

Abstract
Dynorphins, endogenous opioid neuropeptides derived from the prodynorphin gene, are involved in a variety of normative physiologic functions including antinociception and neuroendocrine signaling, and may be protective to neurons and oligodendroglia via their opioid receptor-mediated effects. However, under experimental or pathophysiological conditions in which dynorphin levels are substantially elevated, these peptides are excitotoxic largely through actions at glutamate receptors. Because the excitotoxic actions of dynorphins require supraphysiological concentrations or prolonged tissue exposure, there has likely been little evolutionary pressure to ameliorate the maladaptive, non-opioid receptor mediated consequences of dynorphins. Thus, dynorphins can have protective and/or proapoptotic actions in neurons and glia, and the net effect may depend upon the distribution of receptors in a particular region and the amount of dynorphin released. Increased prodynorphin gene expression is observed in several disease states and disruptions in dynorphin processing can accompany pathophysiological situations. Aberrant processing may contribute to the net negative effects of dysregulated dynorphin production by tilting the balance towards dynorphin derivatives that are toxic to neurons and/or oligodendroglia. Evidence outlined in this review suggests that a variety of CNS pathologies alter dynorphin biogenesis. Such alterations are likely maladaptive and contribute to secondary injury and the pathogenesis of disease.
Keywords: Opioids, neuropeptides, neuropeptides processing, drug abuse, spinal cord injury, neurotrauma, pain, glutamate, N-methyl-D-aspartate, AMPA, apoptosis, neurotoxicity, excitotoxicity, striatum, κ-opioid receptors
2. INTRODUCTION—Physiological and pathophysiological mechanisms of dynorphin action
Dynorphin A [dynorphin A (1–17)], an important posttranslational product of the preprodynorphin gene (figure 1) (1–4), binds with high affinity to kappa opioid receptors (KOR) and is an endogenous ligand that acts preferential at this receptor type. The amino acid sequence of dynorphin A (YGGFLRRIRPKLKWDNQ) is highly conserved and is identical in humans, rat, mouse, bovine, and porcine species (1,3,5), as well as in amphibians (6,7). In addition to dynorphin A, other peptides derived from the preprodynorphin gene (collectively referred to as “dynorphins”), including shorter cleavage products of dynorphin A that lack the first amino acid residue essential for opioid receptor binding, are also biologically active (3,8–10). Moreover, both tissue-specific and age-related differences in the types and ratios of the particular prodynorphin products produced have been reported (3). Dynorphin and other prodynorphin-derived peptides with high affinity for opioid receptors have a defined role as inhibitory neurotransmitters involved in pain reduction in the spinal cord. However, there is growing evidence that injury and disease can lead to pathophysiological changes in the biogenesis, processing, or cellular response to dynorphins, which contribute to the maladaptive neuroplasticity (hyperalgesia and allodynia) and neurotoxicity that characterize these states (11–28). Evidence that dynorphin per se is mediating pathophysiological changes is provided from in vivo studies in which SCI pathophysiology is reduced by anti-dynorphin immunoneutralizing antibodies [e.g., (29–31)] and direct in vitro evidence (discussed below). Studies utilizing dynorphin knockout mice [see (32)] are likely to be an additional strategy to address the role of dynorphins in secondary injury and disease. This review will focus on the pathobiology of dynorphins and the molecular basis for their pathophysiological effects.
Figure 1.
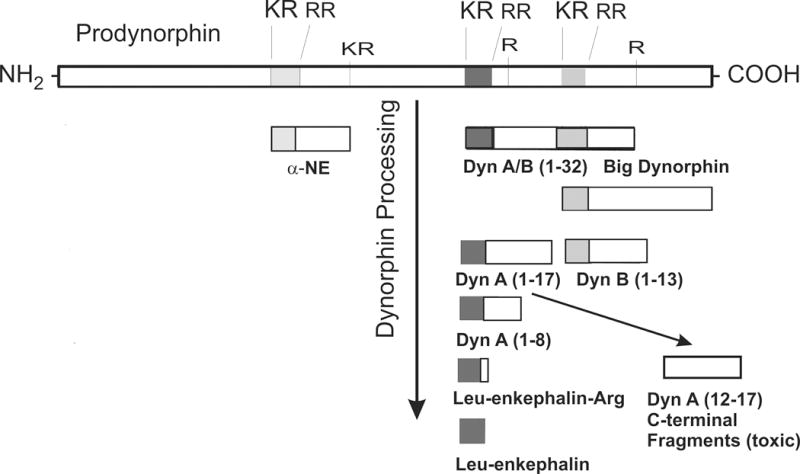
Summary of the major posttranslational products of prodynorphin. Prodynorphin is converted into multiple bioactive peptide fragments (3,224,225), including alpha-Neoendorphin (α-NE), Big Dynorphin (Dyn AB 1–32), Leumorphin (Dyn B 1–29), dynorphin A (Dyn A 1–17), dynorphin B (Dyn B 1–16), Leucine-enkephalin-arginine (Leu-enkephalin-Arg), and potentially Leucine-enkephalin (Leu-enkephalin). Processing is likely to be altered following injury or disease. Additional bioactive derivatives of prodynorphin likely exist besides those shown here. Many of the C-terminal fragments of dynorphin A, which are intrinsically neurotoxic, can be isolated from neural tissue in vivo or in vitro. Prodynorphin is highly basic, containing multiple lysine (K) and arginine (R) residues.
As noted, dynorphin A binds to KOR with somewhat higher affinity than mu (MOR) or delta (DOR) opioid receptors, and is considered to be an endogenous ligand for KOR (4). Consistent with a preferential KOR action, dynorphin A can alter excitatory amino acid levels in the CNS and these changes can be attenuated by the opioid antagonist nalmefene or the selective KOR antagonist, nor-binaltorphimine (33,34). However, it is also apparent that at higher concentrations these peptides activate the NMDA receptor complex and can alter excitatory amino acid neurotransmission (9,12,17,19,20,22–26,35–37,37–42). These effects are clearly not mediated through opioid receptors since they are not reversed by opioid receptor antagonists and are mimicked by peptide fragments that lack the N-terminal opioid Tyr [e.g. dynorphin A (2–17) and dynorphin A (2–13)] (see (22,40,42–44)]. At present, the evolutionary significance of these dual actions is unclear, and the relationship between individual prodynorphin-derived products, particular opioid and non-opioid actions, and cellular pathology is incompletely understood. In general, the activation of opioid receptors by normal extracellular titers of dynorphins is a natural physiological function of benefit to the cell, and, in fact, may serve a neuroprotective role. Recent evidence indicates that high levels of dynorphins, or particular prodynorphin-derived products, can induce cell death through multiple independent pathways involving glutamate receptors—at least one of which involves activation of the caspase-3 apoptotic cascade (45). More novel are recent findings indicating that dynorphins can be cytotoxic through direct protein-protein interactions that seemingly do not involve traditional receptors (10,46). Accumulating evidence suggests that the dynorphins can exert protective or proapoptotic effects depending on whether they activate opioid receptors or non-opioid mechanisms (glutamate receptors or protein-protein interactions). Irrespective of the particular mechanism(s) involved, it appears evident that dynorphins can contribute to nervous system pathology through complex interactions involving multiple receptors and signaling pathways.
2.1 Dynorphin toxicity and/or protection: Opioid receptor mechanisms
Some of the effects of dynorphin which result in neural dysfunction in vivo can be attenuated by broad acting (MOR, DOR, and KOR) antagonists such as naloxone or selective KOR antagonists such as nor-binaltorphimine (21,40,42,47–52). Similarly, opioid antagonists are reported to be beneficial in experimental models of spinal cord injury (SCI) or traumatic brain injury (TBI) suggesting that endogenous dynorphins (or potentially other endogenous opioid peptides) are disrupted and contribute to secondary CNS injury (47,53–57). The components of dynorphin-induced secondary injury or neuropathic pain attributable to opioid receptor activation are complex and somewhat more controversial than the non-opioid mediated neuropathologic effects of dynorphins. Aspects of these studies have been extensively reviewed previously (16,17,19,40,42,44,58–60), and need not be fully reviewed here. Nevertheless, consistent findings that opioid receptor activation contributes to secondary injury was a rationale for clinical trials assessing the effects of high dose, systemic naloxone as a treatment for SCI (61–63). Although naloxone was clinically safe with no real negative consequences, there were fewer beneficial effects noted than had been anticipated (61,64). The positive findings in animal models and relative safety of naloxone, suggest that further assessment of opioid antagonists is warranted [reviewed by (60)].
Though the consequences of opioids following CNS injury have been reported to be deleterious, there are also numerous reports that opioid receptor activation is inconsequential or even beneficial depending on the type of insult and parameter measured (44,65–72). Some inconsistencies undoubtedly arise from the complexity of opioid effects especially in intact animals. Opioid receptor blockade improves blood pressure resulting from hypovolemic (73), experimental endotoxin shock (74), or injury (54). Effects include complex peripheral and central opioid effects on cardiovascular function (21,34,54,75,76), multiple opioid receptor types and reported subtypes (77–79), as well as cell- and tissue-specific CNS responses. While KOR activation may have deleterious cardiovascular effects, in vitro studies suggest that KOR activation can be moderately protective in isolated neurons and oligodendroglia (discussed below). Although possible neuroprotective effects of opioid receptor activation might be inconsequential in the face of an acute cerebrovascular crisis, the complex effects of opioid system activation need to be considered in their entirety. One proposal is that pharmacologically distinct KOR1 and KOR2 subtypes mediate neuroprotective or pathological changes (21), respectively; while MOR activation is neuroprotective (77). Last, as will be discussed later, the non-opioid, excitotoxic effects of dynorphins are exceedingly potent and likely override positive or negative signals originating from opioid receptors.
A number of studies suggest that KOR activation in particular affords protection to neurons subsequent to injury (figure 2). Synthetic KOR agonists as well as dynorphin A (1–13) can improve outcome of some neurological measures of neural injury or stroke (65,66,68,69,71,72). Nerve damage resulting from TBI or SCI in mice is also reduced by KOR activation (80). Highlighting the complexity of these issues within the whole animal, rats receiving drugs with generally opposing effects, i.e., either a KOR agonist (U54,488H) or the opioid antagonist nalmefene, had improved open field behavior after SCI (53). Some of the detrimental effects of dynorphin may stem from KOR-mediated alterations in vascular function subsequent to injury [see (55,58,76,81,82)] or local disruptions in the concentrations of excitatory amino acids including glutamate (33,49,83,84). Circumstantial evidence suggests that dynorphin-induced KOR activation may concurrently protect against the non-opioid, neurotoxic effects of dynorphin (38). administration of the non-selective antagonist naloxone or the KOR selective antagonist nor-binaltorphimine) exacerbates the neurotoxicity induced by high levels (≥ 10 nM) of dynorphin A (1–17) or dynorphin A (1–13) in spinal cord cultures (38).
Figure 2.
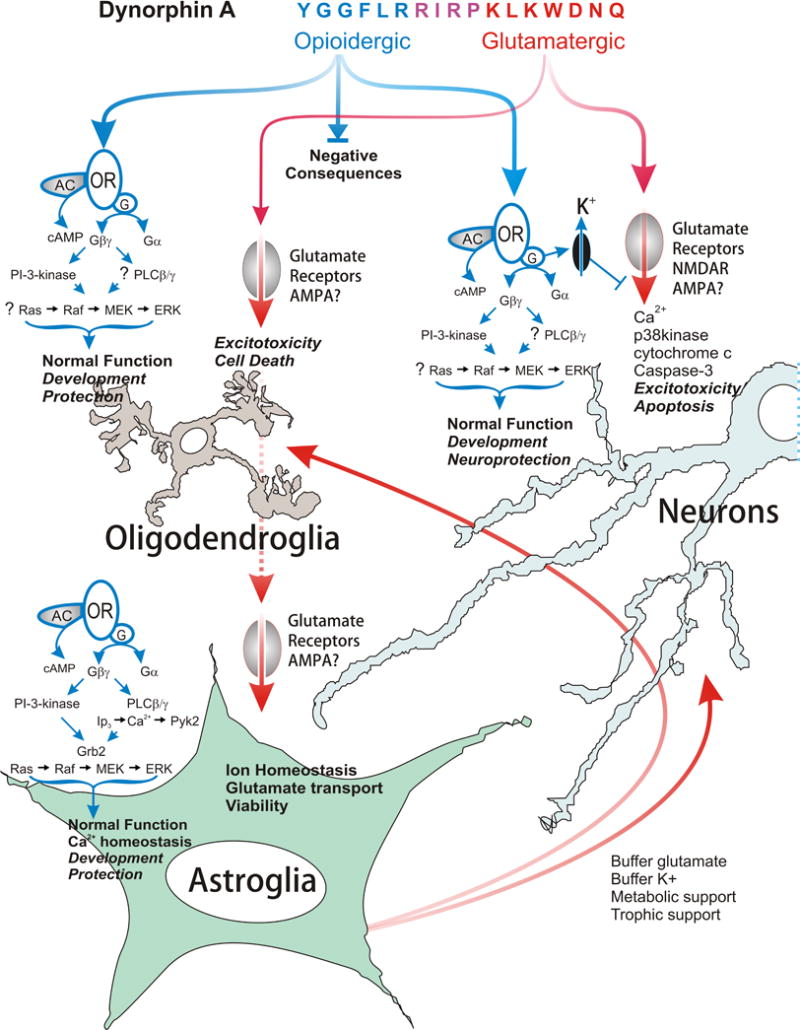
Summary of the neuroprotective and deleterious affects of dynorphin A (1–17) in neurons, astroglia, and oligodendroglia. Dynorphin A has intrinsic activity at both opioid and non-opioid, i.e., glutamatergic receptors. The N-terminal peptide derivatives are active at opioid receptors, whereas C-terminal-derived fragments can activate glutamate receptors. At physiological concentrations, dynorphin functions normally by activating kappa-opioid receptors (KOR). The activation of opioid receptors can have beneficial effects in isolated neurons, but may also have negative consequences depending on the particular neural cell type affected, as well as the nature and timing of the insult. By contrast, at supraphysiological levels, dynorphin A acts via glutamate receptors and has excitotoxic effects in neurons and oligodendroglia, and potentially destabilizes astroglia. Abbreviations: adenylyl cyclase (AC); alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate receptors (AMPA); cyclic AMP (cAMP); extracellular signal-regulated protein kinase (ERK); growth factor receptor-bound protein 2 (Grb2); G-protein complex (G); G-protein-alpha subunit (Gα); G-protein-beta-gamma subunit complex (Gβγ); inositol trisphosphate (IP3); p38 mitogen-activated protein kinase (p38kinase); N-methyl-D-aspartate receptors (NMDAR); phosphatidylinositol 3-kinase (PI-3-kinase), phospholipase C-beta-gamma (PLCβ/γ); proline-rich tyrosine kinase-2 (Pyk2); protein kinase C (PKC);.
