

Abstract
Compstatin peptides are complement inhibitors that bind and inhibit cleavage of complement C3. Peptide binding is enhanced by hydrophobic interactions; however, poor solubility promotes aggregation in aqueous environments. We have designed new compstatin peptides derived from the W4A9 sequence (Ac-ICVWQDWGAHRCT-NH2, cyclized between C2 and C12), based on structural, computational, and experimental studies. Furthermore, we developed and utilized a computational framework for the design of peptides containing non-natural amino acids. These new compstatin peptides contain polar N-terminal extensions and non-natural amino acid substitutions at positions 4 and 9. Peptides with α-modified non-natural alanine analogs at position 9, as well as peptides containing only N-terminal polar extensions, exhibited similar activity compared to W4A9, as quantified via ELISA, hemolytic, and cell-based assays, and showed improved solubility, as measured by UV absorbance and reverse-phase HPLC experiments. Because of their potency and solubility, these peptides are promising candidates for therapeutic development in numerous complement-mediated diseases.
Introduction
The complement system is implicated in the onset and progression of a number of autoinflammatory diseases.1 Despite growing efforts to identify new complement-targeted therapeutics, only one (eculizumab, Alexion) is currently in the clinic.2,3 There is a growing need for new therapeutics to treat chronic inflammatory diseases, which include age-related macular degeneration (AMD), systemic lupus erythematosus, and rheumatoid arthritis, among many others. Most complement therapeutics currently in clinical development are biopharmaceuticals, which are prone to challenges in production, delivery, and bioavailability. Few attempts at developing low-molecular mass complement inhibitors have been successful, largely because of the fact that complement activation cascades are comprised of large protein–protein interfaces and multimolecular complexes.3,4
Compstatin (Table 1, Parent) is a cyclic peptide that inhibits complement activation (reviewed in refs (2, 4−14)). It is one of a small number of low molecular mass complement therapeutics in development. The peptide binds to complement component C3 (as well as its derivatives C3(H2O), C3b, and C3c), the central protein of all complement activation cascades, and prevents its cleavage to C3a and C3b, thus blocking generation of complement effector proteins and complexes. Since its discovery,5 the sequence of compstatin has been optimized to improve its affinity and complement inhibitory activity.8,9,15−30 Numerous sequence modifications led to the development of W4A9 (Table 1), the most active compstatin peptide with only natural amino acids.20 Subsequently, many studies explored incorporation of non-natural amino acids and modifications to the compstatin sequence.20,22,23,26,29,30 Early studies of this type led to development of meW4A9 (Table 1), which is currently being pursued for treatment of AMD (clinicaltrials.gov, identifier numbers NCT00473928 and NCT01157065).22
Table 1. List of Compstatin Peptide Sequencesb.
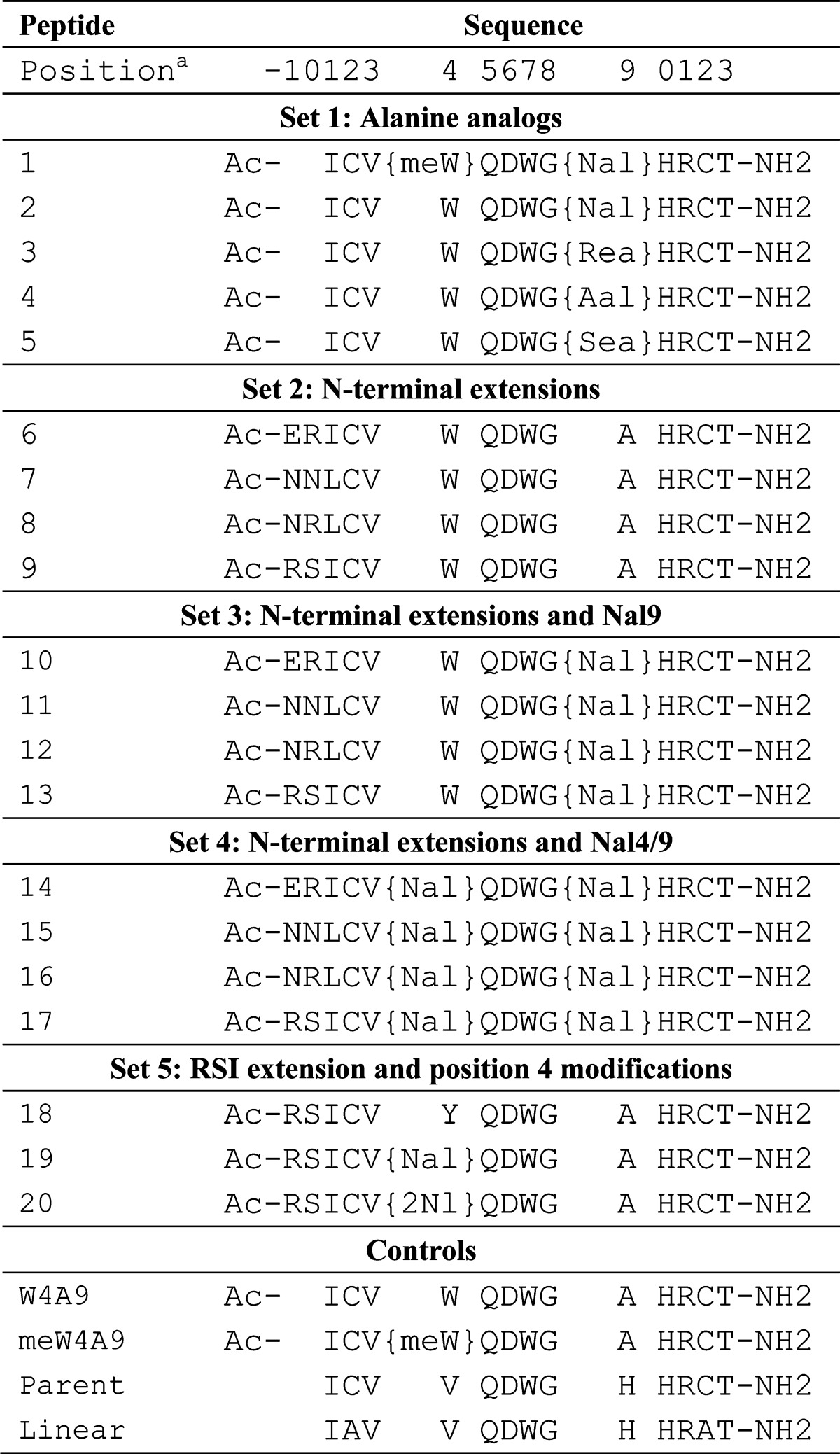
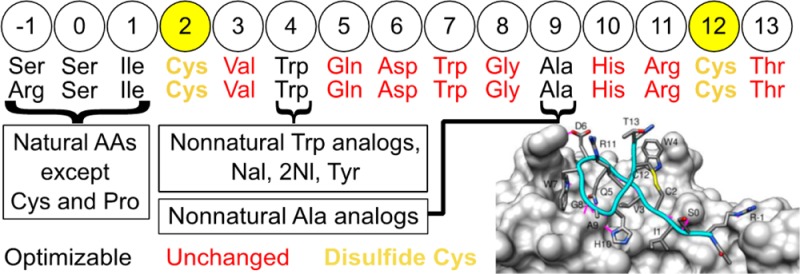
Position refers to residue number within each compstatin sequence. For reference, the Cys residues are always at positions 2 and 12.
Non-natural amino acid abbreviations: meW = l-1-methyltryptophan; Nal = l-1-naphthylalanine; Rea = R-α-ethylalanine; Aal = R-α-allylalanine; Sea = S-α-ethylalanine; 2Nl = l-2-naphthylalanine. All peptides (except linear) are cyclized by a disulfide bond between C2 and C12.
Determination of the structures of free (Parent)15 and C3c-bound (W4A9)31 compstatin peptides paved the way for further structure-based design and optimization of compstatin peptides. The solution NMR structure and subsequent structure–activity studies by NMR identified two opposite faces in compstatin, a hydrophobic face, proposed to be important for binding and inhibitory activity, and a polar face.8,9,15,17,19 The cocrystal structure revealed that compstatin binding is dominated by hydrophobic interactions, accompanied by several hydrogen bonds.31 Recent efforts23−29 have focused on modifying amino acids at positions previously shown to tolerate mutations,8,9,15,17,18,32 but most designed peptides exhibited no significant improvement in inhibitory activity, and some had poor solubility. Consequently, molecular dynamics (MD) simulations were used to explore the possibility of incorporating additional polar amino acids at the compstatin (W4A9) N-terminus and N-terminal extensions.27 Incorporation of hydrophilic amino acids has been recently reported to increase solubility of stapled peptides.33 Indeed, incorporation of Arg at position 1 and Ser-Ser and Arg-Ser extensions at positions −1 and 0 showed similar inhibitory activity and improved solubility compared to W4A9.28 This improvement is attributed to increased polarity at the N-terminus and the potential formation of additional polar contacts (hydrogen bonds and salt bridges) with C3c.27,28
In this study, we sought to identify new compstatin peptides with sequence modifications that promote both potent complement inhibition and improved aqueous solubility. Upon the basis of results from our previous study,28 we designed peptides with varied polar N-terminal extensions. In addition, we introduced a new computational approach for peptide design with non-natural amino acids. We incorporated non-natural amino acids (Figure 1) at positions 4 and 9, which were previously shown to be amenable for inhibitory activity optimization by the incorporation of non-natural amino acids.20−22 These peptides, and combinations thereof, were screened in functional in vitro and cell-based assays for complement inhibitory activity and using absorbance spectroscopy and HPLC for solubility and hydrophobicity. We identified several new compstatin peptides with improved pharmacological properties for potential therapeutic development.
