

Abstract
The human airway is lined with respiratory epithelial cells, which create a critical barrier through the formation of apical tight junctions. To investigate the molecular mechanisms underlying this process, an RNAi screen for guanine nucleotide exchange factors (GEFs) was performed in human bronchial epithelial cells (16HBE). We report that SOS1, acting through the Ras/MEK/ERK pathway, is essential for tight junction formation. Global microarray analysis identifies epithelial membrane protein 1 (EMP1), an integral tetraspan membrane protein, as a major transcriptional target. EMP1 is indispensable for tight junction formation and function in 16HBE cells and in a human airway basal progenitor-like cell line (BCi-NS1.1). Furthermore, EMP1 is significantly downregulated in human lung cancers. Together, these data identify important roles for SOS1/Ras and EMP1 in tight junction assembly during airway morphogenesis.
Keywords: EMP1, lung, Ras, SOS1, tight junctions
Introduction
The human airway is lined with epithelial cells that form the interface between the respiratory system and the outside environment. The airway epithelium acts as a conduit for air and creates a critical barrier against inhaled pathogens, allergens and other xenobiotics 1,2. Proper barrier function depends upon the formation and maintenance of apical tight junctions, which confer selective permeability and segregate the apical and basolateral membrane domains 3–5. Normal epithelial architecture is often disrupted in lung disorders, such as chronic obstructive pulmonary disease, asthma and lung cancer, and by environmental insults such as smoking 1,2,6,7.
Small GTPases of the Rho and Ras families are important regulators of epithelial morphogenesis, controlling polarity establishment and junction formation 8,9. In bronchial epithelia, RhoA and Cdc42 mediate assembly of apical junctions, while in other epithelial cell types, Rac1 and Rap1 also play important roles 10–16. In contrast, Ras is reported to disrupt epithelial polarity and morphogenesis in its activated, oncogenic form 17,18. Spatio-temporal activation of Ras and Rho family GTPases is regulated primarily by guanine nucleotide exchange factors (GEFs) 9,19. Recent studies have identified several GEFs that control distinct aspects of epithelial morphogenesis, including ARHGEF18/p114RhoGEF, ARHGEF17/TEM4 and Tiam1 in junction assembly, ARHGEF36/Tuba in junctional tension, Dbl3 in junction positioning and ARHGEF2/GEF-H1 in junction permeability 20–27.
To investigate the molecular mechanisms controlling tight junction assembly in airway epithelia, we performed an RNAi screen of Rho family GEFs in the human bronchial epithelial cell line, 16HBE. SOS1 was identified and, unexpectedly, shown to act through Ras and the MEK/ERK pathway to control the transcription of several genes, including an integral tetraspan membrane protein, epithelial membrane protein 1 (EMP1). Here, we show that EMP1 is indispensable for tight junction assembly and function in both 16HBE and a human basal progenitor-like cell line, BCi-NS1.1. Furthermore, EMP1 is downregulated in lung tumours. We conclude that EMP1 is a critical component of airway epithelial morphogenesis, which is dysregulated in cancer.
Results and Discussion
To identify Rho family GEFs required for airway epithelial junction formation, an RNAi screen was performed in 16HBE, an immortalised human bronchial line that establishes apical junctions in a RhoA- and Cdc42-dependent manner 10,11,28. Cells were stably infected with the retroviral vector pSUPER, harbouring shRNA hairpins targeting each of 87 predicted human GEFs (3 pooled shRNAs/gene), or a Cdc42 control. Stable pools were seeded sparsely, incubated for 3 days to reach confluence and monolayers and then fixed and stained to visualise tight junctions (ZO-1) and DNA. 16HBE cells infected with control pSUPER vector form mature apical junctions, as visualised by a sharp, continuous ring of ZO-1 at cell–cell contacts (Fig1A) 10,11,22. In contrast, depletion of Cdc42 inhibits junctional assembly, inducing the formation of punctate, “primordial” junctions (Fig1A). Similarly, we found that ARHGEF18/p114RhoGEF depletion disrupts junction formation (Fig1A), consistent with its reported role in epithelial morphogenesis 20–22. Among the other GEFs, depletion of SOS1, which has not previously been implicated in this process, was found to strongly disrupt tight junction formation. To confirm a specific role for SOS1, 3 non-overlapping shRNAs were tested individually to control for possible off-target effects. shSOS1.1 induces a strong depletion of SOS1 protein, as judged by Western blotting (Fig1B), and severely disrupts junction formation (Fig1A and C), while shSOS1.3 promotes a moderate protein depletion and a partial phenotype (Fig1A–C). shSOS1.2 has no significant effect on either protein level or junction formation (Fig1A–C). The close correlation between knock-down and phenotype supports a specific role for SOS1. Depletion of SOS1 also disrupts the localisation of additional junctional markers (Supplementary Fig S1A), including occludin (tight junctions) and E-cadherin (adherens junctions), indicating a broader function for this gene in junctional assembly. Together, these data identify a novel role for the GEF SOS1 in the establishment of apical junctions in bronchial epithelia.
Figure 1. SOS1 and Ras are required for bronchial tight junction formation.

16HBE cells were stably infected with pSUPER, pSUPER expressing shCdc42, shARHGEF18, shSOS1 hairpins 1-3, pQCXIP or pQCXIP expressing dominant-negative myc-HRas N17.
