

Abstract
Hepatitis C virus (HCV) infects an estimated 3% of the global population with the majority of individuals (75–85%) failing to clear the virus without treatment, leading to chronic liver disease. Individuals of African-descent have lower rates of clearance compared to individuals of European-descent and this is not fully explained by social and environmental factors. This suggests that differences in genetic background may contribute to this difference in clinical outcome following HCV infection. Using 473 individuals and 792,721 SNPs from a genome-wide association study (GWAS), we estimated local African ancestry across the genome. Using admixture mapping and logistic regression we identified two regions of interest associated with spontaneous clearance of HCV (15q24, 20p12). A genome-wide significant variant was identified on chromosome 15 at the imputed SNP, rs55817928 (P=6.18×10−8) between the genes SCAPER and RCN. Each additional copy of the African ancestral C allele is associated with 2.4 times the odds of spontaneous clearance. Conditional analysis using this SNP in the logistic regression model explained one-third of the local ancestry association. Additionally, signals of selection in this area suggest positive selection due to some ancestral pathogen or environmental pressure in African, but not in European populations.
Keywords: Hepatitis C, Chronic Infection, Admixture, African Ancestry
Introduction
Hepatitis C virus (HCV) is a major global cause of liver disease, with 350,000 people dying every year from hepatitis C-related disease. (1) HCV is primarily spread through contact with infected blood, leading to chronic infection in 80% of infected individuals. More than half of these chronically infected individuals develop chronic liver disease, including cirrhosis and hepatocellular carcinoma. Twenty percent of individuals spontaneously clear the virus without treatment.
There are marked racial differences in HCV outcomes. In the United States, overall HCV prevalence is more than doubled in African Americans (3.2%) compared to non-Hispanic whites (1.5%). (2) In addition, among those exposed to HCV infection, viral persistence is 5-fold more common among persons of African compared to European descent. (1,3) African-Americans also respond worse to interferon-based treatments for HCV infection. (2,4) In contrast, when compared to European-Americans, African-Americans are less likely to progress to cirrhosis. (5,6) While viral or environmental factors may contribute, these marked divergent clinical outcomes in persons of African compared to European descent suggest a role for host genetics.
Previous human genetic studies have identified susceptibility loci for spontaneous resolution of HCV. The most consistent genetic association has been with variants near IL28B (interleukin-28B), now IFNL3, (7,8) and the Major Histocompatibility Complex (MHC) region, specifically with HLA Class II. (9) Recent genome-wide association studies have identified upstream IL28B variants rs12979860 and rs8099917 to be consistently associated with spontaneous HCV clearance (OR=0.45, 0.43). (7,9) The SNP rs12979860 may explain nearly half of the difference in spontaneous clearance rates between African- and European-Americans, as demonstrated by a conditional analysis. (9) A recent study found a frameshift mutation, ss496415590, in high linkage disequilibrium with rs12979860 that creates a new gene that is similar to IL28B, and stimulates similar cellular pathways. (10) Candidate gene studies have also identified genetic associations with spontaneous HCV clearance in HAVCR1, TNFSF18, TANK, and IL18BP. (11) However, these variants do not fully explain the difference in HCV resolution between individuals of European and African ancestry.
These differences in outcomes of HCV infection between European- and African-Americans makes it an ideal candidate for admixture mapping, which estimates the degree to which an individual’s genome is of African and European origin. (12) When two distinct continental populations mix, in this case Europe and Africa, the resulting population is admixed. After successive generations of mixing, blocks of the genome are inherited intact due to patterns of linkage disequilibrium. These chromosomal segments retain the same structure as their European or African ancestors. An admixed individual’s genomic segments that are African or European in origin, or their “local ancestry”, can then be estimated across the genome. At any given locus, an individual will have 0, 1, or 2 haplotype segments of European/African ancestry. Admixture mapping uses these measures of local ancestry to evaluate differences in phenotype among individuals. The main hypothesis of this study is that some alleles associated with HCV spontaneous clearance or persistence may be more prevalent in individuals with higher local African ancestry compared to European ancestry. Using genome-wide admixture mapping, we tested this hypothesis with 473 individuals of African descent from 7 studies using 792,721 markers to examine associations between local ancestry and HCV spontaneous clearance.
