

Abstract
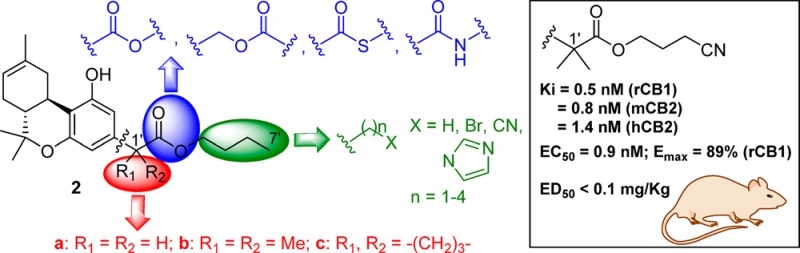
We recently reported on a controlled deactivation/detoxification approach for obtaining cannabinoids with improved druggability. Our design incorporates a metabolically labile ester group at strategic positions within the THC structure. We have now synthesized a series of (−)-Δ8-THC analogues encompassing a carboxyester group within the 3-alkyl chain in an effort to explore this novel cannabinergic chemotype for CB receptor binding affinity, in vitro and in vivo potency and efficacy, as well as controlled deactivation by plasma esterases. We have also probed the chain’s polar characteristics with regard to fast onset and short duration of action. Our lead molecule, namely 2-[(6aR,10aR)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic acid 3-cyano-propyl ester (AM7438), showed picomolar affinity for CB receptors and is deactivated by plasma esterases while the respective acid metabolite is inactive. In further in vitro and in vivo experiments, the compound was found to be a remarkably potent and efficacious CB1 receptor agonist with relatively fast onset/offset of action.
Introduction
(−)-Δ9-Tetrahydrocannabinol1 ((−)-Δ9-THC, 1, Figure 1) and its congeners act at CB1 and CB2,2−4 two Gi/o-protein-coupled cannabinoid receptors that are currently being targeted for various conditions including pain, inflammation, neurodegeneration, glaucoma, eating and mental disorders, as well as cancer.5−14 Owing to the undesirable side effects associated with CB1 receptor activation/deactivation as well as poor pharmacokinetic/pharmacodynamic (PK/PD) properties, only a limited number of cannabinergic drugs have been approved to date.15 Thus, the development of safer THC-based medications with favorable oral bioavailability, consistent efficacy, and predictable time course of action and detoxification remains to be addressed.
Figure 1.
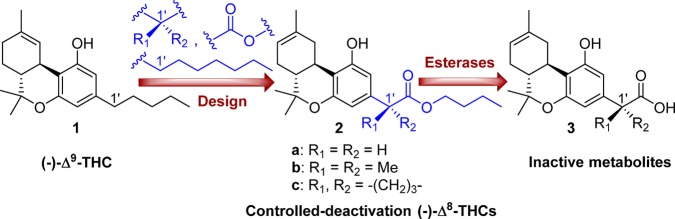
Design of the first-generation side chain carboxylated (−)-Δ8-tetrahydrocannabinols with controllable deactivation and structures of the prototype (−)-Δ9-THC and inactive metabolites.
Toward this end, we recently reported on a controlled deactivation/detoxification approach where the “soft” analogue/drug concept of enzymatic deactivation was combined with a “depot effect” that is commonly observed with Δ9-THC and other lipophilic cannabinoids. In our design, the compound’s systemic half-life is determined by two factors. The first reflects the ability of the compound to sequester in some tissue reservoir such as fatty tissue (depot effect). The tissue sequestration affects the availability of the compound for receptor activation as well as for hydrolytic deactivation through systemic circulation. This process is dependent on the compound’s physicochemical properties and can be modulated by adjusting log P and PSA. Thus, more lipophilic compounds are slowly released in the bloodstream from the depot while more polar compounds are expected to have less of a depot effect. We have also confirmed that, in the compounds discussed here, the hydrolytic metabolic pathway is the most dominant and is considerably faster than other metabolic options such as reactions involving microsomal enzymes. The second is the rate of enzymatic hydrolysis of a metabolically labile ester group (−C(O)-O−) by blood esterases. This can be calibrated by incorporating suitable stereochemical features in the vicinity of the hydrolyzable moiety (enzymatic effect).16 Currently, we have developed two controlled-deactivation cannabinergic templates based on the tricyclic classical cannabinoid prototype. In the first, the C-ring in THC was replaced by a hydrolyzable seven-membered lactone,17 while in the second, the metabolically labile ester group was placed at the 2′-position of the side chain pharmacophore (Figure 1).16
In the present SAR study, we sought to probe the ester side chain pharmacophore in the (−)-Δ8-THC prototype within a novel series of analogues with regard to affinity for the CB1 and CB2 cannabinoid receptors, in vitro and in vivo potency and efficacy, as well as ability to control the half-lives of deactivation through enzymatic action. An additional goal of this work was to explore the polar characteristics of the side chain and gain information related to the modulation of the depot effect and its role in the in vivo pharmacokinetic profile of the analogues. The above considerations led us to design ligands that retain the metabolically labile group at the 2′-position of the side chain and replace the ester group (−C(O)-O−) with the reverse ester (−O–C(O)−) as well as with the corresponding thioester (−C(O)-S−) and the hydrolytically more stable amide group (−C(O)-NH−) (Figure 2). To explore the chain length, we synthesized analogues with four- to nine-atom-long side chains. Additionally, to probe the ligand’s polarity, we incorporated bromo-, cyano-, and imidazolyl-groups at the distal side chain carbon atom. As with our previous work,16 to regulate the rates of enzymatic inactivation while enhancing the compound’s affinities for CB receptors, we introduced benzylic substituents contiguous to the metabolically vulnerable ester group.
Figure 2.
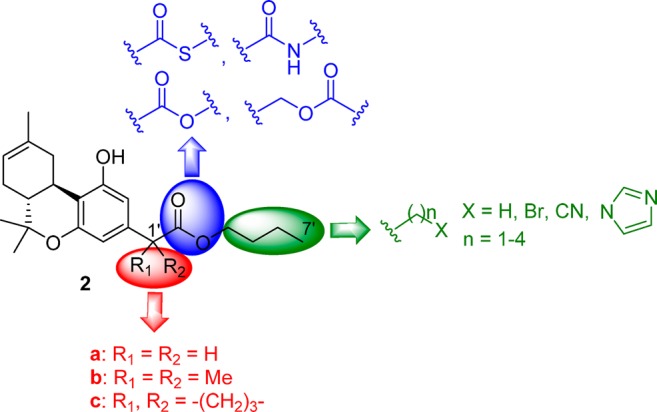
Structure–activity relationship summary of the metabolically vulnerable side chain pharmacophore in (−)-Δ8-tetrahydrocannabinols.
All novel compounds were characterized by determining their in vitro CB1 and CB2 receptor affinities. Of these, the most interesting were assessed for their functional activities and for their in vitro metabolic stabilities toward mouse plasma esterases while a limited representative set of key compounds was evaluated for their hypothermic and analgesic effects in vivo. Our data show that (−)-Δ8-THC analogues carrying five- to nine-atom-long side chains that are substituted with geminal dimethyl and cyclobutyl groups at the C1′-position exhibit remarkably high affinities for the CB1 and CB2 receptors. As predicted, all novel analogues were found to be susceptible to enzymatic deactivation by plasma esterases in a controllable manner, while their metabolites showed no or very low cannabinergic activity. Additionally, all key compounds were shown to be agonists for the CB1 receptor when tested for their abilities to reduce cAMP levels and also produce the characteristic CB1-mediated hypothermia and analgesia in rats and mice. Equally important, in the hypothermia assay, the 6′-cyano-2′-carboxy-Δ8-THC analogue 10a (AM7438) exhibited 10-fold higher potency as well as faster onset and shorter duration of action than the less polar carboxy counterpart 2b. Also, the offset of the analgesic effect was significantly faster for 10a compared to 2b. Congruent with our rational design, these in vivo results suggest that the depot effect of our controlled deactivation analogues can suitably be modulated by enhancing the polar characteristics of the side chain pharmacophore without any significant loss of potency.
Chemistry
Syntheses of the 4′-bromo- and 4′-cyano-butyl esters 6a–6c and 7a–7c are summarized in Scheme 1. The required (−)-Δ8-THC carboxylic acids 5a–5c were synthesized from commercially available (3,5-dimethoxyphenyl)acetonitrile and (+)-cis/trans-p-mentha-2,8-dien-1-ol in three to four steps following our recently reported procedures.16 Alkylation of the respective carboxylate anions with 1,4-dibromobutane under microwave heating led to the corresponding esters 6a–6c in 45–63% yields.16 Treatment of these bromides with sodium cyanide in dimethyl sulfoxide18 produced the respective side chain cyano-substituted analogues 7a–7c in 54–71% yields.
Scheme 1.

In a similar fashion, the tricyclic carboxylic acid 5b was transformed to the side chain homologues 8a, 8b, and 9a–9d in 47–87% yields (Scheme 2). Reaction of the 3-bromo-propyl ester 9b with sodium cyanide or imidazole in the presence of potassium carbonate, in dimethyl sulfoxide, led to the end carbon substituted derivatives 10a and 10b (41–98% yields).
Scheme 2.

Synthesis of the reverse ester analogue 16 involves a Mitsunobu esterification reaction19 as the key step (Scheme 3). Thus, reduction of nitrile 11 with diisobutylaluminum hydride20 at −78 °C led to aldehyde 12 (92% yield), which upon exposure to sodium borohydride in methanol21 afforded the respective alcohol 13 in excellent yield (94%). Cleavage of the methyl ether groups in 13 using boron tribromide18 produced resorcinol 14 in 47% yield. Acid catalyzed condensation of this intermediate with chiral terpenoid alcohol 17 in refluxing chloroform for 4–6 h gave (−)-Δ8-THC alcohol 15 in 21% isolated yield along with unidentified byproducts. We were able to improve this yield (28%) using microwave conditions over a much shorter time period (10 min).16 Subsequently, Mitsunobu esterification of 15 with n-valeric acid using triphenylphosphine and diethyl azodicarboxylate led to the final ester 16 (41% yield).
Scheme 3.

Treatment of the α,α-dimethyl-carboxylic acid 5b with [bis(2-methoxyethyl)amino]sulfur trifluoride and coupling of the in situ generated acyl fluoride22 with n-pentylamine afforded the (−)-Δ8-THC amide 18 in 70% yield (Scheme 4). Exposure of the same starting material (5b) to the benzotriazole/thionyl chloride reagent23 followed by treatment of the intermediate acyl chloride 19 with n-propanethiol led to thioester 20 in 23% yield for the two steps.
Scheme 4.

Cannabinoid Receptor Affinities
The abilities of compounds 6a–6c, 7a–7c, 8a, 8b, 9a–9d, 10a, 10b, 15, 16, 18, and 20 to displace the radiolabeled CB1/CB2 agonist CP-55,940 from membranes prepared from rat brain (source of CB1) and HEK293 cells expressing either mouse CB2 or human CB2 were determined as described earlier,18,20 and inhibition constant values (Ki) from the respective competition binding curves are listed in Table 1 in which our prototype (−)-Δ8-THC, as well as the first-generation carboxy-Δ8-THCs 2a–2c are included for comparison. The rat, mouse, and human CB1 receptors have 97–99% sequence identity across species and, as shown earlier,16 are not expected to exhibit variations in their Ki values. However, mouse CB224 (mCB2) exhibits only 82% sequence identity with the human clone3 (hCB2). This divergent nature of mCB2 and hCB2 receptors was shown in earlier work25,26 to be associated with species-based differences in affinity. For this reason, the analogues were also tested on hCB2.
Table 1. Affinities (Ki) of Side Chain (−)-Δ8-THC Analogues for CB1 and CB2 Cannabinoid Receptors (±95% Confidence Limits) and Half-Lives (t1/2) of Representative Compounds for Mouse Plasma Esterases.
