

Abstract

To investigate the hypothesis that molecules acting as crystallization inhibitors in solution could be transformed into crystallization promoters, additives were synthesized that mimic the pharmaceuticals acetaminophen and mefenamic acid and also possess polymerizable functionality. It was found that, in solution, these additives face-selectively inhibit crystal growth and lead to overall slower crystal appearance. In contrast, when the tailor-made additives were incorporated into an insoluble polymer, the induction time for the onset of crystal formation for both pharmaceuticals was substantially decreased. This approach now allows for the synthesis of tailor-made polymers that decrease the induction time for crystal appearance and may find application in compounds that are resistant to crystallization or in improving the fidelity of heteronucleation approaches to solid form discovery.
Introduction
There is often a large barrier to the formation of an ordered three-dimensional lattice from an isotropic state. The initial stage of crystallization, nucleation, can be accelerated if a surface is present to facilitate the organization of molecules by heteronucleation.1 Among the various methods utilized for heteronucleation,2−6 polymer-induced heteronucleation has proven to be a powerful polymorph discovery method, utilizing hundreds of unique insoluble polymers as crystallization directors for obtaining novel solid forms.7−13 It is well established that functional group interactions at the polymer–crystal interface are responsible for directing and controlling the nucleation of different crystal phases on specific polymer heteronucleants.13−16 However, there are some instances where nucleation from the polymer surface is very slow, allowing alternative pathways to compete. In such cases, it is hypothesized that crystallization is not induced by the polymer heteronucleant because little interaction between the polymer and compound exists; this precludes efficient stabilization of nuclei and subsequent growth into macroscopic crystals. An attractive approach for solving the problem of slow nucleation from polymer heteronucleants is to generate insoluble polymers that are designed to possess complementary interactions for a given compound. To implement this strategy, inspiration was sought from the substantial body of work available on soluble additives. Tailor-made additives are typically designed to adsorb onto specific faces of a growing crystal to slow or block growth perpendicular to that face, often affecting the morphology and the polymorphism of the target compound.17−30 If the strong interactions between a tailor-made additive and a target compound could instead be applied at the surface of an insoluble polymer, it is hypothesized that the additive will act as a crystallization promoter. The nucleation rate should be increased because the polymer possesses functionality complementary to that of the target compound in solution thereby facilitating heteronucleation. Furthermore, the morphology of the resulting crystals should not be affected because an insoluble polymer cannot interact with multiple faces of a growing crystal.
Results and Discussion
Due to the extensive work on the effect of tailor-made additives on the morphology of acetaminophen (ACM) crystals, this compound was used as an initial target in order to determine if polymers could be tailored to accelerate nucleation.31−33 A polymerizable additive, N-hydroxyphenyl methacrylamide, was designed and synthesized34 to mimic ACM (Figure 1).35−38 Whenever designing an inhibitor for a specific compound, the possibility exists that the particular substitution pattern chosen will preclude efficient interaction with the target crystal. Therefore, to verify that N-hydroxyphenyl methacrylamide would act in solution to modify the morphology of the ACM crystals, crystallizations in the presence of the additive were performed. As the concentration of the tailor-made additive was increased, the ACM crystals became more elongated (Figure 2). In spite of this dramatic change in the morphology of the crystals, all were confirmed to be the monoclinic polymorph of ACM by Raman spectroscopy (see Supporting Information). Having determined that N-hydroxyphenyl methacrylamide face-selectively interacts with ACM crystals in solution, the additive was subsequently incorporated into polymers to determine if it possessed the ability to promote crystallization when immobilized. To explore the effect of the concentration of the tailor-made additive present in the polymer heteronucleant on the crystallization rate of the pharmaceutical, binary copolymers were prepared. The requisite properties for the second monomeric component are poor water solubility, a lack of hydrogen-bonding functionality, and a reactivity ratio similar to the additive such that random copolymers would be generated. Thus, three copolymers with styrene and increasing ratios of the tailor-made additive (1, 5, and 10 mol% of additive to total polymer) were synthesized in addition to pure polystyrene (see Supporting Information). In each case the polymer was found to be insoluble in water by UV–vis absorbance spectroscopy implicating a heterogeneous mechanism39,40 for influencing crystallization (see Supporting Information). Crystallizations of ACM in the presence of the three tailor-made additive copolymers and polystyrene as well as in the absence of polymer were carried out in aqueous solution with each crystallization condition performed eight times in triplicate (see Supporting Information). In order to determine the induction time for crystal appearance the crystallizations were checked by optical microscopy every 15 min. On average, the induction time for crystal appearance of ACM in the absence of the synthesized polymers occurred in >6000 min, whereas in the presence of polystyrene this time decreased to 1100 min. These observations are consistent with a decreased induction period resulting from heterogeneous nucleation. More substantial though was the decrease in the induction time for the appearance of crystals in the presence of polymers with incorporated N-hydroxyphenyl methacrylamide. On average crystallizations in the presence of the 1, 5, and 10 mol% N-hydroxyphenyl methacrylamide/styrene copolymers occurred in 243 ± 7, 189 ± 10, and 151 ± 8 min, respectively (times are shown with the standard error) (Figure 3). These results are consistent with the proposition that a soluble tailor-made additive that modifies morphology in solution acts as a crystallization promoter when incorporated into an insoluble polymer.