The idea that KOR activation might be neuroprotective has support from a variety of models (30,31,53,85–91). The underlying mechanism(s) for KOR neuroprotection is not completely understood, though several possible explanations can be envisioned. The activation of postsynaptic KORs typically inhibits the electrical activity of neurons by modulating cAMP, opening K+ channels, and closing Ca2+ channels (92–96). Membrane hyperpolarization may reduce [Ca2+]i and increase the excitotoxic threshold (97,98). Selective KOR agonists (e.g., U50,488H) attenuate both the release of glutamate and increases in [Ca2+]i in synaptosomes in response to K+-induced membrane depolarization (99,100). Although KOR receptor-mediated reductions in [Ca2+]i may result from decreased activity of voltage-dependent Ca2+ channels, KOR stimulation may also increase Ca2+ efflux (101) and/or modulate the organization of NMDA induced-oscillations in [Ca2+]i (97). Interestingly, kainate-induced excitation in sensory spinal cord neurons can be attenuated by activation of MOR and kainate agonists, suggesting interactions between opioid and glutamate systems in spinal cord (102).
Besides the traditional roles ascribed to opioids (neuronal hyperpolarization mentioned above), which might increase the threshold for excitotoxic insult, opioid receptors can couple to signaling pathways involved in trophic support and cell survival (103–107). For example, G-protein coupled opioid receptors (MOR, DOR and KOR) can activate specific phosphotyrosine pathways, such as that involving extracellular signal-regulated protein kinase (ERK), typically associated with trophic factors (104,108–112). Stimulation of traditional opioid signaling pathways versus mitogen-activated protein kinase (MAPK) pathways may explain the varied effects of opioids on cell viability (35,105,113–115). MOR agonists, via G-beta-gamma-protein subunit complexes, have been shown to activate the phosphatidylinositol 3-kinase (PI-3-kinase) pathway to prevent apoptosis (105), and preliminary evidence indicates that dynorphin-induced KOR activation may also stimulate this pathway in striatal neurons (Singh and Hauser, unpublished). Recently, Coscia and coworkers have extended the above findings to show that opioid signaling can converge at both epidermal growth factor (EGF) and fibroblast growth factor (FGF) neurotrophic pathways (106,116,117). Similar opioid and FGF-2 signaling convergence is likely to occur in adult hippocampal, neural progenitors (AHP) (118,119). Although the activation of MEK1/2-ERK1/2 is mediated by MOR or DOR in AHPs (118), it is likely that KOR might similarly activate these MAPKs in other cell types (120,121), which is likely to be neuroprotective (122). Together, the above findings suggest that opioids can be intrinsically neuroprotective.
In addition to neurons, dynorphin A may enhance survival in oligodendroglia, effects that appear to be mediated through KOR. Oligodendrocytes express KOR in a specific developmental timeframe coinciding with the production of myelin basic protein within individual cells. MOR is expressed in extremely immature oligodendrocytes, prior to the time of appearance of KOR (123). Both receptors are constitutively expressed in mature oligodendrocytes in vivo and in culture. Interestingly, oligodendroglia also express proenkephalin and prodynorphin-derived peptides in a transient and coordinated manner during development. Endogenous opioids appear to modulate programmed cell death in oligodendroglia, since KOR blockade is cytotoxic to immature oligodendroglia and exacerbates glutamate-induced toxicity towards mature oligodendrocytes (115). Endogenously produced dynorphin peptides with KOR activity might act as autocrine/paracrine survival factors, at least under in vitro conditions. It is interesting to speculate that similar opioid-evoked protective mechanisms might be operative in oligodendroglia within the CNS and that pathophysiological changes in dynorphins caused by injury or disease might affect oligodendroglial function or viability.
Because a majority of the maladaptive effects of dynorphin A are thought to be mediated through glutamate receptors, and because the excitotoxic effects of sustained actions at glutamate receptors likely override any actions at opioid receptors (38), much of the discussion that follows will focus on non-opioid effects of dynorphins (figure 2).
2.2 Dynorphin toxicity: Non-opioid Mechanisms
2.2.1 Dynorphin Neurotoxicity—role of NMDA receptors
What is the nature of dynorphin interactions with NMDA receptors? Direct evidence that dynorphin A and other prodynorphin-derived peptides can interact with the NMDA receptor has been obtained from radioligand binding and neurophysiological studies (23,25,26,39,94,124,125). Radioligand studies have shown that dynorphin A (1–13) as well as dynorphin A (2–13) bind to the NMDA receptor complex (126). High affinity binding of dynorphin A (2–17) to NMDA receptors has also been shown (125). As noted, the N-terminal tyrosine is critical for activity at opioid receptors. Interestingly, however, both inhibitory and facilitatory actions of dynorphins on NMDA receptor function have been observed. While dynorphins can potentiate binding of competitive antagonists at the glutamate recognition site, they can alternatively inhibit binding of non-competitive antagonists such as MK-801 (16,125,127–129). Depending on the neuronal population under study, both facilitation and inhibition of NMDA receptor channels has been reported (23,24,124,130–133). At present, an explanation for these opposing effects remains unclear. However, the findings that dynorphin peptides inhibit NMDA-receptor gated currents in the presence of high glycine concentrations (e.g. 10 μM) but facilitate NMDA receptor currents under conditions of low glycine suggest that the concentration of this amino acid may determine whether facilitation or inhibition is seen (130). Consistent with this hypothesis, glycine or glycine site antagonists block dynorphin A (1–13)-induced enhancement of NMDA receptor-antagonist binding (128). In this regard, it is noteworthy that in Xenopus oocytes transiently transfected with NR1 and various NR2 subunits (NR2A, NR2B, NR2C, and NR2D), the degree of dynorphin-induced inhibition varies with the subunit composition of the NMDA receptor (134). Since local glycine concentrations, as well as NMDA receptor subunit composition, exhibit regional differences and are altered in response to neuronal injury, it appears likely that the effects of dynorphin may be determined by various physiological factors. Selective NR2B antagonists can reportedly differentiate antinociception from motor impairment at loci in the dorsal spinal cord (135), suggesting that tissue and region-specific differences in the expression of specific NMDA receptor subtypes may determine physiological or pathophysiological outcomes following nerve injury and excitotoxic insults.
What is (are) the site(s) of dynorphin interaction with NMDA receptors? Evidence that dynorphin acts at the polyamine site of the NMDA receptor complex has been obtained (66). An additional provocative observation is that dynorphin interacts with a portion of the NMDA receptor that is conformationally linked to the redox modulatory site (24,136). Work in progress has shown that a specific domain on the NMDA receptor forms a non-covalent complex through selective interactions with a dynorphin epitope that is distinctive from the sequence that interacts with opioid receptors. Blocking the NMDA receptor epitope cancels the dynorphin-mediated neurotoxicity (Woods A. et al., unpublished observations).
Taken together, our studies and others suggest that dynorphin interacts with various factors to modulate the response of NMDA receptors to excitatory stimuli. If dynorphin modulates the cellular response to glutamate itself, then subtle changes in dynorphins could exacerbate injury-induced increases in glutamate. Assuming that the effects of dynorphin at glutamate receptors are aberrant, a negative feedback mechanism that limits dynorphin overproduction may be lacking. Unchecked dynorphin production concurrent with reduced neuroprotection via KOR activation would tend to escalate the excitotoxic events driving secondary injury. In addition, vascular dysfunction has also been reported to result from the glutamatergic effects of dynorphin (34,89,137–139). It is our belief that at some level of increased dynorphin production, the injurious effects of activating NMDA receptors are very likely to outweigh the more modest benefits conferred by KOR mediated protection, so that the net result of dynorphin overproduction will be to drive neuropathology.
2.2.2 Dynorphin Neurotoxicity—role of AMPA/kainate receptors
In addition to modulating NMDA receptor gated channels, dynorphin can modulate AMPA/kainate responses in spinal cord dorsal horn neurons, initially depressing AMPA/kainate currents but then slowing the rate of desensitization (89,140). In certain neurons, the role of AMPA/kainate receptors in signaling dynorphin toxicity may be quite important. For example, Dynorphin A (1–17) is toxic to medium spiny neurons in striatal cultures through an apoptotic mechanism involving cytochrome c release from mitochondria and caspase-3 activation (45). Evidence for caspase-3 involvement is provided by findings that the caspase-3 inhibitor (Asp-Glu-Val-Asp-O-methyl-fluoromethylketone, also known as DEVD-fmk) significantly attenuates dynorphin-induced striatal neuron death in vitro (45). Striatal neurons, unlike many other neuronal types, express AMPA receptors early, while NMDA receptors appear later during ontogeny (141). We have found that, in contrast to spinal cord neurons, striatal toxicity is largely mediated by AMPA/kainate receptors, and only to a lesser extent by NMDA receptors (142). Evidence that AMPA receptors are involved include results showing that dynorphin-induced increases in caspase-3, can be blocked by the AMPA antagonist CNQX, and are mimicked by the ampakine CX546 (142). Moreover, the dying neurons preferentially express GluR2/3 subunits (142). Western-blot analysis showed that dynorphin also significantly elevated the levels of cytochrome c release from mitochondria and that the release was markedly attenuated by CNQX. Importantly, opioid antagonists fail to block, while NMDA antagonists only partially reduce the toxic effects of prolonged dynorphin exposure (72 h), suggesting that NMDA-mediated effects are secondary to AMPA/kainate-induced depolarization and loss of the magnesium block at NMDA receptors. This disproportionately high AMPA/kainate sensitivity is unique to striatal neurons, and may reveal a toxic mechanism normally present in other neuron types that is overshadowed by overwhelming NMDA toxicity (142).
Studies in progress indicate that dynorphin A-induced apoptosis in striatal neurons is mediated by mixed-lineage kinases (MLKs) such as p38-kinase and c-jun-N-terminal kinases 1 and 2 (JNK) (143). In cultured mouse striatal neurons, toxic concentrations of dynorphin A (≥ 10 nM) activate both p38-kinase and JNK. The extent of p38-kinase and JNK levels and their phosphorylation following dynorphin exposure coincided with the activation of caspase-3, as well as the translocation of cytochrome c from mitochondria to the cytosol. Because p38-kinase and JNK can act in a coordinated manner to signal stress-activated programmed cell death (143,144), we used selective inhibitors (SB 203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1 H-imidazole]) to inhibit p38-kinase and SP 600125 (anthrax[1,9-cd]pyrazol-6(2H)-one) to inhibit JNK1,2) to assess their role in dynorphin-induced apoptosis. Our findings indicate that the phosphorylation of p38-kinase, but not JNK-1 and 2, is necessary for dynorphin to activate caspase-3. To explore the event(s) preceding the activation of p38-kinase, we are studying the cascade involving PI-3-kinase, PTEN, and Akt (PKB). Preliminary findings suggest that PI-3-kinase is involved. Ongoing gain of function/loss of function gene targeting studies of PTEN and Akt are assessing the role of these downstream targets of PI-3-kinase in dynorphin-mediated apoptosis.
Certain dynorphin peptides can be toxic to oligodendroglia in culture. Dynorphin toxicity preferentially targets mature cells and is likely to be mediated through a non-NMDA receptor mechanism, since oligodendroglia are generally reported to express AMPA/kainate, but not NMDA, receptors. In support of this idea, the toxicity of dynorphin A (2–17) on cultured oligodendroglia is blocked almost entirely by pretreatment with CNQX (Knapp, unpublished). Alternative targets might also include metabotropic glutamate receptors. mGluR3 and mGluR5 receptors have recently been shown to be expressed on immature oligodendroglia (145,146).
2.2.3 Dynorphin toxicity may reside in the basic amino acid residues
The C-terminal region of dynorphin A, as well as the core region of big dynorphin which contains dynorphin A, are comprised of highly basic amino acid sequences. In fact, dynorphin A is one of the most basic peptides found in nature—effectively remaining positively charged throughout a pH range of 1–12. Dynorphin can directly bind to glutamate non-covalently (147). Structure-activity relationship (SAR) studies in vitro indicate that dynorphin A-derived peptide fragments containing Lys13 and either N-terminal [dynorphin A (3–13)] or C-terminal [dynorphin A (13–17)] flanking positions are neurotoxic (35), while a somewhat different neurotoxic SAR profile has been observed in vivo (148). Conversion of “peptides to bioactive products with retained or modified activities” can be a significant mechanism of peptide action (149).
The dichotomous effects of dynorphin A (opioidergic and glutamatergic) are an inherent feature of the peptide and revealed in structure-activity relationships (SAR) for particular receptors. Dynorphin A toxicity is mediated through C-terminal basic residues. Those fragments that lack basic residues and possess opioid activity alone, such as Leu-enkephalin [dynorphin A (1–5)] or [dynorphin (1–11)], are non-toxic (35). Some metabolites of dynorphin, e.g., dynorphin A (2–13), which are neurotoxic but lack opioid receptor activity, contain Lys and/or Arg residues. C-terminal peptide fragments of dynorphin A that lack the N-terminal tyrosine essential for opioid function can activate NMDA and potentially AMPA/kainate receptors. SAR studies indicate that both dynorphin A (3–13) and dynorphin A (13–17) fragments are toxic (35). While dynorphin A (3–13) toxicity in spinal cord neurons can be attenuated by NMDA antagonists (38), the 13–17 fragment is unique since its toxicity is not blocked by NMDA antagonists in spinal cord neurons (35) or AMPA antagonists in striatal neurons (142). Equimolar amounts of dynorphin A are more toxic than either the (3–13) or (13–17) fragments, suggesting a cumulative toxic effect when both domains are combined on the parent molecule.
Dynorphin A is highly conserved evolutionarily, suggesting that dynorphin’s roles as both an opioid and glutamate agonist are important. Following this logic, dynorphin’s role at glutamate receptors is likely to be quite significant and may not be entirely maladaptive. The bipotent nature of dynorphin underscores the well-established “near-symbiotic” interrelatedness of the opioid and glutamatergic systems. This is revealed not only in the pathophysiology of neurotrauma, neuropathic pain, and drug addiction, but also in the interactions of each of the two systems in normal CNS function [see for example, (86,150–154)].