Figure 1.

Chemical structures of non-natural amino acids in compstatin peptide sequences. The abbreviations used in sequences (Table 1) are shown in parentheses.
Results
Building upon our recent studies,24,25,27,28 we designed new compstatin peptides with the aim of simultaneously improving inhibitory activity and aqueous solubility. Here, we incorporated components of de novo design, including a new method for incorporation of non-natural amino acids, rational design based on molecular dynamics simulations and previous experimental data, and structure–activity relations. The previously known top compstatin peptides, W4A9 and meW4A9, include modifications at positions 4 and 9 (relative to Parent), which enhance inhibitory activity by at least 10-fold in various functional assays. While these modifications enhance peptide inhibitory activity, they contribute to increased peptide hydrophobicity, which has led to aggregation in aqueous solution. Here, we introduced a new computational peptide design method, which allows for the introduction of non-natural amino acids, targeting positions 4 and 9 in order to promote sequence diversity and explore new sets of possible potent and soluble compstatin peptides. Peptides were evaluated based on predicted physical interactions (contacts, clashes, hydrogen bonds) with C3c and screened according to computationally predicted binding affinity (K*), relative to control peptides W4A9 and meW4A9 (Table S1 in Supporting Information). We identified three peptides with higher predicted binding affinity than both W4A9 and meW4A9 (peptides 1, 3, and 4), which were selected for experimental evaluation. In addition, peptides with similar non-natural modifications (peptides 2 and 5) were included in experimental evaluation as well (Table 1, set 1).
Our most recent study showed that the N-terminal sequence Arg-Ser-Ile forms additional polar and nonpolar contacts with C3c, based on results from molecular dynamics simulations.27 Experimental data showed that peptide VI (from Gorham et al., 2013, sequence Ac-RSICV{meW}QDWGAHRCT-NH2, cyclized by disulfide bridge) exhibited potent complement inhibition (in in vitro and cell based assays) and improved solubility (in UV absorption and RP-HPLC experiments).28 Therefore, we screened combinations of polar amino acids in positions −1, 0, and 1 and evaluated predicted binding to C3c relative to control peptides (Table S2). Six peptides were identified with predicted C3c binding similar to or better than W4A9/meW4A9 (Table S3), and three of these were selected based on polarity (Table 1, set 2). In addition, the Arg-Ser-Ile extension was tested without meW at position 4 (Table 1, set 2).
Additionally, we selected components of both sets of peptides into two new sets, N-terminal extensions with Nal9 and N-terminal extensions with Nal4/9 (Table 1, sets 3 and 4). Set 3 peptides were selected in order to balance the potential interaction enhancement of Nal at position 9 with enhanced solubility from polar N-terminal extensions. Set 4 additionally included Nal at position 4, which was thought to improve hydrophobic contacts with C3. In light of our results from the first four sets of peptides and previous studies regarding compstatin modifications,20 we combined selected features to design peptides with optimal binding and solubility (Table 1, set 5), and included Tyr at position 4 to improve solubility (Tyr4 was previously shown to enhance inhibitory activity like other aromatic amino acids at position 4).18,20,32
Complement Inhibition in ELISA and Hemolytic Assays
We evaluated 20 newly designed compstatin peptides (plus four controls) for complement inhibition in vitro. For each peptide, we measured the ability to inhibit formation of both C3b and C5b-9 in ELISA, as well as the ability to inhibit complement-mediated lysis of rabbit erythrocytes in hemolytic assays (Figure 2 and Table S4). We observe that for each peptide, inhibition of the different complement effectors occurs at similar concentration. This is in line with the ability of compstatin peptides to block all complement pathways at the C3 level and subsequent downstream activation. The newly designed peptides exhibit similar or slightly reduced inhibition (IC50 values equal or higher) compared to meW4A9/W4A9 but were approximately an order of magnitude more potent than Parent. Notably, peptides 3–5 from set 1 (Figure 2A–C) and peptides 7–9 from set 2 (Figure 2D–F) show similar inhibitory activity compared to meW4A9/W4A9. This effect is also observed in the bar plots of Figure 3. α-Modified alanine analogs at position 9 (in peptides 3–5) are conservative substitutions, in terms of both C3 interaction and solubility, and thus have similar inhibitory activity to meW4A9/W4A9. Furthermore, polar N-terminal extensions seem to maintain inhibitory activity (as shown previously),28 with the exception of peptide 6, in which the negatively charged Glu residue may destabilize intermolecular salt bridging interactions. All peptides containing Nal at position 9 (peptides 1, 2, and 10–17) required slightly higher concentrations for complement inhibition, likely because of higher aggregation propensity. However, some of the N-terminal extensions (i.e., Asn-Arg-Leu, peptides 12 and 16) improved inhibition of the Nal9 peptides (Figure 2G–L). The final set of peptides (set 5) showed improved complement inhibition (Figure 2M–O) but remained less potent compared to the aforementioned peptides from sets 1 and 2. Indeed, previous work has shown that incorporation of Tyr, Nal, or 2Nl at position 4 increased inhibitory activity relative to Parent but had slightly less activity compared to W4A9.20 We hypothesized that addition of the polar RSI N-terminal extension may compensate for the aforementioned activity loss while simultaneously improving solubility. It seems that the extension neither improves nor diminishes inhibition, and therefore, the slightly reduced activity of set 5 peptides is most likely attributed to the substitutions at position 4. W4A9 has an IC50 value approximately 1 order of magnitude better than Parent in all three assays (Figure 2P–R), which is in agreement with our previous data.25,28 Interestingly, we observe that meW4A9 has only slightly better inhibitory activity than W4A9 (within a 2-fold difference), which differs from previous studies that report nearly an order of magnitude difference in activity.22,23 We expect this discrepancy is due to differences in biochemical and functional assays used, as well as the aggregation propensity of meW4A9.
Figure 2.

Concentration-dependent inhibition curves of compstatin peptides in C3b and C5b-9 ELISAs and hemolytic assays. Data show C3b, C5b-9, and hemolysis inhibition (from left to right) for set 1 (A–C), set 2 (D–F), set 3 (G–I), set 4 (J–L), set 5 (M–O), and control (P–R) peptides. Data points indicate mean percent inhibition ± SEM (standard error of the mean). The intersection of dashed lines shows the IC50 value for meW4A9 in all plots. Curves to the right and left of the intersection point represent peptides with higher IC50 values and lower IC50 values compared to meW4A9, respectively.
Figure 3.

IC50 values for compstatin peptides. Bar plots show IC50 values for compstatin peptides 1–20 and positive control peptides W4A9, meW4A9, and Parent in C3b ELISA (A), C5b-9 ELISA (B), and hemolytic assay (C). Bars show mean IC50 from three independent runs of each experiment (±95% confidence interval). The dashed horizontal line shows the IC50 value for the meW4A9 control peptide, for ease of comparison.
Complement Inhibition in Retinal Pigmented Epithelial Cell Model
A subset of peptides (peptides 5, 8, 12, 18, and 19) was selected for evaluation of complement inhibition in a retinal pigmented epithelial cell model, as described previously.28 Selected peptides represent top-performing peptides from each set (1–5), as determined through ELISAs and hemolytic assays. All tested peptides significantly inhibited complement activation (C5b-9/ApoE fluorescence) at 1 μM concentration, compared to Linear and the positive control (serum without inhibitor), and displayed similar complement inhibition compared to W4A9 (Figure 4, Tables S5 and S6). Notably, there was no significant difference in the levels of complement activation between Parent and the positive control or the linear peptide at 1 μM. This is likely due to the fact that typical IC50 values for Parent range between 4 and 13 μM in biochemical and functional assays (Figures 2 and 3, Table S4), and 1 μM concentration is not sufficient to reduce complement activation in RPE cells. To address this point, we tested higher concentrations of Parent in the in vitro RPE cell assay. While no complement inhibitory effects were noted for Parent at 1 or 10 μM, a significant (p-value of <0.001) inhibitory effect, similar to that of the W4A9 peptide at 1 μM, was observed for Parent at 50 μM (Figure 5). These results are consistent with the results of C3b and C5b-9 ELISA analyses of complement inhibition (Figure 2).
Figure 4.

Effects of compstatin peptides on complement activation in the RPE cell in vitro assay. The ratio of C5b-9/ApoE fluorescence (±SEM, n = 10) is plotted as a percentage of the positive control (POS) for two hfRPE cell lines, 072810 (gray) and 081309 (black). Untreated cells that were not incubated with complement-competent human serum served as negative control (NEG). At 1 μM, the parent compound is not significantly different from the positive or linear peptide controls. All test peptides (W4A9, PEP 5, PEP 8, PEP 12, PEP 18, and PEP 19) displayed significant complement inhibition relative to their corresponding positive control (see Tables S5 and S6).