Figure 1.
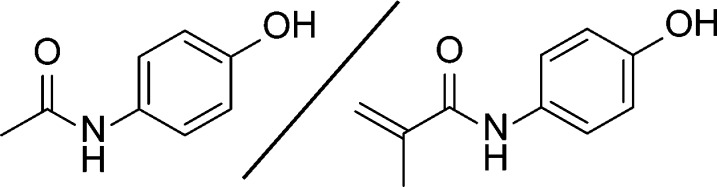
Comparison of the structure of acetaminophen (left) to the tailor-made additive, N-hydroxyphenyl methacrylamide (right).
Figure 2.

Morphology of acetaminophen crystals grown in the presence of N-hydroxyphenyl methacrylamide: (a) no additive, (b) 1 mM additive, (c) 3 mM additive, and (d) 6 mM additive.
Figure 3.
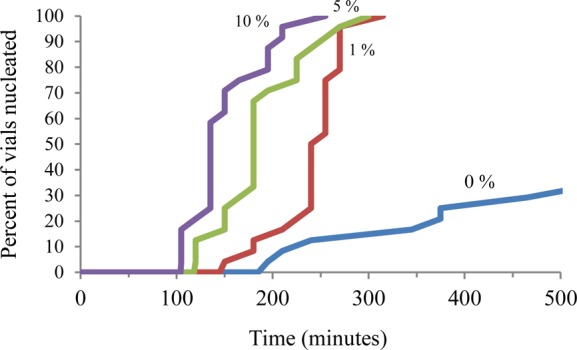
Induction time for crystal appearance for acetaminophen crystallized in the presence of N-hydroxyphenyl methacrylamide/styrene copolymers. The percentages indicated next to each line represent the molar percent of the tailor-made additive in the polymer.
If the strategy of immobilizing a tailor-made additive in a polymer to create a crystallization promoter is generally applicable, then other ACM mimics should yield similar results. To explore this hypothesis, another tailor-made additive, p-acetamidostyrene, was synthesized.34 This tailor-made additive possesses similar amide functionality to that of ACM but also bears a vinyl group for integration into a polymer. Acetaminophen was initially crystallized with p-acetamidostyrene in solution to determine if the additive could affect the morphology of the resulting crystals. Crystals of the monoclinic form of ACM became increasingly elongated as the concentration of the additive was raised from 1 to 6 mM (see Supporting Information). With successful demonstration of face-selective growth inhibition, p-acetamidostyrene was subsequently incorporated into polymers to yield three copolymers with increasing ratios of the tailor-made additive to styrene (1, 5, and 10 mol% of tailor-made additive relative to the total polymer). The crystallizations were conducted and monitored in the same manner as the N-hydroxyphenyl methacrylamide/styrene copolymer system described above. The induction time for crystal appearance was significantly decreased in the presence of the p-acetamidostyrene/styrene copolymers. For crystallizations in the presence of the 10 mol% p-acetamidostyrene/styrene copolymer, crystals appear on average within an hour, one hundredth of the time needed for crystallization to occur in the absence of polymer (Figure 4). Despite this drastic change in the induction time for the appearance of crystals, the morphology of the ACM crystals was not affected by the presence of the tailor-made copolymers (Figure 5). This trend of decreasing induction times can be attributed to an increase in the incorporation of the tailor-made monomer in the copolymers, leading to more efficient organization of molecules on the polymer surface and thus faster heteronucleation.