2.3 Non-receptor mediated actions of dynorphins
Dynorphins may act through protein-protein interactions that are not mediated by cell surface receptors (9,10,46). We have found pronounced cytotoxicity when intracellular levels of big dynorphin are increased artificially (10). Toxicity was evoked by sub-nanomolar concentrations of big dynorphin, and was markedly more toxic than equimolar amounts dynorphin A, inferring that the toxicity was not due to its conversion to dynorphin A. It is uncertain how readily big dynorphin forms smaller dynorphin A or other fragments under these experimental conditions. Nevertheless, since big dynorphin’s toxicity was mimicked by dynorphin A, suggests that a component of parent peptide’s intrinsic toxicity resides within the dynorphin A sequence (10). Big dynorphin appeared to induce toxicity through an apoptotic mechanism that may involve synergistic interactions with the p53 tumor suppressor protein. It was proposed that big dynorphin induces cell death by virtue of its clusters of basic amino acids and resulting net positive charge that mimics (and thereby perhaps interferes with) basic domains involved in protein-protein interactions. We have recently characterized RNA and protein products of the human prodynorphin gene and, in addition to the previously described mRNAs coding for the full-length protein, have identified novel 5′-truncated transcripts (155). These transcripts give rise to N-terminally truncated proteins lacking a signal peptide that are located in the cell nucleus or cytoplasm, suggesting that they have functions other than those of the full-length protein. Interestingly, the shortest prodynorphin consisting of 52 amino acid residues includes the 32 residue big dynorphin fragment as a main component, and when this peptide is delivered into cells by transfection or electroporation, it is located in the cytosol but not in the cell nucleus, similar to big dynorphin. Truncated prodynorphin or its shorter fragment big dynorphin may interfere with intracellular processes by binding to cytoplasmic proteins. The potential for such interference was demonstrated in the study when dynorphins were delivered into the cells. Many human tumor cell lines and tumors naturally express prodynorphin lacking the signal peptide, which therefore does not translocate into the ER but degrades in the cytosol; these degradation products may potentially be involved in the control of tumor cell fate.
Although it is premature to speculate whether big dynorphin might act via this mechanism in neurons and glia, any potential disruptions in dynorphin biogenesis/degradation due to trauma or disease might have disastrous consequences for neuronal and glial function. Few studies have systematically explored whether perturbations in dynorphin posttranslational processing accompany trauma or disease, or whether imbalances in dynorphin products might disrupt neural function.
Many of the maladaptive effects of dynorphin are revealed in experiments in which dynorphin levels are increased beyond the normal physiological range, relying on exposure to large amounts of exogenously administered dynorphins. Although these studies provide compelling evidence for dualistic (beneficial versus maladaptive) roles for dynorphin, they are artificial and do not demonstrate that endogenous dynorphins actually cause pathophysiological changes. Important questions that have been less well addressed are whether “native” dynorphins act in a maladaptive manner, and what regulatory events govern pathophysiological biogenesis of dynorphins. Growing evidence indicates that aberrant dynorphin actions contribute to CNS pathology observed in a wide variety of diseases.
3. DYNORPHINS IN SECONDARY NEURAL INJURY AND THE PATHOGENESIS OF DISEASE
3.1 Neuropathic pain and pathological changes in dynorphins
Pathophysiological levels of dynorphins result in a variety of actions unrelated to the analgesic effects commonly observed at more physiological levels. For example, dynorphin immunoreactivity in the spinal cord is observed in both interneurons and projection neurons. Following nerve injury induced by nerve ligation or constriction, dynorphin immunoreactivity and the percentage of spinal neurons receiving dynorphin-immunoreactive contacts is dramatically increased (156,157). Preprodynorphin gene expression is also elevated (28,158,159). Similar changes are observed in animal models of chronic inflammatory pain (17,160). These increases are temporally correlated with the expression of tactile allodynia and hyperalgesia, suggesting that enhanced dynorphin transmission may contribute to the development of neuropathic pain states. Evidence that peripheral inflammation is associated with increased dynorphin release in the spinal cord has also been obtained (161). The findings that the intrathecal infusion of high doses of dynorphin peptides produces long-lasting tactile allodynia and hyperalgesia, which in contrast to the analgesia produced by low doses of dynorphin A (1–13), cannot be blocked by opioid antagonists. This suggests that the enhanced nociceptive effects are mediated through alternative, non-opioid mechanisms. Because the relationship between dynorphin A-related peptides and pain have been previously reviewed (22,40,42,162–165), we briefly summarize those findings here and discuss some more novel findings concerning nociceptive effects of big dynorphin.
The neurotoxicity produced by intrathecal peptide infusion, as well as the tactile allodynia and hyperalgesia, observed in animal models of neuropathic and chronic inflammatory pain can be reversed by the intrathecal infusion of dynorphin antiserum or antagonists of the NMDA receptor complex. This suggests that many of the deleterious effects are mediated by non-opioid, NMDA receptor-dependent mechanisms, although opioid receptors also participate in this response (22,40,42,162–166). The intrathecal infusion of other dynorphins, including the non-opioid peptides dynorphin A (2–17) and dynorphin A (2–13), also results in hyperalgesia, paralysis and neuronal loss, confirming that the pathophysiological effects of dynorphin peptides involve NMDA-receptor dependent mechanisms. Alternative evidence for dynorphin modulating pain is provided by studies that modulate DREAM, a Ca2++-sensing transcriptional repressor of prodynorphin (167), DREAM knockout mice have increased levels of dynorphin expression and reduced sensitivity to otherwise painful stimuli (168,169). Interestingly, in DREAM null mice, dynorphin increases may be limited to within physiological ranges (rather than the supraphysiological), since preprodynorphin transcription rates are still constrained in the absence of additional enhancers. This might explain why increased dynorphin levels do not result in neuropathic pain in mice lacking DREAM. Interestingly, artemin, a member of the GDNF family which is expressed along blood vessels and may be instrumental in attracting developing sympathetic axons to these sites (170) also reverses pathological increases in dynorphin A levels and prodynorphin immunoreactivity in the dorsal spinal cord (171). In this respect, it is intriguing that artemin has been suggested to be neuroprotective in some adult neuron populations (172,173).
In addition to dynorphin A, other prodynorphin-derived products may modulate pain. For example, big dynorphin can induce nociceptive behavior via NMDA receptors. The NMDA receptor-mediated effects of dynorphin A include neurological deficit and long-lasting mechanical, tactile, and thermal allodynia. These effects may be relevant for neuropathic pain, TBI and SCI. However, they are induced at relatively high micromolar doses. Endogenous dynorphin A appears to be involved in chronic allodynia and hyperalgesia. An important question, which remains is the identity of the most active nociceptive prodynorphin fragment, which likely contains the dynorphin A sequence as a core.
In earlier studies big dynorphin, a 32 amino acid peptide consisting of dynorphin A and dynorphin B (see figures 1 and 3), was identified in the brain as an abundant prodynorphin derived peptide. We reevaluated these observations using the combination of HPLC and RIA with antibodies, which specifically recognize the C-terminal fragment of big dynorphin and dynorphin B and which do not interact with the C-terminally extended versions of these peptides and dynorphin A. After separation of peptide fractions eluted from the SEP-PAC columns on the reverse phase HPLC column, big dynorphin was identified in several structures of human brain including the nucleus accumbens. We then tested whether big dynorphin induces nociceptive behavior similarly to dynorphin A in mice, and compared the effective doses of both peptides (9). In these experiments, intrathecal injection of big dynorphin induces a characteristic behavioral response consisting of biting and licking of the hindpaw and the tail, as well as slight hindlimb scratching directed toward the flank. Importantly, the effects occur at very low doses such as 1–10 fmol/animal. Morphine inhibited this behavior, inferring its nociceptive character. Dynorphin A produced a similar response, although the effective doses were 100-fold higher than morphine; whereas, dynorphin B had no effect even at 3 to 104-fold higher doses. The absence of effects of naloxone on big dynorphin-induced nociceptive behavior indicated that opioid receptors are not involved. The big dynorphin-induced behavior was inhibited by D-APV, a competitive NMDA receptor antagonist, and MK-801, an NMDA ion-channel blocker whereas CNQX, a non-NMDA receptor antagonist, [D-Phe7, D-His9]-substance P (6–11), a specific antagonist for NK1/substance P receptors, and MEN-10,376, a tachykinin NK2 receptor antagonist, had no effect. Big dynorphin-induced behavior may have direct or indirect actions through the NMDA receptor ion-channel complex and does not involve non-NMDA glutamate receptor mechanisms or the tachykinin system. Thus, big dynorphin is a more potent nociceptive peptide than dynorphin A, and it may be involved in the pathophysiology of chronic pain.
Figure 3.

Primary sequence of big dynorphin, dynorphin A and dynorphin B. Big dynorphin has intrinsic biological activity that differs from both dynorphin A and dynorphin B. Big dynorphin’s novel effects are not mediated by opioid receptors. The highly basic nature of the big dynorphin peptide may permit big dynorphin to interact selectively with other target molecules (46).
3.2 Drug abuse and pathological changes in dynorphins
A characteristic effect of various drugs of abuse is their ability to increase dopamine neurotransmission in the nucleus accumbens, a major projection area of dopamine neurons arising in the ventral tegmental area, and in the dorsal striatum, a terminal projection area of mesostriatal dopamine neurons (174,175). These actions are thought to mediate the rewarding effects of these agents and to contribute to the compulsive drug-seeking that characterizes drug addiction (176,177) [see reviews (178,179)]. Evidence that glutamate-dependent neuroplasticity in limbic corticostriatal systems is critical for the maintenance and reinstatement of psychostimulant addiction has also been obtained and is consistent with the documented role of glutamate systems in plasticity and learning (180–182); see review (183).
KORs are located on dopaminergic nerve terminals in the striatum and nucleus accumbens as well as on glutamatergic nerve fibers that synapse on dopamine terminals (184,185). Their activation decreases dopamine release (175,186–188). Extensive colocalization of opioid peptide precursors and dopamine receptors is also observed in these brain regions. Medium spiny neurons, which receive dense dopaminergic input, are rich in preprodynorphin and can modulate dopamine neurotransmission indirectly via efferent projections to the ventral tegmental area, substantia nigra, and ventral pallidum and directly via axon collaterals that feedback on dopamine nerve terminals (184,185,189,190).
The administration of psychostimulants and other drugs that increase dopamine neurotransmission induce preprodynorphin gene expression in both the nucleus accumbens and striatum, an effect that results from the activation of D1 dopamine receptors (190–193). More recent studies have shown that a single injection of methamphetamine is sufficient to increase extracellular levels of dynorphin A and B in the nucleus accumbens (194) (Zangen,, A., et al.; unpublished observations). Increased levels of dynorphin A immunoreactivity precede the induction of gene expression suggesting that enhanced release of prodynorphin-derived peptides and the depletion of releasable peptide pools leads to enhanced peptide synthesis. Synthetic KOR agonists inhibit dopamine release and prevent alterations in behavior and dopamine neurotransmission that occur as a consequence of repeated psychostimulant administration (187,188,195); see review (196). For this reason, it was hypothesized that the induction of prodynorphin gene expression and the ensuing increase in KOR activation is a compensatory mechanism that opposes the effects of psychostimulant administration. To date, however, it remains unclear as to whether the processing of dynorphin peptides is altered in response to drugs of abuse. Furthermore, if, as has been demonstrated in the spinal cord, high concentrations of dynorphin A and its des-tyrosine fragments bind to the NMDA receptor, thereby enhancing excitatory amino acid transmission, then the induction of preprodynorphin gene expression may, in fact, be a pathophysiological response that contributes to the addiction process. Depending upon the concentration and processing of dynorphin, as well as the density of opioid versus NMDA or possibly AMPA receptors, induction of preprodynorphin may oppose or promote neuroplastic changes in the brain that lead to compulsive drug-seeking behavior. As noted previously, the synthesis and release of dynorphin is dramatically increased in response to methamphetamine. Exposure to high doses of this psychostimulant is neurotoxic producing loss of dopamine neurons in the striatum and apoptotic cell death. An important question that arises is whether the activation of dynorphin systems protects against, or promotes, methamphetamine-induced toxicity. In this regard, it is important to note that a dysregulation of striatal dynorphin neurons is associated with various neurological disorders including Parkinson’s disease (197,198), neuroleptic-induced tardive dyskinesias (199), Tourette’s syndrome (200) and Huntington’s disease (201). To what extent the opioid and non-opioid actions of the dynorphins contribute to the pathogenesis and pathophysiology of these disorders has not yet been examined.
3.3 Spinal cord injury (SCI) and pathological changes in dynorphins
A large number of studies have implicated dynorphins in the neuropathology which occurs after TBI and SCI (9,12,17–26,35,37–42,84). Intrathecal dynorphin injections result in a wide spectrum of pathologies including hindlimb paralysis, vascular dysfunction, and neurodegeneration (44,202). As noted in previous sections, the deleterious effects are markedly reduced by MK-801 and/or competitive NMDA receptor antagonists, suggesting that NMDA receptors mediate many of these maladaptive effects (12,22,33,77,84,148,203–205). By contrast, opioid antagonists have fewer effects on injury outcome suggesting a lack of involvement by opioid signaling pathways. To assess whether dynorphins were intrinsically toxic to spinal cord neurons, we isolated sensory neurons from the dorsal spinal cord, which co-expressed KOR and NMDA receptors. Within individual neurons, we examined dynorphin-induced cytotoxicity by assaying intracellular Ca2+ ([Ca2+]i) and cell death using time lapse photomicrography and viability assays (35,38). Interestingly, our findings indicated that dynorphin A (1–13) has paradoxical effects on neurotoxicity through concurrent actions both at opioid and glutamate receptors (38). Dynorphin A (1–13)-induced neurodegeneration was preceded by abnormal increases in [Ca2+]i and accompanied by a loss of cell viability (failure to exclude ethidium ions and loss of esterase activity as measured by calcein-AM incorporation). The neurotoxicity was prevented by the NMDA antagonists MK-801, 2-amino-5-phosphopentanoic acid (AP-5), or 7-chlorokynurenic acid, suggesting the toxic effects were mediated by NMDA receptors. By contrast, KOR blockade exacerbated neurotoxicity, but only in the presence of excitotoxic levels of dynorphin. This provided circumstantial evidence that dynorphin also stimulates KOR and suggests that KOR activation may be moderately neuroprotective, but only in the presence of an excitotoxic insult (38). Thus, dynorphin seemingly modulates neurodegeneration in the spinal cord through complex mechanisms involving KOR and NMDA receptors and multiple signaling pathways.
Dynorphin A and its metabolites are released into the confines of the CNS extracellular space (3,206), and levels of expression can increase significantly during experimentally-induced injury or inflammation (11,15,44,160,207–213). Typical dynorphin increases in whole-CNS extracts following trauma are 2–3-fold, which agrees with increases in Leu-enkephalin-Arg6 [a dynorphin-derived peptide (214)] following SCI (215). Increases in preprodynorphin mRNA following TBI coincide with increases in peptide levels (15). However, depending on the prodynorphin metabolite measured, sustained concentrations greater than 10 micromolar have been proposed within the confined extracellular space of the CNS (24,206). Moreover, C-terminal dynorphin A fragments are not typically examined in normal CNS because almost all antibodies made against dynorphin are by design directed against the N-terminal tyrosine (which imparts opioid activity) to assure selectivity. Smaller dynorphin fragments are present in the CNS and may have longer half lives than dynorphin A (206,216). Many of the C-terminal fragments, which can be toxic, have sustained half-lives in vitro and in vivo (45,217).