- Cells were seeded sparsely on glass coverslips, incubated for 3 days and then fixed and stained for ZO-1 (tight junctions) and DNA (nuclei). Scale bar, 20 μm.
- Cell lysates were analysed by Western blotting for SOS1 and actin; labels as for (C).
- Quantification of tight junction phenotype. > 500 cells were counted per sample/experiment, across n = 3 independent experiments (dots indicate individual data points). Error bars denote mean ± SEM. *P = 0.0199; **P = 0.0041; ****P = 0.0001.
- Cell lysates were analysed by Western blotting for Ras, pERK and total ERK.
- Cells were seeded sparsely on glass coverslips, incubated for 3 days and then fixed and stained for ZO-1 (tight junctions) and DNA. Scale bar, 20 μm.
- Quantification of tight junction phenotype (see C). ***P = 0.0002.
Data information: All data are representative of 3 independent experiments.
SOS1 harbours two distinct catalytic exchange (GEF) domains, a Cdc25-like domain active on Ras and a DH/PH domain active on Rac 29. To explore whether Ras is required for tight junction formation, 16HBE cells were infected with the pQCXIP retroviral vector harbouring cDNA encoding HRas N17; this mutant is dominant negative (DN) with respect to all three Ras isoforms 30. Stable pools were analysed by Western blotting to confirm expression; pERK levels verify its functional activity in suppressing downstream MAPK signalling (Fig1D). Strikingly, expression of DN HRas induces severe junctional defects, with most cells exhibiting immature, primordial junctions as visualised by ZO-1 (Fig1E and F), occludin or E-cadherin (Supplementary Fig S1B). Thus, our screen, which was originally designed to identify relevant Rho GEFs, has uncovered a novel and unexpected role for Ras in bronchial junction formation. We note that our data do not exclude a possible, parallel role for SOS1 Rac GEF activity during junction formation.
Ras mediates its effects through a range of downstream effectors including the RAF/MAP kinase cascade 31,32. To explore the potential role of this pathway in junction assembly, sub-confluent 16HBE cells were incubated with or without either of two, structurally distinct MEK inhibitors (GSK1120212 and PD032590), incubated for 3 days to reach confluence and then fixed and stained to visualise apical junctions (ZO-1) and DNA 33,34. Notably, only a modest reduction in cell number was observed following inhibitor treatment (GSK1120212 reduces cell number by 26 ± 7% at day 4, as compared to DMSO). MEK inhibition was confirmed by Western blotting of its direct substrate pERK and a downstream target of ERK, p-p90RSK (Fig2A). Strikingly, inhibition of MEK phenocopies both SOS1 depletion and expression of DN RasN17, inducing the formation of punctate, primordial junctions, as observed at the endpoint of the assay (Fig2B and C; Supplementary Fig S1C), or following a subsequent calcium switch assay for de novo junction formation (Fig3A, panels 3 and 4). Similarly, direct inhibition of ERK using the small molecule SCH772984 disrupts junctions (Fig2B and C), and ERK inhibition was confirmed using p-p90RSK (Fig2A) 35. Together, these data indicate that SOS1 and Ras control junction formation through activation of MEK and ERK. Consistent with this linear pathway, depletion of SOS1 (Fig2D), or expression of DN RasN17 (Fig1D), inhibits ERK phosphorylation. We conclude that a SOS1/Ras/MEK/ERK cascade controls junction formation in bronchial epithelia. Interestingly, inhibition of this pathway has no obvious effect when added to an established monolayer with mature junctions (Supplementary Fig S2), indicating that while ERK activation is essential for the formation of bronchial junctions, it is dispensable for their maintenance.
Figure 2. MEK and ERK are required for tight junction formation and function.

16HBE cells were seeded sparsely, treated with DMSO, GSK1120212 (500 nM), PD0325901 (500 nM) or SCH772984 (1 μM) and incubated for 3 days.
- Cell lysates were analysed by Western blotting for pERK, total ERK, p-p90RSK or total RSK1.
- Cells were fixed and stained for ZO-1 and DNA. Scale bar, 20 μm.
- Quantification of tight junction phenotype. > 500 cells were counted per sample/experiment, across n = 3 independent experiments (dots indicate individual data points). Error bars denote mean ± SEM. ***P = 0.0002; ****P < 0.0001.
- 16HBE cells were transfected with control siRNA or siSOS1 and incubated for 4 days. Cell lysates were analysed by Western blotting for SOS1, pERK and total ERK.
- 16HBE cells were seeded on transwell filters with DMSO, GSK1120212 (500 nM) or PD0325901 (500 nM). Transepithelial resistance (TER) was measured on day 4 and expressed as % of DMSO control. n = 3 independent experiments (dots indicate individual data points); error bars denote mean ± SEM. ***P = 0.0002; ****P < 0.0001.
- 16HBE cells were seeded on glass-bottomed dishes with DMSO, GSK1120212 (500 nM) or PD0325901 (500 nM). On day 4, FM4-64 dye was applied to the media and confocal z-stacks acquired. Representative images are shown, and the red dotted lines indicate the basal surface.
- 16HBE cells were fixed and stained for MEK or pERK. Scale bar, 20 μm.
Data information: All data are representative of three independent experiments.
Figure 3. Ras, MEK and ERK control bronchial epithelial gene expression.

