

Abstract
Pseudomonas exotoxin (PE) potently blocks protein synthesis by catalyzing the inactivation of elongation factor-2 (EF-2), and PE-cytotoxins have been used as anti-tumor agents. However, their effective clinical translation in solid tumors has been confounded by off-target delivery, systemic toxicity and short chemotherapeutic half-life. To overcome these limitations we have created toxin-resistant stem cells by modifying endogenous EF-2, and engineered them to secrete PE-cytotoxins targeting IL13Rα2 and EGFR expressed by many glioblastomas (GBM). Molecular analysis correlated efficacy of PE-targeted cytotoxins with levels of cognate receptor expression, and optical imaging was applied to simultaneously track the kinetics of protein synthesis inhibition and GBM cell viability in vivo. Stem cell-based delivery of IL13-PE in a clinically-relevant GBM resection model led to increased long-term survival of mice compared to IL13-PE protein infusion. Moreover, multiple patient-derived GBM lines responded to treatment, underscoring its clinical relevance. In sum, integrating stem cell-based engineering, multimodal imaging and delivery of PE-cytotoxins in a clinically-relevant GBM model represents a novel strategy and a potential advancement in GBM therapy.
Keywords: Cytotoxin, stem cell, molecular imaging, Glioblastoma, targeted therapy
INTRODUCTION
Pseudomonas exotoxin (PE) is a single, multi-domain peptide with the ability to enter cells and kill them by catalyzing the inactivation of elongation factor-2 (EF-2), thereby blocking protein synthesis (1). A multitude of antibody variable fragments (Fv) and ligands directed against cancerous cells have been fused to PE (2). Many human cancers, including >50 % of glioblastomas (GBM), express a variant form of the IL-13 receptor called IL-13Rα2, permitting high affinity binding of IL13-PE (3–7). Normal brain cells do not express IL-13Rα2 (8, 9), thus providing a rationale to selectively target and kill GBM cells. Epidermal growth factor receptor (EGFR) is also overexpressed and mutated in a variety of tumors, including GBM, and much effort has been channeled into developing PE-conjugated fusion proteins that target EGFR on malignant cells (10–13).
In the clinic, PE-based cytotoxins have been used with great success to treat a variety of hematologic malignancies including leukemia and Hodgkin’s lymphoma (14–18). Yet, attaining similar results in solid tumors has been hindered by inadequate distribution of the cytotoxin throughout the tumor mass coupled to the relatively short half-life of PE. Preclinical testing demonstrated that IL13-PE was highly toxic in culture and in vivo towards IL-13Rα2-expressing cells (7, 19–21), and early phase clinical trials reported that despite some adverse effects, IL13-PE was well tolerated and appeared to have a favorable risk-benefit profile (6, 21). However, in spite of great expectations, the Phase III PRECISE clinical trial failed to show a significant survival benefit in patients with recurrent GBM (22, 23). The failure of this study was likely due to the short half-life of IL13-PE coupled to ineffective delivery of the toxin to residual GBM cells following surgical resection (22).
To overcome these limitations we have engineered toxin-resistant human somatic cells and human neural stem cells (hNSCs) to robustly secrete two PE-cytotoxins, IL13-PE and EGFR targeted nanobody (ENb)-PE, that target IL13Rα2 or EGFR respectively, expressed by many GBM (3–6, 24). Nanobodies specific to EGFR or mutant EGFR variant (EGFRvIII), have recently been developed that are significantly smaller than conventional antibodies, enabling greater tissue dispersion (25) and the ability to be conjugated to other functional moieties, such as PE (26, 27). We explored the interaction and dynamics of therapeutic hNSCs in culture and in vivo in multiple models of malignant GBM. Furthermore, we tested the efficacy of IL13-PE-secreting hNSCs in a clinically relevant mouse resection model that we have recently developed (28). Cells were encapsulated in a biodegradable synthetic extracellular matrix (sECM) and placed in a resection cavity made by surgically debulking the tumor mass to recapitulate the clinical scenario. The results of this study suggest cell-based delivery of PE-cytotoxins overcome current clinical limitations by prolonging delivery time and eliminating the requirement for multiple invasive administrations. Thus, it represents a novel strategy and a potential advancement in GBM therapy.
MATERIALS AND METHODS
Viral Vector Generation
Recombinant IL13-PE and IL13 were constructed in the previously described Pico2 vector by replacing Firefly luciferase (Fluc) with either IL13-PE or IL13 (29). IL13 was PCR amplified using pORF5-hIL13 (Invitrogen) as a template with primers encoding Nhe1 and PspX1. The PCR fragment was ligated into Nhe1/PspX1-digested Pico2. To create IL13-PE, IL13 was PCR amplified as described above with primers encoding Nhe1 and EcoV. PE was amplified by PCR with primers encoding EcoV and PspX1 using pJH8 (ATCC) as a template. The two fragments were then ligated into Nhe1/ PspX1 digested Pico2. To create ENb-PE, ENb was amplified by PCR as described (26) and ligated into EcoRI/EcoRV-cut pLV-CSC-IG. Additionally, lentiviral vectors (LVs) encoding destabilized luciferase were PCR amplified from pAD-Luc1 (a kind gift from Dr. David Haslam) using primers encoding Nhe1 and Xho1, then ligated into Nhe1/Xho1-digested pLV-CSC-IG that contained an internal ribosomal entry site (IRES) driving eGFP. Construction of LV encoding FLuc-DsRed2, GFP-FLuc, and GFP-Rluc have been described previously (29). All LV constructs were packaged as lentiviral vectors in 293T/17 cells using a helper virus-free packaging system as described previously (30). Stem cells and GBM cells were transduced with LVs at varying multiplicity of infection by incubating virions in culture medium containing 4 μg mL−1 protamine sulfate (Sigma) and cells were visualized for fluorescent protein expression by fluorescence microscopy. After expansion in culture, both stem cells and GBM cells were sorted by fluorescence-activated cell sorting (FACSAria Cell Sorting System, BD Biosciences).
Cell Culture
Established human GBM lines U87, LN229, U251, Gli36vIII and primary GBM4, GBM6, GBM23, GBM64 and BT74 cells were grown as described previously (30–33). 293DT cells were cultured as previously described (34). Melanoma and breast cancer cell lines were grown as previously described (35–37). Human neural stem cells (hNSCs) (38), adipose-derived mesenchymal stem cells (hASCs), bone marrow-derived mesenchymal stem cells (hMSCs; kindly provided by D. Prockop, Tulane University) and multipotent cord blood stem cells (hMCBSCs; Cellular Engineering Technologies) were cultured as previously described (29, 39).
Generation of toxin-resistant stem cell lines
1. Mutation of endogenous EF-2
A single stranded DNA oligonucleotide (ssODN; IDT, Coralville, IA) of 47 bases long was designed to encode the wild-type sequence of EF-2 with a G-to-A transition in the first nucleotide of codon 717 that is known to confer toxin resistance (ssODN-mEF-2) (39). To confirm efficient uptake, a second ssODN was designed that included a 6-carboxyfluorescein (FAM™) on the 3′ end (ssODN-mEF-2-FAM). 293T and hNSCs were transfected with 3 μg of either ssODN-mEF-2 or ssODN-mEF-2-FAM using JET transfection reagent (Polyplus-transfection SA, Illkirch, France) according to manufacturer’s specifications. 24 hrs later, media was changed and transfection efficiency was confirmed by fluorescence microscopy on ssODN-mEF-2-FAM cells. Cells were allowed to proliferate for an additional 72 hrs at which time media was refreshed with culture medium containing 20 ng/ml of diphtheria toxin (DT). Cells were cultured under DT selection for 48 hrs, washed and cultured in normal culture medium. Cells were pulsed three additional times for 24 hrs with media containing DT at 20 ng/ml, 50 ng/ml, and 100 ng/ml. Single clones were then expanded and utilized for future experiments.
2. Introduction of IL13-PE and ENb-PE
Toxin-resistant 293T (293-Oligo) or hNSC (hNSC-Oligo) clones were seeded in 6-well plates at a density of 3x105 cells/well. 24 hrs later, cells were transfected with 1.5 μg of LV-IL13-PE or LV-ENb-PE vector (described above), that contained IL13-PE or ENb-PE cloned upstream of a fluorescence marker and puromycin resistance. 24 hrs post-transfection, growth medium was refreshed and transfection efficiency was confirmed by detection of mCherry or eGFP. 48 hrs later, cells were incubated in culture medium containing puromycin (1 μg/mL) for five days. Single clones were selected, expanded, and characterized.
Western blot analysis
To investigate the expression of IL13 and IL13-PE, 293DT cells were transfected with IL13 or IL13-PE plasmid DNA and 48 hrs later proteins were isolated from harvested cells, resolved by sodium dodecyl sulfate–polycrylamide gel electrophoresis (SDS-PAGE), and immunoblotted with antibodies against IL13 (Abcam, Cambridge, MA). To determine the expression levels of IL13Rα2 or EGFR in various cancer and stem cell lines, cell lysates were collected, resolved by SDS-PAGE and immunoblotted with an antibody against IL13Rα2 (R&D Systems, Minneapolis, MN) or EGFR (BD Biosciences). Blots were developed using enhanced chemiluminescence reagents (Amersham).
