

Abstract
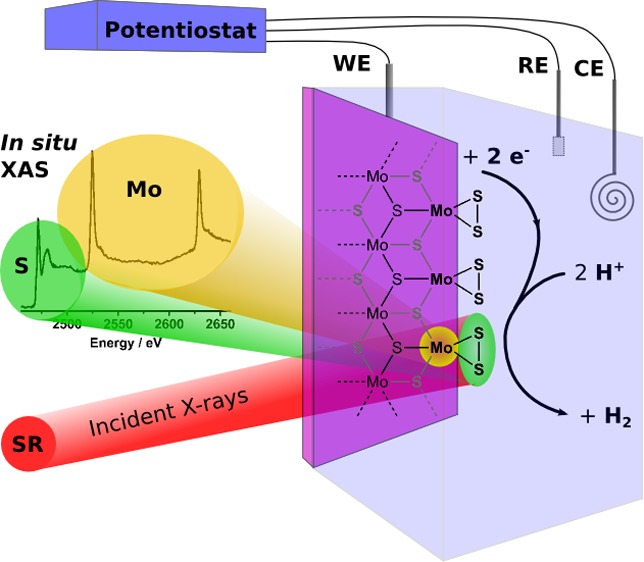
The reduction of protons into dihydrogen is important because of its potential use in a wide range of energy applications. The preparation of efficient and cheap catalysts for this reaction is one of the issues that need to be tackled to allow the widespread use of hydrogen as an energy carrier. In this paper, we report the study of an amorphous molybdenum sulfide (MoSx) proton reducing electrocatalyst under functional conditions, using in situ X-ray absorption spectroscopy. We probed the local and electronic structures of both the molybdenum and sulfur elements for the as prepared material as well as the precatalytic and catalytic states. The as prepared material is very similar to MoS3 and remains unmodified under functional conditions (pH = 2 aqueous HNO3) in the precatalytic state (+0.3 V vs RHE). In its catalytic state (−0.3 V vs RHE), the film is reduced to an amorphous form of MoS2 and shows spectroscopic features that indicate the presence of terminal disulfide units. These units are formed concomitantly with the release of hydrogen, and we suggest that the rate-limiting step of the HER is the reduction and protonation of these disulfide units. These results show the implication of terminal disulfide chemical motifs into HER driven by transition-metal sulfides and provide insight into their reaction mechanism.
1.0. Introduction
One of the most important challenges many societies are currently facing is the supply of fuel for their increasing energy needs.1,2 The extensive use of fossil fuels during the past century is reaching its limits, mostly because of the deleterious effects their combustion is causing to the environment. In this context, the search for alternative energy sources is of critical significance. Among various solutions, solar light is very important, because of its accessibility in most parts of the globe and the power it delivers on the surface of the earth. It is, however, only available during a certain portion of the day and in some places during certain times of the year. An optimal use of this energy source therefore requires its efficient storage. A very promising energy carrier that is able to fulfill this role is hydrogen.3 A tremendous amount of work has been carried out in the past decades to develop systems capable of sustainably producing hydrogen from water electrolysis.4−8 A class of compounds that meets both the technical and economical requirements of large-scale hydrogen production is molybdenum sulfides (MoSx).9 These inorganic materials have been extensively studied and used as natural gas hydrodesulfurization (HDS) catalysts.10,11 More recently, they have been shown to catalyze the hydrogen evolution reaction (HER) in their molecular,12−14 nanoparticulate15−18 and amorphous13,19−21 forms. In particular, some of us have reported on the preparation of electrodeposited amorphous MoSx thin films that are catalytically active for the reduction of protons into hydrogen in acidic water.19 These films have been extensively studied by X-ray photoelectron spectroscopy (XPS) to determine their electronic structure.22 Although very informative, this technique only probes the very first nanometers of a surface and is very difficult to perform under functional conditions. In the current paper, we investigate the structure of an amorphous, HER-active MoSx film observed under catalytic conditions using in situ X-ray spectroscopic techniques. An in situ electrochemical cell was developed and applied in the tender X-ray energy region (∼2–3 keV) to the study of an MoSx HER catalyst under functional conditions. This method allowed us to probe the local and electronic structure of the molybdenum centers and sulfur ligands of the film under precatalytic and catalytic conditions. By comparing the data obtained on the catalytic film poised at constant potentials with those of reference compounds (see Table 1), we propose a structural model for the catalyst film as prepared and under functional conditions in its precatalytic and catalytic states. The results provide evidence for the presence of terminal disulfide units in the MoSx film and demonstrate their involvement in the catalytic cycle. Although the role of disulfide units in the reduction of protons into dihydrogen by MoSx materials has often been suggested,16,23 it is, to the best of our knowledge, the first time that this chemical motif is experimentally observed during the HER. We also suggest on a spectroscopic basis that the rate-limiting step of the reaction is the reductive breaking of the S–S bond. These mechanistic insights will be important in designing hydrogen-evolving catalysts with better performances.
Table 1. Formal Oxidation States, Sulfur Chemical Units, and Structure of the MoSx Reference Compounds Used in This Study.
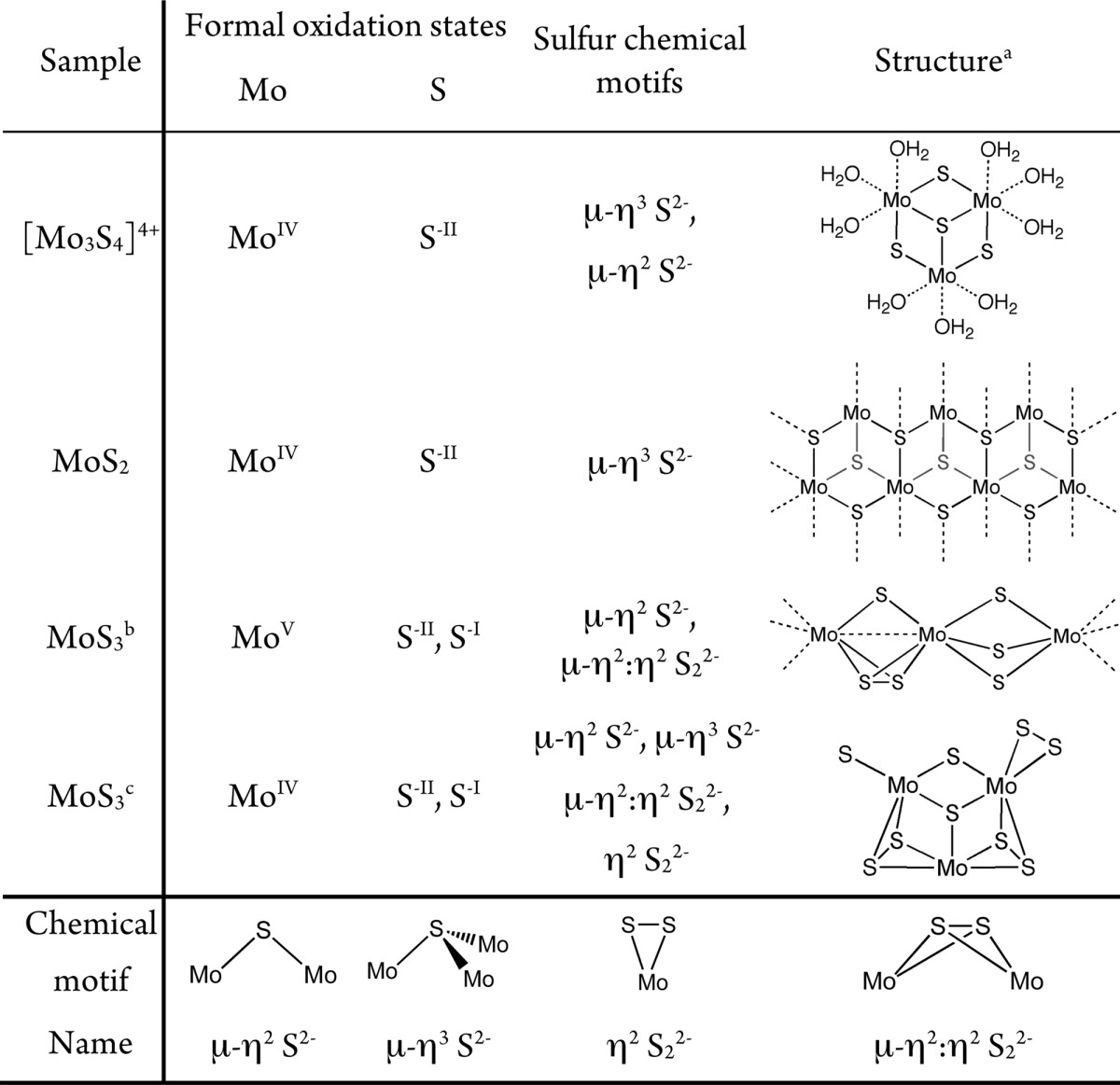
The structures of [Mo3S4(OH2)9]4+ and MoS2 have been determined previously by XRD.
