

Abstract
The role of naturally occurring human α1a-Adrenergic Receptor (α1aAR) genetic variants associated with cardiovascular disorders is poorly understood. Here, we present the novel findings that expression of human α1aAR-247R (247R) genetic variant in cardiomyoblasts leads to transition of cardiomyoblasts into a fibroblast-like phenotype, evidenced by morphology and distinct de novo expression of characteristic genes. These fibroblast-like cells exhibit constitutive, high proliferative capacity and agonist-induced hypertrophy compared with cells prior to transition. We demonstrate that constitutive, synergistic activation of EGFR, Src and ERK kinases is the potential molecular mechanism of this transition. We also demonstrate that 247R triggers two distinct EGFR transactivation-dependent signaling pathways: 1) constitutive Gq-independent βarrestin1/Src/MMP/EGFR/ERK-dependent hyperproliferation and 2) agonist-induced Gq- and EGFR/STAT-dependent hypertrophy. Interestingly, in cardiomyoblasts agonist-independent hyperproliferation is MMP-dependent, but in fibroblast-like cells it is MMP-independent, suggesting that expression of α1aAR genetic variant in cardiomyocytes may trigger extracellular matrix remodeling. Thus, these novel findings demonstrate that EGFR transactivation by α1aAR-247R leads to hyperproliferation, hypertrophy and alterations in cardiomyoblasts, suggesting that these unique genetically-mediated alterations in signaling pathways and cellular function may lead to myocardial fibrosis. Such extracellular matrix remodeling may contribute to the genesis of arrhythmias in certain types of heart failure.
Keywords: Adrenergic receptor, Epidermal growth factor receptor (EGFR), G protein coupled receptors (GPCR), Matrix metalloproteinase (MMP), transactivation, genetic variant
1. Introduction
Activation of Adrenergic Receptors (AR), members of the G protein-coupled receptor (GPCR) superfamily, in response to stimulation of the sympathetic nervous system plays a major role in regulating cardiac function [1]. Cardiomyocytes express both α1ARs and βARs that regulate contractility and growth of the myocardium. Three α1AR subtypes are expressed in human heart (α1a, α1b, α1d), mediating actions of the sympathetic nervous system through binding of endogenous catecholamines epinephrine and norepinephrine. Cardiomyocytes express α1a and α1b subtypes and there is little, if any, expression of α1d in myocardium [2,3]. The role of α1AR signaling in human heart is poorly understood. Under physiological conditions, cardiac inotropy and chronotropy are predominantly regulated by βARs, while α1ARs are thought to become more important during pathological conditions, such as myocardial hypertrophy, heart failure and ischemic heart disease: α1ARs are essential in maintaining or increasing myocardial contractility when βARs are down-regulated during heart failure [4,5]. While previously α1AR antagonists were thought to be useful in treating heart failure by decreasing sympathetic overload, recent studies demonstrate that a nonselective α1AR antagonist increased the risk of heart failure and mortality [6]. In contrast, carvedilol, a β- and α1AR antagonist with higher affinity for α1bAR subtype, provided an effective treatment for chronic heart failure [7], thus promoting a beneficial effect of α1aAR signaling and suggesting that α1AR subtypes may differentially contribute to heart failure. Differential regulation of α1AR subtypes by selective activation has been demonstrated for mouse and rat models and isolated cardiomyocytes [8]. While α1bAR in mouse model is involved in pathogenesis of hypertrophy [9], selective activation of α1aAR in rat neonatal cardiomyocytes is implicated in developmental cardiomyocyte growth and survival [10].
α1ARs also play a major role in regulating vascular tone and in blood vessel repair [11-13]. Activation of α1ARs leads to vasoconstriction, and knockout results in impaired maintenance of arterial blood pressure [14]. α1AR deletion worsens dilated cardiomyopathy after pressure overload by multiple mechanisms, indicating that α1AR signaling is required for cardiac adaptation [10]. While subtype distribution differs between species, α1aAR is the predominant subtype in human resistance vessels [13], and nonselective α1AR and α1aAR-specific antagonists can be used to lower blood pressure in humans.
Canonical signaling of α1ARs involves agonist mediated coupling to Gq/11, activation of PLCβ (1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase beta), and cleavage of PIP2 (phosphatidylinositol 4,5-bisphosphate) to second messengers diacylglycerol and inositol triphosphate. Signal termination results from receptor phosphorylation and desensitization by βarrestins [15]. Induction of cellular signaling through EGFR (epidermal growth factor receptor) transactivation by GPCRs could also be triggered by several distinct mechanisms which may be dependent or independent of G protein activation [16, 17]. The current paradigm of EGFR transactivation involves agonist stimulation of GPCRs leading to not fully defined steps of activation of MMPs (matrix metalloproteinases) and ADAMs (a disintegrins and metalloproteases) and cleavage and extracellular shedding of heparin-binding EGF (HB-EGF) [18]. Recent studies demonstrate that in addition to βarrestins' role in terminating G protein signaling, they also function as adapter molecules for the assembly of signaling complexes with ERKs and tyrosine kinases [19]. EGFR transactivation by GPCRs also mediates proliferative effects [20] and is a potential mechanism underlying changes in vessel structure and myocardial hypertrophy [21,22]. Vascular remodeling as observed in hypertension includes increased cell growth, hypertrophy and extracellular matrix deposition [23].
Polymorphisms in genes involved in regulation of the adrenergic system, including α1aAR gene, have been linked to hypertensive disorders [12,24,25]. Functional effects of α1aAR SNPs might therefore represent a novel mechanism underlying hypertension and heart disease. We reported that the α1aAR SNP in the third intracellular loop, G247R (247R), leads to constitutive hyperproliferation compared to α1aAR-WT (WT) in Rat-1 fibroblasts [26]. It has also been demonstrated that α1aARs are present in cardiac fibroblasts [27,28] and may regulate protein synthesis and secretion for cardiac function. We have demonstrated that 247R genetic variant, identified in a severely hypertensive patient, leads to constitutive hyperproliferation compared not only to WT but also to other α1AR subtypes in Rat-1 fibroblasts [29].
While this work was focused on fibroblasts, the role of α1AR SNPs in cardiovascular cells remains unknown. Here, we elucidated 247R-mediated biological responses and underlying molecular mechanisms of signal transactivation pathways in the H9c2 cardiomyoblast cell line. Derived from embryonic rat heart [30] as an alternative to primary neonatal cardiomyocytes, these cells represent an intriguing experimental model. They have been used as an in vitro model for both cardiac and skeletal muscle, because they exhibit corresponding electrophysiological and biochemical properties and demonstrate morphological characteristics of embryonic cardiac myocytes [31, 32]. Nearly identical hypertrophic responses in the H9c2 cell line compared with primary cardiomyocytes have also been demonstrated, emphasizing the relevance of H9c2 cells for in vitro studies of cardiac hypertrophy and molecular mechanisms regulating heart development and disease [33]. This cell line is therefore widely used as a cardiomyocyte model to study signal transduction pathways of transmembrane receptors.
In this study we present new data demonstrating that cardiomyoblasts expressing 247R genetic variant transition to cells with altered fibroblast-like morphology and phenotype with high proliferative capacity, exhibit increased, constitutive (agonist-independent) proliferation, and undergo hypertrophy upon agonist stimulation. We show that in 247R cells agonist-induced hypertrophy is Gq/EGFR/STAT3-dependent, while basal, constitutive hyperproliferation is mediated by Gq-independent, βarrestin1/Src/MMP-dependent EGFR transactivation and downstream activation of ERK. Our data demonstrate that constitutive, EGFR transactivation-dependent hyperproliferation triggered by 247R genetic variant is not cell type dependent, but generalizable. These novel findings demonstrating that 247R triggers distinct signaling pathways and induces transition of cardiomyoblasts to fibroblast-like cells with very high proliferative capacity suggests that this SNP may trigger detrimental alterations in vessel and heart structure, leading to cardiovascular disease.
2. Materials and Methods
2.1. Cell culture
H9c2 embryonic rat heart-derived cardiomyoblasts (ATCC, Manassas, VA) were cultured in Dulbecco's Modified Eagle Medium (DMEM, Gibco, Auckland, NZ) supplemented with 10% FBS (Hyclone Laboratories, South Logan, UT) and penicillin/streptomycin (Gibco) at 37°C in 5% CO2. Cells were maintained at less than 70% confluence, and experiments were performed in DMEM containing 0%, 0.5%, or 10% FBS as indicated.
2.2. Stable cell lines expressing α1aAR-WT or α1aAR-247R
H9c2 cardiomyoblasts were transfected with pcDNA3 plasmid containing human HA epitope-tagged α1aAR-WT or α1aAR-247R [26] using Lipofectamine 2000 (Invitrogen, Grand Island, NY). Transfection efficiency and expression of the receptors was confirmed by radioligand-binding assays using [125I]-HEAT (Perkin Elmer, Boston, MA) [13]. Cells were selected based on resistance to 800μg/ml G418 (Calbiochem; San Diego, CA) and individual clones were isolated and expanded. Receptor expression level was determined by radioligand-binding assays using [125I]-HEAT, and clones with comparable, low receptor expression levels (≤ 300fmol/mg protein) were used for the experiments.
2.3. Cell proliferation
Proliferation experiments were carried out in DMEM supplemented with 10% or 0.5% FBS, with or without agonist stimulation (10μM phenylephrine, PE, Sigma-Aldrich, St. Louis, MO).
Cells with myoblast morphology were plated at 10×103, 15×103 or 20×103 cells/well in 24- or 12-well plates and cultured for 48h. Tr247R cells were plated at 20×103-60×103 cells/well in 6-, 12- or 24-well plates and cultured for 24, 48 or 72h. At indicated time points, cells were trypsinized and counted using light microscopy. Experiments with prazosin were performed with 1μM prazosin and 1μM PE in 0.5% FBS containing medium. Cell proliferation in the presence of EGFR inhibitor AG1478 (Cell Signaling, Danvers, MA), MMP inhibitor GM6001, or Src inhibitor PP2 (Calbiochem) were evaluated over 24 or 48h in 0.5% FBS. The following concentrations were used: AG1478: 500nM (H9c2, WT, 247R) or 1μM (tr247R); GM6001: 10μM (H9c2, WT, 247R) or 25μM (tr247R); PP2 2.5μM (H9c2, WT, 247R, tr247R). Inhibitors were added 1h prior to agonist treatment.
