

Abstract
Many anecdotal observations exist of a regulatory effect of DNA methylation on gene expression. However, in general, the underlying mechanisms of this effect are poorly understood. In this review, we summarize what is currently known about how this important, but mysterious, epigenetic mark impacts cellular functions. Cytosine methylation can abrogate or enhance interactions with DNA-binding proteins, or it may have no effect, depending on the context. Despite being only a small chemical change, the addition of a methyl group to cytosine can affect base readout via hydrophobic contacts in the major groove and shape readout via electrostatic contacts in the minor groove. We discuss the recent discovery that CpG methylation increases DNase I cleavage at adjacent positions by an order of magnitude through altering the local 3D DNA shape and the possible implications of this structural insight for understanding the methylation sensitivity of transcription factors (TFs). Additionally, 5-methylcytosines change the stability of nucleosomes and, thus, affect the local chromatin structure and access of TFs to genomic DNA. Given these complexities, it seems unlikely that the influence of DNA methylation on protein–DNA binding can be captured in a small set of general rules. Hence, data-driven approaches may be essential to gain a better understanding of these mechanisms.
Keywords: epigenetics, DNA methylation, 5-methylcytosine, protein–DNA interactions, DNase I endonuclease, transcription factors
WIDESPREAD ROLE OF DNA METHYLATION
A key epigenetic mechanism in mammals, DNA methylation plays a central role in development [1], specifically in X-chromosome inactivation [2], genomic imprinting [3] and genome reprogramming [4] during early embryogenesis and gametogenesis. DNA methylation suppresses the expression of repetitive sequences [5] and has been implicated in neural development, neurogenesis, synaptic plasticity, learning and memory, brain function, aging and the immune response [6–8]. Chemically, DNA methylation amounts to the covalent addition of a methyl group at the fifth carbon position of cytosine, forming 5-methylcytosine (5mC). As the methyl group is positioned in the major groove, DNA methylation does not interfere with Watson–Crick base pairing [9]. Another chemical modification of DNA is cytosine hydroxymethylation (5hmC) [10, 11], which recent studies suggest may be a novel epigenetic mark that is relevant to development and disease [12–14]. Despite this relationship, the present review focuses on the 5mC modification.
DNA methylation is mediated by a family of well-studied enzymes, DNA methyltransferases (DNMTs), which catalyze the addition of a methyl group to cytosine [15, 16]. In mammalian adult somatic cells, cytosine methylation typically occurs in a CpG dinucleotide context. However, non-CpG methylation is prevalent in embryonic stem cells [17] and has been implicated in neural development [18, 19]. In plants such as Arabidopsis thaliana, cytosine can be methylated at CpG, CpHpG and CpHpH sites, where ‘H’ represents any nucleotide other than guanine [20, 21]. Also, in plants, an alternative RNA-directed cytosine methylation pathway exists [22, 23]. Although 5mC has not been detected in yeast or Caenorhabditis elegans, and it occurs at a negligible level in Drosophila melanogaster [24], some other fungi such as Neurospora crassa have a well-characterized methylation system [25].
Considerable attention has recently been given to the functional role of DNA methylation in epigenetic inheritance. Several studies have investigated how DNA methylation patterns can be influenced by developmental and environmental factors, such as parental nutritional exposure [26–29]. Moreover, DNA methylation seems to have an effect on subsequent generations [30, 31].
DNA METHYLATION AND DISEASE
Normal cell behavior depends on a precise balance between the various nuclear factors and enzymes involved in DNA methylation. Deregulation of this epigenetic mark often affects posttranslational histone modifications and is a contributing factor in different cancers. Aberrant chromatin structure is a common feature in cancer, and numerous comprehensive reviews have linked aberrant methylation to tumorigenesis [32–35]. Two types of DNA methylation changes are observed in cancer: hypomethylation, which is often linked to chromosomal instability and loss of imprinting [36], and hypermethylation, which can lead to transcriptional silencing [37]. A recent study provided evidence for an increased incidence of spontaneous cancers in mice caused by transcriptional suppression through promoter hypermethylation [38]. Studies related to DNA methylation and cancer have broadly focused on the methionine cycle in cancer cells [35], regions with differential DNA methylation patterns in cancer [39], tumor heterogeneity arising from methylation variability across different tumor types [32] and roles of microRNAs [40] and retrotransposons [41] in establishing aberrant methylation patterns.
DNA methylation plays a role in diseases and disorders other than cancer. Several neurodevelopmental or neurodegenerative disorders (e.g. Rett [42], Rubinstein-Taybi [43] and Fragile X syndromes [44], Alzheimer's [45] and Huntington's diseases [46]) and psychiatric disorders (e.g. depression, anxiety, addiction and schizophrenia) have a DNA methylation component [47, 48]. Atherosclerosis has been attributed to DNA hypomethylation [49], and studies have implicated DNA methylation in obesity [50, 51] and cholesterol biosynthesis [52]. Evidence exists for a correlation between abnormal methylation and pathogenesis of the immune system [53]. In particular, hypomethylation of select DNA promoters in T-cells leads to aberrant development, resulting in autoimmune diseases such as lupus [54]. Finally, a role for increased methylation levels in aging and the estimation of age using DNA methylation levels has been suggested [55]. Taken together, these observations leave little doubt about the functional importance of DNA methylation (Table 1).
Table 1:
Widespread role of DNA methylation in regulation and disease
| Biological processes that involve DNA methylation | Examples |
|---|---|
| Transcription factor binding and nucleosome positioning | [37, 38, 56–60] |
| Development, differentiation, genomic imprinting and X-inactivation | [1–4] |
| Cancer | [32, 33, 35–41, 61, 62] |
| Neurodevelopmental diseases: Rett, Rubinstein-Taybi and Fragile X syndromes | [42–44] |
| Neurodegenerative diseases: Alzheimer’s and Huntington’s diseases | [45, 46] |
| Psychiatric diseases: depression, anxiety, addiction and schizophrenia | [47, 48] |
| Atherosclerosis | [49] |
| Obesity and cholesterol biosynthesis | [50, 51] |
| Aging | [14, 52, 55] |
| Immunity | [8, 54] |
Nevertheless, the molecular mechanisms that explain the function of DNA methylation are poorly understood. Therefore, in this review, we analyze the biophysical mechanisms associated with cytosine methylation and how these mechanisms can potentially explain the functional impact of cytosine methylation. In particular, we discuss the recent discovery that CpG methylation increases DNase I cleavage at adjacent nucleotides by an order of magnitude, through narrowing of the DNA minor groove and increasing the Roll angle in CpG dinucleotides [63]. We examine the possible implications of these structural insights for understanding the methylation sensitivity of transcription factors (TFs). More generally, we discuss open questions in this complex, relatively uncharted and evolving field of research.
