hara M. R.; Juluri K. R.; Nguyen J. V.; Snowman A. M.; Law L.; Hester L. D.; Snyder S. H. Nat. Cell Biol. 2010, 12, 1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ramachandran N.; Root P.; Jiang X. M.; Hogg P. J.; Mutus B. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 9539. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mitchell D. A.; Marletta M. A. Nat. Chem. Biol. 2005, 1, 154. [DOI] [PubMed] [Google Scholar]
- Pacher P.; Beckman J. S.; Liaudet L. Physiol. Rev. 2007, 87, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murad F. J. Clin. Invest. 1986, 78, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nisoli E.; Clementi E.; Paolucci C.; Cozzi V.; Tonello C.; Sciorati C.; Bracale R.; Valerio A.; Francolini M.; Moncada S.; Carruba M. O. Science 2003, 299, 896. [DOI] [PubMed] [Google Scholar]; b Bossy-Wetzel E.; Lipton S. A. Cell Death Differ. 2003, 10, 757. [DOI] [PubMed] [Google Scholar]
- Sarti P.; Arese M.; Bacchi A.; Barone M. C.; Forte E.; Mastronicola D.; Brunori M.; Giuffre A. IUBMB Life 2003, 55, 605. [DOI] [PubMed] [Google Scholar]
- a Pullan S. T.; Gidley M. D.; Jones R. A.; Barrett J.; Stevanin T. M.; Read R. C.; Green J.; Poole R. K. J. Bacteriol. 2007, 189, 1845. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bodenmiller D. M.; Spiro S. J. Bacteriol. 2006, 188, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tucker N. P.; Hicks M. G.; Clarke T. A.; Crack J. C.; Chandra G.; Le Brun N. E.; Dixon R.; Hutchings M. I. PLoS One 2008, 3, e3623. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ding H.; Demple B. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 5146. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Cruz-Ramos H.; Crack J.; Wu G.; Hughes M. N.; Scott C.; Thomson A. J.; Green J.; Poole R. K. EMBO J. 2002, 21, 3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crack J. C.; Smith L. J.; Stapleton M. R.; Peck J.; Watmough N. J.; Buttner M. J.; Buxton R. S.; Green J.; Oganesyan V. S.; Thomson A. J.; Le Brun N. E. J. Am. Chem. Soc. 2011, 133, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A.; Comatas K. E.; Liu L.; Stamler J. S. Science 2003, 301, 657. [DOI] [PubMed] [Google Scholar]
- Kim S. F.; Huri D. A.; Snyder S. H. Science 2005, 310, 1966. [DOI] [PubMed] [Google Scholar]
- Chen C. A.; Lin C. H.; Druhan L. J.; Wang T. Y.; Chen Y. R.; Zweier J. L. J. Biol. Chem. 2011, 286, 29098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wink D. A.; Nims R. W.; Darbyshire J. F.; Christodoulou D.; Hanbauer I.; Cox G. W.; Laval F.; Laval J.; Cook J. A.; Krishna M. C.; DeGraff W. G.; Mitchell J. B. Chem. Res. Toxicol. 1994, 7, 519. [DOI] [PubMed] [Google Scholar]; b Jourd’heuil D.; Jourd’heuil F. L.; Feelisch M. J. Biol. Chem. 2003, 278, 15720. [DOI] [PubMed] [Google Scholar]; c Schrammel A.; Gorren A. C.; Schmidt K.; Pfeiffer S.; Mayer B. Free Radical Biol. Med. 2003, 34, 1078. [DOI] [PubMed] [Google Scholar]; d Keszler A.; Zhang Y.; Hogg N. Free Radical Biol. Med. 2010, 48, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger B. P.; Rich E. N.; Gow A. J.; Williams E. M.; Stamler J. S.; Singel D. J. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichsel A.; Maes E. M.; Andersen J. F.; Valenzuela J. G.; Shokhireva T.; Walker F. A.; Montfort W. R. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige J. S.; Xu G.; Stancevic B.; Jaffrey S. R. Chem. Biol. 2008, 15, 1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran E. E.; Timerghazin Q. K.; Kwong E.; English A. M. J. Phys. Chem. B 2011, 115, 3112. [DOI] [PubMed] [Google Scholar]