2.4. Thymidine incorporation assays
Cells plated as described above were cultured for 48h in 10% or 0.5% FBS-containing medium in the presence or absence of 10μM PE or inhibitors as indicated. Culture medium was refreshed every 24h, and cells were labeled with 1μCi [3H]-thymidine (Perkin Elmer) for 3h as described [29]. Cells were washed with cold PBS, incubated in 5% trichloroacetic acid for 20min and then washed again with PBS. DNA was solubilized with 300μl 0.25N NaOH and mixed with EcoLite (+) liquid scintillation cocktail (MP Biomedicals, Solon, OH). [3H]-thymidine incorporation was quantified using Liquid Scintillation Analyzer Tri-Carb 2810 TR (Perkin Elmer) as described [34]. For evaluation of proliferation of cells with cardiomyoblast morphology upon agonist treatment (PE), cells in parallel wells of the same plate treated similarly with agonist, were trypsinized and the cell numbers were determined using light microscopy. Total genomic DNA was isolated from triplicate wells and used for normalization of [3H]-thymidine incorporation into DNA per 1000 cells.
2.5. Leucine incorporation assays
Cells were plated in 24- or 12-well plates at 10×103 or 20×103 cells/well for myoblast cells and at 20×103 or 40×103 cells/well for tr247R cells. Cells were cultured for 24h in 0.5% FBS-containing medium in the presence or absence of 10μM PE, followed by labeling with 1μCi [3H]-leucine for additional 24h. Cells were washed with ice-cold PBS, 10% trichloroacetic acid, solubilized with 300μl 0.25N NaOH and scintillation counting was performed in EcoLite (+) liquid scintillation cocktail with the Liquid Scintillation Analyzer Tri-Carb 2810 TR. To evaluate hypertrophy in the presence of prazosin (1μM), PE was used at 1μM concentration.
2.6. Evaluation of cell surface area
Cells were plated on glass slides in 0.5% FBS for 72h with or without 10μmol/L PE. Cell membranes were labeled with fluorescein-conjugated wheat germ agglutinin (Alexa Fluor® 488 conjugate, Invitrogen) [35] for 5 min and cells were fixed in 10% neutral buffered formalin solution for 10min at room temperature. To visualize nuclei, cells were covered with mounting medium containing DAPI (Vector Laboratories, Burlingame, CA). Images were captured using Nikon 90i Microscope (Nikon Inc., Melville, NY) with a 20x objective. For each of 3 independent experiments, a minimum of 5 fields per cell type were evaluated using Image J software.
2.7. RNA isolation and RT-PCR analysis
Cells were plated in DMEM with 10% or 0% FBS for 24h in the absence or presence of 10μM PE. RNA was isolated using RNeasy Plus kit (Qiagen, Valencia, CA). cDNA was generated using 1.5μg (MMP-2, MMP-9/ADAMs PCRs) or 3μg RNA (all other PCRs) and SuperScript III First Strand System (Invitrogen). Gene specific primers and PCR conditions used for MMP-2, MMP-9, ADAM-10, ADAM-12, ADAM-17 and GAPDH as internal control were previously described [29]. The following additional primers were used: MMP-7 sense 5′-GTG TCA TGG AGA TAA TGC AGA A-3′; antisense 5′- GAA TGA TCT CCT TGA TAG GTA GG-3′; EGFR sense 5′-TGA AGT GGT CCT TGG AAA CTT G-3′; antisense 5′-GTT GAC ATC CAT CTG GTA CGT-3′. Fibroblast specific protein 1 (FSP-1) sense 5′-CCT GGA TGT AAT AGT GTC CAC CTT C-3′; antisense 5′-ACA TCA TGG CAA TGC AGG ACA G-3′. Discoidin domain receptor 2 (DDR-2) sense 5′-AAT GAT CCC GAT TCC CAG AAT G-3′; antisense 5′-TCC CAT GTC GGT TAC GCC AG-3′. Troponin I sense 5′-CGT GGA AGC AAA AGT CAC CAA-3′; antisense 5′-CCG GTT TTC CTT CTC AAT GTC C-3′. β-MHC (beta myosin heavy chain) sense 5′-GGG CAT CAT GTC CAT CCT-3′; antisense 5′-GTG CAT CAG CTC CAG CAT AGT-3′. PCR conditions were as previously described [29] with the following adjustments: annealing temperature for Troponin I was 62°C and for β-MHC 61°C. PCR conditions for FSP-1 were 94°C 1min, 58°C 1min, 72°C 1 min for 25 cycles. PCR conditions for DDR-2 were 94°C 30sec, 58°C 30sec, 72°C 1 min for 21 cycles. After the last cycle samples were incubated for additional 10min at 72°C. For MMP-7 35 cycles were performed.
2.8. Gelatin zymography for MMP-2 and MMP-9
Cells were serum-deprived for 4h, then cultured with or without 10μM PE for additional 24h. Media was collected and concentrated at 4°C by ultrafiltration using Amicon Ultra columns (Millipore, Tullagreen, CO). Media equivalent to 30×103 cells was run on 10% gelatin zymography gels (Bio-Rad Laboratories, Hercules, CA). Gels were washed in 2.5% Triton X-100 three times 10min each and incubated overnight in developing buffer (0.05M Tris pH 8.2, 5mM CaCl2, 0.5μM ZnCl2) at 37°C, followed by staining with Coomassie brilliant blue and destaining. Gel images were acquired using Gel Doc LT Imaging Systems (BioImaging Systems; Upland, CA).
2.9. Immunostaining
H9c2, WT, 247R, tr247R cells and control Rat1-fibroblasts were plated on glass-bottom plates and fixed for 10 minutes on ice using 1:1 methanol/acetone. Cells were rehydrated, permeabilized with 0.2% Triton X-100 for 5 minutes, blocked with 5% donkey serum, and incubated with anti-vimentin antibody (Dako, Carpinteria, CA, 1:200 dilution) in 1% serum with 0.1% Triton X-100 overnight at 4°C. The next day, cells were washed 3 times 5 minutes in PBS, incubated with secondary antibody (Cy-donkey anti-mouse IgG 1:400 in 1% donkey serum, Jackson Immunoresearch, West Grove, PA) for 45 minutes at room temperature, followed by 3 additional washes with PBS and coverslipping with mounting medium containing DAPI. Images were captured using LSM510Meta scanning confocal microscope (Zeiss, Germany) and a F-Fluar 40x Oil immersion objective (NA = 1.3) under control of Zeiss AIM software, version 4.2, (Zeiss, Germany). The images were created using Zeiss LSM510 software.
2.10. Immunoblotting
For western blot analyses, cells were serum deprived in 0.5% FBS and treated with or without EGFR inhibitor AG1478 (500nM or 1μM), MMP inhibitor GM6001 (25μM) or Src inhibitor PP2 (2.5μM) in the presence or absence of 10μM PE for 48h. DMSO was used as a control vehicle. Cells were lysed in lysis buffer (50mM Tris, pH 7.4, 150mM NaCl, 10mM NaF, 5mM Na3VaO4, 2.5mM β-glycerolphosphate, 1% Triton X-100) containing protease inhibitors (Roche; Indianapolis, IN, USA) and total proteins were separated on SDS-polyacrylamide gel, followed by electroblotting and probing with specific antibodies. After immunoblotting, protein bands were visualized with enhanced chemiluminescence substrates (Perkin Elmer). Membranes were stripped and re-probed with antibodies against total ERK, total AKT, or β-actin to confirm equal protein loading within the same experiments. Densitometric analysis was carried out using Adobe Photoshop Elements 10 (Adobe, San Jose, CA). The following antibodies were used: Phospho-p44/42 Erk1/2 (Thr202/Tyr204) XP rabbit, pan-ERK1/2, phospo-Akt (Ser473), Akt, phospho-Stat3 (Tyr705), Stat3, phospho-Src (Tyr416), Src, EGFR (all Cell Signaling), EGFR pY845 (Invitrogen), EGFR-PY (4G10) (Millipore), β-arrestin-1, FSP-1, β-actin (Abcam, Cambridge, MA).
2.11. Transfection of cells with shRNAs
Control H9c2 cells or cells stably expressing WT, 247R or tr247R were plated in 6-well plates in triplicates then transfected with 4μg βarrestin-1 -specific or scrambled shRNA (SABiosciences; Frederick, MD) per well using Lipofectamine 2000. Target sequences were described previously [29]. Transfection efficiency was determined by fluorescent imaging of EGFP-transfected cells (50-70%). The following day transfected cells were trypsinized, counted and plated in 12-well plates at 20×103 cells/well for H9c2, WT and 247R; and at 40×103 cells/well for tr247R cells. Cells were cultured for 48h in 10% FBS or after 24h changed to 0.5% FBS for an additional 24h. The resultant cells were trypsinized and counted using light microscopy.
2.12. Statistical analysis
The results are graphed as mean±SE (n=3, or more). Statistical comparisons were made with unpaired Student's t-test and statistical significance was set at *p<0.05, **<0.01, ***<0.001.
3. Results
3.1. Expression of 247R triggers constitutive cell proliferation, new fibroblast-like phenotype and agonist-induced hypertrophy
To explore the physiological role of 247R genetic variant in cardiomyoblast cell line, we examined proliferative characteristics of control H9c2 cells and cells expressing comparable levels of 247R or WT receptor, cultured in the presence or absence of agonist phenylephrine (PE). Cells expressing 247R, cultured for 48h in 10% or 0.5% FBS-containing medium, display ∼2-fold increased constitutive proliferation compared to control or WT cells as measured by cell counting (Fig. 1A). Thymidine incorporation assays confirmed increased proliferation triggered by 247R expression (Fig. 1B). Agonist (PE) stimulation significantly reduces proliferation of 247R cells as determined by cell counting in 10% or 0.5% FBS-containing culture medium (Fig. 1A) or by thymidine incorporation (Fig.1B). Agonist stimulation of 247R results not only in inhibition of proliferation, but also in pronounced hypertrophy as quantified by leucine incorporation assays (Fig. 1C), demonstrating a robust ∼2-fold increased protein synthesis for 247R cells compared to their non-stimulated state (209±25% vs 100% in control) and approximately 10-fold higher protein synthesis compared with WT-expressing cells (Fig.1C).
Figure 1.