- A Acute versus chronic MEK inhibition. Acute (left panels): cells were seeded sparsely and incubated for 4 days in normal media and then subjected to a calcium switch and recovery, in the presence of DMSO (panel 1) or 500 nM GSK1120212 (panel 2). Chronic (right panels): cells were seeded sparsely and incubated for 4 days in DMSO (panel 3) or 500 nM GSK1120212 (panel 4). Cells were subjected to a calcium switch and recovery, in the presence of DMSO (panel 3) or 500 nM GSK1120212 (panel 4). Cells were fixed and stained for ZO-1 and DNA. Scale bar, 20 μm.
- B–D Microarray analysis. 16HBE cells were stably infected with pQCXIP (control) or pQCXIP expressing DN HRas. Control cells were treated with DMSO, GSK1120212 (500 nM) or SCH772984 (1 μM) for 4 days. RNA was isolated and analysed using an Illumina gene array; n = 3 independent samples were prepared for each condition. (B) Venn diagram representing genes downregulated by > 1.6-fold versus control, with an unadjusted P-value < 0.05. (C) Relative expression levels of 33 genes downregulated by DN HRas, MEK and ERK inhibition, expressed as % of DMSO control. (D) Relative expression levels of EMP1. Error bars denote mean ± SEM, and dots indicate individual data points. **P < 0.002 (DN Ras = 0.0019, ERKi = 0.0016); ****P < 0.0001.
To analyse the contribution of the MAPK cascade to tight junction-mediated paracellular permeability (gate function), 16HBE cells were seeded on filters, incubated with or without MEK inhibitors for 3 days and then assayed for transepithelial resistance (TER) (Fig2E). Although cells remain confluent and viable throughout the assay, inhibition of MEK significantly reduces TER (e.g. DMSO: 715 ± 139 ohms/cm2; GSK: 48 ± 23 ohms/cm2), indicating a clear defect in barrier function. To analyse effects on the segregation of apical and basolateral membrane domains (fence function), the diffusion of an apically applied, lipophilic, fluorescent dye (FM 4–64) was monitored by live, confocal imaging. When applied to confluent control cells, FM 4–64 fluorescence localises exclusively along the apical surface (Fig2F), but in MEK-inhibited cells, the dye rapidly incorporates into the basal and lateral membranes. The movement of the dye throughout the cell membrane indicates that the tight junction diffusion barrier is defective upon MEK inhibition. Together, these data demonstrate that MEK activity is required to establish both the gate and fence functionality of tight junctions in bronchial epithelia. Consistent with this role, both MEK and pERK localise to cell–cell contacts in 16HBE cells (Fig2G), similar to what has been reported in keratinocytes 36.
ERK could control apical junction formation through direct phosphorylation of cytosolic substrates or through changes in gene expression 37. To investigate the mechanism of ERK function further, the kinetics of pathway inhibition were manipulated. To explore the effects of acute MEK inhibition, a calcium switch assay was performed (Fig3A, panels 1 and 2). 16HBE cells were cultured to confluence and then deprived of calcium to disrupt cell–cell contacts. Rapid, synchronous junction reformation was initiated by the re-addition of calcium for 4 h, with or without the MEK inhibitor (GSK1120212). Under these conditions, junctions form normally in both control and MEK-inhibited cells (Fig3A). This contrasts with the chronic treatment of cells, seeded in the presence of MEK inhibitor and incubated for 4 days, which dramatically inhibits junction assembly both at the endpoint (Fig2B) and following a subsequent calcium switch (Fig3A, panels 3 and 4). We conclude that chronic inhibition of MEK is required to disrupt bronchial tight junction formation and reason that this likely reflects an effect on gene expression.
To investigate the contribution of the SOS/Ras/MEK/ERK pathway to bronchial epithelial gene expression, microarray analysis was performed using an Illumina array to analyse 47,000 transcripts. To increase stringency, three distinct modes of pathway inhibition were compared: DN HRas expression and chronic treatment with either MEK (GSK1120212) or ERK (SCH772984) inhibitors. Control cells were compared to each experimental group to identify all genes downregulated, by 1.6-fold or more, after pathway inhibition (Fig3B; Supplementary Fig S3). 33 genes were significantly downregulated by all three treatments (Fig3C). Importantly, these include several known transcriptional targets of Ras/MEK/ERK, including DUSP5, EGR1 and PHLDA1 38, thus validating the analysis. The list also identifies several other proteins of potential significance in the context of epithelial morphogenesis (see Supplementary Fig S3). Among these hits, epithelial membrane protein 1 (EMP1) represents an intriguing candidate. It is significantly downregulated by inhibition of Ras, MEK and ERK (Fig3D), as well as by depletion of SOS1 (Fig4A), consistent with a linear pathway signalling from exchange factor to transcriptional target. The EMP1 gene encodes a little-studied integral tetraspan membrane protein, belonging to the PMP-22/EMP/MP20/Claudin family 3. Notably, EMP1 shares some sequence and structural homology with claudins, which are well-known components of the tight junction 4,5,39,40. However, these relatives are evolutionarily distant (e.g. 27% identity between EMP1 and claudin 1) and whether they perform related or distinct functions is currently unresolved 39. EMP1 has been localised to the region of cell–cell contacts in rat liver and can interact with ZO-1 and occludin in mouse brain endothelial cells 40,41. However, the functional role of the EMP proteins (EMP1-3) has yet to be investigated directly.