- Doulias P. T.; Greene J. L.; Greco T. M.; Tenopoulou M.; Seeholzer S. H.; Dunbrack R. L.; Ischiropoulos H. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cho D. H.; Nakamura T.; Fang J.; Cieplak P.; Godzik A.; Gu Z.; Lipton S. A. Science 2009, 324, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chung K. K.; Thomas B.; Li X.; Pletnikova O.; Troncoso J. C.; Marsh L.; Dawson V. L.; Dawson T. M. Science 2004, 304, 1328. [DOI] [PubMed] [Google Scholar]; c Uehara T.; Nakamura T.; Yao D.; Shi Z. Q.; Gu Z.; Ma Y.; Masliah E.; Nomura Y.; Lipton S. A. Nature 2006, 441, 513. [DOI] [PubMed] [Google Scholar]; d Nott A.; Watson P. M.; Robinson J. D.; Crepaldi L.; Riccio A. Nature 2008, 455, 411. [DOI] [PubMed] [Google Scholar]; e Benhar M.; Forrester M. T.; Hess D. T.; Stamler J. S. Science 2008, 320, 1050. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Tada Y.; Spoel S. H.; Pajerowska-Mukhtar K.; Mou Z.; Song J.; Wang C.; Zuo J.; Dong X. Science 2008, 321, 952. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Gu Z.; Kaul M.; Yan B.; Kridel S. J.; Cui J.; Strongin A.; Smith J. W.; Liddington R. C.; Lipton S. A. Science 2002, 297, 1186. [DOI] [PubMed] [Google Scholar]; h Foster M. W.; Forrester M. T.; Stamler J. S. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonov V. G.; Sundquist A. R.; Sharma V. S. J. Biol. Chem. 1995, 270, 28158. [DOI] [PubMed] [Google Scholar]
- Keshive M.; Singh S.; Wishnok J. S.; Tannenbaum S. R.; Deen W. M. Chem. Res. Toxicol. 1996, 9, 988. [DOI] [PubMed] [Google Scholar]
- a Liu M.; Hou J.; Huang L.; Huang X.; Heibeck T. H.; Zhao R.; Pasa-Tolic L.; Smith R. D.; Li Y.; Fu K.; Zhang Z.; Hinrichs S. H.; Ding S. J. Anal. Chem. 2010, 82, 7160. [DOI] [PubMed] [Google Scholar]; b Pawloski J. R.; Hess D. T.; Stamler J. S. Nature 2001, 409, 622. [DOI] [PubMed] [Google Scholar]
- Marino S. M.; Gladyshev V. N. J. Mol. Biol. 2010, 395, 844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao G.; Derakhshan B.; Shi L.; Campagne F.; Gross S. S. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedospasov A.; Rafikov R.; Beda N.; Nudler E. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Stamler J. S.; Jia L.; Eu J. P.; McMahon T. J.; Demchenko I. T.; Bonaventura J.; Gernert K.; Piantadosi C. A. Science 1997, 276, 2034. [DOI] [PubMed] [Google Scholar]; b Sun J.; Xin C.; Eu J. P.; Stamler J. S.; Meissner G. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhar M.; Forrester M. T.; Stamler J. S. Nat. Rev. Mol. Cell Biol. 2009, 10, 721. [DOI] [PubMed] [Google Scholar]
- a Liu L.; Hausladen A.; Zeng M.; Que L.; Heitman J.; Stamler J. S. Nature 2001, 410, 490. [DOI] [PubMed] [Google Scholar]; b Bateman R. L.; Rauh D.; Tavshanjian B.; Shokat K. M. J. Biol. Chem. 2008, 283, 35756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann J.; Haendeler J.; Zeiher A. M.; Dimmeler S. J. Biol. Chem. 2001, 276, 41383. [DOI] [PubMed] [Google Scholar]