Results
A total of 473 individuals of African-descent from the following 7 studies were included: AIDS Linked to the Intravenous Experience (ALIVE), Boston Area HCV Study Transmission, Immunity, Outcomes Network (BAHSTION), Baltimore Before and After Acute Study in Hepatitis (BBAASH), Hemophilia Growth & Development Study (HGDS), Multicenter Hemophilia Cohort Studies (MHCS-I and MHCS-II), and the Women’s Interagency HIV Study (WIHS). Spontaneous clearance occurred in 177 individuals, and chronic infection was found in 296 individuals. (Table 1) Nearly half of all participants were HIV positive. The predominant mode of transmission was non-transfusion (injection drug use or sexual activity). There was no difference in the underlying global ancestry between the spontaneous clearance and chronic infection groups. (Supplemental Figure 1) Local ancestry was estimated at 792,721 SNP locations genome-wide using the statistical program LAMP-LD. (13) (Figure 1) Each location is represented by a single SNP. The average African ancestry proportion across the genome was 79.49%, which is consistent with prior estimates of African ancestry for African-Americans. (14,15)
Table 1.
Demographics for Study Individuals.
There was no statistically significant (P<0.05) difference between groups for any of these variables.
| Clearance | Persistence | Total | ||
|---|---|---|---|---|
| HIV Positive (%) | 54.8 | 47.0 | 49.9 | |
| Average African Ancestry (%) | 80.3 | 78.3 | 79.1 | |
| Female (%) | 48.6 | 56.5 | 51.6 | |
| Route of Transmission | Non-Transfusion | 153 | 258 | 411 |
| Transfusion | 22 | 35 | 57 | |
| Unknown | 2 | 3 | 5 | |
| Study | ALIVE | 79 | 168 | 247 |
| BAHSTION | 3 | 7 | 10 | |
| BBAASH | 6 | 13 | 19 | |
| HGDS | 2 | 7 | 9 | |
| MHCS | 14 | 20 | 34 | |
| MHCS2 | 6 | 8 | 14 | |
| WIHS | 67 | 73 | 140 | |
| TOTAL | 177 | 296 | 473 | |
Figure 1.
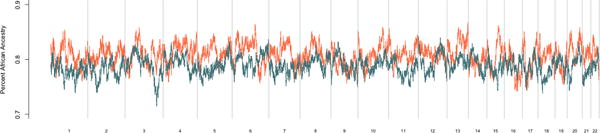
Genome-wide average local ancestry by chromosome and position on the x-axis Spontaneous clearance is in orange and chronic infection is in blue.
Local ancestry estimates were analyzed for association with spontaneous clearance versus chronic or persistent infection of HCV using an unadjusted simple model of logistic regression. Local ancestry was represented in an additive format, with individuals having 0, 1, or 2 haplotype segments of African-ancestry for each location on the genome-wide SNP array. The strongest association with local African ancestry occurred at 15q24 (P=3.19×10−4) (Figure 2) with additional signals occurring at 1p31.1 (P=8.43×10−4) and 20p12 (P=8.68×10−4). (Table 2) The 15q24 and 20p12 regions were confirmed using an alternative method (P< 0.01). Each SNP location within these two regions was further analyzed in an extended model logistic regression that assessed the association of local ancestry in an additive format, controlling for HIV status and genotype (0, 1, or 2 copies of the SNP’s minor allele). (Table 2) This extended model controls for signals due to a single ancestry-informative marker, while improving the resolution and significance of the findings.
Figure 2.
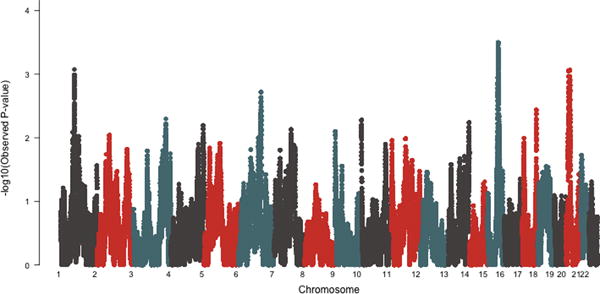
Genome-wide results for association of local ancestry with spontaneous clearance versus chronic infection of HCV by chromosome (x-axis). Significance is indicated by the y-axis using the −log10 P-value.
Table 2.