Figure 4. EMP1 is required for bronchial epithelial tight junction formation and function.

- 16HBE cells were stably infected with lentiviral vector pLKO alone or expressing EMP1 shRNAs (1 or 2). Total RNA was isolated and analysed for EMP1 expression using TaqMan/qPCR with a GAPDH control. Wild-type cells treated with 500 nM GSK1120212 or siSOS1 for 4 days were also analysed. Error bars denote mean ± SEM, and dots indicate individual data points. ***P < 0.0002 (GSK = 0.0002, siSOS1 = 0.0001); ****P < 0.0001.
- Cells as in (A) were fixed and stained for ZO-1, E-cadherin and DNA. Scale bar, 20 μm.
- Quantification of tight junction phenotype of cells as in (A). > 500 cells were counted per sample/experiment, across n = 3 independent experiments (dots indicate individual data points). Error bars denote mean ± SEM. *P = 0.0148; **P = 0.006.
- Transepithelial resistance (TER) was measured in 16HBE cells as in (A) on day 4 post-seeding. Error bars denote mean ± SEM, and dots indicate individual data points. **P = 0.0036; ***P = 0.0005.
- Cells as in (A) were seeded on glass-bottomed dishes for 4 days. FM 4–64 dye was applied to the media and confocal z-stacks acquired.
- 16HBE cells were fixed and stained for endogenous EMP1 and DNA using two different commercial antibodies. Scale bar, 20 μm.
- 16HBE cells were seeded on glass coverslips for 10 days to form a mature, polarised monolayer. Cells were fixed and costained for EMP1 and ZO-1 and then analysed by confocal microscopy. A representative z-stack is presented; arrows indicate ZO-1-positive tight junctions.
Data information: All data are representative of n = 3 independent experiments.
We hypothesised that EMP1 may play a role in epithelial junction formation. To test this, 16HBE cells were infected with pLKO.1 lentiviral vectors harbouring EMP1-specific shRNAs; the extent of depletion was determined by qPCR (Fig4A). Two distinct and non-overlapping shRNA hairpins were identified that knock-down EMP1 mRNA by 80–90%; this is similar to the level of depletion observed after chronic MEK inhibition (Fig4A). Strikingly, both shRNA hairpins severely disrupt both adherens and tight junction formation, as measured by E-cadherin and ZO-1 staining (Fig4B and C). Furthermore, depletion of EMP1 significantly impairs barrier function, as judged by a decrease in TER (Fig4D), and abrogates fence function, as shown by diffusion of FM 4–64 through the basolateral membrane (Fig4E). Consistent with a junctional role, EMP1 localises to cell–cell contacts (Fig4F), as visualised using two different antibodies against the endogenous protein. Analysis of protein recruitment during de novo junction formation in a calcium switch assay indicates that EMP1 is not obviously recruited to E-cad/ZO-1-positive primordial puncta (Supplementary Fig S4). Instead, it gradually accumulates at cell–cell contacts with a more continuous, linear pattern, similar to its relative claudin-1. Confocal imaging reveals that EMP1 is apically enriched in polarised monolayers and colocalises with ZO-1 (Fig4G), suggesting it resides at tight junctions. We conclude that EMP1 is a novel and essential regulator of bronchial apical junction formation and function. Similar to ZO-1, EMP1 influences both adherens and tight junction formation, but ultimately localises to the tight junction.
To confirm the wider importance of EMP1 in respiratory cells, a more physiologically relevant model of the human airway epithelium was exploited 42. BCi-NS1.1 cells retain key characteristics of primary basal cells, including a multi-potent capacity to differentiate into various airway cell types and the ability to form intact tight junctions when cultured under air–liquid interface (ALI) conditions. Lentiviral shRNA-mediated depletion of EMP1 was performed in BCi-NS1.1 cells and confirmed using qPCR (Fig5A). To assay tight junction formation, filter-grown cells were stained for ZO-1 and analysed by confocal microscopy. The BCi-NS1.1 epithelium is multi-layered, complicating a standard junctional assay, and so the number of intact ZO-1 rings was scored per field of view (Fig5B and C). EMP1 depletion induces a significant decrease in tight junction formation and, importantly, also abrogates barrier function, as judged by TER (Fig5D; pLKO: 1055 ± 416 ohms/cm2; shEMP1.1: 98 ± 37 ohms/cm2). Together, these data identify EMP1 as an important regulator of epithelial morphogenesis and function in the human respiratory airway.
Figure 5. EMP1 in human airway basal progenitor-like cells and lung cancer.
