

Abstract
Background
Calmodulin (CaM) mutations have been identified recently in subjects with congenital long QT syndrome (LQTS) or catecholaminergic polymorphic ventricular tachycardia (CPVT), but the mechanisms responsible for these divergent arrhythmia‐susceptibility syndromes in this context are unknown. We tested the hypothesis that LQTS‐associated CaM mutants disrupt Ca2+ homeostasis in developing cardiomyocytes possibly by affecting either late Na current or Ca2+‐dependent inactivation of L‐type Ca2+ current.
Methods and Results
We coexpressed CaM mutants with the human cardiac Na channel (NaV1.5) in tsA201 cells, and we used mammalian fetal ventricular cardiomyocytes to investigate LQTS‐ and CPVT‐associated CaM mutations (LQTS‐ and CPVT‐CaM). LQTS‐CaM mutants do not consistently affect L‐type Na current in heterologous cells or native cardiomyocytes, suggesting that the Na channel does not contribute to LQTS pathogenesis in the context of CaM mutations. LQTS‐CaM mutants (D96V, D130G, F142L) impaired Ca2+‐dependent inactivation, whereas the CPVT‐CaM mutant N54I had no effect on Ca2+‐dependent inactivation. LQTS‐CaM mutants led to loss of Ca2+‐transient entrainment with the rank order from greatest to least effect: CaM‐D130G~CaM‐D96V>>CaM‐F142L. This rank order follows measured Ca2+‐CaM affinities for wild‐type and mutant CaM. Acute isoproterenol restored entrainment for CaM‐130G and CaM‐D96V but caused irreversible cytosolic Ca2+ overload for cells expressing a CPVT‐CaM mutant.
Conclusions
CaM mutations associated with LQTS may not affect L‐type Na+ current but may evoke defective Ca2+‐dependent inactivation of L‐type Ca2+ current.
Keywords: calcium, calmodulin, cardiomyocyte, long QT Syndrome, L‐type Ca2+ channel
Introduction
Congenital long QT syndrome (LQTS) and catecholaminergic polymorphic ventricular tachycardia (CPVT) are 2 genetic disorders of heart rhythm that may present during childhood and cause life‐threatening cardiac arrhythmias.1 The molecular mechanisms underlying arrhythmia susceptibility have been inferred from studies of mutant gene products, which are largely ion channels or channel regulators. In LQTS, loss of function in potassium channels or gain of function in either Na or Ca2+ channels represent the major molecular mechanisms. In contrast to disturbances in plasma membrane ion channel function, CPVT arises from disordered intracellular Ca2+ regulation, most frequently because of mutation of the ryanodine receptor Ca2+‐release channel of the sarcoplasmic reticulum (SR). These advances have improved our understanding of arrhythmogenesis in general and have revealed new therapeutic targets.
Recently, recurrent cardiac arrest in infants with a severe form of LQTS was associated with mutations in CALM1 (D130G, F142L) or CALM2 (D96V), 2 genes encoding the ubiquitous Ca2+‐signaling protein calmodulin (CaM).2 Independently, 2 distinct CaM mutations (N54I, N98S) were discovered in subjects with a CPVT‐like syndrome although one of these mutations (N98S) has also been identified in a subject with LQTS.3 Most of the mutations affect conserved amino acid residues that are known to participate in Ca2+ binding or in energetic coupling of ion binding to CaM activation. Biochemical studies revealed that most CaM mutations, except N54I, cause reduced Ca2+‐binding affinity.2,4 However, the cellular basis for arrhythmogenesis in the setting of CaM mutations has not been elucidated.
CaM controls a large number of enzymes, ion channels, and other proteins.5 In cardiomyocytes, key CaM‐interacting proteins include those critically involved in beat‐to‐beat Ca2+ homeostasis. CaM is prebound to the cardiac L‐type Ca2+ channel (LTCC), where it senses local Ca2+ influx and cytosolic Ca2+. The complex of Ca2+‐CaM promotes rapid inactivation of LTCC Ca2+ current (ICa,L).6 This process, known as Ca2+‐dependent inactivation (CDI), is a major determinant of cardiac excitability.7 Engineered CaM with carboxyl lobe (C‐lobe) mutations abrogating C‐lobe Ca2+ binding attenuated CDI and prolonged action potential (AP) duration.7 The majority of the z‐line CaM also binds to ryanodine receptor 2 (RyR2),8 and CaM inhibits RyR2 openings independent of Ca2+ concentration.9–10 Disrupting CaM‐RyR2 interaction has multiple effects including increased opening of RyR2, diminished ICa,L, and increased cytosolic Ca2+ at negative membrane potentials, creating a molecular substrate for arrhythmogenic triggers.11 Consequently, at the cellular level, we anticipate that CaM mutants targeting CDI will have distinct effects from those disrupting CaM‐RyR2 interactions. Most recently, CPVT‐CaMs were shown to cause arrhythmogenic Ca2+ disturbances in permeabilized cardiomyocytes that were distinct from LQT‐CaM–provoked alterations.12
In this study, we examined the effects of LQTS‐associated CaM mutations on Na and Ca2+ channel currents to elucidate cellular and molecular mechanisms underlying arrhythmia predisposition. We found that CaM mutations had inconsistent effects on Na channels but caused impaired CDI of ICa,L in cardiomyocytes and dysregulation of cytosolic Ca2+. In parallel, we examined the impact of the CPVT‐associated mutation, N54I, on ICa,L and observed no direct effects on LTCC current in cardiomyocytes; however, this mutation caused significant abnormalities in Ca2+ homeostasis. Our findings implicate different molecular substrates for arrhythmogenesis associated with CaM mutations.