It is challenging to measure the exact concentrations of dynorphins present within the extracellular compartment following SCI, and measurements in whole tissue homogenates will greatly underestimate actual dynorphin concentrations within the extracellular compartment (38). Concentrations of native dynorphin A within dense core vesicles are estimated to be 1 mM (218), while native- or partially processed “cryptic” dynorphin (held within prodynorphin precursors, protein aggregates and/or oligomers) are present at 3–100-fold higher concentrations than native levels and can be liberated with resulting increased activity (Yakovleva et al., in preparation). Concentrations of other, non-prodynorphin-derived aggregate peptides in dense core vesicles have been estimated to be ~42 mM (219). Only transient exposure to dynorphin A may be required to set off toxic cascades and a “hit and run” phenomenon may be operative (38).
The volume and geometry of the extracellular space of the CNS is highly dynamic. Following excitotoxic injury or trauma the volume and tortuosity of the extracellular space may decrease 10-fold, effectively concentrating substances within this compartment, while altering ion homeostasis and the kinetics of enzymes that might otherwise degrade dynorphins (220–223). Up to 10-fold SCI-induced decreases in extracellular space would act synergistically with increased dynorphin production to increase the concentration of dynorphins within the extracellular compartment. We recently found in cultured cortical and striatal neurons that K+-depolarization stimulated 2–6-fold increases in the release of dynorphin B and Leu-enkephalin-Arg6 into the medium (Yakovleva et al., in preparation). Peptide levels in the medium at times exceeded the levels in neurons by ~100-fold. Furthermore, the amounts of mature processed peptides released by depolarization were substantially higher (3 to100-fold) than their amounts in cells, suggesting activity-dependent processing (Yakovleva et al., in preparation) which might be greatly exaggerated following SCI.
4. Therapeutic Intervention
Since the pathophysiological effects of dynorphin are complex and can be manifest through multiple receptor and non-receptor mechanisms, multifaceted approaches may be needed to ameliorate the negative effects of dynorphin. Although opioid receptors mediate some aspects of dynorphin-mediated neural dysfunction in vivo and the net effect is seemingly harmful—opioid actions following trauma may be tissue specific. The potentially beneficial effects of KOR activation in some neurons and oligodendroglia would be lost with opioid receptor blockade, though this is seemingly offset by limiting the more disruptive effects of opioids on cardiovascular function. As noted earlier, we propose that any negative or positive KOR-mediated effects of dynorphin will be largely overridden by more potent and irrecoverable consequences via glutamatergic receptors. For example, spinal cord neurons continuously exposed to lethal concentrations of dynorphin sufficient to activate glutamate receptors will not survive—despite chronic activation of KOR on the same cells (38).
In general, the most severe toxicity is seen via activation of excitotoxic events signaled through glutamate (NMDA and/or AMPA/kainate) receptors. For this reason, broad strategies to reduce the negative consequences of excitotoxicity and oxidative stress, such as free radical scavengers or antioxidants, would be likely to attenuate the bulk of the deleterious effects of dynorphins. An alternative strategy might consider blocking dynorphin action at NMDA and/or AMPA/kainate receptors through specific receptor blockade. The NMDA antagonist memantine, which is in clinical trials, would likely be efficacious in attenuating the negative effects of elevated levels of dynorphins. In instances where a glutamate receptor is the therapeutic target, little or no distinction can be made between the two ligands, dynorphin, and glutamate. Since this strategy interferes with normal glutamate signaling, which is essential for neural functioning, it has distinct disadvantages as an ongoing therapeutic measure. An alternative strategy might be to target dynorphins directly.
Dynorphins are intriguing targets for therapeutic intervention since dynorphins can modulate excitotoxic glutamate signaling, either via an additive or perhaps synergistic manner, without modulating glutamate itself. Because of the profound ability of dynorphins to modify/mimic glutamate, modulating the bioactivity of dynorphins may be an excellent strategy for modulating excitatory amino acid neurotransmission and for ameliorating excitotoxic CNS damage resulting from trauma or disease. The unique-highly basic structure of dynorphin A might make it an exceptionally good target for peptide scavengers that interact and neutralize dynorphin through selective peptide-peptide interactions (Woods A. and T. Shippenberg, unpublished observations).
5. PERSPECTIVE
Dynorphins are multi-functional peptides with the potential to act via multiple receptor-effector pathways in either a protective or a toxic manner. The types of interactions that dynorphins will have in any particular biological context are ultimately defined and guided by the bioavailability of the different peptides based on the manner in which dynorphins are processed, degraded, and trafficked between subcellular compartments. Although normal processing and production of dynorphins must have a net adaptive effect, these processing and trafficking events are likely to be disrupted following disease or injury. Altered receptor expression within CNS tissues in pathological situations could also drive the net effects of dynorphin signaling in a negative direction.
Because of the pleiotropic nature of dynorphin effects, it is critically important to understand the changes in processing of dynorphin A, which accompany injury or disease states. This information will be predictive in determining not only the toxic mechanisms involved in aberrant dynorphin signaling, but also will aid in identifying potential therapeutic approaches. For example, changes in processing might result in accumulation of glutamatergic, C-terminal dynorphin fragments to a level where they drive significant losses of neurons and oligodendroglia. Alternatively, an acutely injured cell that is energetically compromised might be unlikely to expend the energy needed to undertake extensive posttranslational processing of prodynorphin. In that scenario, intact or partly processed and larger peptides such as big dynorphin might accumulate or redistribute into inappropriate subcellular compartments leading to anomalous protein-protein interactions with deleterious consequences for cell survival. Either of these situations might converge with other proapoptotic or excitotoxic events occurring within injured tissues, and the additive effects might differ according to the insult and the brain regions involved.
Dynorphin peptides are undoubtedly important modulators of disease / injury outcome in the CNS. The emerging picture from a number of laboratories suggests that effective therapeutic strategies to target dynorphins’ pathophysiological effects are likely to require a comprehensive approach that evaluates the effect of aberrant dynorphin production in the context of overlapping and converging pathological processes in a given CNS region.
Acknowledgments
This work was supported by the Kentucky Spinal Cord and Head Injury Research Trust, the NIDA Intramural Research Program and NIH grants AA13486-03, DA15097, and DA13359.
ABBREVIATIONS
- SB 203580
[4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1 H-imidazole]
- SP 600125
anthrax[1,9-cd]pyrazol-6(2H)-one
- PKB or Akt
protein kinase B
- AP-5
2-amino-5-phosphopentanoic acid
- AMPA
alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate
- CX546
ampakine
- JNK
c-jun-N-terminal kinase
- CNQX
7-chlorokynurenic acid
- p38
p38-kinase
- D-APV
D-(−)-2-amino-5-phosphonovaleric acid
- Dyn A
dynorphin A
- Dyn B
dynorphin B
- any peptide derived from the preprodynorphin gene
dynorphins
- EGF
epidermal growth factor
- ERK
extracellular signal-regulated protein kinase
- FGF
fibroblast growth factor
- [Ca2+]i
intracellular calcium
- MAPK
mitogen-activated protein kinase
- MLKs
mixed-ligand kinases
- MK-801
dozacilpine
- NMDA
N-methyl-D-aspartate
- PI-3-kinase
phosphoinositide-3-kinase
- PKC
protein kinase C
- PKB or Akt
protein kinase B
- PTEN
phosphatase and tensin homolog on chromosome 10
- SCI
spinal cord injury
- TBI
traumatic brain injury
References
- 1.Kakidani H, Furutani Y, Takahashi H, Noda M, Morimoto Y, Hirose T, Asai M, Inayama S, Nakanishi S, Numa S. Cloning and sequence analysis of cDNA for porcine beta-neo- endorphin/dynorphin precursor. Nature. 1982;298:245–249. doi: 10.1038/298245a0. [DOI] [PubMed] [Google Scholar]
- 2.Goldstein A, Tachibana S, Lowney LI, Hunkapiller M, Hood L. Dynorphin-(1–13), an extraordinarily potent opioid peptide. Proc Natl Acad Sci U S A. 1979;76:6666–6670. doi: 10.1073/pnas.76.12.6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans CJ, Hammond DL, Pasternak GW, editors. The Opiate Receptors. Humana; Clifton, New Jersey: pp. 23–71. [Google Scholar]
- 4.Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein A. Binding selectivity profiles for ligands of multiple receptor types: focus on opioid receptors. Trends Pharmacol Sci. 1987;8:456–459. [Google Scholar]
- 6.Pattee P, Ilie AE, Benyhe S, Toth G, Borsodi A, Nagalla SR. Cloning and characterization of Xen-dorphin prohormone from Xenopus laevis: A new opioid-like prohormone distinct from proenkephalin and prodynorphin. J Biol Chem. 2003 doi: 10.1074/jbc.M306724200. [DOI] [PubMed] [Google Scholar]
- 7.Danielson P, Walker D, Alrubaian J, Dores RM. Identification of a fourth opioid core sequence in a prodynorphin cDNA cloned from the brain of the amphibian, Bufo marinus: deciphering the evolution of prodynorphin and proenkephalin. Neuroendocrinology. 2002;76:55–62. doi: 10.1159/000063684. [DOI] [PubMed] [Google Scholar]
- 8.Day R, Schafer MK, Collard MW, Watson SJ, Akil H. Atypical prodynorphin gene expression in corticosteroid-producing cells of the rat adrenal gland. Proc Natl Acad Sci U S A. 1991;88:1320–1324. doi: 10.1073/pnas.88.4.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan-No K, Esashi A, Nakagawasai O, Niijima F, Tadano T, Sakurada C, Sakurada T, Bakalkin G, Terenius L, Kisara K. Intrathecally administered big dynorphin, a prodynorphin-derived peptide, produces nociceptive behavior through an N-methyl-D-aspartate receptor mechanism. Brain Res. 2002;952:7–14. doi: 10.1016/s0006-8993(02)03180-3. [DOI] [PubMed] [Google Scholar]
- 10.Tan-No K, Cebers G, Yakovleva T, Hoon GB, Gileva I, Reznikov K, Aguilar-Santelises M, Hauser KF, Terenius L, Bakalkin G. Cytotoxic effects of dynorphins through nonopioid intracellular mechanisms. Exp Cell Res. 2001;269:54–63. doi: 10.1006/excr.2001.5309. [DOI] [PubMed] [Google Scholar]
- 11.McIntosh TK, Head VA, Faden AI. Alterations in regional concentrations of endogenous opioids following traumatic brain injury in the cat. Brain Res. 1987;425:225–233. doi: 10.1016/0006-8993(87)90505-1. [DOI] [PubMed] [Google Scholar]
- 12.Bakshi R, Ni R-X, Faden AI. N-Methyl-D-aspartate (NMDA) and opioid receptors mediate dynorphin-induced spinal cord injury: Behavioral and histological studies. Brain Res. 1992;580:255–264. doi: 10.1016/0006-8993(92)90952-6. [DOI] [PubMed] [Google Scholar]
- 13.Cox BM, Molineaux CJ, Jacobs TP, Rosenberger JG, Faden AI. Effects of traumatic injury on dynorphin immunoreactivity in spinal cord. Neuropeptides. 1985;5:571–574. doi: 10.1016/0143-4179(85)90082-4. [DOI] [PubMed] [Google Scholar]
- 14.Sharma HS, Nyberg F, Olsson Y. Dynorphin A content in the rat brain and spinal cord after a localized trauma to the spinal cord and its modification with p-chlorophenylalanine. An experimental study using radioimmunoassay technique. Neurosci Res. 1992;14:195–203. doi: 10.1016/0168-0102(92)90080-v. [DOI] [PubMed] [Google Scholar]
- 15.Redell JB, Moore AN, Dash PK. Expression of the prodynorphin gene after experimental brain injury and its role in behavioral dysfunction. Exp Biol Med (Maywood) 2003;228:261–269. doi: 10.1177/153537020322800304. [DOI] [PubMed] [Google Scholar]
- 16.Shukla VK, Lemaire S. Central non-opioid physiological and pathophysiological effects of dynorphin A and related peptides. J Psychiatry Neurosci. 1992;17:106–119. [PMC free article] [PubMed] [Google Scholar]
- 17.Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Huang L-YM. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- 19.Faden AI, Salzman S. Pharmacological strategies in CNS trauma. Trends Pharmacol Sci. 1992;13:29–35. doi: 10.1016/0165-6147(92)90013-v. [DOI] [PubMed] [Google Scholar]