DNase I CLEAVAGE IS DEPENDENT ON THE DNA METHYLATION STATE
The classic footprinting endonuclease DNase I has been exploited to probe chromatin structure on a genome-wide scale across many cell types [64]. With the advent of massively parallel sequencing, it has become possible to achieve single-nucleotide resolution. Several of the authors of this review recently characterized the intrinsic sequence biases of DNase I when cleaving naked DNA [63]. When analyzing deeply sequenced DNase I digests of purified genomic DNA, the authors discovered that the rate at which individual phosphodiester bonds were nicked varied dramatically with local sequence context. Specifically, the rates at the most and least cleavable hexamers differed by no less than a factor of a thousand. A later study confirmed this observation and emphasized the importance of considering these strong biases when analyzing in vivo DNase I footprinting data at single-nucleotide resolution [65].
In addition to the dramatic dependence of the DNase I cleavage rate on the local primary sequence context, Lazarovici et al. discovered that direct 5′ cleavage of CpG base pair steps was enhanced by an order of magnitude when the two cytosine bases were methylated [63]. Moreover, they were able to provide a unified quantitative structural explanation for the sequence and the methylation dependence of DNA nicking by DNase I. Briefly, variations in sequence lead to variations in the detailed 3D geometry of the DNA molecule. For each primary sequence, the corresponding DNA duplex assumes a B-form configuration. However, local geometric parameters, such as Roll and minor groove width (MGW), vary over a sufficiently large range to influence the interaction with DNase I. The effect of these DNA shape features on DNase I cleavage is due to the recognition of electrostatic potential in the DNA minor groove through arginine residues (Figure 1) [68].
Figure 1:
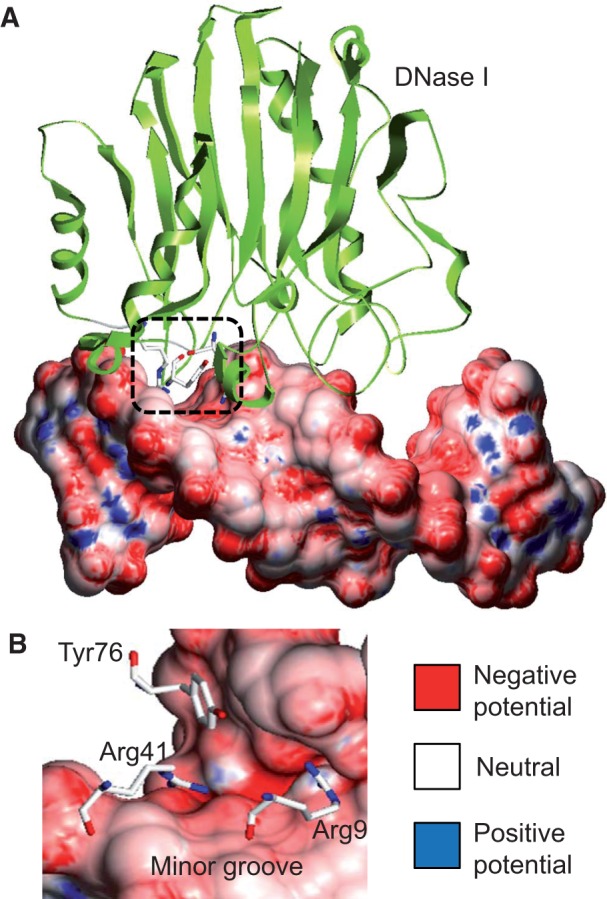
Complex of DNase I endonuclease bound to DNA. (A) Cocrystal structure of the complex (PDB ID 2DNJ) illustrates that DNase I binds DNA through contacts to the DNA minor groove. (B) A rotated view of the region contacted by DNase I shows that positively charged Arg9 and Arg41 residues recognize the negative electrostatic potential in the DNA minor groove, while Tyr76 stacks with a hydrophobic sugar moiety. Electrostatic potential was calculated for only the DNA molecule taken from the complex at physiologic ionic strength (0.145 M, based on a previously described protocol [66, 67]). Electrostatic potential is shown at the molecular surface of the DNA, using blue for +10 kT/e, red for −10 kT/e and white for neutral potentials.
Figure 2A illustrates the construction of the ‘shape-to-affinity model’ used by Lazarovici et al. [63]. Each hexamer sequence is converted into a set of position-specific Roll and MGW values, which serve as the independent (‘predictor’) variables in a multiple linear regression. The negative logarithm of the relative cleavage rate serves as the dependent (‘response’) variable, which can be interpreted as the binding free energy ΔΔG relative to that for the most cleavable sequence (for which, by definition, ΔΔG = 0). Thus, a value of ΔΔG/RT = 1 corresponds to a relative cleavage rate of 37% and ΔΔG/RT = 2 corresponds to a relative rate of 14%, etc. Analyzing the fraction of the total variance of ΔΔG among all unmethylated sequences of type NNN|CGN (where ‘|’ indicates the cleavage site) that can be explained by different variants of the shape-based model (Figure 2B) shows that although Roll is more predictive than MGW, the two variables complement each other.
Figure 2:

Intrinsic DNA methylation sensitivity of DNase I. This figure illustrates the original analysis performed by Lazarovici et al. [63]. (A) Schematic diagram illustrating construction of the ‘shape-to-affinity model’ for predicting the binding free energy ΔΔG (logarithm of the relative cleavage rate) from DNA structural features, such as Roll and MGW. (B) Fraction of the variance in binding free energy explained by the different variants of the shape-based model when fit to all 256 unmethylated DNA sequences containing a CpG dinucleotide. (C) Effect of cytosine methylation on each shape parameter (distribution of difference for Roll and MGW values, along the leading strand of the hexamer), derived from all-atom Monte Carlo simulations [69, 70] of methylated DNA fragments. Five Roll values describe the base pair steps in a hexamer, whereas each of the six base pairs can be assigned a MGW value, as previously described [71]. (D) Change in binding free energy for each sequence, predicted from changes in shape features in (C), using the model constructed in (A). (E) Empirically observed change in binding free energy obtained from parallel methylation and DNase I profiling on genomic DNA from the same cell line. (F) Effect of methylation across all 256 nucleotide sequences, quantified as the change in ΔΔG, as empirically observed and as predicted using Roll, MGW or both.