Methods
All experimental procedures and protocols involving animals were approved by the animal care and use committee of the University of Kentucky and conformed to the NIH Guide for Care and Use of Laboratory Animals. Isolated fetal ventricular cardiomyocytes (FVM) were prepared as previously described,13 except embryonic day 18 to 19 pregnant dams were used.
Mutagenesis of Calmodulin
Mutant CaMs were engineered by making one of the following amino acid substitutions: N54I, D96V, N98S, D130G, or F142L. A mammalian expression plasmid was used to drive expression of wild‐type (WT) or mutant CaM by the immediate early CMV promoter along with the coding region for either green fluorescent protein or CD8 preceded by the IRES2 element as cotransfection markers.
Heterologous Expression of Human NaV1.5 With Calmodulin
Plasmids encoding recombinant human cardiac Na channel isoforms (NaV1.5) were described previously.14 For studies of Na channel function, WT cardiac Na channel cDNA (0.5 μg) and WT or mutant CaM (D130G, D96V, and F142L) cDNA (0.5 μg) in vectors encoding the cotransfection marker CD8 were transiently transfected into tsA201 cells using FuGene HD (Roche Diagnostics, Indianapolis, IN), then incubated for 48 hours at 37°C prior to electrophysiological measurements. Dynabeads‐CD8 (Invitrogen Life Technologies, Carlsbad, CA) were added to cell culture media according to manufacturer's instructions to mark cells transfected successfully. Cells selected for patch clamp recordings exhibited CD8 expression with Dynabeads attached to the cell surface and were easily distinguishable from non–CD8‐expressing cells.
Fetal Ventricular Myocyte Tissue Harvest or Cell Culture
Hearts of embryonic day 18 to 19 (a fetal stage) mouse (ICR outbred strain; Harlan Laboratories) were dissected free of connective tissues, and ventricles were separated from atria. Cells were enzymatically dispersed and cultured as previously described.15 Briefly, about 10 fetuses were minced and quickly transferred to nominally Ca2+‐free digestion buffer containing 0.5 mg/mL collagenase (type II; Worthington Biochemical Corporation) and 1 mg/mL pancreatin for two 15‐minute cycles. Digested tissue yielded a large fraction of single spontaneously beating cells in culture media consisting of DMEM plus 10% FBS. Cells were transfected in 24‐well plates using Lipofectamine transfection reagent (Invitrogen Life Technologies). Cells were transfected with plasmids given above and used 24 to 48 hours after transfection. FVM were selected for study based on enhanced green fluorescent protein fluorescence unless otherwise noted.
Electrophysiology
Na currents were recorded at room temperature using the whole‐cell patch clamp technique as described previously.14,16 The extracellular bath solution contained the following (in millimoles per liter): 140 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.35 (adjusted with NaOH). The low‐Ca2+ intracellular pipette solution contained the following (in millimoles per liter): 10 NaF, 100 CsF, 20 CsCl, 20 BAPTA, 10 HEPES, 10 glucose, pH 7.35 (adjusted with CsOH). The high‐Ca2+ (~1‐μmol/L free) intracellular pipette solution contained the following (in millimoles per liter): 10 NaF, 100 CsF, 20 CsCl, 1 BAPTA, 0.9 CaCl2, 10 HEPES, 10 glucose, pH 7.35 (adjusted with CsOH).17 Osmolarity was adjusted to 310 mOsm/L with sucrose for the bath solution and 300 mOsm/L for pipette solution. Data were acquired with an Axopatch 200B patch clamp amplifier and pClamp 10.2 software. Tetrodotoxin‐sensitive persistent current was determined with 200‐ms depolarization to −30 mV as the average current recorded between 190 and 200 ms and reported as a percentage of peak current following digital subtraction of currents recorded in the presence and absence of 30 μmol/L tetrodotoxin (Tocris Biosciences, Ellisville, MO). All data were analyzed with pClamp 10.2 (Axon Instruments, Inc, Sunnyvale, CA) and plotted using SigmaPlot 10 (IBM Corp, Armonk, NY) software. Unless otherwise noted, statistical comparisons were made by using a 1‐way ANOVA and Bonferroni correction to WT NaV1.5 coexpressed with WT CaM. Statistical significance was assumed for P<0.05. Results are presented as mean±SEM for all pooled data.
FVM ICa,L recordings were recorded in the whole‐cell configuration of the patch clamp technique as previously described.18 All recordings were performed at room temperature (20 to 22°C). The pipette solution consisted of (in millimoles per liter): 125 Cs‐methanesulfonate, 15 TEA‐Cl, 1 MgCl2, 0.5 EGTA, 5 HEPES, pH 7.2. Bath solution contained (in millimoles per liter):150 NMDG, 2.5 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, 5 4‐aminopyridine, pH7.2. Activation voltage dependence parameters were obtained by fitting the current‐voltage slope conductance transform to a Boltzmann distribution of the form G(V)=Gmax/(1+exp(V½/k)) in which Gmax is the maximal conductance and V½ is the activation midpoint. Results are presented as mean±SEM for all pooled data.