- 20.Gentile NT, McIntosh TK. Antagonists of excitatory amino acids and endogenous opioid peptides in the treatment of experimental central nervous system injury. Ann Emer Med. 1993;22:1028–1034. doi: 10.1016/s0196-0644(05)82746-5. [DOI] [PubMed] [Google Scholar]
- 21.McIntosh TK, Fernyak S, Yamakami I, Faden AI. Central and systemic kappa-opioid agonists exacerbate neurobehavioral response to brain injury in rats. Am J Physiol. 1994;267:R665–72. doi: 10.1152/ajpregu.1994.267.3.R665. [DOI] [PubMed] [Google Scholar]
- 22.Shukla VK, Lemaire S. Non-opioid effects of dynorphins: Possible role of the NMDA receptor. Trends Pharmacol Sci. 1994;15:420–424. doi: 10.1016/0165-6147(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Gu Y, Huang L-YM. The opioid peptide dynorphin directly blocks NMDA receptor channels in the rat. J Physiol (Lond) 1995;482:575–581. doi: 10.1113/jphysiol.1995.sp020541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Gu Y, Huang L-YM. The mechanism of action for the block of NMDA receptor channels by the opioid peptide dynorphin. J Neurosci. 1995;15:4602–4611. doi: 10.1523/JNEUROSCI.15-06-04602.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanderah TW, Laughlin T, Lashbrook JM, Nichols ML, Wilcox GL, Ossipov MH, Malan TPJ, Porreca F. Single intrathecal injections of dynorphin A or des-Tyr-dynorphins produce long-lasting allodynia in rats: blockade by MK-801 but not naloxone. Pain. 1996;68:275–281. doi: 10.1016/s0304-3959(96)03225-3. [DOI] [PubMed] [Google Scholar]
- 26.Laughlin TM, Vanderah TW, Lashbrook J, Nichols ML, Ossipov M, Porreca F, Wilcox GL. Spinally administered dynorphin A produces long-lasting allodynia: involvement of NMDA but not opioid receptors. Pain. 1997;72:253–260. doi: 10.1016/s0304-3959(97)00046-8. [DOI] [PubMed] [Google Scholar]
- 27.Bian D, Ossipov MH, Ibrahim M, Raffa RB, Tallarida RJ, Malan TP, Jr, Lai J, Porreca F. Loss of antiallodynic and antinociceptive spinal/supraspinal morphine synergy in nerve-injured rats: restoration by MK-801 or dynorphin antiserum. Brain Res. 1999;831:55–63. doi: 10.1016/s0006-8993(99)01393-1. [DOI] [PubMed] [Google Scholar]
- 28.Malan TP, Ossipov MH, Gardell LR, Ibrahim M, Bian D, Lai J, Porreca F. Extraterritorial neuropathic pain correlates with multisegmental elevation of spinal dynorphin in nerve-injured rats. Pain. 2000;86:185–194. doi: 10.1016/s0304-3959(00)00243-8. [DOI] [PubMed] [Google Scholar]
- 29.Winkler T, Sharma HS, Gordh T, Badgaiyan RD, Stalberg E, Westman J. Topical application of dynorphin A (1–17) antiserum attenuates trauma induced alterations in spinal cord evoked potentials, microvascular permeability disturbances, edema formation and cell injury: an experimental study in the rat using electrophysiological and morphological approaches. Amino Acids. 2002;23:273–281. doi: 10.1007/s00726-001-0138-y. [DOI] [PubMed] [Google Scholar]
- 30.Nichols ML, Lopez Y, Ossipov MH, Bian D, Porreca F. Enhancement of the antiallodynic and antinociceptive efficacy of spinal morphine by antisera to dynorphin A (1–13) or MK-801 in a nerve-ligation model of peripheral neuropathy. Pain. 1997;69:317–322. doi: 10.1016/S0304-3959(96)03282-4. [DOI] [PubMed] [Google Scholar]
- 31.Ossipov MH, Kovelowski CJ, Wheeler-Aceto H, Cowan A, Hunter JC, Lai J, Malan TP, Jr, Porreca F. Opioid antagonists and antisera to endogenous opioids increase the nociceptive response to formalin: demonstration of an opioid kappa and delta inhibitory tone. J Pharmacol Exp Ther. 1996;277:784–788. [PubMed] [Google Scholar]
- 32.Sharifi N, Diehl N, Yaswen L, Brennan MB, Hochgeschwender U. Generation of dynorphin knockout mice. Brain Res Mol Brain Res. 2001;86:70–75. doi: 10.1016/s0169-328x(00)00264-3. [DOI] [PubMed] [Google Scholar]
- 33.Bakshi R, Newman AH, Faden AI. Dynorphin A-(1–17) induces alterations in free fatty acids, excitatory amino acids, and motor function through an opiate- receptor-mediated mechanism. J Neurosci. 1990;10:3793–3800. doi: 10.1523/JNEUROSCI.10-12-03793.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Long JB, Rigamonti DD, Oleshansky MA, Wingfield CP, Martinez-Arizala A. Dynorphin A-induced rat spinal cord injury: evidence for excitatory amino acid involvement in a pharmacological model of ischemic spinal cord injury. J Pharmacol Exp Ther. 1994;269:358–366. [PubMed] [Google Scholar]
- 35.Hauser KF, Knapp PE, Turbek CS. Structure-activity analysis of dynorphin A toxicity in spinal cord neurons: Intrinsic neurotoxicity of dynorphin A and its carboxyl-terminal, nonopioid metabolites. Exp Neurol. 2001;168:78–87. doi: 10.1006/exnr.2000.7580. [DOI] [PubMed] [Google Scholar]
- 36.Koetzner L, Hua XY, Lai J, Porreca F, Yaksh T. Nonopioid actions of intrathecal dynorphin evoke spinal excitatory amino acid and prostaglandin E2 release mediated by cyclooxygenase-1 and -2. J Neurosci. 2004;24:1451–1458. doi: 10.1523/JNEUROSCI.1517-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faden AI, Jacobs TP. Dynorphin-related peptides cause motor dysfunction in the rat through a non-opiate action. Br J Pharmacol. 1984;81:271–276. doi: 10.1111/j.1476-5381.1984.tb10074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hauser KF, Foldes JK, Turbek CS. Dynorphin A (1–13) neurotoxicity in vitro: Opioid and non-opioid mechanisms in mouse spinal cord neurons. Exp Neurol. 1999;160:361–375. doi: 10.1006/exnr.1999.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, Huang LY. Dynorphin block of N-methyl-D-aspartate channels increases with the peptide length. J Pharmacol Exp Ther. 1998;284:826–831. [PubMed] [Google Scholar]
- 40.Caudle RM, Mannes AJ. Dynorphin: friend or foe? Pain. 2000;87:235–239. doi: 10.1016/S0304-3959(00)00360-2. [DOI] [PubMed] [Google Scholar]
- 41.Caudle RM, Isaac L. A novel interaction between dynorphin(1–13) and an N-methyl-D-aspartate site. Brain Res. 1988;443:329–332. doi: 10.1016/0006-8993(88)91628-9. [DOI] [PubMed] [Google Scholar]
- 42.Laughlin TM, Larson AA, Wilcox GL. Mechanisms of induction of persistent nociception by dynorphin. J Pharmacol Exp Ther. 2001;299:6–11. [PubMed] [Google Scholar]
- 43.Walker JM, Moises HC, Coy DH, Baldrighi G, Akil H. Nonopiate effects of dynorphin and des-Tyr-dynorphin. Science. 1982;218:1136–1138. doi: 10.1126/science.6128791. [DOI] [PubMed] [Google Scholar]
- 44.Long JB, Martinez-Arizala A, Petras JM, Holaday JW. Endogenous opioids in spinal cord injury: a critical evaluation. Cent Nerv Syst Trauma. 1986;3:295–315. doi: 10.1089/cns.1986.3.295. [DOI] [PubMed] [Google Scholar]
- 45.Singh IN, Goody RJ, Goebel SM, Martin KM, Knapp PE, Bakalkin G, Hauser KF. Dynorphin A (1–17) induces apoptosis in striatal neurons thorugh α-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor-mediated cytochrome c release and caspase-3 activation. Neurosci. 2003;122:1013–1023. doi: 10.1016/j.neuroscience.2003.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yakovleva T, Pramanik A, Kawasaki T, Tan-No K, Gileva I, Lindegren H, Langel U, Ekstrom TJ, Rigler R, Terenius L, Bakalkin G. p53 Latency. C-terminal domain prevents binding of p53 core to target but not to nonspecific DNA sequences. J Biol Chem. 2001;276:15650–15658. doi: 10.1074/jbc.M100482200. [DOI] [PubMed] [Google Scholar]
- 47.Faden AI, Takemori AE, Portoghese PS. Kappa-selective opiate antagonist nor-binaltorphimine improves outcome after traumatic spinal cord injury in rats. Cent Nerv Syst Trauma. 1987;4:227–237. doi: 10.1089/cns.1987.4.227. [DOI] [PubMed] [Google Scholar]
- 48.Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Terman GW, Chavkin C. Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci. 2004;24:4576–4584. doi: 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Faden AI, Shirane R, Chang L-H, James TL, Lemke M, Weinstein PR. Opiate-receptor antagonist improves metabolic recovery and limits neurochemical alterations associated with reperfusion after global brain ischemia in rats. J Pharmacol Exp Ther. 1990;255:451–458. [PubMed] [Google Scholar]
- 50.Benzel EC, Khare V, Fowler MR. Effects of naloxone and nalmefene in rat spinal cord injury induced by the ventral compression technique. J Spinal Disord. 1992;5:75–77. doi: 10.1097/00002517-199203000-00009. [DOI] [PubMed] [Google Scholar]
- 51.Puniak MA, Freeman GM, Agresta CA, Van Newkirk L, Barone CA, Salzman SK. Comparison of a serotonin antagonist, opioid antagonist, and TRH analog for the acute treatment of experimental spinal trauma. J Neurotrauma. 1991;8:193–203. doi: 10.1089/neu.1991.8.193. [DOI] [PubMed] [Google Scholar]
- 52.Yum SW, Faden AI. Comparison of the neuroprotective effects of the N-methyl-D-aspartate antagonist MK-801 and the opiate-receptor antagonist nalmefene in experimental spinal cord ischemia. Arch Neurol. 1990;47:277–281. doi: 10.1001/archneur.1990.00530030043014. [DOI] [PubMed] [Google Scholar]
- 53.Behrmann DL, Bresnahan JC, Beattie MS. A comparison of YM-14673, U-50488H, and nalmefene after spinal cord injury in the rat. Exp Neurol. 1993;119:258–267. doi: 10.1006/exnr.1993.1028. [DOI] [PubMed] [Google Scholar]
- 54.McIntosh TK, Hayes RL, DeWitt DS, Agura V, Faden AI. Endogenous opioids may mediate secondary damage after experimental brain injury. Am J Physiol. 1987;253:E565–E574. doi: 10.1152/ajpendo.1987.253.5.E565. [DOI] [PubMed] [Google Scholar]
- 55.DeWitt DS, Prough DS, Uchida T, Deal DD, Vines SM. Effects of nalmefene, CG3703, tirilazad, or dopamine on cerebral blood flow, oxygen delivery, and electroencephalographic activity after traumatic brain injury and hemorrhage. J Neurotrauma. 1997;14:931–941. doi: 10.1089/neu.1997.14.931. [DOI] [PubMed] [Google Scholar]
- 56.Vink R, McIntosh TK, Rhomhanyi R, Faden AI. Opiate antagonist nalmefene improves intracellular free Mg2+, bioenergetic state, and neurologic outcome following traumatic brain injury in rats. J Neurosci. 1990;10:3524–3530. doi: 10.1523/JNEUROSCI.10-11-03524.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baskin DS, Simpson RK, Jr, Browning JL, Dudley AW, Rothenberg F, Bogue L. The effect of long-term high-dose naloxone infusion in experimental blunt spinal cord injury. J Spinal Disord. 1993;6:38–43. [PubMed] [Google Scholar]
- 58.Lyeth BG, Hayes RL. Cholinergic and opioid mediation of traumatic brain injury. J Neurotrauma. 1992;9(Suppl 2):S463–S474. [PubMed] [Google Scholar]
- 59.Faden AI. Opioid and nonopioid mechanisms may contribute to dynorphin’s pathophysiological actions in spinal cord injury. Ann Neurol. 1990;27:67–74. doi: 10.1002/ana.410270111. [DOI] [PubMed] [Google Scholar]
- 60.Hall ED, Springer JE. Neuroprotection and Acute Spinal Cord Injury: A Reappraisal. NeuroRx. 2004;1:80–100. doi: 10.1602/neurorx.1.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bracken MB, Shepard MJ, Collins WF, Jr, Holford TR, Baskin DS, Eisenberg HM, Flamm E, Leo-Summers L, Maroon JC, Marshall LF, Perot PL, Jr, Piepmeier J, Sonntag VKH, Wagner FC, Jr, Wilberger JL, Winn HR, Young W. Methylprednisolone or naloxone treatment after acute spinal cord injury: 1-Year follow-up data. Results of the second National Acute Spinal Cord Injury Study. J Neurosurg. 1992;76:23–31. doi: 10.3171/jns.1992.76.1.0023. [DOI] [PubMed] [Google Scholar]
- 62.Flamm ES, Young W, Collins WF, Piepmeier J, Clifton GL, Fischer B. A phase I trial of naloxone treatment in acute spinal cord injury. J Neurosurg. 1985;63:390–397. doi: 10.3171/jns.1985.63.3.0390. [DOI] [PubMed] [Google Scholar]
- 63.Bracken MB, Holford TR. Effects of timing of methylprednisolone or naloxone administration on recovery of segmental and long-tract neurological function in NASCIS 2. J Neurosurg. 1993;79:500–507. doi: 10.3171/jns.1993.79.4.0500. [DOI] [PubMed] [Google Scholar]
- 64.Bracken MB. Pharmacological treatment of acute spinal cord injury: current status and future projects. J Emerg Med. 1993;11(Suppl 1):43–8. 43–48. [PubMed] [Google Scholar]
- 65.Hall ED, Pazara KE. Quantitative analysis of effects of kappa-opioid agonists on postischemic hippocampal CA1 neuronal necrosis in gerbils. Stroke. 1988;19:1008–1012. doi: 10.1161/01.str.19.8.1008. [DOI] [PubMed] [Google Scholar]
- 66.Caudle RM, Dubner R. Ifenprodil blocks the excitatory effects of the opioid peptide dynorphin 1–17 on NMDA receptor-mediated currents in the CA3 region of the guinea pig hippocampus. Neuropeptides. 1998;32:87–95. doi: 10.1016/s0143-4179(98)90022-1. [DOI] [PubMed] [Google Scholar]
- 67.Long JB, Petras JM, Mobley WC, Holaday JW. Neurological dysfunction after intrathecal injection of dynorphin A (1– 13) in the rat. II. Nonopioid mechanisms mediate loss of motor, sensory and autonomic function. J Pharmacol Exp Ther. 1988;246:1167–1174. [PubMed] [Google Scholar]