- Ozawa K.; Whalen E. J.; Nelson C. D.; Mu Y.; Hess D. T.; Lefkowitz R. J.; Stamler J. S. Mol. Cell 2008, 31, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.; Eu J. P.; Meissner G.; Stamler J. S. Science 1998, 279, 234. [DOI] [PubMed] [Google Scholar]
- Singel D. J.; Stamler J. S. Annu. Rev. Physiol. 2005, 67, 99. [DOI] [PubMed] [Google Scholar]
- a Selvakumar B.; Huganir R. L.; Snyder S. H. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 16440. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shahani N.; Sawa A. Antioxid. Redox Signaling 2011, 14, 1493. [DOI] [PubMed] [Google Scholar]
- a Delledonne M.; Xia Y.; Dixon R. A.; Lamb C. Nature 1998, 394, 585. [DOI] [PubMed] [Google Scholar]; b Delledonne M.; Zeier J.; Marocco A.; Lamb C. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 13454. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Torres M. A.; Dangl J. L.; Jones J. D. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi K.; Brennan L. A.; Levic S.; Ross P. A.; Black S. M. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidge T. C.; Urvil P.; Oezguen N.; Ali K.; Choudhury A.; Acharya V.; Pinchuk I.; Torres A. G.; English R. D.; Wiktorowicz J. E.; Loeffelholz M.; Kumar R.; Shi L.; Nie W.; Braun W.; Herman B.; Hausladen A.; Feng H.; Stamler J. S.; Pothoulakis C. Nat. Med. 2011, 17, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M.; Charlesworth P.; Dempster M.; Webster L. C.; Watabe A. M.; Makhinson M.; He Y.; Ramsay M. F.; Morris R. G.; Morrison J. H.; O’Dell T. J.; Grant S. G. Nature 1998, 396, 433. [DOI] [PubMed] [Google Scholar]
- Christopherson K. S.; Hillier B. J.; Lim W. A.; Bredt D. S. J. Biol. Chem. 1999, 274, 27467. [DOI] [PubMed] [Google Scholar]
- a El-Husseini Ael D.; Schnell E.; Dakoji S.; Sweeney N.; Zhou Q.; Prange O.; Gauthier-Campbell C.; Aguilera-Moreno A.; Nicoll R. A.; Bredt D. S. Cell 2002, 108, 849. [DOI] [PubMed] [Google Scholar]; b Topinka J. R.; Bredt D. S. Neuron 1998, 20, 125. [DOI] [PubMed] [Google Scholar]
- a Chen L.; Chetkovich D. M.; Petralia R. S.; Sweeney N. T.; Kawasaki Y.; Wenthold R. J.; Bredt D. S.; Nicoll R. A. Nature 2000, 408, 936. [DOI] [PubMed] [Google Scholar]; b Schnell E.; Sizemore M.; Karimzadegan S.; Chen L.; Bredt D. S.; Nicoll R. A. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler J. S.; Meissner G. Physiol. Rev. 2001, 81, 209. [DOI] [PubMed] [Google Scholar]
- Kakizawa S.; Yamazawa T.; Chen Y.; Ito A.; Murayama T.; Oyamada H.; Kurebayashi N.; Sato O.; Watanabe M.; Mori N.; Oguchi K.; Sakurai T.; Takeshima H.; Saito N.; Iino M. EMBO J. 2011, 31, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T.; Inoue R.; Morii T.; Takahashi N.; Yamamoto S.; Hara Y.; Tominaga M.; Shimizu S.; Sato Y.; Mori Y. Nat. Chem. Biol. 2006, 2, 596. [DOI] [PubMed] [Google Scholar]
- Wei W.; Li B.; Hanes M. A.; Kakar S.; Chen X.; Liu L. Sci. Transl. Med. 2010, 2, 19ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kettenhofen N. J.; Broniowska K. A.; Keszler A.; Zhang Y.; Hogg N. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2007, 851, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang H.; Xian M. Curr. Opin. Chem. Biol. 2011, 15, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wink D. A.; Kim S.; Coffin D.; Cook J. C.; Vodovotz Y.; Chistodoulou D.; Jourd’heuil D.; Grisham M. B. Methods Enzymol. 1999, 301, 201. [DOI] [PubMed] [Google Scholar]