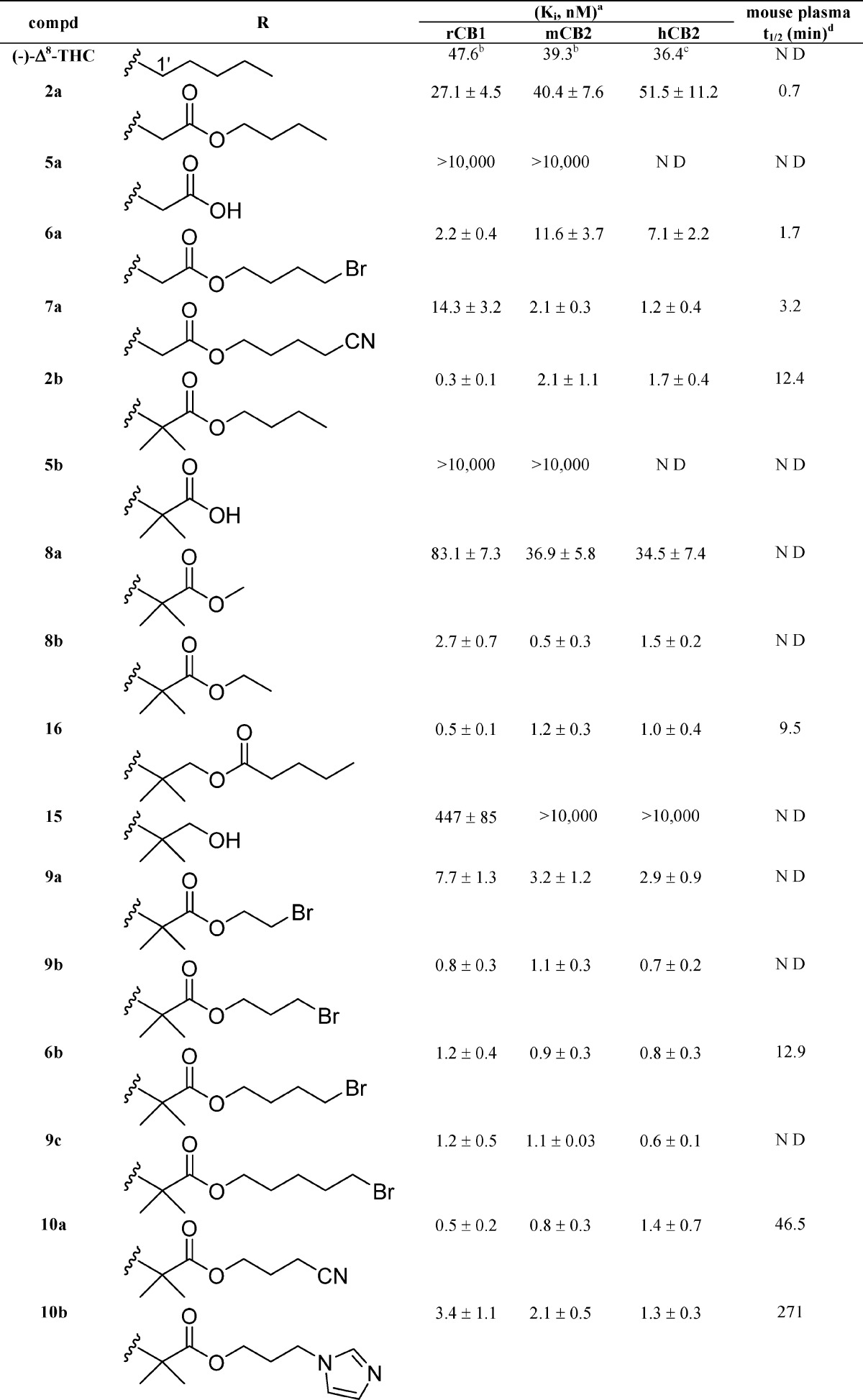
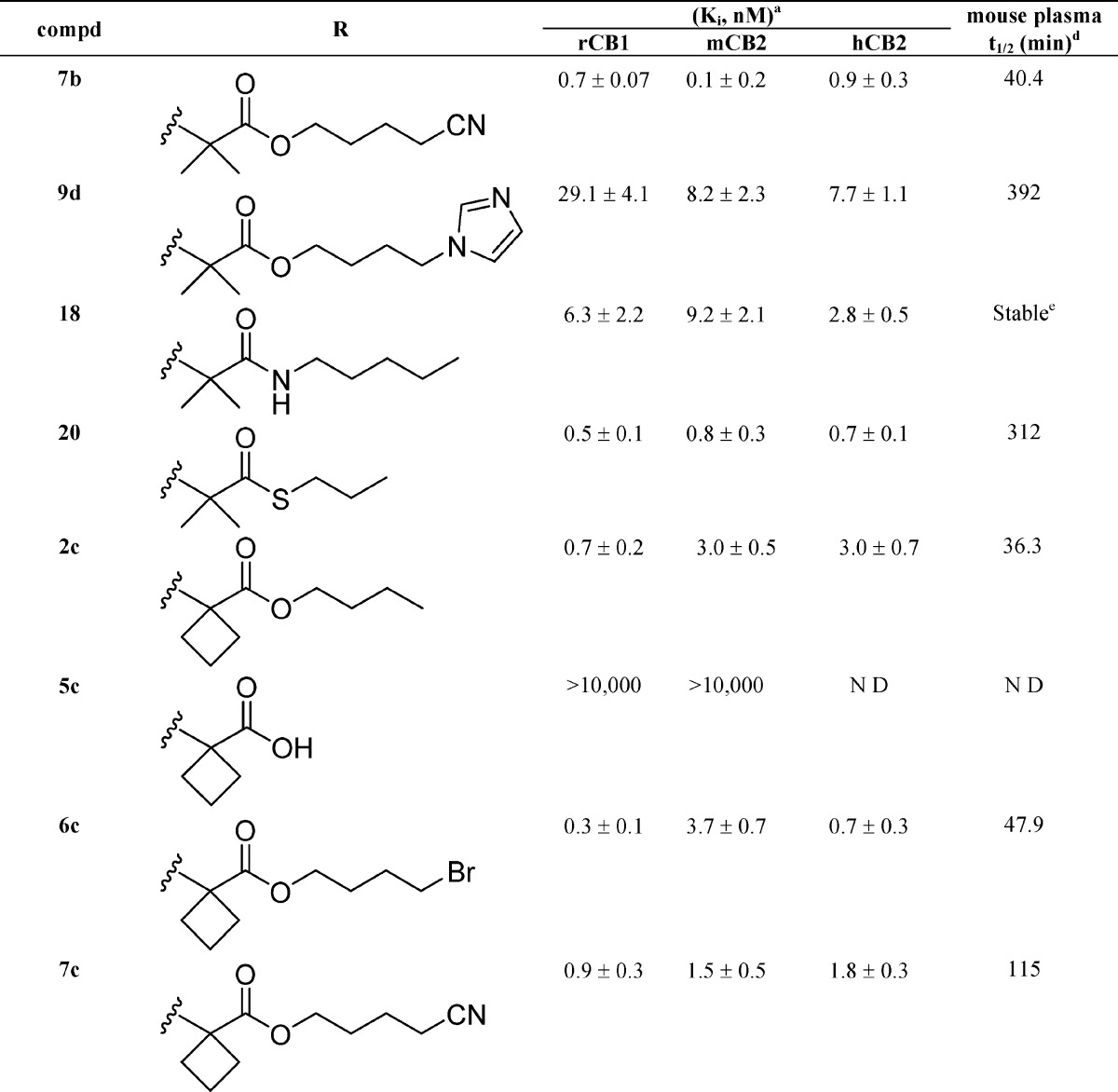
Affinities for CB1 and CB2 were determined using rat brain (CB1) or membranes from HEK293 cells expressing mouse or human CB2 and [3H]CP-55,940 as the radioligand following previously described procedures.18,20,51 Data were analyzed using nonlinear regression analysis. Ki values were obtained from three independent experiments performed in triplicate and are expressed as the mean of the three values.
Reported previously.33
Reported previously.14
Half-lives (t1/2) for mouse plasma were determined as described under Experimental Section.
No observable hydrolysis within 5 h. ND: not determined.
The compounds included in this study are (−)-Δ8-THC analogues in which a four- to nine-atom-long side chain with or without 1′-substituents incorporates an enzymatically vulnerable group at the 2′- or 3′-positions. Optimization of the novel carboxy or thiocarboxy-ester side chains was probed through the synthesis of analogues carrying bromo-, cyano-, and imidazolyl-substituents at the distal carbon atom. As predicted based on our earlier results, the hydrolytic metabolites 5a–5c and 15 have no significant affinities for CB1 and CB2 receptors, thus minimizing the possibility of undesirable cannabinoid receptor related side effects. Comparison of the binding data of (−)-Δ8-THC and its carboxyester congeners 2a, 6a, and 7a suggests that extension of the linear chain from five to seven–nine atoms, along with incorporation of an ester group at the 2′,3′-positions, enhances the binding affinities of these analogues for both the CB1 and CB2 receptors. We also observed that incorporation of the bromo as well as the more polar cyano substituents at the terminal carbon of the side chain is well tolerated. Comparison of the binding data of our prototype (−)-Δ8-THC and compound 8b demonstrates the remarkable effects of 1′,1′-dimethyl-2′-carboxyester substitution (−C(CH3)2–C(O)O−) on the side chain pharmacophore. Thus, 8b with the five-atom-long side chain exhibits 10-, 78-, and 24-fold higher binding affinities for rCB1, mCB2, and hCB2 receptors, respectively, when compared to (−)-Δ8-THC. The one-carbon shorter homologue 8a has significantly reduced affinities for both CB receptors, indicating that a minimum requirement for substantial affinity for both receptors in this series is a five-atom-long side chain. This high affinity can be maintained all through side chain lengths of nine atoms with or without terminal bromine substituents (analogues 2b, 6b, and 9a–c). Interestingly, this holds true when the terminal two or three atoms of the chain are replaced by the polar cyano group or the bulkier and amphiprotic imidazole ring (compounds 10a, 10b and 7b). Notably, the cyano analogues 10a and 7b exhibit remarkably high affinities for the CB1 and CB2 receptors. However, extension of the ω-substituted imidazolyl chain of 10b by one methylene group (compound 9d) results in a reduction of the ligand’s affinity for both cannabinoid receptors, an effect more accentuated in CB1 (∼8-fold reduction). Taken together, these data suggest that the pharmacophoric limits for an ω-substituted 1′,1′-dimethyl-2′-carboxyester chain could not be extended beyond the nine atoms.
A comparison of the binding affinities of the nonsubstituted analogues 6a and 7a with their respective gem-dimethyl counterparts 6b and 7b shows that introduction of two methyl substituents at the 1′-position of an ω-substituted carboxylated chain leads to an enhancement (20-fold maximum) in CB1 and CB2 receptor affinities. This increase in the ligand’s affinities for both CB1 and CB2 receptors holds true when the gem-dimethyl substitution is modified to the bulkier cyclobutyl ring (analogues 6c and 7c). An examination of the binding data of the 2′,3′-carboxyester analogue 2b and its sulfur and nitrogen congeners 18 and 20 shows that the ester moiety (−C(O)-O−) can be replaced by the respective thioester (−C(O)-S−) and amide (−C(O)-NH−) groups. Likewise, analogue 16 incorporating the sterically less hindered retro ester group (−O–C(O)−) at the 3′,4′-position maintains high affinity for both the CB1 and CB2 receptors.
In summary, the detailed SAR reported here shows that a five- to nine-atom-long side chain with gem-dimethyl or cyclobutyl substituents at the 1′-position and an enzymatically vulnerable group within the 2′ or 3′ chain segment results in analogues with remarkably high affinities for both CB1 and CB2 receptors. Importantly, addition of the bromo- or cyano-groups as well as the bulky and amphiprotic imidazole ring at the terminal carbon maintains or enhances the affinity of the ligand for the CB receptors.
In Vitro Plasma Stability Studies
Representative analogues within this series were assessed for their in vitro plasma stability toward mouse plasma esterases as detailed in the Experimantal Section.16,17 It should be noted that blood contains various esterases which play the major role in the hydrolysis of compounds carrying ester, carbamate, or phosphate bonds. These esterases include acetylcholinesterases (ACHE), butyrylcholinesterases (BCHE), paraoxonases, and carboxyesterases (in mice and rat but not in human plasma). Esterase activity can be found mainly in plasma, with less activity in red blood cells. Plasma albumin itself may also act as an esterase under certain conditions. For example, albumin contributes about 20% of the total hydrolysis of aspirin to salicylic acid in human plasma. The esterase activity in blood seems to be more extensive in small animals such as rats than in large animals and humans.27 A comparison of the half-lives (t1/2, Table 1) of the alkyl bromide and nitrile having no substitution at the 1′-position (6a, 7a) with their 1′-gem-dimethyl (6b, 7b) and 1′-cyclobutyl (6c, 7c) counterparts shows that the plasma esterase stabilities of the analogues correlate well with the presence and the size of 1′-substituents. Thus, the order of metabolic stabilities for the bromo- and cyano-substituted analogues is 6a < 7a < 6b < 7b < 6c < 7c, with the compounds carrying the bulkier cyclobutyl group being the most hydrolytically stable. This trend with the ω-substituted eight- to nine-atom-long side chain analogues parallels our earlier observations with the shorter (seven-atoms) and unsubstituted side chain congeners 2a, 2b, and 2c.16 Another general observation through this structure–stability relationship study is that regardless of the 1′-substituent, addition of a bromo- or cyano-group at the terminal carbon of a seven-atom-long chain increases its stability toward esterases with the cyano-substituted chain exhibiting the highest stability (comparison of 2a with 6a and 7a, 2b with 6b and 7b, and 2c with 6c and 7c).
The effect of ω-substitution on the chain’s enzymatic stability profile was studied in more detail in the 1′-gem-dimethyl series through the assessment of the half-lives of the cyano- and imidazolyl-substituted analogues 10a, 10b, 7b, and 9d. We observe that presence of a terminal cyano group or the larger imidazole ring reduce the enzymatic lability of the 1′-gem-dimethyl chain (analogue 2b) by 3- to 31-fold. As expected, the sterically less hindered retro ester analogue 16 was more prone to enzymatic hydrolysis when compared to the α,α-dimethyl-carboxylated counterpart 2b, while the amide group (18) was shown to have remarkable plasma stability. In agreement with earlier work on the rates of hydrolysis of ethyl butyrate and ethyl thiobutyrate by pig liver esterases,28 the thioester 20 exhibits higher stability in mouse plasma esterases than the respective oxoester 2b. Overall, our data show that the rate of enzymatic inactivation of our side chain carboxyester Δ8-THCs can be controlled by (1) structural features in the vicinity of the hydrolyzable group and (2) substituents at the terminal carbon.
Functional Characterization
Functional characterization of key compounds for the rCB1 receptor was carried out by measuring the decrease in forskolin-stimulated cAMP, as detailed earlier.16,18 Compounds were initially screened in three different concentrations (three-point data), and the approximate EC50 values were calculated (Table 2). Subsequently, the accurate EC50 value of the most potent analogue 10a was determined from two independent experiments (eight-point data) and listed in Table 2 along with data for the related compounds 2b and 2c for comparison. We observe that all tested analogues are full agonists at the CB1 receptor while their EC50 values correlate well with their respective binding affinities. Of the compounds tested, the carboxyester Δ8-THC analogues 2b and 2c as well as their ω-substituted cyanide congener 10a were found to be the most potent and efficacious compounds within the series with EC50s of 0.5, 0.4, and 0.9 nM, respectively.
Table 2. Functional Potencies (EC50) of Selected (−)-Δ8-THC Ester Analogues for the rCB1 Cannabinoid Receptor.
| rCB1 (EC50, nM)a |
|||
|---|---|---|---|
| compd | 3-pointsb | 8-pointsc | E(max) (%)d |
| 7a | 10–100 | ||
| 2b | <1 | 0.5 (0.1–1.2)e | 92e |
| 8b | 1–10 | ||
| 10a | <1 | 0.9 (0.3–1.5) | 89 |
| 10b | 1–10 | ||
| 7b | 1–10 | ||
| 18 | 1–10 | ||
| 2c | <1 | 0.4 (0.2–1.2)e | 90e |
| 7c | 1–10 | ||
Functional potencies at rCB1 receptor were determined by measuring the decrease in forskolin-stimulated cAMP levels, as described in Experimental Section.16,18
Three point data were obtained from one experiment (3 points) run in duplicate (less accurate EC50 values).
Data are average of two independent experiments (8 points) run in triplicate, and 95% confidence intervals for the EC50 values are given in parentheses. EC50 values were calculated using nonlinear regression analysis.
Forskolin stimulated cAMP levels were normalized to 100% and E(max) is the maximum inhibition of forskolin stimulated cAMP levels and is presented as the percentage of CP-55,940 response at 500 nM.
Reported previously.16
Molecular Modeling
As an aid in the interpretation of the data, we carried out conformational analyses and docking studies (detailed procedures are given under Experimental Section) involving our lead analogue 10a, the nonhydrolyzable counterpart (−)-Δ8-THC-DMH as well as the parent compound (−)-Δ8-THC.