Structural and electronic information according to the model of Hibble et al.24
2.0. Experimental Section
2.1. Model Compounds
HNO3 (90%), MoS2 (99%), MoS4(NH4)2 (>99.9%), and NaClO4 (>98%) were purchased from Aldrich, and MoS3·2H2O was purchased from Alfa-Aesar. [Mo3S4(H2O)9]Cl4 was synthesized according to a published procedure.26 All compounds were used as received without further purification. Deionized water was used for all experiments.
2.2. Electrodeposition of MoSx Electrocatalyst
Prior to electrodeposition of the MoSx catalyst film, a 500 nm thick silicon nitride (Si3N4, purchased from Silson Ltd.) membrane was sputter coated with a 150 nm layer of indium-doped tin oxide (ITO) to produce a suitable X-ray transparent conductive surface. The ITO surface was connected from the front with copper tape to ensure electrical contact. The deposition of MoSx was carried out in a glovebox under nitrogen. The ITO electrode was immersed into a 2 mM solution of (NH4)2MoS4 in 0.1 M NaClO4 in water (8 mL). Consecutive cyclic voltammograms (typically 25) were carried out using an Ivium Stat potentiostat (Ivium Technologies) with a saturated silver/silver chloride reference electrode (separated by a porous Vycor tip) and a titanium wire counter electrode. The cyclic voltammograms were performed between +0.7 and −0.4 V vs reversible hydrogen electrode (RHE) and a scan rate of 0.05 V/s was employed. The modified electrode was rinsed with distilled water.
2.3. X-ray Absorption Data Collection
X-ray absorption data were collected at the Stanford Synchrotron Radiation Lightsource (SSRL) on beamline 7-3 (Mo K-edge) and 4-3 (S K-edge and Mo L-edge) at an electron energy of 3.0 GeV with an average current of 300 mA. At beamline 7-3, the radiation was monochromatized by a Si(220) double-crystal monochromator. The intensity of the incident X-ray was monitored by an Ar-filled ion chamber (I0) in front of the sample. Solid samples were diluted in boron nitride (1% w/w) and placed in an aluminum sample holder sealed with kapton tape. Data were collected as fluorescence excitation spectra with a Ge 30 element detector (Canberra). Energy was calibrated by the first peak maximum of the first derivative of a molybdenum foil (20003.9 eV), placed between two Ar-filled ionization chambers (I1 and I2) after the sample. All data were collected at room temperature. At beamline 4-3, the incoming X-rays were monochromatized by a Si(111) double-crystal monochromator. The intensity of the incident X-rays was monitored by a He-filled ion chamber (I0) in front of the sample. Solid sample spectra were collected from a thin layer of the sample smeared on sulfur-free tape. Data were collected as fluorescence excitation spectra with a Vortex 4 element silicon drift detector (SII NanoTechnology). Monochromator energy was calibrated to the first peak of thiosulfate reference sample, which was assigned at 2470.8 eV. The sample environment was kept under He gas atmosphere with an He-filled bag to reduce air absorption of incoming X-rays and fluorescence signals. The data were collected at room temperature. Additional data on reference samples were also collected at the LUCIA Beam line at SOLEIL, at an electron energy of 2.7 GeV and an average ring current of 430 mA. The incoming photons were selected with a Si (111) double crystal monochromator. Samples were compressed as pellets in 1:10 (w/w) mixture with cellulose and fixed on a copper sample holder with conductive carbon tape. The S K-edge and Mo L-edges were measured as fluorescence spectra with a grazing angle (<2°) for the outgoing photons in order to avoid self-absorption phenomena. Energy ranges for the first moment calculation are as follow: S K-edge: 2465–2475 eV; Mo L3-edge: 2520–2535 eV; Mo L2-edge: 2623–2640 eV.
2.4. In Situ Electrochemical XAS Measurements
A glass electrochemical cell consisting of two compartments separated by a porous frit was employed for in situ spectroscopic experiments, as previously reported.27,28 The working compartment has flat walls (∼1.5 cm wide) with a single circular hole of 0.8 cm in diameter. An MoSx-coated ITO/Si3N4 membrane as described above was in contact with a slip of copper tape and fixed with epoxy glue to the exterior of the wall of the cell, over the 0.8 cm hole, with the MoSx layer facing inward. A nitric acid solution adjusted to pH 2 was placed in both compartments. The solutions were not stirred during the experiment. The cell was connected to a potentiostat by making electrical contact to the copper tape slip that protruded from the side of the working compartment. A Teflon cap fitted with a reference electrode (Ag/AgCl) was used to cover the working compartment and to ensure a fixed distance between working and reference electrodes for all experiments. A platinum gauze was placed in the second compartment separated by a frit from the first one and used as a counter electrode. X-ray absorption spectra were recorded at different positions on the electrode to check the materials for homogeneity. The same electrodes were used to measure the Mo K-edge (BL 7-3) and L-edges (BL 4-3) and the S K-edge (BL 4-3) spectra. For Mo K-edge measurements, the spectra were recorded in air. For S K-edge and Mo L-edge measurements, the electrochemical cell was placed in a He-filled bag with a polypropylene membrane placed as close as possible to the Si3N4 membrane to maximize the penetration of X-rays into and from the MoSx film. Spectra were recorded on the dry electrodeposited MoSx films at first and then in pH 2 nitric acid solution at 0.3, 0.1, −0.1, and −0.3 V vs RHE. All references hereafter are given vs RHE. The experiments were conducted at pH = 2 since the ITO layer deposited on the Si3N4 membrane could not withstand lower pH values. The open-circuit potential measured prior to applying a potential was 0.8 V. At each potential, 10 scans were recorded for the Mo K-edge and 4 scans for the S K-edge and Mo L-edge. After each potential change, the system was allowed to equilibrate for 10 min before recording a spectrum. No noticeable change was observed between the first and last spectra recorded at a given potential. The current density measured for the potentials of interest (see Figure S1) was similar to those recorded on a regular ITO electrode under identical conditions (room temperature, pH 2 in nitric acid). A polarization curve of the film on a rotating disk carbon electrode at pH = 2 is also shown in Figure S2.