- Faccenda A.; Bonham C. A.; Vacratsis P. O.; Zhang X.; Mutus B. J. Am. Chem. Soc. 2010, 132, 11392. [DOI] [PubMed] [Google Scholar]
- a Berti P. J.; Storer A. C. J. Mol. Biol. 1995, 246, 273. [DOI] [PubMed] [Google Scholar]; b Mavridou D. A.; Stevens J. M.; Ferguson S. J.; Redfield C. J. Mol. Biol. 2007, 370, 643. [DOI] [PubMed] [Google Scholar]
- a Bartberger M. D.; Liu W.; Ford E.; Miranda K. M.; Switzer C.; Fukuto J. M.; Farmer P. J.; Wink D. A.; Houk K. N. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 10958. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shafirovich V.; Lymar S. V. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jaffrey S. R.; Erdjument-Bromage H.; Ferris C. D.; Tempst P.; Snyder S. H. Nat. Cell Biol. 2001, 3, 193. [DOI] [PubMed] [Google Scholar]; b Jaffrey S. R.; Snyder S. H. Sci. Signal Tranduction Knowl. Environ. 2001, 2001, pl1. [DOI] [PubMed] [Google Scholar]
- a Forrester M. T.; Foster M. W.; Stamler J. S. J. Biol. Chem. 2007, 282, 13977. [DOI] [PubMed] [Google Scholar]; b Wang X.; Kettenhofen N. J.; Shiva S.; Hogg N.; Gladwin M. T. Free Radical Biol. Med. 2008, 44, 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang Y.; Keszler A.; Broniowska K. A.; Hogg N. Free Radical Biol. Med. 2005, 38, 874. [DOI] [PubMed] [Google Scholar]
- a Huang B.; Chen C. Free Radical Biol. Med. 2006, 41, 562. [DOI] [PubMed] [Google Scholar]; b Landino L. M.; Koumas M. T.; Mason C. E.; Alston J. A. Biochem. Biophys. Res. Commun. 2006, 340, 347. [DOI] [PubMed] [Google Scholar]
- Kallakunta V. M.; Staruch A.; Mutus B. Biochim. Biophys. Acta 2010, 1800, 23. [DOI] [PubMed] [Google Scholar]
- Derakhshan B.; Wille P. C.; Gross S. S. Nat. Protoc. 2007, 2, 1685. [DOI] [PubMed] [Google Scholar]
- Forrester M. T.; Thompson J. W.; Foster M. W.; Nogueira L.; Moseley M. A.; Stamler J. S. Nat. Biotechnol. 2009, 27, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Han P.; Zhou X.; Huang B.; Zhang X.; Chen C. Anal. Biochem. 2008, 377, 150. [DOI] [PubMed] [Google Scholar]; b Santhanam L.; Gucek M.; Brown T. R.; Mansharamani M.; Ryoo S.; Lemmon C. A.; Romer L.; Shoukas A. A.; Berkowitz D. E.; Cole R. N. Nitric Oxide 2008, 19, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tello D.; Tarin C.; Ahicart P.; Breton-Romero R.; Lamas S.; Martinez-Ruiz A. Proteomics 2009, 9, 5359. [DOI] [PubMed] [Google Scholar]
- Sinha V.; Wijewickrama G. T.; Chandrasena R. E.; Xu H.; Edirisinghe P. D.; Schiefer I. T.; Thatcher G. R. ACS Chem. Biol. 2010, 5, 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Xian M. Angew. Chem., Int. Ed. 2008, 47, 6598. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Li S.; Zhang D.; Wang H.; Whorton A. R.; Xian M. Org. Lett. 2010, 12, 4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Downing J. A.; McHale J. L.; Xian M. Mol. Biosyst. 2009, 5, 918. [DOI] [PubMed] [Google Scholar]
- Bechtold E.; Reisz J. A.; Klomsiri C.; Tsang A. W.; Wright M. W.; Poole L. B.; Furdui C. M.; King S. B. ACS Chem. Biol. 2010, 5, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles G. I.; Tasker K. M.; Jacob C. Free Radical Biol. Med. 2001, 31, 1279. [DOI] [PubMed] [Google Scholar]
- a Wardman P.; von Sonntag C. Methods Enzymol. 1995, 251, 31. [DOI] [PubMed] [Google Scholar]; b Bindoli A.; Fukuto J. M.; Forman H. J. Antioxid. Redox Signaling 2008, 10, 1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Nat. Rev. Drug Discovery 2007, 6, 917. [DOI] [PubMed] [Google Scholar]