Conformational Analysis
Initial conformational analyses of (−)-Δ8-THC, (−)-Δ8-THC-DMH, and 10a revealed a difference in the conformation of the side chains in their global minimum energy conformers. Both (−)-Δ8-THC-DMH and 10a have their side chains oriented orthogonal to the phenol ring, while for (−)-Δ8-THC, the five-carbon side chain extends in the same plane as its phenol ring. Initial in vacuo conformational analyses of 10a identified an intramolecular hydrogen bond between the cyano nitrogen and the phenolic hydroxyl. The 10a phenolic hydroxyl to cyano nitrogen hydrogen bond heteroatom distance (N–O) and hydrogen bond (O–H– −N) angle were 3.22 Å and 144°, respectively. The intramolecular hydrogen bond in 10a was not found in the global minimum energy conformation from a second conformational search performed using the OPSL2005 force field with an implicit GB/SA solvent model for water. Preliminary results from a 70 ns NAMD molecular dynamics simulation of 10a in a fully hydrated POPC lipid bilayer found a very low incidence of this internal hydrogen bond as well (unpublished data). For this reason, the nearest low energy conformer without an intramolecular hydrogen bond (which was 1.77 kcal/mol above the initial global min) was used for the calculation of conformational energy costs for 10a. The first and second side chain dihedrals of this conformer compare well with the global minimum energy conformer of (−)-Δ8-THC-DMH, shown in Table 3. Finally, the phenolic hydroxyls of all three ligands were found to prefer the proton directed toward the C2 phenyl ring position nearer the side chain and with the value of the C2–C1–O1–H1 dihedral near zero. A comparison of the lowest energy conformation of 10a without the internal hydrogen bond to those of (−)-Δ8-THC-DMH and (−)-Δ8-THC is shown in Figure 3.
Table 3. Comparison of Side Chain Dihedral Values for Global Minimum Energy Conformers and Binding Affinities (Ki) of Key Analogues.
|
Ki = 0.5 nM (rCB1), = 0.8 nM (mCB2) (deg) |
Ki = 0.9 nM (rCB1), = 1.4 nM (mCB2) (deg) | Ki = 47.6 nM (rCB1), = 39.3 nM (mCB2) (deg) | ||
|---|---|---|---|---|
| side chain dihedral | 10a | no int H-bond 10a | (−)-Δ8-THC-DMH | (−)-Δ8-THC |
| C2–C3–C1′–C2′ | –38.3 | –54.5 | –52.3 | –85.6 |
| C3–C1′–C2′–(C3′, O3′) | –60.2 | –68.7 | –60.0 | 179.9 |
| C1′–C2′–(C3′, O3′)–C4′ | 174.6 | 176.1 | 175.2 | –179.9 |
| C2′–(C3′, O3′)–C4′–C5′ | –164.7 | –174.5 | 179.5 | 180.0 |
| (C3′, O3′)–C4′–C5′–C6′ | 54.4 | 61.4 | 179.1 | |
| C4′–C5′–C6′–C7′ | 54.3 | 179.1 | 180.0 | |
Figure 3.
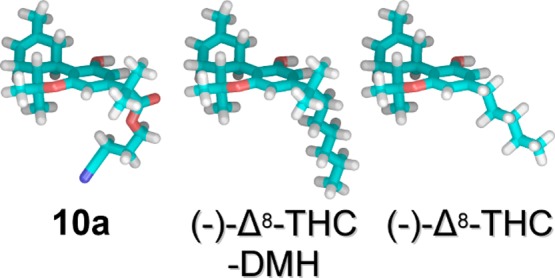
10a is the lowest energy conformer that does not have the internal H-bond and is shown on the left. The (−)-Δ8-THC-DMH global minimum energy conformer is in the middle and (−)-Δ8-THC is on the right. All carbons atoms are shown in cyan.
Docking Studies
To provide a representation for the interaction of the above three compounds with the hCB1 receptor, we carried out docking studies with an activated form of a CB1 receptor model and calculated relative ligand/receptor interaction energies. Glide docking studies of 10a in the activated CB1 receptor revealed a significant role for the ester group at the 2′ position. The carbonyl oxygen within the ester group has a hydrogen bonding interaction with T3.33(197) as shown in Figure 4A. The hydrogen bond heteroatom distance (O–O) and hydrogen bond (O–H– −O) angle are 2.56 Å and 165°, respectively. The T3.33(197) hydrogen bonding interaction is not available to the (−)-Δ8-THC-DMH counterpart or the parent compound (−)-Δ8-THC as shown in parts B and C of Figure 4. However, all three compounds modeled in the Glide docking studies have a phenolic hydroxyl that interacts directly with K3.28(192). The K3.28(192) and 10a phenolic hydroxyl hydrogen bond heteroatom distance (N–O) and hydrogen bond (N–H– −O) angle are 2.73 Å and 170°, respectively. The K3.28(192) and (−)-Δ8-THC-DMH phenolic hydroxyl hydrogen bond heteroatom distance (N–O) and hydrogen bond (N–H– −O) angle are 2.69 Å and 171°, respectively. Finally, the K3.28(192) and (−)-Δ8-THC phenolic hydroxyl hydrogen bond heteroatom distance (N–O) and hydrogen bond (N–H– −O) angle are 2.64 Å and 168°, respectively.
Figure 4.
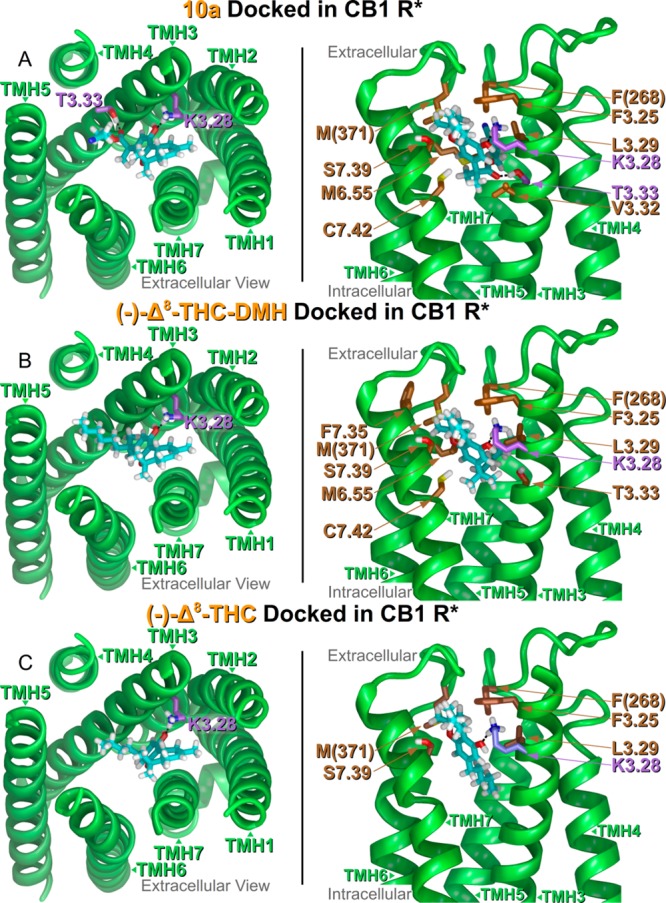
(A–C) (Left Panel, A–C) Extracellular viewpoint of each ligand/receptor complex. Termini and loops are not shown in this panel. Ligand carbons are colored cyan and hydrophilic interactions are displayed with amino acid carbons in violet. (Right Panel, A–C) Side view of ligand/receptor complexes as if looking through TMH1, 2 (not displayed). Hydrophobic interacting residues with better than −2.0 kcal/mol ligand interaction energies are displayed with brown carbons. Hydrophilic ligand interactions are displayed with the amino acid carbons in violet.
Glide XP scores adjusted for ligand strain reflect the rank order of Kis (Table 3) in rCB1 with (10a = −5.7 kcal/mol) ≈ ((−)-Δ8-THC-DMH = −5.4 kcal/mol) < ((−)-Δ8-THC = −4.1 kcal/mol). For a list of Glide XP scores in rank order and adjusted by ligand conformational cost, see Supporting Information, Tables S2–S4. A breakdown of ligand/receptor interaction energies revealed the importance of van der Waals interactions for each of ligands and how the two 1′,1′-dimethyl analogues (10a and (−)-Δ8-THC-DMH) have far more of these interactions than (−)-Δ8-THC. The (−)-Δ8-THC-DMH and 10a have nine and eight van der Waals interactions better than −2.0 kcal/mol, respectively, while (−)-Δ8-THC has five such interactions. The most important van der Waals interactions in common for the 1′,1′-dimethyl analogues are F3.25(189), L3.29(193), F(268), M6.55(363), M(371), S7.39(383), and C7.42(386). The (−)-Δ8-THC parent interacts in a likewise manner with only F3.25(189), L3.29(193), F(268), M(371), and S7.39(383). Of these residues, L3.29(193), F(268), M6.55(363), and M(371) form part of a tunnel-shaped hydrophobic enclosure between TMHs 3 and 6 for the side chain of each analogue to occupy. Unique to 10a is the van der Waals interaction with V3.32(196), and unique to (−)-Δ8-THC-DMH is F7.35(329). A full listing of all ligand/receptor interaction energies is given in the Supporting Information, Tables 5–7.
Total ligand/receptor interaction energies also follow the trend of the Glide XP scores and Kis in rCB1 with (10a = −42.9 kcal/mol) ≈ ((−)-Δ8-THC-DMH = −44.4 kcal/mol) < ((−)-Δ8-THC = −35.6 kcal/mol). The docked conformational cost for each ligand after the post Glide minimization (10a, 0.18 kcal/mol; (−)-Δ8-THC-DMH, 0.28 kcal/mol; and (−)-Δ8-THC, 0.13 kcal/mol) was small. Compound 10a did not have a statistically significant lower Ki compared to (−)-Δ8-THC-DMH, even though Glide docking studies identified two hydrogen bonds for 10a compared to one hydrogen bond for (−)-Δ8-THC-DMH. This may be due to the energetic cost for 10a to place its hydrophilic ester and cyano moieties in a generally hydrophobic pocket in the CB1 receptor. This cost likely causes the two Kis to be essentially equivalent.
In Vivo Behavioral Characterization
Representative analogues within this series were initially screened using the hypothermia test in rats (data not shown), and the order of the potency was found to be 10a > 7b ≥ 6b > 2b ≈ 20 ≥ 16 ≈ 8b ≥ 18. Subsequently, the most promising compound 10a was studied in more detail and its in vivo hypothermic and antinociceptive profiles were compared to those of the earlier analogue 2b as well as with the nonhydrolyzable parent compound (−)-Δ8-THC-DMH.
Hypothermia Testing
Body temperature was measured in isolated rats over a 6 h period following drug injection (detailed procedures are given in the Experimental Section). Compound 10a decreased core body temperature in a dose-dependent manner, with a dose of 0.1 mg/kg reducing body temperature up to 4.5 ± 0.5 °C from an average baseline of 37.8 ± 0.1 °C (Figure 5). Its ED50 value (i.e., the dose required to reduce temperature by 3 °C) was 0.034 mg/kg (95% CI: 0.026, 0.041 mg/kg). For comparison, the effects of our first-generation analogue 2b and those of the (−)-Δ8-THC-DMH are also shown. The ED50 values were 0.37 (0.14, 0.73 with 95% CI) for 2b and 0.29 (0.18, 0.45 with 95% CI) for (−)-Δ8-THC-DMH. Thus, 10a is approximately 8–10-fold more potent than (−)-Δ8-THC-DMH and 2b. Limited extended studies comparing hypothermia induced by equipotent doses of the 6′-cyano-2′-carboxy-Δ8-THC analogue 10a and its structurally related analogues 2b and (−)-Δ8-THC-DMH for 12 h provided an estimate of their in vivo times of action. Thus, at a dose of 0.03 mg/kg, near its ED50 value, compound 10a reduced body temperature by 2 °C within 3 h of injection and these effects were not observed 6 h after injection (see Supporting Information, Figure S1). This was compared with the effects of equivalent doses (near their ED50 values) of 2b and (−)-Δ8-THC-DMH. Thus, for 2b, a dose of 0.3 mg/kg produced effects that persisted for at least 6 h after injection while a dose of 0.3 mg/kg of (−)-Δ8-THC-DMH produced an equivalent reduction in temperature beyond 12 h after injection. It should be noted that in order to compare equiactive doses of the most potent compound 10a with those of the in vivo less potent (−)-Δ8-THC-DMH and 2b, we have used different doses of the three drugs that are consistent with their ED50 values. Our data indicate that the depot effect as reflected in the log P and PSA values (Table 4) plays the most dominant role in determining the in vivo hypothermic half-lives of compounds 10a, 2b, and the nonhydrolyzable (−)-Δ8-THC-DMH. Thus, notwithstanding the shorter in vitro hydrolytic half-life of compound 2b, when compared to 10a, this compound has a longer in vivo half-life. These results are represented in Table 4.
Figure 5.

Effects of 10a, 2b, and Δ8-THC-DMH or vehicle (above V) on body temperature. Symbols represent the group mean ± SEM (n = 6 rats). Abscissa, dose in mg/kg; ordinate, change in body temperature from an average baseline of 37.8 ± 0.1 °C. Data with compound 2b and Δ8-THC-DMH published previously.16
Table 4. Calculated log P and tPSA Values for (−)-Δ8-THC-DMH, 2b, and 10a, and Duration of Their Hypothermic Effects in Rats.
| compd | clogPa | tPSAa | duration of hypothermic effects in ratsb |
|---|---|---|---|
| (−)-Δ8-THC-DMH | 9.1 | 29.5 | t > 12 h |
| 2b | 6.6 | 55.8 | t = 6–12 h |
| 10a | 5.0 | 79.5 | t < 6 h |
Calculations were performed using ChemBioDraw Ultra 14.0 software.
Hypothermic effects were determined using equiactive doses of the test compounds.
Analgesia Testing
The pharmacokinetic profiles of analogues bearing the carboxyester (2b) and the more polar cyano-carboxyester (10a) side chains were further studied in the CB1 receptor-characteristic analgesia test in mice, and the results are depicted in Figure 6 (the dose response graph for four doses of 10a are given under Supporting Information, Figure S2). A mixed model repeated measures ANOVA (IBM software package SPSS, v.21) applied to the tail-flick latency data produced by compounds 10a and 2b during the descending phase (180 and 360 min) showed significant main effects for drug (D) (F1.44 = 8.98; p ≤ 0.004), dose-level (L) (F1.44 = 40.95; p ≤ 0.001), and time (T) (F1.44 = 28.46; p ≤ 0.001). Of more interest, however, is that pairwise comparisons within these parameters (D, L, and T) using Sidak multiple comparison t test procedure suggested significant differences for all three parameters (p = 0.05). Thus, the tail-flick latencies were dose- and time-dependent for both compounds in a similar manner, and because the pairwise comparison for drug was also significant, the offset of the analgesia effect was significantly faster for 10a compared to 2b. The average (±SEM) baseline tail-flick withdrawal latency for all mice (N = 24) examined with compound 10a was 0.99 ± 0.05 s.