Figure 4.
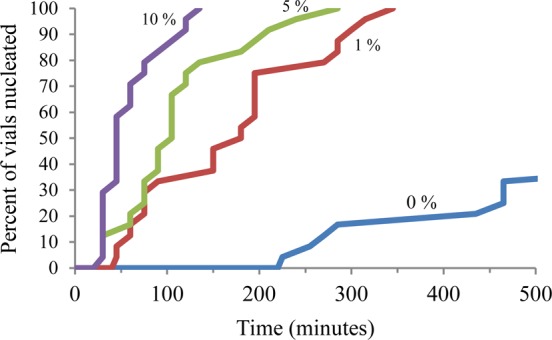
Induction time for crystal appearance for acetaminophen crystallized in the presence of p-acetamidostyrene/styrene copolymers. The percentages indicated next to each line represent the molar percent of the tailor-made additive in the polymer.
Figure 5.

Morphology of acetaminophen crystals grown in the presence of 10 mol% p-acetamidostyrene/styrene.
In order to expand the capabilities of this method to crystallizations in organic solvents and eliminate any issues due to polymer solubility, cross-linked tailor-made polymers were also explored as crystallization promoters. The anti-inflammatory compound mefenamic acid was utilized as an initial target compound. A tailor-made additive, 2-((4-vinylphenyl)amino)benzoic acid, was synthesized, which is structurally similar to mefenamic acid but bears a vinyl group to enable polymerization (Figure 6; see Supporting Information).41 Mefenamic acid was initially crystallized with 2-((4-vinylphenyl)amino)benzoic acid in solution to determine if the additive would affect the morphology of the resulting crystals (1, 5, and 10 mol% relative to the total amount of mefenamic acid). As the concentration of the tailor-made additive was increased, the mefenamic acid crystals became increasingly elongated and the induction time for crystal appearance was significantly increased (see Supporting Information). However, with the highest amount of the tailor-made additive the crystal growth was inhibited so strongly that the crystals, although still blade-like, lacked a distinct morphology. Despite this drastic change in the morphology, all of the crystals were confirmed to be form I of mefenamic acid by Raman spectroscopy (see Supporting Information). Having determined that 2-((4-vinylphenyl)amino)benzoic acid face-selectively interacts with mefenamic acid crystals in solution, the tailor-made additive was copolymerized with divinylbenzene (DVB), in increasing molar ratios, to create cross-linked copolymers (see Supporting Information). Similar to the ACM studies each crystallization condition was performed eight times in triplicate. In order to determine the induction time for crystal appearance, the crystallizations were monitored by time-lapse photography (photos were taken every 60 s). The induction time for the appearance of crystals was considerably decreased for crystallizations in the presence of the 2-((4-vinylphenyl)amino)benzoic acid/DVB copolymers, and the copolymer with the highest incorporation of the tailor-made additive yielded a 10-fold decrease in induction time for crystal appearance (Figure 7; see Supporting Information). To determine if the observed decrease in the induction period primarily stemmed from changes in the surface energy of the tailored copolymers, the induction time for crystal appearance was determined for four copolymers: the 10 mol% 2-((4-vinylphenyl)amino)benzoic acid/DVB copolymer with an advancing water contact angle (CA) of 63.4°, the 1 mol% 2-((4-vinylphenyl)amino)benzoic acid/DVB copolymer (CA = 86.7°), the 10 mol% hydroxyethyl methacrylate/DVB copolymer (CA = 32.2°), and 1 mol% hydroxyethyl methacrylate/DVB copolymer (CA = 60.3°). The two copolymers with similar surface energies, the 10 mol% 2-((4-vinylphenyl)amino)benzoic acid/DVB copolymer and the 1 mol% hydroxyethyl methacrylate/DVB copolymer, did not exhibit similar induction times for crystal appearance. This result demonstrates that the decrease in the induction time is not dictated by surface energy alone (see Supporting Information).
Figure 6.