- Li L.; Rose P.; Moore P. K. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169. [DOI] [PubMed] [Google Scholar]
- a Vorobets V. S.; Kovach S. K.; Kolbasov G. Y. Russ. J. Appl. Chem 2002, 75, 229. [Google Scholar]; b Kabil O.; Banerjee R. J. Biol. Chem. 2010, 285, 21903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N.; Paul B. D.; Gadalla M. M.; Mustafa A. K.; Sen T.; Xu R.; Kim S.; Snyder S. H. Mol. Cell 2012, 45, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Caliendo G.; Cirino G.; Santagada V.; Wallace J. L. J. Med. Chem. 2010, 53, 6275. [DOI] [PubMed] [Google Scholar]; b Liu Y. Y.; Sparatore A.; Del Soldato P.; Bian J. S. Neuroscience 2011, 193, 80. [DOI] [PubMed] [Google Scholar]
- Baskar R.; Bian J. Eur. J. Pharmacol. 2011, 656, 5. [DOI] [PubMed] [Google Scholar]
- Whiteman M.; Moore P. K.. Is hydrogen sulfide a regulator of nitric oxide bioavailability in the vasculature? In Redox Signaling and Regulation in Biology and Medicine; Jacob C., Winyard P. G., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp 293–314. [Google Scholar]
- Whiteman M.; Moore P. K. J. Cell Mol. Med. 2009, 13, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M.; Armstrong J. S.; Chu S. H.; Jia-Ling S.; Wong B. S.; Cheung N. S.; Halliwell B.; Moore P. K. J. Neurochem. 2004, 90, 765. [DOI] [PubMed] [Google Scholar]
- Chang L.; Geng B.; Yu F.; Zhao J.; Jiang H.; Du J.; Tang C. Amino Acids 2008, 34, 573. [DOI] [PubMed] [Google Scholar]
- Geng B.; Yang J.; Qi Y.; Zhao J.; Pang Y.; Du J.; Tang C. Biochem. Biophys. Res. Commun. 2004, 313, 362. [DOI] [PubMed] [Google Scholar]
- Shatalin K.; Shatalina E.; Mironov A.; Nudler E. Science 2011, 334, 986. [DOI] [PubMed] [Google Scholar]
- Xiao D.; Herman-Antosiewicz A.; Antosiewicz J.; Xiao H.; Brisson M.; Lazo J. S.; Singh S. V. Oncogene 2005, 24, 6256. [DOI] [PubMed] [Google Scholar]
- Borkowska A.; Sielicka-Dudzin A.; Herman-Antosiewicz A.; Wozniak M.; Fedeli D.; Falcioni G.; Antosiewicz J. Eur. J. Nutr. 2011, 51, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P.; Winterbourn C. C. Chem. Res. Toxicol. 2010, 23, 1541. [DOI] [PubMed] [Google Scholar]
- a Ashby M. T.; Aneetha H. J. Am. Chem. Soc. 2004, 126, 10216. [DOI] [PubMed] [Google Scholar]; b Nagy P.; Wang X.; Lemma K.; Ashby M. T. J. Am. Chem. Soc. 2007, 129, 15756. [DOI] [PubMed] [Google Scholar]
- Giles G. I.; Jacob C. Biol. Chem. 2002, 383, 375. [DOI] [PubMed] [Google Scholar]
- a Gadalla M. M.; Snyder S. H. J. Neurochem. 2010, 113, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kimura H. Amino Acids 2011, 41, 113. [DOI] [PubMed] [Google Scholar]
- a Enokido Y.; Suzuki E.; Iwasawa K.; Namekata K.; Okazawa H.; Kimura H. FASEB J. 2005, 19, 1854. [DOI] [PubMed] [Google Scholar]; b Ichinohe A.; Kanaumi T.; Takashima S.; Enokido Y.; Nagai Y.; Kimura H. Biochem. Biophys. Res. Commun. 2005, 338, 1547. [DOI] [PubMed] [Google Scholar]
- a Diwakar L.; Ravindranath V. Neurochem. Int. 2007, 50, 418. [DOI] [PubMed] [Google Scholar]; b Hosoki R.; Matsuki N.; Kimura H. Biochem. Biophys. Res. Commun. 1997, 237, 527. [DOI] [PubMed] [Google Scholar]; c Kaneko Y.; Kimura Y.; Kimura H.; Niki I. Diabetes 2006, 55, 1391. [DOI] [PubMed] [Google Scholar]; d Patel P.; Vatish M.; Heptinstall J.; Wang R.; Carson R. J. Reprod. Biol. Endocrinol. 2009, 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Vitvitsky V.; Thomas M.; Ghorpade A.; Gendelman H. E.; Banerjee R. J. Biol. Chem. 2006, 281, 35785. [DOI] [PubMed] [Google Scholar]