2.5. XAS Data Reduction and Analysis
Data reduction of the Mo K-edge extended X-ray absorption fine structure (EXAFS) spectra was performed using EXAFSPAK (Drs. Graham George and Ingrid Pickering, SSRL). Pre-edge and post-edge backgrounds were subtracted from the XAS spectra, and the results were normalized with respect to edge height. Background removal in k-space was achieved through a five-domain cubic spline. Curve fitting was performed with Artemis and IFEFFIT software using ab initio calculated phases and amplitudes from the program FEFF 8.2.29,30 The details of curve fitting are discussed in the SI. For the S K-edge spectra, an E0 value of 2470.8 eV was used, and normalization was performed over the range 2488.8–2510.8 eV. For the Mo L3-edge and L2-edge, E0 values of 2523.6 and 2627.0 eV were used, and normalization was performed over the ranges 2545.3–2591.6 eV and 2648.7–2666.7 eV, respectively.
3.0. Results
We use the notation precatalytic and catalytic states for the MoSx film poised at 0.3 and −0.3 V in a nitric acid solution at pH = 2, respectively. These states were measured in situ, while the as prepared film was measured ex situ.
3.1. MoSx Film Deposition
The deposition and characterization of MoSx films under various conditions were described in a recent publication.19 The as prepared MoSx film studied in this work corresponds to the MoS3-CV film before activation in ref (19).
3.2. Mo K-edge XANES of MoSx Films and Reference Compounds
The films deposited did not show any X-ray diffraction (XRD) signal, which indicates that they are amorphous. We therefore used X-ray absorption spectroscopy to study the structure of these films. The Mo K-edge X-ray absorption near edge spectroscopy (XANES) spectra were recorded ex situ on the MoSx film as prepared and in situ in the precatalytic and catalytic states (Figure 1A) using a custom-made X-ray spectroelectrochemical cell described in the Electrodeposition of MoSx Electrocatalyst Section. For comparison, the XANES spectra of MoS3, MoS2, and [Mo3S4(H2O)9]Cl4 are shown in Figure 1B. No clear trend reflecting the formal oxidation state of Mo is observed in the Mo rising edge energy of the reference compounds, which is largely due to the charge delocalization between the molybdenum ion and the sulfur ligands. For the MoSx film, changes at the peak top are observed, while the rising edge energy remains identical when changing the potential applied to the film. None of these spectra present a good match with those of the reference compounds, suggesting a mixed composition for the MoSx film.
Figure 1.
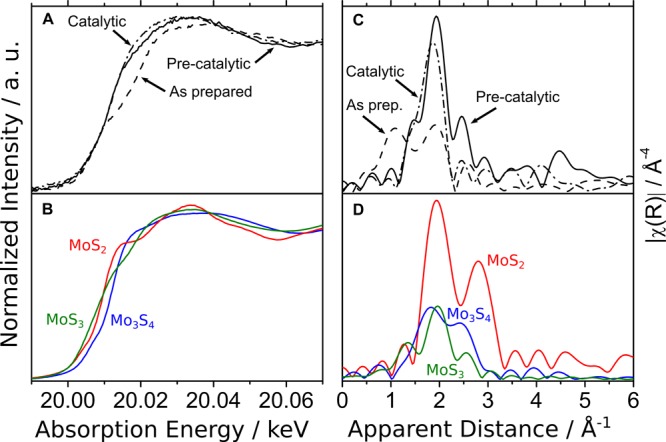
(A) Mo K-edge XANES spectra of the MoSx film as prepared and in the precatalytic and catalytic states. (B) Mo K-edge XANES spectra of Mo3S4, MoS2 and MoS3. (C) Mo K-edge Fourier transform EXAFS (k3-weighted) of the MoSx film as prepared and in the precatalytic and catalytic states. (D) Mo K-edge Fourier transform EXAFS (k3-weighted) of Mo3S4, MoS2, and MoS3.
3.3. Mo K-edge EXAFS of MoSx Films and Reference Compounds
The Mo K-edge EXAFS spectra were recorded ex situ on the MoSx film (as prepared) and in situ in the precatalytic and catalytic states (Figure 1C). For comparison, the EXAFS spectra of the MoS3, MoS2, and [Mo3S4(H2O)9]Cl4 are shown in Figure 1D. The k-space spectra of these compounds are also compared on Figure S3. The EXAFS spectrum of as prepared film shows two peaks at R′ = 1.2 and 1.9 Å (R′ being the apparent distance and R the actual distance), corresponding to Mo–O and Mo–S interactions, respectively (vide infra). When poised at 0.3 V, the short-distance peak (R′ = 1.2 Å) disappears, and the spectrum of the precatalytic state shows similarities with that of MoS3, with a high intensity peak at ca. R′ = 2.0 Å and a small intensity peak at ca. R′ = 2.5 Å. Under catalytic conditions, the main peak shifts to a lower apparent distance (R′ = 1.86 Å), suggesting a shortening of the Mo–S bond. In addition, the small intensity peak at R′ = 2.5 Å disappears, suggesting the breaking of the Mo–Mo bond.
Prior to fitting the EXAFS spectra of MoSx films, the EXAFS spectra of three model compounds were fit using structural data derived from their crystal structures to extract the EXAFS fitting parameters. The final fitting parameters are listed in Table 2. In the case of MoS3, two distances are required to fit the spectrum, one accounting for 6 Mo–S bonds at 2.42 Å and another one for 1 Mo–Mo bond at 2.77 Å. Two competing structural models have been proposed for MoS3 by Hibble et al.24,31 and Weber et al.25 The Hibble structure for MoS3 consists of chains of molybdenum ions alternatively bridged by three sulfides (μ-η2 S2–) or one sulfide and one disulfide (μ-η2:η2 S22–) ligands (see Table 1).32 The formula for this material is thus MoV(S2–)2(S22–)0.5. Several authors33,34 have shown that the EXAFS spectrum of MoS3 is indeed best fit with a single Mo–Mo interaction because Mo ions only interact with neighboring Mo through the short bridge containing a disulfide unit but not through the long one containing sulfide groups only. The Weber structure consists of a random arrangement of four isostructural Mo3S9 clusters, which are linked to each other by one or two μ-η2 sulfide bridge(s). Each cluster contains three sulfide and three disulfide ligands, which can be either bridging or terminal. The formulation for this structure is thus MoIV(S2–)(S22–). The fitting parameters are in agreement with the literature for both proposed structures, and we cannot discriminate one from the other. In the Weber structure, each molybdenum center is surrounded by 5, 6, or 7 sulfur ligands. This is compatible with the quite large Debye–Waller factor (0.010) found for the Mo–S interaction in the MoS3 EXAFS fit (see Table 2). The spectrum of MoS2 (which is microcrystalline) shows two well-separated peaks at ca. R′ = 1.95 and R′ = 2.80 Å that can be fit with Mo–S and Mo–Mo interactions at 2.41 and 3.17 Å with N values of 6 for each interaction. These values are in very good agreement with the XRD distances reported for MoS2 (2.40 and 3.15 Å).35 The spectrum of [Mo3S4 (OH2)9]4+ shows two peaks at ca. R′ = 1.80 and R′ = 2.40 Å. The low R′ peak was fit with Mo–O and Mo–S interactions at 2.13 and 2.23 Å, respectively, with N values of 3 for both interactions. The higher R′ peak is due to a single Mo–Mo vector at R = 2.72 Å with an N value of 2. These values are in very good agreement with the XRD structure of [Mo3S4(H2O)9]Cl4.36
Table 2. EXAFS Fitting Parameters for MoS3, MoS2, Mo3S4, and the MoSx Film As Prepared, Under Pre-Catalytic (0.3 V), and Catalytic State (−0.3 V)a.