RNA extraction and reverse transcription-PCR Analysis
Total RNA was extracted from cells using the RNeasy RNA extraction kit (Qiagen) as per manufacturer’s instructions. The optimal RT-PCR conditions for human IL13Rα2 chain amplification have been previously described (40) using the primer pair (sense: 5′-ATGGCTTTCGTTTGCTTGGCTAT-3′, antisense: 5′-TCATGTATCACAGAAAAATTCTGG-3′), generating a 1130 base pair (bp) product. A human GAPDH primer pair (sense: 5′-GTCAGTGGTGGACCTGACCT-3′, antisense: 5′-TGCTGTAGCCAAATTCGTTG-3′) generating a 245 bp fragment was used as a positive control. A portion of PE was amplified using the primer pair (sense: 5′-GAACCCGACGCACGCGGCCGG -3′, antisense: 5′-CCGCTCGAGCTTCAGGTCCTC GCG CGG CG-3′) that generated a 445 bp product. The human PAX6 primer pair (sense: 5′-GAATCAGAGAAGACAGGCCA-3′, antisense: 5′-GTGTAGGTATCATAACTCCG-3′) generated a 303 bp product.
Dot Blot Analysis
To determine the expression of IL13 and IL13-PE, 293DT cells were transfected with IL13 or IL13-PE. After 24 hrs of incubation, conditioned medium was collected, spotted on filter paper adjacent to purified IL13 (Chemicon, Billerica, MA; 100 ng/μL), and immunoblotted with antibodies against IL13 (Abcam). The blots were quantified with NIH ImageJ and concentrations of IL13-PE were calculated by comparison with purified IL13.
In vitro Protein Synthesis and Cell Viability Dual bioluminescence Assays
To investigate the efficacy of PE-cytotoxins, various GBM lines were co-transduced with the reporters LV-Dest-luc (protein synthesis) and LV-Rluc (cell viability) and plated in 96 well plates (Matrical Bioscience). GBM lines were treated with conditioned medium containing known concentrations of PE-cytotoxin. At defined time points, protein synthesis was determined by incubation of cells with 150 μg/mL of D-luciferin (Biotium, Hayward, CA) and cell viability was measured by incubation of cells with 1 μg/mL coelenterazine (Nanolight). In non-transduced primary GBM lines, cell viability was determined in separate wells by measuring aggregate metabolic activity using an ATP-dependent luminescent reagent (CellTiter-Glo, Promega, Madison, WI). For all in vitro assays, photon emission was measured using a cryogenically cooled high efficiency CCD camera system (Roper Scientific, Trenton, New Jersey).
Cell cycle analysis
U251 GBM cells were treated with IL13-PE or control conditioned medium. 96 hrs after treatment, cells were pulsed for 1 h with bromodeoxyuridine and propidium iodide (PI)(Invitrogen) according to manufacturer’s instructions. Cells were harvested, stained, and cell cycle progression was processed by FACS and results were analyzed using FlowJo software.
Co-culture Studies
1. Basic co-culture
To investigate the effect of stem cell-produced IL13 and IL13-PE on GBM cell viability in co-culture analysis, GBM cells (1x103 cells/well) transduced with bimodal LV virus were seeded in a 96-well plate (Matrical Bioscience). 24 hrs later, WT or therapeutic stem cells (1x103 cells/well) were overlaid on the seeded GBM cells in triplicate. 120 hrs after the addition of stem cells (96 hrs with Gli36vIII co-cultures), fluorescence images were taken and GBM cell viability was determined by Fluc imaging following the addition of 150 μg/mL of D-luciferin (Biotium) to each well.
2. sECM-encapsulated hNSC cell viability and therapeutic efficacy co-culture
The sECM components, Hystem and Extralink (Glycosan Hystem-C, Biotime Inc.), were reconstituted according to manufacturer’s instructions. hNSCs were resuspended in Hystem and the matrix was cross-linked by adding half the volume of Extralink. Typically, 4.5 μL drops were placed in the center of glass-bottomed 96-well plates (Matrical Bioscience). After 20 min gelation time the drops were overlaid in triplicate with hNSC media containing GBM cells expressing Fluc, and cultured under standard conditions. GBM cell viability was determined by Fluc imaging as previously described (28, 39). To determine the cell viability of encapsulated hNSCs, hNSCs co-expressing mCherry and RLuc were encapsulated in sECM (500/1000/5000 cells/4.5 μL drop) and imaged 1, 3 and 5 days post-encapsulation as previously described (28).
In Vivo Studies
All in vivo procedures were approved by the subcommittee on Research Animal Care at Massachusetts General Hospital.
1. Kinetic Studies
To simultaneously investigate the effects of stem cell-mediated delivery of PE-cytotoxin on GBM cell viability and protein synthesis in vivo, GBM cells were engineered to express Dest-luc (protein synthesis) and Rluc (cell viability) by LV transduction. GBM-Dest-luc-Rluc cells (2x105 cells/animal) were mixed 2:1 with WT/therapeutic hNSC cells (1x105 cells/animal) or unmodified/IL13-PE-transfected 293DT cells and implanted subcutaneously into SCID mice (3 weeks of age, Massachusetts General Hospital, Boston; n≥3/group). On days 0, 1, and 2, Rluc imaging was performed by i.p. injection of coelenterazine (CaliperLS, Xenolight; 3.3 μg/g body weight) to determine tumor volume. To determine protein synthesis in the same tumors, Rluc imaging was followed 8 hrs later by Fluc imaging performed by administration of D-luciferin (1 mg/animal in 100 μL saline).
2. Assessing retention of encapsulated hNSCs
To determine the retention of encapsulated hNSCs in the resection cavity, U87-eGFP-FLuc tumor cells (0.5x106 cells/animal) were superficially implanted into SCID mice (n=3) through established cranial windows as previously described (28). Three days later, tumors were resected and 2x106 hNSCs transduced to express mCherry-Fluc were encapsulated in sECM and seeded in the resection cavity. FLuc imaging was performed on days 1 and 3 by intraperitoneal injection of D-Luciferin (1 mg/mouse).
3. Therapeutic Efficacy in GBM resection model
To determine the effects of hNSC-IL13-PE or infusion of IL13-PE protein on GBM progression, U87-GFP-FLuc cells (0.5x106 cells/animal) were implanted into SCID mice through established cranial windows (n=25). 6 days later, mice underwent surgical tumor resection as previously described (28). hNSC-IL13-PE (n=7) or hNSC-mCherry (n=7) cells were encapsulated in sECM (2x106 cells) and seeded in the resection cavity, or 10 μls of concentrated conditioned media (n=5) containing 40 ng IL13-PE was infused into the cavity boarder. There was also a resection alone group (n=4) where the tumor was resected and the cavity left untreated. Tumor progression was monitored by serial Fluc imaging performed between days 1 to 85 following intraperitoneal injection of D-Luciferin (1 mg/mouse).
Tissue processing
Mice bearing resected tumors were perfused with formalin and brains were extracted. After 24 hours in formalin, brains were transferred to 30 % w/v sucrose. 24 hours later, brains were sectioned on a vibratome at a thickness of 20 μm. Photomicrographs and fluorescence images of brain sections were acquired using a Nikon E400 light microscope attached to a SPOT CCD digital camera (Diagnostic Instruments) and processed using ImageJ software.
Statistical analysis
Data were analyzed by Student t-test when comparing two groups and expressed as mean ± s.e.m. Differences were considered significant at P<0.05 (*); P<0.01 (#); P<0.001 (§). Survival times of mouse groups were compared using a Mantel Cox log-rank test.
RESULTS
Construction of toxin resistant stem cells that secrete PE-cytotoxin
Elongation factor-2 (EF-2) can be specifically inactivated by ADP-ribosylating toxins such as diphtheria toxin (DT) and pseudomonas exotoxin (PE), thereby inhibiting protein synthesis and killing the cell (41–43). To engineer human cell lines capable of delivering PE-based cytotoxins, we first rendered them resistant to PE (Fig. 1A). Toxin resistance was conferred by utilizing single-stranded oligonucleotides that converted endogenous EF-2 into a toxin-resistant variant in both human somatic cells (SO-Oligo) and human neural stem cells (hNSC-Oligo) (Fig. 1A). The mutation of EF-2 had no marked impact on the proliferation rates, or other cellular characteristics of either Oligo cell line (Fig. 1B, Fig. S1). Both SO-Oligo and hNSC-Oligo cell lines displayed resistance to purified DT up to a concentration of 1000 ng/mL (Fig. 1C), an effect not observed in parental lines. A number of plasmids encoding IL13-PE, ENb-PE, and non-PE containing variants were constructed (Fig. S2A) and initially characterized in SO-Oligo cells. SO-Oligo cells were able to transiently express IL13-PE, unlike parental lines which were only able to express the non-toxic IL13 version (Fig. S2B). Furthermore, unmodified SO cells displayed a marked reduction in cell viability upon treatment with conditioned medium containing IL13-PE, whilst toxin-resistant SO-Oligo cells were unaffected (Fig. S2C). To quantify the secretory capacity of SO-Oligo cells, they were transfected with IL13-PE or IL13 plasmids (Fig. S2D), and analysis of conditioned medium revealed proteins were secreted at >10 ng/ml/106 cells (Fig. S2E). To create therapeutic stem cell lines that stably secreted PE-cytotoxins, hNSC-Oligo lines were transfected with plasmids encoding PE-cytotoxins upstream of a fluorescent reporter, to enable identification of stably expressing clones (Fig. 1D). hNSC-Oligo cells were engineered to stably express IL13-PE (Fig. 1E+F) and EGFR Nanobody (ENb)-PE (Fig. 1G+H). These results confirm that human somatic and hNSCs can be modified to display resistance to EF-2-ADP-ribosylating toxins and engineered to secrete functional PE-cytotoxins.