Figure 5.

Effects of varying concentrations of Parent on complement activation in the RPE cell in vitro assay. The ratio of C5b-9/ApoE fluorescence (±SEM, n = 10) is plotted as a percentage of the positive control. Parent was tested at concentrations of 1, 10, and 50 μM (PAR1, PAR10, and PAR50). The concentration of W4A9 was 1 μM. All values are expressed relative to the positive control. Parent shows no significant difference from the positive control at 1 μM or 10 μM concentrations. At 50 μM the effect of Parent is equivalent to that of 1 μM W4A9. Both Parent at 50 μM and W4A9 at 1 μM are significantly different than the positive control (p-value of <0.001, two-tailed Mann–Whitney U test).
Solubility of Compstatin Peptides
Newly designed compstatin peptides were tested for solubility via absorbance measurements at 280 nm. The peptides showed a wide range of solubility, ranging from 0.1 to >5 mg/mL (Table S7). Control peptide meW4A9 showed moderate solubility in this assay (1.9 mg/mL), significantly lower than W4A9 and Parent, which exhibited apparent solubilities of 3.2 and 4.5 mg/mL, respectively. This result is consistent with the propensity of meW4A9 to aggregate in aqueous environments.29,34,35 Peptides 1 and 2, which contain Nal at position 9, exhibited the poorest solubility (∼0.1 mg/mL), much lower than all control peptides. Addition of polar N-terminal extensions (peptides 10–17) improved solubility only slightly (<0.4 mg/mL). Peptides with α-modified alanine analogs at position 9 (peptides 3–5) showed much improved solubility, with values near the detection limit in this assay (and similar to W4A9 and Parent). These results show the importance of position 9 to compstatin solubility. Indeed, solubility ranking follows the trend Parent > W4A9 ∼ peptides 3–5 > peptides 1–2 ∼ peptides 10–17 and, in turn, His > Ala ∼ Rea ∼ Aal ∼ Sea > Nal at position 9. Thus, increased hydrophobicity of residues at position 9 strongly influences the solubility of compstatin peptides. As in the case of complement inhibition, set 5 peptides showed intermediate solubility. There is likely a balancing effect between the polar Arg-Ser-Ile N-terminal extension and the hydrophobic residue (Tyr, Nal, or 2Nl) at position 4. The importance of position 4 is also evidenced by decreased solubility of peptides containing meW at position 4 (peptide 2 and meW4A9). Interestingly, while peptide 18 was initially highly soluble (∼5 mg/mL), we observed a time-dependent decrease in concentration when stored in a polypropylene tube (Figure 6, Table S7). Thus, it is unclear whether the change in solubility of peptide 18 is due to time-dependent aggregation or simply interaction with the container. It should be noted that while complement inhibitory activity of compstatin peptides has been associated with increased hydrophobicity in past studies,25,28,29 we observed that solubility is in fact associated with inhibitory activity (Figure 6). This is a direct result of our peptide design, as we engineered polar residues in regions of compstatin that should not affect binding. A clear exception to the aforementioned trend is Parent, in which solubility-enhancing substitutions are at positions that strongly influence binding.
Figure 6.

Relation between activity and solubility of compstatin peptides. IC50 values for compstatin peptides in C3b ELISA (A), C5b-9 ELISA (B), and hemolytic assay (C) are plotted against solubility. Horizontal and vertical error bars represent the standard deviation and 95% confidence intervals of solubility and IC50, respectively. Points inside the ellipse (lower right corner) have a favorable balance between activity and solubility. Note the peptide 8 solubility was measured using less starting material compared to the rest, and thus, its solubility is not directly comparable to that of other peptides.
Relative Lipophilicity of Compstatin Peptides
Modifications of peptide amino acid content can have a significant effect on peptide inhibitory activity, solubility, and lipophilicity.36 The inhibitory activity of the compstatin peptides depends in part on hydrophobic interactions with C3, which can be mimicked by the interactions of hydrophobic amino acid side chains of the peptides with the C18 HPLC stationary phase. The retention time of each peptide on the HPLC column can be used to calculate logarithm of the retention factor (log(k)), which is related to its lipophilicity (and hydrophobicity). Figure 7 shows log(k) values for compstatin peptides. A full summary of RP-HPLC retention times, retention factors (k), and log(k) values are summarized in Table S8, with the peptides organized according to substitution patterns. The peptides are listed in order of increasing relative lipophilicity in Table S9. As expected, peptide lipophilicity is inversely related to solubility with an R2 = 0.72 (Figure S1).
Figure 7.

RP-HPLC retention factors for compstatin peptides. Bar plots show log(k) values for compstatin peptides 1–20 and positive control peptides W4A9, meW4A9, and Parent in C3b ELISA. The dashed horizontal line shows the log(k) value for the meW4A9 control peptide, for ease of comparison.
Modifications of peptide sequence can have a large effect on their RP-HPLC retention behavior. Comparison of the HPLC results for peptides 2 and 10–13 demonstrates that addition of the charged and polar amino acids at positions −1, 0, and 1 reduces the retention time while substitution of the hydrophobic l-1-naphthylalanine (Nal) for Trp increases the interaction with the stationary phase. Peptides 14–17 are more retained than peptides 10–13, which differ in structure only by a Nal substitution at position 4. Peptides 1 and 2 also contain Nal at position 9 in place of Ala in structurally similar peptides meW4A9 and W4A9, respectively, and are more highly retained than these analogs. A similar observation can be made when comparing peptides 9 and 19, which differ at position 4 by the substitution of Nal. Substitution of Tyr for Trp in peptide 18 reduces its interaction with the stationary phase as reflected by its smaller value of log(k).
Structural modifications of amino acid residues can also affect the interactions of peptides with the hydrophobic stationary phase. For example, the addition of a methyl group to a Trp residue at position 4 increases the retention compared with the Trp analogs. Peptides 1 and meW4A9 are more highly retained than peptides 2 and W4A9, respectively. The position of the naphthyl addition on alanine also affects the interaction of the peptide with the stationary phase. Peptide 19, which contains l-1-naphthylalanine, is less retained than peptide 20, which contains l-2-naphthylalanine at the same position. Similarly, peptides 3 and 5, which differ only by substitution of R- and S-α-ethylalanine, respectively, can be resolved by HPLC with peptide 3 being more highly retained than peptide 5.
Discussion
Although continual efforts to improve compstatin have yielded peptides with potent complement inhibition, many are characterized by poor solubility in aqueous environments containing physiological ionic strength.22,25,28,29 The inhibitor meW4A9 (AL-78898A, Alcon)29 has recently completed phase II clinical trials (clinicaltrials.gov, identifier numbers NCT00473928 and NCT01157065) for AMD; however, the results showed that this peptide did not yield reduced retinal thickness in AMD patients in comparison to ranibizumab (Genentech/Novartis),14 a current AMD therapeutic. It is likely that the low aqueous solubility of meW4A9 contributes to its poor performance.29,34,35 Thus, recent studies have aimed at improving solubility of compstatin peptides while simultaneously maintaining or improving complement inhibitory activity.
In this study, we designed and tested new compstatin peptides with diverse polar N-terminal substitutions and extensions. We also employed a new computational design framework for non-natural amino acids to identify non-natural amino acids at positions 4 and 9 predicted to improve C3 binding. Furthermore, we tested peptides containing combinations of these features, selected using rational design arguments. We note that all of our designed peptides have inhibitory activities similar to controls meW4A9 and W4A9 and significantly better than Parent (Figures 2–5). Most importantly, we found that peptides containing either α-modified non-natural alanine analogs at position 9 (peptides 3, 4, and 5) or N-terminal natural amino acid extensions (peptides 7, 8, and 9) have inhibitory activity similar to meW4A9 while exhibiting greatly improved aqueous solubility. Given the clinical interest in meW4A9 for treatment of a variety of complement-mediated disorders, in conjunction with problems recently encountered in clinical trials, we believe that our newly designed compstatin peptides (i.e., peptides 3, 4, 5, 7, 8, and 9) represent more promising therapeutic alternatives.
Several recent studies have focused on improving affinity of compstatin peptides.23,25,26,28,29 Indeed, some compstatin peptides bind to C3 with affinities of <1 nM, with correspondingly low IC50 values in complement assays.29 While improved affinities are attractive for therapeutic design when taken at face value, we must remain aware that human plasma C3 concentrations are at least 5 μM, and thus high compstatin concentrations are required for therapeutic efficacy despite high binding affinity. Consequently, we conclude that (a) it is unnecessary to further improve compstatin binding affinity and inhibitory activity and (b) compstatin must be highly soluble to inhibit C3 cleavage in vivo. It is now crucial to focus on improving pharmacological properties of compstatin peptides. Recent studies have addressed several of these properties, including solubility, half-life, and degradation.28−30,37 We have identified several highly soluble compstatin peptides with potent inhibitory activity. In the past, it was thought that a high degree of hydrophobicity was required for potent compstatin activity. We illustrate here that the incorporation of N-terminal extensions can help to circumvent this activity–hydrophobicity/solubility paradigm. Our inhibitory activity versus solubility data (Figure 6) denote peptides with similar inhibitory activity compared to highly active controls (i.e., meW4A9) but with improved solubility characteristics. Furthermore, several peptides (peptides 6–9 and 18) are notably less lipophilic than both meW4A9 and W4A9 (Figure 7 and Table S9). These peptides, as well as additional peptides containing alternative combinations of top-performing modifications, represent promising candidates for therapeutic development, for treatment of a wide variety of complement-mediated autoinflammatory diseases.