Figure 6.
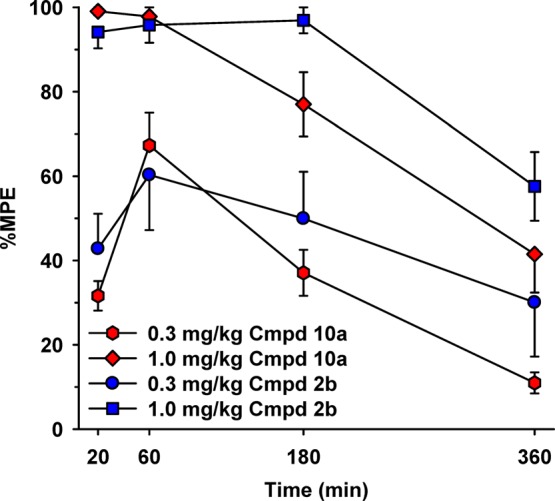
Tail-flick latencies in a hot water bath (52 °C) after administration of 0.3 and 1.0 mg/kg of compounds 10a and 2b at four time-points (20, 60, 180, and 360 min postadministration) using male CD-1 mice. Abscissa, time (min) after injection; ordinate, tail-flick withdrawal latencies expressed as a percentage of maximum possible effect (% MPE; group mean ± SEM). Data for compound 2b are reproduced from our earlier work.16
Overall, our in vivo experiments show that in rats, compound 10a is approximately a 10–30-fold more potent as a cannabinoid agonist when compared to 2b and (−)-Δ8-THC-DMH. Also, this more potent analogue has a faster onset and shorter duration of action when compared to the less polar counterpart 2b and the lipophilic and nonhydrolizable parent compound (−)-Δ8-THC-DMH. Our antinociception data in mice clearly show that the cyano-carboxyl-analogue 10a has a shorter duration of action than its congener with no cyano substitution (2b).
Conclusions
In summary, as a continuation of our earlier work on the controlled deactivation/detoxification ligand development project, we sought to probe the novel carboxyester side chain pharmacophore in (−)-Δ8-THCs for CB receptor binding affinity, in vitro and in vivo potency and efficacy, as well as its effects on the half-lives of deactivation through enzymatic activity. We have also explored the chain’s polar characteristics that are associated with the depot effect, in an effort to produce cannabinoids with faster onset/offset and shorter duration of action than the currently existing (−)-Δ8-THC analogues. In connection with our earlier work where we focused on the chain’s benzylic position and the related subsite within the receptor’s binding domain,18,20,29−33 the current SAR study extends the mapping of the chain’s pharmacophoric space beyond the 1′-carbon and argues that both CB receptors can tolerate polar groups and atoms within the second and the fourth position of the chain. We are hopeful that these observations will provide us with new opportunities for the design of high affinity/efficacy novel analogues with improved selectivities for the two cannabinoid receptors. The most successful compound identified through this careful study, namely 2-[(6aR,10aR)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic acid 3-cyano-propyl ester (10a), is a remarkably potent and efficacious CB1 receptor agonist with relatively shorter duration of action than its less polar 2′-carboxy-(−)-Δ8-THC-DMH counterpart and the highly lipophilic and long lasting (−)-Δ8-THC-DMH. Our data support the hypothesis that the pharmacological half-lives of our selectively detoxified analogues can be controlled by the joint modulation of their relative stabilities for plasma esterases as well as through variation of their polar characteristics and thus to the depot effects.
Experimental Section
Materials
All reagents and solvents were purchased from Aldrich Chemical Co., unless otherwise specified, and used without further purification. All anhydrous reactions were performed under a static argon atmosphere in flame-dried glassware using scrupulously dry solvents. Flash column chromatography employed silica gel 60 (230–400 mesh). All compounds were demonstrated to be homogeneous by analytical TLC on precoated silica gel TLC plates (Merck, 60 F245 on glass, layer thickness 250 μm), and chromatograms were visualized by phosphomolybdic acid staining. Melting points were determined on a micromelting point apparatus and are uncorrected. IR spectra were recorded on a PerkinElmer Spectrum One FT-IR spectrometer. NMR spectra were recorded in CDCl3, unless otherwise stated, on a Bruker Ultra Shield 400 WB plus (1H at 400 MHz, 13C at 100 MHz) or on a Varian INOVA-500 (1H at 500 MHz, 13C at 125 MHz) spectrometers, and chemical shifts are reported in units of δ relative to internal TMS. Multiplicities are indicated as br (broadened), s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet), and coupling constants (J) are reported in hertz (Hz). Low and high-resolution mass spectra were performed in School of Chemical Sciences, University of Illinois at Urbana–Champaign. Mass spectral data are reported in the form of m/z (intensity relative to base = 100). Elemental analyses were obtained in Baron Consulting Co, Milford, CT, and were within ±0.4% of the theoretical values (see Supporting Information). Purities of the tested compounds were determined by elemental analysis or by HPLC (using Waters Alliance HPLC system, 4.6 mm × 250 mm, Supelco Discovery column, acetonitrile/water with 8.5% o-phosphoric acid) or by LC/MS analysis using a Waters MicroMass ZQ system (electrospray ionization (ESI) with Waters-2525 binary gradient module coupled to a photodiode array detector (Waters-2996) and ELS detector (Waters-2424) using a XTerra MS C18 (5 μm, 4.6 mm × 50 mm column and acetonitrile/water) and were >95%.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]acetic Acid 4-Bromo-butyl Ester (6a)
A stirred mixture of 5a (175 mg, 0.58 mmol), dibromobutane (313 mg, 1.45 mmol), and sodium bicarbonate (73 mg, 0.87 mmol) in DMF (2 mL) was heated at 165 °C for 12 min using microwave irradiation. The reaction mixture was cooled to room temperature and diluted with water and ethyl acetate. The organic layer was separated, and the aqueous phase was extracted with ethyl acetate. The combined organic layer was washed with brine, dried (MgSO4), and concentrated under reduced pressure. Purification by flash column chromatography on silica gel gave 6a (159 mg, 63% yield) as a light-yellow gum. IR (CHCl3): 3407, 2967, 2928, 1712 (s, >C=O), 1621, 1583, 1431, 1254 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.33 (d, J = 1.0 Hz, 1H, 4-H), 6.24 (d, J = 1.0 Hz, 1H, 2-H), 5.42 (m as d, J = 4.0 Hz, 1H, 8-H), 5.23 (s, 1H, OH), 4.12 (t, J = 6.5 Hz, 2H, −OCH2−), 3.45 (s, 2H, −CH2–C(O)O−), 3.39 (t, J = 6.5 Hz, 2H, −CH2Br), 3.19 (dd, J = 15.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.69 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.19–2.10 (m, 1H, 7α-H), 1.94–1.76 (m, 7H, 10β-H, 7β-H, 6a-H, −CH2-CH2-CH2-Br, −CH2-CH2-CH2-Br), 1.69 (s, 3H, 9-CH3), 1.37 (s, 3H, 6β-CH3), 1.09 (s, 3H, 6α-CH3). Mass spectrum (EI) m/z (relative intensity) 438 (M++ 2, 97), 436 (M+, 97), 395 (38), 393 (38), 355 (95), 353 (95), 330 (60), 316 (38), 287 (46), 273 (48), 257 (45), 247 (98), 233 (60), 213 (100).Exact mass (EI) calculated for C22H29O4Br (M+), 436.1249; found, 436.1252. HPLC (4.6 mm × 250 mm, Supelco discovery column, acetonitrile/water) showed purity 97.5% and retention time 12.6 min for the title compound. LC/MS analysis (Waters MicroMass ZQ system) showed purity 97% and retention time 7.3 min for the title compound.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic Acid 4-Bromo-butyl Ester (6b)
The synthesis was carried out as described for 6a using 5b (200 mg, 0.61 mmol), dibromobutane (330 mg, 1.53 mmol), and sodium bicarbonate (77 mg, 0.92 mmol) in DMF and gave 6b (148 mg, 53% yield) as a light-yellow gum. IR (neat): 3403, 2970, 2917, 1727, and 1703 (s, >C=O), 1620, 1577, 1416, 1256 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.41 (d, J = 2.0 Hz, 1H, 4-H), 6.25 (d, J = 2.0 Hz, 1H, 2-H), 5.42 (m as d, J = 5.0 Hz, 1H, 8-H), 5.18 (s, 1H, OH), 4.09 (t, J = 6.5 Hz, 2H, −OCH2−), 3.32 (t, J = 6.5 Hz, 2H, −CH2Br), 3.19 (dd, J = 15.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.69 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.18–2.10 (m, 1H, 7α-H), 1.85–1.68 (m and s overlapping, 10H, 10β-H, 7β-H, 6a-H, −CH2–CH2– of the side chain and 9-CH3, especially 1.70 s, 9-CH3), 1.51 (s, 6H, −C(CH3)2−), 1.38 (s, 3H, 6β-CH3), 1.10 (s, 3H, 6α-CH3). 13C NMR (125 MHz, CDCl3) δ 177.8 (>C=O), 155.7 (C-1 or C-5), 155.1 (C-5 or C-1), 144.0, 134.9, 119.4, 112.0, 107.1, 105.3, 77.0 (C-6), 64.3 (−OCH2−), 46.3, 45.0, 35.9, 33.3, 31.7, 29.3, 28.0, 27.7, 27.2, 26.2, 26.2, 23.8, 18.7. Mass spectrum (ESI) m/z (relative intensity) 467 (M++ 2 + H, 100), 465 (M+ + H, 100), 285 (30). Exact mass (ESI) calculated for C24H34O4Br (M+ + H), 465.1640; found, 465.1647. LC/MS analysis (Waters MicroMass ZQ system) showed purity 98% and retention time 7.4 min for the title compound. Anal. (C24H33BrO4) C, H.
1-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-cyclobutanecarboxylic Acid 4-Bromo-butyl Ester (6c)
The synthesis was carried out as described for 6a using 5c (145.0 mg, 0.42 mmol), dibromobutane (136 mg, 0.63 mmol), and sodium bicarbonate (39 mg, 0.46 mmol) in DMF and gave 6c (90 mg, 45% yield) as a light-yellow gum. IR (neat): 3407, 2968, 2915, 1728, and 1705 (s, >C=O), 1622, 1416, 1258 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.39 (d, J = 2.0 Hz, 1H, 4-H), 6.24 (d, J = 2.0 Hz, 1H, 2-H), 5.85 (s, 1H, −OH), 5.42 (m as d, J = 4.5 Hz, 1H, 8-H), 4.10 (m, 2H, −OCH2−), 3.31 (t, J = 6.5 Hz, 2H, −CH2Br), 3.24 (dd, J = 16.0 Hz, J = 4.0 Hz, 1H, 10α-H), 2.79–2.66 (m, 3H, 10a-H, 2H of the cyclobutane ring, overlapping), 2.46 (m as qt, J = 9.0 Hz, 2H of the cyclobutane ring), 2.19–2.10 (m, 1H, 7α-H), 1.99–1.90 (m, 1H of the cyclobutane ring), 1.89–1.64 (m and s overlapping, 11H, 10β-H, 7β-H, 6a-H, 1H of the cyclobutane ring, −CH2CH2CH2Br, 9-CH3, especially 1.69 s, 9-CH3), 1.38 (s, 3H, 6β-CH3), 1.09 (s, 3H, 6α-CH3). Mass spectrum (ESI) m/z (relative intensity) 501 (M+ + 2 + Na, 33), 499 (M+ + Na, 33), 479 (M+ + 2 + H, 100), 477 (M+ + H, 100), 297 (30). Exact mass (ESI) calculated for C25H34O4Br (M+ + H), 477.1640; found, 477.1636. LC/MS analysis (Waters MicroMass ZQ system) showed purity 97% and retention time 5.6 min for the title compound.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]acetic Acid 4-Cyano-butyl Ester (7a)
A solution of bromide 6a (80 mg, 0.18 mmol) and sodium cyanide (88 mg, 1.8 mmol) in anhydrous DMSO (5 mL) was stirred at 50 °C for 3 h under argon. The reaction mixture was cooled to room temperature and diluted with water and ethyl acetate. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layer was washed with brine, dried (MgSO4), and evaporated under reduced pressure. Purification by flash column chromatography on silica gel (20% ethyl acetate in hexane) afforded 7a (50 mg, 71% yield) as a light-yellow gum. IR (neat): 3405, 2926, 2249 (CN), 1732 (s, >C=O), 1621, 1583, 1431, 1262, 1182 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.31 (d, J = 2.0 Hz, 1H, 4-H), 6.27 (d, J = 2.0 Hz, 1H, 2-H), 5.72 (s, 1H, OH), 5.42 (m as d, J = 4.0 Hz, 1H, 8-H), 4.14 (t, J = 6.0 Hz, 2H, −OCH2−), 3.46 (s, 2H, −CH2–C(O)O−), 3.22 (dd, J = 15.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.69 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.35 (t, J = 7.0 Hz, 2H, −CH2CN), 2.17–2.09 (m, 1H, 7α-H), 1.85–1.70 (m, 7H, 10β-H, 7β-H, 6a-H, −CH2–CH2– of the side chain), 1.68 (s, 3H, 9-CH3), 1.37 (s, 3H, 6β-CH3), 1.09 (s, 3H, 6α-CH3). Mass spectrum (EI) m/z (relative intensity) 383 (M+, 32), 368 (10), 340 (12), 315 (8), 300 (40), 279 (13), 213 (38), 205 (29), 97 (38), 83 (41), 69 (71), 57 (100). Exact mass (EI) calculated for C23H29O4N (M+), 383.2097; found, 383.2092. LC/MS analysis (Waters MicroMass ZQ system) showed purity 96.5% and retention time 6.8 min for the title compound.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic Acid 4-Cyano-butyl Ester (7b)
The synthesis was carried out as described for 7a using 6b (20 mg, 0.04 mmol) and sodium cyanide (21 mg, 0.43 mmol) in anhydrous DMSO and gave 7b (9.5 mg, 54% yield) as light-yellow gum. IR (CHCl3): 3405, 2973, 2249 (CN), 1727 and 1705 (s, >C=O), 1620, 1578, 1417, 1258, 1184 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.41 (d, J = 2.0 Hz, 1H, 4-H), 6.26 (d, J = 2.0 Hz, 1H, 2-H), 5.42 (m as d, J = 5.0 Hz, 1H, 8-H), 5.26 (s, 1H, OH), 4.11 (t, J = 6.5 Hz, 2H, −OCH2−), 3.20 (dd, J = 15.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.69 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.28 (t, J = 6.5 Hz, 2H, −CH2CN), 2.18–2.10 (m, 1H, 7α-H), 1.87–1.72 (m, 5H, 10β-H, 7β-H, 6a-H, −CH2– of the side chain), 1.70 (s, 3H, 9-CH3), 1.68–1.58 (m, 2H, −CH2– of the side chain), 1.51 (s, 3H, −C(CH3)2−), 1.50 (s, 3H, −C(CH3)2−), 1.38 (s, 3H, 6β-CH3), 1.10 (s, 3H, 6α-CH3). Mass spectrum (ESI) m/z (relative intensity) 434 (M+ + Na, 10), 412 (M+ + H, 100), 285 (5). Exact mass (ESI) calculated for C25H34NO4 (M+ + H), 412.2488; found, 412.2480. LC/MS analysis (Waters MicroMass ZQ system) showed purity 98% and retention time 6.8 min for the title compound.