|
R, Å |
||||||
|---|---|---|---|---|---|---|
| sample | shell | XRD | EXAFS | N | σ2 | R, % |
| MoS3b | Mo–S | – | 2.42 | 6 | 0.010 | 0.06 |
| Mo–Mo | – | 2.77 | 1 | 0.005 | ||
| MoS2c | Mo–S | 2.41 | 2.41 | 6 | 0.001 | 0.4 |
| Mo–Mo | 3.15 | 3.17 | 6 | 0.003 | ||
| Mo3S4d | Mo–O | 2.18 | 2.13 | 3 | 0.002 | 1.3 |
| Mo–S | 2.30 | 2.23 | 3 | 0.010 | ||
| Mo–Mo | 2.73 | 2.72 | 2 | 0.003 | ||
| MoSx as preparede | Mo–O | – | 1.74 | 1.2 | 0.002 | 0.01 |
| Mo–S | – | 2.44 | 3.0 | 0.010 | ||
| Mo–Mo | – | 2.76 | 0.8 | 0.005 | ||
| MoSx 0.3 Vb | Mo–S | – | 2.40 | 6.4 | 0.010 | 1.6 |
| Mo–Mo | – | 2.74 | 1.1 | 0.003 | ||
| MoSx 1 | Mo–S | – | 2.36 | 6.2 | 0.010 | 2.2 |
| -0.3 Vb 2 | Mo–S | – | 2.37 | 6.1 | 0.010 | 2.1 |
| Mo–Mo | – | 2.69 | 0.2 | 0.003 | ||
Bold numbers indicate fixed values. R is the apparent distance in Å from the central atom to the scatterer, k is the wave vector describing the trajectory of the scattered photoelectron, N is the number of scatterers, R2 is the Debye–Waller factor, and R is the goodness of fit in %.
Fitting range: 3.10 ≤ k (/Å) ≤ 11.5 and 1.57 ≤ R (Å) ≤ 2.82.
Fitting range: 3.10 ≤ k (/Å) ≤ 11.5 and 1.15 ≤ R (Å) ≤ 3.20.
Fitting range: 3.10 ≤ k (/Å) ≤ 11.5 and 1.15 ≤ R (Å) ≤ 2.82.
Fitting range: 3.10 ≤ k (/Å) ≤ 10.5 and 1.15 ≤ R (Å) ≤ 2.82.
The EXAFS curve fitting results of the MoSx film as prepared and in the precatalytic and catalytic states are summarized in Table 2, and the k-space curves are displayed in Figure S4. For the as prepared film, an Mo–O interaction with a distance of R = 1.74 Å and an N value of 1.2 had to be included to model the peak at short R′ value. The peak at R′ = 1.90 Å was fit with an Mo–S interaction (R = 2.44 Å, N = 3.0), while the low intensity one at R′ = 2.61 Å was fit with an Mo–Mo interaction (R = 2.76 Å, N = 0.8). For the film in the precatalytic state (poised at 0.3 V), the best fit was obtained with an Mo–S interaction at 2.40 Å with an N value close to 6 and an Mo–Mo interaction at 2.74 Å with an N value of 1.1. Under these conditions, the parameters of the Mo–S and Mo–Mo interaction are in good agreement with those of MoS3. We therefore considered the film in the precatalytic state to be predominantly composed of MoS3. When the potential was lowered to −0.3 V (catalytic state), the Mo–Mo peak is barely present in the EXAFS spectrum. Fitting the data with a single shell (fit #1), yields an Mo–S interaction with R = 2.36 Å and N = 6. This distance is slightly shorter (0.05 Å) than that of the film poised at 0.3 V. We also considered the presence of an Mo–Mo second shell (fit #2). In this fit, the N value for the Mo–Mo interaction dropped to 0.2, while it did not show a significant improvement in the fit quality. We therefore conclude that the Mo–Mo bond is significantly diminished in the MoSx film in the catalytic state.
3.4. In Situ Sulfur K-edge XANES
Since the sulfur K-edge energy (ca. 2472 eV) is just below that of the molybdenum L-edges (ca. 2526 and 2631 eV for the L3- and L2-edges, respectively), we recorded spectra with both edges in one single scan (see Figure S5). Figure 2A shows the S K-edge spectra collected on the MoSx film as prepared and in the precatalytic and catalytic states as well as the reference compounds listed in Table 1. The spectrum of Mo3S4 shows several features, with a main peak at 2471.6 eV (measured at peak top), a shoulder at 2472.8 eV, and an intense low energy peak at 2469.8 eV. In MoS2, a single, sharp peak is observed at ca. 2471.4 eV, and a shoulder is observed at 2473.6 eV. The spectrum of MoS3 shows a single, broad feature at 2472.5 eV.
Figure 2.