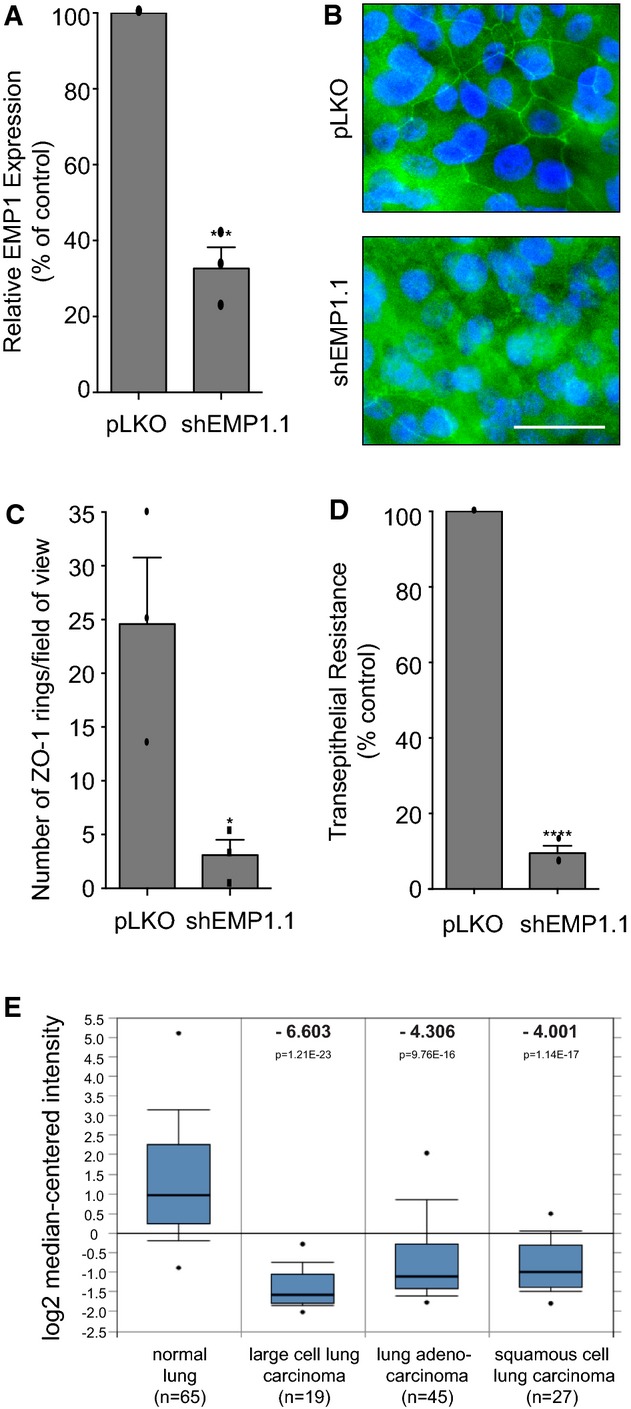
- BCi-NS1.1 cells were stably infected with pLKO control or shEMP1.1. Total RNA was isolated and analysed for EMP1 expression using TaqMan/qPCR with a GAPDH control. Error bars denote mean ± SEM, ***P = 0.0003.
- Filter-grown cells as in (A) were fixed and stained for ZO-1 and DNA and analysed by confocal imaging.
- Quantification of tight junction phenotype of cells as in (A). > 500 cells were counted per sample/experiment, across n = 3 independent experiments (dots indicate individual data points). Error bars denote mean ± SEM. *P = 0.0276.
- Cells as in (A) were cultured on filters under ALI conditions, and transepithelial resistance (TER) was measured on day 21 post-seeding. Error bars denote mean ± SEM. ****P < 0.0001.
- EMP1 mRNA expression in normal lung versus cancer tissue. The Hou Lung data set was analysed using Oncomine software and the 213895_at reporter 45. Box-whisker plots are presented. The points represent the minimum/maximum values, bars denote the 10th–90th percentiles, boxes show the 25th–75th percentiles, and the horizontal line marks the median. The number of samples analysed (n), the fold change (bold) and the P-value are indicated.
Data information: Data are representative of n = 3 independent experiments.
Consistent with its role in airway morphogenesis, EMP1 is abundantly expressed in the lung and represents one of the signature genes identified in human airway basal cells 43,44. To determine whether changes in EMP1 transcription may be associated with diseases of the airway, we investigated EMP1 expression in normal lung versus tumour samples. Oncomine analysis of the Hou Lung data set reveals that EMP1 is downregulated by 4- to 6-fold in three different classes of lung cancer (Fig5E) 45. Similarly, decreased EMP1 expression has been reported in oral squamous cell carcinoma and nasopharyngeal cancer 46,47. We conclude that loss of EMP1 is associated with certain types of tumour.
In the current study, we identify a role for the SOS/Ras/MEK/ERK cascade in airway morphogenesis. Unusually, inactivation of this pathway does not significantly impair proliferation in 16HBE cells, providing an opportunity to observe and interrogate this novel function under conditions of chronic ERK suppression. We find that pathway inhibition impairs both adherens and tight junction formation and disrupts the establishment of gate and fence function in bronchial epithelial cells. However, the effects of Ras/MEK/ERK signalling on epithelial morphogenesis are pleiotropic and likely to be cell type and stimulus specific. Oncogenic Ras, for example, can disrupt epithelial polarity, while the ERK MAP kinase pathway has been implicated in cytokine induced junction disassembly 17,48. In the current study, we identify EMP1, a little-studied integral tetraspan membrane protein, as an important transcriptional target of the Ras/MAPK pathway in bronchial epithelia, with a critical role in airway morphogenesis. EMP1 is a member of the PMP-22/EMP/MP20/Claudin superfamily and bears structural and sequence homology to known tight junction components. The claudins are thought to comprise the major components of tight junction strands, defining selective permeability by forming size- and charge-selective pores 3,39,49, while PMP-22 localises to epithelial junctions and modulates barrier function in MDCK cells 50,51. Our current data reveal the distantly related EMP subgroup can also contribute to the formation and function of tight junctions, opening up a new avenue for investigation.