Logistic regression analysis of HCV spontaneous clearance on local African ancestry for the three most significant regions. The top association within the simple model using an unadjusted logistic regression on local ancestry estimates is denoted simple. The extended label is the top ten associations in that same region, using the extended model of logistic regression on local ancestry estimates, adjusting for HIV status and genotype.
| Chromosomal Region | Analysis | Chromosomal Position | Local Ancestry Odds Ratio | Local Ancestry P-Value |
|---|---|---|---|---|
| 20p12.1 | Simple | 8306421–8370369 | 1.7949 | 8.6821E-04 |
| Extended | 8567519 | 2.0934 | 1.5217E-04 | |
| 8397222 | 1.9052 | 5.7424E-04 | ||
| 8324256 | 1.9190 | 5.7846E-04 | ||
| 8340373 | 1.8762 | 6.3315E-04 | ||
| 8564915 | 2.0477 | 6.6330E-04 | ||
| 8601707 | 2.0531 | 7.4682E-04 | ||
| 8386087 | 1.8501 | 7.9500E-04 | ||
| 8488087 | 1.9291 | 8.1494E-04 | ||
| 8355437 | 1.8593 | 8.1561E-04 | ||
| 8269583 | 1.8185 | 8.3978E-04 | ||
| 20p12.3 | Simple | 13078275–13115148 | 1.7989 | 8.8622E-04 |
| Extended | 13080648 | 2.0676 | 1.6933E-04 | |
| 13199299 | 1.9789 | 2.5846E-04 | ||
| 13082281 | 2.0037 | 3.0541E-04 | ||
| 13892158 | 2.0377 | 3.0996E-04 | ||
| 13111323 | 1.9468 | 3.2714E-04 | ||
| 13095232 | 1.9134 | 3.3491E-04 | ||
| 13281689 | 1.9508 | 3.4468E-04 | ||
| 13112699 | 1.9134 | 3.7582E-04 | ||
| 13260252 | 1.9390 | 4.0612E-04 | ||
| 12898678 | 1.9159 | 4.7388E-04 | ||
| 15q24 | Simple | 76462575–76678692 | 1.8723 | 3.1932E-05 |
| Extended | 77241542 | 2.2330 | 2.2593E-05 | |
| 76678692 | 2.2585 | 2.5100E-05 | ||
| 76772062 | 2.2240 | 2.5778E-05 | ||
| 76490547 | 2.1550 | 3.5743E-05 | ||
| 76930805 | 2.1271 | 4.1500E-05 | ||
| 77176491 | 2.1353 | 5.8441E-05 | ||
| 76521770 | 2.1241 | 6.1589E-05 | ||
| 76965922 | 2.1166 | 6.6862E-05 | ||
| 77087785 | 2.0784 | 6.9310E-05 | ||
| 76470755 | 2.0311 | 7.7342E-05 |
The strongest signal on chromosome 20 for the association of local ancestry with spontaneous clearance using the unadjusted model was in a 36.87 kilobase (kb) region on 20p12.1 (P=8.68×10−4). (Supplemental Figure 2) This region is within the gene SPTLC3, serine palmytoyltransferase long chain base subunit 3. When this region was adjusted for HIV status and local genotype in the extended logistic regression model, the strongest local ancestry association localized within an intronic region of SPTLC3. For each copy of the African ancestral haplotype there is a 2-fold increased association with HCV spontaneous clearance (OR=2.07, P=1.69×10−4). Within the extended model adjusting for local ancestry and HIV status, the local genotype at this location (SNP rs6041855) was not significant (P=0.05). Although the mean African-ancestry at this location was 79.37%, consistent with the genome-wide average (79.49%), the cases with HCV spontaneous clearance had higher African-ancestry (85.31%), while the controls with HCV persistent infection had lower African-ancestry (75.84%) (P=0.019).
An additional local ancestry association signal from the unadjusted model was identified for a 63.95 kb region on 20p12.3 within the gene PLCB1, phospholipase C beta 1 (P=8.86×10−4). Adjusted for HIV status and local genotype in the extended model, the strongest local ancestry association within this region was found in an intronic region of PLCB1 (OR=2.09, P=1.52×10−4). For every additional ancestral African haplotype at this location, an individual had 2 times the odds of clearance. The local ancestry signal in this region was independent of both rs4273729 (HLA) and rs12979860 (IL28B). The SNP at this location, rs6086518, was not significantly associated (P=0.01). No genome-wide significant SNPs were found (P<5×10−7) after imputation in either of the chromosome 20 regions.