- Abe K.; Kimura H. J. Neurosci. 1996, 16, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto K.; Ogasawara M.; Umemura K.; Nagai Y.; Kimura H. J. Neurosci. 2002, 22, 3386. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Miles E. W.; Kraus J. P. J. Biol. Chem. 2004, 279, 29871. [DOI] [PubMed] [Google Scholar]
- Zhao W.; Zhang J.; Lu Y.; Wang R. EMBO J. 2001, 20, 6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiku T.; Padovani D.; Zhu W.; Singh S.; Vitvitsky V.; Banerjee R. J. Biol. Chem. 2009, 284, 11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Wu L.; Jiang B.; Yang W.; Qi J.; Cao K.; Meng Q.; Mustafa A. K.; Mu W.; Zhang S.; Snyder S. H.; Wang R. Science 2008, 322, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean K. N.; Janosik M.; Kraus E.; Kozich V.; Allen R. H.; Raab B. K.; Kraus J. P. J. Cell Physiol. 2002, 192, 81. [DOI] [PubMed] [Google Scholar]
- Eto K.; Kimura H. J. Biol. Chem. 2002, 277, 42680. [DOI] [PubMed] [Google Scholar]
- a Cooper A. J. Annu. Rev. Biochem. 1983, 52, 187. [DOI] [PubMed] [Google Scholar]; b Frendo J.; Wrobel M. Acta Biochim. Pol. 1997, 44, 771. [PubMed] [Google Scholar]; c Kuo S. M.; Lea T. C.; Stipanuk M. H. Biol. Neonate 1983, 43, 23. [DOI] [PubMed] [Google Scholar]; d Nagahara N.; Ito T.; Kitamura H.; Nishino T. Histochem. Cell Biol. 1998, 110, 243. [DOI] [PubMed] [Google Scholar]
- a Shibuya N.; Mikami Y.; Kimura Y.; Nagahara N.; Kimura H. J. Biochem. 2009, 146, 623. [DOI] [PubMed] [Google Scholar]; b Shibuya N.; Tanaka M.; Yoshida M.; Ogasawara Y.; Togawa T.; Ishii K.; Kimura H. Antioxid. Redox Signaling 2009, 11, 703. [DOI] [PubMed] [Google Scholar]
- Jacob C.; Battaglia E.; Burkholz T.; Peng D.; Bagrel D.; Montenarh M. Chem. Res. Toxicol. 2011, 25, 588. [DOI] [PubMed] [Google Scholar]
- Benavides G. A.; Squadrito G. L.; Mills R. W.; Patel H. D.; Isbell T. S.; Patel R. P.; Darley-Usmar V. M.; Doeller J. E.; Kraus D. W. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson K. R. Biochim. Biophys. Acta 2009, 1787, 856. [DOI] [PubMed] [Google Scholar]
- a Li L.; Bhatia M.; Zhu Y. Z.; Zhu Y. C.; Ramnath R. D.; Wang Z. J.; Anuar F. B.; Whiteman M.; Salto-Tellez M.; Moore P. K. FASEB J. 2005, 19, 1196. [DOI] [PubMed] [Google Scholar]; b Yusuf M.; Kwong Huat B. T.; Hsu A.; Whiteman M.; Bhatia M.; Moore P. K. Biochem. Biophys. Res. Commun. 2005, 333, 1146. [DOI] [PubMed] [Google Scholar]
- Furne J.; Saeed A.; Levitt M. D. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1479. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A.; Pyriochou A.; Altaany Z.; Yang G.; Marazioti A.; Zhou Z.; Jeschke M. G.; Branski L. K.; Herndon D. N.; Wang R.; Szabo C. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 21972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert A. R.; New E. J.; Chang C. J. J. Am. Chem. Soc. 2011, 133, 10078. [DOI] [PubMed] [Google Scholar]