Fig. 1. Engineering toxin-resistant stem cells that secrete PE-cytotoxins.
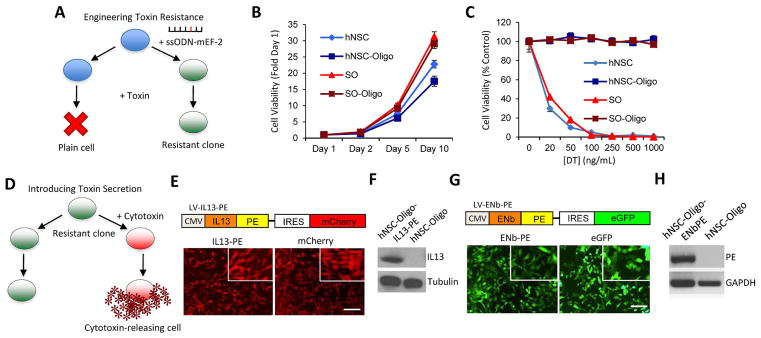
(A) Schematic representation of the approach used to make cells resistant to PE-immunotoxins. Wild-type cells (blue) were transfected with ssODN-mEF-2 to introduce a mutation in the endogenous EF-2 gene. Cells were cultured in toxin-containing medium, and single resistant clones were selected and expanded. (B) Summary data showing the growth rates of SO, hNSC, SO-Oligo, and hNSC-Oligo cells at day 1, 2, 5 and 10 days. (C) Summary graph demonstrating the viability of SO, hNSC, SO-Oligo, and hNSC-Oligo cells treated with DT at increasing concentrations (0–1000 ng/mL). (D) Schematic representation of the approach for introducing cytotoxins into toxin-resistant cells. Resistant clones (green) were transfected with a vector encoding IL13-PE cloned upstream of a fluorescence marker and puromycin selection cassette. Cells were cultured in the presence of puromycin (1 μg/mL) and positive clones (red) were selected, expanded and characterized. (E) Toxin resistant hNSC-Oligo cells were engineered to stably express IL13-PE-mCherry or mCherry alone. Fluorescence images showing mCherry expression in both hNSC lines (inset x10 magnification). (F) Western blot analysis demonstrating IL13-PE protein expression in the lysates of hNSC-Oligo cells stably expressing IL13-PE. (G) Toxin resistant hNSC-Oligo cells were engineered to stably express ENb-PE-eGFP or eGFP alone. Fluorescence images showing eGFP expression in both hNSC lines (inset x10 magnification). (H) RT-PCR demonstrating the presence of PE transcript expression in hNSC-ENb-PE cells versus an absence in unmodified hNSC cells. Scale bars, 100 μm. Data are expressed as mean ± s.e.m. Scale bars, 100 μm.
Stem cell-delivered PE-cytotoxins reduce cell viability in multiple GBMs
To establish potential sensitivity of GBMs towards cell-delivered IL13-PE, levels of their cognate receptor, IL13Rα2, were determined in multiple established GBM lines (Fig. 2A). Variation in receptor protein levels corresponded to the degree of response to IL13-PE treatment in co-culture experiments (Fig. 2B+C). Furthermore, ectopic over-expression of IL13Rα2 in a GBM line with low endogenous levels of receptor (Gli36vIII-IL13Rα2 or Gli36vIII-IL13Rα2-RLuc; Fig. 2D–F) displayed the greatest degree of sensitivity to hNSC-IL13-PE, indicating the requirement of cognate receptor for cytotoxin binding (Fig. 2G+H). To assess the potential sensitivity of GBMs to hNSC-ENb-PE, EGFR expression was analyzed in a panel of GBMs (Fig. 2I). Again, a decrease in GBM viability was correlated to the level of EGFR expressed, with the line expressing constitutively active EGFR (Gli36vIII) showing the greatest efficacy (Fig. 2J+K). These results demonstrate that stem cell-delivered PE-cytotoxins reduce the viability of GBM lines in a response that is consistent to the level of receptor expressed by the target GBM line.
Fig. 2. Stem cell-delivered PE-cytotoxins reduce cell viability of GBMs.

(A) Western blot analysis of IL13Rα2 expression from the lysates of established human GBM lines. (B) Representative fluorescence images from final day of co-culture. GBM cells (green) and hNSCs (red). (C) Cell viability of human GBM cells expressing eGFP-Fluc, co-cultured with hNSC-IL13-PE-mCherry or hNSC-mCherry. (D) Lentiviral vectors were constructed consisting of IL13Rα2 cloned upstream of IRES-eGFP or as a direct fusion to eGFP-RLuc. (E) Representative fluorescence images (inset x10 magnification) and (F) Western blot analysis revealing the expression of IL13Rα2 and IL13Rα2-eGFP-RLuc in unmodified and LV-tranduced Gli36vIII cells. (G) Representative fluorescence images from final day of co-culture. Gli36vIII-IL13Rα2 cells (green) and hNSCs (red). (H) Cell viability of Gli36vIII-IL13Rα2 GBM cells expressing eGFP-Fluc, co-cultured with hNSC-IL13-PE-mCherry or hNSC-mCherry. (I) Western blot analysis of EGFR expression from the lysates of established human GBM lines. (J) Representative fluorescence images from final day of co-culture. GBM cells (red) and hNSCs (green). (K) Cell viability of human GBM cells expressing mCherry-Fluc, co-cultured with hNSC-ENb-PE-eGFP or hNSC-eGFP. Scale bars, 100 μm. Data are expressed as mean ± s.e.m. Significance of unpaired t test, * P < 0.05; # P <0.01; § P < 0.001; treated versus control for each GBM line.
Imaging the kinetics of IL13-PE action on GBMs in vitro and in vivo
Given the GBM-specific expression of IL13Rα2 versus the widespread distribution of EGFR, coupled to our previous data indicating that stem cell-delivered IL13-PE was more efficacious than ENb-PE in the GBM lines tested, we largely focused on this IL13-PE cytotoxin in subsequent experiments. To investigate the molecular mechanisms that mediate IL13-PE toxicity and to define the kinetics of cytotoxin action, three GBM lines expressing low (Gli36vIII), intermediate (U251) and high (Gli36vIII-IL13Rα2) levels of receptor were transduced to express the novel protein synthesis reporter destabilized luciferase (dsluc), in addition to Renilla luciferase (Rluc) to assess cell viability (Fig. 3A, Fig. S3). Dual bioluminescence imaging (BLI) was performed daily to simultaneously assess the extent of protein synthesis and cell viability in GBMs treated with control or IL13-PE-containing conditioned medium (Fig. 3A). IL13-PE treatment did not inhibit protein synthesis or affect cell viability in Gli36vIII that lack IL13Rα2, indicating the requirement of this receptor for therapeutic efficacy (Fig. 3A). Response to IL13-PE was most rapid and profound in Gli36vIII-IL13Rα2 cells overexpressing the receptor. In addition, inhibition of protein synthesis preceded the reduction in cell viability providing evidence that PE-induced toxicity was via inhibition of protein synthesis (Fig. 3A). Cell cycle analysis revealed that protein synthesis inhibition was associated with a marked reduction in the number of cells in S-phase and an accumulation of cells in G2/M phase (Fig. 3B+C). These results demonstrate that binding of IL13-PE to IL13Rα2 causes inhibition of protein synthesis, induces cell cycle arrest and ultimately reduces GBM cell viability in culture.
Fig. 3. IL13-PE decreases GBM viability by blocking protein synthesis and inducing cell cycle arrest.

(A) Plot of cell viability and protein synthesis in three GBM lines treated with IL13-PE or control conditioned medium and followed daily by simultaneous Fluc and Rluc imaging. (B) Scatter plots and summary data (C) of cell cycle analysis performed on U251 GBM cells treated with IL13-PE or control conditioned media. Data are expressed as percentage of total cell population in G1, S, or G2-M. (D) U251 GBM cells were engineered to co-express the protein synthesis marker, dsluc, and cell viability marker Rluc. These cells were mixed with either hNSC-Oligo-IL13-PE or unmodified hNSC cells and implanted subcutaneously in SCID mice. Bioluminescence imaging was performed daily to assess protein synthesis and GBM viability. Representative visible light plus superimposed bioluminescence images of tumors are shown (color scale units, photons min−1 cm−2; here and in subsequent figures) and quantified. Data are expressed as mean ± s.e.m. Significance of unpaired t test, * P < 0.05; # P <0.01; § P < 0.001; treated versus control for each GBM line.