Lazarovici et al. [63] applied computationally intensive Monte Carlo simulations of free DNA to obtain the Roll and MGW values across the DNA molecule [69, 70], which were used as predictors in the shape-to-affinity model. Simulations were performed with or without a methyl group added to each cytosine base of CpG steps in the DNA molecule. This approach enabled the authors to build a shape-to-affinity model from the shape parameters derived from unmethylated sequences of the form NNN|CGN, as well as to predict, for each DNA sequence, the change in binding free energy associated with methylation of the CpG dinucleotide downstream of the cleavage site. Strikingly, the simulations revealed that a change in Roll owing to the substitution of C by 5mC was largely independent of the identity of the other four bases within the NNN|CGN hexamer (Figure 2C). Moreover, a linear model was trained on shape parameters derived from unmethylated sequences. When only the shape changes in the sequences were used to transfer the model to methylated DNA, the model predicted an increased binding free energy that was independent of the sequence context and indicated increased binding in the presence of 5mC (Figure 2D).
The qualitative observation that methylation increases the DNase I cleavage rate by a fixed multiplicative factor (corresponding to the shift in binding free energy) and the quantitative prediction of the magnitude of this effect agreed well with direct empirical observations made by using parallel DNase I footprinting and methylome data for genomic DNA from the same cell line (Figure 2E). Interestingly, Roll seemed to be more useful than MGW for predicting the effect of CpG methylation on binding free energy (Figure 2F). Analysis of the empirical data revealed an ∼9-fold change in cleavage rate (and, presumably, in protein–DNA binding affinity) associated with CpG methylation (median ΔΔΔG/RT = −2.2). However, this value should be interpreted as a lower bound, because it was obtained by constructing hexamer cleavage tables from the top and bottom 20% of genomic locations in terms of observed cytosine methylation levels. Consistent with this finding, comparisons of simulations of strictly unmethylated and fully methylated DNA molecules with a model driven by the empirical cleavage rate for unmethylated sequences [63] predicted a change in the cleavage rate of up to 16-fold on methylation (median ΔΔΔG/RT = −2.8).
IMPLICATIONS FOR DNA BINDING OF TFS
Mechanisms explaining the change in the DNase I cleavage rate on CpG methylation [63] suggest that 5mC could have an impact on protein–DNA interactions in general, and on the binding specificity of TFs in particular. In the case of DNase I, it is important that two arginine side chains be present in the minor groove (Figure 1). Thus, the magnitude of the enhancement of an interaction based on a similar mechanism is expected to depend strongly on the DNA recognition mechanism used by the specific protein [72–74]. More generally, the hydrophobic methyl group can influence direct contacts in the major groove (base readout), CpG methylation can alter the 3D structure of the DNA binding site (shape readout) and methylated cytosines can modify the nucleosome stability (and, thus, chromatin structure).
Any of these effects can occur in isolation or combination; hence, it is unlikely that the effect of CpG methylation on TF-DNA binding can be summarized in a set of simple rules. Moreover, DNA methylation interacts with other epigenetic marks in a complex manner to affect transcription and regulate gene expression [21, 75]. The exact mechanisms are still not understood, although several have been proposed. TFs might be subjected to a physical barrier created by DNA methylation, which hinders access to TF binding sites (TFBSs). TFBSs that are stably bound by TFs are highly resistant to de novo methylation [76]. Mechanical properties of DNA, such as stiffness [77] and strand separation [78], change upon cytosine methylation, with possible effects on TF binding.
EFFECTS OF DNA METHYLATION ON DNA BASE AND SHAPE READOUT
Three major families of methyl-CpG–binding proteins can be distinguished on the basis of the domain structure that they use to interact with DNA: methyl-CpG–binding domain (MBD) proteins, SET and RING-finger associated domain proteins, and Kaiso-like C2H2 zinc-finger proteins [79–81]. The most intuitive effect of CpG methylation on TF–DNA binding is the addition of a methyl group at the major groove edge of the cytosine base (Figure 3A). The 5-methyl group is present in 5mC and thymine. Thus, the 5mC-G base pair can be contacted through hydrophobic contacts, similar to how thymine is contacted in unmethylated DNA. Cytosine methylation alters functional group signatures in the major groove, but not in the minor groove (Figure 3B). Addition of a methyl group at the major groove edge of 5mC can be specifically recognized through hydrophobic base contacts (base readout).
Figure 3:

Base and shape readout of methylated DNA. (A) Chemical configurations of C-G (left), 5mC-G (center) and T-A (right) base pairs. The methyl group (indicated by the C5M carbon atom) is present at the major groove edge of the 5mC-G and T-A base pairs. (B) Signatures of functional groups at the major groove (left) and minor groove (right) edges of C-G (top), 5mC-G (center) and T-A (bottom) base pairs. The methyl group (yellow) changes the signature of functional groups at the major groove edge of the C-G base pair, but base readout at the minor groove edge is not affected. (C) Presence of a 5mCpG dinucleotide (C5M carbon atom of the methyl groups shown in red) in methylated DNA (top) can affect the widths of the major (left) and minor grooves (right) compared with unmethylated DNA (bottom) as a function of its sequence context. (D) The methyl group of the 5mC nucleotide is in close proximity to the sugar moiety and phosphate group of the adjacent nucleotide in 5′ direction (here, thymine). Structures in this figure are derived from all-atom Monte Carlo simulations of naked DNA, using a previously described protocol [69, 70].
Of the few crystal structures of DNA oligonucleotides with 5mC bases that have been reported, some of these structures suggest that the steric hindrance of the methyl group in the major groove counters DNA bending and twisting [82]. The presence of a bulky methyl group might lead to a subtle widening of the major groove and, consequently, a subtle narrowing of the minor groove (Figure 3C), owing to the close proximity of the methyl group to the phosphodiester backbone, which might lead to steric hindrance (Figure 3D).
DNA-binding proteins recognize the 3D DNA structure (shape readout) [66, 83], which is highly sequence dependent [84–86]. As discussed above, Lazarovici et al. [63] showed that the impact of DNA methylation on DNA structure can explain the methylation state-dependent DNase I cleavage rate. Local DNA shape features were powerful predictors of the effect of cytosine methylation on DNA shape [63]. The effect was strongly sequence dependent in that it only occurred for protein–DNA binding events that led to cleavage of the phosphate immediately 5′ of the CpG dinucleotide. However, the favorable (negative) contribution of methylation to the overall protein–DNA binding free energy was largely independent of the base identity at other nucleotide positions near the cleavage site. Whereas the DNase I cleavage rates of unmethylated CpG-containing sequences varied widely, the fold increase in these cleavage rates because of methylation was the same for all sequences.