- 68.Silvia RC, Slizgi GR, Ludens JH, Tang AH. Protection from ischemia-induced cerebral edema in the rat by U-50488H, a kappa opioid receptor agonist. Brain Res. 1987;403:52–57. doi: 10.1016/0006-8993(87)90121-1. [DOI] [PubMed] [Google Scholar]
- 69.Baskin DS, Hosobuchi Y, Loh HH, Lee NM. Dynorphin(1–13) improves survival in cats with focal cerebral ischaemia. Nature. 1984;312:551–552. doi: 10.1038/312551a0. [DOI] [PubMed] [Google Scholar]
- 70.Long JB, Rigamonti DD, de Costa B, Rice KC, Martinez-Arizala A. Dynorphin A-induced rat hindlimb paralysis and spinal cord injury are not altered by the kappa opioid antagonist nor- binaltorphimine. Brain Res. 1989;497:155–162. doi: 10.1016/0006-8993(89)90982-7. [DOI] [PubMed] [Google Scholar]
- 71.Itoh J, Ukai M, Kameyama T. Dynorphin A-(1–13) potently prevents memory dysfunctions induced by transient cerebral ischemia in mice. Eur J Pharmacol. 1993;234:9–15. doi: 10.1016/0014-2999(93)90699-i. [DOI] [PubMed] [Google Scholar]
- 72.Itoh J, Ukai M, Kameyama T. U-50,488H, a kappa-opioid receptor agonist, markedly prevents memory dysfunctions induced by transient cerebral ischemia in mice. Brain Res. 1993;619:223–228. doi: 10.1016/0006-8993(93)91615-y. [DOI] [PubMed] [Google Scholar]
- 73.Faden AI, Holaday JW. Opiate antagonists: A role in the treatment of hypovolemic shock. Science. 1979;205:317–318. doi: 10.1126/science.451606. [DOI] [PubMed] [Google Scholar]
- 74.Faden AI, Holaday JW. Experimental endotoxin shock: the pathophysiologic function of endorphins and treatment with opiate antagonists. J Infect Dis. 1980;142:229–238. doi: 10.1093/infdis/142.2.229. [DOI] [PubMed] [Google Scholar]
- 75.Faden AI. Role of thyrotropin-releasing hormone and opiate receptor antagonists in limiting central nervous system injury. Adv Neurol. 1988;47:531–46. 531–546. [PubMed] [Google Scholar]
- 76.Armstead WM, Kurth CD. The role of opioids in newborn pig fluid percussion brain injury. Brain Res. 1994;660:19–26. doi: 10.1016/0006-8993(94)90834-6. [DOI] [PubMed] [Google Scholar]
- 77.Faden AI. Neurotoxic versus neuroprotective actions of endogenous opioid peptides: implications for treatment of CNS injury. NIDA Res Monogr. 1996;163:318–330. [PubMed] [Google Scholar]
- 78.Eliav E, Herzberg U, Caudle RM. The kappa opioid agonist GR89,696 blocks hyperalgesia and allodynia in rat models of peripheral neuritis and neuropathy. Pain. 1999;79:255–264. doi: 10.1016/s0304-3959(98)00177-8. [DOI] [PubMed] [Google Scholar]
- 79.Caudle RM, Mannes AJ, Iadarola MJ. GR89,696 is a kappa-2 opioid receptor agonist and a kappa-1 opioid receptor antagonist in the guinea pig hippocampus. J Pharmacol Exp Ther. 1997;283:1342–1349. [PubMed] [Google Scholar]
- 80.Hall ED, Wolf DL, Althaus JS, Von Voigtlander PF. Beneficial effects of the kappa opioid receptor agonist U-50488H in experimental acute brain and spinal cord injury. Brain Res. 1987;435:174–180. doi: 10.1016/0006-8993(87)91599-x. [DOI] [PubMed] [Google Scholar]
- 81.Armstead WM. Role of opioids in the physiologic and pathophysiologic control of the cerebral circulation. Proc Soc Exp Biol Med. 1997;214:210–221. doi: 10.3181/00379727-214-44089. [DOI] [PubMed] [Google Scholar]
- 82.La Torre BP, Favalli L, Rozza A, Lanza E, Scavini C, Racagni G, Savoldi F. Ischemic cerebral pathologies and K opioid receptors in rabbits. Ital J Neurol Sci. 1991;12:7–10. [PubMed] [Google Scholar]
- 83.Graham SH, Shimizu H, Newman A, Weinstein P, Faden AI. Opioid receptor antagonist nalmefene stereospecifically inhibits glutamate release during global cerebral ischemia. Brain Res. 1993;632:346–350. doi: 10.1016/0006-8993(93)91175-r. [DOI] [PubMed] [Google Scholar]
- 84.Faden AI. Dynorphin increases extracellular levels of excitatory amino acids in the brain through a non-opioid mechanism. J Neurosci. 1992;12:425–429. doi: 10.1523/JNEUROSCI.12-02-00425.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hudson CJ, Von Voigtlander PF, Althaus JS, Scherch HM, Means ED. The kappa opioid-related anticonvulsants U-50488H and U-54494A attenuate N-methyl-D-aspartate induced brain injury in the neonatal rat. Brain Res. 1991;564:261–267. doi: 10.1016/0006-8993(91)91462-a. [DOI] [PubMed] [Google Scholar]
- 86.Solbrig MV, Koob GF. Epilepsy, CNS viral injury and dynorphin. Trends Pharmacol Sci. 2004;25:98–104. doi: 10.1016/j.tips.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 87.Przewlocka B, Machelska H, Lason W. Kappa opioid receptor agonists inhibit the pilocarpine-induced seizures and toxicity in the mouse. Eur Neuropsychopharmacol. 1994;4:527–533. doi: 10.1016/0924-977x(94)90302-6. [DOI] [PubMed] [Google Scholar]
- 88.Genovese RF, Moreton JE, Tortella FC. Evaluation of neuroprotection and behavioral recovery by the kappa-opioid, PD117302 following transient forebrain ischemia. Brain Res Bull. 1994;34:111–116. doi: 10.1016/0361-9230(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 89.Tortella FC, DeCoster MA. Kappa opioids: therapeutic considerations in epilepsy and CNS injury. Clin Neuropharmacol. 1994;17:403–416. [PubMed] [Google Scholar]
- 90.Sheng WS, Hu S, Gekker G, Zhu S, Peterson PK, Chao CC. Immunomodulatory role of opioids in the central nervous system. Arch Immunol Ther Exp (Warsz) 1997;45:359–366. [PubMed] [Google Scholar]
- 91.Kong L, Jeohn G, Hudson PM, Du L, Liu B, Hong J. Reduction of lipopolysaccharide-induced neurotoxicity in mouse mixed cortical neuron/glia cultures by ultralow concentrations of dynorphins. J Biomed Sci. 2000;7:241–247. doi: 10.1007/BF02255472. [DOI] [PubMed] [Google Scholar]
- 92.Macdonald RL, Werz MA. Dynorphin A decreases voltage-dependent calcium conductance of mouse dorsal root ganglion neurones. J Physiol (Lond) 1986;377:237–249. doi: 10.1113/jphysiol.1986.sp016184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rusin KI, Giovannucci DR, Stuenkel EL, Moises HC. Kappa-opioid receptor activation modulates Ca2+ currents and secretion in isolated neuroendocrine nerve terminals. J Neurosci. 1997;17:6565–6574. doi: 10.1523/JNEUROSCI.17-17-06565.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tseng LF, editor. The Pharmacology of Opioid Peptides. Harwood Academic Publishers; pp. 131–149. [Google Scholar]
- 95.Surprenant A, Shen K-Z, North RA. Inhibiton of calcium currents by noradrenaline, somatostatin and opioids in guinea-pig submucosal neurons. J Physiol. 1990;431:585–608. doi: 10.1113/jphysiol.1990.sp018349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xiang JZ, Adamson P, Brammer MJ, Campbell IC. The kappa-opiate agonist U50488H decreases the entry of 45Ca into rat cortical synaptosomes by inhibiting N- but not L-type calcium channels. Neuropharmacology. 1990;29:439–444. doi: 10.1016/0028-3908(90)90165-n. [DOI] [PubMed] [Google Scholar]
- 97.Gruol DL, Parsons K, Sweeney D, Przewlocki R, Netzeband J, Siggins GR, Trotter C. Involvement od NMDA receptors in opiate regulationof Ca2+ oscillations in cultured hippocampal neurons. :30. doi: 10.1523/JNEUROSCI.19-22-09705.1999. (Abstract) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wagner JJ, Caudle RM, Chavkin C. Kappa-opioids decrease excitatory transmission in the dentate gyrus of the guinea pig hippocampus. J Neurosci. 1992;12:132–141. doi: 10.1523/JNEUROSCI.12-01-00132.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gannon RL, Terrian DM. Presynaptic modulation of glutamate and dynorphin release by excitatory amino acids in the guinea-pig hippocampus. Neuroscience. 1991;41:401–410. doi: 10.1016/0306-4522(91)90336-m. [DOI] [PubMed] [Google Scholar]
- 100.Gannon RL, Terrian DM. Kappa opioid agonists inhibit transmitter release from guinea pig hippocampal mossy fiber synaptosomes. Neurochem Res. 1992;17:741–747. doi: 10.1007/BF00969007. [DOI] [PubMed] [Google Scholar]
- 101.Olson KG, Welch SP. The effects of dynorphin A (1–13) and U50, 488H on free intracellular calcium in guinea pig cerebellar synaptosomes. Life Sci. 1991;48:575–581. doi: 10.1016/0024-3205(91)90473-o. [DOI] [PubMed] [Google Scholar]
- 102.Li P, Wilding TJ, Kim SJ, Calejesan AA, Huettner JE, Zhuo M. Kainate-receptor-mediated sensory synaptic transmission in mammalian spinal cord. Nature. 1999;397:161–164. doi: 10.1038/16469. [DOI] [PubMed] [Google Scholar]
- 103.Hauser KF, Mangoura D. Diversity of the endogenous opioid system in development: novel signal transduction translates multiple extracellular signals into neural cell growth and differentiation. Perspect Dev Neurobiol. 1998;5:437–449. [PubMed] [Google Scholar]
- 104.Mangoura D, Dawson G. Opioid peptides activate phospholipase D and protein kinase C-ɛ in chicken embryo neuron cultures. Proc Natl Acad Sci USA. 1993;90:2915–2919. doi: 10.1073/pnas.90.7.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Polakiewicz RD, Schieferl SM, Gingras AC, Sonenberg N, Comb MJ. mu-Opioid receptor activates signaling pathways implicated in cell survival and translational control. J Biol Chem. 1998;273:23534–23541. doi: 10.1074/jbc.273.36.23534. [DOI] [PubMed] [Google Scholar]
- 106.Belcheva MM, Szucs M, Wang D, Sadee W, Coscia CJ. mu-Opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J Biol Chem. 2001;276:33847–33853. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- 107.Bohn LM, Belcheva MM, Coscia CJ. Mu-opioid agonist inhibition of kappa-opioid receptor-stimulated extracellular signal-regulated kinase phosphorylation is dynamin-dependent in C6 glioma cells. J Neurochem. 2000;74:574–581. doi: 10.1046/j.1471-4159.2000.740574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Burt AR, Carr IC, Mullaney I, Anderson NG, Milligan G. Agonist activation of p42 and p44 mitogen-activated protein kinases following expression of the mouse δ opioid receptor in Rat-1 fibroblasts: Effects of receptor expression levels and comparisons with G-protein activation. Biochem J. 1996;320:227–235. doi: 10.1042/bj3200227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mangoura D. mu-Opioids activate tyrosine kinase focal adhesion kinase and regulate cortical cytoskeleton proteins cortactin and vinculin in chick embryonic neurons. J Neurosci Res. 1997;50:391–401. doi: 10.1002/(SICI)1097-4547(19971101)50:3<391::AID-JNR5>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 110.Wilson MA, Burt AR, Milligan G, Anderson NG. Mitogenic signalling by delta opioid receptors expressed in rat-1 fibroblasts involves activation of the p70s6k/p85s6k S6 kinase. Biochem J. 1997;325:217–222. doi: 10.1042/bj3250217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Milligan G, MacEwan DJ, Mercouris M, Mullaney I. Inverse agonism at adrenergic and opioid receptors: studies with wild type and constitutively active mutant receptors. Receptors Channels. 1997;5:209–213. [PubMed] [Google Scholar]
- 112.Georgoussi Z, Merkouris M, Mullaney I, Megaritis G, Carr C, Zioudrou C, Milligan G. Selective interactions of mu-opioid receptors with pertussis toxin- sensitive G proteins: involvement of the third intracellular loop and the c-terminal tail in coupling. Biochim Biophys Acta. 1997;1359:263–274. doi: 10.1016/s0167-4889(97)00097-9. [DOI] [PubMed] [Google Scholar]
- 113.Wang P, Gao H, Ni Y, Wang B, Wu Y, Ji L, Qin L, Ma L, Pei G. Beta-arrestin 2 functions as a G-protein-coupled receptor-activated regulator of oncoprotein Mdm2. J Biol Chem. 2003;278:6363–6370. doi: 10.1074/jbc.M210350200. [DOI] [PubMed] [Google Scholar]
- 114.Law PY, Bergsbaken C. Properties of delta opioid receptor in neuroblastoma NS20Y: receptor activation and neuroblastoma proliferation. J Pharmacol Exp Ther. 1995;272:322–332. [PubMed] [Google Scholar]
- 115.Knapp PE, Itkis OS, Zhang L, Spruce BA, Bakalkin G, Hauser KF. Endogenous opioids and oligodendroglial function: possible autocrine/paracrine effects on cell survival and development. Glia. 2001;35:156–165. doi: 10.1002/glia.1080. [DOI] [PubMed] [Google Scholar]
- 116.Belcheva MM, Haas PD, Tan Y, Heaton VM, Coscia CJ. The fibroblast growth factor receptor is at the site of convergence between mu-opioid receptor and growth factor signaling pathways in rat C6 glioma cells. J Pharmacol Exp Ther. 2002;303:909–918. doi: 10.1124/jpet.102.038554. [DOI] [PubMed] [Google Scholar]
- 117.Belcheva MM, Tan Y, Heaton VM, Clark AL, Coscia CJ. {micro} Opioid Transactivation and Down-Regulation of the Epidermal Growth Factor Receptor in Astrocytes: Implications for Mitogen-Activated Protein Kinase Signaling. Mol Pharmacol. 2003;64:1391–1401. doi: 10.1124/mol.64.6.1391. [DOI] [PubMed] [Google Scholar]
- 118.Persson AI, Thorlin T, Bull C, Eriksson PS. Opioid-induced proliferation through the MAPK pathway in cultures of adult hippocampal progenitors. Mol Cell Neurosci. 2003;23:360–372. doi: 10.1016/s1044-7431(03)00061-7. [DOI] [PubMed] [Google Scholar]