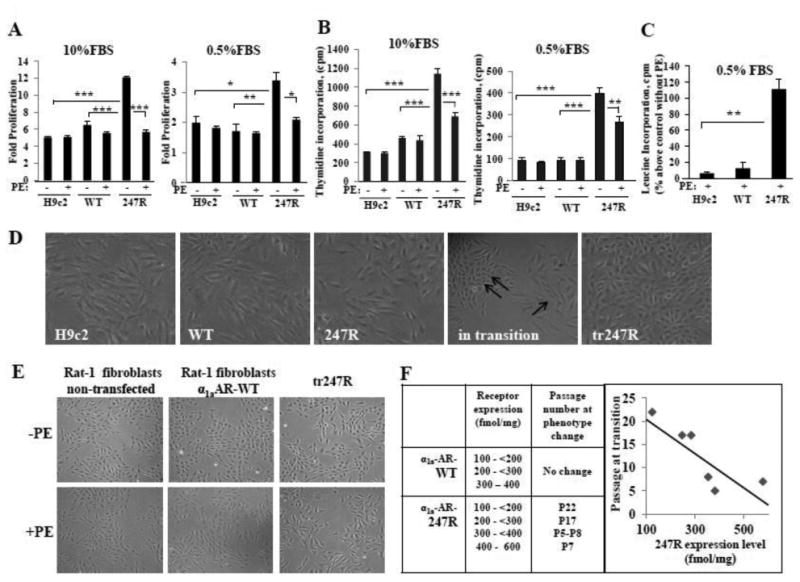
Expression of 247R triggers increased proliferation of cardiomyoblasts and their transition into fibroblast-like cells. (A,B) 247R triggers increased cell proliferation compared with control H9c2 cells or cells expressing WT; agonist (PE) treatment inhibits cell proliferation. Cell counts (A) are displayed as fold-proliferation, counts per minute (CPM) of incorporated [3H]-thymidine (B) are normalized to DNA per 1,000 cells. (C) Agonist stimulation induces pronounced hypertrophy in 247R cells as determined by leucine incorporation. Cells were cultured in the presence or absence of 10μM PE for 24h, followed by labeling with 1μCi [3H]-leucine for additional 24h. In (A,B,C) representative data from one of three independent experiments with mean values ± SE are shown. (D) 247R induces morphological and phenotypic changes. Morphology of control and WT cells or 247R cells before, during and after transition (tr247R). Double arrow indicates cell clusters with altered morphology compared with cardiomyoblast morphology (single arrow). (E) Tr247R cells exhibit morphological similarities to fibroblasts. Images of cells cultured for 24h in the presence or absence of PE taken at 20X magnification. (F) Morphology and phenotype changes of H9c2 cardiomyoblast induced by 247R correlate with receptor expression level.
3.1.1. Expression of 247R induces morphological and phenotypic changes of cardiomyoblasts resulting in fibroblast-like cells
We found that cells expressing 247R cultured in 10% FBS-containing medium transition to a different cell type with altered fibroblast-like morphology and phenotype, while control or WT receptor-expressing cells retain their cardiomyoblast morphology (Fig. 1D). Morphological changes in 247R cells were initially observed in small clusters with fibroblast-like cells (Fig. 1D, “in transition”) that rapidly expanded, becoming predominantly fibroblast-like cells within 2-3 days (Fig. 1D, “tr247R”). For convenience, morphologically-altered 247R-expressing cells are referred to as tr247R and cells before transition as 247R. Tr247R cells cultured under basal or stimulated conditions exhibit morphological appearance of fibroblasts (Fig. 1E, tr247R compared with Rat-1 fibroblasts or Rat-1 fibroblasts stably expressing α1aAR-WT). Clones with higher 247R receptor levels (e.g. 400-600 fmol/mg) acquire these changes at earlier passages, while clones with low expression levels (100-200 fmol/mg) transition at later passages. No phenotypic changes were observed for cardiomyoblasts with corresponding α1aAR-WT expression levels and passage numbers (Fig. 1F). After transition, the morphology and phenotype of the cells is stable, and the receptor expression levels increased significantly from 269±47 to 941±217 fmol/mg and from 238±29 to 997±282 fmol/mg for two independent clones tested. These data suggest that phenotypic changes are initiated by 247R.
3.2. Transition to new phenotype results in even more pronounced agonist-independent hyperproliferation and agonist-mediated hypertrophy in tr247R cells
Interestingly, tr247R cells cultured under serum-deprived conditions for 48h exhibit ∼4-fold increased proliferation compared with WT or control cells (Fig. 2A). Similar to 247R cells, agonist stimulation of tr247R cells results in inhibition of proliferation (Fig. 2B) and distinct hypertrophy as quantified by staining with wheat germ agglutinin (Fig. 2C). Cell surface area measurements after PE stimulation (Fig.2D) revealed the following cell sizes relative to their non-stimulated state (100%): H9c2 117±3.0%; WT 119.3±8.4%; 247R 133.0±5.5%; tr247R 232.4±30.4%. Thus, agonist stimulation of 247R and tr247R cells results in inhibition of proliferation and increased hypertrophy compared to control or WT cells. Treatment of cells with 10-5M PE for 48h did not cause any detectable toxicity to the cells as determined by cell viability.
Figure 2.
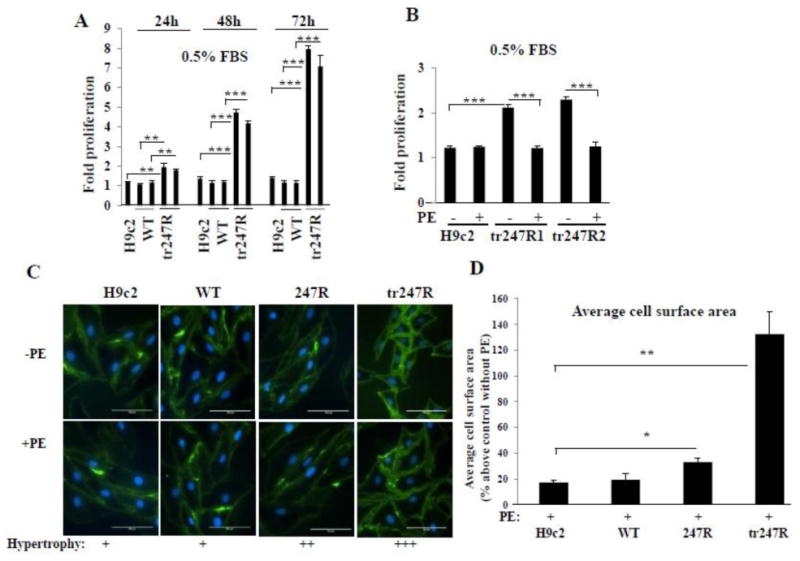
(A) Tr247R cells are highly proliferative. Tr247R cell proliferation at different time points compared with control or WT cells. Data from two different WT and tr247R clones are shown. (B) Agonist (PE) treatment inhibits tr247R cell proliferation. Two different tr247R clones are presented. Cell counts are displayed as fold-proliferation (mean±SE, n=3). (C) 247R and tr247R cells treated with PE are larger compared with non-treated cells. Cell membranes were stained with wheat germ agglutinin-specific antibody. Representative images from one of three independent experiments are shown. (D) Average cell surface area was evaluated with Image J software at 20x magnification. Data (mean±SE, n=3) presented as percentage above corresponding non-stimulated cells (control).
3.3. Characterization of tr247R cells
To further characterize tr247R cells, we performed RT-PCR analyses which revealed that expression levels of fibroblast markers such as fibroblast-specific protein 1 (FSP1) and discoidin domain receptor 2 (DDR2) are substantially higher in tr247R cells than in control, WT or 247R cells, and are comparable to those in Rat-1 fibroblasts (Fig. 3A). In contrast, expression levels of cardiac markers β-MHC or Troponin I are low in tr247R (Fig. 3B). Western blot for FSP-1 confirmed RT-PCR findings (Fig. 3C). While for tr247R and Rat-1 fibroblasts very short exposure times showed FSP1 protein bands, longer exposure time was needed to demonstrate FSP1 protein bands in cells with cardiomyoblast morphology (H9c2, WT, 247R, Fig. 3C). Similar to Rat-1 fibroblasts expressing α1aAR-WT, tr247R cells also express comparably high levels of MMP2 as evaluated by gelatin zymography (Fig. 3D). Immunostaining for vimentin (Fig. 3E) also demonstrates increased expression of vimentin in tr247R comparable to that in Rat-1 fibroblasts, in contrast to relatively lower expression levels in control, WT or 247R cells. These data further substantiate the fibroblast-like phenotype of tr247R cells.
Figure 3.
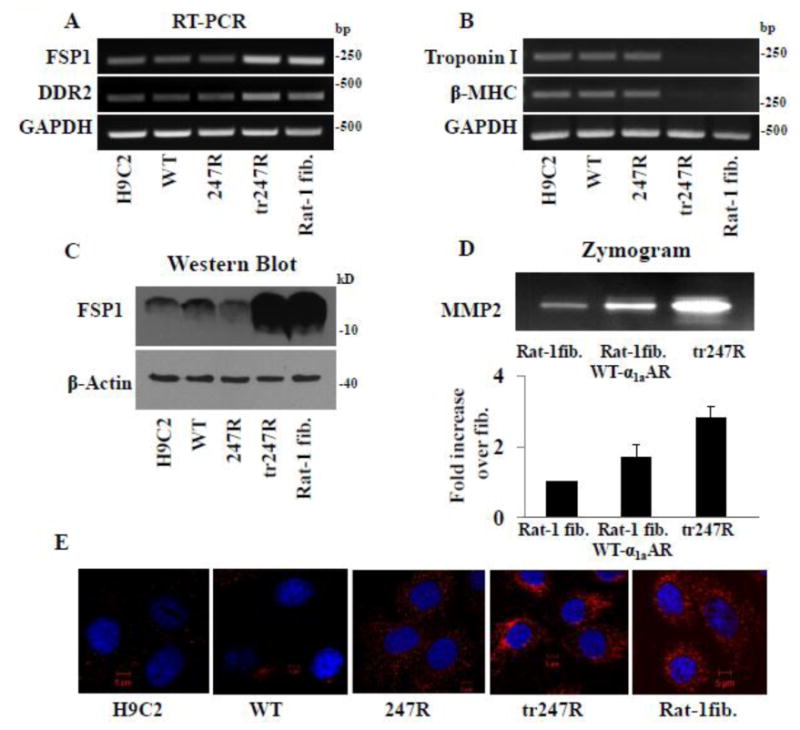
Tr247R cells exhibit fibroblast-like phenotype and express fibroblast markers. (A) Expression of fibroblast markers is upregulated in tr247R cells while expression of cardiac markers (B) is downregulated as determined by RT-PCR using gene specific primers and GAPDH as internal control. (C) FSP1 protein levels in tr247R cells are comparable to those in Rat-1 fibroblasts (Rat-1 fib) as determined by Western blot analyses with FSP1 specific antibody; β-actin was used as a loading control. Western blot with longer exposure time (30min) for FSP1 protein is shown to demonstrate expression levels in cells with myoblast morphology. (D) MMP2 levels are elevated in tr247R cells compared with fibroblasts as determined by zymograms. Representative image is presented. Lower panel depicts relative band intensities, values are presented as mean±SE, n=4. (E) Vimentin levels in tr247R cells are comparable to that in Rat-1 fibroblasts as determined by immunocytochemistry using vimentin antibody (red) and DAPI for nuclear staining (blue). Rat-1 fibroblasts or Rat-1 fibroblasts expressing WT-α1aAR were used to confirm fibroblast-like phenotype. Representative images are presented, n=3.
3.4. Increased proliferation of 247R cells is MMP-, Src- and EGFR-dependent
To evaluate signaling pathways responsible for constitutive hyperproliferation of 247R expressing cardiomyoblasts and new tr247R cells and to determine whether MMP-dependent EGFR transactivation is cell type dependent, we evaluated proliferation in the presence or absence of EGFR and MMP inhibitors. We used 500nM AG1478 for all cell lines with myoblast morphology and 1μM for tr247R cells with higher proliferative capacity.