1-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]- cyclobutanecarboxylic Acid 4-Cyano-butyl Ester (7c)
The synthesis was carried out as described for 7a using 6c (180 mg, 0.38 mmol) and sodium cyanide (186 mg, 3.8 mmol) in anhydrous DMSO and gave 7c (100 mg, 63% yield) as light-yellow gum. IR (CHCl3): 3400, 2970, 2250 (CN), 1723 and 1703 (s, >C=O), 1619, 1577, 1417, 1268, 1183 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.37 (d, J = 1.5 Hz, 1H, 4-H), 6.26 (d, J = 1.5 Hz, 1H, 2-H), 5.95 (s, 1H, −OH), 5.42 (m as d, J = 4.0 Hz, 1H, 8-H), 4.11 (t, J = 5.5 Hz, 2H, −OCH2−), 3.26 (dd, J = 16.5 Hz, J = 5.0 Hz, 1H, 10α-H), 2.79–2.67 (m, 3H, 10a-H, 2H of the cyclobutane ring, overlapping), 2.46 (m, 2H of the cyclobutane ring), 2.26 (t, J = 7.0 Hz, 2H, −CH2CN), 2.18–2.10 (m, 1H, 7α-H), 2.00–1.90 (m, 1H of the cyclobutane ring), 1.89–1.71 (m, 6H, 10β-H, 7β-H, 6a-H, 1H of the cyclobutane ring, −CH2– of the side chain), 1.69 (s, 3H, 9-CH3), 1.63–1.54 (m, 2H, −CH2– of the side chain), 1.38 (s, 3H, 6β-CH3), 1.09 (s, 3H, 6α-CH3). Mass spectrum (ESI) m/z (relative intensity) 446 (M+ + Na, 50), 424 (M+ + H, 100), 297 (5). Exact mass (ESI) calculated for C26H34NO4 (M+ + H), 424.2488; found, 424.2491. LC/MS analysis (Waters MicroMass ZQ system) showed purity 98% and retention time 4.9 min for the title compound. Anal. (C26H33NO4) C, H.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic Acid Methyl Ester (8a)
The synthesis was carried out as described for 6a using 5b (240 mg, 0.73 mmol), iodomethane (260 mg, 1.83 mmol), and sodium bicarbonate (92 mg, 1.09 mmol) in DMF and gave 8a (160 mg, 64% yield) as a light-yellow gum. IR (neat): 3412, 2962, 2932, 1728, and 1702 (s, >C=O), cm–1. 1H NMR (500 MHz, CDCl3) δ 6.41 (d, J = 2.0 Hz, 1H, 4-H), 6.26 (d, J = 2.0 Hz, 1H, 2-H), 5.75 (s, 1H, OH), 5.42 (m as d, J = 5.0 Hz, 1H, 8-H), 3.66 (s, 3H, −OCH3), 3.22 (dd, J = 15.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.69 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.19–2.10 (m, 1H, 7α-H), 1.86–1.74 (m, 3H, 10β-H, 7β-H, 6a-H), 1.69 (s, 3H, 9-CH3), 1.51 (s, 6H, −C(CH3)2−), 1.38 (s, 3H, 6β-CH3), 1.11 (s, 3H, 6α-CH3). 13C NMR (125 MHz, CDCl3) δ 177.8 (>C=O), 155.4 (C-1 or C-5), 155.1 (C-5 or C-1), 144.4, 135.0, 119.4, 111.9, 107.5, 105.4, 77.1 (C-6), 52.6 (−OCH3), 46.3, 44.9, 35.9, 31.7, 28.1, 27.8, 26.5, 26.4, 23.8, 18.8. Mass spectrum (EI) m/z (relative intensity) 344 (M+, 91), 329 (15), 301 (38), 285 (22), 276 (18), 261 (100), 241 (22), 223 (19). Exact mass (EI) calculated for C21H28O4 (M+), 344.1988; found, 344.1992. HPLC (4.6 mm × 250 mm, Supelco Discovery column, acetonitrile/water) showed purity 96.5% and retention time 12 min for the title compound. LC/MS analysis (Waters MicroMass ZQ system) showed purity 96.5% and retention time 6.6 min for the title compound.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic Acid Ethyl Ester (8b)
The synthesis was carried out as described for 6a using 5b (300 mg, 0.91 mmol), iodoethane (356 mg, 2.28 mmol), and sodium bicarbonate (115 mg, 1.37 mmol) in DMF and gave 8b (283 mg, 87% yield) as a light-yellow gum. IR (neat): 3410, 2959, 2931, 1728, and 1702 (s, >C=O), cm–1. 1H NMR (500 MHz, CDCl3) δ 6.41 (d, J = 1.0 Hz, 1H, 4-H), 6.28 (d, J = 1.0 Hz, 1H, 2-H), 5.74 (s, 1H, OH), 5.42 (m as d, J = 5.0 Hz, 1H, 8-H), 4.12 (q,, J = 7.0 Hz, 2H, −OCH2−), 3.22 (dd, J = 15.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.69 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.19–2.10 (m, 1H, 7α-H), 1.88–1.75 (m, 3H, 10β-H, 7β-H, 6a-H), 1.69 (s, 3H, 9-CH3), 1.50 (s, 6H, −C(CH3)2−), 1.38 (s, 3H, 6β-CH3), 1.19 (t, J = 7.0 Hz, 3H, −OCH2CH3), 1.10 (s, 3H, 6α-CH3). Mass spectrum (EI) m/z (relative intensity) 358 (M+, 92), 343 (17), 315 (39), 285 (48), 275 (100), 241 (35). Exact mass (EI) calculated for C22H30O4 (M+), 358.2144; found, 358.2145. LC/MS analysis (Waters MicroMass ZQ system) showed purity 97% and retention time 6.9 min for the title compound.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic Acid 2-Bromo-ethyl Ester (9a)
The synthesis was carried out as described for 6a using 5b (200 mg, 0.61 mmol), 1,2-dibromoethane (287 mg, 1.53 mmol), and sodium bicarbonate (77 mg, 0.92 mmol) in DMF and gave 9a (127 mg, 48% yield) as a light-yellow gum. IR (neat): 3405, 2972, 2918, 1725, and 1701 (s, >C=O), 1621, 1577, 1415, 1262 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.42 (d, J = 1.5 Hz, 1H, 4-H), 6.27 (d, J = 1.5 Hz, 1H, 2-H), 5.42 (m as d, J = 4.0 Hz, 1H, 8-H), 5.04 (br s, 1H, OH), 4.36 (m as td, J = 5.5 Hz, J = 2.0 Hz, 2H, −OCH2−), 3.46 (t, J = 6.0 Hz, 2H, −CH2Br), 3.19 (dd, J = 15.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.69 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.18–2.10 (m, 1H, 7α-H), 1.89–1.72 (m, 3H, 10β-H, 7β-H, 6a-H), 1.70 (s, 3H, 9-CH3), 1.53 (s, 6H, −C(CH3)2−), 1.38 (s, 3H, 6β-CH3), 1.10 (s, 3H, 6α-CH3). Mass spectrum (EI) m/z (relative intensity) 438 (M+ + 2, 68), 436 (M+, 68), 395 (22), 393 (22), 355 (67), 353 (67), 285 (44), 241 (45), 59 (100). Exact mass (EI) calculated for C22H29O4Br (M+), 436.1249; found, 436.1250. LC/MS analysis (Waters MicroMass ZQ system) showed purity 98% and retention time 8.5 min for the title compound.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic Acid 3-Bromo-propyl Ester (9b)
The synthesis was carried out as described for 6a using 5b (405 mg, 1.23 mmol), 1,3-dibromopropane (622 mg, 3.08 mmol), and sodium bicarbonate (155 mg, 1.85 mmol) in DMF and gave 9b (282 mg, 51% yield) as a light-yellow gum. IR (neat): 3401, 2973, 2917, 1727, and 1703 (s, >C=O), 1621, 1576, 1416, 1260 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.41 (d, J = 1.5 Hz, 1H, 4-H), 6.27 (d, J = 1.5 Hz, 1H, 2-H), 5.89 (s, 1H, OH), 5.42 (m as d, J = 5.0 Hz, 1H, 8-H), 4.19 (m as td, J = 6.5 Hz, J = 1.5 Hz, 2H, −OCH2−), 3.30–3.18 (t and dd overlapping, 3H, −CH2Br, 10α-H, especially 3.24, t, J = 6.5 Hz, 2H, −CH2Br), 2.69 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.18–2.10 (m, 1H, 7α-H), 2.09 (qt, J = 6.5 Hz, 2H, −CH2CH2Br), 1.87–1.74 (m, 3H, 10β-H, 7β-H, 6a-H), 1.69 (s, 3H, 9-CH3), 1.51 (s, 6H, −C(CH3)2−), 1.39 (s, 3H, 6β-CH3), 1.11 (s, 3H, 6α-CH3). Mass spectrum (ESI) m/z (relative intensity) 453 (M+ + 2 + H, 55), 450 (M+ + H, 55), 285 (100). LC/MS analysis (Waters MicroMass ZQ system) showed purity 96% and retention time 7.6 min for the title compound. Anal. (C23H31BrO4) C, H.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic Acid 5-Bromo-pentyl Ester (9c)
The synthesis was carried out as described for 6a using 5b (350 mg, 1.06 mmol), 1,5-dibromopentane (609 mg, 2.65 mmol), and sodium bicarbonate (134 mg, 1.59 mmol) in DMF and gave 9c (239 mg, 47% yield) as a light-yellow gum. IR (CHCl3): 3408, 2973, 2931, 1728, and 1702 (s, >C=O), 1621, 1578, 1417, 1261 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.42 (d, J = 2.0 Hz, 1H, 4-H), 6.25 (d, J = 2.0 Hz, 1H, 2-H), 5.42 (m as d, J = 5.0 Hz, 1H, 8-H), 5.07 (s, 1H, OH), 4.03 (t, J = 6.5 Hz, 2H, −OCH2−), 3.29 (t, J = 6.5 Hz, 2H, −CH2Br), 3.19 (dd, J = 15.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.69 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.18–2.10 (m, 1H, 7α-H), 1.84–1.67 (m and s overlapping, 10H, 10β-H, 7β-H, 6a-H, −CH2-CH2– of the side chain and 9-CH3, especially 1.70 s, 9-CH3), 1.51 (s, 6H, −C(CH3)2−), 1.38 (s, 3H, 6β-CH3), 1.28–1.23 (m, 2H, −CH2– of the side chain), 1.10 (s, 3H, 6α-CH3). Mass spectrum (EI) m/z (relative intensity) 480 (M+ + 2, 2), 478 (M+, 2), 398 (M+ – Br, 82), 355 (19), 330 (21), 315 (100), 285 (60), 241 (39). Exact mass (EI) calculated for C25H35O4Br (M+), 478.1719; found, 478.1715. LC/MS analysis (Waters MicroMass ZQ system) showed purity 96% and retention time 7.5 min for the title compound.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic Acid 4-(1H-Imidazol-1-yl)butyl Ester (9d)
The synthesis was carried out as described for 6a using 5b (900 mg, 2.72 mmol), 4-(1H-imidazol-1-yl)butyl bromide (1.12 g, 5.44 mmol), and sodium bicarbonate (343 mg, 4.08 mmol) in DMF and gave 9d (579 mg, 47% yield) as a light-yellow solid, mp 73–75 °C. IR (CHCl3): 2971, 2932, 1727 (s, >C=O), 1618, 1578, 1512, 1417, 1251 cm–1. 1H NMR (500 MHz, CDCl3) δ 7.50 (s, 1H, imidazole), 7.10 (s, 1H, imidazole), 6.86 (s, 1H, imidazole), 6.35 (d, J = 2.0 Hz, 1H, 4-H), 6.23 (d, J = 2.0 Hz, 1H, 2-H), 5.41 (m as d, J = 5.0 Hz, 1H, 8-H), 3.95 (m as td, J = 6.5 Hz, J = 2.0 Hz, 2H, −OCH2−), 3.88 (m as td, J = 7.5 Hz, J = 3.0 Hz, 2H, −CH2–N< ), 3.37 (dd, J = 15.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.72 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.18–2.09 (m, 1H, 7α-H), 1.84–1.68 (m and s overlapping, 10H, 10β-H, 7β-H, 6a-H, −CH2-CH2– of the side chain and 9-CH3, especially 1.70 s, 9-CH3), 1.47 (s, 3H, −C(CH3)2−), 1.46 (s, 3H, −C(CH3)2−), 1.38 (s, 3H, 6β-CH3), 1.09 (s, 3H, 6α-CH3). Mass spectrum (EI) m/z (relative intensity) 452 (M+, 100), 437 (20), 402 (48), 367 (56), 318 (23), 303 (38), 265 (31), 91 (18). Exact mass (EI) calculated for C27H36N2O4 (M+), 452.2675; found, 452.2666. Anal. (C27H36N2O4) C, H, N.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic Acid 3-Cyano-propyl Ester (10a)
The synthesis was carried out as described for 7a using 9b (110 mg, 0.24 mmol) and sodium cyanide (118 mg, 2.4 mmol) in anhydrous DMSO and gave 10a (95 mg, 98% yield) as light-yellow gum. IR (CHCl3): 3403, 2971, 2248 (CN), 1727 and 1704 (s, >C=O), 1621, 1576, 1421, 1261, 1185 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.40 (d, J = 2.0 Hz, 1H, 4-H), 6.29 (d, J = 2.0 Hz, 1H, 2-H), 5.82 (s, 1H, OH), 5.42 (m as d, J = 4.0 Hz, 1H, 8-H), 4.17 (m, AB system, 2H, −OCH2−), 3.22 (dd, J = 15.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.70 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.21 (t, J = 7.5 Hz, 2H, −CH2CN), 2.18–2.10 (m, 1H, 7α-H), 1.93 (m as qt, J = 6.0 Hz, 2H, −CH2– of the side chain), 1.87–1.75 (m, 3H, 10β-H, 7β-H, 6a-H), 1.70 (s, 3H, 9-CH3), 1.52 (s, 6H, −C(CH3)2−), 1.38 (s, 3H, 6β-CH3), 1.10 (s, 3H, 6α-CH3). Mass spectrum (EI) m/z (relative intensity) 397 (M+, 79), 382 (12), 354 (9), 329 (13), 314 (100), 285 (27), 276 (21), 241 (78), 149 (21), 70 (41). Exact mass (EI) calculated for C24H31NO4 (M+), 397.2253; found, 397.2254. LC/MS analysis (Waters MicroMass ZQ system) showed purity 98% and retention time 11.4 min for the title compound. Anal. (C24H31NO4) C, H, N.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methyl-propanoic Acid 3-(1H-Imidazol-1-yl)propyl Ester (10b)
To a stirred suspension of 9b (196.0 mg, 0.43 mmol) and potassium carbonate (594 mg, 4.30 mmol) in DMSO (5 mL) was added imidazole (146 mg, 2.15 mmol) at room temperature under an argon atmosphere. Stirring was continued for 14 h, and then the mixture was diluted with water and ethyl acetate. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layer was washed with water and brine, dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography on silica gel (50% acetone in hexane) gave 10b (78 mg, 41% yield) as light-yellow gum. IR (CHCl3): 2971, 2932, 1727 (s, >C=O), 1618, 1578, 1512, 1417, 1251 cm–1. 1H NMR (500 MHz, CDCl3) δ 7.33 (s, 1H, imidazole), 7.04 (s, 1H, imidazole), 6.44 (s, 1H, imidazole), 6.40 (d, J = 2.0 Hz, 1H, 4-H), 6.32 (d, J = 2.0 Hz, 1H, 2-H), 5.40 (m as d, J = 5.0 Hz, 1H, 8-H), 3.93 (td, J = 17.0 Hz, J = 5.5 Hz, 1H, −OCH2−), 3.85 (td, J = 17.0 Hz, J = 5.5 Hz, 1H, −OCH2−), 3.72 (t, J = 7.0 Hz, 2H, −CH2–N< ), 3.41 (dd, J = 16.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.73 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.17–2.09 (m, 1H, 7α-H), 2.01–1.93 (m, 2H, −OCH2-CH2−), 1.85–1.71 (m, 3H, 10β-H, 7β-H, 6a-H), 1.60 (s, 3H, 9-CH3), 1.50 (s, 3H, −C(CH3)2−), 1.49 (s, 3H, −C(CH3)2−), 1.38 (s, 3H, 6β-CH3), 1.08 (s, 3H, 6α-CH3). 13C NMR (100 MHz, CDCl3) δ 176.8 (>C=O), 157.4 (C-1 or C-5), 155.1 (C-5 or C-1), 143.8, 137.5 (imidazole), 135.4, 128.7 (imidazole), 119.4, 119.2, 112.6, 106.0 (C-2 or C-4), 105.7 (C-4 or C-2), 77.0 (C-6), 60.9 (−OCH2−), 46.2, 45.2, 43.8, 36.0, 31.9, 29.8, 28.1, 27.8, 26.1, 25.8, 23.7, 18.7. Mass spectrum (EI) m/z (relative intensity) 438 (M+, 62), 423 (12), 395 (80, 355 (7), 317 (6), 304 (10), 285 (7), 201 (6), 149 (10, 88 (18), 56 (100). Exact mass (EI) calculated for C26H34N2O4 (M+), 438.2519; found, 438.2518. HPLC (4.6 mm × 250 mm, Supelco discovery column, acetonitrile/water) showed purity 98% and retention time 7 min for the title compound. LC/MS analysis (Waters MicroMass ZQ system) showed purity 98% and retention time 5.5 min for the title compound.