To test if IL13-PE secreted by hNSCs could cause a similar response to GBMs in vivo, we applied non-invasive BLI to track protein synthesis and cell viability in sub-cutaneous tumors made by mixing U251-dsluc-Rluc cells with either hNSC-IL13-PE or unmodified hNSCs (Fig. 3D). Stem cell-secreted IL13-PE reduced protein synthesis in U251 cells by over 90 % as early as 24 hours post-treatment that persisted through 48 hours (Fig. 3D). This was accompanied by a 70 % reduction in cell viability after 24 hours that increased to >90 % by 48 hours (Fig. 3D). Similar results were obtained by using human somatic cells expressing IL13-PE (Fig. S4) and hNSC-ENb-PE (Fig. S5). Together, these results indicate that cellular delivery of PE-cytotoxins can efficiently and robustly reduce GBM viability in vivo by inhibiting protein synthesis, and that these anti-tumor effects can be tracked non-invasively using BLI.
Stem cell-delivered IL13-PE kills residual tumor and prolongs survival of mice in a GBM resection model
One of the major limitations in current GBM therapies is the inadequate distribution of chemotherapeutic agents towards residual GBM cells following surgical resection. To investigate the efficacy of stem cell-delivered IL13-PE on residual tumor cells in vivo, they were encapsulated in a synthetic extracellular matrix (sECM) and applied to a mouse tumor resection model (Fig. 4A). In culture, encapsulated hNSCs remained viable and could escape the sECM (Fig. S6A+B). Furthermore, U87 GBM cells were shown to be sensitive to IL13-PE secreted by encapsulated hNSC-IL13-PE cells in vitro (Fig. S6C+D), indicating they would be an ideal GBM line to use in this resection experiment. U87-GFP-Fluc tumors were surgically debulked under a fluorescence microscope (Fig. 4B–E), and encapsulated hNSCs were injected into the resection cavity (Fig. 4F+G). The extent of surgical resection was determined by comparing Fluc signal pre- and post-resection, with >95 % of the tumor typically resected (Fig. 4H, Fig. S6E). Mice were followed longitudinally for changes in tumor volume by serial BLI (Fig. 4I+J). 21 days after resection variable tumor masses had developed in the control sECM-hNSC and IL13-PE infusion groups, whilst no tumor could be detected in the sECM-hNSC-IL13-PE group (Fig. 4J). This was most likely due to the initial retention of encapsulated therapeutic stem cells in the resection cavity, versus the transient exposure of infused IL13-PE (Fig. S6F + S7). Indeed, this group conferred a statistically significant survival benefit with a median survival of 79 days versus 48 days in the IL13-PE infusion group and 26 days in both the resection alone and encapsulated control hNSC groups (P=0.0003 versus hNSC group; P=0.0093 versus IL13-PE infusion group; Fig. 4K). These results demonstrate that encapsulated hNSCs secreting IL13-PE significantly increase anti-GBM efficacy compared to direct injection of IL13-PE protein in a preclinical model of GBM resection.
Fig. 4. Stem cell-delivered IL13-PE kills residual tumor and prolongs survival of mice in a GBM resection cavity.

(A) Schematic showing how the resection experiment was performed. (B) U87 GBM cells were transduced with LV-Fluc-eGFP and imaged 48 hours later for eGFP expression. (C) A cranial window was established in mice and 2 x 105 U87-Fluc-eGFP cells/mouse were superficially implanted through the cranial window. Dashed circle demarcates the established tumor in the cranial window. (D) Fluorescence photomicrograph showing an established U87-Fluc-eGFP GBM (green) in the cranial window. (E) Light image of cranial window following tumor resection. (F) Fluorescence photomicrograph showing hNSC-mCherry cells (red) encapsulated in sECM and placed in the tumor resection cavity. (G) Light image showing encapsulated hNSCs in tumor resection cavity. (H) Light image and fluorescent micrograph of a coronal brain section following GBM resection. U87-Fluc-eGFP (green), DAPI-stained nuclei (blue). Mean Fluc signal intensity was quantified and plotted before and following surgical resection in both stem cell groups to determine the extent of resection. (I) Plot of Fluc signal intensity before and after tumor resection in treatment groups. (J) Representative visible light plus superimposed bioluminescence images (color scale units, photons min−1 cm−2) before and at various time points following tumor resection. Four treatment groups correspond to resection alone; resection plus hNSC-mCherry in sECM; resection plus hNSC-IL13PE in sECM and resection plus infusion of IL13-PE conditioned medium (40 ng/mouse) into the resection cavity. Tumor recurrence was determined 21 days post- tumor resection in the four treatment groups, assessed histologically and by correlative fluorescence imaging of serial coronal brain sections. U87-Fluc-eGFP (green), DAPI-stained nuclei (blue). Dashed white boxes indicate region of interest. (K) Kaplan-Meier survival curves of mice bearing resected U87-Fluc-eGFP tumors in the four treatment groups. Significance of comparison groups assessed by Mantel Cox Log rank test and tabulated. Scale bars, 100 μm (B,H (right), J (far right)) and 400 μm (H (left), J (brightfield images)). Data are expressed as mean ± s.e.m.
IL13-PE has anti-tumor effects in patient-derived GBMs
To investigate the clinical potential and wider applicability of IL13-PE as a therapeutic agent, the presence of IL13Rα2 transcript was assessed in five patient-derived GBM lines and a panel of cancer and stem cell lines (Fig. 5A). The efficacy of IL13-PE was once again correlated to IL13Rα2 transcript levels expressed by the cancer cells (Fig. 5B, Fig. S8). Patient-derived GBM lines expressing robust IL13Rα2 transcript (GBM23, GBM64 and BT74) displayed a significant reduction in cell viability upon IL13-PE treatment (Fig. 5B). This correlation was also observed in established GBM and melanoma lines (Fig. 5B). None of the stem cell lines tested responded to IL13-PE treatment confirming the cancer-selective nature of IL13-PE (Fig. 5B). To test if stem cell-delivered IL13-PE could also act on primary patient-derived GBMs, hNSC-IL13-PE or unmodified hNSCs were encapsulated in sECM and surrounded by primary GBMs (Fig. 5C). GBM23 and BT74 displayed a profound decrease in viability compared to controls (P<0.0001; Fig. 5D). These results demonstrate that sECM-encapsulated hNSCs expressing IL13-PE have therapeutic efficacy against primary patient-derived GBMs that express IL13Rα2. Furthermore, IL13-PE can act on non-GBM cancers, indicating broader therapeutic potential.
Fig. 5. IL13-PE has anti-tumor effects on primary human GBMs.

(A) Semi-quantitative RT-PCR analysis of IL13Rα2 expression from a variety of cancer and stem cell lines. (B) Cell viability of these cancer and stem cell lines following treatment with 25 ng/mL IL13-PE or control conditioned medium. (C) hNSCs expressing mCherry or IL13-PE were encapsulated in sECM and co-cultured with primary human GBM cells. Representative photomicrographs of GBM neurospheres and encapsulated hNSCs. Black dashed line indicates edge of sECM. (D) Plot showing GBM cell viability following 5 days culture with encapsulated hNSCs expressing mCherry or IL13-PE. Scale bars, 100 μm. Data are expressed as mean ± s.e.m. Significance of unpaired t test, * P < 0.05; # P <0.01; § P < 0.001; treated versus control for each cell line. Scale bar, 100 μm.
DISCUSSION
In this study we engineered toxin-resistant somatic and human neural stem cells (hNSCs) to secrete two PE-cytotoxins, IL13-PE and ENb-PE, that target IL13Rα2 and EGFR respectively, expressed by many GBMs. We show that both PE-cytotoxins impaired cell viability in multiple GBM lines via protein synthesis inhibition and cell cycle arrest, and that these events could be non-invasively followed in vivo. Furthermore, we show that IL13-PE-secreting sECM-encapsulated hNSCs transplanted in the surgical resection cavity significantly delayed tumor regrowth and increased survival of mice bearing established GBMs (Fig. 6). Finally we demonstrated efficacy of IL13-PE on patient-derived GBMs and melanoma lines, underscoring its therapeutic relevance and wider therapeutic applicability.
Fig. 6. Summary scheme outlining the action of stem cell-delivered PE-cytotoxins in the tumor resection cavity.