Cytosolic Ca2+ Imaging
Cardiac myocytes were loaded with 2 μmol/L fura‐2‐AM for 10 minutes in a 5% CO2 incubator and then de‐esterified in Tyrodes (140 NaCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES [free acid], 5.4 KCl, 10 glucose, pH 7.4, NaOH) solution for ~20 minutes. Field‐stimulated Ca2+ transients were recorded from the annulus of the photometry tube focused on a single cell or a cell cluster. All recordings were performed at 20 to 22°C. The cells were excited with light of 340‐nm and 380‐nm wavelengths. The images obtained at 340 and 380 nm were divided pixel by pixel, and the ratio data were reported. Data were collected and analyzed with IonOptix (Milton, MA) hardware and software. Additional offline transient analysis was performed with custom routines written in MatLab (Northampton, MA). Exponential fitting was performed using pClamp 9.2 (Axon Instruments, Union City, CA). The function used for exponential fitting of caffeine‐induced Ca2+ transient relaxations was f(t)=Ai×exp (−t/τi)+C.
Statistical Analysis
Data were analyzed for Figures 5C, 6B, 7B, and 7D with the chi‐square test. Figure 9A was analyzed with 1‐way ANOVA with Dunn's multiple comparison test. Figure 9A and the remaining data analysis passed the D'Agostino and Pearson test and the Shapiro‐Wilk test for normality, and tests of statistical difference for the remaining data were performed using the t test with statistical significance correction of multiple comparisons and α=0.05 using the Holm‐Sidak method in GraphPad Prism version 6.0 for Windows (GraphPad Software, La Jolla, CA).
Figure 5.
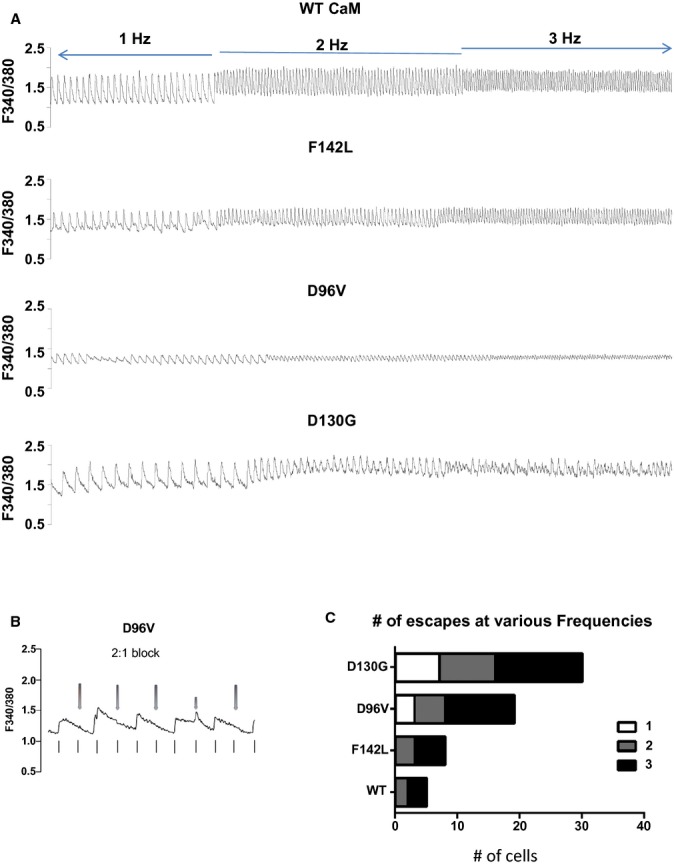
Long QT syndrome (LQTS) CaM mutant expression induces loss of field‐stimulation entrainment in rank order with Ca2+ affinity. A, Representative Ca2+ transients for pacing frequencies of 0.5, 1, and 2 Hz. B, Representative Ca2+ transient for a cardiomyocyte expressing D96V‐CaM showing 2:1 block. The stimulus frequency is 1 Hz, and individual stimuli are denoted by vertical lines below the Ca2+ transients. The red arrows above the Ca2+ transient show the stimuli that did not elicit a Ca2+ transient. C, Summary plot of the prevalence of escape from entrainment events in cardiomyocytes expressing exogenous WT CaM or LQTS‐CaM mutants. Higher stimulus frequency evoked a greater prevalence of escape events in rank order with Ca2+‐CaM affinity. For statistical testing, we lumped together escape events at 2 and 3 Hz, P<10−4. CaM indicates calmodulin; WT, wild type.
Figure 6.