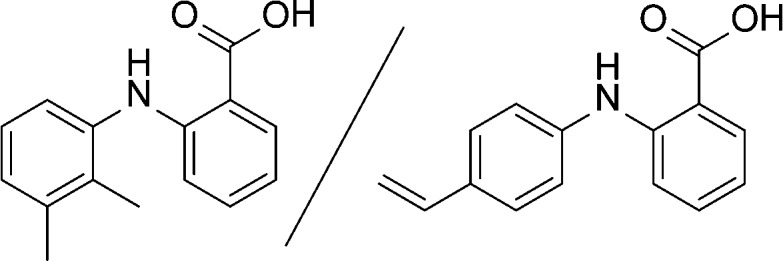
Comparison of the structure of mefenamic acid (left) to the tailor-made additive, 2-((4-vinylphenyl)amino)benzoic acid (right).
Figure 7.
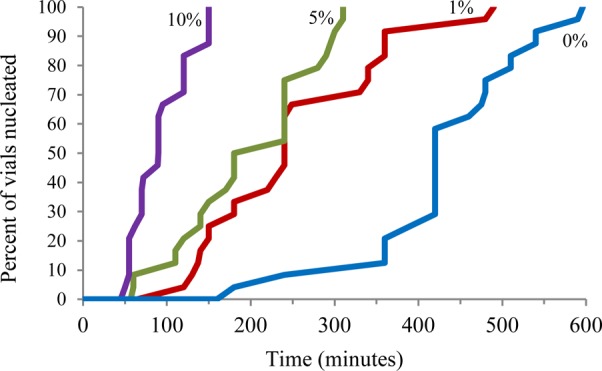
Induction time for crystal appearance for mefenamic acid crystallized in the presence of 2-((4-vinylphenyl)amino)benzoic acid/DVB copolymers. The percentages indicated next to each line represent the molar percent of the tailor-made additive in the polymer.
Although the molecular-level events leading to the induction of crystal growth from polymer surfaces cannot be directly observed, rate acceleration can arise either from the polymer stabilizing subcritically sized nuclei of the target compound in solution or through organization of molecules on the polymer surface leading to aggregates of critical dimensions. In either case, it is hypothesized that the face-selectivity of crystal growth results from preferential interaction with the surface of pre-nuclear aggregates mediated by intermolecular interactions between the polymer and the forming nucleus.14,15 In order to determine how the tailored copolymers are interacting with the target compound, ACM was crystallized on three distinct types of polymer films: polystyrene, the 10 mol% N-hydroxyphenyl methacrylamide/styrene copolymer, and the 10 mol% p-acetamidostyrene/styrene copolymer. The resulting crystals were then analyzed for preferred growth orientation by face indexing via single-crystal X-ray diffractometry. Crystals grown from the 10 mol% p-acetamidostyrene/styrene and 10 mol% N-hydroxyphenyl methacrylamide/styrene copolymer films both exhibited preferred orientation along the (001) face of form I (monoclinic) of ACM (Figure 8; see Supporting Information). For the monoclinic form of ACM the hydroxyl and the amide carbonyl functionalities are oriented perpendicular to the (001) face, suggesting that the tailored copolymers may be preferentially interacting with these groups through hydrogen bonding. Conversely, ACM was found to be oriented along the (10–1) face on the polystyrene films (Figure 9; see Supporting Information). For the monoclinic form of ACM the benzene rings and amide N–H functionalities are present perpendicular to this face, meaning that polystyrene may be interacting with the ACM molecules through π–π interactions.
Figure 8.

Acetaminophen crystals grown in the presence of the 10 mol% p-acetamidostyrene/styrene copolymer film.
Figure 9.

Acetaminophen crystals grown in the presence of the polystyrene film.
In order to determine if there was any preferential interaction between the functionality on the tailor-made copolymer surface and the mefenamic acid molecules in solution, mefenamic acid was crystallized on polymer films comprised of the 10 mol% 2-((4-vinylphenyl)amino)benzoic acid/DVB copolymer. Powder X-ray diffraction (PXRD) analysis of the crystals present on the tailor-made copolymer films revealed that there are two reflections at 6.3° (100) and 12.7° (200); these correspond to mefenamic acid form I crystals oriented along {100} (see Supporting Information). In form I, carboxylic acid groups are oriented perpendicular to the (100) face,42 suggesting that the tailor-made copolymer is preferentially interacting with these groups through hydrogen bonding.14,15 An intriguing question that can test the proposed mechanism of interaction is if adsorption occurs in the same orientation when an additive is in solution as when it is anchored to a polymer. In order to test this, mefenamic acid crystals grown in the presence of 2-((4-vinylphenyl)amino)benzoic acid in solution were indexed. It was found that the additive in solution was in fact adsorbing onto the (100) face, showing that the mechanism of interaction is not changed when the additive is incorporated into a polymer (see Supporting Information).