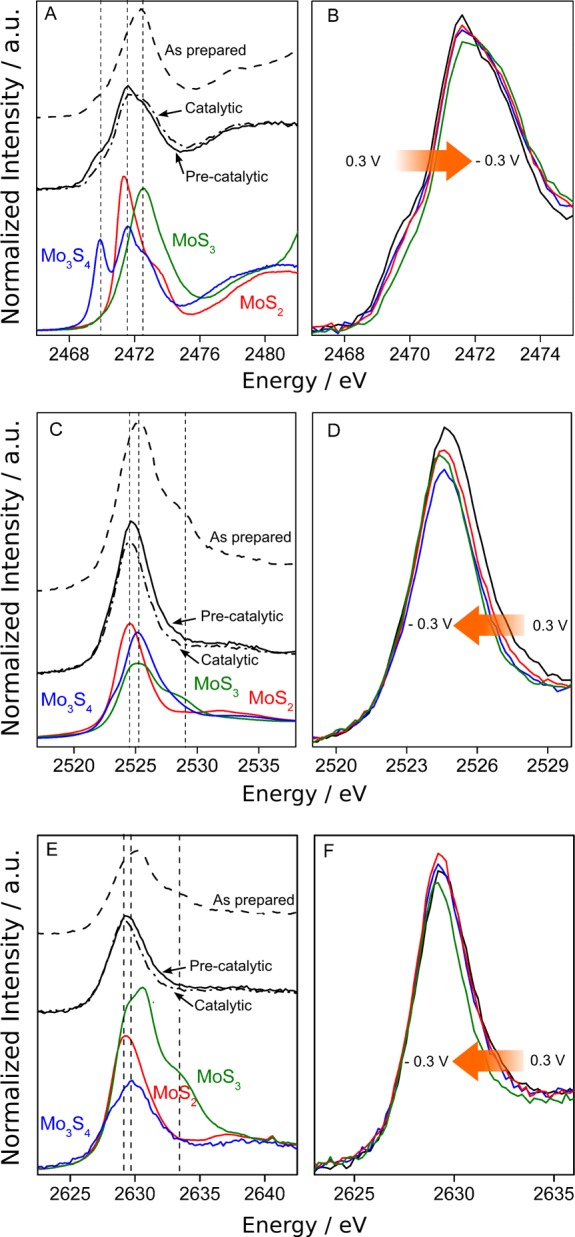
Top: Sulfur K-edge spectra of (A) the MoSx film as prepared, in the precatalytic and catalytic states, together with Mo3S4, MoS2 and MoS3 reference spectra, and (B) the MoSx film poised at 0.3 (black), 0.1 (blue), −0.1 (red), and −0.3 V (green) in nitric acid at pH = 2. Middle: Molybdenum L3-edge spectra of (C) the MoSx film as prepared, in the precatalytic and catalytic states, together with Mo3S4, MoS2, and MoS3 and (D) the MoSx film poised at 0.3 (black), 0.1 (blue), −0.1 (red), and −0.3 V (green) in nitric acid at pH = 2. Bottom: Molybdenum L2-edge spectra of (E) the MoSx film as prepared, in the precatalytic and catalytic states, together with Mo3S4, MoS2, and MoS3 and (F) the MoSx film poised at 0.3 (black), 0.1 (blue), −0.1 (red), and −0.3 V (green) in nitric acid at pH = 2.
The S K-edge spectra of the MoSx film poised at 0.3, 0.1, −0.1, and −0.3 V are shown in Figure 2B, and the corresponding first moment energies are listed in Table 3. In the as prepared MoSx film, the sulfur K-edge main peak energy (2472.1 eV) and shape are similar to that of MoS3, although a shoulder is present at low energy (2469.7 eV) as in Mo3S4, but with a much smaller intensity. When the sample is set in a pH 2 nitric acid solution and the potential is poised to 0.3 V, the first moment energy of the main peak remains at 2472.0 eV, while the shoulder at lower energy (2469.8 eV) increases and the one at higher energy (2472.4 eV) decreases. When the potential is gradually decreased to −0.3 V, the first moment energy of the main peak shifts to a slightly higher energy (from 2472.0 to 2472.3 eV), which indicates an overall oxidation of the sulfur ligands. Concomitantly, the lower energy shoulder that is present in Mo3S4 decreases.
Table 3. Sulfur K-Edge and Molybdenum L2,3-Edge Peak First Moment Energies (in eV) for the MoSx Film As Prepared and Poised at 0.3, 0.1, −0.1, and −0.3 V in pH 2 Nitric Acid.
| Mo |
|||
|---|---|---|---|
| sample | S K-edge | L3-edge | L2-edge |
| as prepared | 2472.15 | 2526.95 | 2631.43 |
| 0.3 V | 2472.00 | 2526.54 | 2631.00 |
| 0.1 V | 2472.10 | 2526.51 | 2630.90 |
| –0.1 V | 2472.13 | 2526.49 | 2630.86 |
| –0.3 V | 2472.27 | 2526.48 | 2630.80 |
3.5. In Situ Molybdenum L-edge XANES
Figure 2C shows the molybdenum L3-edge spectra recorded on the MoSx film as prepared and in the precatalytic and catalytic states as well as the reference compounds listed in Table 1. The Mo L3-edge spectrum of Mo3S4 shows one main, broad feature, centered at 2525.1 eV (measured at peak top), with a shoulder at 2523.25 eV. The spectrum of MoS2 shows a single feature at 2524.7 eV, while that of MoS3 has a main peak at 2525.8 eV with a shoulder at higher energy (2528.4 eV). The spectrum recorded on the as prepared MoSx film is very similar to that of MoS3. For instance, it has a shoulder at 2528.7 eV, which is a strong component of the MoS3 spectrum. This is even more visible in the L2-edge spectrum (see Figure 2E), where the as prepared spectrum shows the double peak feature (2629.3 and 2629.6 eV) that is also present in MoS3.
The Mo L3-edge spectra of the MoSx film poised at 0.3, 0.1, −0.1, and −0.3 V are shown in Figure 2D, and the corresponding first moment energies (as well as those of the Mo L2-edge spectra) are reported in Table 3. Under precatalytic conditions (pH 2 nitric acid, 0.3 V), the L3-edge peak first moment energy shifts to a lower energy by ca. 0.4 eV as compared to the as prepared film, and the higher energy component (2528.4 eV) characteristic of MoS3 disappears. When progressively decreasing the potential to −0.3 V, the first moment energy of the Mo L3-edge peak shifts to lower values by ca. 0.06 eV, indicating a reduction of the Mo center. The same trend is observed for the L2-edge part of the spectra (see Figure 2D) with an even more pronounced shift of the first moment to lower energies (by ca. 0.2 eV) as the potential decreases.
4.0. Discussion
The MoSx films studied in this paper were prepared by electrochemical cycling of an ITO electrode in an [NH4]2MoS4 aqueous solution. This procedure was shown to involve anodic and cathodic deposition processes, as well as cathodic corrosion. Eventually, the final composition of the film was estimated from XPS and Electrochemical Quartz Crystal Microbalance (EQCM) analysis as a mixture of MoS2 and MoS3 phases.22 In line with this first analysis, the X-ray spectroscopic data of the as prepared sample presented in this paper show features associated with several compounds, clearly indicating that the MoSx electrocatalyst is a mixture of species. Table 4 summarizes the spectroscopic information collected on the as prepared, precatalytic and catalytic states. It also indicates the structural and electronic changes observed when the potential is gradually decreased from 0.3 (precatalytic state) to −0.3 V (catalytic state).
Table 4. Summary of the Spectroscopic Features Observed for the MoSx Film As Prepared and Under Pre-Catalytic (0.3 V) and Catalytic (−0.3 V) Conditions.
| Mo K-edge
EXAFS |
S K-edge XANES | Mo L-edge XANES | ||
|---|---|---|---|---|
| sample | number and nature of interaction | R, Å | sulfur oxidation state and binding mode | formal oxidation states of molybdenum |
| dry (as prepared) | 1 Mo–O | 1.74 | μ-η2 S2–, μ-η3 S2–, μ-η2:η2 S22– | Mo(V) or Mo(IV)a |
| 3 Mo–S | 2.44 | |||
| 1 Mo–Mo | 2.75 | |||
| ↓ | shortening of Mo–S bond, increased number of Mo–Mo bonds | reduction | reduction | |
| 0.3 V (precatalytic) | 6 Mo–S | 2.40 | μ-η2 S2–, μ-η3 S2–, μ-η2:η2 S22– | Mo(IV) |
| 1 Mo–Mo | 2.74 | |||
| ↓ | shortening of Mo–S bond, diminished Mo–Mo interactions | oxidation | reduction | |
| –0.3 V (catalytic) | 6 Mo–S | 2.36 | μ-η2 S2–, μ-η3 S2–, η2 S22– | Mo(IV), Mo(III) |
In MoS3, the molybdenum ion is described as Mo(V) in the Hibble model and as Mo(IV) in the Weber model.