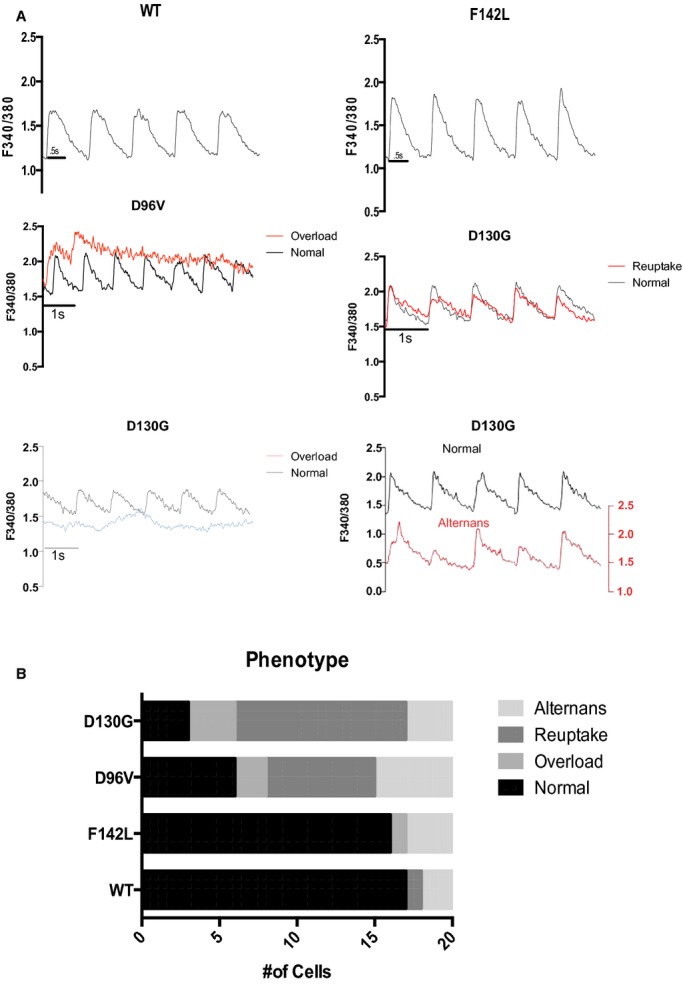
Loss of Ca2+ transient entrainment is dominated by faulty reuptake in myocytes expressing the lowest Ca2+‐affinity calmodulin (CaM) mutants. A, Representative Ca2+ transients for 1‐Hz stimulation frequency. WT CaM and F142L‐CaM expressing cardiomyocyte Ca2+ transients were entrained to 1‐Hz stimuli. Pathological loss of entrainment categorized as overload (middle and lower left for D96V‐CaM and D130G, respectively), reuptake defective (middle right for D130G‐CaM), and alternans (lower right). The lower left D130G trace displaying overload (blue) is at a level greater than the WT diastolic level (upper left panel). Note that the representative alternans trace (lower right, red) is displaced by 1 fluorescent unit to illustrate an alternating pattern of relatively large‐ and small‐amplitude transients. B, Summary of pathological Ca2+‐transient phenotypes. For statistical testing, we tested the hypothesis that long QT syndromeh CaM mutants induced any pathological phenotype. N=20 cells for each group; P<10−4. WT indicates wild type.
Figure 7.
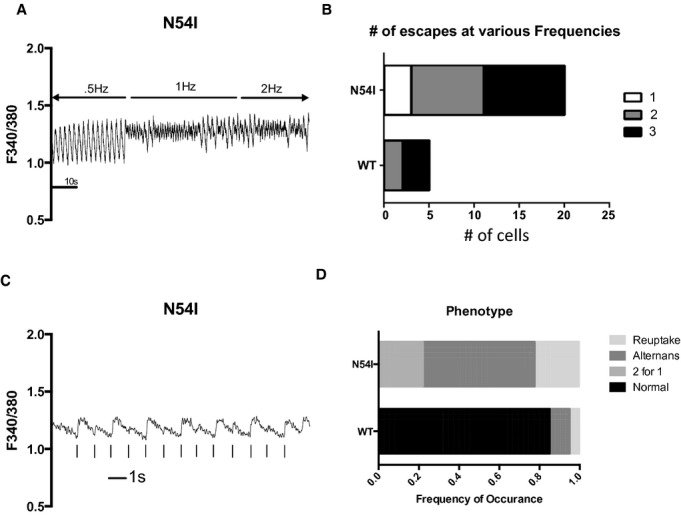
Catecholaminergic polymorphic ventricular tachycardia calmodulin induces abnormal Ca2+ transients. A, Representative Ca2+ transients in cardiomyocytes expressing calmodulin‐N54I. B, Loss of entrainment for frequencies ≥1 Hz; for 3 Hz, no entrainment was possible. P<10−4. C, Representative Ca2+ transient displaying alternans at 1 Hz. D, For 1 Hz, all calmodulin‐N54I cardiomyocytes displayed abnormal Ca2+‐transient phenotypes; the majority of cells displayed alternans. P<10−4. WT indicates wild type.
Figure 9.
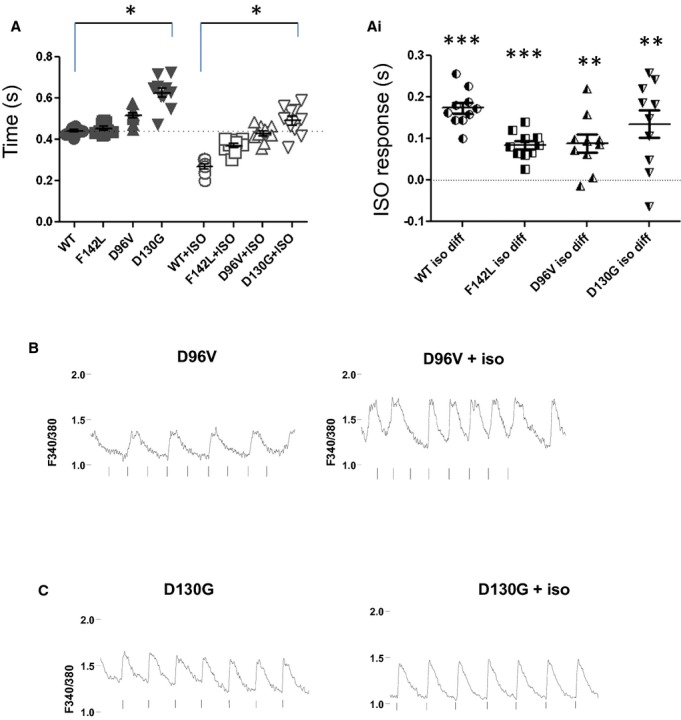
β‐adrenergic stimulation speeds twitch duration and restores entrained rhythmicity in cardiomyocytes expressing relatively low Ca2+‐affinity long QT syndrome calmodulin (CaM) mutants. A, Stimulated twitch decay (1 Hz) at baseline and following 100 nmol/L ISO. *P<0.05. Ai, Regardless of exogenous CaM expression, the twitch decay rate was faster following ISO. Data from (A) replotted as cell‐by‐cell response to ISO.**P<0.001; ***P<10−4; n=10 or 11. B, Representative Ca2+ transient for D96V‐CaM expressing fetal ventricular myocyte. Left panel shows 1‐Hz stimulation to illustrate baseline 2:1 block. Right panel shows an example of partial restoration of entrainment of Ca2+ transient to field stimulation. With ISO, the 2:1 activity spontaneously reverts to 1:1 activity. n=10. C, Representative Ca2+ transient for D130G‐CaM expressing fetal ventricular myocyte. Left panel shows 0.5‐Hz stimulation to illustrate baseline alternans. Right panel shows an example of recovery of entrainment following ISO. n=10. ISO indicates isoproterenol, WT, wild type.