Conclusions
The studies outlined here demonstrate that tailor-made additives, which alter crystal morphology in solution, can be incorporated into insoluble polymers to promote crystallization. This approach has the potential to impact a problem of considerable importance in the pharmaceutical industry: the emergence of compounds which, for purely kinetic reasons, under all growth conditions are resistant to crystallization.43 This can severely complicate purification and form identification. In these cases, tailoring substrates to decrease the time needed for crystals to appear is an attractive approach for creating appropriate seed crystals; studies examining this approach are currently underway.
Acknowledgments
Thanks are extended to a number of graduate and undergraduate researchers who explored the concept of tailor-made monomers in controlling pharmaceutical crystallization. This work was supported by the National Institutes of Health Grant No. RO1 GM106180.
Supporting Information Available
Crystallization procedure for each pharmaceutical with its respective tailor-made additive, Raman spectra for acetaminophen and mefenamic acid crystals, tailor-made additive copolymer preparation, crystallization procedure for each pharmaceutical with its respective tailor-made copolymer, UV–vis studies on the solubility of the tailor-made copolymers in water, PXRD data for mefenamic acid crystallized in the presence of tailor-made copolymers, and procedure for indexing mefenamic acid crystals grown in the presence of 2-((4-vinylphenyl)amino)benzoic acid in solution. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Liu X. Y. Langmuir 2000, 16, 7337. [Google Scholar]
- Capacci-Daniel C.; Gaskell K. J.; Swift J. A. Cryst. Growth Des. 2010, 10, 952. [Google Scholar]
- Mitchell C. A.; Yu L.; Ward M. D. J. Am. Chem. Soc. 2001, 123, 10830. [DOI] [PubMed] [Google Scholar]
- Kang J. F.; Zaccaro J.; Ulman A.; Myerson A. Langmuir 2000, 16, 3791. [Google Scholar]
- Lee A. Y.; Ulman A.; Myerson A. S. Langmuir 2002, 18, 5886. [Google Scholar]
- Ulman A.; Kang J. F.; Shnidman Y.; Liao S.; Jordan R.; Choi G. Y.; Zaccaro J.; Myerson A. S.; Rafailovich M.; Sokolov J.; Fleischer C. J. Biotechnol. 2000, 74, 175. [DOI] [PubMed] [Google Scholar]
- Price C. P.; Grzesiak A. L.; Matzger A. J. J. Am. Chem. Soc. 2005, 127, 5512. [DOI] [PubMed] [Google Scholar]
- Lopez-Mejias V.; Kampf J. W.; Matzger A. J. J. Am. Chem. Soc. 2009, 131, 4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mejias V.; Kampf J. W.; Matzger A. J. J. Am. Chem. Soc. 2012, 134, 9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudha C.; Nandhini R.; Srinivasan K. Cryst. Growth Des. 2014, 14, 705. [Google Scholar]
- Chen J. H.; Shao M.; Xiao K.; He Z. R.; Li D. W.; Lokitz B. S.; Hensley D. K.; Kilbey S. M.; Anthony J. E.; Keum J. K.; Rondinone A. J.; Lee W. Y.; Hong S. Y.; Bao Z. A. Chem. Mater. 2013, 25, 4378. [Google Scholar]
- Lee M. K.; Lee H.; Kim I. W.; Lee J. Pharmazie 2011, 66, 766. [PubMed] [Google Scholar]
- Diao Y.; Helgeson M. E.; Myerson A. S.; Hatton T. A.; Doyle P. S.; Trout B. L. J. Am. Chem. Soc. 2011, 133, 3756. [DOI] [PubMed] [Google Scholar]