Experimental Section
De Novo Peptide Design with Natural Amino Acids
Sequence selection, fold specificity, and approximate binding affinity calculations for natural amino acid design were performed following the procedure described previously in Protein WISDOM and are described below.38 The purpose of the design was to produce novel natural amino acid sequence extensions using the previously designed sequences Ac-RSICVWQDWGAHRCT-NH2 (sequence of peptide 9 in Table 1, called RSI hereafter) and Ac-SSICVWQDWGAHRCT-NH227,28 (called SSI hereafter) as templates. Both sequences are identical to the W4A9 variant of compstatin with two positions added to the N-terminus, termed positions −1 and 0. These sequences were used as input to further design these positions for C3c binding. RSI was chosen as a design template, as it previously yielded improved IC50 values relative to W4A9 in C3b ELISA and hemolytic assays, as well as significant complement inhibition according to a human retinal pigmented epithelial cell-based model mimicking drusen biogenesis.28 SSI had near W4A9 calculated affinity for human C3 and simultaneously high calculated affinity for non-primate C3.27 Because of the previous designs of both of these sequences, MD simulations were previously performed and therefore were utilized as design templates in this next generation of design.
Sequence Selection
The peptide structure template for sequence selection was constructed based on the structure of the W4A9 analog (PDB code 2QKI).31 Molecular dynamics simulations of the template compstatin extensions were performed previously and were used as a flexible template for this design.28 In all cases, both extended positions, −1 and 0, as well as the adjacent position 1, were allowed to mutate. Because of the relatively small sequence space in this study (three mutatable positions only), the three positions were allowed to mutate to all possible amino acids except for proline and cysteine, which were excluded because of their unique chemical and geometric properties. The centroid–centroid potential energy function was used as the input force field,39 which serves as a look-up table of energies of all combinations of amino acid pairs at a fixed set of distance bins.
The flexible distance–bin sequence selection method38 was used to solve for a rank-ordered list of low energy sequences according to the following model.
| 1a |
subject to
| 1b |
| 1c |
| 1d |
| 1e |
| 1f |
The set i = 1, ..., n defines each residue position in the design template. At each position i, mutations are represented by j{i} = 1, ..., mi, where mi = 20 if the sequence position is allowed to mutate to any of the 20 natural amino acids. Alias sets k ≡ i and l ≡ j, with k >i, are used to represent the pairwise interactions between all residue positions and amino acid types. The decision variables yij and yk are solved for in order to determine the amino acid types in each position. The yij variable will assume the value of 1 if the model assigns amino acid j to position i and assume the value of zero otherwise (similarly for yk). The objective function represents the sum of all pairwise energy interactions in the design template.38 The model incorporates the distance information from the template structures by introducing a binary variable bikd, which equals 1 if the distance between residues i and k falls within distance bin d and equals zero otherwise. The parameter disbin(xi, xk, d) equals 1 if the distance between residue positions i and k in any of the input template structures falls into distance bin d and equals zero otherwise, which forces only one distance bin per amino acid pair to contribute to the total energy.38
Since we started with template structures with sequences of both RSI and SSI, three separate runs were performed for the design. Run 1 used only the RSI structures as the input template. Run 2 used only SSI structures as the input template, and run 3 used a combined RSI and SSI structure set as the input template. For each run, 500 low-energy sequences were generated using the flexible distance–bin sequence selection method. From runs 1–3, a consensus set of sequences was constructed from the 1500 total sequences generated. This resulted in a total of 1019 sequences, which were then validated using the fold specificity metric.38
Fold Specificity
The aim of the fold specificity calculation is to determine the stability of a designed sequence in the target template structure as compared to the stability of the native sequence. The flexible template structure defined bounds on the minimum and maximum Cα–Cα distances and φ and ψ angles experienced. Ensembles are next generated using a torsion angle dynamics simulated annealing containing 500 conformers using CYANA 2.1.40,41 A local energy minimization is performed on every conformer in TINKER 3.642 using the AMBER force field,43 and the final potential energy of each conformer is tabulated. The energetics of the native sequence is similarly tabulated. The fold specificity calculation is next performed as
| 2 |
with more positive values being favorable.38
Three separate runs of fold specificity were performed, based on which template structures were used in the calculation: run 1 using RSI structures only, run 2 using the SSI structures only, and run 3 using the combined RSI and SSI structure set. The sequences were then rank-ordered by fold specificity for each set, and a consensus set of sequences was determined for validation using approximate binding affinity (Table S2). Sequences were chosen for validation to maximize fold specificity across runs, as well as to cover a large chemical space. This resulted in 15 sequences with mutations in positions −1, 0, and 1 and in 7 sequences with mutations in positions −1 and 0 only.
Approximate Binding Affinity
The approximate binding affinity calculation in Protein WISDOM consists of several steps, which include diverse ligand ensemble generation, clustering, docking, and final ensemble generation. The goal is to generate a diverse ensemble of docked ligand poses to the receptor to assess the relative energetics of the complex, receptor, and ligand through calculation of approximations of their partition functions. The approximate binding affinity44 is defined as K* = qPL/(qPqL), where qPL = ∑b∈B e–[Eb/(RT)], qP = ∑f∈F e–[Ef/(RT)], and qL = ∑l∈Le–[Ei/(RT)] are the partition functions of the protein–ligand complex, the free protein, and ligand, respectively. The sets B, F, and L, contain rotamerically based conformations of the bound complex, free protein, and free ligand. En is the energy of each conformation. R is the gas constant, and T is the temperature.
Two-thousand structures for each candidate compstatin extension sequence are generated using the Rosetta AbRelax function as part of the Rosetta 3.4 package.45−47 Monte Carlo sampling is used to replace local protein structures with structural fragments derived from homologous sequences. The structures are clustered based on their φ and ψ angles using OREO.48,49 The average structure from the 10 largest clusters and the lowest energy structure are chosen for docking to the target protein. This yields 11 structures with unique backbones which are next docked using RosettaDock.50 The 10 lowest energy docked poses for each of the 11 ligands are passed to RosettaDesign51 to generate 200 rotamer conformations per starting complex structure (22 000 total states representing the bound set). The ligand ensemble is constructed by taking the 10 lowest energy structures from each of the 10 largest clusters plus the 10 lowest overall energy peptide conformations and generating 200 rotamer conformations (22 000 free ligand structures) for each. The protein ensemble is generated directly by assembling 2000 rotamer structures from the single starting C3c structure. With these ensembles and their corresponding Rosetta energies, the K* is calculated.
The 22 sequences from fold specificity were run through the approximate binding affinity protocol38 in order to determine which designed peptides were predicted to bind more strongly than the W4A9 peptide. Of the 22 tested peptides, six peptides were predicted to have higher binding affinity than W4A9 and peptide 9. The sequences of these peptides are provided in Table S3. Of these six peptides, three were selected for further experimental testing.
De Novo Peptide Design with Non-Natural Amino Acids
Recently, efforts have expanded to create methods for designing non-natural amino acids into proteins.52−60 We have created new force fields based on quantum chemical calculations for post-translational modifications and unnatural amino acids for AMBER.61,62 Thus, we developed a novel framework combining our force fields, integer linear optimization, and molecular dynamics simulations to aid us in generating lead compounds for experimental exploration. The first step in the procedure is to derive candidate sequences containing post-translational modifications and non-natural amino acids. The overall idea is that we want to introduce modified amino acids to enhance the number of contacts and hydrogen bonds relative to the starting peptide while minimizing the clashes introduced by the modified amino acid. In addition, we want to preserve the local electrostatic landscape and any salt-bridges that are formed between the original or designed sequence scaffold and the target receptor. This is modeled as the following integer linear optimization problem:
| 3a |
subject to
| 3b |
| 3c |
| 3d |
| 3e |
| 3f |
| 3g |
| 3h |
with the following sets, parameters, and binary decision variables.
Sets:
p ∈ (1, 2, ..., Npositions); this is the set of positions in the amino acid sequence.
Np ⊆ (Ala, Arg, Asn, ..., Tyr); this is the native amino acid in each position p.
M ∈ (Np, ..., PTMs, ..., Non-naturalAminoAcids); this is the universe of modification types under consideration for which we have derived parameters for. This universe comes from a limited set of modifications parametrized early on for Forcefield_PTM61 and Forcefield_NCAA.62
C ∈ (1, 2, 3, ..., Ncomplexes); this is the set of complex configurations.
Ω is the set of complex positions that correspond to the lowest energy configurations from a molecular dynamics simulation, an NMR structural ensemble, or a single crystal structure. Note that the set M contains all the post-translational modifications and unnatural amino acids for which we have parameters for, as well as the native amino acid in position p being designed. Modified amino acid parameters were taken from Forcefield_PTM61 and Forcefield_NCAA.62
Parameters:
Cmp, charge of modification m at position p.
Dp, charge of original amino acid on template sequence at position p.
Hmp, hydrophobicity of modification m at position p.