Results
Effect of CaM Mutants on Cardiac Na Channels
Because CaM is known to regulate cardiac Na channel inactivation and a specific defect in Na channel inactivation (increased persistent Na current) occurs in LQTS,19 we investigated the effects of LQTS‐CaM mutants on heterologously expressed NaV1.5 as well as on native Na current. We performed heterologous coexpression of WT or mutant CaM with human NaV1.5 in tsA201 cells and then measured electrophysiological properties of the expressed channels. When recording Na current with a nominally Ca2+‐free intracellular solution, we did not observe differences in the level of persistent Na current (expressed as a percentage of peak current) for any of the CaM mutations compared with WT CaM. Consequently, we repeated these experiments with elevated intracellular calcium (~1 μmol/L free Ca2+) to provide a saturating concentration of Ca2+ to promote binding of CaM to NaV1.5.2,17 Furthermore, because of the early age of onset of LQTS in the reported probands, we also tested the effects of CaM mutations on a fetal and neonatal‐expressed NaV1.5 splice variant.14 Cells coexpressing CaM‐D130G with fetal NaV1.5 exhibited 7.5‐fold larger persistent Na current (1.5±0.4%) compared with cells coexpressing the fetal splice variant with WT CaM (0.2±0.1%; P<0.05) when recordings were made with high intracellular calcium (Figure 1A). This greater level of persistent current was not observed when CaM‐D130G was coexpressed with the canonical (adult expressed) splice isoform of NaV1.5 or with low intracellular calcium (Table 1). In contrast, cells coexpressing fetal or canonical NaV1.5 splice isoforms with either CaM‐D96V or CaM‐F142L did not exhibit abnormal levels of persistent Na current under high calcium conditions (Table 1). Other Na channel properties including conductance‐voltage relationship, voltage dependence of steady‐state inactivation, inactivation kinetics, and recovery from inactivation were largely unaffected by CaM mutants (Table 2). Furthermore, expression of CaM‐D130G in mouse FVM did not evoke detectable differences in the level of persistent Na current compared with cells transfected with WT CaM (Figure 1B). Given the inconsistent effect of CaM mutants on heterologous and native Na current, we concluded that NaV1.5 dysfunction was not the major cause of LQTS in the setting of CaM mutations.
Figure 1.
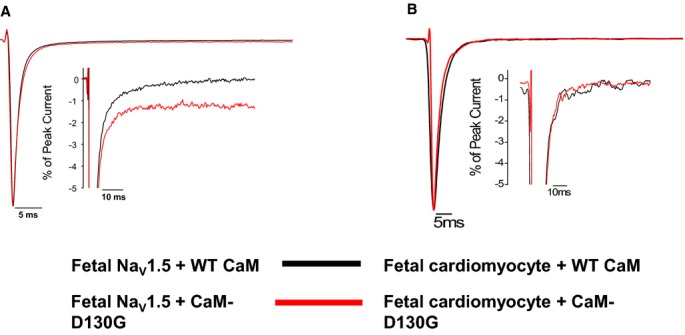
Long QT syndrome (LQTS) CaM mutation effects on persistent Na current are limited to a single splice variant of NaV1.5 and a single CaM mutation. A, CaM‐D130G evoked increased persistent sodium current with fetal NaV1.5. Averaged tetrodotoxin‐sensitive currents were normalized to the peak current measured at −30 mV during a 200‐ms depolarization recorded under high intracellular Ca2+ concentration (see Methods). The inset represents the same data plotted on an expanded vertical scale. Summary data are provided in Table 1. B, CaM‐D130G has no significant effect on persistent Na current in fetal ventricular cardiomyocytes. Tetrodotoxin‐sensitive currents were normalized to the peak current measured at −30 mV during a 300‐ms depolarization recorded under high intracellular Ca2+ concentration (see Methods). The inset represents the same data plotted on an expanded vertical scale. CaM indicates calmodulin; WT, wild type.
Table 1.
Persistent Sodium Current in Cells Expressing WT or Mutant CaM
| Persistent Current % | n | |
|---|---|---|
| NaV1.5+apo WT* CaM | 0.3±0.2 | 4 |
| NaV1.5+apo CaM‐D130G | 0.2±0.1 | 4 |
| NaV1.5+WT CaM | 0.3±0.1 | 5 |
| NaV1.5+CaM‐D130G | 0.2±0.1 | 5 |
| NaV1.5+CaM‐D96V | 0.2±0.1 | 7 |
| NaV1.5+CaM‐F142L | 0.1±0.1 | 4 |
| Fetal NaV1.5+apo WT CaM | 0.4±0.1 | 6 |
| Fetal NaV1.5+apo CaM‐D130G | 0.6±0.2 | 7 |
| Fetal NaV1.5+WT CaM | 0.2±0.1 | 9 |
| Fetal NaV1.5+CaM‐D130G | 1.5±0.4*,* | 13 |
| Fetal NaV1.5+CaM‐D96V | 0.4±0.1 | 8 |
| Fetal NaV1.5+CaM‐F142L | 0.6±0.5 | 8 |
CaM indicates calmodulin; WT, wild type.
apo WT CaM is Apocalmodulin (apo WT CaM is Ca2+‐free CaM; WT refers to the disease‐associated sites identified in the probands2).