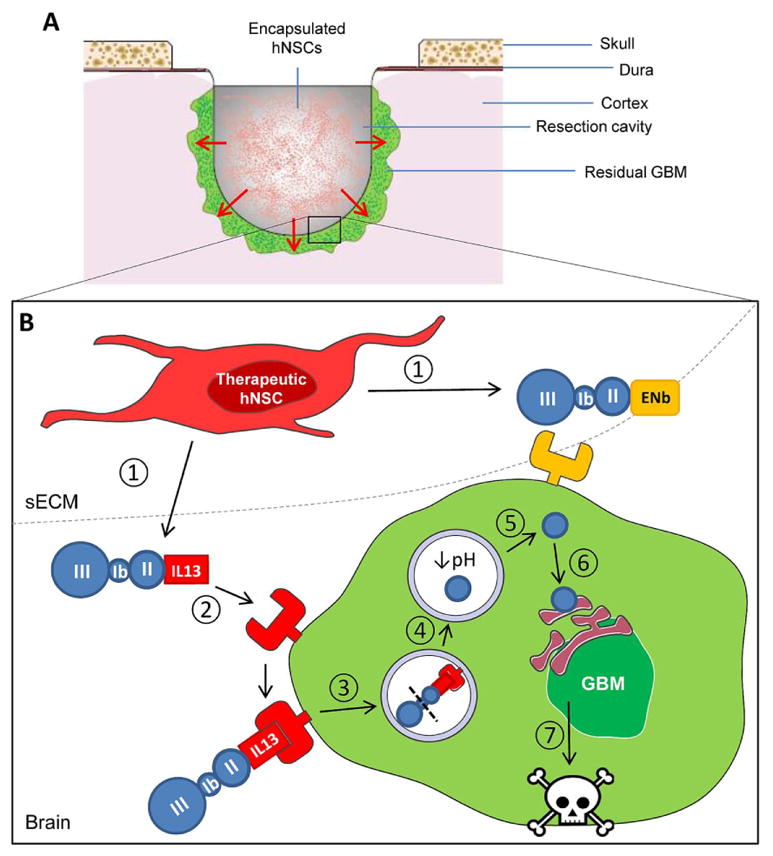
(A) A cross section of the tumor resection cavity. Residual tumor cells (green) are depicted in the resection margins. The cavity has been filled with therapeutic hNSCs encapsulated in sECM. Secreted PE-cytotoxins pass through the sECM matrix where they can act on remaining tumor cells in the resection border. (B) An enlarged projection of the resection cavity highlighting the mechanism of action of the PE-cytotoxin strategy. 1. IL13-PE and ENb-PE cytotoxins are secreted from toxin-resistant hNSCs that are encapsulated in the resection cavity. 2. The PE-cytotoxins bind to their cognate receptor at high affinity. In this case IL13-PE is binding to IL13Rα2 expressed on the cell surface of the GBM cell. 3. Toxin-bound receptor is internalized. Domain II of PE mediates the translocation of the complex into the endosome. 4. Once in the endosome, the protease furin cleaves PE and activates catalytic domain III. 5. The low pH of the endosome compartment causes the toxin to translocate into the cytosol. 6. The catalytic domain traverses the endoplasmic reticulum and inhibits protein synthesis by binding to elongation factor-2. 7. Inhibition of protein synthesis leads to GBM cell death.
Pioneering work by Pastan and colleagues has helped to propel targeted cytotoxin therapy into the clinical arena, and a multitude of Fv antibody fragments and ligands directed against cancerous cells have been fused to PE and tested in multiple malignancies (2). Despite promising preclinical results, translating PE into humans has been problematic due to a combination of the short half-life of protein formulations and its ineffective delivery throughout the tumor mass. Indeed the Phase III PRECISE clinical trial of IL13-PE on recurrent GBM failed to demonstrate a significant improvement compared to the current standard of care because of these therapeutic limitations (22). It was reported that only 68 % of catheters were positioned in accordance with protocol guidelines suggesting that IL13-PE was inadequately distributed to the residual GBM at sufficient concentrations to have a therapeutic effect (22). Recent evidence from our lab and others have shown stem cells can be utilized as unique vehicles for highly effective local delivery of anti-tumor therapies (44–46). In this study we proposed that stem cell delivery of PE-cytotoxins could circumvent current limitations by allowing the continuous release of therapeutic agent. Previous studies have demonstrated that a mutant form of elongation factor-2 (EF-2) confers resistance to EF-2-ADP-ribosylating toxins (39, 47), and that mammalian cells can be modified to secrete diphtheria-fused toxin (34). We used a similar strategy to engineer a toxin-resistant stem cell line able to stably secrete PE-cytotoxins by utilizing single-stranded oligonucleotides encoding mutant EF-2 to convert endogenous EF-2 into a toxin-resistant variant. Encapsulation of these therapeutic stem cells in the surgical resection cavity permitted retention and local delivery of therapeutic proteins directly into the resection margins, which could act on residual cancerous cells.
The first stage of treatment for patients suffering from GBM typically consists of surgical debulking of the tumor mass where possible, with substantial resection corresponding to prolonged survival (48, 49). In the vast majority of preclinical GBM studies, therapeutic agents are tested on intact solid tumors. In light of the critical role tumor resection has in GBM management, we felt it was essential to use a clinically relevant model to test GBM therapies, in this case one that incorporated debulking of the GBM mass to recapitulate the clinical scenario. Previously a small number of preclinical studies have incorporated surgical resection of GBM (28, 50, 51). We extended this model by incorporating non-invasive bioluminescence imaging (BLI), an approach we have used in previous studies (28, 29, 31). The expression of biomodal (bioluminescent and fluorescent) imaging markers in tumor cells and therapeutic stem cells allowed us to assess multiple processes in vivo including determining the degree of surgical resection, assessing the therapeutic success of IL13-PE by following the growth of residual tumor cells, tracking the kinetics of protein synthesis inhibition and non-invasively confirming the retention of encapsulated hNSCs in the resection cavity. Regarding the last point, BLI indicated that encapsulated hNSCs were efficiently retained in the resection cavity for at least 24 hours. However, we were unable to detect viable cells 72 hours after encapsulation, perhaps as a result of the species mismatch between the human stem cells and albeit immunocompromised murine host. If mouse stem cells were engineered to secrete PE-toxins, their in vivo survival in a syngeneic context should vastly improve, along with the efficacy of treatment. This should enable greater insight into the complete response of PE-based cytotoxins in models which more accurately recapitulate the human situation. This is an avenue we are actively pursuing.
In this study we have tested the therapeutic efficacy of two PE-cytotoxins, directed against different receptor targets expressed by malignant cells. In the case of IL13-PE, many human cancers, including over half of GBMs, express a variant form of the IL-13 receptor called IL13Rα2, permitting high affinity binding of IL13-PE (3–6). The EGFR pathway is also highly overactive in gliomagenesis, where gene amplification of EGFR and activating mutations in EGFR can be found in up to 70 % of all GBMs (24). We show that the therapeutic efficacy of PE-cytotoxins is correlated to the levels of cognate receptor expressed on the GBM cell. We opted to test IL13-PE therapy in the resection model as it showed a somewhat greater efficacy towards the GBM lines we tested compared to ENb-PE. In addition, the expression of IL13Rα2 is largely restricted to malignant cells whereas EGFR is widely expressed by somatic cells (52), thus potentially compromising ENb-PE’s cancer-specific mode of action. The choice of U87 GBM in the resection model was three fold: it is a well characterized established GBM line, it expresses moderate levels of IL13RαR2 to represent a more pathophysiological scenario, and it grows as a nodular mass enabling a greater degree of precision during resection, thus facilitating a more tractable resection model. One criticism is that U87 tumors do not recapitulate every aspect of GBM, such as its highly invasive nature (53). We are in the process of testing encapsulated therapeutic stem cells in a resection model incorporating highly invasive primary-derived GBMs, although this represents a significant technical challenge.
An additional concern is GBMs that do not express the target receptor or comprise a heterogeneous receptor population will be refractory to the cytotoxin therapy. Indeed approximately half of the cancer lines that were tested in this study did not express IL13Rα2 transcript and were recalcitrant to IL13-PE cytotoxin. A recent report demonstrated that treatment of IL13Rα2-negative pancreatic cancer cells with HDAC inhibitors were able to upregulate IL13Rα2 expression, rendering them sensitive to IL13-PE therapy (54). Strategies to sensitize resistant cancer populations, such as treating with epigenetic modifiers, are required to optimize a cytotoxins’ therapeutic effectiveness. It is also conceivable to engineer toxin-resistant stem cells that secrete multiple PE-cytotoxins or sensitizing agents in parallel, so heterogeneous cancer populations can be sensitized and targeted in situ. Despite this caveat, it was encouraging to confirm the response of patient-derived GBMs to IL13-PE and its efficacy in melanoma lines, underscoring its clinical relevance and broader therapeutic potential.
It is tempting to speculate the prospect of tailoring a therapeutic response towards individual GBMs in the clinic. Resected tumor could be molecularly profiled and toxin-resistant human stem cell lines subsequently engineered to secrete the most efficacious combination of cytotoxins. As with most preclinical studies, translation into human patients would need to be adapted to tackle the challenges imposed by the new host. For example the volume of the resection cavity is significantly larger in humans compared to mice necessitating an unfeasibly large number of encapsulated therapeutic stem cells. One strategy to mitigate this problem might be to ‘coat’ the resection margins with encapsulated cells and pack the core of the cavity with factors that might attract GBM cells, thus enhancing the overall therapeutic effect.