Using all-atom Monte Carlo simulations [69, 70], we observed that the Roll angle of CpG dinucleotides consistently increased upon cytosine methylation [63]. This effect occurred for both fully and hemi-methylated CpG steps, as only the size of the increase in Roll depended on sequence context. Roll decreased at the two base pair steps surrounding a CpG dinucleotide, to compensate for the increased Roll at the CpG step. This observation was in agreement with all-atom molecular dynamics simulations, in which the increase in Roll at CpG steps was the most pronounced effect of cytosine methylation on DNA structure [77].
INSIGHTS FROM STRUCTURES OF PROTEIN–DNA COMPLEXES
Few structures containing 5mC bases have been solved by X-ray crystallography or nuclear magnetic resonance spectroscopy. Several structural studies have demonstrated the importance of the 5mC methyl group for contacts with hydrophobic patches on the protein surface of MBDs [87, 88] or with water molecules [89, 90]. Understanding the role of hydrophobic contacts with the 5mC methyl group in the major groove for methylation state-specific binding ideally requires 3D structures of a protein bound to methylated and unmethylated copies of its DNA target. However, this information is only available for a few TFs, including the zinc-finger proteins Kaiso [91] and Kruppel-like factor 4 (Klf4) [92, 93].
Recognition processes for methylated and unmethylated DNA use similar overall geometries. When Kaiso or Klf4 is in contact with methylated DNA [92, 94], arginine and glutamate form hydrophobic contacts with the cytosine methyl groups. For Kaiso, a hydrophobic pocket built of threonine and cysteine was observed to contact another methyl group [91]. Interestingly, the binding affinities of Klf4 to methylated DNA and to its unmethylated target are similar [92]. In the case of the zinc-finger protein Zfp57, structural information is only available for binding to methylated DNA. The cytosine methyl group of the Zfp57 target site engages in hydrophobic contacts with arginine [95]. Taken together, these data support the hypothesis that hydrophobic contacts with methyl groups merely fine-tune the binding specificities of TFs.
If the presence of a methyl group confer binding specificity, then in principle, 5mC should mimic the presence of a thymine. This possibility has been experimentally proven for the binding site of the lac repressor. In this case, replacement of a thymine by cytosine caused loss of activity, which was restored when thymine was replaced by 5mC [96]. Another example is the complex of the P22 c2 repressor and its operator. In this case, four thymine methyl groups form a hydrophobic binding pocket for a specific valine contact [97]. Contacts with the thymine methyl group were described as intermediates between weak hydrogen bonds and strong van der Waals interactions [98].
A TF or other DNA-binding protein physically interacts via the electrostatic potential at the molecular surface of the DNA [66]. DNA is a polyanion, and its surface is dominated by a negative electrostatic potential. Whereas DNA phosphate groups and bases carry negative charges, the sugar moieties, thymine and 5mC methyl groups of DNA are positively charged [72]. Thus, the electrostatic potential at the molecular surface of these hydrophobic groups is less negative than at the remainder of the DNA surface. We analyzed variations in the electrostatic potential on molecular surfaces of DNA with and without methyl groups using Poisson–Boltzmann calculations at physiologic ionic strength [66]. Comparison of the Klf4 binding sites with and without cytosine methylation [92, 93] clearly showed the impact of the cytosine methyl group on hydrophobic contacts (Figure 4A and B). The same effect was seen for the four thymine-methyl groups, which formed a binding site for a valine of the P22 c2 repressor (Figure 4C) [78].
Figure 4:
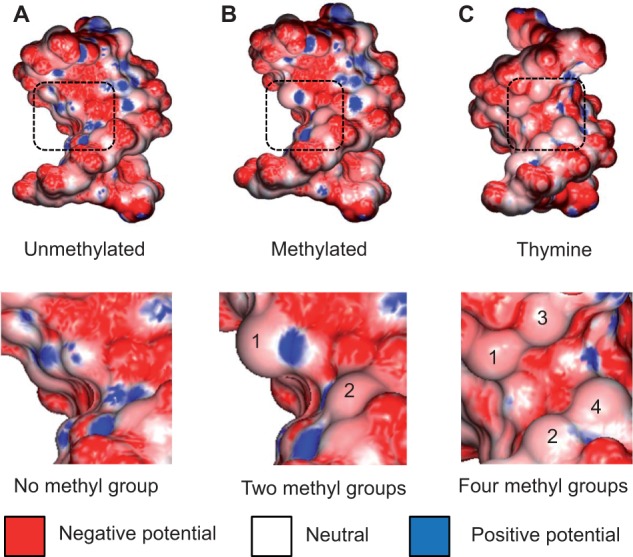
Effect of methyl groups on electrostatic potential of DNA. Hydrophobic methyl groups affect the electrostatic potential in the DNA major groove, as shown for the comparison of the (A) unmethylated and (B) methylated DNA targets of Klf4 based on cocrystal structures (PDB IDs 2WBU and 4M9E, respectively). A similar effect is observed for a (C) DNA target with four thymine methyl groups, which form a binding site for a valine residue of the P22 c2 repressor in the cocrystal structure of the complex (PDB ID 2R1J). The electrostatic potential was calculated at physiologic ionic strength (0.145 M, based on a previously described protocol [66, 67]). The electrostatic potential is shown at the molecular surface of the DNA, using blue for +10 kT/e, red for −10 kT/e, and white for neutral potentials.
LARGE-SCALE BINDING ASSAYS REVEAL METHYLATION-DEPENDENT BINDING
The hypothesis that DNA methylation leads to transcriptional inhibition is still being debated. Despite studies on the effects of cytosine methylation on gene expression [99], the underlying mechanisms are still not understood. A recent high-throughput study of TF binding [56], which combined in vitro binding assays with in vivo validations, offered a rich perspective on this question. This study used protein microarrays to probe the binding of 1321 human TFs and 210 cofactors to 154 TF binding motifs containing at least one CpG dinucleotide. In general, DNA methylation did not inhibit the binding of TFs from different families. The in vitro assay indicated interactions of at least one, and a median of eight, TFs with each of the studied CpG-containing motifs. Moreover, 5mC did not inhibit binding per se. A subset of 41 TFs and 6 cofactors from different protein families exhibited specific or nonspecific 5mC-dependent binding. Further analysis of methylated consensus TFBSs with known binding motifs showed almost no correlation, suggesting that cytosine methylation created completely different binding sites for some TFs [56].