Genome-wide, the strongest association of local ancestry using the simple model a 216.12 kb region on 15q24 (P=3.19×10−4). (Supplemental Figure 3) This region contained 5 different genes (C15orf27, ETFA, TYRO3P, ISL2, and SCAPER), and was centered over ETFA. After adjustment for HIV status and genotype in the extended model logistic regression, the strongest local ancestry association lay within RCN2 (OR=2.23, P=2.26×10−5), a gene next to SCAPER. The local ancestry signal in this region was independent of both HLA and IL28B variants (P>0.05) as assessed by interaction analyses using logistic regression. The genotyped SNP at this location, rs15939, did not reach genome-wide significance (P=0.004) with HCV clearance. We imputed SNPs across this region using 1000 Genomes as a reference, identifying a genome-wide significantly associated SNP between SCAPER and RCN2 at rs55817928 (OR=2.38, P=6.18×10−8), which conferred more than twice the odds of spontaneous clearance with every ancestral allele. (Figure 3) The major allele (C), which was associated with clearance, is found in lower frequencies within European populations (40%) than within African populations (76%). This is consistent with African-ancestry being associated with spontaneous clearance in this region. Conditioning on the minor allele of this SNP attenuated the local ancestry signal in this region below 10−2, (Figure 4) suggesting that rs55817928, or likely a variant in strong linkage disequilibrium to this SNP, is largely responsible for the local ancestry association in this location.
Figure 3.
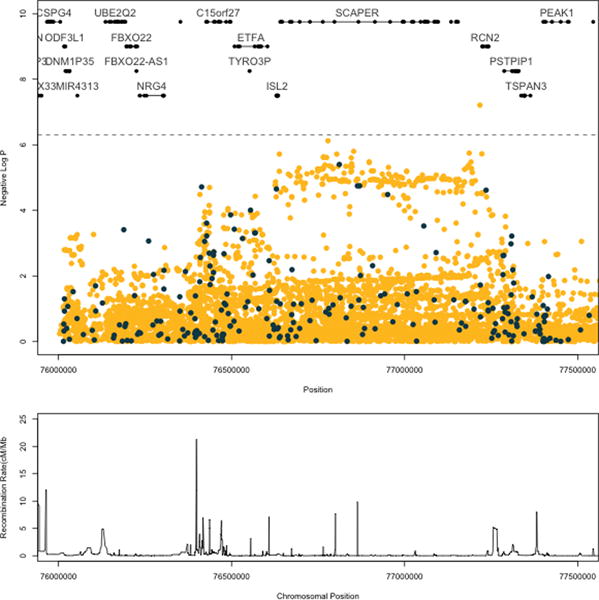
Imputation results for SCAPER region. The top panel shows the SNPs by position (x-axis). Dark blue indicates the genotyped SNPs; yellow indicates the imputed SNPs. Significance is shown as a negative log p-value on the y-axis. The dotted grey line (p = 5 × 10−7) is the threshold for genome-wide significance. Genes and their exonic regions are shown above the SNPs. The bottom panel shows the recombination rate in this region by chromosomal position.
Figure 4.
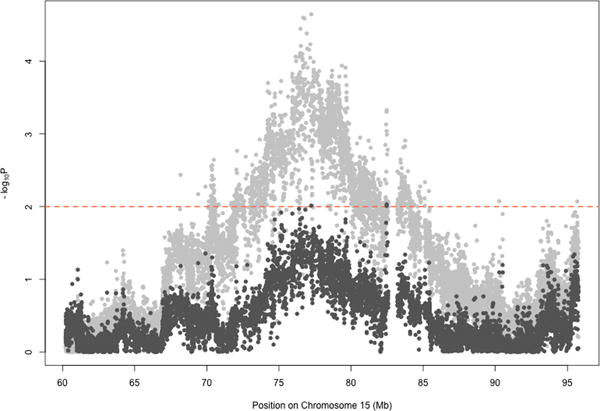
Conditional analysis of SCAPER region on rs55817928. The original results from the extended model for local ancestry association are shown in light grey. After adjusting for the genotype of imputed SNP rs55817928 within the extended model, the association results of local ancestry are shown in dark gray. P-values are significantly decreased after inclusion of the one imputed SNP in the model, indicating that some of the local ancestry signal was due to rs55817928, or another SNP in strong linkage disequilibrium with this SNP.