CONCLUSION
Our studies reveal the creation and application of stem cell-delivered PE-cytotoxins directed against brain tumors. To our knowledge, this is the first report to describe the engineering and utilization of toxin-resistant therapeutic stem cells. We show that stem cell-based delivery of IL13-PE in a novel GBM resection model led to increased long-term survival of mice compared to IL13-PE protein infusion. Moreover, multiple patient-derived GBM lines responded to treatment, underscoring its clinical relevance. In sum, stem cell-based delivery of PE-cytotoxins can potentiate anti-tumor response by prolonging delivery time and eliminating the requirement for multiple invasive administrations, and thus represents a novel strategy and a potential advancement in GBM therapy.
Supplementary Material
Fig. S1. Characterizing the hNSC-Oligo cell line
(A) Immunocytochemistry on parental (hNSC) and toxin-resistant (hNSC-Oligo) lines using an anti-nestin antibody (red) and DAPI nuclear counterstain (blue). Scale bar 100 μm (B) RT-PCR transcript analysis on cDNA derived from unmodified hNSCs and hNSC-Oligo lines. Primer pairs amplify Pax6 (an hNSC marker) and GAPDH products from cDNA. (C) Genomic profiling of parental and modified hNSC lines. (D) Proliferation assay performed on hNSC and hNSC-IL13-PE lines at day 1, 3 and 5. Data are expressed as mean ± s.e.m. Scale bar, 100 μm.
Fig. S2. Secretion and activity of IL13-PE produced by PE-resistant somatic cells
(A) Schematic of vectors encoding the cytotoxins IL13-PE and ENb-PE and the non-toxic variants IL13 and ENb conjugated to either IRES-eGFP or IRES-mCherry. (B) Fluorescence images of 293T (SO) or 293-Oligo (SO-Oligo) cells transiently transfected with vectors encoding IL13-PE or IL13. Expression of eGFP was determined 24 hours post-transfection. (C) Plot of cell viability of toxin sensitive (293T) and toxin resistant (293DT) cells towards purified PE, and conditioned medium containing IL13 or the toxic IL13-PE variant. Representative visible light plus superimposed bioluminescence images of treated cells in 96 well plates are shown (color scale units, photons min−1 cm−2). (D) Western blot and dot blot (E) analysis using anti-IL13, demonstrating the expression and secretion of IL13-PE from SO-Oligo cells transfected with a vector encoding IL13-PE. Scale bar, 100 μm.
Fig. S3. Ectopic expression of IL13Rα2 confers IL13-PE sensitivity to resistant GBMs
(A) Gli36vIII-IL13Rα2-eGFP-RLuc cells were plated at increasing numbers and bioluminescence imaging was performed. The plot reveals a direct correlation between cell number and Rluc activity and a representative bioluminescence image of increasing cell numbers in a 96 well plate is shown (color scale units, photons min−1 cm−2). (B–E) Gli36vIII cells transduced with LV encoding IL13Rα2 (B) or IL13Rα2-eGFP-Rluc (C) were treated with control or IL13-PE conditioned medium at increasing concentrations (0–750 ng/ml), and cell viability was determined 48 hrs after treatment. Gli36vIII-IL13Rα2 (D) or Gli36vIII-IL13Rα2-eGFP-RLuc (E) cell lines were co-transduced with LV-destabilized-luciferase and treated with 100 ng of IL13-PE or control medium. Levels of protein synthesis were measured by luciferase-based assay 0, 24, 48, or 72 hrs post-treatment.
Fig. S4. IL13-PE-expressing somatic cells reduce GBM viability by inhibiting protein synthesis in vivo
(A) Schematic showing how the experiment was performed. (B+C) Gli36vIII-IL13Rα2 (B) or U251 (C) GBM cells were co-transduced to express eGFP-Rluc (Rluc) and destabilized-luciferase (dsluc), and implanted subcutaneously in a mix with control- or IL13-PE-293DT cells. Dual Fluc and Rluc bioluminescence imaging was performed 0, 1, and 2 days post-implantation to track protein synthesis (PS) and tumor volume (TV) respectively. Representative visible light plus superimposed bioluminescence images of tumors are shown (color scale units, photons min−1 cm−2).
Fig. S5. Stem cell secretion of ENb-PE reduces GBM viability by inhibiting protein synthesis
(A) GBM cell lines were transduced with LV-destabilized-luciferase (dsluc) and co-cultured with hNSC or hNSC-ENb-PE cells. Aggregate levels of protein synthesis were measured by luciferase-based assay after five days of culture. (B) Schematic showing how the dual bioluminescence in vivo assay was performed. (C) LN229 GBM cells were engineered to co-express the protein synthesis marker, dsluc, and cell viability marker Rluc. These cells were mixed with either hNSC-Oligo-ENb-PE or plain hNSC cells and implanted subcutaneously in SCID mice. Bioluminescence imaging was performed daily to assess protein synthesis and GBM viability. Representative visible light plus superimposed bioluminescence images of tumors are shown (color scale units, photons min−1 cm−2) and quantified. Data are expressed as mean ± s.e.m. Significance of unpaired t test, * P < 0.05; # P <0.01; § P < 0.001; ENb-PE-treated versus control for each GBM line.
Fig. S6. Viability and therapeutic capacity of encapsulated hNSCs
(A+B) Summary data (A) and representative fluorescence images (B) demonstrating the cell viability of hNSC-mCherry-RLuc cells encapsulated in sECM and cultured for up to five days at different encapsulation numbers. White line in (B) demarcates edge of sECM. Cells start to escape sECM after three days. (C) hNSCs or hNSCs secreting IL13-PE were encapsulated in sECM and placed in a culture dish containing U87-eGFP-Fluc. Plot shows tumor cell viability after five days culture as determined by luciferase-based assay. (D) Representative photomicrographs showing encapsulated hNSCs and surrounding U87 GBM cells. Black line demarcates edge of sECM. (E) Plot revealing a direct correlation between U87-eGFP-Fluc cell number and Fluc activity. (F) Representative images and summary data showing retention and viability of sECM-encapsulated hNSC-Fluc cells seeded in the post-resection surgical cavity. Fluc imaging revealing retention of encapsulated hNSCs after 24 hours as seen in the superimposed bioluminescence images (color scale units, photons min−1 cm−2). Scale bars, 100 μm. Data are expressed as mean ± s.e.m. Significance of unpaired t test, § P < 0.001; treated versus control.
Fig. S7. Characterizing IL13-PE secretion using a diagnostic variant
(A) Schematic of lentiviral vector encoding a fusion of IL13, Gluc and PE. (B) hNSCs engineered to secrete IL13-Gluc-PE were encapsulated in sECM and cultured for up to seven days. Plot shows secretion of IL13-Gluc-PE as determined by a Gluc-based assay. (C) Concentrated medium containing IL13-GLuc-PE was infused into the borders of the resection cavity and analyzed by serial Gluc imaging. (D) Cells secreting IL13-Gluc-PE, encapsulated in sECM and seeded in the resection cavity were analyzed by serial Gluc imaging. Representative visible light plus superimposed bioluminescence images of mice at various time points (color scale units, photons min−1 cm−2).
Fig. S8. GBM modifications do not affect their response to IL13-PE
(A) Cell viability of unmodified and transduced (LV-eGFP-Fluc expression) GBMs, following treatment with 25 ng/mL IL13-PE conditioned medium. Data are expressed as mean ± s.e.m; ns, not significant.
Acknowledgments
We thank G. Prestwich (University of Utah) and T. Zarembinski (Biotime Inc.) for providing us with synthetic extracellular matrix gels, G. Mohapatra (Massachusetts General Hospital) for genome sequencing analysis and Sara Pignatta (Biosciences Laboratory, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori) for experimental assistance during the review process. This work was supported by US National Institutes of Health grants R01CA138922 (K.S.), R01CA173077 (K.S.)
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
Authors’ contributions: D.W.S.: Conception and design, Provision of study material, Collection and assembly of data, Data analysis and interpretation, Manuscript writing, Final approval of manuscript; S.D.H.: Conception and design, Provision of study material, Collection and assembly of data, Data analysis and interpretation, Manuscript writing, Final approval of manuscript; N.K.: Collection and assembly of data, Data analysis and interpretation, Final approval of manuscript; B.E.R.: Data analysis and interpretation, Provision of study material, Final approval of manuscript; K.S.: Conception and design, Financial support, Provision of study material, Assembly of data, Data analysis and interpretation, Manuscript writing, Final approval of manuscript.