- Lopez-Mejias V.; Knight J. L.; Brooks C. L.; Matzger A. J. Langmuir 2011, 27, 7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland A. A.; Lopez-Mejias V.; Matzger A. J.; Chen Z. Langmuir 2011, 27, 2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio E.; López-Mejías V.; Di Profio G.; Fontananova E.; Drioli E.; Trout B. L.; Myerson A. S. Cryst. Growth Des. 2013, 14, 678. [Google Scholar]
- Weissbuch I.; Leisorowitz L.; Lahav M. Adv. Mater. 1994, 6, 952. [Google Scholar]
- Davey R. J.; Blagden N.; Potts G. D.; Docherty R. J. Am. Chem. Soc. 1997, 119, 1767. [Google Scholar]
- Staab E.; Addadi L.; Leiserowitz L.; Lahav M. Adv. Mater. 1990, 2, 40. [Google Scholar]
- Kitamura M.; Ishizu T. J. Cryst. Growth 1998, 192, 225. [Google Scholar]
- Blagden N. Powder Technol. 2001, 121, 46. [Google Scholar]
- He X. R.; Stowell J. G.; Morris K. R.; Pfeiffer R. R.; Li H.; Stahly G. P.; Byrn S. R. Cryst. Growth Des. 2001, 1, 305. [Google Scholar]
- Thallapally P. K.; Jetti R. K. R.; Katz A. K.; Carrell H. L.; Singh K.; Lahiri K.; Kotha S.; Boese R.; Desiraju G. R. Angew. Chem., Int. Ed. 2004, 43, 1149. [DOI] [PubMed] [Google Scholar]
- Cashell C.; Corcoran D.; Hodnett B. K. Cryst. Growth Des. 2005, 5, 593. [Google Scholar]
- Ward M. D. Chem. Rev. 2001, 101, 1697. [DOI] [PubMed] [Google Scholar]
- Shtukenberg A. G.; Lee S. S.; Kahr B.; Ward M. D. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 77. [DOI] [PubMed] [Google Scholar]
- van Enckevort W. J. P.; Los J. H. J. Phys. Chem. C 2008, 112, 6380. [Google Scholar]
- Farmanesh S.; Ramamoorthy S.; Chung J. H.; Asplin J. R.; Karande P.; Rimer J. D. J. Am. Chem. Soc. 2014, 136, 367. [DOI] [PubMed] [Google Scholar]
- Rimer J. D.; An Z. H.; Zhu Z. N.; Lee M. H.; Goldfarb D. S.; Wesson J. A.; Ward M. D. Science 2010, 330, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbuch I.; Lahav M.; Leiserowitz L. Cryst. Growth Des. 2003, 3, 125. [Google Scholar]
- Li T. L.; Wen H.; Park K.; Morris K. R. Cryst. Growth Des. 2002, 2, 185. [Google Scholar]
- Li T.; Morris K.; Park K. Pharm. Res. 2001, 18, 398. [DOI] [PubMed] [Google Scholar]
- Li T.; Park K.; Morris K. R. Cryst. Growth Des. 2002, 2, 177. [Google Scholar]
- Tessier T. G.; Fréchet J. M. J.; Willson C. G.; Ito H. In Materials for Microlithography; Thompson L. F., Willson C. G., Fréchet J. M. J., Eds.; ACS Symposium Series266; American Chemical Society: Washington, DC, 1985. [Google Scholar]
- Haisa M.; Kashino S.; Kawai R.; Maeda H. Acta Crystallogr., Sect. B 1976, 32, 1283. [Google Scholar]
- Haisa M.; Kashino S.; Maeda H. Acta Crystallogr., Sect. B 1974, 30, 2510. [Google Scholar]
- DiMartino P.; Conflant P.; Drache M.; Huvenne J. P.; GuyotHermann A. M. J. Therm. Anal. 1997, 48, 447. [Google Scholar]
- Peterson M. L.; Morissette S. L.; McNulty C.; Goldsweig A.; Shaw P.; LeQuesne M.; Monagle J.; Encina N.; Marchionna J.; Johnson A.; Gonzalez-Zugasti J.; Lemmo A. V.; Ellis S. J.; Cima M. J.; Almarsson O. J. Am. Chem. Soc. 2002, 124, 10958. [DOI] [PubMed] [Google Scholar]
- Bernstein J.Polymorphism in Molecular Crystals; Oxford University Press: New York, 2002. [Google Scholar]
- Davey R.; Garside J.. From Molecules to Crystallizers; Oxford University Press: New York, 2000. [Google Scholar]
- Wolf C.; Liu S. L.; Mei X. F.; August A. T.; Casimir M. D. J. Org. Chem. 2006, 71, 3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell J. F. Cryst. Struct. Commun. 1976, 5, 861. [Google Scholar]
- Dunitz J. D.; Bernstein J. Acc. Chem. Res. 1995, 28, 193. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.