4.1. Initial Structure of the as prepared MoSx Film
In the Mo K-edge EXAFS Fourier transform spectrum of the as prepared sample, the peak observed at low R′ values (1.05 Å) indicates the presence of an Mo–O interaction. The Mo–O distance obtained from the fits (R = ca. 1.75 Å) is much shorter than usually observed in most MoxOySz oxysulfide compounds (typically 2.0 ≤ R ≤ 2.2 Å)37 or in Mo3S4(OH2)9 (vide supra), and is longer than the Mo–O distances observed in molybdenum oxides.38,39 The presence of oxygen has also been observed previously in the XPS spectra recorded ex situ on the as prepared MoSx film.22 Given that the latter technique is only sensitive to the surface, we estimated that a layer of molybdenum oxide MoOx covers the bulk MoS3 material in the as prepared film, as a result of air oxidation during transfer from the synthesis laboratory to the synchrotron facility. Since the film is highly porous, the actual surface exposed to air and converted into an oxide could be significant. The EXAFS fits indicate the presence of 3 Mo–S interactions at R = 2.44 Å and one Mo–Mo interaction at R = 2.76 Å. All parameters corresponding to an MoS3 structure, except for the lower number of Mo–S vectors.
This deviation can be explained by the presence of the outer-shell MoOx layer in the as prepared film that decreases the average Mo–S bonds per Mo center. This analysis is consistent with the previous XPS observations showing the presence of Mo(IV) and of S2– and S22– ligands.8 The presence of peaks characteristic of MoS3 at 2528.7 and 2633.5 eV in the Mo L3- and L2-edge spectra, respectively, confirms an MoS3 structure. The small intensity feature at 2470 eV that is characteristic of Mo3S4 suggests the presence of similar chemical motifs in the as prepared material. Indeed, the Mo3S9 trinuclear units proposed by Weber as constituents of MoS3 are closely related to Mo3S4, since they share the same geometrical arrangement as well as μ-η2 and μ-η3 bridging S2– ligands. It is, however, very difficult to distinguish small contributions of Mo3S4 within the MoS3 K-edge EXAFS, since the Mo–S and Mo–Mo distances of these two materials are almost identical (see Table 2). The starting material is thus best described as an amorphous film of MoS3 in the bulk, with a surface layer of MoOx. The oxidation state of molybdenum centers is predominantly +IV, while the sulfur ligands are present as bridging sulfides (μ-η2 and μ-η3 S2–) and disulfides (μ-η2:η2 S22–).
4.2. Structure of the MoSx Film in its Precatalytic State
When the MoSx film is set in a nitric acid solution at pH = 2 and poised at 0.3 V, the Mo–O interaction in the Mo K-edge EXAFS spectrum of the as prepared material disappears, indicating that the molybdenum oxide fraction is hydrolyzed. This phenomenon was already observed by XPS on the surface of a similar system.22 The current data extend these structural considerations to the bulk of the material. Under precatalytic conditions, the EXAFS fits indicate 6 Mo–S interaction at R = 2.40 Å and 1 Mo–Mo interactions at R = 2.74 Å, which correspond to the fitting parameters of MoS3. However, the Mo L2,3-edge and S K-edge XAS show that the material formed cannot be considered as pure MoS3. In the S K-edge spectrum, the shoulder at 2470 eV characteristic of Mo3S4 is more intense than in the as prepared material, which suggests a larger fraction of trinuclear Mo3Sx units as compared to the as prepared material. Hence, the overall S K-edge first moment energy is shifted to lower energy by 0.15 eV, indicating a reduction of the disulfide units originally present in the as prepared material. The features characteristic of MoS3 in the Mo L3- and L2-edge spectra (at 2528.7 and 2633.5 eV, respectively) disappeared, and the first moment of these peaks are shifted to lower energy by ca. 0.4 eV, indicating a reduction of the molybdenum centers as well. These data indicate that, when placed in an acidic media, the as prepared material is reduced, both from the sulfur and the molybdenum side, into an amorphous material where molybdenum and sulfur are in the +IV and −II oxidation state, respectively. Although MoS2 fits best with this description, the spectral features of the S K and Mo L2,3-edge XAS suggest that a fraction of MoS3 and/or Mo3S4 is still present. It is likely that the outer shell, which is accessible to the solvent, is converted into MoS2. We suggest that the reduction under acidic conditions of the disulfide ligands in MoS3 produces sulfide ligands, which in turn displaces the oxygen ligands in MoOx to yield MoS2. We note that the strong peak at R = 3.15 Å corresponding to the Mo–Mo interaction in microcrystalline MoS2 is not observed, which can be explained by the amorphous nature of the MoS2 layer.
4.3. Structure of the MoSx Film in its Catalytic State
When the potential is decreased to −0.3 V, the Mo–Mo interaction observed at ca. R = 2.75 Å in the Mo K-edge EXAFS diminishes. The disappearance of this peak, corresponding to Mo–Mo interactions in Mo3S4 and MoS3, can be interpreted as the formation of MoS2 from MoS3 and the corresponding disappearance of the bridging disulfide units in the bulk of the film. This hypothesis is supported by the Mo–S distance at −0.3 V, which is longer than that in Mo3S4 and matches the one found in MoS2. As the potential is decreased from 0.3 to −0.3 V, the S K-edge XANES spectra of the MoSx film shows a progressive increase in their first moment energy (Figure 2B), which indicates an overall oxidation of the sulfur ligands. The difference in peak position between the highest and lowest potentials is only of 0.27 eV (compared to the 1.1 eV shift observed in the sulfur oxidation state change from MoS2 to MoS3), indicating a partial fraction of sulfur being oxidized in the sample. This small shift can be explained by a modification of the material only at the surface in contact with the electrolyte. Since the Mo–Mo interaction disappears in the Mo K-edge EXAFS, the increase in the oxidation state of the sulfur ligands is likely due to the presence of terminal disulfide ligands (η2 S22–) rather than bridging ones (μ-η2:η2 S22–). When the potential is decreased from 0.3 to −0.3 V, the Mo L-edge first moment concomitantly shifts to lower energies (−0.06 and −0.2 eV for the Mo L3- and L2-edges, respectively), which indicates that the molybdenum centers are reduced to a lower oxidation state. As for the S K-edge XANES, the energy shift is limited as compared to what is observed for pure, bulk compounds (−0.48 and −0.55 eV from MoS3 to MoS2 for the L3- and L2-edges, respectively). Namely, the S K-edge and Mo L-edge shifts are correlated to each other, and it suggests a change in oxidation state for the molybdenum centers located only at the solid–liquid interface.