- 119.Persson AI, Thorlin T, Bull C, Zarnegar P, Ekman R, Terenius L, Eriksson PS. Mu- and delta-opioid receptor antagonists decrease proliferation and increase neurogenesis in cultures of rat adult hippocampal progenitors. Eur J Neurosci. 2003;17:1159–1172. doi: 10.1046/j.1460-9568.2003.02538.x. [DOI] [PubMed] [Google Scholar]
- 120.Bohn LM, Belcheva MM, Coscia CJ. Mitogenic signaling via endogenous kappa-opioid receptors in C6 glioma cells: evidence for the involvement of protein kinase C and the mitogen-activated protein kinase signaling cascade. J Neurochem. 2000;74:564–573. doi: 10.1046/j.1471-4159.2000.740564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Belcheva MM, Coscia CJ. Diversity of G protein-coupled receptor signaling pathways to ERK/MAP kinase. Neurosignals. 2002;11:34–44. doi: 10.1159/000057320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 123.Knapp PE, Maderspach K, Hauser KF. Endogenous opioid system in developing normal and jimpy oligodendrocytes: μ and κ opioid receptors mediate differential mitogenic and growth responses. Glia. 1998;22:189–201. doi: 10.1002/(sici)1098-1136(199802)22:2<189::aid-glia10>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 124.Lai SL, Gu Y, Huang LY. Dynorphin uses a non-opioid mechanism to potentiate N-methyl-D- aspartate currents in single rat periaqueductal gray neurons [In Process Citation] Neurosci Lett. 1998;247:115–118. doi: 10.1016/s0304-3940(98)00293-6. [DOI] [PubMed] [Google Scholar]
- 125.Tang Q, Gandhoke R, Burritt A, Hruby VJ, Porreca F, Lai J. High-affinity interaction of (des-Tyrosyl)dynorphin A (2–17) with NMDA receptors. J Pharmacol Exp Ther. 1999;291:760–765. [PubMed] [Google Scholar]
- 126.Massardier D, Hunt PF. A direct non-opiate interaction of dynorphin-(1–13) with the N-methyl-D-aspartate (NMDA) receptor. Eur J Pharmacol. 1989;170:125–126. doi: 10.1016/0014-2999(89)90149-0. [DOI] [PubMed] [Google Scholar]
- 127.Shukla VK, Lemaire S, Ibrahim IH, Cyr TD, Chen Y, Michelot R. Design of potent and selective dynorphin A related peptides devoid of supraspinal motor effects in mice. Can J Physiol Pharmacol. 1993;71:211–216. doi: 10.1139/y93-033. [DOI] [PubMed] [Google Scholar]
- 128.Dumont M, Lemaire S. Dynorphin potentiation of [3H]CGP-39653 binding to rat brain membranes. Eur J Pharmacol. 1994;271:241–244. doi: 10.1016/0014-2999(94)90288-7. [DOI] [PubMed] [Google Scholar]
- 129.Shukla VK, Prasad JA, Lemaire S. Nonopioid motor effects of dynorphin A and related peptides: structure dependence and role of the N-methyl-D-aspartate receptor. J Pharmacol Exp Ther. 1997;283:604–610. [PubMed] [Google Scholar]
- 130.Zhang L, Peoples RW, Oz M, Harvey-White J, Weight FF, Brauneis U. Potentiation of NMDA receptor-mediated responses by dynorphin at low extracellular glycine concentrations. J Neurophysiol. 1997;78:582–590. doi: 10.1152/jn.1997.78.2.582. [DOI] [PubMed] [Google Scholar]
- 131.Jarvis CR, Xiong ZG, Plant JR, Churchill D, Lu WY, MacVicar BA, MacDonald JF. Neurotrophin modulation of NMDA receptors in cultured murine and isolated rat neurons. J Neurophysiol. 1997;78:2363–2371. doi: 10.1152/jn.1997.78.5.2363. [DOI] [PubMed] [Google Scholar]
- 132.Zhang FX, Rubin R, Rooney TA. N-Methyl-D-aspartate inhibits apoptosis through activation of phosphatidylinositol 3-kinase in cerebellar granule neurons. A role for insulin receptor substrate-1 in the neurotrophic action of n-methyl-D-aspartate and its inhibition by ethanol. J Biol Chem. 1998;273:26596–26602. doi: 10.1074/jbc.273.41.26596. [DOI] [PubMed] [Google Scholar]
- 133.Hu WH, Zhang CH, Yang HF, Zheng YF, Liu N, Sun XJ, Jen J, Jen MF. Mechanism of the dynorphin-induced dualistic effect on free intracellular Ca2+ concentration in cultured rat spinal neurons. Eur J Pharmacol. 1998;342:325–332. doi: 10.1016/s0014-2999(97)01492-1. [DOI] [PubMed] [Google Scholar]
- 134.Brauneis U, Oz M, Peoples RW, Weight FF, Zhang L. Differential sensitivity of recombinant N-methyl-D-aspartate receptor subunits to inhibition by dynorphin. J Pharmacol Exp Ther. 1996;279:1063–1068. [PubMed] [Google Scholar]
- 135.Boyce S, Wyatt A, Webb JK, O’Donnell R, Mason G, Rigby M, Sirinathsinghji D, Hill RG, Rupniak NM. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology. 1999;38:611–623. doi: 10.1016/s0028-3908(98)00218-4. [DOI] [PubMed] [Google Scholar]
- 136.Laughlin TM, Kitto KF, Wilcox GL. Redox manipulation of NMDA receptors in vivo: alteration of acute pain transmission and dynorphin-induced allodynia. Pain. 1999;80:37–43. doi: 10.1016/s0304-3959(98)00191-2. [DOI] [PubMed] [Google Scholar]
- 137.Long JB, Kinney RC, Malcolm DS, Graeber GM, Holaday JW. Intrathecal dynorphin A (1–13) and (3–13) reduce spinal cord blood flow by non-opioid mechanisms. NIDA Res Monogr. 1986;75:524–6. 524–526. [PubMed] [Google Scholar]
- 138.Long JB, Kinney RC, Malcolm DS, Graeber GM, Holaday JW. Intrathecal dynorphin A 1–13 and dynorphin A 3–13 reduce rat spinal cord blood flow by non-opioid mechanisms. Brain Res. 1987;436:374–379. doi: 10.1016/0006-8993(87)91683-0. [DOI] [PubMed] [Google Scholar]
- 139.Herz A, Akil H, Simon EJ, editors. Opioids II. Springer-Verlag; Berlin: Epilepsy and neuroprotection; pp. 343–360. [Google Scholar]
- 140.Kolaj M, Cerne R, Randic M. The opioid peptide dynorphin modulates AMPA and kainate responses in acutely isolated neurons from the dorsal horn. Brain Res. 1995;671:227–244. doi: 10.1016/0006-8993(94)01333-d. [DOI] [PubMed] [Google Scholar]
- 141.Martin LJ, Furuta A, Blackstone CD. AMPA receptor protein in developing rat brain: glutamate receptor-1 expression and localization change at regional, cellular, and subcellular levels with maturation. Neuroscience. 1998;83:917–928. doi: 10.1016/s0306-4522(97)00411-9. [DOI] [PubMed] [Google Scholar]
- 142.Goody RJ, Martin KM, Goebel SM, Hauser KF. Dynorphin A toxicity in striatal neurons via an alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/Kainate receptor mechanism. Neurosci. 2003;116:807–816. doi: 10.1016/s0306-4522(02)00563-8. [DOI] [PubMed] [Google Scholar]
- 143.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 144.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 145.Luyt K, Varadi A, Molnar E. Functional metabotropic glutamate receptors are expressed in oligodendrocyte progenitor cells. J Neurochem. 2003;84:1452–1464. doi: 10.1046/j.1471-4159.2003.01661.x. [DOI] [PubMed] [Google Scholar]
- 146.Furuta A, Martin LJ. Laminar segregation of the cortical plate during corticogenesis is accompanied by changes in glutamate receptor expression. J Neurobiol. 1999;39:67–80. [PubMed] [Google Scholar]
- 147.Woods A, Zangen A. A direct chemical interaction between dynorphin and excitatory amino acids. Neurochem Res. 2001;26:395–400. doi: 10.1023/a:1010903215566. [DOI] [PubMed] [Google Scholar]
- 148.Long JB, Martinez-Arizala A, Echevarria EE, Tidwell RE, Holaday JW. Hindlimb paralytic effects of prodynorphin-derived peptides following spinal subarachnoid injection in rats. Eur J Pharmacol. 1988;153:45–54. doi: 10.1016/0014-2999(88)90586-9. [DOI] [PubMed] [Google Scholar]
- 149.Hallberg M, Nyberg F. Neuropeptide conversion to bioactive fragments–an important pathway in neuromodulation. Curr Protein Pept Sci. 2003;4:31–44. doi: 10.2174/1389203033380313. [DOI] [PubMed] [Google Scholar]
- 150.Koob GF. Neurobiology of addiction. Toward the development of new therapies. Ann N Y Acad Sci. 2000;909:170–185. doi: 10.1111/j.1749-6632.2000.tb06682.x. [DOI] [PubMed] [Google Scholar]
- 151.Bonham AC. Neurotransmitters in the CNS control of breathing. Respir Physiol. 1995;101:219–230. doi: 10.1016/0034-5687(95)00045-f. [DOI] [PubMed] [Google Scholar]
- 152.Ossipov MH, Lai J, Malan TP, Jr, Porreca F. Spinal and supraspinal mechanisms of neuropathic pain. Ann N Y Acad Sci. 2000;909:12–24. 12–24. doi: 10.1111/j.1749-6632.2000.tb06673.x. [DOI] [PubMed] [Google Scholar]
- 153.McGinty JF. Introduction to the role of excitatory amino acids in the actions of abused drugs: A symposium presented at the 1993 annual meeting of the College on Problems of Drug Dependence. Drug Alcohol Depend. 1995;37:91–94. doi: 10.1016/0376-8716(94)01062-p. [DOI] [PubMed] [Google Scholar]
- 154.Zhang WQ, Mundy WR, Thai L, Hudson PM, Gallagher M, Tilson HA, Hong JS. Decreased glutamate release correlates with elevated dynorphin content in the hippocampus of aged rats with spatial learning deficits. Hippocampus. 1991;1:391–397. doi: 10.1002/hipo.450010407. [DOI] [PubMed] [Google Scholar]
- 155.Nikoshkov T, Yakovleva T, Pasikova I, Usynin G, Cebers G, Terenius L, Bakalkin G, Hurd YL. Prodynorphin expression in the human brain. :241. (Abstract) [Google Scholar]
- 156.Nahin RL, Hylden JL, Humphrey E. Demonstration of dynorphin A 1–8 immunoreactive axons contacting spinal cord projection neurons in a rat model of peripheral inflammation and hyperalgesia. Pain. 1992;51:135–143. doi: 10.1016/0304-3959(92)90254-9. [DOI] [PubMed] [Google Scholar]
- 157.Wagner JJ, Terman GW, Chavkin C. Endogenous dynorphins inhibit excitatory neurotransmission and block LTP induction in the hippocampus. Nature. 1993;363:451–454. doi: 10.1038/363451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Iadarola MJ, Douglass J, Civelli O, Naranjo JR. Increased spinal cord dynorphin mRNA during peripheral inflammation. NIDA Res Monogr. 1986;75:406–9. 406–409. [PubMed] [Google Scholar]
- 159.Kajander KC, Sahara Y, Iadarola MJ, Bennett GJ. Dynorphin increases in the dorsal spinal cord in rats with a painful peripheral neuropathy. Peptides. 1990;11:719–728. doi: 10.1016/0196-9781(90)90187-a. [DOI] [PubMed] [Google Scholar]
- 160.Iadarola MJ, Brady LS, Draisci G, Dubner R. Enhancement of dynorphin gene expression in spinal cord following experimental inflammation: stimulus specificity, behavioral parameters and opioid receptor binding. Pain. 1988;35:313–326. doi: 10.1016/0304-3959(88)90141-8. [DOI] [PubMed] [Google Scholar]
- 161.Riley RC, Zhao ZQ, Duggan AW. Spinal release of immunoreactive dynorphin A (1–8) with the development of peripheral inflammation in the rat. Brain Res. 1996;710:131–142. doi: 10.1016/0006-8993(95)01394-6. [DOI] [PubMed] [Google Scholar]
- 162.Vanderah TW, Ossipov MH, Lai J, Malan TP, Jr, Porreca F. Mechanisms of opioid-induced pain and antinociceptive tolerance: descending facilitation and spinal dynorphin. Pain. 2001;92:5–9. doi: 10.1016/s0304-3959(01)00311-6. [DOI] [PubMed] [Google Scholar]
- 163.Stanfa L, Dickenson A. Spinal opioid systems in inflammation. Inflamm Res. 1995;44:231–241. doi: 10.1007/BF01782974. [DOI] [PubMed] [Google Scholar]
- 164.Przewlocki R, Przewlocka B. Opioids in chronic pain. Eur J Pharmacol. 2001;429:79–91. doi: 10.1016/s0014-2999(01)01308-5. [DOI] [PubMed] [Google Scholar]
- 165.Ossipov MH, Lai J, Vanderah TW, Porreca F. Induction of pain facilitation by sustained opioid exposure: relationship to opioid antinociceptive tolerance. Life Sci. 2003;73:783–800. doi: 10.1016/s0024-3205(03)00410-7. [DOI] [PubMed] [Google Scholar]
- 166.Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Terman GW, Chavkin C. Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci. 2004;24:4576–4584. doi: 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Carrion AM, Link WA, Ledo F, Mellstrom B, Naranjo JR. DREAM is a Ca2+-regulated transcriptional repressor [see comments] Nature. 1999;398:80–84. doi: 10.1038/18044. [DOI] [PubMed] [Google Scholar]
- 168.Cheng HY, Penninger JM. When the DREAM is gone:from basic science to future prospectives in pain management and beyond. Expert Opin Ther Targets. 2003;7:249–263. doi: 10.1517/14728222.7.2.249. [DOI] [PubMed] [Google Scholar]
- 169.Cheng HY, Pitcher GM, Laviolette SR, Whishaw IQ, Tong KI, Kockeritz LK, Wada T, Joza NA, Crackower M, Goncalves J, Sarosi I, Woodgett JR, Oliveira-dos-Santos AJ, Ikura M, van der KD, Salter MW, Penninger JM. DREAM is a critical transcriptional repressor for pain modulation. Cell. 2002;108:31–43. doi: 10.1016/s0092-8674(01)00629-8. [DOI] [PubMed] [Google Scholar]
- 170.Honma Y, Araki T, Gianino S, Bruce A, Heuckeroth R, Johnson E, Milbrandt J. Artemin is a vascular-derived neurotropic factor for developing sympathetic neurons. Neuron. 2002;35:267–282. doi: 10.1016/s0896-6273(02)00774-2. [DOI] [PubMed] [Google Scholar]
- 171.Gardell LR, Wang R, Ehrenfels C, Ossipov MH, Rossomando AJ, Miller S, Buckley C, Cai AK, Tse A, Foley SF, Gong B, Walus L, Carmillo P, Worley D, Huang C, Engber T, Pepinsky B, Cate RL, Vanderah TW, Lai J, Sah DW, Porreca F. Multiple actions of systemic artemin in experimental neuropathy. Nat Med. 2003;9:1383–1389. doi: 10.1038/nm944. [DOI] [PubMed] [Google Scholar]
- 172.Takahashi M. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev. 2001;12:361–373. doi: 10.1016/s1359-6101(01)00012-0. [DOI] [PubMed] [Google Scholar]