Hp, hydrophobicity of original amino acid on template sequence at position p.
goodcontacts(C,m,p), number of contacts between modification m at position p and the receptor with van der Waals radii overlap of atoms (i,j) ≥ −2 in configuration C.
clashes(C,m,p), number of contacts between modification m at position p and adjacent residues in the peptide or the receptor with a van der Waals radii overlap of atoms (i,j) ≥ 0 or ≥ 0.4 if atoms (i,j) can potentially hydrogen bond in configuration C.63
hbonds(C,m,p), number of valid hydrogen bonds (distance and angle orientation) between modification m at position p and the receptor in configuration C.63,64
IE(C,m,p), energy of binding between protein and peptide in configuration C as a result of modifying position p to modification type m, where this energy is defined as
sp(p) = 1 if position p is allowed to be modified, 0 otherwise.
S(C,m,p) = 0.1 goodcontacts(C,m,p) – 5 clashes(C,m,p) + 1 hbonds(C,m,p), weighted contact score of modification m at position p in configuration C. This score is not an energy that is physically derived but a tabulation of quantities of interactions.
Natural amino acids are classified as hydrophobic or hydrophilic if their relative side chain solvent-accessible surface areas (SASAs) calculated with the program NACCESS65 in their position in three-dimensional space within the context of the complex are ≤20 or ≥50, in accordance with work by Bellows et al.24 Introducing hydrophobicity constraints in previous designs using only natural amino acids was observed to be a critical component to successful design.24,66−69 Next, we define Mp ⊆ M, which is the subset of modifications meeting the charge and hydrophobicity constraints below at position p, ∀ positions p. This set, derived from the following constraints, reduces the search space.
Constraints That Must be Met for Inclusion in Set Mp
Local charge/salt-bridges must be conserved in each position p
| 4 |
Local hydrophobicity must be conserved at each p
| 5 |
We further want to limit the modifications to those that are of the same type as the native amino acid in position p. For example, if the native amino acid in position 5 were a tyrosine, we would allow only for modifications of tyrosine as design choices and not modifications of alanine. Therefore, we define MpNp ⊆ Mp as the subset of modifications in position p that are allowed given the native amino acid in position p, Np. Utilizing this subset dramatically reduces the search space.
There are geometric conditions required for a contact, clash, and hydrogen bond to occur. These conditions reflect the fact that two atoms that hydrogen bond may come closer to each other than their van der Waals radii would normally allow. Contacts, clashes, and hydrogen bonds were evaluated between the modified position of the compstatin analog and C3c using custom scripts interfacing with UCSF Chimera.64 Intermolecular contacts were defined with an overlap cutoff of −2 and a hydrogen bond allowance of 0. Both inter- and intramolecular clashes were calculated with an overlap cutoff of 0.4 and a hydrogen bond allowance of 0.4. Intermolecular hydrogen bonds were calculated between compstatin analogs and C3c with a 0.4 Å distance relaxation and a 20° angle relaxation from the default hydrogen bond criteria.
Binary Decision Variables
ymp = 1 if modification m is assigned to position p, 0 otherwise.
The objective function, eq 3a, aims to maximize a Boltzmann-weighted contact score. Equation 3b defines the weights of the contact score as a weighted sum of goodcontacts, clashes, and hbonds. The goal of the contact score is to improve affinity/specificity through the optimization of shape-complementarity at the individual amino acid level. It rewards good contacts and hydrogen bonds while strongly penalizing unfavorable clashes. Equation 3c defines a position-specific partition function. This quantity becomes the denominator for the Boltzmann-weighted contact score so that for each position, a probability can be derived based on the sum over the allowed modification types m ∈ MpNp and complex configurations C. Equation 3d defines the Boltzmann factor that is the ratio of the exponent of the weighted contact score, eq 3b divided by the position-specific partition function, eq 3c. The constraint 3e states that the average energy of binding across all complex configurations must remain favorable for the modification m to be chosen in position p. This is important to preserve the local electrostatic interactions of the template sequence if any are present. Equation 3f states that only one amino acid is allowed to be selected in each position. Equation 3g is the integer cut constraints that generate a rank-ordered list of optimal solutions. Equation 3h indicates that the decision variables ym are binary.
A computational pipeline to design proteins and peptides by introducing non-natural amino acids was created. The pipeline takes as input design positions, as well as a set of complex configurations corresponding to a peptide with natural amino acids bound to a receptor protein. These configurations were the single state of the C3c:W4A9 crystal structure (PDB code 2QKI).31 Trp4 and Ala9 were selected as design positions. Next, the initial energy of binding, charge, and hydrophobicity constraints are populated using AMBER 1170 and Chimera.64 Then for each complex configuration and each allowed modification in each design position, the complex is modified at the specified design position, a local energy minimization is performed, and the contacts, clashes, hydrogen bonds, and binding energies are populated. The AMBER program tleap was used to construct the modified side chains according to their orientations defined in Forcefield_PTM61 and Forcefield_NCAA.62 In position 4, tryptophan, 5-hydroxytryptophan, 5-methyltryptophan, N-methyltryptophan, 1-methyltryptophan, and 7-hydroxytrptophan were evaluated. In position 9, alanine, 1-naphthylalanine, N-methylalanine, pyrenylalanine, (R)-α-ethylalanine, (S)-α-ethylalanine, and R(+)-α-allylalanine were evaluated. These were chosen as they met the criteria to be included in each of their respective sets MPNP, and their parameters were included in Forcefield_PTM or Forcefield_NCAA. For each modification upon construction, 1500 steps of steepest descent followed by 500 steps of conjugate gradient minimization were performed to relax the new non-natural side chain placement in the context of compstatin and the C3c receptor. The implicit solvent model by Onufriev71,72 (igb = 5) was utilized in all AMBER calculations. After population of this information for all complex configurations, the integer linear optimization model presented above is solved to global optimality, generating a rank-ordered list of modified amino acid substitutions.
The sequence solutions were next simulated in AMBER 11 to assess the approximate binding affinity (K*) with a procedure described previously.62 Specifically, three independent simulations are carried out of the complex, protein, and peptide. A single simulation of the protein was used to assess its energetic contributions. Structures were minimized with 600 steps of steepest descent with 400 steps of conjugate gradient minimization to relax the new side chains. The structures were heated in six stages over 30 ps from 0 to 300 K. Shake constraints were used for all simulation steps on the heavy-atom to hydrogen bonds, and a nonbonded interaction cutoff of 16 Å was used. Each C3c:compstatin peptide candidate sequence underwent short 0.5 ns production simulations using a 1 fs time step at 300 K. The theoretical foundation for this calculation was previously described in Khoury et al.62 and Lilien et al.44 Those sequences with the highest K* values relative to the control peptide W4A9 were proposed for experimental testing (Table S1). We note that the approximate binding affinity is used as a metric to select which peptides may be promising62 candidates for testing based on physics-based potentials, and we do not attempt to compare the calculated values to the exact experimental values. Instead, we aim to utilize metrics that increase our probability that top ranked peptides are indeed inhibitors of C3.
Peptide Synthesis
Compstatin peptides 1–9, 14–20, W4A9, meW4A9, and Parent were synthesized by WuXi AppTec (Shanghai, China). Peptides 10–13 were synthesized by Genscript, Inc. (Piscataway, NJ, USA). Linear (negative control) was obtained from either Tocris Bioscience (Bristol, U.K.) or WuXi AppTec. All peptides were of >95% purity, as determined by HPLC and MS. Non-natural amino acids used in this study included l-1-methyltryptophan, l-1-naphthylalanine, l-2-naphthylalanine, R-α-ethylalanine, R-α-allylalanine, and S-α-ethylalanine (Figure 1). Peptides containing l-1-methyltryptophan were synthesized using Fmoc-1-methyl-dl-tryptophan (commercially available preparation) and subsequently purified by HPLC.
Solubility Measurements
Approximately 1 mg of each tested peptide was dissolved in 200 μL of PBS, pH 7.4. It should be noted that the resulting concentration yields saturated solutions for only some peptides, and the maximum solubility reported here is thus 5 mg/mL. Samples were vortexed for 30 s each, then centrifuged at 13000g for 5 min. Supernatant was collected and measured spectrophotometrically at 280 nm, using a NanoDrop Lite spectrophotometer. Absorbance measurements were converted to concentration using the Beer–Lambert law. Molar extinction coefficients were calculated for each peptide based on the number of Trp and Tyr residues in their sequences. Because of their aromatic properties, l-1-methyltryptophan, l-1-naphthylalanine, and l-2-naphthylalanine also contribute to absorbance at 280 nm. Molar extinction coefficients used for these amino acids were 5470 M–1 cm–1,28 3936 M–1 cm–1,20 and 3936 M–1 cm–1, respectively (the coefficient for l-2-naphthylalanine was assumed to be equal to that of l-1-naphthylalanine). Dissolved samples were stored at 4 °C, and measurements of the same samples were repeated at 24 and 48 h time points. Measurements represent the mean and standard deviation of three measurements performed on the each sample.