2-(3,5-Dimethoxyphenyl)-2-methylpropanal34 (12)
To a solution of 11 (3.0 g, 14.6 mmol) in dry CH2Cl2 (50 mL) at −78 °C under an argon atmosphere was added dropwise DIBAL-H (37 mL, 37 mmol, 1 M solution in CH2Cl2). The reaction mixture was stirred at the same temperature for 1 h and then quenched by dropwise addition of potassium sodium tartrate (10% solution in water). The resulting mixture was warmed to room temperature, stirred vigorously for 40 min, and then diluted with ethyl acetate. The organic phase was separated, and the aqueous phase was extracted with ethyl acetate. The combined organic layer was washed with brine, dried (MgSO4), and concentrated under reduced pressure. Purification by flash column chromatography on silica gel (20% ethyl acetate in hexane) gave 12 (2.8 g, 94%) as a colorless oil. IR (neat): 2970, 2937, 1725 (s, >C=O), 1592, 1456, 1423, 1204, 1159, 1048 cm–1. 1H NMR (500 MHz, CDCl3) δ 9.46 (s, −CH=O), 6.40 (d, J = 2.0 Hz, 2H, ArH), 6.39 (t, J = 2.0 Hz, 1H, ArH), 3.79 (s, 6H, −OCH3), 1.43 (s, 6H, −C(CH3)2−). Mass spectrum (EI) m/z (relative intensity) 208 (M+, 25), 196 (16), 179 (M+ – CHO), 165 (25), 151 (14), 139 (39), 91 (20), 77 (20). Exact mass (EI) calculated for C12H16O3 (M+), 208.1099; found, 208.1108.
2-(3,5-Dimethoxyphenyl)-2-methylpropan-1-ol (13)
To a solution of 12 (1.70 g, 8.16 mmol) in anhydrous methanol (40 mL) at room temperature under an argon atmosphere was added sodium borohydride (1.39 g, 36.7 mmol) portionwise. The reaction mixture was stirred for 1 h and then quenched by the addition of aqueous 1 N HCl and extracted with ethyl acetate. The organic layer was washed with brine, dried (MgSO4), and evaporated. Purification by flash column chromatograph on silica gel (20% ethyl acetate in hexane) gave 13 (1.6 g, 94%) as a colorless oil. IR (neat): 3433 (br, OH), 2961, 2836, 1592, 1456, 1422, 1202, 1159, 1048 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.53 (d, J = 2.0 Hz, 2H, ArH), 6.34 (t, J = 2.0 Hz, 1H, ArH), 3.80 (s, 6H, −OCH3), 3.58 (s, 2H, −CH2OH), 1.30 (s, 6H, −C(CH3)2−). Mass spectrum (ESI) m/z (relative intensity) 211 (M+ + H, 100).
5-(1-Hydroxy-2-methylpropan-2-yl)resorcinol (14)
To a stirred solution of 13 (400 mg, 1.9 mmol) in dry CH2Cl2 (20 mL) at −78 °C, under an argon atmosphere, was added boron tribromide (6.6 mL, 6.6 mmol, 1 M solution in CH2Cl2). Following this addition, the reaction temperature was gradually raised over a period of 3 h to 25 °C, and the stirring was continued at that temperature until the reaction was completed (4 h). Unreacted boron tribromide was destroyed by the addition of methanol and ice at 0 °C. The resulting mixture was warmed to room temperature, and volatiles were removed in vacuo. The residue was dissolved in diethyl ether and washed with water and brine and dried (MgSO4). Solvent evaporation and purification by flash column chromatography on silica gel (40% diethyl ether in hexane) gave 14 (165 mg, 47%) as light-yellow oil. IR (neat): 3443 (br, OH), 2959, 1591, 1458, 1420, 1201, 1048 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.43 (d, J = 2.0 Hz, 2H, ArH), 6.22 (t, J = 2.0 Hz, 1H, ArH), 4.84 (s, 2H, ArOH), 3.56 (d, J = 6.0 Hz, 2H, −CH2OH), 1.28 (s, 6H, −C(CH3)2−). Mass spectrum (ESI) m/z (relative intensity) 183 (M+ + H, 35), 165 (100).
(6aR,10aR)-3-(1-Hydroxy-2-methylpropan-2-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-1-ol (15)
To a stirred solution of 14 (165 mg, 0.9 mmol) and (+)-cis/trans-p-mentha-2,8-dien-1-ol (166 mg, 1.09 mmol) in anhydrous CHCl3 (3 mL), under an argon atmosphere, was added p-toluenesulfonic acid (52 mg, 0.3 mmol). The reaction mixture was heated at 150 °C for 10 min using microwave irradiation, and then it was cooled to room temperature and diluted with water and CHCl3. The organic layer was separated, and the aqueous phase was extracted with CHCl3. The combined organic layer was washed with water and brine and dried (MgSO4). Solvent evaporation and purification by flash column chromatography on silica gel (15% ethyl acetate in hexane) gave 15 (80 mg, 28%) as an orange solid, mp 91–93 °C. IR (CHCl3): 3341 (br, OH), 2968, 1621, 1575, 1415, 1329, 1185 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.42 (d, J = 1.5 Hz, 1H, 4-H), 6.28 (d, J = 1.5 Hz, 1H, 2-H), 5.43 (m as d, J = 4.5 Hz, 1H, 8-H), 3.54 (s, 2H, −CH2OH), 3.22 (dd, J = 15.5 Hz, J = 4.5 Hz, 1H, 10α-H), 2.70 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.19–2.11 (m, 1H, 7α-H), 1.90–1.71 (m, 3H, 10β-H, 7β-H, 6a-H), 1.70 (s, 3H, 9-CH3), 1.38 (s, 3H, 6β-CH3), 1.24 (s, 6H, −C(CH3)2−), 1.08 (s, 3H, 6α-CH3). Mass spectrum (ESI) m/z (relative intensity) 317 (M+ + H, 100). LC/MS analysis (Waters MicroMass ZQ system) showed purity 97% and retention time 5.7 min for the title compound.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]- 2-methyl propylpentanoate (16)
To a stirred solution of 15 (180 mg, 0.57 mmol), valeric acid (1.0 mL, 0.8 mmol) and triphenylphosphine (223 mg, 0.85 mmol) in anhydrous THF (10 mL) at 0 °C, under an argon atmosphere, was added diethyl azodicarboxylate (0.15 mL, 0.8 mmol) dropwise. The reaction temperature was raised to 25 °C, and stirring was continued for 20 h. The reaction mixture was quenched with aqueous 1 N HCl solution and extracted with ethyl acetate. The organic phase was washed with water and brine and dried (MgSO4). Solvent evaporation and purification by flash column chromatography on silica gel (25% ethyl acetate in hexane) gave 16 (93 mg, 41%) as light-yellow gum. IR (CHCl3): 3389 (br, OH), 2968, 2891, 1710 (>C=O), 1620, 1578, 1417, 1332, 1186 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.42 (d, J = 2.0 Hz, 1H, 4-H), 6.26 (d, J = 2.0 Hz, 1H, 2-H), 5.43 (m as d, J = 5.0 Hz, 1H, 8-H), 4.94 (s, 1H, OH), 4.05 (s, 2H, −CH2O−), 3.19 (dd, J = 15.5 Hz, J = 5.5 Hz, 1H, 10α-H), 2.69 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.28 (t, J = 7.0 Hz, 2H, −CH2-C(O)−), 2.19–2.11 (m, 1H, 7α-H), 1.89–1.74 (m, 3H, 10β-H, 7β-H, 6a-H), 1.70 (s, 3H, 9-CH3), 1.55 (qt, J = 7.0 Hz, 2H, −CH2– of the side chain), 1.38 (s, 3H, 6β-CH3), 1.32–1.24 (m and s overlapping, 8H, −C(CH3)2– and −CH2– of the side chain, especially 1.28, s, −C(CH3)2−), 1.10 (s, 3H, 6α-CH3), 0.88 (t, J = 7.0 Hz, 3H, −CH2–CH3). Mass spectrum (EI) m/z (relative intensity) 400 (M+, 100), 385 (9), 357 (11), 327 (35), 317 (27), 285 (26), 255 (17), 217 (9). Exact mass (EI) calculated for C25H36O4 (M+), 400.2614; found, 400.2611. Anal. (C25H36O4) C, H.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]- 2-methyl-N-pentylpropanamide (18)
Carboxylic acid intermediate 5b (200 mg, 0.61 mmol) was dissolved in dry CH2Cl2 (3 mL) under an argon atmosphere and cooled to 0 °C. To the cold solution, triethylamine (0.12 mL, 0.92 mmol) was added followed by the dropwise addition of bis(2-methoxyethyl)aminosulfur trifluoride (0.13 mL, 0.72 mmol). After stirring for 15 min, a solution of amylamine (80 mg, 0.92 mmol) in dry CH2Cl2 (6 mL) was added. The resulting mixture was stirred at 0 °C for 15 min and at 25 °C for 2 h and then diluted with CH2Cl2 and saturated aqueous sodium bicarbonate solution. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layer was washed with water and brine, dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography on silica gel (12% ethyl acetate in hexane) afforded 18 (170 mg, 70% yield) as light-yellow gum. IR (neat): 3274, 2930, 1643, and 1618 (>C=O), 1577, 1518, 1415, 1264, 1183, 1083 cm–1. 1H NMR (500 MHz, CDCl3) δ 8.53 (s, 1H, >NH), 6.40 (d, J = 1.5 Hz, 1H, 4-H), 6.25 (d, J = 1.5 Hz, 1H, 2-H), 5.46–5.35 (m as d, J = 5.0 Hz, and br s overlapping, 2H, 8-H, OH), 3.41 (dd, J = 16.0 Hz, J = 4.5 Hz, 1H, 10α-H), 3.12 (m as q, J = 7.0 Hz, 2H, −CH2-N< ), 2.73 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.20–2.15 (m, 1H, 7α-H), 1.88–1.76 (m, 3H, 10β-H, 7β-H, 6a-H), 1.70 (s, 3H, 9-CH3), 1.49 (s, 6H, −C(CH3)2−), 1.40 (s, 3H, 6β-CH3),1.36 (qt, J = 7.0 Hz, 2H, −CH2– of the side chain), 1.18 (qt, J = 7.0 Hz, 2H, −CH2– of the side chain), 1.16–1.08 (m and s overlapping, 5H, 6α-CH3, −CH2– of the side chain, especially 1.14, s, 6α-CH3), 0.81 (t, J = 7.0 Hz, 3H, −CH2–CH3). Mass spectrum (EI) m/z (relative intensity) 399 (M+, 25), 286 (100), 220 (21), 205 (82), 164 (15), 149 (18), 88 (23). Exact mass (EI) calculated for C25H37NO3 (M+), 399.2773; found, 399.2777. LC/MS analysis (Waters MicroMass ZQ system) showed purity 96% and retention time 5.7 min for the title compound.