- 173.Paveliev M, Airaksinen MS, Saarma M. GDNF family ligands activate multiple events during axonal growth in mature sensory neurons. Mol Cell Neurosci. 2004;25:453–459. doi: 10.1016/j.mcn.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 174.Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Spanagel R, Herz A, Shippenberg TS. The effects of opioid peptides on dopamine release in the nucleus accumbens: an in vivo microdialysis study. J Neurochem. 1990;55:1734–1740. doi: 10.1111/j.1471-4159.1990.tb04963.x. [DOI] [PubMed] [Google Scholar]
- 176.Ito R, Dalley JW, Robbins TW, Everitt BJ. Dopamine release in the dorsal striatum during cocaine-seeking behavior under the control of a drug-associated cue. J Neurosci. 2002;22:6247–6253. doi: 10.1523/JNEUROSCI.22-14-06247.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wise RA. Drug-activation of brain reward pathways. Drug Alcohol Depend. 1998;51:13–22. doi: 10.1016/s0376-8716(98)00063-5. [DOI] [PubMed] [Google Scholar]
- 178.De Vries TJ, Shippenberg TS. Neural systems underlying opiate addiction. J Neurosci. 2002;22:3321–3325. doi: 10.1523/JNEUROSCI.22-09-03321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Weiss F, Porrino LJ. Behavioral neurobiology of alcohol addiction: recent advances and challenges. J Neurosci. 2002;22:3332–3337. doi: 10.1523/JNEUROSCI.22-09-03332.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cornish JL, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. 2000;20:RC89. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hotsenpiller G, Giorgetti M, Wolf ME. Alterations in behaviour and glutamate transmission following presentation of stimuli previously associated with cocaine exposure. Eur J Neurosci. 2001;14:1843–1855. doi: 10.1046/j.0953-816x.2001.01804.x. [DOI] [PubMed] [Google Scholar]
- 182.McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Everitt BJ, Wolf ME. Psychomotor stimulant addiction: a neural systems perspective. J Neurosci. 2002;22:3312–3320. doi: 10.1523/JNEUROSCI.22-09-03312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Meshul CK, Mcginty JF. Kappa opioid receptor immunoreactivity in the nucleus accumbens and caudate-putamen is primarily associated with synaptic vesicles in axons. Neuroscience. 2000;96:91–99. doi: 10.1016/s0306-4522(99)90481-5. [DOI] [PubMed] [Google Scholar]
- 185.Svingos AL, Colago EE, Pickel VM. Cellular sites for dynorphin activation of kappa-opioid receptors in the rat nucleus accumbens shell. J Neurosci. 1999;19:1804–1813. doi: 10.1523/JNEUROSCI.19-05-01804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci (USA) 1992;89:2046–2050. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chefer VI, Moron JA, Hope B, Rea W, Shippenberg TS. Kappa-opioid receptor activation prevents alterations in mesocortical dopamine neurotransmission that occur during abstinence from cocaine. Neuroscience. 2000;101:619–627. doi: 10.1016/s0306-4522(00)00417-6. [DOI] [PubMed] [Google Scholar]
- 188.Thompson AC, Zapata A, Justice JB, Jr, Vaughan RA, Sharpe LG, Shippenberg TS. Kappa-opioid receptor activation modifies dopamine uptake in the nucleus accumbens and opposes the effects of cocaine. J Neurosci. 2000;20:9333–9340. doi: 10.1523/JNEUROSCI.20-24-09333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Van Bockstaele EJ, Gracy KN, Pickel VM. Dynorphin-immunoreactive neurons in the rat nucleus accumbens: ultrastructure and synaptic input from terminals containing substance P and/or dynorphin. J Comp Neurol. 1995;351:117–133. doi: 10.1002/cne.903510111. [DOI] [PubMed] [Google Scholar]
- 190.Pickel VM, Chan J, Sesack SR. Cellular substrates for interactions between dynorphin terminals and dopamine dendrites in rat ventral tegmental area and substantia nigra. Brain Res. 1993;602:275–289. doi: 10.1016/0006-8993(93)90693-h. [DOI] [PubMed] [Google Scholar]
- 191.Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Turchan J, Przewlocka B, Lason W, Przewlocki R. Effects of repeated psychostimulant administration on the prodynorphin system activity and kappa opioid receptor density in the rat brain. Neuroscience. 1998;85:1051–1059. doi: 10.1016/s0306-4522(97)00639-8. [DOI] [PubMed] [Google Scholar]
- 193.Mathieu-Kia AM, Besson MJ. Repeated administration of cocaine, nicotine and ethanol: effects on preprodynorphin, preprotachykinin A and preproenkephalin mRNA expression in the dorsal and the ventral striatum of the rat. Brain Res Mol Brain Res. 1998;54:141–151. doi: 10.1016/s0169-328x(97)00338-0. [DOI] [PubMed] [Google Scholar]
- 194.Bustamante D, You ZB, Castel MN, Johansson S, Goiny M, Terenius L, Hokfelt T, Herrera-Marschitz M. Effect of single and repeated methamphetamine treatment on neurotransmitter release in substantia nigra and neostriatum of the rat. J Neurochem. 2002;83:645–654. doi: 10.1046/j.1471-4159.2002.01171.x. [DOI] [PubMed] [Google Scholar]
- 195.Heidbreder CA, Babovic-Vuksanovic D, Shoaib M, Shippenberg TS. Development of behavioral sensitization to cocaine: influence of kappa opioid receptor agonists. J Pharmacol Exp Ther. 1995;275:150–163. [PubMed] [Google Scholar]
- 196.Shippenberg TS, Maldonado R, editors. Molecular biology of drug addiction. Humana Press; New Jersey: pp. 107–133. [Google Scholar]
- 197.Newman DD, Rajakumar N, Flumerfelt BA, Stoessl AJ. A kappa opioid antagonist blocks sensitization in a rodent model of Parkinson’s disease. Neuroreport. 1997;8:669–672. doi: 10.1097/00001756-199702100-00018. [DOI] [PubMed] [Google Scholar]
- 198.Henry B, Brotchie JM. Potential of opioid antagonists in the treatment of levodopa-induced dyskinesias in Parkinson’s disease. Drugs Aging. 1996;9:149–158. doi: 10.2165/00002512-199609030-00001. [DOI] [PubMed] [Google Scholar]
- 199.Meredith GE, De Souza IE, Hyde TM, Tipper G, Wong ML, Egan MF. Persistent alterations in dendrites, spines, and dynorphinergic synapses in the nucleus accumbens shell of rats with neuroleptic-induced dyskinesias. J Neurosci. 2000;20:7798–7806. doi: 10.1523/JNEUROSCI.20-20-07798.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Leckman JF, Riddle MA, Berretini WH, Anderson GM, Hardin M, Chappell P, Bissette G, Nemeroff CB, Goodman WK, Cohen DJ. Elevated CSF Dynorphin A [1–8] in Tourette’s Syndrome. Life Sci. 1988;43:2015–2023. doi: 10.1016/0024-3205(88)90575-9. [DOI] [PubMed] [Google Scholar]
- 201.Seizinger BR, Liebisch DC, Kish SJ, Arendt RM, Hornykiewicz O, Herz A. Opioid peptides in Huntington’s disease: alterations in prodynorphin and proenkephalin system. Brain Res. 1986;378:405–408. doi: 10.1016/0006-8993(86)90946-7. [DOI] [PubMed] [Google Scholar]
- 202.Long JB, Martinez-Arizala A, Rigamonti DD, Holaday JW. Hindlimb paralytic effects of arginine vasopressin and related peptides following spinal subarachnoid injection in the rat. Peptides. 1988;9:1335–1344. doi: 10.1016/0196-9781(88)90200-8. [DOI] [PubMed] [Google Scholar]
- 203.Faden AI, Jacobs TP. Dynorphin induces partially reversible paraplegia in the rat. Eur J Pharmacol. 1983;91:321–324. doi: 10.1016/0014-2999(83)90487-9. [DOI] [PubMed] [Google Scholar]
- 204.Isaac L, O’Malley TVZ, Ristic H, Stewart P. MK-801 blocks dynorphin A (1–13)-induced loss of the tail-flick reflex in the rat. Brain Res. 1990;531:83–87. doi: 10.1016/0006-8993(90)90760-9. [DOI] [PubMed] [Google Scholar]
- 205.Skilling SR, Sun X, Kurtz HJ, Larson AA. Selective potentiation of NMDA-induced activity and release of excitatory amino acids by dynorphin: possible roles in paralysis and neurotoxicity. Brain Res. 1992;575:272–278. doi: 10.1016/0006-8993(92)90090-v. [DOI] [PubMed] [Google Scholar]
- 206.Prokai L, Kim HS, Zharikova A, Roboz J, Ma L, Deng L, Simonsick WJ., Jr Electrospray ionization mass spectrometric and liquid chromatographic- mass spectrometric studies on the metabolism of synthetic dynorphin A peptides in brain tissue in vitro and in vivo. J Chromatogr A. 1998;800:59–68. doi: 10.1016/s0021-9673(97)01295-8. [DOI] [PubMed] [Google Scholar]
- 207.Faden AI, Molineaux CJ, Rosenberger JG, Jacobs TP, Cox BM. Endogenous opioid immunoreactivity in rat spinal cord following traumatic injury. Ann Neurol. 1985;17:386–390. doi: 10.1002/ana.410170414. [DOI] [PubMed] [Google Scholar]
- 208.Weihe E, Millan MJ, Hollt V, Nohr D, Herz A. Induction of the gene encoding pro-dynorphin by experimentally induced arthritis enhances staining for dynorphin in the spinal cord of rats. Neuroscience. 1989;31:77–95. doi: 10.1016/0306-4522(89)90031-6. [DOI] [PubMed] [Google Scholar]
- 209.Noguchi K, Kowalski K, Traub R, Solodkin A, Iadarola MJ, Ruda MA. Dynorphin expression and Fos-like immunoreactivity following inflammation induced hyperalgesia are colocalized in spinal cord neurons. Mol Brain Res. 1991;10:227–233. doi: 10.1016/0169-328x(91)90065-6. [DOI] [PubMed] [Google Scholar]
- 210.Duggan AW, Riley RC. Studies of the release of immunoreactive galanin and dynorphin A (1–8) in the spinal cord of the rat. Prog Brain Res. 1996;110:137–47. 137–147. doi: 10.1016/s0079-6123(08)62571-6. [DOI] [PubMed] [Google Scholar]
- 211.Millan MJ, Czlonkowski A, Morris B, Stein C, Arendt R, Huber A, Hollt V, Herz A. Inflammation of the hind limb as a model of unilateral, localized pain: influence on multiple opioid systems in the spinal cord of the rat. Pain. 1988;35:299–312. doi: 10.1016/0304-3959(88)90140-6. [DOI] [PubMed] [Google Scholar]
- 212.Tachibana T, Miki K, Fukuoka T, Arakawa A, Taniguchi M, Maruo S, Noguchi K. Dynorphin mRNA expression in dorsal horn neurons after traumatic spinal cord injury: temporal and spatial analysis using in situ hybridization. J Neurotrauma. 1998;15:485–494. doi: 10.1089/neu.1998.15.485. [DOI] [PubMed] [Google Scholar]
- 213.Messersmith DJ, Kim DJ, Iadarola MJ. Transcription factor regulation of prodynorphin gene expression following rat hindpaw inflammation. Brain Res Mol Brain Res. 1998;53:260–269. doi: 10.1016/s0169-328x(97)00308-2. [DOI] [PubMed] [Google Scholar]
- 214.Christensson-Nylander I, Nyberg F, Ragnarsson U, Terenius L. A general procedure for analysis of proenkephalin B derived opioid peptides. Regul Pept. 1985;11:65–76. doi: 10.1016/0167-0115(85)90032-1. [DOI] [PubMed] [Google Scholar]
- 215.Sharma HS, Nyberg F, Olsson Y. Dynorphin A content in the rat brain and spinal cord after a localized trauma to the spinal cord and its modification with p-chlorophenylalanine. An experimental study using radioimmunoassay technique. Neurosci Res. 1992;14:195–203. doi: 10.1016/0168-0102(92)90080-v. [DOI] [PubMed] [Google Scholar]
- 216.Young EA, Walker JM, Houghten R, Akil H. The degradation of dynorphin A in brain tissue in vivo and in vitro. Peptides. 1987;8:701–707. doi: 10.1016/0196-9781(87)90046-5. [DOI] [PubMed] [Google Scholar]
- 217.Reed B, Zhang Y, Chait BT, Kreek MJ. Dynorphin A (1–17) biotransformation in striatum of freely moving rats using microdialysis and matrix-assisted laser desorption/ionization mass spectrometry. J Neurochem. 2003;86:815–823. doi: 10.1046/j.1471-4159.2003.01859.x. [DOI] [PubMed] [Google Scholar]
- 218.Thureson-Klein AK, Klein RL. Exocytosis from neuronal large dense-cored vesicles. Int Rev Cytol. 1990;121:67–126. 67–126. doi: 10.1016/s0074-7696(08)60659-2. [DOI] [PubMed] [Google Scholar]
- 219.Dannies PS. Protein hormone storage in secretory granules: mechanisms for concentration and sorting. Endocr Rev. 1999;20:3–21. doi: 10.1210/edrv.20.1.0354. [DOI] [PubMed] [Google Scholar]
- 220.Sykova E. Extracellular space volume and geometry of the rat brain after ischemia and central injury. Adv Neurol. 1997;73:121–35. [PubMed] [Google Scholar]
- 221.Ziak D, Chvatal A, Sykova E. Glutamate-, kainate- and NMDA-evoked membrane currents in identified glial cells in rat spinal cord slice. Physiol Res. 1998;47:365–375. [PubMed] [Google Scholar]
- 222.Zoli M, Jansson A, Sykova E, Agnati LF, Fuxe K. Volume transmission in the CNS and its relevance for neuropsychopharmacology. Trends Pharmacol Sci. 1999;20:142–150. doi: 10.1016/s0165-6147(99)01343-7. [DOI] [PubMed] [Google Scholar]
- 223.Sykova E, Vargova L, Prokopova S, Simonova Z. Glial swelling and astrogliosis produce diffusion barriers in the rat spinal cord. Glia. 1999;25:56–70. doi: 10.1002/(sici)1098-1136(19990101)25:1<56::aid-glia6>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 224.Day R, Lazure C, Basak A, Boudreault A, Limperis P, Dong W, Lindberg I. Prodynorphin processing by proprotein convertase 2. Cleavage at single basic residues and enhanced processing in the presence of carboxypeptidase activity. J Biol Chem. 1998;273:829–836. doi: 10.1074/jbc.273.2.829. [DOI] [PubMed] [Google Scholar]
- 225.Berman Y, Mzhavia N, Polonskaia A, Furuta M, Steiner DF, Pintar JE, Devi LA. Defective prodynorphin processing in mice lacking prohormone convertase PC2. J Neurochem. 2000;75:1763–1770. doi: 10.1046/j.1471-4159.2000.0751763.x. [DOI] [PubMed] [Google Scholar]