Using Phase II data from the International HapMap (16), measures of selection were determined using Haplotter (17), which maps the integrated haplotype score (iHS), a measure of positive selection that examines extended homozygosity of haplotypes around a positively selected allele. (17) An iHS score >2 indicates the haplotypes on the ancestral allele are longer compared to the derived allele and an iHS score <-2 indicates the haplotypes are longer on the derived allele compared to the ancestral allele. We expect the region around a derived (non-ancestral) allele that emerged under strong selection to have higher haplotype homozygosity than a neutral model, and this is reflected in the iHS. An iHS score can be standardized by other locations with the same derived (non-ancestral) allele frequency.
Both regions on chromosomes 15 and 20 contain evidence for selection, specifically in the Yoruba (YRI), a Nigerian population. The chromosome 15 region, centered within SCAPER, has the strongest signal at rs280026 (position 77,031,930) with an iHS of −4.6 in YRI (standardized iHS= −2.58). The same region in CEU (a European population) does not contain these signals of selection. The chromosome 20 region of interest near SPTLC3 at rs3898706 (position 12,901,895) had a strong signal of selection with standardized iHS of 3.37 in YRI and a standardized iHS of only 0.24 in CEU. This SNP is upstream of the gene SPTLC3. Both indicate that selection is unique to a West African population in these regions.
Discussion
We used admixture mapping to identify two regions, 20p12.3 (SPTLC3) and 15q24 (SCAPER, RCN) that were suggestive of African-descent conferring a higher odds of spontaneous clearance compared to European-descent. This is surprising, as African-Americans clear the virus at a much lower rate than European-Americans. Our findings highlight specific regions that may reveal a more complex picture of genetic susceptibility to HCV chronic infection. Measures of selection in the Yoruba in Nigeria show evidence for selection in these two regions that are absent in European populations. Although unlikely that HCV has exerted this selective pressure since infection typically occurs later in life and does not alter reproduction, the selective pressures and resulting variants in these regions are associated with clearance of HCV among those who should have been predisposed to chronic infection. This mutation on an ancestral African haplotype may allow individuals to spontaneously clear the virus, despite the virus having evolved to persist as an asymptomatic infection.
There is a potential biological basis for these findings. The associated region on chromosome 20 is in SPTLC3, serine palmitoyltransferase long chain base subunit 3. This gene is an isoform of the third component of SPT, serine palmitoyltransferase. SPT is responsible for the formation of ceramide from palmitoyl-CoA and serine. (18) This gene was previously implicated in the circulating levels of sphingolipid concentrations. (19) Prior molecular evidence points to the importance of sphingolipids, SPTLC3, and chronic HCV infection. (20) Use of a hepatotropic serine palmitoyltransferase inhibitor, NA808, was found to suppress HCV-RNA production, also interfered with sphingolipid metabolism. SPTLC3, when overexpressed, resulted in a 2- to 3-fold increase in the expression of SPT. This gene is expressed only in the liver and is involved in the rate-limiting step of de novo synthesis of sphingomyelin and ceramide. (21) Decreased SPT activity results in decreased levels of ceramide in the liver, which in turn could affect HCV replication. (22)
The most significant association was on chromosome 15 and the region includes the gene SCAPER, an S-phase Cyclin A. SCAPER expression is known to be cell cycle independent and it interacts with cyclin A as well as Cdk2. (23) HCV non-structural protein 5-B (NS5B) has been shown to interact with cyclin A2 (CycA2), the somatic form of cyclin A, for the propagation of HCV. (24) Because the top associated region on chromosome 15 is between SCAPER and RCN2, we must also consider the potential role of RCN2 (reticulocalbin 2, EF-hand calcium binding domain). A part of the EF-hand superfamily, RCN2 contains conserved regions with similarity to a high-affinity Ca2+ binding motif. HCV proteins have been shown to induce oxidative stressmediated Ca2+ homeostasis alterations. (25) Due to the biological plausibility of both genes, it is not possible to know which contributed to the signal detected in this analysis.
This analysis was conducted in a relatively small sample size of African-Americans, which could lead to spurious results. However, infectious diseases tend to have higher effect sizes than many chronic diseases due to selective pressures, so it is not surprising to see a significant association even with this smaller sample size. (26) By design, admixture mapping like linkage analysis has relatively low-resolution and thus it is not feasible to identify causal alleles without additional sequencing of the regions. Future directions should include sequencing and phasing individuals of African-descent in these regions to provide higher resolution of the underlying haplotypes. These findings highlight the potential role of African ancestry in chronic HCV infection.