EFFECT OF DNA METHYLATION ON CHROMATIN ACCESSIBILITY AND NUCLEOSOME STABILITY
Another set of putative mechanisms for gene inactivation through DNA methylation involves MBD proteins [100, 101], which establish a positive feedback loop between DNA methylation and specific histone modifications [102]. MBD proteins are capable of acting as histone deacetylase (HDAC)-dependent transcriptional repressors [103]. Repression is achieved by association with methylated DNA sequences [104]. The functional consequences of HDAC–DNMT interactions are still not completely understood, although extensive evidence exists that the catalytically active DNMTs interact with HDACs [61]. DNA methylation enables the DNA binding of MBDs [81], which, in turn, recruit HDACs and other chromatin remodeling proteins. Thus, chromatin becomes compacted and inactive, resulting in gene silencing [62]. DNA methylation can also influence chromatin structure and cause gene silencing [105].
Several studies have addressed the interplay of DNA methylation and nucleosome positioning [57, 58], including one study that used nucleosome mapping in A. thaliana [58]. In the latter study, DNase I digestion revealed a consistent 10 bp periodicity, with the minor groove at WW (W = A or T) dinucleotides facing the histone core and the major groove at SS (S = C or G) dinucleotides facing the histone core a half-period (5 bp) away. Interestingly, cytosine methylation of different types (i.e. CpG, CpHpG or CpHpH, where H = A, C or T) showed the same 10 bp periodicity in nucleosomal DNA in phase with the WW dinucleotides, which are regions where the major groove is accessible. This study also revealed a higher methylation level in nucleosome-occupied DNA than in its flanking regions, supporting the argument that DNMTs preferentially bind to nucleosomes. The observed periodicity for all types of cytosine methylation suggested that nucleosomes control access to the DNA for DNMTs [58]. These observations support the hypothesis that DNA is first organized in nucleosomes before being methylated.
Conversely, epigenetic marks (e.g. cytosine methylation) can affect nucleosome positioning. Accurate predictions of nucleosome occupancies are required to study complex regulatory mechanisms. However, most available tools for nucleosome occupancy predictions are knowledge based, fail to account for epigenetic marks and perform poorly on DNA sequences with unknown nucleosome occupancy characteristics. A recently developed physics-based method by Minary and Levitt [59] used the all-atom energy of the 3D nucleosome structure to predict the nucleosome occupancy of a given genomic sequence. Advantages of an ab initio method are that it does not require any training, and it can make accurate predictions in the presence of epigenetic marks on a DNA. Minary and Levitt found that the sequence-dependent nucleosome formation energy was anticorrelated with in vitro nucleosome occupancy. When all of the cytosine bases in a given genomic sequence were methylated, the dependence of the nucleosome formation energy on the sequence was more moderate. Specifically, the formation of weakly or strongly bound nucleosomes was strengthened or weakened, respectively [59].
The capability of cytosine methylation to moderate the sequence dependence of nucleosome occupancy likely promotes the relocation of nucleosomes to hypermethylated sites within promoter regions. It is interesting to imagine how cytosine methylation could affect the positioning of nucleosomes to change on a fine-grained scale, causing subtle shifts that could expose or block TFBSs. The effect of CpG methylation on nucleosome stability was also evaluated by molecular dynamics simulations [60]. Different nucleosome models were designed with single and multiple CpG steps facing the histone core with either the minor or major groove. Cytosine methylation destabilized nucleosome formation, and multiple steps amplified the destabilization. The stability of nucleosomes correlated with the base-stacking interaction, suggesting that CpG methylation reduced the ability of DNA to tolerate deformation when wrapped around the histone core.
OPEN QUESTIONS
The biological importance of DNA methylation in development and disease is well established, but a thorough mechanistic understanding of these processes is still lacking. On one hand, the few available experimental structures of TFs bound to methylated versus unmethylated DNA targets suggest that the cytosine methyl group plays only a marginal role in refining hydrophobic contacts, with small effects on binding affinity [91–93]. On the other hand, protein microarray studies suggest that some TFs select novel binding motifs containing methylated CpG dinucleotides [56]. Whereas the exact impact of cytosine methylation on TF binding is an open question, it is known that the DNase I cleavage directly adjacent to CpG dinucleotides strongly depends on the CpG methylation status [63]. Together, these findings suggest that the extent to which TF–DNA binding relies on cytosine methylation likely depends on the readout mode used by the TF under consideration [106]. Base readout can be affected by hydrophobic contacts in the major groove (Figure 5A), and shape readout can vary with the sequence-dependent impact of CpG methylation on DNA structure (Figure 5B).
Figure 5:
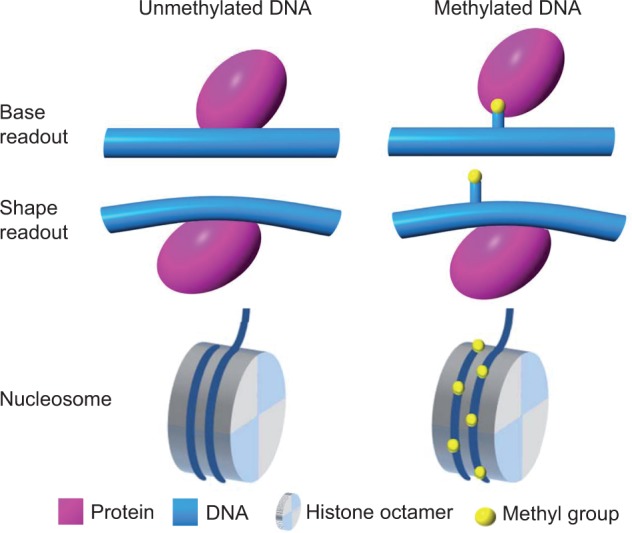
DNA methylation can affect base readout, shape readout and nucleosome stability. Presence of cytosine methyl groups (yellow) can (A) affect direct interactions between the protein (purple) and its DNA binding site (blue; base readout), (B) cause an indirect effect on DNA structure (blue), which can be recognized by the DNA-binding protein (purple; shape readout) and (C) alter nucleosome stability by modifying interactions between the histone core octamer (gray) and the DNA that is wrapped around it (blue).
Another open question regards the effect of DNA methylation on nucleosome positioning and stability (Figure 5C) and, in turn, their impact on TF binding [107]. Some studies have reported generally decreased nucleosome stability on cytosine methylation, which they attributed to the rigidity introduced by methyl groups [77, 60]. Another study suggested that weakly positioned nucleosomes are stabilized, whereas strongly positioned nucleosomes are destabilized [59]. The latter study also proposed a context-dependent effect of cytosine methylation on nucleosome positioning.