C3b and C5b-9 ELISA
Nunc Maxisorp 96-well microtiter plates were coated with 20 μg/mL lipopolysaccharides (LPS) from Salmonella enterica serotype enteritidis (Sigma-Aldrich, St. Louis, MO, USA) for 16 h at ambient temperature. Plates were washed three times with PBS (containing 0.05% Tween-20; PBS-T) between each incubation step. Plates were blocked with 4% BSA in PBS-T for 1 h at 37 °C. Lyophilized compstatin peptides were initially dissolved in PBS, pH 7.4, and concentration was measured spectrophotometrically at 280 nm. Serial dilutions of each peptide were prepared in veronal-buffered saline containing 0.1% gelatin, 5 mM MgCl2, and 10 mM EGTA (GVBS-MgEGTA). Normal human serum (Complement Technology, Inc., Tyler, TX, USA) was dissolved in GVBS-MgEGTA and was preincubated with peptide dilutions (or buffer) for 15 min at ambient temperature. Samples were subsequently incubated in blocked plates for 1 h at 37 °C. After incubation, plates were incubated with either horseradish peroxidase (HRP) conjugated anti-C3 (MP Biomedicals, Solon, OH, USA) or anti-C5b-9 aE11 (Abcam, Cambridge, MA, USA) for 1 h at 37 °C. C5b-9 detection additionally involved incubation with an HRP-conjugated secondary antibody, also for 1 h at 37 °C. Levels of C3b and C5b-9 deposition were measured using a 3,3′,5,5′-tetramethylbenzidine substrate solution containing urea hydrogen peroxide in 0.11 M sodium acetate buffer, followed by addition of 1 N H2SO4. Plates were subsequently measured spectrophotometrically at 450 nm.
Hemolytic Assays
Rabbit erythrocytes (Complement Technology, Inc., Tyler, TX, USA) were washed and resuspended in veronal-buffered saline containing 5 mM MgCl2 and 10 mM EGTA (VBS-MgEGTA). Normal human serum and compstatin peptides (serial dilutions) were diluted in VBS-MgEGTA and preincubated together in round-bottom 96-well plates for 15 min at ambient temperature. 5 × 106 rabbit erythrocytes were added to each well, and plates were incubated for 20 min at 37 °C. Erythrocytes in either deionized water or normal human serum (dissolved in VBS-MgEGTA) were used as positive controls for lysis, and erythrocytes in either VBS-MgEGTA or normal human serum (dissolved in VBS-EDTA) were used as negative controls for lysis. Reactions were quenched by adding ice cold VBS (containing 50 mM EDTA) to each well. Plates were centrifuged at 1000g for 5 min, and supernatant was diluted 1:1 in deionized water in new plates. Lysis was quantified spectrophotometrically at 405 nm.
RPE Cell Culture
The RPE cell-based in vitro model of drusen formation73 was employed as previously described.28 Human fetal RPE cells (Advanced Bioscience Resources, Alameda, CA) were cultured on Millipore HA porous supports (Millipore, catalog no. PIHA 01250) in Miller medium supplemented with 5% fetal calf serum (FCS). RPE cell cultures derived from two different donor eyes (line no. 081309 and line no. 072810) were used. Samples were rinsed in PBS and then individually exposed to an experimental peptide in serum-free Miller medium containing 10% human complement serum (Innovative Research, catalog no. IPLA-CSER AB, lot no. L12402). The 1 μM peptide concentration employed was previously shown to be in the linear range of inhibitory concentrations during titrations.28 One experiment employed the parent compound at 1, 10, and 50 μM. Negative control cells were exposed to Miller medium + 5% FCS; positive control cells were exposed to Miller medium + 10% human complement serum. Experimental and control solutions (1 mL) were mixed on a rocker at room temperature for 30 min, then warmed to 37 °C, before sample exposure and overnight incubation at 37 °C in a 7.0% CO2 incubator. Following this period, the samples were rinsed with warm, sterile PBS, fixed in cold 4% paraformaldehyde (PFA) in PBS for 20 min, and stored in 0.4% PFA until use in immunohistochemical assays.
Immunohistochemistry
After rinsing the fixed samples in PBS, the inset membrane was excised with a scalpel and cut into ∼4 mm2 squares. Duplicate samples from each condition were embedded in 10% agarose (Type XI, Sigma-Aldrich, catalog no. A3038) and sectioned at 100 μm using a vibratome. Sections were blocked with normal donkey serum (1/20 in PBT/PBS containing 0.5% bovine serum albumin and 0.1% Triton X-100) overnight at 4 °C. The sections were co-incubated with two primary antibodies (polyclonal goat anti-ApoE, Millipore catalog no. AB947, 1/1000 in PBT, and mouse monoclonal anti-C5b-9, Dako catalog no. MO777, 1/200 in PBT) overnight at 4 °C, then rinsed in PBT, and then co-incubated in secondary antibodies (Alexa Fluor 546-conjugated donkey anti-goat IgG and Alexa Fluor 488-conjugated donkey anti-mouse IgG, both 1/200 in PBT, Life Technologies catalog no. A-11056 and no. A-21202) overnight at 4 °C. The immunolabeled sections were then rinsed in PBT, stained with Hoechst 33342 (Life Technologies catalog no. H3570), and mounted on slides with Prolong Gold (Life Technologies catalog no. P36930).
Confocal Imaging and Analysis
Samples were imaged with an Olympus FV1000 confocal laser scanning microscope. Ten single-plane images, captured at a resolution of 1024 × 768 pixels and saved as 24-bit tiff files, were acquired for each sample. Digital image files were analyzed using MetaMorph software (Molecular Devices, Sunnyvale, CA). For image quantification, the area of C5b-9 specific fluorescence was normalized to the area of ApoE specific fluorescence and expressed as the C5b-9/ApoE ratio. Statistical significance was determined using the Mann–Whitney U test with p-value of ≤0.05 considered significant.
RP-HPLC Study
The relative lipophilicity of compstatin peptides was determined using RP-HPLC.28,74 This method has been used to evaluate the retention factor, k, of peptides between the hydrophobic stationary phase and hydrophilic mobile phase. The logarithm of the retention factor was determined based on the peptide retention time, tR, and column dead time, t0.28 These peptides are structurally similar, some differing by only one site or position of substitution. The isocratic RP-HPLC method used in this work allows for comparison of the interaction of the structurally similar peptides with the hydrophobic stationary phase.
The separations were performed using an Agilent 1100 HPLC with UV detection at 280 nm and a Waters (Milford, MA, USA) 4.6 mm × 150 mm XTerra MS C18 column with 5 μm particles and a 125 Å pore size. The peptides were eluted using a mobile phase A, consisting of 10 mM phosphate buffer at pH of 7.4 with 1% TFA, and acetonitrile as mobile phase B. The peptides were first evaluated using method 1, running 32% B isocratically at a flow rate of 1.0 mL/min at 25 °C. The less hydrophobic peptides 3–9 and 18–20 were better resolved using isocratic method 2 which used 28% B. Triplicate 10 μL injections were performed for each peptide at concentrations ranging from 9 to 16 μM. The column dead time was determined to be 1.418 ± 0.004 (method 1) and 1.425 ± 0.006 (method 2) using a 5 mg/mL solution of aspartic acid as the unretained analyte.28
Dibasic sodium phosphate, aspartic acid, and acetonitrile were purchased from Fisher Scientific (Pittsburgh, PA, USA). Trifluoroacetic acid (TFA) was purchased from Acros (Geel, Belgium). HPLC-grade water was obtained from Burdick and Jackson (Muskegon, MI, USA).
Acknowledgments
D.M. and M.J.R. acknowledge donors to Macular Degeneration Research, a program of the BrightFocus Foundation, for support of this research (Grant M2013106). D.M. is the recipient of the Carolyn K. McGillvray Memorial Award for Macular Degeneration Research, administered by the BrightFocus Foundation. D.L.F. was supported by the Garland Initiative for Vision at University of California, Santa Barbara, and the BrightFocus Foundation (Grant M2013106). C.A.F. acknowledges support from the National Institutes of Health (Grant R01GM052032). G.A.K. is grateful for support by a National Science Foundation Graduate Research Fellowship under Grant DGE-1148900. The authors gratefully acknowledge that the design calculations were performed at the TIGRESS high performance computing center at Princeton University, NJ, which is supported by the Princeton Institute for Computational Science and Engineering (PICSciE) and the Princeton University Office of Information Technology.