P<0.05 compared to fetal NaV1.5+WT CaM.
P<0.05 compared to fetal NaV1.5+WT CaM with 10 μmol/L KN‐93.
Data compared using Kruskal‐Wallis 1‐way ANOVA and Dunn's Test.
Table 2.
CaM Mutants Do Not Exhibit Major Biophysical Effects on Peak INa
| Voltage‐Dependence of Activation | Steady‐State Availability | Recovery From Inactivation | ||||||
|---|---|---|---|---|---|---|---|---|
| V1/2 (mV) | k (mV) | n | V1/2 (mV) | k (mV) | n | τ (ms) | n | |
| Biophysical parameters | ||||||||
| NaV1.5+WT CaM | −45.0±2.1 | 6.6±0.5 | 9 | −102.9±3.2 | −8.4±0.2 | 10 | 10.9±1.3 | 6 |
| NaV1.5+CaM‐D130G | −43.1±2.3 | 6.6±0.5 | 11 | −98.5±1.7 | −8.1±0.1 | 10 | 8.8±1.1 | 8 |
| NaV1.5+CaM‐D96V | −42.6±1.1 | 7.8±0.3 | 10 | −105.2±1.0 | −8.7±0.4 | 13 | 11.2±0.5 | 11 |
| NaV1.5+CaM‐F142L | −41.8±2.0 | 6.7±0.8 | 10 | −96.4±2.2 | −8.5±0.4 | 11 | 8.7±0.6 | 10 |
| Fetal NaV1.5+WT CaM | −38.4±1.7 | 7.5±0.5 | 11 | −103.3±2.2 | −9.7±1.0 | 9 | 9.1±0.8 | 10 |
| Fetal NaV1.5+CaM‐D130G | −41.0±1.7 | 7.2±0.5 | 10 | −98.6±1.2 | −8.9±0.3 | 8 | 11.0±1.3 | 9 |
| Fetal NaV1.5+CaM‐D96V | −35.2±2.1* | 9.4±0.4* | 9 | −99.1±1.4 | −13.1±2.3** | 9 | 12.1±1.6 | 7 |
| Fetal NaV1.5+CaM‐F142L | −35.4±1.7* | 8.3±0.6 | 6 | −96.3±1.7 | −8.4±0.3 | 7 | 8.9±1.4 | 6 |
All recordings were performed with high intracellular calcium concentration. CaM indicates calmodulin; WT, wild type.
P<0.05 compared to NaV1.5+WT CaM.
P<0.05 compared to fetal NaV1.5+WT CaM.
P<0.005 compared to NaV1.5+WT CaM.
CDI is Slowed by LQTS‐CaM But Not by a CPVT‐Associated CaM Mutant
We tested the hypothesis that LQTS‐CaM mutations with impaired C‐domain Ca2+ affinity will slow ICa,L decay in a native cardiomyocyte environment due to impaired CDI.7 Figure 2A illustrates representative current sweeps normalized to peak current for FVM expressing exogenous WT CaM superimposed on traces from cells expressing each of the LQTS‐CaM mutants. All LQTS‐CaM mutants slow ICa,L decay with the 2 relatively low Ca2+ Kd CaM‐mutants with altered highly conserved aspartic acids that directly chelate the Ca2+ ion, showing relatively greater effect2 (Figure 2). Current density and voltage dependence of current activation were not different between FVM expressing WT or LQTS‐CaM mutants (Figure 3; Table 3). In contrast, the CPVT‐association CaM mutation N54I did not significantly affect CDI (Figure 4). These data suggest that attenuated CDI and resulting slower ICa,L decay can contribute to mutant CaM‐associated LQTS but not CPVT.
Figure 2.
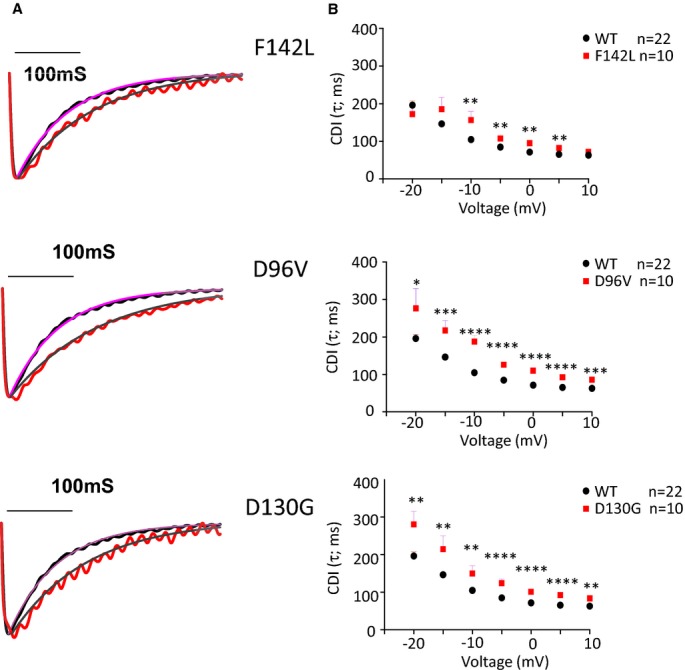
Long QT syndrome (LQTS) calmodulin (CaM) mutants slow CDI. A, Representative traces recorded during a depolarization to 0 mV. The single exponential fit of a representative WT CaM current sweep (dotted line) is illustrated for reference. The smooth line through the current traces is a single exponential fit of the form: I(t)=Ipeak*e−t/τ. B, Inactivation time constant (τ) plotted as a function of voltage. For clarity, the WT CaM data are illustrated along with each LQTS‐CaM mutation plot. **P<0.01; ****P<10−4. CDI indicates Ca2+‐dependent inactivation; WT, wild type.