Finally, it remains unclear whether the nucleosome core particle provides a scaffold for DNMTs to contact the DNA while bound to the histone octamer [58] or whether the core particle protects DNA from cytosine methylation [108]. Context dependence can probably explain some of these apparently contradictory observations, as suggested by other comprehensive reviews discussing the effect of DNA methylation on nucleosome positioning [109], TF binding [110] and gene regulation [111]. This context may include the DNA sequence environment, cooperativity, chromatin accessibility or other higher-order effects on protein–DNA binding [74, 112]. Understanding how DNA methylation can affect so many biological processes will require further studies and the continued development of new experimental and computational approaches.
Key points.
DNA methylation has many known functions, both in normal cell function and disease.
Mechanisms by which 5mC is recognized by the regulatory machinery of the cell remain largely obscure.
5mC readout can occur both directly (by base readout) and indirectly (by shape readout).
DNA shape changes in response to cytosine methylation could be a general mechanism for modulating protein–DNA interactions.
FUNDING
This work was supported by the National Institutes of Health (grants R01HG003008 to H.J.B. and R.R., R01GM106056 and U01GM103804 to R.R. and U54CA121852 to H.J.B.) and the Alfred P. Sloan Foundation (to R.R.). The open-access publication charges are defrayed through the National Science Foundation (grant MCB-1413539 to R.R.) and the National Institutes of Health (grant R01HG003008 to H.J.B.).
Biographies
Ana Carolina Dantas Machado is a graduate student in the Molecular Biology Ph.D. Program at the University of Southern California.
Tianyin Zhou is a graduate student in the Computational Biology and Bioinformatics Ph.D. Program at the University of Southern California.
Satyanarayan Rao is a graduate student in the Computational Biology and Bioinformatics Ph.D. Program at the University of Southern California.
Pragya Goel is a graduate student in the Computational Biology and Bioinformatics Ph.D. Program at the University of Southern California.
Chaitanya Rastogi is a graduate student in the Applied Physics Ph.D. Program at Columbia University.
Allan Lazarovici is a graduate student in the Electrical Engineering Ph.D. Program at Columbia University.
Harmen J. Bussemaker is a Professor of Biological Sciences and Systems Biology at Columbia University.
Remo Rohs is an Assistant Professor of Biological Sciences, Chemistry, Physics and Computer Science and a Member of the Norris Comprehensive Cancer Center at the University of Southern California.
References
- 1.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 2.Augui S, Nora EP, Heard E. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat Rev Genet. 2011;12:429–42. doi: 10.1038/nrg2987. [DOI] [PubMed] [Google Scholar]
- 3.Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011;3:a002592. doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee HJ, Hore TA, Reik W. Reprogramming the methylome: erasing memory and creating diversity. Cell Stem Cell. 2014;14:710–9. doi: 10.1016/j.stem.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nature. 1998;20:116–7. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 6.Dulac C. Brain function and chromatin plasticity. Nature. 2010;465:728–35. doi: 10.1038/nature09231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winnefeld M, Lyko F. The aging epigenome: DNA methylation from the cradle to the grave. Genome Biol. 2012;13:165. doi: 10.1186/gb-2012-13-7-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suarez-Alvarez B, Rodriguez RM, Fraga MF, et al. DNA methylation: a promising landscape for immune system-related diseases. Trends Genet. 2012;28:506–14. doi: 10.1016/j.tig.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem. 2002;3:274–93. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 10.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2012;13:7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- 12.Song CX, Szulwach KE, Fu Y, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2010;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yildirim O, Li R, Hung JH, et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498–510. doi: 10.1016/j.cell.2011.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szulwach KE, Li X, Li Y, et al. 5-hmC–mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14:1607–16. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okano M, Bell DW, Haber DA, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 16.Chedin F, Lieber MR, Hsieh CL. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci USA. 2002;99:16916–21. doi: 10.1073/pnas.262443999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramsahoye BH, Biniszkiewicz D, Lyko F, et al. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA. 2000;97:5237–42. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lister R, Mukamel EA, Nery JR, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo JU, Su Y, Shin JH, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17:215–22. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Yazaki J, Sundaresan A, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell. 2006;126:1189–201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 21.Zilberman D, Gehring M, Tran RK, et al. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2006;39:61–9. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 22.Wassenegger M, Heimes S, Riedel L. RNA-directed de novo methylation of genomic sequences in plants. Cell. 1994;76:567–76. doi: 10.1016/0092-8674(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 23.Henderson IR, Jacobsen SE. Epigenetic inheritance in plants. Nature. 2007;447:418–24. doi: 10.1038/nature05917. [DOI] [PubMed] [Google Scholar]
- 24.Takayama S, Dhahbi J, Roberts A, et al. Genome methylation in D. melanogaster is found at specific short motifs and is independent of DNMT2 activity. Genome Res. 2014;24:821–30. doi: 10.1101/gr.162412.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selker EU, Tountas NA, Cross SH, et al. The methylated component of the Neurospora crassa genome. Nature. 2003;422:893–7. doi: 10.1038/nature01564. [DOI] [PubMed] [Google Scholar]
- 26.Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. 2008;105:17046–9. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014;157:95–109. doi: 10.1016/j.cell.2014.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dominguez-Salas P, Moore SE, Baker MS, et al. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun. 2014;5:3746. doi: 10.1038/ncomms4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Radford EJ, Ito M, Shi H, et al. In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science. 2014;345:1255903. doi: 10.1126/science.1255903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skinner MK, Guerrero-Bosagna C. Role of CpG deserts in the epigenetic transgenerational inheritance of differential DNA methylation regions. BMC Genomics. 2014;15:692. doi: 10.1186/1471-2164-15-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McRae AF, Powell JE, Henders AK, et al. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 2014;15:R73. doi: 10.1186/gb-2014-15-5-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 33.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 35.Montenegro MF, Sanchez-Del-Campo L, Fernandez-Perez MP, et al. Targeting the epigenetic machinery of cancer cells. Oncogene. 2014 doi: 10.1038/onc.2013.605. doi: 10.1038/onc.2013.605. [DOI] [PubMed] [Google Scholar]