References
- 1.Hwang J, Fitzgerald DJ, Adhya S, Pastan I. Functional domains of Pseudomonas exotoxin identified by deletion analysis of the gene expressed in E. coli. Cell. 1987 Jan 16;48:129. doi: 10.1016/0092-8674(87)90363-1. [DOI] [PubMed] [Google Scholar]
- 2.Weldon JE, Pastan I. A guide to taming a toxin--recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. The FEBS journal. 2011 Dec;278:4683. doi: 10.1111/j.1742-4658.2011.08182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okada H, et al. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. Journal of neuro-oncology. 2008 Jul;88:245. doi: 10.1007/s11060-008-9566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wykosky J, Gibo DM, Stanton C, Debinski W. Interleukin-13 receptor alpha 2, EphA2, and Fos-related antigen 1 as molecular denominators of high-grade astrocytomas and specific targets for combinatorial therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008 Jan 1;14:199. doi: 10.1158/1078-0432.CCR-07-1990. [DOI] [PubMed] [Google Scholar]
- 5.Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin-13 receptor alpha2 in glioblastoma multiforme: implications for targeted therapies. Cancer research. 2007 Sep 1;67:7983. doi: 10.1158/0008-5472.CAN-07-1493. [DOI] [PubMed] [Google Scholar]
- 6.Debinski W, Slagle B, Gibo DM, Powers SK, Gillespie GY. Expression of a restrictive receptor for interleukin 13 is associated with glial transformation. Journal of neuro-oncology. 2000 Jun;48:103. doi: 10.1023/a:1006446426611. [DOI] [PubMed] [Google Scholar]
- 7.Candolfi M, et al. Gene therapy-mediated delivery of targeted cytotoxins for glioma therapeutics. Proceedings of the National Academy of Sciences of the United States of America. 2010 Nov 16;107:20021. doi: 10.1073/pnas.1008261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Debinski W, Gibo DM. Molecular expression analysis of restrictive receptor for interleukin 13, a brain tumor-associated cancer/testis antigen. Mol Med. 2000 May;6:440. [PMC free article] [PubMed] [Google Scholar]
- 9.Liu H, et al. Interleukin-13 sensitivity and receptor phenotypes of human glial cell lines: non-neoplastic glia and low-grade astrocytoma differ from malignant glioma. Cancer immunology, immunotherapy : CII. 2000 Aug;49:319. doi: 10.1007/s002620000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theuer CP, FitzGerald D, Pastan I. A recombinant form of Pseudomonas exotoxin directed at the epidermal growth factor receptor that is cytotoxic without requiring proteolytic processing. The Journal of biological chemistry. 1992 Aug 25;267:16872. [PubMed] [Google Scholar]
- 11.Lorimer IA, et al. Recombinant immunotoxins specific for a mutant epidermal growth factor receptor: targeting with a single chain antibody variable domain isolated by phage display. Proceedings of the National Academy of Sciences of the United States of America. 1996 Dec 10;93:14815. doi: 10.1073/pnas.93.25.14815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu TF, et al. Diphtheria toxin-epidermal growth factor fusion protein and Pseudomonas exotoxin-interleukin 13 fusion protein exert synergistic toxicity against human glioblastoma multiforme cells. Bioconjugate chemistry. 2003 Nov-Dec;14:1107. doi: 10.1021/bc034111+. [DOI] [PubMed] [Google Scholar]
- 13.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. The New England journal of medicine. 2008 Mar 13;358:1160. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 14.Kreitman RJ, Pastan I. Immunotoxins in the treatment of hematologic malignancies. Current drug targets. 2006 Oct;7:1301. doi: 10.2174/138945006778559139. [DOI] [PubMed] [Google Scholar]
- 15.Kreitman RJ, et al. Responses in refractory hairy cell leukemia to a recombinant immunotoxin. Blood. 1999 Nov 15;94:3340. [PubMed] [Google Scholar]
- 16.Kreitman RJ, et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2000 Apr;18:1622. doi: 10.1200/JCO.2000.18.8.1622. [DOI] [PubMed] [Google Scholar]
- 17.Kreitman RJ, et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005 Sep 20;23:6719. doi: 10.1200/JCO.2005.11.437. [DOI] [PubMed] [Google Scholar]
- 18.Kreitman RJ, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009 Jun 20;27:2983. doi: 10.1200/JCO.2008.20.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debinski W, Obiri NI, Pastan I, Puri RK. A novel chimeric protein composed of interleukin 13 and Pseudomonas exotoxin is highly cytotoxic to human carcinoma cells expressing receptors for interleukin 13 and interleukin 4. The Journal of biological chemistry. 1995 Jul 14;270:16775. doi: 10.1074/jbc.270.28.16775. [DOI] [PubMed] [Google Scholar]
- 20.Debinski W, Obiri NI, Powers SK, Pastan I, Puri RK. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clinical cancer research : an official journal of the American Association for Cancer Research. 1995 Nov;1:1253. [PubMed] [Google Scholar]
- 21.Husain SR, Joshi BH, Puri RK. Interleukin-13 receptor as a unique target for anti-glioblastoma therapy. International journal of cancer Journal international du cancer. 2001 Apr 15;92:168. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1182>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 22.Kunwar S, et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro-oncology. 2010 Aug;12:871. doi: 10.1093/neuonc/nop054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Jiang T. Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer letters. 2013 May 1;331:139. doi: 10.1016/j.canlet.2012.12.024. [DOI] [PubMed] [Google Scholar]
- 24.Huang PH, Xu AM, White FM. Oncogenic EGFR signaling networks in glioma. Science signaling. 2009;2:re6. doi: 10.1126/scisignal.287re6. [DOI] [PubMed] [Google Scholar]
- 25.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nature biotechnology. 2005 Sep;23:1126. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 26.van de Water JA, et al. Therapeutic stem cells expressing variants of EGFR-specific nanobodies have antitumor effects. Proceedings of the National Academy of Sciences of the United States of America. 2012 Oct 9;109:16642. doi: 10.1073/pnas.1202832109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roovers RC, et al. Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR Nanobodies. Cancer immunology, immunotherapy : CII. 2007 Mar;56:303. doi: 10.1007/s00262-006-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kauer TM, Figueiredo JL, Hingtgen S, Shah K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nature neuroscience. 2012 Feb;15:197. doi: 10.1038/nn.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shah K, et al. Bimodal viral vectors and in vivo imaging reveal the fate of human neural stem cells in experimental glioma model. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008 Apr 23;28:4406. doi: 10.1523/JNEUROSCI.0296-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kock N, Kasmieh R, Weissleder R, Shah K. Tumor therapy mediated by lentiviral expression of shBcl-2 and S-TRAIL. Neoplasia. 2007 May;9:435. doi: 10.1593/neo.07223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasportas LS, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proceedings of the National Academy of Sciences of the United States of America. 2009 Mar 24;106:4822. doi: 10.1073/pnas.0806647106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wakimoto H, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer research. 2009 Apr 15;69:3472. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bagci-Onder T, Wakimoto H, Anderegg M, Cameron C, Shah K. A dual PI3K/mTOR inhibitor, PI-103, cooperates with stem cell-delivered TRAIL in experimental glioma models. Cancer research. 2011 Jan 1;71:154. doi: 10.1158/0008-5472.CAN-10-1601. [DOI] [PubMed] [Google Scholar]
- 34.Shulga-Morskoy S, Rich BE. Bioactive IL7-diphtheria fusion toxin secreted by mammalian cells. Protein engineering, design & selection : PEDS. 2005 Jan;18:25. doi: 10.1093/protein/gzi007. [DOI] [PubMed] [Google Scholar]
- 35.Haq R, et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2013 Mar 12;110:4321. doi: 10.1073/pnas.1205575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gazdar AF, et al. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. International journal of cancer Journal international du cancer. 1998 Dec 9;78:766. doi: 10.1002/(sici)1097-0215(19981209)78:6<766::aid-ijc15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 37.Lasfargues EY, Coutinho WG, Redfield ES. Isolation of two human tumor epithelial cell lines from solid breast carcinomas. Journal of the National Cancer Institute. 1978 Oct;61:967. [PubMed] [Google Scholar]
- 38.Villa A, Snyder EY, Vescovi A, Martinez-Serrano A. Establishment and properties of a growth factor-dependent, perpetual neural stem cell line from the human CNS. Experimental neurology. 2000 Jan;161:67. doi: 10.1006/exnr.1999.7237. [DOI] [PubMed] [Google Scholar]
- 39.Kohno K, Uchida T. Highly frequent single amino acid substitution in mammalian elongation factor 2 (EF-2) results in expression of resistance to EF-2-ADP-ribosylating toxins. The Journal of biological chemistry. 1987 Sep 5;262:12298. [PubMed] [Google Scholar]