Materials and Methods
Cloning
DN HRas N17 was subcloned into pQCXIP using BamHI/EcoRI and fully sequenced.
RNAi
Rho family GEF and Cdc42 shRNAs were cloned in pSUPERpuro, EMP1 shRNAs in pLKO.1. siRNAs were obtained from Dharmacon. Sequences are shown in the Supplementary Materials and Methods.
Cell culture and treatment
16HBE cells were cultured as described previously 22,28. BCi-NS1.1 cells were grown in Bronchial Epithelial Growth Media (Lonza, CA). For calcium switch experiments, confluent cells were washed in PBS and incubated in calcium-free medium for 4 h, and normal growth media was then replaced for a further 4 h. siRNA transfections were performed using Lipofectamine LTX. Inhibitor treatments were as follows: 500 nM GSK1120212 (Selleck), 500 nM PD0325901 (Selleck) or 1 μM Sch772984 (provided by Neal Rosen); DMSO (1:20,000) was used as a carrier control. More detail can be found in the Supplementary Materials and Methods.
Virus production and preparation of stable cells
Retroviral or lentiviral particles were prepared to deliver cDNA or shRNA for stable expression, as described previously 52. 16HBE or BCi-NS1.1 cells were infected and selected with puromycin to yield stable pools. More detail can be found in the Supplementary Materials and Methods.
qPCR
RNA was isolated using the RNeasy kit (Qiagen). cDNA was synthesised using Superscript III First-Strand Synthesis Supermix (Invitrogen). qPCR was performed using Taqman Universal PCR mastermix, with probes for EMP1 and GAPDH (Applied Biosystems). Samples were analysed using a Bio-Rad iQ5 Multicolor RT–PCR Detection System. Further details can be found in the Supplementary Materials and Methods.
Western blotting
Total cell extracts were prepared, and Western blotting was performed as described previously 53. Antibodies are listed in the Supplementary Materials and Methods.
Immunofluorescence and imaging
Fixation conditions, antibodies, microscopes, objectives, cameras and software are listed in the Supplementary Materials and Methods.
Tight junction formation assays
A detailed protocol can be found in the Supplementary Materials and Methods. Briefly, 16HBE were seeded on coverslips and imaged by epifluorescence, and BCi-NS1.1 cells were seeded on transwell inserts and imaged by confocal microscopy. All cells were fixed and stained for ZO-1/DNA. Multiple random, non-overlapping images were acquired. For 16HBE, cells with a continuous ring of ZO-1 at cell–cell contacts were scored as having intact apical junctions, cells with punctate, discontinuous or absent ZO-1 at cell–cell contacts were defined as not having apical junctions. For BCi-NS1.1, apical junction formation was quantified by counting continuous rings of ZO-1 staining/field of view.
Transepithelial resistance assay
5 × 104 16HBE cells were seeded on collagen-coated, 0.4-μm PTFE membrane filters in 6.5-mm inserts (Costar, 3495), on 24-well plates (Corning). Transepithelial resistance was measured using an EVOM voltohmmeter and STX2 electrode (World Precision Instruments). For BCi-NS1.1 cells, air–liquid interface (ALI) cultures were analysed on day 21 post-seeding, as described previously 42.
FM4-64 dye assay
Cells were seeded on glass-bottomed, 35-mm dishes (MatTek). FM 4–64 (5 μg/ml; Invitrogen) was added to the media for 10 min at 37°C. Confocal z-stacks were acquired from 10 fields of view; 3 x/z slices per image were analysed using ImageJ software. Line profiles were drawn over apical and basolateral surfaces to calculate average fluorescent intensities.
Microarray analysis
Microarray analysis was performed using an Illumina gene expression array (Human HT-12, 47,000 transcripts). The data were analysed using Partek software. More detail can be found in the Supplementary Materials and Methods.
Statistics
Unpaired t-tests were performed in Prism, with two-tailed P-values and 95% confidence intervals.
Acknowledgments
We thank Dieter Gruenert for providing 16HBE cells, Rona Cameron for advice on MEK/ERK inhibitors and Jeffrey Zhao for assistance with Partek software. We are grateful to members of the Hall laboratory for helpful discussions and to Teodoro Pulvirenti for critical reading of the manuscript. The work was supported by National Institutes of Health (NIH) grants GM081435 and CA008748 (AH) and HL107882 (RC). JD is funded by a Marie Curie fellowship (624161), and OF is funded by Cancer Research UK (C47718/A16337).