- 36.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 37.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–7. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 38.Yu DH, Waterland RA, Zhang P, et al. Targeted p16Ink4a epimutation causes tumorigenesis and reduces survival in mice. J Clin Invest. 2014;124:3708–12. doi: 10.1172/JCI76507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–75. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saito Y, Liang G, Egger G, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 41.Estecio MR, Gallegos J, Vallot C, et al. Genome architecture marked by retrotransposons modulates predisposition to DNA methylation in cancer. Genome Res. 2010;20:1369–82. doi: 10.1101/gr.107318.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 43.Petrij F, Giles RH, Dauwerse HG, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–51. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 44.Sutcliffe JS, Nelson DL, Zhang F, et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1:397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- 45.Chen KL, Wang SS, Yang YY, et al. The epigenetic effects of amyloid-beta(1-40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem Biophys Res Commun. 2009;378:57–61. doi: 10.1016/j.bbrc.2008.10.173. [DOI] [PubMed] [Google Scholar]
- 46.Graff J, Mansuy IM. Epigenetic dysregulation in cognitive disorders. Eur J Neurosci. 2009;30:1–8. doi: 10.1111/j.1460-9568.2009.06787.x. [DOI] [PubMed] [Google Scholar]
- 47.Renthal W, Nestler EJ. Chromatin regulation in drug addiction and depression. Dialogues Clin Neurosci. 2009;11:257–68. doi: 10.31887/DCNS.2009.11.3/wrenthal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA. 2005;102:9341–6. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lund G, Andersson L, Lauria M, et al. DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem. 2004;279:29147–54. doi: 10.1074/jbc.M403618200. [DOI] [PubMed] [Google Scholar]
- 50.Wang X, Zhu H, Snieder H, et al. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 2010;8:87. doi: 10.1186/1741-7015-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soubry A, Murphy SK, Wang F, et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int J Obes (Lond) 2013 doi: 10.1038/ijo.2013.193. doi: 10.1038/ijo.2013.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carone BR, Fauquier L, Habib N, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–96. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang Y, Wang P, Zhao M, et al. Demethylation of the FCER1G promoter leads to FcepsilonRI overexpression on monocytes of patients with atopic dermatitis. Allergy. 2012;67:424–30. doi: 10.1111/j.1398-9995.2011.02760.x. [DOI] [PubMed] [Google Scholar]
- 54.Gorelik G, Richardson B. Key role of ERK pathway signaling in lupus. Autoimmunity. 2010;43:17–22. doi: 10.3109/08916930903374832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gonzalo S. Epigenetic alterations in aging. J Appl Physiol. 2010;109:586–97. doi: 10.1152/japplphysiol.00238.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu S, Wan J, Su Y, et al. DNA methylation presents distinct binding sites for human transcription factors. eLife. 2013;2:e00726. doi: 10.7554/eLife.00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smalla EC, Xi L, Wang JP, et al. Single-cell nucleosome mapping reveals the molecular basis of gene expression heterogeneity. Proc Natl Acad Sci USA. 2014;111:E2462–71. doi: 10.1073/pnas.1400517111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chodavarapu RK, Feng S, Bernatavichute YV, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–92. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minary P, Levitt M. Training-free atomistic prediction of nucleosome occupancy. Proc Natl Acad Sci USA. 2014;111:6293–8. doi: 10.1073/pnas.1404475111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Portella G, Battistini F, Orozco M. Understanding the connection between epigenetic DNA methylation and nucleosome positioning from computer simulations. PLoS Comput Biol. 2013;9:e1003354. doi: 10.1371/journal.pcbi.1003354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robertson KD. DNA methylation and chromatin – unraveling the tangled web. Oncogene. 2002;21:5361–79. doi: 10.1038/sj.onc.1205609. [DOI] [PubMed] [Google Scholar]
- 62.Baylin SB, Esteller M, Rountree MR, et al. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet. 2001;10:687–92. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- 63.Lazarovici A, Zhou T, Shafer A, et al. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc Natl Acad Sci USA. 2013;110:6376–81. doi: 10.1073/pnas.1216822110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neph S, Vierstra J, Stergachis AB, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.He HH, Meyer CA, Hu SS, et al. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods. 2014;11:73–8. doi: 10.1038/nmeth.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rohs R, West SM, Sosinsky A, et al. The role of DNA shape in protein-DNA recognition. Nature. 2009;461:1248–53. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harris R, Mackoy T, Dantas Machado AC, et al. Opposites attract: shape and electrostatic complementarity in protein–DNA complexes. In: Schlick T, editor. Innovations in Biomolecular Modeling and Simulation. Vol. 2. London: Royal Society of Chemistry; 2012. pp. 53–80. [Google Scholar]
- 68.Lahm A, Suck D. DNase I-induced DNA conformation. 2 A structure of a DNase I-octamer complex. J Mol Biol. 1991;222:645–67. doi: 10.1016/0022-2836(91)90502-w. [DOI] [PubMed] [Google Scholar]
- 69.Rohs R, Sklenar H, Shakked Z. Structural and energetic origins of sequence-specific DNA bending: Monte Carlo simulations of papillomavirus E2-DNA binding sites. Structure. 2005;13:1499–509. doi: 10.1016/j.str.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 70.Zhang X, Dantas Machado AC, Ding Y, et al. Conformations of p53 response elements in solution deduced using site-directed spin labeling and Monte Carlo sampling. Nucleic Acids Res. 2014;42:2789–97. doi: 10.1093/nar/gkt1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou T, Yang L, Lu Y, et al. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 2013;41:W56–62. doi: 10.1093/nar/gkt437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rohs R, Jin X, West SM, et al. Origins of specificity in protein-DNA recognition. Annu Rev Biochem. 2010;79:233–69. doi: 10.1146/annurev-biochem-060408-091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levo M, Segal E. In pursuit of design principles of regulatory sequences. Nat Rev Genet. 2014;15:453–68. doi: 10.1038/nrg3684. [DOI] [PubMed] [Google Scholar]
- 74.Slattery M, Zhou T, Yang L, et al. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci. 2014;39:381–99. doi: 10.1016/j.tibs.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feldmann A, Ivanek R, Murr R, et al. Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions. PLoS Genet. 2013;9:e1003994. doi: 10.1371/journal.pgen.1003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gebhard C, Benner C, Ehrich M, et al. General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res. 2010;70:1398–407. doi: 10.1158/0008-5472.CAN-09-3406. [DOI] [PubMed] [Google Scholar]