Glossary
Abbreviations Used
- C3
complement component 3
- Ac
acetyl group
- C3(H2O)
hydrolyzed C3
- C3a
complement component 3a
- C3b
complement component 3b
- C3c
complement component 3c
- C5b-9
membrane attack complex comprising complement proteins C5b, C6, C7, C8, and polymeric C9
- meW
l-1-methyltryptophan
- Nal
l-1-naphthylalanine
- Rea
R-α-ethylalanine
- Aal
R-α-allylalanine
- Sea
S-α-ethylalanine
- 2Nl
l-2-naphthylalanine
- Nmw
N-methyltryptophan
- Nma
N-methylalanine
- Pal
pyrenylalanine
- ELISA
enzyme-linked immunosorbent assay
- IC50
half maximal inhibitory concentration
- UV
ultraviolet
- HPLC
high-performance liquid chromatography
- MD
molecular dynamics
- RP-HPLC
reverse phase high-performance liquid chromatography
- RPE
retinal pigmented epithelium
- AMD
age-related macular degeneration
Supporting Information Available
Tables and figures of peptide design rankings, compstatin peptide inhibitory activity (from ELISAs, hemolytic assays, and cell based RPE assays), and solubility/lipophilicity data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Dimitrios Morikis, Ronald D. Gorham, Jr., Christodoulos A. Floudas, George A. Khoury, Georgios Archontis, and Phanourios Tamamis have a patent application on the peptides reported in this paper.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Walport M. J. Complement. Second of two parts. N. Engl. J. Med. 2001, 344, 1140–1144. [DOI] [PubMed] [Google Scholar]
- Ricklin D.; Lambris J. D. Complement-targeted therapeutics. Nat. Biotechnol. 2007, 25, 1265–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D.; Lambris J. D. Complement in immune and inflammatory disorders: therapeutic interventions. J. Immunol. 2013, 190, 3839–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu H.; Ricklin D.; Lambris J. D. Recent developments in low molecular weight complement inhibitors. Mol. Immunol. 2009, 47, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu A.; Kay B. Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J. Immunol. 1996, 157, 884–891. [PubMed] [Google Scholar]
- Morikis D.; Sahu A.; Moore W. T.; Lambris J. D.. Design, structure, function and application of compstatin. In Bioactive Peptides in Drug Discovery and Design: Medical Aspects; Matsoukas J., Mavromoustakos T., Eds.; IOS Press: Amsterdam, The Netherlands, 1999; pp 235–246. [Google Scholar]
- Sahu A.; Morikis D.; Lambris J. D.. Complement inhibitors targeting C3, C4, and C5. In Therapeutic Interventions in the Complement System; Lambris J. D., Holers V. M., Eds.; Humana Press: Totowa, NJ, U.S., 2000; pp 75–112. [Google Scholar]
- Morikis D.; Lambris J. D. Structural aspects and design of low-molecular-mass complement inhibitors. Biochem. Soc. Trans. 2002, 30, 1026–1036. [DOI] [PubMed] [Google Scholar]
- Morikis D.; Soulika A. M.; Mallik B.; Klepeis J. L.; Floudas C. A.; Lambris J. D. Improvement of the anti-C3 activity of compstatin using rational and combinatorial approaches. Biochem. Soc. Trans. 2004, 32, 28–32. [DOI] [PubMed] [Google Scholar]
- Holland M. C. H.; Morikis D.; Lambris J. D. Synthetic small-molecule complement inhibitors. Curr. Opin. Invest. Drugs 2004, 5, 1164–1173. [PubMed] [Google Scholar]
- Floudas C.; Klepeis J.; Lambris J. D.; Morikis D.. De novo protein design: an interplay of global optimization, mixed-integer optimization and experiments. In FOCAPD 2004, Sixth International Conference on Foundations of Computer-Aided Process Design, Discovery through Product and Process Design; Floudas C. A., Agrawal R., Eds.; CACHE Corporation: Austin, TX, U.S., 2004; pp 133–146. [Google Scholar]
- Morikis D.; Floudas C.; Lambris J. D.. Structure-based integrative computational and experimental approach for the optimization of drug design. In ICCS 2005, Lecture Notes in Computer Science: Computational Science; Sunderam V. S.; van Albada G. D.; Sloot P. M.; Dongarra J., Eds.; Springer-Verlag: Berlin, Germany, 2005; pp 680–688. [Google Scholar]
- Morikis D.; Lambris J. D.. Structure, dynamics, activity, and function of compstatin and design of more potent analogs. In Structural Biology of the Complement System; Morikis D., Lambris J. D., Eds.; CRC Press: Boca Raton, FL, U.S., 2005; pp 317–340. [Google Scholar]
- Ricklin D.; Lambris J. D.. Compstatin: a complement inhibitor on its way to clinical application. In Current Topics in Complement II: Advances in Experimental Medicine and Biology; Springer US: New York, NY, U.S., 2008; pp 262–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikis D.; Assa-Munt N.; Sahu A.; Lambris J. D. Solution structure of compstatin, a potent complement inhibitor. Protein Sci. 1998, 7, 619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu A.; Soulika A. M.; Morikis D.; Spruce L.; Moore W. T.; Lambris J. D. Binding kinetics, structure-activity relationship, and biotransformation of the complement inhibitor compstatin. J. Immunol. 2000, 165, 2491–2499. [DOI] [PubMed] [Google Scholar]
- Morikis D.; Roy M.; Sahu A.; Troganis A.; Jennings P. A.; Tsokos G. C.; Lambris J. D. The structural basis of compstatin activity examined by structure-function-based design of peptide analogs and NMR. J. Biol. Chem. 2002, 277, 14942–14953. [DOI] [PubMed] [Google Scholar]
- Klepeis J. L.; Floudas C. A.; Morikis D.; Tsokos C. G.; Argyropoulos E.; Spruce L.; Lambris J. D. Integrated computational and experimental approach for lead optimization and design of compstatin variants with improved activity. J. Am. Chem. Soc. 2003, 125, 8422–8423. [DOI] [PubMed] [Google Scholar]
- Soulika A. M.; Morikis D.; Sarrias M.-R.; Roy M.; Spruce L. A.; Sahu A.; Lambris J. D. Studies of structure-activity relations of complement inhibitor compstatin. J. Immunol. 2003, 171, 1881–1890. [DOI] [PubMed] [Google Scholar]
- Mallik B.; Katragadda M.; Spruce L. A.; Carafides C.; Tsokos C. G.; Morikis D.; Lambris J. D. Design and NMR characterization of active analogues of compstatin containing non-natural amino acids. J. Med. Chem. 2005, 48, 274–286. [DOI] [PubMed] [Google Scholar]
- Katragadda M.; Lambris J. D. Expression of compstatin in Escherichia coli: incorporation of unnatural amino acids enhances its activity. Protein Expression Purif. 2006, 47, 289–295. [DOI] [PubMed] [Google Scholar]
- Katragadda M.; Magotti P.; Sfyroera G.; Lambris J. D. Hydrophobic effect and hydrogen bonds account for the improved activity of a complement inhibitor, compstatin. J. Med. Chem. 2006, 49, 4616–4622. [DOI] [PubMed] [Google Scholar]
- Magotti P.; Ricklin D.; Qu H.; Wu Y.-Q.; Kaznessis Y. N.; Lambris J. D. Structure-kinetic relationship analysis of the therapeutic complement inhibitor compstatin. J. Mol. Recognit. 2009, 22, 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows M. L.; Fung H. K.; Taylor M. S.; Floudas C. A.; de Victoria A. L.; Morikis D. New compstatin variants through two de novo protein design frameworks. Biophys. J. 2010, 98, 2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López de Victoria A.; Gorham R. D.; Bellows-Peterson M. L.; Ling J.; Lo D. D.; Floudas C. A.; Morikis D. A new generation of potent complement inhibitors of the compstatin family. Chem. Biol. Drug Des. 2011, 77, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu H.; Magotti P.; Ricklin D.; Wu E. L.; Kourtzelis I.; Wu Y.-Q.; Kaznessis Y. N.; Lambris J. D. Novel analogues of the therapeutic complement inhibitor compstatin with significantly improved affinity and potency. Mol. Immunol. 2011, 48, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamis P.; López de Victoria A.; Gorham R. D.; Bellows-Peterson M. L.; Pierou P.; Floudas C. A.; Morikis D.; Archontis G. Molecular dynamics in drug design: new generations of compstatin analogs. Chem. Biol. Drug Des. 2012, 79, 703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorham R. D.; Forest D. L.; Tamamis P.; López de Victoria A.; Kraszni M.; Kieslich C. A.; Banna C. D.; Bellows-Peterson M. L.; Larive C. K.; Floudas C. A.; Archontis G.; Johnson L. V.; Morikis D. Novel compstatin family peptides inhibit complement activation by drusen-like deposits in human retinal pigmented epithelial cell cultures. Exp. Eye Res. 2013, 116C, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu H.; Ricklin D.; Bai H.; Chen H.; Reis E. S.; Maciejewski M.; Tzekou A.; DeAngelis R. A.; Resuello R. R. G.; Lupu F.; Barlow P. N.; Lambris J. D. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology 2013, 218, 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risitano A. M.; Ricklin D.; Huang Y.; Reis E. S.; Chen H.; Ricci P.; Lin Z.; Pascariello C.; Raia M.; Sica M.; del Vecchio L.; Pane F.; Lupu F.; Notaro R.; Resuello R. R. G.; Deangelis R. A.; Lambris J. D. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood 2014, 123, 2094–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen B. J. C.; Halff E. F.; Lambris J. D.; Gros P. Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition. J. Biol. Chem. 2007, 282, 29241–29247. [DOI] [PubMed] [Google Scholar]
- Klepeis J. L.; Floudas C. A.; Morikis D.; Tsokos C. G.; Lambris J. D. Design of peptide analogues with improved activity using a novel de novo protein design approach. Ind. Eng. Chem. Res. 2004, 43, 3817–3826. [Google Scholar]