In summary, the material’s bulk structure at −0.3 V is modified from amorphous MoS3 to amorphous MoS2+x, in which bridging disulfides are absent and the predominant oxidation state of molybdenum is +IV. As mentioned before, the small changes observed at the S K- and Mo L-edges strongly suggest that these changes occur at the solid–liquid interface only, where the MoSx material is in contact with the acidic media. At −0.3 V, this interface presents a significant amount of oxidized sulfur ligands (as terminal disulfide units) as well as reduced molybdenum (formally Mo(III)) centers. These conclusions are in line with the previous XPS and electrochemical quartz crystal microbalance (EQCM) analysis of the same film after the first reductive voltametric scan.22 According to these techniques, which probe the surface and mass composition of the film, the surface of the material was converted upon immersion in acid from MoS3 to a material very similar to MoS2, with a slight excess of sulfur ligands. The material was thus formulated as MoS2+x, which corresponds to a loss of sulfur ligands. We did not, however, observe any decrease in the sulfur X-ray fluorescence intensity when the potential was reduced from +0.3 to −0.3 V. This indicates that the loss of sulfur occurs before setting the potential to the precatalytic conditions.
4.4. Implications for the Hydrogen Evolution Mechanism
Scheme 1 shows the proposed phase changes of the MoSx film under the various conditions studied in this paper, and proposes catalytic intermediates for the proton reduction reaction. The Cat species is the one predominantly observed at −0.3 V, while Cat–H stands for a putative species formed during the HER catalytic cycle. It is important to note that the species observed spectroscopically at a given potential represent the kinetically most populated state under these conditions. Therefore, in the case of a catalytic reaction, it also probes the rate-limiting step of the process, which involves the disappearance of the species observed spectroscopically.
Scheme 1. Proposed Changes and Catalytic Cycle for the MoSx Film As Prepared and at pH = 2 under Pre-Catalytic and Catalytic Conditions.
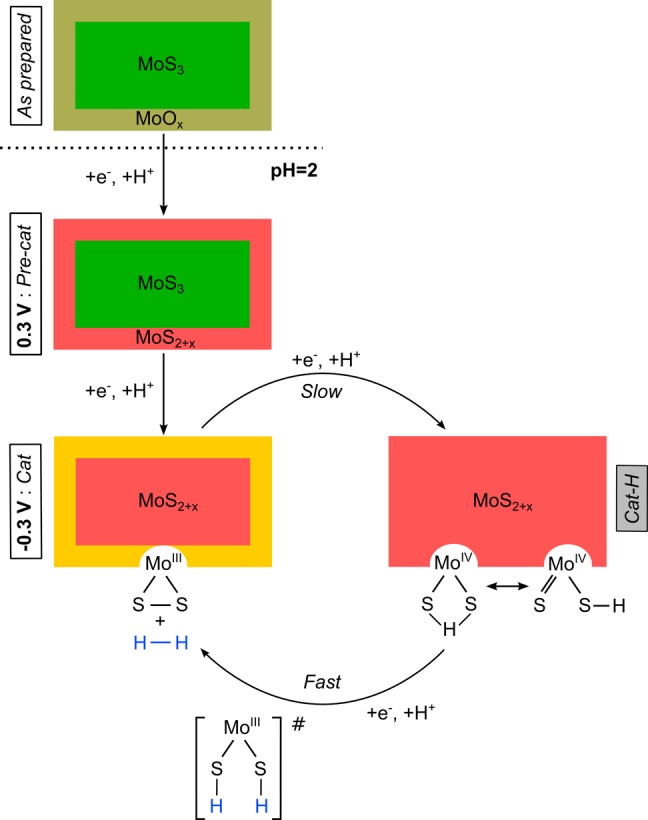
The Cat–H species is putative and has not been observed experimentally.
In the MoSx film, the interfacial Mo centers are in the +IV oxidation state when the potential is poised to 0.3 V, and the sulfur is protonated due to the highly acidic conditions. Since the potential is maintained at a quite high value, no hydrogen evolution is observed. When the potential is decreased, the driving force for the reduction of MoS3 into Cat increases, leading to the reduction of the molybdenum centers from +IV to +III, the release of dihydrogen and the formation of terminal disulfide units. Although the Cat species may not be stable in the long-term under such acidic and reductive conditions (the terminal disulfides are likely to be reduced and/or protonated), it is longer-lived than Cat–H, which is consumed as soon as it is reformed. Consequently, the rate-limiting step under HER conditions appears to be the protonation and reduction of the interfacial MoIII(S2) units formed together with the release of H2.
The addition of a hydrogen atom to Cat–H is therefore favored (and thus faster) than it is to Cat, which accumulates and can be observed spectroscopically. DuBois et al. have also proposed12,40 that dimeric, bis-μ-hydrosulfido Mo2(III) complexes can eliminate H2 when reacting with alkenes or alkynes in 2 + 2 additions. This type of reaction parallels the HER studied here, where electrons are provided by the electrode in place of the unsaturated olefins. By analogy with this addition–elimination mechanism and on the basis of our own observations, we suggest that the last intermediate before the release of H2 is an Mo(III) species with two terminal hydrosulfide ligands, generated by the facile addition of a hydrogen atom to the MoIV(S)(SH) species in Cat–H.
5.0. Conclusion
By combining in situ X-ray spectroscopies with electrochemistry, we have investigated the structural changes that occur in an MoSx hydrogen-evolving electrocatalyst under functional conditions. The starting material before catalysis is identified as predominantly MoS3. When set in the acidic conditions used for catalysis, the material remains essentially the same, except for the outermost layer. As the potential is decreased to induce the production of H2, a progressive reduction of the molybdenum centers is observed concomitantly with the oxidation of the sulfur ligands. We therefore suggest that Mo(III) units with terminal disulfide ligands (η2 S22–) are transiently formed at the interface with the solution when H2 is released. As a corollary, the catalytic species prior to the release of H2 is very similar to MoS2, with terminal hydrosulfide ligands at the interface with the liquid. Following this analysis, we propose that the rate-limiting step under HER conditions is the protonation and reduction of interfacial MoIII(S2) sites. These results show experimental evidence for the direct involvement of a disulfide unit in molybdenum sulfide-based HER and proposes new hypotheses for the mechanism of hydrogen evolution in these materials. Further investigations are currently underway to understand in more detail the structure of these films under functional conditions and their reaction mechanism.
Acknowledgments
Dr. Delphine Vantelon of the LUCIA beamline at SOLEIL is greatly acknowledged for the collection of S K-edge and Mo L-edge spectra on reference samples. XAS data collection was carried out at the Stanford Synchrotron Radiation Lightsource (SSRL) beamlines 4-3, operated by Stanford University for the U.S. DOE Office of Science and supported by the DOE Office of Biological and Environmental Research and by the NIH (including P41GM103393). XAS work was funded by the Director, Office of Science, Office of Basic Energy Sciences (OBES), Division of Chemical Sciences, Geosciences, and Biosciences of the Department of Energy (DOE) under contract DE-AC02-05CH11231 (J.Y. and V.K.Y). The work at EPFL is supported by a starting grant from the European Research Council (no. 257096).