- Peng H.; Cheng Y.; Dai C.; King A. L.; Predmore B. L.; Lefer D. J.; Wang B. Angew. Chem., Int. Ed. 2011, 50, 9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Pan J.; Li S.; Zhao Y.; Wu L. Y.; Berkman C. E.; Whorton A. R.; Xian M. Angew. Chem., Int. Ed. 2011, 50, 10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y.; Karpus J.; Kabil O.; Zhang S. Y.; Zhu H. L.; Banerjee R.; Zhao J.; He C. Nat. Commun. 2011, 2, 495. [DOI] [PubMed] [Google Scholar]
- a Goubern M.; Andriamihaja M.; Nubel T.; Blachier F.; Bouillaud F. FASEB J. 2007, 21, 1699. [DOI] [PubMed] [Google Scholar]; b Powell M. A.; Somero G. N. Science 1986, 233, 563. [DOI] [PubMed] [Google Scholar]
- Hildebrandt T. M.; Grieshaber M. K. FEBS J. 2008, 275, 3352. [DOI] [PubMed] [Google Scholar]
- Zanardo R. C.; Brancaleone V.; Distrutti E.; Fiorucci S.; Cirino G.; Wallace J. L. FEBS J. 2006, 20, 2118. [DOI] [PubMed] [Google Scholar]
- a Pan T. T.; Feng Z. N.; Lee S. W.; Moore P. K.; Bian J. S. J. Mol. Cell. Cardiol. 2006, 40, 119. [DOI] [PubMed] [Google Scholar]; b Sivarajah A.; McDonald M. C.; Thiemermann C. Shock 2006, 26, 154. [DOI] [PubMed] [Google Scholar]
- a Elrod J. W.; Calvert J. W.; Morrison J.; Doeller J. E.; Kraus D. W.; Tao L.; Jiao X.; Scalia R.; Kiss L.; Szabo C.; Kimura H.; Chow C. W.; Lefer D. J. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 15560. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang R. Kidney Int. 2009, 76, 700. [DOI] [PubMed] [Google Scholar]
- Tiranti V.; Viscomi C.; Hildebrandt T.; Di Meo I.; Mineri R.; Tiveron C.; Levitt M. D.; Prelle A.; Fagiolari G.; Rimoldi M.; Zeviani M. Nat. Med. 2009, 15, 200. [DOI] [PubMed] [Google Scholar]
- Klentz R. D.; Fedde M. R. Respir. Physiol. 1978, 32, 355. [DOI] [PubMed] [Google Scholar]
- a Geng B.; Cui Y.; Zhao J.; Yu F.; Zhu Y.; Xu G.; Zhang Z.; Tang C.; Du J. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1608. [DOI] [PubMed] [Google Scholar]; b Kubo S.; Kurokawa Y.; Doe I.; Masuko T.; Sekiguchi F.; Kawabata A. Toxicology 2007, 241, 92. [DOI] [PubMed] [Google Scholar]; c Oh G. S.; Pae H. O.; Lee B. S.; Kim B. N.; Kim J. M.; Kim H. R.; Jeon S. B.; Jeon W. K.; Chae H. J.; Chung H. T. Free Radical Biol. Med. 2006, 41, 106. [DOI] [PubMed] [Google Scholar]
- Muzaffar S.; Shukla N.; Bond M.; Newby A. C.; Angelini G. D.; Sparatore A.; Del Soldato P.; Jeremy J. Y. J. Vasc. Res. 2008, 45, 521. [DOI] [PubMed] [Google Scholar]
- Blackstone E.; Morrison M.; Roth M. B. Science 2005, 308, 518. [DOI] [PubMed] [Google Scholar]
- Kimura Y.; Goto Y.; Kimura H. Antioxid. Redox Signaling 2010, 12, 1. [DOI] [PubMed] [Google Scholar]
- Mueller E. G. Nat. Chem. Biol. 2006, 2, 185. [DOI] [PubMed] [Google Scholar]
- a Kurihara T.; Mihara H.; Kato S.; Yoshimura T.; Esaki N. Biochim. Biophys. Acta 2003, 1647, 303. [DOI] [PubMed] [Google Scholar]; b Li K.; Tong W. H.; Hughes R. M.; Rouault T. A. J. Biol. Chem. 2006, 281, 12344. [DOI] [PubMed] [Google Scholar]
- Abdolrasulnia R.; Wood J. L. Biochim. Biophys. Acta 1979, 567, 135. [DOI] [PubMed] [Google Scholar]
- a Francoleon N. E.; Carrington S. J.; Fukuto J. M. Arch. Biochem. Biophys. 2011, 516, 146. [DOI] [PubMed] [Google Scholar]; b Sorbo B. Biochim. Biophys. Acta 1957, 23, 412. [DOI] [PubMed] [Google Scholar]; c Truong D. H.; Eghbal M. A.; Hindmarsh W.; Roth S. H.; O’Brien P. J. Drug Metab. Rev. 2006, 38, 733. [DOI] [PubMed] [Google Scholar]