2-[(6aR,10aR)-6a,7,10,10a-Tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]- 2-methyl-propanoyl Chloride (19)
To a stirred solution of acid 5b (90 mg, 0.27 mmol) in dry CH2Cl2 (3.4 mL), at room temperature under an argon atmosphere, was added the SOCl2–BTA reagent (0.23 mL (0.35 mmol) of a 1.5 M solution in CH2Cl2, which was prepared by dissolving 0.54 mL (7.5 mmol) of SOCl2 and 0.893 g (7.5 mmol) of BTA in 5 mL CH2Cl2). Stirring was continued for 20 min, and insoluble materials were filtered off. The filtrate was washed with aqueous 1 N HCl, water, and brine, and then dried (MgSO4). Solvent evaporation under reduced pressure afforded the title compound (95 mg) which was used in the next step without further purification.
S-Propyl-2-[(6aR,10aR)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6,9-trimethyl-6H-dibenzo[b,d]pyran-3-yl]-2-methylpropanethioate (20)
To a solution of 19 (95 mg, 0.27 mmol) and propanethiol (54 mg, 64 μL, 0.71 mmol) in dry CH2Cl2 (5.4 mL) at room temperature, under an argon atmosphere, was added anhydrous pyridine (0.3 mL). The reaction mixture was stirred for 3 h and then diluted with water and diethyl ether. The organic layer was separated, and the aqueous layer was extracted with diethyl ether. The combined organic layer was washed with water and brine, dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography on silica gel (5–15% diethyl ether in hexane) gave 20 (24 mg, 23% yield for the two steps) as a pale-yellow gum. IR (CHCl3): 3435, 2966, 2927, 1678, and 1652 (s, >C=O), 1620, 1579, 1417, 1264 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.45 (d, J = 2.0 Hz, 1H, 4-H), 6.24 (d, J = 2.0 Hz, 1H, 2-H), 5.42 (m as d, J = 5.0 Hz, 1H, 8-H), 5.02 (br s, 1H, OH), 3.19 (dd, J = 16.0 Hz, J = 4.5 Hz, 1H, 10α-H), 2.78 (m as t, J = 7.5 Hz, 1H, −SCH2−), 2.77 (m as t, J = 7.5 Hz, 1H, −SCH2−), 2.70 (td, J = 11.0 Hz, J = 4.5 Hz, 1H, 10a-H), 2.18–2.10 (m, 1H, 7α-H), 1.90–1.74 (m, 3H, 10β-H, 7β-H, 6a-H), 1.70 (s, 3H, 9-CH3), 1.58–1.50 (m and s overlapping, 8H, −C(CH3)2–, −CH2– of the side chain, especially 1.54, s, −C(CH3)2−), 1.38 (s, 3H, 6β-CH3), 1.09 (s, 3H, 6α-CH3), 0.92 (t, J = 7.5 Hz, 3H, −CH2-CH3). Mass spectrum (EI) m/z (relative intensity) 388 (M+, 19), 285 (100). Exact mass (EI) calculated for C23H32O3S (M+), 388.2072; found, 388.2075. LC/MS analysis (Waters MicroMass ZQ system) showed purity 98% and retention time 7.8 min for the title compound.
Radioligand Binding Assays
Rat brain CB1 receptor, mouse and human CB2 receptor binding assays: Compounds were tested for their affinities for the CB1 and CB2 receptors using membrane preparations from rat brain or HEK293 cells expressing either mCB2 or hCB2 receptors, respectively, and [3H]CP-55,940, as previously described.18,20,32 Results from the competition assays were analyzed using nonlinear regression to determine the IC5035 values for the ligand; Ki values were calculated from the IC50 (Prism by GraphPad Software, Inc.). Each experiment was performed in triplicate, and Ki values determined from three independent experiments and are expressed as the mean of the three values.
cAMP Assay16,18
HEK293 cells stably expressing rCB1 receptor were used for the studies. The cAMP assay was carried out using PerkinElmer’s Lance ultra cAMP kit following the protocol of the manufacturer. Briefly, the assays were carried out in 384-well plates using 1000–1500 cells/well. The cells were harvested with nonenzymatic cell dissociation reagent Versene and were washed once with HBSS and resuspended in the stimulation buffer. The various concentrations of the test compound (5 μL) in forskolin (2 μM final concentration) containing stimulation buffer were added to the plate followed by the cell suspension (5 μL). The cells were stimulated for 30 min at room temperature. Then Eu-cAMP tracer working solution (5 μL) and Ulight-anti-cAMP working solution (5 μL) were added to the plate and incubated at room temperature for 60 min. The data were collected on a PerkinElmer Envision instrument. The EC50 values were determined by nonlinear regression analysis using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA).
Plasma Stability16,36
Compounds or their proposed products were diluted (200 μM) in mouse plasma and incubated at 37 °C, 100 rpm. At various time points, samples were taken, diluted 1:4 in acetonitrile, and centrifuged to precipitate the proteins. The resulting supernatant was analyzed by HPLC. In vitro plasma half-lives were determined using exponential decay calculations in Prism (GraphPad).
HPLC Analysis
Chromatographic separation was achieved using a Supelco Discovery C18 (4.6 mm × 250 mm) column on a Waters Alliance HPLC system. Mobile phase consisted of acetonitrile (A) and a mixture of 60% water (acidified with 8.5% o-phosphoric acid) and 40% acetonitrile (B). Gradient elution started with 5% A, transitioning to 95% A over 10 min and holding for 5 min before returning to starting conditions; run time was 15 min, the flow rate was 1 mL/min, and UV detection was used at each compound’s maximal absorbance (204 and 230 nM).
Molecular Modeling
Ballesteros–Weinstein Nomenclature
The Ballesteros–Weinstein numbering system for GPCR amino acid residues is used here.37 In this numbering system, the label 0.50 is assigned to the most highly conserved class A residue in each transmembrane helix (TMH). This is preceded by the TMH number. In this system, for example, the most highly conserved residue in TMH6 is P6.50. The residue immediately before this would be labeled 6.49, and the residue immediately after this would be labeled 6.51. When referring to a specific CB1 residue, the Ballesteros–Weinstein name is followed by the absolute sequence number given in parentheses (e.g., K3.28(192)); however, when referring to a highly conserved residue among class A GPCRs (and not a specific residue in CB1), only the Ballesteros–Weinstein name is given.
Conformational Searches
The structures of the ligands (−)-Δ8-THC, (−)-Δ8-THC-dimethylheptyl, and 10a were built in Spartan’08 (Wave function, Inc., Irvine, CA). Initial conformational analyses of these compounds were performed in vacuo using the semiempirical method RM1 encoded in Spartan’08. Conformational searches were performed (using 3–8-fold rotations) for each rotatable bond. All unique conformers identified were then optimized with ab initio Hartree–Fock calculations at the 6-31G* level. To calculate the difference in energy between the global minimum energy conformer of each compound and its final docked conformation, rotatable bonds in the global minimum energy conformer were driven to their corresponding value in the final docked conformation and the single-point energy of the resultant structure was calculated at the HF 6-31G*. A duplicate conformational search of 10a was conducted at high dielectric using the MCMM (Monte Carlo multiple minimum) protocol in MacroModel with the OPLS_2005 force field in a GB/SA water model with an extended cutoff.38,39 An energy window of 21.0 kJ/mol (5 kcal/mol) was employed, and the redundant conformers of 10a were eliminated using a rmsd cutoff of 0.5 Å for all atoms.
CB1R Active State Model
The studies reported here used our previously published CB1 activated state model.40 Below, we describe briefly the construction of both the inactive and active states of this model. Because no crystal structure of the CB1 receptor has been published, the CB1 model uses the crystal structure of the class A GPCR rhodopsin in the dark state as its template.41 This template was chosen because no mutations or modifications were made to its structure for crystallization and because the cannabinoid receptors and rhodopsin share some unusual sequence motifs that have important spatial relevance. For example, these receptors share a TMH4 GWNC motif at their extracellular ends. Here a TRP forms an aromatic stacking interaction with Y5.39, influencing the extracellular (EC) positions of TMH3–4–5. Changes to the general Rho structure that were necessitated by sequence divergences included the absence of helix kinking proline residues in TMH1 and TMH5, the lack of a GG motif in TMH2 (at position 2.56 and 2.57), as well as the presence of extra flexibility in the TMH6 CWXP motif because of the presence of G6.49 immediately before P6.50.42 An activated state (R*) CB1 model was created by modification of our inactive state model. This R* model construction was guided by the biophysical literature on the R-to-R* transition in rhodopsin (Rho) and included a TMH6 conformer derived from our Conformational Memories study of CB1 TMH6 that is straightened, breaking the ionic lock interaction between R3.50 and D6.30.43 Extracellular (EC-1, H181–S185; EC-2, C257–E273; EC-3, D366–K376) and intracellular (IC-1, S146–R150; IC-2, P221–V228; IC-3, A301–P332) loops, as well as portions of the N (K90–N112) and C termini (S414–G427), were then added to the refined model of the CB1 R* bundle. The Modeler program was then used to refine loop structures.44,45 Specific conformations of the EC-2 and EC-3 loop were refined to reflect mutagenesis data showing that EC-2 loop residue F(268) should be available to the binding crevice and that EC-3 loop residue K(373) should interact in a salt bridge with TMH2 residue D2.63(176).46,47
Ligand/CB1R* Complexes
The automatic docking program, Glide v6.4 (Schrödinger, LLC, NY 2014) was used to explore possible binding conformations or receptor site interactions for subject ligands. Because K3.28(192) has been shown to be a critical residue for classical cannabinoid binding,48 K3.28(192) was defined as a required interaction during the docking procedure. Glide was used to generate a grid based on the centroid of the ligand in the binding site. Any hydrophobic region defined in the grid generation that contacted the ligand was selected as important to the docking procedure. The box for Glide docking was defined to be 22 Å in the x, y, and z dimensions. The lowest energy conformations (<4 kcal/mol above the global min) of each ligand were docked using Glide. Extra precision (XP) was selected with no scaling of VdW radii and rigid docking invoked. Glide scoring functions do not take into account the conformational cost, or internal strain energy, of each generated pose of a ligand inside the binding pocket of the receptor. To determine the best ligand/receptor complex to proceed with a postdocking minimization for the acquisition of pairwise interaction energies, the conformational cost for each pose was subtracted from the Glide score for all poses (Supporting Information, Tables S2–4). The pose with the final best score was then subjected to a post docking minimization in two stages using the OPLS2005 all atom force field in Macromodel 10.5 (Schrödinger, LLC, NY 2014). An 8.0 Å nonbonded cutoff (updated every 10 steps), a 20.0 Å electrostatic cutoff, and a 4.0 Å hydrogen bond cutoff were used in each stage of the calculation. The first stage consisted of Polak–Ribier conjugate gradient minimization using a distance-dependent dielectric function with a base constant of 2. No harmonic constraints were placed on the side chains, but 1000 kJ/mol fixed atom constraints were applied to hold all the backbone atoms in place. The termini and loops were not allowed to move during this part of the procedure. The minimization was continued until the bundle reached the 0.05 kJ/mol·A2 gradient. To relax the loops, a second stage Polak–Ribier conjugate gradient minimization of the loop regions was performed until the 0.05 kJ/mol·A2 gradient was reached. The loop and termini regions were left free, while the transmembrane regions and ligand were not allowed to move during this final stage. An 8.0 Å extended nonbonded cutoff (updated every 10 steps), 20.0 Å electrostatic cutoff, and 4.0 Å hydrogen bond cutoff were used in this calculation, and the generalized Born/surface area (GB/SA) continuum solvation model for water available in Macromodel was employed.
Assessment of Pairwise Interaction and Total Energies
Interaction energies between each bound ligand and residue in the CB1R* complex were calculated using Macromodel, as described previously.49 Specifically, after defining the atoms of the ligand as one group (group 1) and the atoms corresponding to a residue that lines the binding site in the final ligand-CB1R* complex as another group (group 2), Macromodel was used to output the pairwise interaction energy (Coulombic and van der Waals) for a given pair.
Methods for Characterization of in Vivo Effects18,50
Subjects
For hypothermia testing, female Sprague–Dawley rats (n = 6/group), weighing between 250 and 350 g (Charles River, Wilmington MA) were used. Rats were tested repeatedly with at least 5 days intervening between drug sessions. Experiments occurred at approximately the same time (10:00 a.m.–5:00 p.m.) during the light portion of the daily light/dark cycle. Outside of experimental sessions, rats were pair housed (2/cage) in a climate controlled vivarium with unrestricted access to food and water. For tail-flick withdrawal (analgesia) testing, male CD-1 mice (n = 6/group), weighing between 30 and 35 g (Charles River, Wilmington MA), were used. Mice were housed 4/cage in a climate controlled vivarium with unrestricted access to food and water and acclimated to these conditions for at least a week before any experimental manipulations occurred. Analgesia testing took place between 11:00 a.m. and 7:00 p.m. Mice were used once.
Procedures
Temperature was recorded using a thermistor probe (Model 401, Measurement Specialties, Inc., Dayton, OH) inserted to a depth of 6 cm and secured to the tail with micropore tape. Rats were minimally restrained and isolated in 38 × 50 × 10 cm3 plastic stalls. Temperature was read to the nearest 0.01 °C using a thermometer (model 4000A, Measurement Specialties, Inc.).
Two baseline temperature measures were recorded at 15 min intervals, and drugs were injected immediately after the second baseline was recorded. After injection, temperature was recorded every 30 min for 3 h and every hour thereafter for a total of 6 h. In some studies, temperature readings at later time points were obtained by inserting the probe 6 cm and holding it in place for at least 1 min before taking a reading. The change in temperature was determined for each rat by subtracting temperature readings from the average of the two baseline measures. Analgesia testing utilized a thermostatically controlled 2 L water bath commercially available from VWR International where the water temperature was set at 52 °C (±0.5 °C). The tail was immersed into the water at a depth of 2 cm and the withdrawal latency recorded by a commercially available stopwatch (Fisher Scientific), allowing measurements in seconds and 1/100 s. Cut-off was set at 10 s to minimize the risk of tissue damage. A test session consisted of five recordings, the first of which constituted the baseline recording. Injections occurred immediately after the baseline recording, and the remaining recordings took place 20, 60, 180, and 360 min post administration. Prior to this testing, the animals had been accustomed to the procedure for three consecutive “mock” sessions where the water was held at 38 °C, i.e., average body temperature of mice; no tail-flicks were elicited by this water temperature. The third “mock” session also included an ip injection of vehicle (10 mL/kg). The tail-flick withdrawal latencies are expressed as a percentage of maximum possible effect (%MPE), according to the formula: %MPE = [(test latency – baseline latency)/(10 – baseline latency)] × 100.