- 40.Wu AH, Low WC. Molecular cloning and identification of the human interleukin 13 alpha 2 receptor (IL-13Ra2) promoter. Neuro-oncology. 2003 Jul;5:179. doi: 10.1215/S1152-8517-02-00051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gill DM, Pappenheimer AM, Jr, Brown R, Kurnick JT. Studies on the mode of action of diphtheria toxin. VII. Toxin-stimulated hydrolysis of nicotinamide adenine dinucleotide in mammalian cell extracts. The Journal of experimental medicine. 1969 Jan 1;129:1. doi: 10.1084/jem.129.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iglewski BH, Kabat D. NAD-dependent inhibition of protein synthesis by Pseudomonas aeruginosa toxin. Proceedings of the National Academy of Sciences of the United States of America. 1975 Jun;72:2284. doi: 10.1073/pnas.72.6.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamaizumi M, Mekada E, Uchida T, Okada Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell. 1978 Sep;15:245. doi: 10.1016/0092-8674(78)90099-5. [DOI] [PubMed] [Google Scholar]
- 44.Corsten MF, Shah K. Therapeutic stem-cells for cancer treatment: hopes and hurdles in tactical warfare. The lancet oncology. 2008 Apr;9:376. doi: 10.1016/S1470-2045(08)70099-8. [DOI] [PubMed] [Google Scholar]
- 45.Ahmed AU, Alexiades NG, Lesniak MS. The use of neural stem cells in cancer gene therapy: predicting the path to the clinic. Current opinion in molecular therapeutics. 2010 Oct;12:546. [PMC free article] [PubMed] [Google Scholar]
- 46.Aboody KS, Najbauer J, Danks MK. Stem and progenitor cell-mediated tumor selective gene therapy. Gene therapy. 2008 May;15:739. doi: 10.1038/gt.2008.41. [DOI] [PubMed] [Google Scholar]
- 47.Foley BT, Moehring JM, Moehring TJ. Mutations in the elongation factor 2 gene which confer resistance to diphtheria toxin and Pseudomonas exotoxin A. Genetic and biochemical analyses. The Journal of biological chemistry. 1995 Sep 29;270:23218. doi: 10.1074/jbc.270.39.23218. [DOI] [PubMed] [Google Scholar]
- 48.Asthagiri AR, Pouratian N, Sherman J, Ahmed G, Shaffrey ME. Advances in brain tumor surgery. Neurologic clinics. 2007 Nov;25:975. doi: 10.1016/j.ncl.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 49.Lacroix M, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. Journal of neurosurgery. 2001 Aug;95:190. doi: 10.3171/jns.2001.95.2.0190. [DOI] [PubMed] [Google Scholar]
- 50.Altaner C, et al. Complete regression of glioblastoma by mesenchymal stem cells mediated prodrug gene therapy simulating clinical therapeutic scenario. International journal of cancer. Journal international du cancer. 2013 Aug 26; doi: 10.1002/ijc.28455. [DOI] [PubMed] [Google Scholar]
- 51.Bello L, et al. Suppression of malignant glioma recurrence in a newly developed animal model by endogenous inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002 Nov;8:3539. [PubMed] [Google Scholar]
- 52.Herbst RS. Review of epidermal growth factor receptor biology. International journal of radiation oncology, biology, physics. 2004;59:21. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 53.Jacobs VL, Valdes PA, Hickey WF, De Leo JA. Current review of in vivo GBM rodent models: emphasis on the CNS-1 tumour model. ASN neuro. 2011;3:e00063. doi: 10.1042/AN20110014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujisawa T, Joshi BH, Puri RK. Histone modification enhances the effectiveness of IL-13 receptor targeted immunotoxin in murine models of human pancreatic cancer. Journal of translational medicine. 2011;9:37. doi: 10.1186/1479-5876-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Characterizing the hNSC-Oligo cell line
(A) Immunocytochemistry on parental (hNSC) and toxin-resistant (hNSC-Oligo) lines using an anti-nestin antibody (red) and DAPI nuclear counterstain (blue). Scale bar 100 μm (B) RT-PCR transcript analysis on cDNA derived from unmodified hNSCs and hNSC-Oligo lines. Primer pairs amplify Pax6 (an hNSC marker) and GAPDH products from cDNA. (C) Genomic profiling of parental and modified hNSC lines. (D) Proliferation assay performed on hNSC and hNSC-IL13-PE lines at day 1, 3 and 5. Data are expressed as mean ± s.e.m. Scale bar, 100 μm.
Fig. S2. Secretion and activity of IL13-PE produced by PE-resistant somatic cells
(A) Schematic of vectors encoding the cytotoxins IL13-PE and ENb-PE and the non-toxic variants IL13 and ENb conjugated to either IRES-eGFP or IRES-mCherry. (B) Fluorescence images of 293T (SO) or 293-Oligo (SO-Oligo) cells transiently transfected with vectors encoding IL13-PE or IL13. Expression of eGFP was determined 24 hours post-transfection. (C) Plot of cell viability of toxin sensitive (293T) and toxin resistant (293DT) cells towards purified PE, and conditioned medium containing IL13 or the toxic IL13-PE variant. Representative visible light plus superimposed bioluminescence images of treated cells in 96 well plates are shown (color scale units, photons min−1 cm−2). (D) Western blot and dot blot (E) analysis using anti-IL13, demonstrating the expression and secretion of IL13-PE from SO-Oligo cells transfected with a vector encoding IL13-PE. Scale bar, 100 μm.
Fig. S3. Ectopic expression of IL13Rα2 confers IL13-PE sensitivity to resistant GBMs
(A) Gli36vIII-IL13Rα2-eGFP-RLuc cells were plated at increasing numbers and bioluminescence imaging was performed. The plot reveals a direct correlation between cell number and Rluc activity and a representative bioluminescence image of increasing cell numbers in a 96 well plate is shown (color scale units, photons min−1 cm−2). (B–E) Gli36vIII cells transduced with LV encoding IL13Rα2 (B) or IL13Rα2-eGFP-Rluc (C) were treated with control or IL13-PE conditioned medium at increasing concentrations (0–750 ng/ml), and cell viability was determined 48 hrs after treatment. Gli36vIII-IL13Rα2 (D) or Gli36vIII-IL13Rα2-eGFP-RLuc (E) cell lines were co-transduced with LV-destabilized-luciferase and treated with 100 ng of IL13-PE or control medium. Levels of protein synthesis were measured by luciferase-based assay 0, 24, 48, or 72 hrs post-treatment.
Fig. S4. IL13-PE-expressing somatic cells reduce GBM viability by inhibiting protein synthesis in vivo
(A) Schematic showing how the experiment was performed. (B+C) Gli36vIII-IL13Rα2 (B) or U251 (C) GBM cells were co-transduced to express eGFP-Rluc (Rluc) and destabilized-luciferase (dsluc), and implanted subcutaneously in a mix with control- or IL13-PE-293DT cells. Dual Fluc and Rluc bioluminescence imaging was performed 0, 1, and 2 days post-implantation to track protein synthesis (PS) and tumor volume (TV) respectively. Representative visible light plus superimposed bioluminescence images of tumors are shown (color scale units, photons min−1 cm−2).
Fig. S5. Stem cell secretion of ENb-PE reduces GBM viability by inhibiting protein synthesis
(A) GBM cell lines were transduced with LV-destabilized-luciferase (dsluc) and co-cultured with hNSC or hNSC-ENb-PE cells. Aggregate levels of protein synthesis were measured by luciferase-based assay after five days of culture. (B) Schematic showing how the dual bioluminescence in vivo assay was performed. (C) LN229 GBM cells were engineered to co-express the protein synthesis marker, dsluc, and cell viability marker Rluc. These cells were mixed with either hNSC-Oligo-ENb-PE or plain hNSC cells and implanted subcutaneously in SCID mice. Bioluminescence imaging was performed daily to assess protein synthesis and GBM viability. Representative visible light plus superimposed bioluminescence images of tumors are shown (color scale units, photons min−1 cm−2) and quantified. Data are expressed as mean ± s.e.m. Significance of unpaired t test, * P < 0.05; # P <0.01; § P < 0.001; ENb-PE-treated versus control for each GBM line.
Fig. S6. Viability and therapeutic capacity of encapsulated hNSCs
(A+B) Summary data (A) and representative fluorescence images (B) demonstrating the cell viability of hNSC-mCherry-RLuc cells encapsulated in sECM and cultured for up to five days at different encapsulation numbers. White line in (B) demarcates edge of sECM. Cells start to escape sECM after three days. (C) hNSCs or hNSCs secreting IL13-PE were encapsulated in sECM and placed in a culture dish containing U87-eGFP-Fluc. Plot shows tumor cell viability after five days culture as determined by luciferase-based assay. (D) Representative photomicrographs showing encapsulated hNSCs and surrounding U87 GBM cells. Black line demarcates edge of sECM. (E) Plot revealing a direct correlation between U87-eGFP-Fluc cell number and Fluc activity. (F) Representative images and summary data showing retention and viability of sECM-encapsulated hNSC-Fluc cells seeded in the post-resection surgical cavity. Fluc imaging revealing retention of encapsulated hNSCs after 24 hours as seen in the superimposed bioluminescence images (color scale units, photons min−1 cm−2). Scale bars, 100 μm. Data are expressed as mean ± s.e.m. Significance of unpaired t test, § P < 0.001; treated versus control.
Fig. S7. Characterizing IL13-PE secretion using a diagnostic variant
(A) Schematic of lentiviral vector encoding a fusion of IL13, Gluc and PE. (B) hNSCs engineered to secrete IL13-Gluc-PE were encapsulated in sECM and cultured for up to seven days. Plot shows secretion of IL13-Gluc-PE as determined by a Gluc-based assay. (C) Concentrated medium containing IL13-GLuc-PE was infused into the borders of the resection cavity and analyzed by serial Gluc imaging. (D) Cells secreting IL13-Gluc-PE, encapsulated in sECM and seeded in the resection cavity were analyzed by serial Gluc imaging. Representative visible light plus superimposed bioluminescence images of mice at various time points (color scale units, photons min−1 cm−2).
Fig. S8. GBM modifications do not affect their response to IL13-PE
(A) Cell viability of unmodified and transduced (LV-eGFP-Fluc expression) GBMs, following treatment with 25 ng/mL IL13-PE conditioned medium. Data are expressed as mean ± s.e.m; ns, not significant.