Supporting Information Available
Polarization curve for the film at pH = 2, current density as a function of potential for the MoSx film during in situ XAS measurements, k-space EXAFS spectra and corresponding fits for the MoSx film and reference compounds and combined sulfur K-edge and molybdenum L2,3-edge spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Armaroli N.; Balzani V. Angew. Chem., Int. Ed. 2007, 46, 52. [DOI] [PubMed] [Google Scholar]
- Nocera D. G. Inorg. Chem. 2009, 48, 10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J. A. Science 1999, 285, 687. [DOI] [PubMed] [Google Scholar]
- Merki D.; Hu X. Energy Environ. Sci. 2011, 4, 3878. [Google Scholar]
- Nguyen M. T. D.; Ranjbari A.; Catala L.; Brisset F.; Millet P.; Aukauloo A. Coord. Chem. Rev. 2012, 256, 2435. [Google Scholar]
- Cobo S.; Heidkamp J.; Jacques P.-A.; Fize J.; Fourmond V.; Guetaz L.; Jousselme B.; Ivanova V.; Dau H.; Palacin S.; Fontecave M.; Artero V. Nat. Mater. 2012, 11, 802. [DOI] [PubMed] [Google Scholar]
- Di Giovanni C.; Wang W.-A.; Nowak S.; Greneche J.-M.; Lecoq H.; Mouton L.; Giraud M.; Tard C. ACS Catal. 2014, 4, 681. [Google Scholar]
- Popczun E. J.; Read C. G.; Roske C. W.; Lewis N. S.; Schaak R. E. Angew. Chem., Int. Ed. 2014, 53, 5427. [DOI] [PubMed] [Google Scholar]
- Laursen A. B.; Kegnaes S.; Dahl S.; Chorkendorff I. Energy Environ. Sci. 2012, 5, 5577. [Google Scholar]
- Chianelli R. R.; Daage M.; Ledoux M. J. In Advances in Catalysis; Academic Press: San Diego, CA, 1994; Vol. 40, pp 177–232. [Google Scholar]
- Riaz U.; Curnow O. J.; Curtis M. D. J. Am. Chem. Soc. 1994, 116, 4357. [Google Scholar]
- Appel A. M.; DuBois D. L.; DuBois M. R. J. Am. Chem. Soc. 2005, 127, 12717. [DOI] [PubMed] [Google Scholar]
- Jaramillo T. F.; Bonde J.; Zhang J.; Ooi B.-L.; Andersson K.; Ulstrup J.; Chorkendorff I. J. Phys. Chem. C 2008, 112, 17492. [Google Scholar]
- Kibsgaard J.; Jaramillo T. F.; Besenbacher F. Nat. Chem. 2014, 6, 248. [DOI] [PubMed] [Google Scholar]
- Hinnemann B.; Moses P. G.; Bonde J.; Jorgensen K. P.; Nielsen J. H.; Horch S.; Chorkendorff I.; Norskov J. K. J. Am. Chem. Soc. 2005, 127, 5308. [DOI] [PubMed] [Google Scholar]
- Jaramillo T. F.; Jorgensen K. P.; Bonde J.; Nielsen J. H.; Horch S.; Chorkendorff I. Science 2007, 317, 100. [DOI] [PubMed] [Google Scholar]
- Tang M. L.; Grauer D. C.; Lassalle-Kaiser B.; Yachandra V. K.; Amirav L.; Long J. R.; Yano J.; Alivisatos A. P. Angew. Chem., Int. Ed. 2011, 50, 10203. [DOI] [PubMed] [Google Scholar]
- Li Y.; Wang H.; Xie L.; Liang Y.; Hong G.; Dai H. J. Am. Chem. Soc. 2011, 133, 7296. [DOI] [PubMed] [Google Scholar]
- Merki D.; Fierro S.; Vrubel H.; Hu X. Chem. Sci. 2011, 2, 1262. [Google Scholar]
- Kibsgaard J.; Chen Z.; Reinecke B. N.; Jaramillo T. F. Nat. Mater. 2012, 11, 963. [DOI] [PubMed] [Google Scholar]
- Vrubel H.; Merki D.; Hu X. Energy Environ. Sci. 2012, 5, 6136. [Google Scholar]
- Vrubel H.; Hu X. ACS Catal. 2013, 3, 2002. [Google Scholar]
- Karunadasa H. I.; Montalvo E.; Sun Y.; Majda M.; Long J. R.; Chang C. J. Science 2012, 335, 698. [DOI] [PubMed] [Google Scholar]
- Hibble S. J.; Rice D. A.; Pickup D. M.; Beer M. P. Inorg. Chem. 1995, 34, 5109. [Google Scholar]
- Weber T.; Muijser J. C.; Niemantsverdriet J. W. J. Phys. Chem. 1995, 99, 9194. [Google Scholar]
- Shibahara T.; Kuroya H. Polyhedron 1986, 5, 357. [Google Scholar]
- Bediako D. K.; Lassalle-Kaiser B.; Surendranath Y.; Yano J.; Yachandra V. K.; Nocera D. G. J. Am. Chem. Soc. 2012, 134, 6801. [DOI] [PubMed] [Google Scholar]
- Gorlin Y.; Lassalle-Kaiser B.; Benck J. D.; Gul S.; Webb S. M.; Yachandra V. K.; Yano J.; Jaramillo T. F. J. Am. Chem. Soc. 2013, 135, 8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newville M. J. Synchrotron Radiat. 2001, 8, 96. [DOI] [PubMed] [Google Scholar]
- Rehr J. J.; Albers R. C. Rev. Mod. Phys. 2000, 72, 621. [Google Scholar]
- Afanasiev P.; Jobic H.; Lorentz C.; Leverd P.; Mastubayashi N.; Piccolo L.; Vrinat M. J. Phys. Chem. C 2009, 113, 4139. [Google Scholar]
- Hibble S. J.; Walton R. I.; Pickup D. M.; Hamon A. C. J. Non. Cryst. Solids 1998, 232, 434. [Google Scholar]
- Cramer S. P.; Liang K. S.; Jacobson A. J.; Chang C. H.; Chianelli R. R. Inorg. Chem. 1984, 23, 1215. [Google Scholar]
- Hedoire C. E.; Cadot E.; Villain F.; Davidson A.; Louis C.; Breysse M. Appl. Catal., A 2006, 306, 165. [Google Scholar]
- Dickinson R. G.; Pauling L. J. Am. Chem. Soc. 1923, 45, 1466. [Google Scholar]
- Akashi H.; Shibahara T.; Kuroya H. Polyhedron 1990, 9, 1671. [Google Scholar]
- Genuit D.; Bezverkhyy I.; Afanasiev P. J. Solid State Chem. 2005, 178, 2759. [Google Scholar]
- Ijima K.; Ohminami Y.; Suzuki S.; Asakura K. Top. Catal. 2002, 18, 125. [Google Scholar]
- Menard M. C.; Ishii R.; Nakatsuji S.; Chan J. Y. Inorg. Chem. 2011, 50, 8767. [DOI] [PubMed] [Google Scholar]
- Casewit C. J.; Coons D. E.; Wright L. L.; Miller W. K.; Dubois M. R. Organometallics 1986, 5, 951. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.