Author contributions
JD and AH conceived the project and wrote the manuscript. JD carried out the experiments unless otherwise indicated. GT performed SOS1 and EMP1 depletions and assisted with the microarray. MSW, VA and RGC provided and cultured the BCi-NS1.1 cells. OF performed confocal microscopy. AS constructed the shRNA library. NR provided the small-molecule inhibitors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Review Process File
References
- Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME. Airway epithelial cells: current concepts and challenges. Proc Am Thorac Soc. 2008;5:772–777. doi: 10.1513/pats.200805-041HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8:432–446. doi: 10.1046/j.1440-1843.2003.00493.x. [DOI] [PubMed] [Google Scholar]
- Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22:207–235. doi: 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soini Y. Claudins in lung diseases. Respir Res. 2011;12:70. doi: 10.1186/1465-9921-12-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici-Barel Y, Salit J, Harvey BG, Crystal RG. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci. 2011;68:877–892. doi: 10.1007/s00018-010-0500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga VM, Yap AS. The challenges of abundance: epithelial junctions and small GTPase signalling. Curr Opin Cell Biol. 2005;17:466–474. doi: 10.1016/j.ceb.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Wallace SW, Magalhaes A, Hall A. The Rho target PRK2 regulates apical junction formation in human bronchial epithelial cells. Mol Cell Biol. 2011;31:81–91. doi: 10.1128/MCB.01001-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SW, Durgan J, Jin D, Hall A. Cdc42 regulates apical junction formation in human bronchial epithelial cells through PAK4 and Par6B. Mol Biol Cell. 2010;21:2996–3006. doi: 10.1091/mbc.E10-05-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga VM, Machesky LM, Hall A, Hotchin NA. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J Cell Biol. 1997;137:1421–1431. doi: 10.1083/jcb.137.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou TS, Schneeberger EE, Nelson WJ. Structural and functional regulation of tight junctions by RhoA and Rac1 small GTPases. J Cell Biol. 1998;142:101–115. doi: 10.1083/jcb.142.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratheesh A, Priya R, Yap AS. Coordinating Rho and Rac: the regulation of Rho GTPase signaling and cadherin junctions. Prog Mol Biol Transl Sci. 2013;116:49–68. doi: 10.1016/B978-0-12-394311-8.00003-0. [DOI] [PubMed] [Google Scholar]
- Mack NA, Porter AP, Whalley HJ, Schwarz JP, Jones RC, Khaja AS, Bjartell A, Anderson KI, Malliri A. beta2-syntrophin and Par-3 promote an apicobasal Rac activity gradient at cell-cell junctions by differentially regulating Tiam1 activity. Nat Cell Biol. 2012;14:1169–1180. doi: 10.1038/ncb2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price LS, Hajdo-Milasinovic A, Zhao J, Zwartkruis FJ, Collard JG, Bos JL. Rap1 regulates E-cadherin-mediated cell-cell adhesion. J Biol Chem. 2004;279:35127–35132. doi: 10.1074/jbc.M404917200. [DOI] [PubMed] [Google Scholar]
- Magudia K, Lahoz A, Hall A. K-Ras and B-Raf oncogenes inhibit colon epithelial polarity establishment through up-regulation of c-myc. J Cell Biol. 2012;198:185–194. doi: 10.1083/jcb.201202108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenenberger CA, Zuk A, Kendall D, Matlin KS. Multilayering and loss of apical polarity in MDCK cells transformed with viral K-ras. J Cell Biol. 1991;112:873–889. doi: 10.1083/jcb.112.5.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- Terry SJ, Zihni C, Elbediwy A, Vitiello E, Leefa Chong San IV, Balda MS, Matter K. Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat Cell Biol. 2011;13:159–166. doi: 10.1038/ncb2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Tanoue T. Lulu2 regulates the circumferential actomyosin tensile system in epithelial cells through p114RhoGEF. J Cell Biol. 2011;195:245–261. doi: 10.1083/jcb.201104118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Jin D, Durgan J, Hall A. LKB1 controls human bronchial epithelial morphogenesis through p114RhoGEF-dependent RhoA activation. Mol Cell Biol. 2013;33:2671–2682. doi: 10.1128/MCB.00154-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngok SP, Geyer R, Kourtidis A, Mitin N, Feathers R, Der C, Anastasiadis PZ. TEM4 is a junctional Rho GEF required for cell-cell adhesion, monolayer integrity and barrier function. J Cell Sci. 2013;126(15):3271–3277. doi: 10.1242/jcs.123869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malliri A, van Es S, Huveneers S, Collard JG. The Rac exchange factor Tiam1 is required for the establishment and maintenance of cadherin-based adhesions. J Biol Chem. 2004;279:30092–30098. doi: 10.1074/jbc.M401192200. [DOI] [PubMed] [Google Scholar]
- Otani T, Ichii T, Aono S, Takeichi M. Cdc42 GEF Tuba regulates the junctional configuration of simple epithelial cells. J Cell Biol. 2006;175:135–146. doi: 10.1083/jcb.200605012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zihni C, Munro PM, Elbediwy A, Keep NH, Terry SJ, Harris J, Balda MS, Matter K. Dbl3 drives Cdc42 signaling at the apical margin to regulate junction position and apical differentiation. J Cell Biol. 2014;204:111–127. doi: 10.1083/jcb.201304064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benais-Pont G, Punn A, Flores-Maldonado C, Eckert J, Raposo G, Fleming TP, Cereijido M, Balda MS, Matter K. Identification of a tight junction-associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. J Cell Biol. 2003;160:729–740. doi: 10.1083/jcb.200211047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10:38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- Nimnual AS, Yatsula BA, Bar-Sagi D. Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science. 1998;279:560–563. doi: 10.1126/science.279.5350.560. [DOI] [PubMed] [Google Scholar]