EGFR-specific inhibitor AG1478 and MMP inhibitor GM6001 had no effect on basal proliferation of H9c2 and WT cells whereas AG1478 inhibited proliferation of 247R and tr247R cells (Fig. 4A,C), confirming EGFR involvement in hyperproliferation. While we chose to work with clones with low receptor expression levels (≤ 300 fmol/mg) to be close to physiological levels of the receptor, in newly formed tr247R cells upon transition of cardiomyoblasts to fibroblast-like cells the receptor expression levels increased to ∼ 1pmol/mg as described above. These new tr247R cells are not cardiac cells anymore and they exhibit about 4-8-fold increased proliferation compared to WT or control cardiomyoblasts (Fig. 2A). Dose response experiments revealed that treatment with 1μM AG1478 results in a statistically significant inhibition of tr247R cell proliferation while 500nM did not (Fig. 4D), which suggests that the higher the proliferative potential of the cells the higher dose of AG1478 inhibitor is needed. In cardiac 247R cells, 500nM AG1478 effectively decreases the proliferation rate to that in WT or control cells (Fig. 4A). Interestingly, GM6001 inhibited proliferation of 247R cells (Fig. 4A), but had no effect on proliferation of tr247R cells (Fig. 4C) regardless if 25μM or 50μM was used (Fig. 4E) suggesting activation of two different EGFR transactivation pathways: MMP-dependent EGFR transactivation in 247R cells and MMP-independent EGFR transactivation in tr247R cells. These data also indicate that 247R-triggered EGFR transactivation is generalizable and is not cell type dependent.
Figure 4.
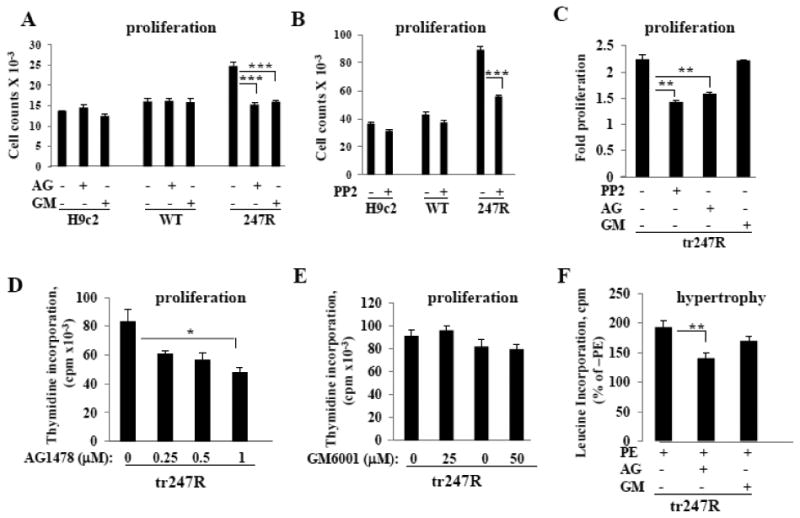
247R expression triggers EGFR transactivation in cardiomyoblasts. (A,B) EGFR- transactivation is MMP/Src-dependent in 247R cells and (C,D,E) MMP-independent but Src-dependent in tr247R cells. (A,C) Cells were cultured in the presence or absence of EGFR-specific inhibitor AG1478, MMP inhibitor GM6001, or (B,C) Src inhibitor PP2. Proliferation was evaluated by cell counting using light microscopy. (D) Tr247R cells were cultured in the presence or absence of different concentrations of EGFR-specific inhibitor AG1478 or (E) MMP inhibitor GM6001 and cell proliferation was evaluated using thymidine incorporation assays. Data are mean±SE of 3 independent experiments for AG1478 or representative from one of two independent experiments for GM6001 performed in triplicates. (F) Hypertrophy in tr247R cells is EGFR-transactivation-dependent. Hypertrophy was evaluated by leucine incorporation normalized to 1000 cells. Representative data are presented (mean±SE, n=3).
EGFR could be transactivated in Src-dependent or -independent manner [36]. To assess involvement of Src-kinase, we examined cell proliferation in the presence of Src-kinase-specific inhibitor PP2. Inhibition of Src significantly reduces proliferation of 247R (Fig. 4B) and tr247R cells (Fig. 4C), indicating constitutive hyperproliferation through activation of Src-EGFR pathway. Interestingly, in tr247R cells EGFR is transactivated in an MMP-independent, but Src-dependent manner. Importantly, hypertrophy in tr247R cells is also significantly reduced by EGFR-specific inhibitor, whereas inhibition of MMPs does not significantly reduce PE-induced hypertrophy (Fig. 4F).
3.5. EGFR-induced ERK phosphorylation is responsible for induction of cell cycle progression in 247R and tr247R cells
For some GPCRs, PI3K (phosphatidylinositide 3-kinase), AKT and ERK are key signaling molecules downstream of agonist-induced EGFR transactivation [37]. Therefore, we evaluated the levels of P-ERK and P-AKT by Western blotting. Basal levels of P-ERK are increased in 247R and tr247R cells compared to WT (Fig. 5A, lanes 3,5vs1). PE treatment significantly reduced levels of P-ERK (Fig. 5A, lanes 4vs3, 6vs5) whereas it had no effect on WT cells (lanes 2vs1). P-AKT levels are reduced in tr247R cells under basal conditions and nearly abolished in response to PE (Fig. 5B, lanes 5,6).
Figure 5.
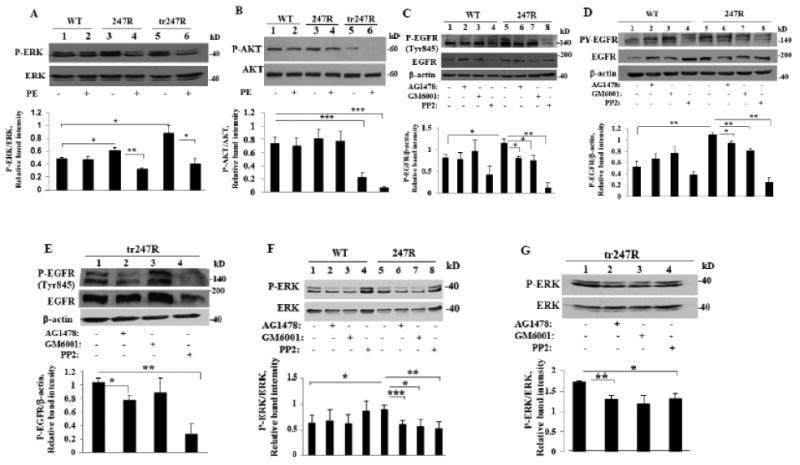
247R-triggered hyperproliferation is due to EGFR transactivation and ERK activation. (A,B) ERK, but not AKT, is activated in 247R cells; PE stimulation inhibits ERK and AKT phosphorylation. Cells were cultured for 48h in the presence or absence of PE and cell lysates were examined by Western blotting. (C,D) EGFR transactivation is MMP/Src-dependent in 247R expressing cells, but not in WT cells. (E) EGFR transactivation is MMP-independent but Src-dependent in tr247R cells. MMP inhibitor GM6001 does not reduce levels of P-EGFR (Tyr845) in tr247R cells (lane 3vs1) in contrast to EGFR inhibitor AG1478 (lane 2vs1) or Src inhibitor PP2 (lane 4vs1). (F) ERK is constitutively activated and regulated by EGFR/Src/MMP pathway in 247R cells or (G) by EGFR/Src pathway in tr247R cells. Cells were cultured in the presence or absence of specific inhibitors and cell lysates were examined by Western blotting. Lower panels depict relative intensities of phospho-protein bands normalized to corresponding total protein bands as determined by densitometric analysis. (C,D,E) β-actin was used as a loading control. Data are mean±SE, n=3 (A,B,C,D,G), n=4 (E), or n=6 (F).
To determine if basal ERK activation in 247R and tr247R cells is downstream of Src- and MMP-dependent EGFR transactivation, the cells were treated with Src, MMP or EGFR inhibitors and cell lysates were immunoblotted with antibody specific for Src phosphorylation site of EGFR (Tyr845) or with phosphotyrosine antibody (anti-PY). Compared with WT, 247R cells exhibit increased basal EGFR phosphorylation (Fig. 5C, lanes 5vs1 and 5D lanes 5vs1), which is significantly reduced by inhibitor treatment of 247R cells (Fig. 5C lanes 6,7,8vs5 and 5D, lanes 6,7,8vs5). Slight upregulation of P-EGFR levels in lane 3vs1 in WT cells is not statistically significant (Fig. 5C,D). These findings confirm that 247R triggers agonist-independent (constitutive) transactivation of Src/MMP/EGFR pathway. MMP inhibitor did not reduce levels of P-Tyr845-EGFR in tr247R cells (Fig. 5E, lane 3vs1) or hyperproliferation of tr247R cells at 25μM or 50μM (Fig. 4E), whereas the levels of P-Tyr845-EGFR in tr247R cells decreased in response to AG1478 (Fig. 5E, lane 2vs1) or PP2 (Fig. 5E, lane 4vs1). EGFR and Src inhibition also significantly reduced P-ERK in tr247R cells (Fig. 5G, lanes 2,4vs1), while MMP inhibitor did not have statistically significant effect (Fig. 5G, lane 3vs1). Fig. 5F demonstrates that all 3 inhibitors reduce P-ERK in 247R compared with WT cells (Fig. 5F, lanes 6,7,8vs5). These data indicate that increased proliferation in 247R cells before and after phenotype change is EGFR/ERK-dependent and confirm that Src and MMP are involved in EGFR transactivation before transition, but only Src is involved in EGFR transactivation after transition.
3.6. Src and EGFR are constitutively activated in cells expressing 247R
It has been reported that synergistic activation of EGFR, Src, and ERK-kinases induced by agonist stimulation of angiotensin II type 1 receptor leads to endothelial-mesenchymal transition [38]. We examined Src and EGFR activation in 247R and tr247R cells. To determine involvement of Src in 247R-mediated EGFR-transactivation, we analyzed Src phosphorylation in basal conditions. As shown in Fig. 6A, P-Src levels are significantly increased in 247R and tr247R cells compared with control or WT cells (lanes 3,4vs1,2), while total Src levels remain unchanged. Phosphorylation of Tyr845-EGFR, a Src-mediated and EGFR ligand-dependent phosphorylation site, is also upregulated in 247R and tr247R cells (Fig. 6B, lanes 3,4vs1,2). Expression levels of total EGFR, as evaluated by Western blotting, are also elevated in 247R and tr247R cells compared to WT cells (Fig. 6B, lanes 3,4vs2). Importantly, the level of active P-EGFR, that is critical for downstream signaling, is significantly upregulated in 247R or tr247R cells compared with control or WT cells (Fig.6B, lanes 3.4vs1,2) emphasizing the role of EGFR transactivation in hyperproliferation of 247R and tr247R cells. Thus, 247R expression triggers statistically significant increase in the levels of activated EGFR and Src-kinases, leading to hyperproliferation.