Methods
Study Population and Quality Control
Individuals were selected from a previously published genome-wide association study of spontaneous Hepatitis C Virus clearance. (9) A principal components analysis was conducted on the diverse study population using EIGENSTRAT, (27) and a subgroup of individuals of African-descent was selected from the larger study. A total of 473 individuals were included in all further analyses. These individuals were drawn from 7 different cohorts: ALIVE (N=247), BAHSTION (N=10), BBAASH (N=19), HGDS (N=9), MHCS (N=34), MHCSII (N=14), and WIHS (N=140). Cohort descriptions can be found in the a previous publication of the genome-wide association study. (9) Of these individuals, 177 had spontaneously cleared, while 296 remained persistently infected. These individuals have been screened for individual missingness <2%, heterozygosity within the range of the study population, as well as cryptic relatedness. Genotypic data had previously gone through standard GWAS quality control measures as well. SNPs were filtered for a minor allele frequency > 1%, genotypic missingness < 5%, and Hardy-Weinberg Equilibrium p-value > 10E-5. A total of 792,721 SNPs were included for analysis.
Local Admixture Estimation
Local ancestry was estimated using LAMP-LD (Local Ancestry in adMixed Populations). (13) Each chromosome was estimated separately for all 473 individuals. Two populations were assumed, consistent with most individuals of mixed African ancestry. The 1000 Genomes Project’s European (CEU, TSI, FIN, GBR, and IBS) and African (YRI and LWK) populations were used as reference populations. Average African Ancestry was calculated by weighting each individual’s chromosomal average by the number of markers used for that chromosome. These weighted chromosomal means were then averaged across all chromosomes.
ANCESTRYMAP
To confirm findings with another analytical program, ANCESTRYMAP (28) was used to estimate areas of the genome associated with clearance. Markers that are informative to the individual’s ancestry were subsetted from the entire GWAS. These markers were drawn from a previously published high-density admixture map (12), as well as a commercially available Illumina admixture panel [http://support.illumina.com/documents/MyIllumina/e5c05695-bf53-4be8-85ab-f9612839dfdc/African_American_Admixture.zip]. A total of 1,748 SNPs were used in this analysis. A Markov Chain Monte Carlo run using 1000 burn-in and 2000 follow-up iterations was used. The Case Control Statistic (CCS), a modified t-statistic, was used to control for possible fluctuations in ancestry estimates that occurred in both the cases and controls.
Association Testing
Genome-wide association results were calculated for the association of local ancestry with spontaneous clearance. An additive logistic regression model was used within R. Signals that were consistent between ANCESTRYMAP and the associations of local ancestry were followed up. These included regions on chromosomes 5, 15, and 20. They were followed up with an additive logistic regression model, adjusted for the number of minor alleles (genotype) at that location, as well as individual HIV status. This provided a finer resolution to the association mapping. To control for small numbers, the top locus for each region was then followed up with a recessive model for local ancestry, and with a dominant model for the genotype. Results remained consistent. A possible interaction with IL28B risk variant rs12979860 was conducted by introducing an interaction term between the SNP and the local ancestry at the location of interest within a logistic regression model and evaluating for significance of the interaction term. This process was repeated for the HLA variant rs4273729. The conditional analysis with rs55817928 was conducted using a dominant model for the SNP, which was then used as a covariate with logistic regression in R.
Imputation
Imputation was conducted in the top regions including 73.5–75.5 Mb on chromosome 5, 76–78 Mb on chromosome 15, and 10–14 Mb on chromosome 20 using IMPUTE2 (29) with a multi-ethnic reference panel from 1000G. Imputed genotypes were subject to quality control measures, such as minor allele frequency >1%, and information content >80%. The chromosome 5 region had 448 genotyped SNPs and 5,712 imputed SNPs that passed quality control. For the chromosome 15 region, there were 385 originally genotyped SNPs and 7,656 imputed SNPs. Chromosome 20 region had 2,366 genotyped SNPs and 27,170 imputed variants. Association testing was conducted using a Frequentist EM algorithm, and an additive model within SNPTEST V2 (29) controlling for HIV status and the first principal component.
Measures of Selection
Data including iHS scores, as well as ancestral states, for three HapMap populations using Phase II data were downloaded from Haplotter (http://haplotter.uchicago.edu/). These populations include the Yoruba from Nigeria (YRI), Caucasians from the United States (CEU), as well as a combined Chinese and Japanese population (CHB+JPT). The iHS (integrated haplotype score) is a statistic designed to detect recent positive selection. An extremely high iHS > 2 is indicative of the ancestral haplotypes being longer than the derived allele background. On the other hand, if there is an extremely low iHS below -2, the derived allele background is longer than the ancestral haplotypes. Both extremes are suggestive of selection.