- a Heiss E.; Herhaus C.; Klimo K.; Bartsch H.; Gerhauser C. J. Biol. Chem. 2001, 276, 32008. [DOI] [PubMed] [Google Scholar]; b Horton N. D.; Biswal S. S.; Corrigan L. L.; Bratta J.; Kehrer J. P. J. Biol. Chem. 1999, 274, 9200. [DOI] [PubMed] [Google Scholar]; c Lambert C.; Li J.; Jonscher K.; Yang T. C.; Reigan P.; Quintana M.; Harvey J.; Freed B. M. J. Biol. Chem. 2007, 282, 19666. [DOI] [PubMed] [Google Scholar]; d Pande V.; Ramos M. J. Bioorg. Med. Chem. Lett. 2005, 15, 4057. [DOI] [PubMed] [Google Scholar]
- Seiner D. R.; LaButti J. N.; Gates K. S. Chem. Res. Toxicol. 2007, 20, 1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kong A. N.; Owuor E.; Yu R.; Hebbar V.; Chen C.; Hu R.; Mandlekar S. Drug Metab. Rev. 2001, 33, 255. [DOI] [PubMed] [Google Scholar]; b Zhang H.; Liu H.; Iles K. E.; Liu R. M.; Postlethwait E. M.; Laperche Y.; Forman H. J. Am. J. Respir. Cell Mol. Biol. 2006, 34, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hinman A.; Chuang H. H.; Bautista D. M.; Julius D. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 19564. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Macpherson L. J.; Dubin A. E.; Evans M. J.; Marr F.; Schultz P. G.; Cravatt B. F.; Patapoutian A. Nature 2007, 445, 541. [DOI] [PubMed] [Google Scholar]; c Salazar H.; Llorente I.; Jara-Oseguera A.; Garcia-Villegas R.; Munari M.; Gordon S. E.; Islas L. D.; Rosenbaum T. Nat. Neurosci. 2008, 11, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gong P.; Stewart D.; Hu B.; Li N.; Cook J.; Nel A.; Alam J. Antioxid. Redox Signaling 2002, 4, 249. [DOI] [PubMed] [Google Scholar]; b Hong F.; Sekhar K. R.; Freeman M. L.; Liebler D. C. J. Biol. Chem. 2005, 280, 31768. [DOI] [PubMed] [Google Scholar]; c Hu R.; Xu C.; Shen G.; Jain M. R.; Khor T. O.; Gopalkrishnan A.; Lin W.; Reddy B.; Chan J. Y.; Kong A. N. Life Sci. 2006, 79, 1944. [DOI] [PubMed] [Google Scholar]; d Ishii T.; Itoh K.; Ruiz E.; Leake D. S.; Unoki H.; Yamamoto M.; Mann G. E. Circ. Res. 2004, 94, 609. [DOI] [PubMed] [Google Scholar]; e Kensler T. W.; Wakabayashi N.; Biswal S. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89. [DOI] [PubMed] [Google Scholar]; f Satoh T.; Okamoto S. I.; Cui J.; Watanabe Y.; Furuta K.; Suzuki M.; Tohyama K.; Lipton S. A. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreier S. M.; Muellner M. K.; Steinkellner H.; Hermann M.; Esterbauer H.; Exner M.; Gmeiner B. M.; Kapiotis S.; Laggner H. Neurotox. Res. 2010, 17, 249. [DOI] [PubMed] [Google Scholar]
- a Hayes J. D.; Pulford D. J. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445. [DOI] [PubMed] [Google Scholar]; b Kaplowitz N.; Fernandez-Checa J. C.; Kannan R.; Garcia-Ruiz C.; Ookhtens M.; Yi J. R. Biol. Chem. Hoppe–Seyler 1996, 377, 267. [DOI] [PubMed] [Google Scholar]
- Cavallini D.; Federici G.; Barboni E. Eur. J. Biochem. 1970, 14, 169. [DOI] [PubMed] [Google Scholar]
- Toohey J. I. Anal. Biochem. 2011, 413, 1. [DOI] [PubMed] [Google Scholar]
- Ikeuchi Y.; Shigi N.; Kato J.; Nishimura A.; Suzuki T. Mol. Cell 2006, 21, 97. [DOI] [PubMed] [Google Scholar]
- Mustafa A. K.; Gadalla M. M.; Sen N.; Kim S.; Mu W.; Gazi S. K.; Barrow R. K.; Yang G.; Wang R.; Snyder S. H. Sci. Signaling 2009, 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N.; Fu C.; Pappin D. J.; Tonks N. K. Sci. Signaling 2011, 4, ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]