Figure 6.
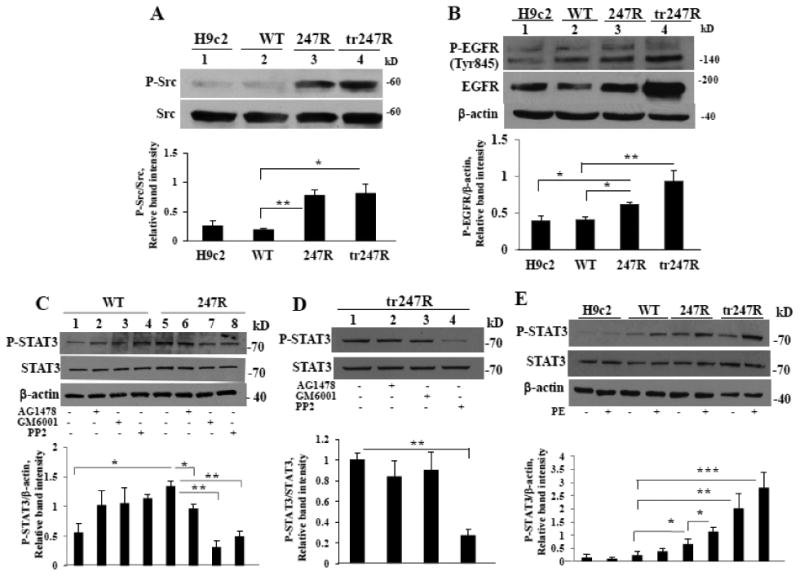
247R expression triggers Src, EGFR and STAT3 activation. (A) Src kinase is constitutively activated in 247R and tr247R cells. (B) P-EGFR levels are upregulated in 247R and tr247R cells. Cell lysates from control, WT, 247R or tr247R cells were examined by Western blot analysis. (C) Constitutive activation of STAT3 is EGFR/Src/MMP-dependent in 247R cells and only (D) Src-dependent in tr247R cells. (E) STAT3 is activated in 247R and tr247R cells in basal conditions and is upregulated upon agonist stimulation compared with WT. Cells were cultured in the presence or absence of specific inhibitors for 48h and cell lysates were examined by Western blotting. β-actin was used as loading control. Lower panels depict relative intensities of phospho-protein bands normalized to corresponding total protein or to β-actin protein bands as determined by densitometric analysis. Data represent mean±SE (A,B,C,D, n=3; E, n=5).
3.6.1. STAT3 is activated in 247R expressing cells
Because Src is implicated in STAT3 activation [39], we examined if STAT3 is activated in 247R cells with constitutively activated Src. Western blot analyses demonstrate that basal levels of P-STAT3 and STAT3 are upregulated in 247R cells compared with WT cells (Fig. 6C, lane 5vs1). EGFR, MMP and Src inhibitors significantly reduce P-STAT3 levels in 247R, but not in WT cells (Fig. 6C), indicating that in 247R cells STAT3 is downstream of the EGFR transactivation pathway. Activation of STAT3 in tr247R cells was affected only by Src inhibitor suggesting that in these cells STAT3 is activated downstream of Src-kinase (Fig. 6D, lane 4vs1). Our findings reveal that PE-induced hypertrophy in tr247R cells is EGFR transactivation-dependent (Fig. 4F). PE stimulation results in a significant increase of P-STAT3 levels in 247R and tr247R cells compared with P-STAT3 level in WT cells (Fig. 6E), indicating involvement of P-STAT3 in agonist-induced hypertrophy in these cells.
3.7. MMPs and ADAMs are differentially expressed in 247R and tr247R cells
In fibroblasts, 247R induces MMP7/ADAM12-dependent transactivation of EGFR [29]. Therefore, we evaluated expression of MMPs and ADAMs at RNA and protein levels in control, WT, 247R and tr247R cells (Fig. 7). In tr247R cells, MMP2, MMP7 and MMP9 expression levels are increased, while in 247R cells the levels of MMP2 are reduced as evidenced by zymograms or RT-PCRs (Fig. 7A,B). The weak upper band of MMP2 in tr247R cells (Fig. 7A) corresponds to latent (inactive) MMP2, while the lower more intense band is the active form, which is consistent with previous reports on MMP2 [40,41]. The basal MMP7 levels appear to be slightly upregulated in 247R cells compared with WT cells. In contrast, expression of ADAM12 (but not ADAM10 or ADAM17) was decreased in tr247R cells (Fig. 7B). Agonist stimulation does not appear to have an effect on MMP or ADAM levels.
Figure 7.
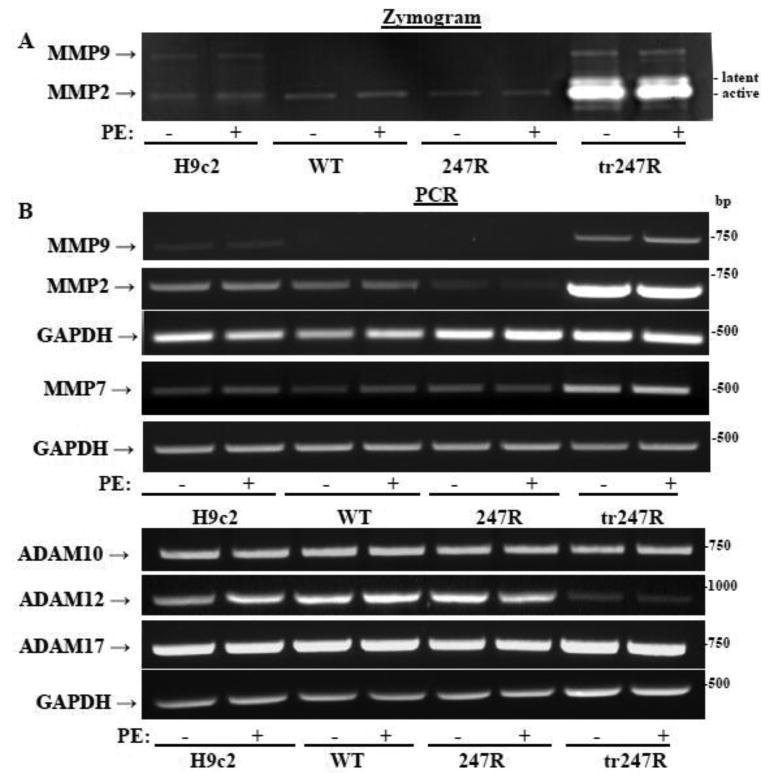
Differential expression of specific MMPs and ADAMs in cardiomyoblasts expressing WT or 247R as determined by (A) zymograms or (B) RT-PCR using gene-specific primers. GAPDH was used as an internal control. Data are representatives of three independent experiments.
3.8. α1AR inverse agonist prazosin inhibits agonist-induced hypertrophy, but not constitutive hyperproliferation of 247R or tr247R cells
Agonist binding to GPCRs activates canonical G protein-coupled intracellular signaling cascades with subsequent receptor phosphorylation leading to recruitment of βarrestins which may result in receptor desensitization and internalization [15]. In addition, βarrestins can activate G protein-independent signaling pathways. To elucidate whether 247R triggers Gq- or βarrestin-mediated signaling pathways in cardiomyoblasts, we performed proliferation and hypertrophy assays in the presence of α1AR inverse agonist prazosin. Similar to stimulation with 10μM PE (Fig. 1C, 4F), 247R and tr247R cells stimulated with 1μM PE exhibit approximately 60% and 85% increased leucine incorporation, indicating enhanced protein synthesis consistent with hypertrophy (Fig. 8A). Treatment with 1μM prazosin alone does not alter leucine incorporation, but completely reverses the hypertrophic effect of PE (Fig. 8A), indicating that hypertrophy in 247R and tr247R cells is Gq-dependent. In control or WT cells, PE or prazosin do not impact leucine incorporation. In contrast to hypertrophy, constitutive hyperproliferation of 247R and tr247R cells is not altered by prazosin (Fig. 8B), indicating a Gq-independent mechanism.
Figure 8.
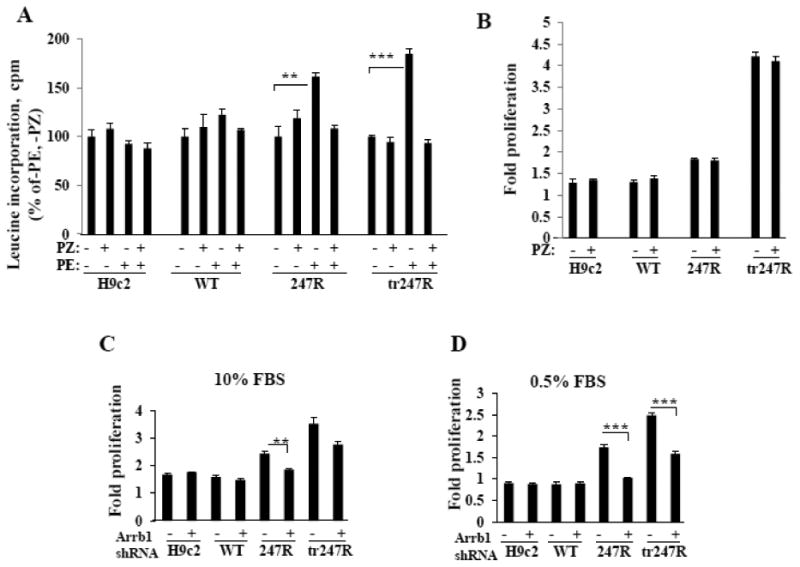
(A) The α1-AR inverse agonist prazosin (PZ) inhibits agonist-induced hypertrophy, but (B) not agonist-independent hyperproliferation. Hypertrophy was evaluated by leucine incorporation normalized to 1000 cells and cell proliferation was evaluated by cell counting using light microscopy. (C,D) 247R-triggered cell proliferation is βarrestin1-dependent. Cells were transfected with scrambled or βarrestin1 specific shRNA and cultured for 48h. Representative data from one of three independent experiments, each done in triplicates, are presented as mean±SE.
3.8.1. 247R-triggered hyperproliferation is βarrestin1-dependent
To assess if EGFR transactivation in 247R cells is βarrestin-dependent, we transfected cells with βarrestin1-specific shRNAs. In serum-containing medium, knockdown of βarrestin1 reduces proliferation of 247R and tr247R cells by ∼20%, but has no effect in WT or control cells (Fig. 8C). Under serum-deprived conditions, knockdown of βarrestin1 resulted in a highly significant reduction of proliferation in 247R cells to near normal levels and by ∼40% in tr247R cells (Fig. 8D). These data demonstrate that the 247R-triggered cell proliferation is βarrestin1-dependent.