Drugs
For hypothermia testing, (−)-Δ8-THC-DMH and compounds 2b and 10a were initially dissolved in a solution of 20% ethanol, 20% alkamuls, and 60% saline and were further diluted with saline. Injections were administered sc in a volume of 1.0 mL/kg. For tail-flick withdrawal (analgesia) testing, 10a and 2b were initially dissolved in 2% dimethyl sulfoxide, 4% Tween-80, and 4% propylene glycol before saline was slowly added just prior to the 10 mL/kg ip administration. All suspensions were freshly prepared for analgesia testing.
Data Analysis
Time-effect functions for hypothermia testing were analyzed using two-way repeated measures ANOVA procedures followed by Bonferroni’s posthoc test. Hypothermia dose–effect functions for compounds (−)-Δ8-THC-DMH, 2b, and 10a were analyzed using one-way repeated measures ANOVA procedures followed by the Holm–Sidak multiple comparisont-test; p was set at ≤0.05, and statistical analyses were performed using the software package GraphPad Prism 5.03 (GraphPad Software, San Diego, CA). A linear mixed model repeated measures ANOVA (IBM software package SPSS, v.21) was applied to tail-flick latency data depicted in Figure 5.
Acknowledgments
This work was supported by grants from the National Institute on Drug Abuse to A.M., DA009158, DA007215, and DA09064.
Glossary
Abreviations Used
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- (−)-Δ9-THC
(−)-Δ9-tetrahydrocannabinol
- CNS
central nervous system
- PK/PD
pharmacokinetic/pharmacodynamic
- SC
side chain
- SAR
structure–activity relationship
- HEK293
human embryonic kidney cell line
- log P
octanol–water partition coefficient
- PSA
polar surface area
- NMR
nuclear magnetic resonance
- HPLC
high-performance liquid chromatography
Supporting Information Available
Elemental analysis results for compounds 6b, 7c, 9b, 9d, 10a, and 16, tables for molecular modeling, hypothermic effects of approximately equivalent doses of 10a, 2b, and Δ8-THC-DMH at different times after injection, and tail flick latencies in a hot water bath after administration of 10a. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ (For R.S.) Rishi Sharma Microconstants Inc., 10191 Caminito Volar, San Diego California 92126, United States; E-mail, sharmarishi2004@yahoo.co.in.
Author Contributions
# S.P.N. and R.S. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mechoulam R.; Hanus L. A historical overview of chemical research on cannabinoids. Chem. Phys. Lipids 2000, 108, 1–13. [DOI] [PubMed] [Google Scholar]
- Devane W. A.; Dysarz F. A. III; Johnson M. R.; Melvin L. S.; Howlett A. C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [PubMed] [Google Scholar]
- Munro S.; Thomas K. L.; Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [DOI] [PubMed] [Google Scholar]
- Pavlopoulos S.; Thakur G. A.; Nikas S. P.; Makriyannis A. Cannabinoid receptors as therapeutic targets. Curr. Pharm. Des. 2006, 12, 1751–1769. [DOI] [PubMed] [Google Scholar]
- Buchwald A.; Derendorf H.; Ji F.; Nagaraja N. Y.; Wu W. M.; Bodor N. Soft cannabinoid analogues as potential anti-glaucoma agents. Pharmazie 2002, 57, 108–114. [PubMed] [Google Scholar]
- Hwang J.; Adamson C.; Butler D.; Janero D. R.; Makriyannis A.; Bahr B. A. Enhancement of endocannabinoid signaling by fatty acid amide hydrolase inhibition: a neuroprotective therapeutic modality. Life Sci. 2010, 86, 615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvinen T.; Pate D. W.; Laine K. Cannabinoids in the treatment of glaucoma. Pharmacol. Ther. 2002, 95, 203–220. [DOI] [PubMed] [Google Scholar]
- Karst M.; Wippermann S.; Ahrens J. Role of cannabinoids in the treatment of pain and (painful) spasticity. Drugs 2010, 70, 2409–2438. [DOI] [PubMed] [Google Scholar]
- Lu D.; Vemuri V. K.; Duclos R. I. Jr.; Makriyannis A. The cannabinergic system as a target for anti-inflammatory therapies. Curr. Top. Med. Chem. 2006, 6, 1401–1426. [DOI] [PubMed] [Google Scholar]
- Marco E. M.; Romero-Zerbo S. Y.; Viveros M. P.; Bermudez-Silva F. J. The role of the endocannabinoid system in eating disorders: pharmacological implications. Behav. Pharmacol. 2012, 23, 526–536. [DOI] [PubMed] [Google Scholar]
- Pacher P.; Batkai S.; Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee R. G. Cannabinoid receptors and pain. Prog. Neurobiol. 2001, 63, 569–611. [DOI] [PubMed] [Google Scholar]
- Pertwee R. G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.; Thatte J.; Buzard D. J.; Jones R. M. Therapeutic utility of cannabinoid receptor type 2 (CB2) selective agonists. J. Med. Chem. 2013, 56, 8224–8256. [DOI] [PubMed] [Google Scholar]
- Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin. Pharmacokinet. 2003, 42, 327–360. [DOI] [PubMed] [Google Scholar]
- Sharma R.; Nikas S. P.; Paronis C. A.; Wood J. T.; Halikhedkar A.; Guo J. J.; Thakur G. A.; Kulkarni S.; Benchama O.; Raghav J. G.; Gifford R. S.; Jarbe T. U.; Bergman J.; Makriyannis A. Controlled-deactivation cannabinergic ligands. J. Med. Chem. 2013, 56, 10142–10157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R.; Nikas S. P.; Guo J. J.; Mallipeddi S.; Wood J. T.; Makriyannis A. C-Ring cannabinoid lactones: a novel cannabinergic chemotype. ACS Med. Chem. Lett. 2014, 5, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikas S. P.; Alapafuja S. O.; Papanastasiou I.; Paronis C. A.; Shukla V. G.; Papahatjis D. P.; Bowman A. L.; Halikhedkar A.; Han X.; Makriyannis A. Novel 1′,1′-chain substituted hexahydrocannabinols: 9beta-hydroxy-3-(1-hexyl-cyclobut-1-yl)-hexahydrocannabinol (AM2389) a highly potent cannabinoid receptor 1 (CB1) agonist. J. Med. Chem. 2010, 53, 6996–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsunobu O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981, 1–28. [Google Scholar]
- Papahatjis D. P.; Nahmias V. R.; Nikas S. P.; Andreou T.; Alapafuja S. O.; Tsotinis A.; Guo J.; Fan P.; Makriyannis A. C1′-Cycloalkyl side chain pharmacophore in tetrahydrocannabinols. J. Med. Chem. 2007, 50, 4048–4060. [DOI] [PubMed] [Google Scholar]
- Nikas S. P.; Thakur G. A.; Makriyannis A. Regiospecifically deuterated (−)-delta(9)-tetrahydrocannabivarins. J. Chem. Soc., Perkin Trans. 1 2002, 2544–2548. [Google Scholar]
- White J. M.; Tunoori A. R.; Turunen B. J.; Georg G. I. [Bis(2-methoxyethyl)amino]sulfur trifluoride, the Deoxo-Fluor reagent: application toward one-flask transformations of carboxylic acids to amides. J. Org. Chem. 2004, 69, 2573–2576. [DOI] [PubMed] [Google Scholar]
- Chaudhari S. S.; Akamanchi K. G. Thionyl chloride–benzotriazole in methylene chloride: A convenient solution for conversion of alcohols and carboxylic acids expeditiously into alkyl chlorides and acid chlorides by simple titration. Synlett 1999, 1763–1765. [Google Scholar]
- Shire D.; Calandra B.; RinaldiCarmona M.; Oustric D.; Pessegue B.; BonninCabanne O.; LeFur G.; Caput D.; Ferrara P. Molecular cloning, expression and function of the murine CB2 peripheral cannabinoid receptor. Biochim. Biophys. Acta 1996, 1307, 132–136. [DOI] [PubMed] [Google Scholar]
- Khanolkar A. D.; Lu D.; Ibrahim M.; Duclos R. I. Jr.; Thakur G. A.; Malan T. P. Jr.; Porreca F.; Veerappan V.; Tian X.; George C.; Parrish D. A.; Papahatjis D. P.; Makriyannis A. Cannabilactones: a novel class of CB2 selective agonists with peripheral analgesic activity. J. Med. Chem. 2007, 50, 6493–6500. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Adams M.; Whiteaker K.; Daza A.; Kage K.; Cassar S.; Meyer M.; Yao B. B. Species comparison and pharmacological characterization of rat and human CB2 cannabinoid receptors. Eur. J. Pharmacol. 2004, 505, 1–9. [DOI] [PubMed] [Google Scholar]
- Fukami T.; Yokoi T. The emerging role of human esterases. Drug Metab. Pharmacokinet. 2012, 27, 466–477. [DOI] [PubMed] [Google Scholar]
- Um P. J.; Drueckhammer D. G. Dynamic enzymatic resolution of thioesters. J. Am. Chem. Soc. 1998, 120, 5605–5610. [Google Scholar]
- Durdagi S.; Kapou A.; Kourouli T.; Andreou T.; Nikas S. P.; Nahmias V. R.; Papahatjis D. P.; Papadopoulos M. G.; Mavromoustakos T. The application of 3D-QSAR studies for novel cannabinoid ligands substituted at the C1′ position of the alkyl side chain on the structural requirements for binding to cannabinoid receptors CB1 and CB2. J. Med. Chem. 2007, 50, 2875–2885. [DOI] [PubMed] [Google Scholar]
- Nikas S. P.; Grzybowska J.; Papahatjis D. P.; Charalambous A.; Banijamali A. R.; Chari R.; Fan P.; Kourouli T.; Lin S.; Nitowski A. J.; Marciniak G.; Guo Y.; Li X.; Wang C. L.; Makriyannis A. The role of halogen substitution in classical cannabinoids: a CB1 pharmacophore model. AAPS J. 2004, 6, e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papahatjis D. P.; Nikas S. P.; Andreou T.; Makriyannis A. Novel 1′,1′-chain substituted delta(8)-tetrahydrocannabinols. Bioorg. Med. Chem. Lett. 2002, 12, 3583–3586. [DOI] [PubMed] [Google Scholar]
- Papahatjis D. P.; Nikas S. P.; Kourouli T.; Chari R.; Xu W.; Pertwee R. G.; Makriyannis A. Pharmacophoric requirements for the cannabinoid side chain. Probing the cannabinoid receptor subsite at C1′. J. Med. Chem. 2003, 46, 3221–3229. [DOI] [PubMed] [Google Scholar]
- Thakur G. A.; Nikas S. P.; Li C.; Makriyannis A. Structural requirements for cannabinoid receptor probes. Handb Exp. Pharmacol. 2005, 209–246. [DOI] [PubMed] [Google Scholar]
- Shih N. Y.; Mangiaracina P.; Green M. J.; Ganguly A. K. Synthesis of a carbocyclic analog of quercetin via a Barbier reaction. Tetrahedron Lett. 1989, 30, 5563–5566. [Google Scholar]
- Cheng Y.; Prusoff W. H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Wood J. T.; Smith D. M.; Janero D. R.; Zvonok A. M.; Makriyannis A. Therapeutic modulation of cannabinoid lipid signaling: metabolic profiling of a novel antinociceptive cannabinoid-2 receptor agonist. Life Sci. 2013, 92, 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H.. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Stuart C. S., Ed.; Academic Press: San Diego, 1995; Vol. 25, pp 366–428. [Google Scholar]
- Chang G.; Guida W. C.; Still W. C. An internal-coordinate Monte Carlo method for searching conformational space. J. Am. Chem. Soc. 1989, 111, 4379–4386. [Google Scholar]
- Saunders M.; Houk K. N.; Wu Y. D.; Still W. C.; Lipton M.; Chang G.; Guida W. C. Conformations of cycloheptadecane—a comparison of methods for conformational searching. J. Am. Chem. Soc. 1990, 112, 1419–1427. [Google Scholar]
- Kapur A.; Hurst D. P.; Fleischer D.; Whitnell R.; Thakur G. A.; Makriyannis A.; Reggio P. H.; Abood M. E. Mutation studies of Ser7.39 and Ser2.60 in the human CB1 cannabinoid receptor: evidence for a serine-induced bend in CB1 transmembrane helix 7. Mol. Pharmacol. 2007, 71, 1512–1524. [DOI] [PubMed] [Google Scholar]
- Palczewski K.; Kumasaka T.; Hori T.; Behnke C. A.; Motoshima H.; Fox B. A.; Le Trong I.; Teller D. C.; Okada T.; Stenkamp R. E.; Yamamoto M.; Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [DOI] [PubMed] [Google Scholar]
- Barnett-Norris J.; Hurst D. P.; Buehner K.; Ballesteros J. A.; Guarnieri F.; Reggio P. H. Agonist alkyl tail interaction with cannabinoid CB1 receptor V6.43/I6.46 groove induces a helix 6 active conformation. Int. J. Quantum Chem. 2002, 88, 76–86. [Google Scholar]
- Barnett-Norris J.; Hurst D. P.; Lynch D. L.; Guarnieri F.; Makriyannis A.; Reggio P. H. Conformational memories and the endocannabinoid binding site at the cannabinoid CB1 receptor. J. Med. Chem. 2002, 45, 3649–3659. [DOI] [PubMed] [Google Scholar]
- Fiser A.; Do R. K.; Sali A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Ahn K. H.; Bertalovitz A. C.; Mierke D. F.; Kendall D. A. Dual role of the second extracellular loop of the cannabinoid receptor 1: ligand binding and receptor localization. Mol. Pharmacol. 2009, 76, 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertalovitz A. C.; Ahn K. H.; Kendall D. A. Ligand binding sensitivity of the extracellular loop two of the cannabinoid receptor 1. Drug Dev. Res. 2010, 71, 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z. H.; Bonner T. I. A lysine residue of the cannabinoid receptor is critical for receptor recognition by several agonists but not WIN55212-2. Mol. Pharmacol. 1996, 49, 891–896. [PubMed] [Google Scholar]
- Marcu J.; Shore D. M.; Kapur A.; Trznadel M.; Makriyannis A.; Reggio P. H.; Abood M. E. Novel insights into CB1 cannabinoid receptor signaling: a key interaction identified between the extracellular-3 loop and transmembrane helix 2. J. Pharmacol. Exp. Ther. 2013, 345, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronis C. A.; Nikas S. P.; Shukla V. G.; Makriyannis A. Delta(9)-Tetrahydrocannabinol acts as a partial agonist/antagonist in mice. Behav. Pharmacol. 2012, 23, 802–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon D. D.; Sethumadhavan D.; Benneche T.; Banaag A. R.; Tius M. A.; Thakur G. A.; Bowman A.; Wood J. T.; Makriyannis A. Heteroadamantyl cannabinoids. J. Med. Chem. 2010, 53, 5656–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.