- Walensky L. D.; Bird G. H. Hydrocarbon-stapled peptides: principles, practice, and progress. J. Med. Chem. 2014, 57, 6275–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Z.-L.; Yoshida T.; Lambris J. D.; Iwata T. Suppression of drusen formation by compstatin, a peptide inhibitor of complement C3 activation, on cynomolgus monkey with early-onset macular degeneration. Adv. Exp. Med. Biol. 2010, 703, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehoshua Z.; Rosenfeld P. J.; Albini T. A. Current clinical trials in dry AMD and the definition of appropriate clinical outcome measures. Semin. Ophthalmol. 2011, 26, 167–180. [DOI] [PubMed] [Google Scholar]
- Hilbich C.; Kisters-Woike B.; Reed J.; Masters C. L.; Beyreuther K. Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer’s disease beta A4 peptides. J. Mol. Biol. 1992, 228, 460–473. [DOI] [PubMed] [Google Scholar]
- Knerr P. J.; Tzekou A.; Ricklin D.; Qu H.; Chen H.; van der Donk W. A.; Lambris J. D. Synthesis and activity of thioether-containing analogues of the complement inhibitor compstatin. ACS Chem. Biol. 2011, 6, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smadbeck J.; Peterson M. B.; Khoury G. A.; Taylor M. S.; Floudas C. A. Protein WISDOM: a workbench for in silico de novo design of biomolecules. J. Visualized Exp. 2013, e50476–e50476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajgaria R.; McAllister S. R.; Floudas C. A. Distance dependent centroid to centroid force fields using high resolution decoys. Proteins 2008, 70, 950–970. [DOI] [PubMed] [Google Scholar]
- Güntert P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [DOI] [PubMed] [Google Scholar]
- Güntert P.; Mumenthaler C.; Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997, 273, 283–298. [DOI] [PubMed] [Google Scholar]
- Ponder J. W.TINKER, Software Tools for Molecular Design. Jay Ponder Lab, Washington Unversity: St. Louis, MO, 2004. [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar]
- Lilien R. H.; Stevens B. W.; Anderson A. C.; Donald B. R. A novel ensemble-based scoring and search algorithm for protein redesign and its application to modify the substrate specificity of the gramicidin synthetase a phenylalanine adenylation enzyme. J. Comput. Biol. 2005, 12, 740–761. [DOI] [PubMed] [Google Scholar]
- Lee M. R.; Baker D.; Kollman P. A. 2.1 and 1.8 Å average Cα rmsd structure predictions on two small proteins, HP-36 and S15. J. Am. Chem. Soc. 2001, 123, 1040–1046. [DOI] [PubMed] [Google Scholar]
- Rohl C. A.; Baker D. De novo determination of protein backbone structure from residual dipolar couplings using Rosetta. J. Am. Chem. Soc. 2002, 124, 2723–2729. [DOI] [PubMed] [Google Scholar]
- Rohl C. A.; Strauss C. E. M.; Misura K. M. S.; Baker D.. Protein structure prediction using Rosetta. In Methods in Enzymology; Elsevier: San Diego, CA, 2004; Vol. 383, pp 66–93. [DOI] [PubMed] [Google Scholar]
- DiMaggio P. A.; McAllister S. R.; Floudas C. A.; Feng X.-J.; Rabinowitz J. D.; Rabitz H. A. Biclustering via optimal re-ordering of data matrices in systems biology: rigorous methods and comparative studies. BMC Bioinf. 2008, 9, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaggio P. A. Jr; McAllister S. R.; Floudas C. A.; Feng X.-J.; Rabinowitz J. D.; Rabitz H. A. A network flow model for biclustering via optimal re-ordering of data matrices. J. Global Optim. 2010, 47, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. J.; Moughon S.; Wang C.; Schueler-Furman O.; Kuhlman B.; Rohl C. A.; Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [DOI] [PubMed] [Google Scholar]
- Kuhlman B.; Baker D. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 10383–10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew K.; Renfrew P. D.; Craven T. W.; Butterfoss G. L.; Chou F.-C.; Lyskov S.; Bullock B. N.; Watkins A.; Labonte J. W.; Pacella M.; Kilambi K. P.; Leaver-Fay A.; Kuhlman B.; Gray J. J.; Bradley P.; Kirshenbaum K.; Arora P. S.; Das R.; Bonneau R. Adding diverse noncanonical backbones to Rosetta: enabling peptidomimetic design. PLoS One 2013, 8, e67051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfrew P. D.; Choi E. J.; Bonneau R.; Kuhlman B. Incorporation of noncanonical amino acids into Rosetta and use in computational protein-peptide interface design. PLoS One 2012, 7, e32637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills J. H.; Khare S. D.; Bolduc J. M.; Forouhar F.; Mulligan V. K.; Lew S.; Seetharaman J.; Tong L.; Stoddard B. L.; Baker D. Computational design of an unnatural amino acid dependent metalloprotein with atomic level accuracy. J. Am. Chem. Soc. 2013, 135, 13393–13399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Daura X.; van Gunsteren W. F. Molecular dynamics simulations of peptides containing an unnatural amino acid: dimerization, folding, and protein binding. Proteins 2004, 54, 116–127. [DOI] [PubMed] [Google Scholar]
- Daura X.; van Gunsteren W. F.; Mark A. E. Folding-unfolding thermodynamics of a beta-heptapeptide from equilibrium simulations. Proteins 1999, 34, 269–280. [DOI] [PubMed] [Google Scholar]
- Daura X.; Gademann K.; Schäfer H.; Jaun B.; Seebach D.; van Gunsteren W. F. The β-peptide hairpin in solution: conformational study of a β-hexapeptide in methanol by NMR spectroscopy and MD simulation. J. Am. Chem. Soc. 2001, 123, 2393–2404. [DOI] [PubMed] [Google Scholar]
- Schäfer H.; Daura X.; Mark A. E.; van Gunsteren W. F. Entropy calculations on a reversibly folding peptide: changes in solute free energy cannot explain folding behavior. Proteins 2001, 43, 45–56. [DOI] [PubMed] [Google Scholar]
- Rathore N.; Gellman S. H.; de Pablo J. J. Thermodynamic stability of β-peptide helices and the role of cyclic residues. Biophys. J. 2006, 91, 3425–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern M.; Abbott N.; de Pablo J. J. Dimerization of helical β-peptides in solution. Biophys. J. 2012, 102, 1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G. A.; Thompson J. P.; Smadbeck J.; Kieslich C. A.; Floudas C. A. Forcefield_PTM: ab initio charge and AMBER forcefield parameters for frequently occurring post-translational modifications. J. Chem. Theory Comput. 2013, 9, 5653–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G. A.; Smadbeck J.; Tamamis P.; Vandris A. C.; Kieslich C. A.; Floudas C. A.. Forcefield_NCAA: ab initio charge parameters to aid in the discovery and design of therapeutic proteins and peptides with unnatural amino acids and their application to complement inhibitors of the compstatin family. ACS Synth. Biol. [Online early access]. DOI: 10.1021/sb400168u. Published Online: January 6, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills J. E.; Dean P. M. Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J. Comput.-Aided Mol. Des. 1996, 10, 607–622. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Hubbart S. J.; Thornton J. M.. NACCESS (computer program); Department of Biochemistry and Molecular Biology, University College London: London, 1993.
- Bellows M. L.; Taylor M. S.; Cole P. A.; Shen L.; Siliciano R. F. Discovery of entry inhibitors for HIV-1 via a new de novo protein design framework. Biophys. J. 2010, 99, 3445–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows M. L.; Floudas C. A. Computational methods for de novo protein design and its applications to the human immunodeficiency virus 1, purine nucleoside phosphorylase, ubiquitin specific protease 7, and histone demethylases. Curr. Drug Targets 2010, 11, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows-Peterson M. L.; Fung H. K.; Floudas C. A.; Kieslich C. A.; Zhang L.; Morikis D.; Wareham K. J.; Monk P. N.; Hawksworth O. A.; Woodruff T. M. De novo peptide design with C3a receptor agonist and antagonist activities: theoretical predictions and experimental validation. J. Med. Chem. 2012, 55, 4159–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows M. L.; Fung H. K.; Floudas C. A.. Recent advances in de novo protein design. In Molecular Systems Engineering; Adjiman C. S., Galindo A., Eds.; Process Systems Engineering, Vol. 6; Wiley-VCH: Weinheim, Germany, 2010; pp 207–232. [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Wang B.; Hayik S.; Roitberg A.; Seabra G.; Kolossváry I.; Wong K. F.; Paesani F.; Vanicek J.; Liu J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Wang J.; Hsieh M.-J.; Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. A.. AMBER 11; University of California: San Francisco, CA, 2010.
- Onufriev A.; Bashford D.; Case D. A. Modification of the generalized Born model suitable for macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar]
- Onufriev A.; Bashford D.; Case D. A. Exploring protein native states and large-scale conformational changes with a modified generalized Born model. Proteins 2004, 55, 383–394. [DOI] [PubMed] [Google Scholar]
- Johnson L. V.; Forest D. L.; Banna C. D.; Radeke C. M.; Maloney M. A.; Hu J.; Spencer C. N.; Walker A. M.; Tsie M. S.; Bok D.; Radeke M. J.; Anderson D. H. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 2011, 1, 18277–18282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchoz Y.; Bard B.; Guillarme D.; Carrupt P.-A.; Veuthey J.-L.; Martel S. Analytical tools for the physicochemical profiling of drug candidates to predict absorption/distribution. Anal. Bioanal. Chem. 2009, 394, 707–729. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.