Thus, our findings reveal that 247R expression in cardiomyoblasts leads to synergistic, constitutive activation of EGFR, Src and ERK kinases which appears to lead to transition of cardiomyoblasts to fibroblast-like cells and activation of two distinct signaling pathways: constitutive Gq-independent βarrestin1/Src/MMP/EGFR/ERK-dependent hyperproliferation and agonist-induced Gq-dependent EGFR/STAT-dependent hypertrophy. In new tr247R fibroblast-like cells activation of these pathways appears to be MMP-independent (Schematic, Fig. 9).
Figure 9.
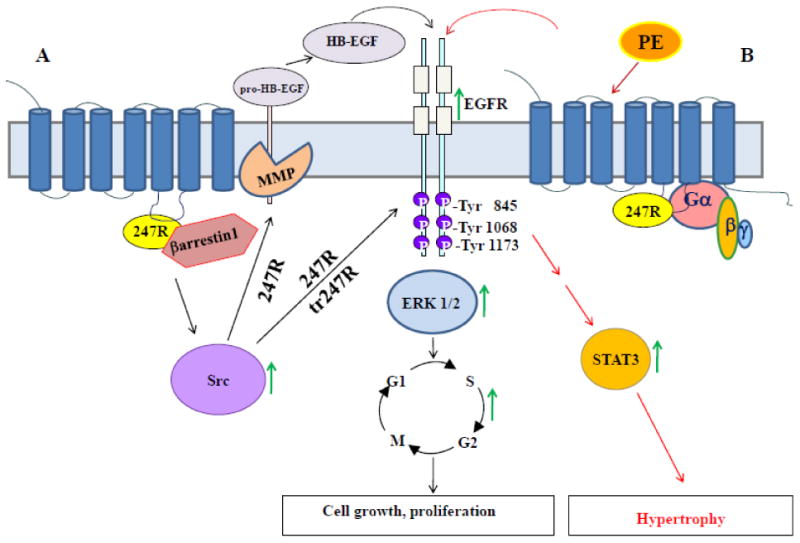
Schematic of 247R-induced serum- and agonist-independent cell proliferation and agonist-dependent cell hypertrophy in cardiomyoblasts. (A) 247R triggers constitutive, βarrestin1/Src/MMP-dependent EGFR transactivation, while tr247R cells exhibit βarrestin1/Src-dependent, but MMP-independent EGFR transactivation leading to ERK activation, cell cycle progression and cell proliferation. (B) Agonist stimulation of 247R or tr247R cells triggers Gq-dependent EGFR/STAT3 activation and hypertrophy.
4. Discussion
4.1. 247R-induced transition of cardiomyoblasts to fibroblast-like cell phenotype
In this study we report a novel finding that constitutive transactivation of EGFR by naturally occurring human α1aAR-247R genetic variant triggers transition of cardiomyoblasts to fibroblast-like cells. Cells expressing higher levels of 247R receptor undergo this transition earlier than cells with lower expression levels, while control or WT cells do not change the phenotype, thus confirming that the transition is 247R-dependent. The newly formed tr247R cells exhibit increased levels of fibroblast-specific protein 1 (FSP1) and DDR2, which is expressed in cardiac fibroblasts, but not in myocytes or vascular smooth muscle [42]. In addition, other fibroblast markers are highly expressed in tr247R cells (vimentin, MMP2) while expression of cardiomyocyte markers Troponin I, β-MHC, normally expressed in H9c2 cells [43], are downregulated indicating that tr247R cells no longer have mRNA/protein expression profiles matching cardiac muscle cells. These new cells, originated from cardiomyoblasts expressing 247R, exhibit a fibroblast-like phenotype, constitutive hyperproliferation and agonist-induced hypertrophy. It is known that fibroblasts are responsible for deposition of fibrotic extracellular matrix and their activation may cause cardiomyocyte hypertrophy via paracrine mechanisms further impairing cardiac function [4]. It is therefore plausible that expression of 247R in human cardiomyocytes leads to myocardial fibrosis, a condition known to lead to arrhythmias by altering mechano-electric coupling, and to increase stiffness of cardiac chambers, ultimately leading to cardiac failure. Our novel data revealing 247R-dependent transition of cardiomyoblasts to fibroblast-like cells, with expression of characteristic fibroblast markers and highly proliferative capacity could represent a novel mechanism for myocardial fibrosis.
4.2. 247R-induced hyperproliferation
We recently reported that in fibroblasts the naturally occurring human α1aAR-247R genetic variant, originally identified in a hypertensive patient, triggers βarrestin1/MMP7/EGFR transactivation pathway in an agonist-independent manner, leading to constitutive hyperproliferation [29]. In the fibroblasts EGFR transactivation dependent hyperproliferation is agonist independent and PE treatment does not induce hypertrophy, whereas in cardiomyoblasts two distinct signaling pathways are activated by 247R. First, 247R-expression triggers Gq-independent, βarrestin1/Src/MMP-dependent constitutive, intrinsic, serum-independent EGFR transactivation and ∼2-fold increased cell proliferation compared with WT or control cells, which is inhibited by EGFR-, MMP-, and Src-specific inhibitors, as well as βarrestin1-specific shRNAs. Second, we demonstrate that agonist stimulation of 247R and tr247R but not WT cells leads to hypertrophy via Gq-dependent EGFR/Src/STAT3 transactivation pathway. Thus, our data reveal that agonist- and serum-independent EGFR transactivation by 247R is not cell type dependent, but generalizable.
Recent studies suggest that some GPCRs may require receptor tyrosine kinases such as PDGF, VEGF, EGF receptors as intermediate molecules to mediate downstream effects of GPCR signaling via transactivation pathways [45]. Cell cycle progression can be mediated by GPCR-induced EGFR transactivation and RAS (rat sarcoma)/MEK/ERK and PI3K/AKT pathway activation [37]. In many cases EGFR transactivation is mediated by endogenous ligands such as HB-EGF [46], which is synthesized as a transmembrane protein, and then is proteolytically processed into a soluble form which activates EGFR. HB-EGF is a potent mitogen for cardiomyocytes. Mice carrying a constitutively active form of HB-EGF show cardiac hyperplasia [47], and high levels of HB-EGF secreted from embryonic cardiac fibroblasts stimulate proliferation in embryonic cardiomyocytes [48]. Previous studies demonstrate that MMPs/ADAMs are responsible for shedding of HB-EGF and subsequent transactivation of EGFR [29]. However, contribution of different MMPs/ADAMs appears highly dependent on specific stimulus and cellular environment. Our new data in cardiomyoblasts demonstrate that MMP/ADAM inhibitor GM6001 inhibits 247R-induced EGFR transactivation, indicating that MMP/ADAM-dependent EGFR transactivation is an essential mechanism for 247R-triggered proliferation. We identified MMP7 as one of the MMPs involved in this process. MMP2, that has reduced levels in 247R cells, is thought to play a key role in degradation of fibrillar collagens. Low levels of MMP2 in 247R cells may suggest extracellular matrix deposition and subsequent loss of myocardial contractility. Interestingly, after phenotype change, in tr247R cells MMP2 and MMP7 levels are increased and ADAM12 levels are downregulated. It has been demonstrated that cardiac hypertrophy in agonist-induced hypertension depends on transcriptional regulation of MMP7 and ADAM12 [49]. It is possible that the MMP7/ADAM12 signaling axis is controlled with specific mediators in 247R cells and MMP7/ADAM12-dependent signaling might represent a new therapeutic target for drug resistant hypertensive disorders and cardiac disease.
Recently, the classic paradigm of GPCR-G protein signaling has been challenged with increasing evidence that GPCR signaling is not always mediated by G proteins. βarrestin-dependent, G protein-independent EGFR transactivation by β1ARs in response to agonist stimulation was reported as cardioprotective in mice [50]. Angiotensin receptor AT1A activates ERK by both βarrestin2 and G protein-mediated signaling pathways in parallel [51]. Our results reveal that α1AR inverse agonist prazosin does not inhibit proliferation of 247R cells, thus indicating Gq-independent transactivation of the 247R/MMP/EGFR signaling pathway. We hypothesize that G247R SNP in the 3rd intracellular loop is changing the receptor conformation to be unfavorable for G protein coupling and demonstrate that 247R-triggered hyperproliferation in these cells is βarrestin1-dependent.
247R-mediated Gq-independent EGFR transactivation is Src-dependent. Src is a non-receptor tyrosine kinase involved in regulation of many physiological responses, including cell proliferation, survival and differentiation [52]. GPCR agonists induce transient and rapid activation of Src-family members and EGFR phosphorylation at Tyr845, a site phosphorylated directly by Src-kinase. In 247R cardiomyoblasts the general MMP inhibitor GM6001 inhibited phosphorylation of EGFR at Tyr845 as effectively as it blocked the phosphorylation at known EGFR autophosphorylation sites [36], indicating Src involvement in EGFR ligand generation and EGFR transactivation processes by MMP/ADAM-family members. We demonstrate that Src-specific inhibitor PP2 inhibits proliferation of 247R cells, but not WT or control cells, indicating the key role of Src in the 247R/EGFR transactivation pathway. Src-kinase is activated in 247R cells in basal conditions and triggers phosphorylation of Tyr845-EGFR which is sensitive to GM6001. These data demonstrate key involvement of Src in βarrestin/MMP/ADAM-dependent shedding of EGFR ligand(s). Interestingly, Src inhibitor PP2 also inhibited MMP-independent proliferation of tr247R cells, suggesting that in tr247R cells Src may directly phosphorylate Tyr845-EGFR (see schematic, Fig. 9).
Prior reports suggest that Src cooperates with EGFR in cell transformation and mediates MAPK/ERK activation pathway through GPCRs [53]. The MAPK/ERK signaling pathway regulates multiple cellular activities, such as proliferation, apoptosis and activation of MAPK cascades to regulate cardiac function [54]. Nevertheless, association between GPCR-induced EGFR transactivation and MAPK/ERK signaling pathway in cardiomyocytes remains elusive. 247R expression in H9c2 cells triggers ERK activation that is blocked by EGFR, MMP and Src inhibitors. Thus, our data demonstrate that expression of 247R triggers constitutive transactivation of EGFR and activation of Src and ERK. Activation of these three kinases may trigger the observed phenotype change in 247R-expressing cardiomyoblasts.