- Matallanas D, Arozarena I, Berciano MT, Aaronson DS, Pellicer A, Lafarga M, Crespo P. Differences on the inhibitory specificities of H-Ras, K-Ras, and N-Ras (N17) dominant negative mutants are related to their membrane microlocalization. J Biol Chem. 2003;278:4572–4581. doi: 10.1074/jbc.M209807200. [DOI] [PubMed] [Google Scholar]
- Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- Warne PH, Viciana PR, Downward J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature. 1993;364:352–355. doi: 10.1038/364352a0. [DOI] [PubMed] [Google Scholar]
- Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold WR, et al. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg Med Chem Lett. 2008;18:6501–6504. doi: 10.1016/j.bmcl.2008.10.054. [DOI] [PubMed] [Google Scholar]
- Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–750. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- Iden S, van Riel WE, Schafer R, Song JY, Hirose T, Ohno S, Collard JG. Tumor type-dependent function of the par3 polarity protein in skin tumorigenesis. Cancer Cell. 2012;22:389–403. doi: 10.1016/j.ccr.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- Tullai JW, Schaffer ME, Mullenbrock S, Kasif S, Cooper GM. Identification of transcription factor binding sites upstream of human genes regulated by the phosphatidylinositol 3-kinase and MEK/ERK signaling pathways. J Biol Chem. 2004;279:20167–20177. doi: 10.1074/jbc.M309260200. [DOI] [PubMed] [Google Scholar]
- Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol. 2006;68:403–429. doi: 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- Bangsow T, Baumann E, Bangsow C, Jaeger MH, Pelzer B, Gruhn P, Wolf S, von Melchner H, Stanimirovic DB. The epithelial membrane protein 1 is a novel tight junction protein of the blood-brain barrier. J Cereb Blood Flow Metab. 2008;28:1249–1260. doi: 10.1038/jcbfm.2008.19. [DOI] [PubMed] [Google Scholar]
- Lee HS, Sherley JL, Chen JJ, Chiu CC, Chiou LL, Liang JD, Yang PC, Huang GT, Sheu JC. EMP-1 is a junctional protein in a liver stem cell line and in the liver. Biochem Biophys Res Commun. 2005;334:996–1003. doi: 10.1016/j.bbrc.2005.06.194. [DOI] [PubMed] [Google Scholar]
- Walters MS, Gomi K, Ashbridge B, Moore MA, Arbelaez V, Heldrich J, Ding BS, Rafii S, Staudt MR, Crystal RG. Generation of a human airway epithelium derived basal cell line with multipotent differentiation capacity. Respir Res. 2013;14:135. doi: 10.1186/1465-9921-14-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobsiger CS, Magyar JP, Taylor V, Wulf P, Welcher AA, Program AE, Suter U. Identification and characterization of a cDNA and the structural gene encoding the mouse epithelial membrane protein-1. Genomics. 1996;36:379–387. doi: 10.1006/geno.1996.0482. [DOI] [PubMed] [Google Scholar]
- Hackett NR, Shaykhiev R, Walters MS, Wang R, Zwick RK, Ferris B, Witover B, Salit J, Crystal RG. The human airway epithelial basal cell transcriptome. PLoS ONE. 2011;6:e18378. doi: 10.1371/journal.pone.0018378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Cao W, Xu Q, Chen WT. The expression of EMP1 is downregulated in oral squamous cell carcinoma and possibly associated with tumour metastasis. J Clin Pathol. 2011;64:25–29. doi: 10.1136/jcp.2010.082404. [DOI] [PubMed] [Google Scholar]
- Sun GG, Lu YF, Fu ZZ, Cheng YJ, Hu WN. EMP1 inhibits nasopharyngeal cancer cell growth and metastasis through induction apoptosis and angiogenesis. Tumour Biol. 2013;35:3185–3193. doi: 10.1007/s13277-013-1416-5. [DOI] [PubMed] [Google Scholar]
- Petecchia L, Sabatini F, Usai C, Caci E, Varesio L, Rossi GA. Cytokines induce tight junction disassembly in airway cells via EGFR-dependent MAPK/ERK1/2 pathway. Lab Invest. 2012;92:1140–1148. doi: 10.1038/labinvest.2012.67. [DOI] [PubMed] [Google Scholar]
- Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol. 2010;2:a002907. doi: 10.1101/cshperspect.a002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notterpek L, Roux KJ, Amici SA, Yazdanpour A, Rahner C, Fletcher BS. Peripheral myelin protein 22 is a constituent of intercellular junctions in epithelia. Proc Natl Acad Sci USA. 2001;98:14404–14409. doi: 10.1073/pnas.251548398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Amici SA, Fletcher BS, Notterpek L. Modulation of epithelial morphology, monolayer permeability, and cell migration by growth arrest specific 3/peripheral myelin protein 22. Mol Biol Cell. 2005;16:1142–1151. doi: 10.1091/mbc.E04-07-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durgan J, Kaji N, Jin D, Hall A. Par6B and atypical PKC regulate mitotic spindle orientation during epithelial morphogenesis. J Biol Chem. 2011;286:12461–12474. doi: 10.1074/jbc.M110.174235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durgan J, Cameron AJ, Saurin AT, Hanrahan S, Totty N, Messing RO. Parker PJ. The identification and characterization of novel PKClunate epsilon phosphorylation sites provide evidence for functional cross-talk within the PKC superfamily. Biochem J. 2008;411:319–331. doi: 10.1042/bj20071348. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Review Process File