- 77.Perez A, Castellazzi CL, Battistini F, et al. Impact of methylation on the physical properties of DNA. Biophys J. 2012;102:2140–8. doi: 10.1016/j.bpj.2012.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Severin PM, Zou X, Gaub HE, et al. Cytosine methylation alters DNA mechanical properties. Nucleic Acids Res. 2011;39:8740–51. doi: 10.1093/nar/gkr578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sasai N, Nakao M, Defossez PA. Sequence-specific recognition of methylated DNA by human zinc-finger proteins. Nucleic Acids Res. 2010;38:5015–22. doi: 10.1093/nar/gkq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fournier A, Sasai N, Nakao M, et al. The role of methyl-binding proteins in chromatin organization and epigenome maintenance. Brief Funct Genomics. 2012;11:251–64. doi: 10.1093/bfgp/elr040. [DOI] [PubMed] [Google Scholar]
- 81.Zou X, Ma W, Solov'yov IA, et al. Recognition of methylated DNA through methyl-CpG binding domain proteins. Nucleic Acids Res. 2012;40:2747–58. doi: 10.1093/nar/gkr1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tippin DB, Sundaralingam M. Nine polymorphic crystal structures of d(CCGGGCCCGG), d(CCGGGCCm5CGG), d(Cm5CGGGCCm5CGG) and d(CCGGGCC(Br)5CGG) in three different conformations: effects of spermine binding and methylation on the bending and condensation of A-DNA. J Mol Biol. 1997;267:1171–85. doi: 10.1006/jmbi.1997.0945. [DOI] [PubMed] [Google Scholar]
- 83.Rohs R, West SM, Liu P, et al. Nuance in the double-helix and its role in protein-DNA recognition. Curr Opin Struct Biol. 2009;19:171–7. doi: 10.1016/j.sbi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gordân R, Shen N, Dror I, et al. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 2013;3:1093–104. doi: 10.1016/j.celrep.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang L, Zhou T, Dror I, et al. TFBSshape: a motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2014;42:D148–55. doi: 10.1093/nar/gkt1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dror I, Zhou T, Mandel-Gutfreund Y, et al. Covariation between homeodomain transcription factors and the shape of their DNA binding sites. Nucleic Acids Res. 2014;42:430–41. doi: 10.1093/nar/gkt862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ohki I, Shimotake N, Fujita N, et al. Solution structure of the methyl-CpG binding domain of human MBD1 in complex with methylated DNA. Cell. 2001;105:487–97. doi: 10.1016/s0092-8674(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 88.Scarsdale JN, Webb HD, Ginder GD, et al. Solution structure and dynamic analysis of chicken MBD2 methyl binding domain bound to a target-methylated DNA sequence. Nucleic Acids Res. 2011;39:6741–52. doi: 10.1093/nar/gkr262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mayer-Jung C, Moras D, Timsit Y. Hydration and recognition of methylated CpG steps in DNA. EMBO J. 1998;17:2709–18. doi: 10.1093/emboj/17.9.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ho KL, McNae IW, Schmiedeberg L, et al. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. 2008;29:525–31. doi: 10.1016/j.molcel.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 91.Buck-Koehntop BA, Stanfield RL, Ekiert DC, et al. Molecular basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc Natl Acad Sci USA. 2012;109:15229–34. doi: 10.1073/pnas.1213726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu Y, Olanrewaju YO, Zheng Y, et al. Structural basis for Klf4 recognition of methylated DNA. Nucleic Acids Res. 2014;42:4859–67. doi: 10.1093/nar/gku134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schuetz A, Nana D, Rose C, et al. The structure of the Klf4 DNA-binding domain links to self-renewal and macrophage differentiation. Cell Mol Life Sci. 2011;68:3121–31. doi: 10.1007/s00018-010-0618-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu Y, Zhang X, Blumenthal RM, et al. A common mode of recognition for methylated CpG. Trends Biochem Sci. 2013;38:177–83. doi: 10.1016/j.tibs.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu Y, Toh H, Sasaki H, et al. An atomic model of Zfp57 recognition of CpG methylation within a specific DNA sequence. Genes Dev. 2012;26:2374–9. doi: 10.1101/gad.202200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Razin A, Riggs AD. DNA methylation and gene function. Science. 1980;210:604–10. doi: 10.1126/science.6254144. [DOI] [PubMed] [Google Scholar]
- 97.Watkins D, Hsiao C, Woods KK, et al. P22 c2 repressor-operator complex: mechanisms of direct and indirect readout. Biochemistry. 2008;47:2325–38. doi: 10.1021/bi701826f. [DOI] [PubMed] [Google Scholar]
- 98.Mandel-Gutfreund Y, Margalit H, Jernigan RL, et al. A role for CH…O interactions in protein-DNA recognition. J Mol Biol. 1998;277:1129–40. doi: 10.1006/jmbi.1998.1660. [DOI] [PubMed] [Google Scholar]
- 99.Gutierrez-Arcelus M, Lappalainen T, Montgomery SB, et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife. 2013;2:e00523. doi: 10.7554/eLife.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lewis JD, Meehan RR, Henzel WJ, et al. Purification, sequence, and cellular localization of a novel chromosomal to methylated DNA. Cell. 1992;69:905–14. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 101.Jones PL, Veenstra GJ, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nature. 1998;19:187–91. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 102.Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–9. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 103.Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269–77. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- 104.Wade PA, Gegonne A, Jones PL, et al. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 1999;23:62–6. doi: 10.1038/12664. [DOI] [PubMed] [Google Scholar]
- 105.Zilberman D, Coleman-Derr D, Ballinger T, et al. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature. 2008;456:125–9. doi: 10.1038/nature07324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dantas Machado AC, Saleebyan SB, Holmes BT, et al. Proteopedia: 3D visualization and annotation of transcription factor-DNA readout modes. Biochem Mol Biol Educ. 2012;40:400–1. doi: 10.1002/bmb.20650. [DOI] [PubMed] [Google Scholar]
- 107.Barozzi I, Simonatto M, Bonifacio S, et al. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell. 2014;54:844–57. doi: 10.1016/j.molcel.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Felle M, Hoffmeister H, Rothammer J, et al. Nucleosomes protect DNA from DNA methylation in vivo and in vitro. Nucleic Acids Res. 2011;39:6956–69. doi: 10.1093/nar/gkr263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pennings S, Allan J, Davey CS. DNA methylation, nucleosome formation and positioning. Brief Funct Genomic Proteomic. 2005;3:351–61. doi: 10.1093/bfgp/3.4.351. [DOI] [PubMed] [Google Scholar]
- 110.Blattler A, Farnham PJ. Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem. 2013;288:34287–94. doi: 10.1074/jbc.R113.512517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Baubec T, Schübeler D. Genomic patterns and context specific interpretation of DNA methylation. Curr Opin Genet Dev. 2014;25:85–92. doi: 10.1016/j.gde.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 112.Lelli KM, Slattery M, Mann RS. Disentangling the many layers of eukaryotic transcriptional regulation. Annu Rev Genet. 2012;46:43–68. doi: 10.1146/annurev-genet-110711-155437. [DOI] [PMC free article] [PubMed] [Google Scholar]