4.3. 247R-induced hypertrophy
Increased levels of catecholamines leading to activation of ARs are believed to be a primary mediator of cardiac hypertrophy [55,56]. The role of α1ARs in cardiac hypertrophy is mainly through activated Gq-proteins and their downstream signaling, such as the MAPK pathway [57]. Recent evidence suggests that G protein-independent signal transduction pathways also contribute to hypertrophic growth responses induced by α1AR [58]. Yet, the mechanisms of α1AR-induced hypertrophy are complicated and unclear. In previous studies of other GPCRs, the generally accepted mechanism is that transactivated EGFR could activate ERK kinase, which subsequently phosphorylates other protein kinases or substrates to promote hypertrophy [59]. Recently, involvement of novel molecules, such as phosphorylation of p66Shc, was proposed in α 1 AR-induced hypertrophic pathway via transactivation of EGFR [60]. Interestingly, angiotensin II type-1 receptor stimulation induces EGFR transactivation, leading to activation of ERK2 and cardiac hypertrophy in cultured cardiac myocytes in vitro, while a receptor mutant lacking EGFR transactivation does not [61]. To date, it remains unknown whether EGFR transactivation is involved in α1aAR-mediated hypertrophy in cardiomyoblasts. In this study we demonstrate that agonist stimulation of 247R cells induces distinct hypertrophy prior to and after phenotype change that is sensitive to EGFR specific inhibitor, indicating that 247R-triggered hypertrophy is EGFR transactivation-dependent. Interestingly, hypertrophy, but not cell proliferation, is inhibited by the α1aAR inverse agonist prazosin, supporting the notion that increased proliferation and hypertrophy observed in 247R and tr247R cells are two independent pathways triggered by 247R (Fig. 9).
STAT3 impacts a broad range of molecular and cellular mechanisms involved in remodeling processes in normal cardiac physiology and during myocardial infarction, pressure overload, and ischemia/reperfusion [62]. STAT3 is constitutively activated by cytokines, growth factors, mechanical load, and ischemia [63]. It is implicated in cardiomyocyte hypertrophy, and could affect neighboring non-myocyte cells via paracrine pathways. Our data reveal that in basal conditions expression of 247R genetic variant of α1aAR leads to STAT3 activation that is inhibited by EGFR- and MMP- specific inhibitors, demonstrating EGFR transactivation-dependent STAT3 activation. Furthermore, agonist stimulation of 247R and tr247R cells induces STAT3 activation and hypertrophy, indicating that 247R-initiated hypertrophy is Src/EGFR/STAT3-dependent. α 1aAR-WT does not activate this pathway.
In summary, our data demonstrate that the naturally occurring human α1aAR-247R genetic variant constitutively triggers a novel signaling mechanism that leads to transition of cardiomyoblasts to fibroblast-like cells with hyperproliferation in a Gq-independent manner via βarrestin1/MMP7/ADAM/Src-dependent EGFR transactivation pathway, or to agonist- and Gq-dependent hypertrophy in cardiomyoblasts.
Conclusions
Our previous findings in fibroblasts [29] suggest a potentially novel mechanism underlying some forms of sympathetically-mediated hypertension. Here we explored the role of naturally occurring α1aAR genetic variant (247R versus WT) in cardiomyoblasts. Our findings reveal that the presence of 247R triggers “transition” of cardiomyoblasts to fibroblast-like cells accompanied by alterations in expression of various matrix metalloproteinases. Together these findings suggest that 247R leads to extracellular matrix remodeling in myocardial tissue and myocardial fibrosis. Thus, one can extrapolate that the molecular and cellular alterations triggered by 247R may potentially lead to myocardial fibrosis, arrhythmias, and ultimately heart failure. The constellation of these 3 findings is commonly seen in humans after myocardial ischemia and infarction, yet the mechanism for this constellation has never been elucidated in full. Thus our findings, while speculative, offer the first cohesive clue to examining these mechanisms. Indeed, the fact that this totally unexpected finding, with such potential for clinical relevance in myocardial tissue, is very exciting. Future studies in human cardiac cells should provide more comprehensive data on the role of α1aARs and its genetic variants in cardiac pathology.
Highlights.
α1aAR genetic variant triggers cardiomyoblast-to-fibroblast-like cell transition
α1aAR-247R triggers constitutive hyperproliferation and agonist-induced hypertrophy
α1aAR-247R-triggered hyperproliferation is Src/MMP/EGFR-transactivation dependent
α1aAR-247R triggers synergistic activation of EGFR, Src, ERK kinases
α1aAR-247R-induced changes may lead to myocardial fibrosis and arrhythmias
Acknowledgments
Source of Funding: This work was supported by NIH HL49103 (DAS) and Swiss National Science Foundation PBSKP3_134335; PBSKP3_140095 (MKB).
Abbreviations used are
- PLCβ
1-Phosphatidylinositol-4,5-bisphosphate phosphodiesterase beta
- AR
Adrenergic Receptor
- HB-EGF
Heparin-binding EGF-like growth factor
- EGFR
Epidermal growth factor receptor
- EGFP
Enhanced green fluorescent protein
- GPCR
G-protein-coupled receptor
- MHC
Myosin heavy chain
- PIP2
Phosphatidylinositol 4,5-bisphosphate
- PI3K
Phosphatidylinositide 3-kinase
- ADAMs
A disintegrin and metalloproteinase domain-containing proteins
- DDR2
Discoidin domain-containing receptor 2
- FSP
Fibroblast specific protein
- MMP
Matrix metalloproteinase
- PE
Phenylephrine
- RAS
Rat sarcoma
Footnotes
Disclosures: None.
Author contributions: MKB, IG and EB performed the experiments, contributed to analysis and design, and wrote the article; DAS contributed to design, data review and writing the article; AO designed the research, performed some of the experiments, analyzed the data, and wrote the article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Woodcock EA, Du XJ, Reichelt ME, Graham RM. Cardiac alpha 1-adrenergic drive in pathological remodelling. Cardiovasc Res. 2008;77:452–462. doi: 10.1093/cvr/cvm078. [DOI] [PubMed] [Google Scholar]
- 2.Perez DM, Doze VA. Cardiac and neuroprotection regulated by alpha 1-adrenergic receptor subtypes. J Recept Signal Transduct Res. 2011;31:98–110. doi: 10.3109/10799893.2010.550008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen BC, O'Connell TD, Simpson PC. Alpha-1-adrenergic receptors: Targets for agonist drugs to treat heart failure. J Moll Cell Cardiol. 2011;51:518–528. doi: 10.1016/j.yjmcc.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landzberg JS, Parker JD, Gauthier DF, Colucci WS. Effects of myocardial alpha 1-adrenergic receptor stimulation and blockade on contractility in humans. Circulation. 1991;84:1608–1614. doi: 10.1161/01.cir.84.4.1608. [DOI] [PubMed] [Google Scholar]
- 5.Skomedal T, Borthne K, Aass H, Geiran O, Osnes JB. Comparison between alpha-1 adrenoceptor-mediated and beta adrenoceptor-mediated inotropic components elicited by norepinephrine in failing human ventricular muscle. J Pharmacol Exp Ther. 1997;280:721–729. [PubMed] [Google Scholar]
- 6.ALLHAT Collaborative Research Group. Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT) JAMA. 2000;283:1967–1975. [PubMed] [Google Scholar]
- 7.Koshimizu TA, Tsujimoto G, Hirasawa A, Kitagawa Y, Tanoue A. Carvedilol selectively inhibits oscillatory intracellular calcium changes evoked by human alpha1D- and alpha1B-adrenergic receptors. Cardiovasc Res. 2004;63:662–672. doi: 10.1016/j.cardiores.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 8.Woodcock EA. Roles of alpha1A- and alpha1B-adrenoceptors in heart: insights from studies of genetically modified mice. Clin Exp Pharmacol Physiol. 2007;34:884–888. doi: 10.1111/j.1440-1681.2007.04707.x. [DOI] [PubMed] [Google Scholar]
- 9.Milano CA, Dolber PC, Rockman HA, Bond RA, Venable ME, Allen LF, Lefkowitz RJ. Myocardial expression of a constitutively active alpha 1B-adrenergic receptor in transgenic mice induces cardiac hypertrophy. Proc Natl Acad Sci U S A. 1994;91:10109–10113. doi: 10.1073/pnas.91.21.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Connell TD, Swigart PM, Rodrigo MC, Ishizaka S, Joho S, Turnbull L, Tecott LH, Baker AJ, Foster E, Grossman W, Simpson PC. Alpha1-adrenergic receptors prevent a maladaptive cardiac response to pressure overload. J Clin Invest. 2006;116:1005–1015. doi: 10.1172/JCI22811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Autelitano DJ, Woodcock EA. Selective activation of alpha1A-adrenergic receptors in neonatal cardiac myocytes is sufficient to cause hypertrophy and differential regulation of alpha1-adrenergic receptor subtype mRNAs. J Mol Cell Cardiol. 1998;30:1515–1523. doi: 10.1006/jmcc.1998.0717. [DOI] [PubMed] [Google Scholar]
- 12.Reder NP, Tayo BO, Salako B, Ogunniyi A, Adeyemo A, Rotimi C, Cooper RS. Adrenergic alpha-1 pathway is associated with hypertension among Nigerians in a pathway-focused analysis. PLoS One. 2012;7:e37145. doi: 10.1371/journal.pone.0037145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rudner XL, Berkowitz DE, Booth JV, Funk BL, Cozart KL, D'Amico EB, El-Moalem H, Page SO, Richardson CD, Winters B, Marucci L, Schwinn DA. Subtype specific regulation of human vascular alpha(1)-adrenergic receptors by vessel bed and age. Circulation. 1999;100:2336–2343. doi: 10.1161/01.cir.100.23.2336. [DOI] [PubMed] [Google Scholar]
- 14.Rokosh DG, Simpson PC. Knockout of the alpha 1A/C-adrenergic receptor subtype: the alpha 1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc Natl Acad Sci U S A. 2002;99:9474–9479. doi: 10.1073/pnas.132552699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 16.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 17.Werry TD, Sexton PM, Christopoulos A. “Ins and outs” of seven-transmembrane receptor signalling to ERK. Trends Endocrinol Metab. 2005;16:26–33. doi: 10.1016/j.tem.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 18.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 19.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffold. Proc Natl Acad Sci U S A. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhuang S, Kinsey GR, Rasbach K, Schnellmann RG. Heparin-binding epidermal growth factor and Src family kinases in proliferation of renal epithelial cells. Am J Physiol Renal Physiol. 2008;294:F459–468. doi: 10.1152/ajprenal.00473.2007. [DOI] [PubMed] [Google Scholar]