Figure 3.
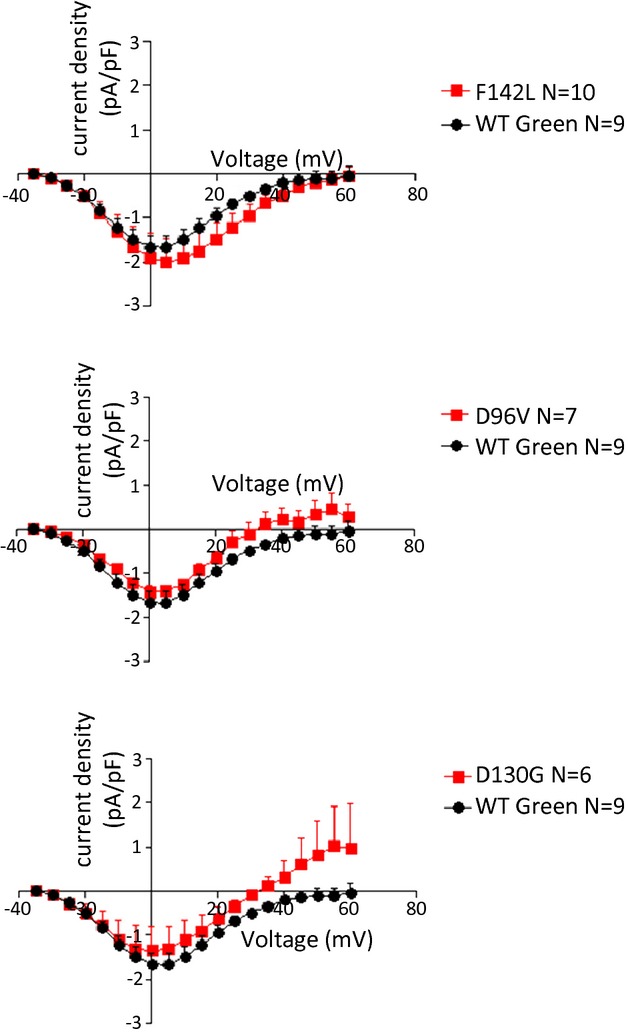
Peak current‐voltage relationships for ICa,L in fetal ventricular cardiomyocytes. WT calmodulin data are repeated in each panel for clarity of presentation. No significant differences were noted in voltage dependence or amplitude. See Table 3 for Boltzmann distribution fitted parameters. WT indicates wild type.
Table 3.
Gmax, V½, and k in Fetal Cardiac Ventricular Myocytes Expressing WT or long QT CaM
| Gmax | V1/2 | k | n | |
|---|---|---|---|---|
| WT CaM nongreen | 0.109±0.018 | −7.2±0.6 | 6.6±0.2 | 13 |
| WT CaM green | 0.055±0.008* | −8.8±0.4 | 6.6±0.2 | 9 |
| WT CaM combination | 0.087±0.012 | −7.9±0.4 | 6.6±0.1 | 22 |
| CaM‐F142L | 0.059±0.014* | −5.6±1.7 | 7.2±0.3 | 10 |
| CaM‐D96V | 0.057±0.013 | −6.3±0.5 | 6.1±0.3 | 6 |
| CaM‐D130G | 0.054±0.024 | −9.8±1.5 | 6.3±0.1 | 6 |
CaM indicates calmodulin; WT, wild type.
P<0.05 compared with WT CaM nongreen.
Figure 4.
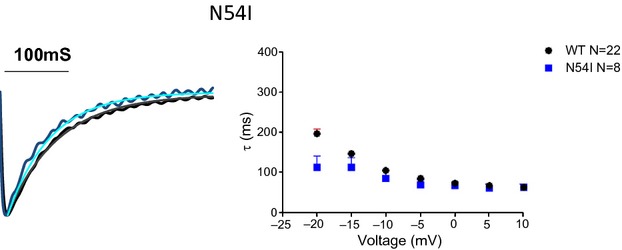
Calmodulin (CaM) N54I does not significantly alter Ca2+‐dependent inactivation of L‐type Ca2+ channel Ca2+ current. Left, representative L‐type Ca2+ channel Ca2+ current for WT CaM and CaM‐N54I superimposed. Smooth line is a single exponential function fit. Right, pooled data for the time constant of decay of the fitted exponential decay as a function of voltage. For Vtest −20 mV, CDI is significantly faster (P<0.05). For all other potentials, there is no significant difference in rate of decay. WT indicates wild type.