Ethics Statement
Each individual study obtained consent for genetic testing as approved by the governing institutional review board and provided DNA without identifiers to Johns Hopkins University School of Medicine, where DNA samples were prepared for testing. This process was approved by the Johns Hopkins Medicine Institutional Review Board.
Supplementary Material
Acknowledgments
Primary data are available through the central NIH GWAS data repository at the National Center for Biotechnology Information (NCBI), National Library of Medicine (http://grants.nih.gov/grants/guide/notice-files/NOT-OD-07-088.html).
This project was funded in whole or in part by the Office of AIDS Research through the Center for Inherited Diseases at Johns Hopkins University, the National Institute on Drug Abuse (R01DA013324, DA033541, DA012568, and DA04334), the National Institute of Allergy and Infectious Diseases (U19AI088791 and AI082630), and the Frederick National Laboratory for Cancer Research (contract HHSN261200800001E). This research was supported in part by the Intramural Research Programs of the National Institutes of Health and the Frederick National Laboratory for Cancer Research. The WIHS is funded by the National Institute of Allergy and Infectious Diseases (UO1-AI-35004, UO1-AI-31834, UO1-AI-34994, UO1-AI-34989, UO1-AI-34993, and UO1-AI-42590) and by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (UO1-HD-32632). The study is co-funded by the National Cancer Institute, the National Institute on Drug Abuse, and the National Institute on Deafness and Other Communication Disorders. Funding is also provided by the National Center for Research Resources (UCSF-CTSI Grant Number UL1 RR024131).
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research.
Footnotes
Author contributions follow: GW, PD, CT, WK, and DT lead the writing team; GW and PD performed the primary analysis in conjunction with DT, WK, and CT. DNA and key data for discovery panel was contributed by JG, SM, GK, MP, AC, AK, and RC. All the authors reviewed and approved the final manuscript.
References
- 1.World Health Organization. Hepatitis C Fact Sheet. 2012 Jul 1;:1–4. Available from: http://www.who.int/mediacentre/factsheets/fs164/en/index.html#.
- 2.Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006 May 16;144(10):705–14. doi: 10.7326/0003-4819-144-10-200605160-00004. [DOI] [PubMed] [Google Scholar]
- 3.Thomas DL, Astemborski J, Rai RM, Anania FA, Schaeffer M, Galai N, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA. 2000 Jul 26;284(4):450–6. doi: 10.1001/jama.284.4.450. [DOI] [PubMed] [Google Scholar]
- 4.Muir AJ, Bornstein JD, Killenberg PG, Atlantic Coast Hepatitis Treatment Group Peginterferon alfa-2b and ribavirin for the treatment of chronic hepatitis C in blacks and non-Hispanic whites. N Engl J Med. 2004 May 27;350(22):2265–71. doi: 10.1056/NEJMoa032502. [DOI] [PubMed] [Google Scholar]
- 5.Sarkar M, Bacchetti P, French AL, Tien P, Glesby MJ, Nowicki M, et al. Lower liver-related death in African-American women with human immunodeficiency virus/hepatitis C virus coinfection, compared to Caucasian and Hispanic women. Hepatology. 2012 Aug 27;56(5):1699–705. doi: 10.1002/hep.25859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hepburn MJ, Hepburn LM, Cantu NS, Lapeer MG, Lawitz EJ. Differences in treatment outcome for hepatitis C among ethnic groups. The American Journal of Medicine. 2004 Aug;117(3):163–8. doi: 10.1016/j.amjmed.2004.02.043. [DOI] [PubMed] [Google Scholar]
- 7.Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, Mueller T, et al. Genetic Variation in IL28B Is Associated With Chronic Hepatitis C and Treatment Failure: A Genome-Wide Association Study. Gastroenterology. 2010 Apr 1;138(4):1338–1345.e7. doi: 10.1053/j.gastro.2009.12.056. Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- 8.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’hUigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009 Aug 10;461(7265):798–801. doi: 10.1038/nature08463. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duggal P, Thio CL, Wojcik G, Goedert JJ, Mangia A, Latanich R, et al. Genome-wide association study of spontaneous resolution of hepatitis C virus infection: data from multiple cohorts. Ann Intern Med. 2013 Feb 19;158(4):235–45. doi: 10.7326/0003-4819-158-4-201302190-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013 Jan 6; doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mosbruger TL, Duggal P, Goedert JJ, Kirk GD, Hoots WK, Tobler LH, et al. Large-Scale Candidate Gene Analysis of Spontaneous Clearance of Hepatitis C Virus. J INFECT DIS. 2010 May;201(9):1371–80. doi: 10.1086/651606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith MW, Patterson N, Lautenberger JA, Truelove AL, McDonald GJ, Waliszewska A, et al. A high-density admixture map for disease gene discovery in african americans. Am J Hum Genet [Internet] 2007 Oct 12;74(5):1001–13. doi: 10.1086/420856. Available from: http://ac.els-cdn.com/S000292970764364X/1-s2.0-S000292970764364X-main.pdf?_tid=e75a17a2-aab6-11e2-843a-00000aab0f01&acdnat=1366571511_9aa7d8486a4d44f8f2008f2386e65687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baran Y, Pasaniuc B, Sankararaman S, Torgerson DG, Gignoux C, Eng C, et al. Fast and accurate inference of local ancestry in Latino populations. Bioinformatics. 2012 May 9;28(10):1359–67. doi: 10.1093/bioinformatics/bts144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003 Aug;164(4):1567–87. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian C, Hinds DA, Shigeta R, Kittles R, Ballinger DG, Seldin MF. A genomewide single-nucleotide-polymorphism panel with high ancestry information for African American admixture mapping. Am J Hum Genet. 2006 Oct;79(4):640–9. doi: 10.1086/507954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007 Oct 18;449(7164):851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A Map of Recent Positive Selection in the Human Genome. PLoS Biol. 2006;4(3):e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakamoto H, Okamoto K, Aoki M, Kato H, Katsume A, Ohta A, et al. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat Chem Biol. 2005 Oct 16;1(6):333–7. doi: 10.1038/nchembio742. [DOI] [PubMed] [Google Scholar]
- 19.Hicks AA, Pramstaller PP, Johansson Å, Vitart V, Rudan I, Ugocsai P, et al. Genetic Determinants of Circulating Sphingolipid Concentrations in European Populations. PLoS Genetics. 2009 Oct 2;5(10):e1000672. doi: 10.1371/journal.pgen.1000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hornemann T, Richard S, Rütti MF, Wei Y, Eckardstein von A. Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. Journal of Biological Chemistry. 2006;281(49):37275–81. doi: 10.1074/jbc.M608066200. ASBMB. [DOI] [PubMed] [Google Scholar]
- 21.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008 Feb;9(2):139–50. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 22.Hojjati MR, Li Z, Jiang X-C. Serine palmitoyl-CoA transferase (SPT) deficiency and sphingolipid levels in mice. Biochimica et Biophysica Acta (BBA) – Molecular and Cell Biology of Lipids. 2005 Oct;1737(1):44–51. doi: 10.1016/j.bbalip.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Tsang WY, Dynlacht BD. Double identity of SCAPER: a substrate and regulator of cyclin A/Cdk2. Cell Cycle. 2008 Mar 15;7(6):702–5. doi: 10.4161/cc.7.6.5611. [DOI] [PubMed] [Google Scholar]
- 24.Pham LV, Ngo HTT, Lim Y-S, Hwang SB. Hepatitis C virus non-structural 5B protein interacts with cyclin A2 and regulates viral propagation. Journal of Hepatology. 2012 Nov 1;57(5):960–6. doi: 10.1016/j.jhep.2012.07.006. European Association for the Study of the Liver. [DOI] [PubMed] [Google Scholar]
- 25.Dionisio N, Garcia-Mediavilla MV, Sanchez-Campos S, Majano PL, Benedicto I, Rosado JA, et al. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. Journal of Hepatology. 2009 May;50(5):872–82. doi: 10.1016/j.jhep.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 26.Hill AVS. Evolution, revolution and heresy in the genetics of infectious disease susceptibility. Philosophical Transactions of the Royal Society B: Biological Sciences. 2012 Feb 6;367(1590):840–9. doi: 10.1098/rstb.2011.0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006 Jul 23;38(8):904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 28.Patterson N, Hattangadi N, Lane B, Lohmueller KE, Hafler DA, Oksenberg JR, et al. Methods for high-density admixture mapping of disease genes. Am J Hum Genet. 2004 May;74(5):979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007 Jun 17;39(7):906–13. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.