- 21.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 22.Shah BH, Catt KJ. Matrix metalloproteinase-dependent EGF receptor activation in hypertension and left ventricular hypertrophy. Trends Endocrinol Metab. 2004;15:241–243. doi: 10.1016/j.tem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 23.Mulvany MJ. Small artery remodeling in hypertension. Curr Hypertens Rep. 2002;4:49–55. doi: 10.1007/s11906-002-0053-y. [DOI] [PubMed] [Google Scholar]
- 24.Thomsen M, Dahl M, Tybjaerg-Hansen A, Nordestgaard BG. beta2 -adrenergic receptor Thr164IIe polymorphism, blood pressure and ischaemic heart disease in 66 750 individuals. J Intern Med. 2012;271:305–314. doi: 10.1111/j.1365-2796.2011.02447.x. [DOI] [PubMed] [Google Scholar]
- 25.Kelsey RM, Alpert BS, Dahmer MK, Krushkal J, Quasney MW. Alpha-adrenergic receptor gene polymorphisms and cardiovascular reactivity to stress in Black adolescents and young adults. Psychophysiology. 2012;49:401–412. doi: 10.1111/j.1469-8986.2011.01319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei B, Morris DP, Smith MP, Svetkey LP, Newman MF, Rotter JI, Buchanan TA, Beckstrom-Sternberg SM, Green ED, Schwinn DA. Novel human alpha1a-adrenoceptor single nucleotide polymorphisms alter receptor pharmacology and biological function. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:229–239. doi: 10.1007/s00210-005-1019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burger A, Benicke M, Deten A, Zimmer HG. Catecholamines stimulate interleukin-6 synthesis in rat cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2001;281:H14–21. doi: 10.1152/ajpheart.2001.281.1.H14. [DOI] [PubMed] [Google Scholar]
- 28.Lai KB, Sanderson JE, Yu CM. Suppression of collagen production in norepinephrine stimulated cardiac fibroblasts culture: differential effect of alpha and beta-adrenoreceptor antagonism. Cardiovasc Drugs Ther. 2009;23:271–280. doi: 10.1007/s10557-009-6183-6. [DOI] [PubMed] [Google Scholar]
- 29.Oganesian A, Yarov-Yarovoy V, Parks WC, Schwinn DA. Constitutive coupling of a naturally occurring human alpha1a-adrenergic receptor genetic variant to EGFR transactivation pathway. Proc Natl Acad Sci U S A. 2011;108:19796–19801. doi: 10.1073/pnas.1116271108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimes BW, Brandt BL. Properties of a clonal muscle cell line from rat heart. Exp Cell Res. 1976;98:367–381. doi: 10.1016/0014-4827(76)90447-x. [DOI] [PubMed] [Google Scholar]
- 31.Hescheler J, Meyer R, Plant S, Krautwurst D, Rosenthal W, Schultz G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ Res. 1991;69:1476–1486. doi: 10.1161/01.res.69.6.1476. [DOI] [PubMed] [Google Scholar]
- 32.Mejia-Alvarez R, Tomaselli GF, Marban E. Simultaneous expression of cardiac and skeletal muscle isoforms of the L-type Ca2+ channel in a rat heart muscle cell line. J Physiol. 1994;478(Pt 2):315–329. doi: 10.1113/jphysiol.1994.sp020252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watkins SJ, Borthwick GM, Arthur HM. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. In Vitro Cell Dev Biol Anim. 2011;47:125–131. doi: 10.1007/s11626-010-9368-1. [DOI] [PubMed] [Google Scholar]
- 34.Oganesian A, Armstrong LC, Migliorini MM, Strickland DK, Bornstein P. Thrombospondins use the VLDL receptor and a nonapoptotic pathway to inhibit cell division in microvascular endothelial cells. Mol Biol Cell. 2008;19:563–571. doi: 10.1091/mbc.E07-07-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delgado-Olguin P, Huang Y, Li X, Christodoulou D, Seidman CE, Seidman JG, Tarakhovsky A, Bruneau BG. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet. 2012;44:343–347. doi: 10.1038/ng.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boerner JL, Biscardi JS, Silva CM, Parsons SJ. Transactivating agonists of the EGF receptor require Tyr 845 phosphorylation for induction of DNA synthesis. Mol Carcinog. 2005;44:262–273. doi: 10.1002/mc.20138. [DOI] [PubMed] [Google Scholar]
- 37.Chen H, Ma N, Xia J, Liu J, Xu Z. beta2-Adrenergic receptor-induced transactivation of epidermal growth factor receptor and platelet-derived growth factor receptor via Src kinase promotes rat cardiomyocyte survival. Cell Biol Int. 2012a;36:237–244. doi: 10.1042/CBI20110162. [DOI] [PubMed] [Google Scholar]
- 38.Chen J, Chen JK, Harris RC. Angiotensin II induces epithelial-to-mesenchymal transition in renal epithelial cells through reactive oxygen species/Src/caveolin-mediated activation of an epidermal growth factor receptor-extracellular signal-regulated kinase signaling pathway. Mol Cell Biol. 2012b;32:981–991. doi: 10.1128/MCB.06410-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE., Jr Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol. 1998;18:2553–2558. doi: 10.1128/mcb.18.5.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toth M, Sohail A, Fridman R. Assessment of gelatinases (MMP-2 and MMP-9) by gelatin zymography. Methods Mol Biol. 2012;878:121–135. doi: 10.1007/978-1-61779-854-2_8. [DOI] [PubMed] [Google Scholar]
- 41.Aica Dell', Donà M, Sartor L, Pezzato E, Garbisa S. (-)Epigallocatechin-3-gallate directly inhibits MT1-MMP activity, leading to accumulation of nonactivated MMP-2 at the cell surface. Lab Invest. 2002;82:1685–1693. doi: 10.1097/01.lab.0000043122.00384.91. [DOI] [PubMed] [Google Scholar]
- 42.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 43.Chen ZC, Yu BC, Chen LJ, Cheng KC, Lin HJ, Cheng JT. Characterization of the mechanisms of the increase in PPARdelta expression induced by digoxin in the heart using the H9c2 cell line. Br J Pharmacol. 2011;163:390–398. doi: 10.1111/j.1476-5381.2011.01212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weber KT. Fibrosis and hypertensive heart disease. Curr Opin Cardiol. 2000;15:264–272. doi: 10.1097/00001573-200007000-00010. [DOI] [PubMed] [Google Scholar]
- 45.Grisanti LA, Talarico JA, Carter RL, Yu JE, Repas AA, Radcliffe SW, Tang HA, Makarewich CA, Houser SR, Tilley DG. Beta-Adrenergic receptor-mediated transactivation of epidermal growth factor receptor decreases cardiomyocyte apoptosis through differential subcellular activation of ERK1/2 and Akt. J Mol Cell Cardiol. doi: 10.1016/j.yjmcc.2014.02.009. Epub (2014) Feb 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, Ohmoto H, Node K, Yoshino K, Ishiguro H, Asanuma H, Sanada S, Matsumura Y, Takeda H, Beppu S, Tada M, Hori M, Higashiyama S. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat Med. 2002;8:35–40. doi: 10.1038/nm0102-35. [DOI] [PubMed] [Google Scholar]
- 47.Yamazaki S, Iwamoto R, Saeki K, Asakura M, Takashima S, Yamazaki A, Kimura R, Mizushima H, Moribe H, Higashiyama S, Endoh M, Kaneda Y, Takagi S, Itami S, Takeda N, Yamada G, Mekada E. Mice with defects in HB-EGF ectodomain shedding show severe developmental abnormalities. J Cell Biol. 2003;163:469–475. doi: 10.1083/jcb.200307035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong TT, Shaw RM, Srivastava D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell. 2009;16:233–244. doi: 10.1016/j.devcel.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, Chow FL, Oka T, Hao L, Lopez-Campistrous A, Kelly S, Cooper S, Odenbach J, Finegan BA, Schulz R, Kassiri Z, Lopaschuk GD, Fernandez-Patron C. Matrix metalloproteinase-7 and ADAM-12 (a disintegrin and metalloproteinase-12) define a signaling axis in agonist-induced hypertension and cardiac hypertrophy. Circulation. 2009;119:2480–2489. doi: 10.1161/CIRCULATIONAHA.108.835488. [DOI] [PubMed] [Google Scholar]
- 50.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, Rockman HA. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest. 2007;117:2445–2458. doi: 10.1172/JCI31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma YC, Huang XY. Novel regulation and function of Src tyrosine kinase. Cell Mol Life Sci. 2002;59:456–462. doi: 10.1007/s00018-002-8438-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1999;96:1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gutkind JS, Offermanns S. A new G(q)-initiated MAPK signaling pathway in the heart. Dev Cell. 2009;16:163–164. doi: 10.1016/j.devcel.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 55.Jensen BC, O'Connell TD, Simpson PC. Alpha-1-Adrenergic Receptors in Heart Failure: The Adaptive Arm of the Cardiac Response to Chronic Catecholamine Stimulation. J Cardiovasc Pharmacol. doi: 10.1097/FJC.0000000000000032. Epub (2013) October. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Y, Zhang H, Liao W, Song Y, Ma X, Chen C, Lu Z, Li Z, Zhang Y. Transactivated EGFR mediates alpha(1)-AR-induced STAT3 activation and cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2011;301:H1941–1951. doi: 10.1152/ajpheart.00338.2011. [DOI] [PubMed] [Google Scholar]
- 57.Zhang W, Anger T, Su J, Hao J, Xu X, Zhu M, Gach A, Cui L, Liao R, Mende U. Selective loss of fine tuning of Gq/11 signaling by RGS2 protein exacerbates cardiomyocyte hypertrophy. J Biol Chem. 2006;281:5811–5820. doi: 10.1074/jbc.M507871200. [DOI] [PubMed] [Google Scholar]
- 58.Sun Y, McGarrigle D, Huang XY. When a G protein-coupled receptor does not couple to a G protein. Mol Biosyst. 2007;3:849–854. doi: 10.1039/b706343a. [DOI] [PubMed] [Google Scholar]
- 59.Williams-Pritchard G, Knight M, Hoe LS, Headrick JP, Peart JN. Essential role of EGFR in cardioprotection and signaling responses to A1 adenosine receptors and ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2011;300:H2161–2168. doi: 10.1152/ajpheart.00639.2010. [DOI] [PubMed] [Google Scholar]
- 60.Guo J, Gertsberg Z, Ozgen N, Steinberg SF. p66Shc links alpha1-adrenergic receptors to a reactive oxygen species-dependent AKT-FOXO3A phosphorylation pathway in cardiomyocytes. Circ Res. 2009;104:660–669. doi: 10.1161/CIRCRESAHA.108.186288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhai P, Galeotti J, Liu J, Holle E, Yu X, Wagner T, Sadoshima J. An angiotensin II type 1 receptor mutant lacking epidermal growth factor receptor transactivation does not induce angiotensin II-mediated cardiac hypertrophy. Circ Res. 2006;99:528–536. doi: 10.1161/01.RES.0000240147.49390.61. [DOI] [PubMed] [Google Scholar]
- 62.Hilfiker-Kleiner D, Hilfiker A, Drexler H. Many good reasons to have STAT3 in the heart. Pharmacol Ther. 2005;107:131–137. doi: 10.1016/j.pharmthera.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 63.Willey CD, Palanisamy AP, Johnston RK, Mani SK, Shiraishi H, Tuxworth WJ, Zile MR, Balasubramanian S, Kuppuswamy D. STAT3 activation in pressure-overloaded feline myocardium: role for integrins and the tyrosine kinase BMX. Int J Biol Sci. 2008;4:184–199. doi: 10.7150/ijbs.4.184. [DOI] [PMC free article] [PubMed] [Google Scholar]