Rate Dependence of Entrainment to Field Stimulation
Field stimulation of FVM causes a Ca2+‐induced Ca2+ release that follows a fire‐diffusion‐fire paradigm qualitatively similar to that of mature cardiomyocytes.20 Figure 5A illustrates representative Ca2+ transients from FVMs expressing either WT or mutant CaM continuously paced from quiescence to 2 Hz. For cells expressing WT CaM, there was a synchronous entrainment of field stimulation to Ca2+ transient (eg, 1:1 ratio; Figure 5A, top panel). Expression of CaM‐F142L does not compromise entrainment of Ca2+ transients at low stimulation frequencies. The lower Ca2+‐affinity mutations CaM‐D96V and CaM‐D130G show frequent loss of entrainment, as shown by the presence of 2:1 pacing block (eg, 2 stimuli for 1 transient; Figure 5B). The loss of entrainment observed for LQTS‐CaM mutants summarized as “number of escapes” follows the rank order of Ca2+‐CaM affinity, whereby cardiomyocytes expressing the weaker Ca2+‐affinity CaM mutants escaped from pacing at progressively lower frequencies (Figure 5C).
We categorized the Ca2+ transient phenotypes into those displaying alternans, and those exhibiting slower Ca2+ reuptake manifested as smaller amplitude, slower decaying, and elevated diastolic levels (Figure 6). The prevalence of pathological Ca2+‐transient phenotypes tracks with the rank order of Ca2+‐CaM affinity. The lowest affinity CaM mutations, D96V and D130G, uniquely displayed overload phenotypes (defined as diastolic Ca2+ >1 standard deviation above the WT‐CaM diastolic fluorescence level), and cells expressing D96V and D130G also show a preponderance of reuptake anomalies (Figure 6). In summary, LQTS‐CaM mutants induce arrhythmogenic activity in developing ventricular cardiomyocytes. The severity of the dysfunction induced by various LQTS‐CaM mutations tracks with reported Ca2+‐CaM affinities.2 In contrast, the CPVT‐CaM mutant N54I has near normal Ca2+ affinity,4 but myocytes expressing this mutant exhibited a marked inability to be paced by field stimulation (Figure 7A and 7B). All N54I cardiomyocytes tested displayed pathological Ca2+ transients with 1‐Hz pacing, with the majority showing alternans (Figure 7C and 7D). These findings indicate that disrupted Ca2+‐transient entrainment is a common disease‐associated end point, despite the unique effect of LQTS‐CaM slowing of CDI.
LQTS‐CaM Mutations Perturb Ca2+ Handling
We investigated whether elevation of cytosolic Ca2+ could drive the loss of entrainment induced by LQTS‐CaM mutants (Figure 6). FVM expressing LQTS‐CaM mutations have significantly higher twitch amplitude than cells expressing WT CaM (Figure 8A). Notably, the mean twitch amplitude for D130G is less than that for F142L and D96V. Similarly, diastolic Ca2+ is higher in cells expressing the lower Ca2+‐affinity CaM mutations D96V and D130G (Figure 8B). CaM has multiple sites of action with respect to Ca2+ homeostasis: CaM modulation of RyR2 and Ca‐CaMKII regulation of SR Ca2+ stores. Consequently, we probed the contribution of LQTS‐CaM mutation to SR Ca2+ load by assessing caffeine‐releasable Ca2+ stores (Figure 8C). The mean SR Ca2+ appears greater in LQTS‐CaM mutant expressing cardiomyocytes compared with cells expressing WT CaM, but the differences did not reach statistical significance (Figure 8D). Examination of the decay of caffeine‐induced Ca2+ transient reveals no significant difference for any of the LQTS‐CaM mutants (Figure 8E). This suggests that Na+/Ca2+ exchange (NCX) function is not altered by LQTS‐CaM mutations. These results are consistent with the model that LQTS‐CaM mutant promoter increased trigger Ca2+ from the slowed decay of ICa,L. NCX is the main surface membrane efflux pathway, thus more LTCC Ca2+ influx paired with unchanged NCX‐based efflux could lead to elevated cytosolic Ca2+.
Figure 8.
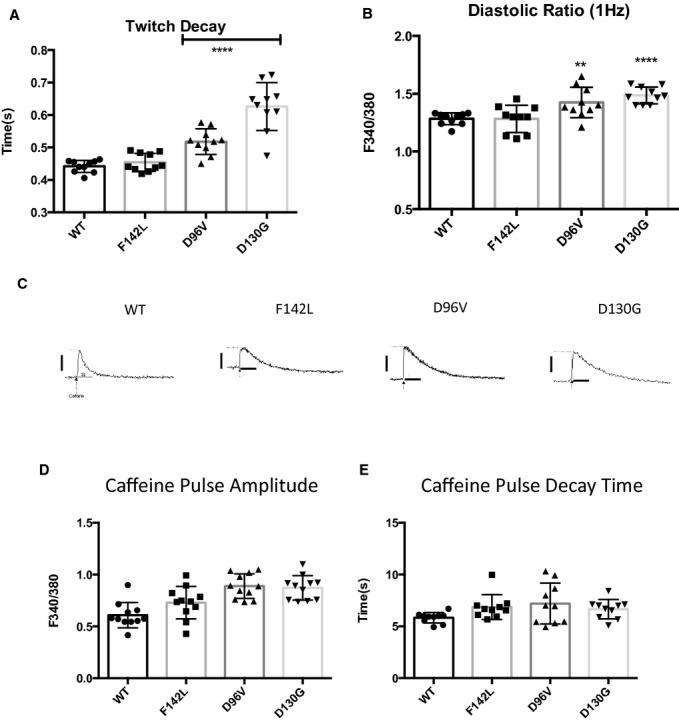
LQT‐CaM Long QT calmodulin (CaM) slows Ca2+ reuptake but does not alter sarcoplasmic reticulum Ca2+ load. A, The mean stimulated twitch decay rate is progressively slowed in rank order of Ca2+‐CaM affinity. Relatively low Ca2+‐affinity D96V and D130G‐CaM show a significantly slower decay rate for 1‐Hz stimulation. ****P<10−4. n=10. B, The mean diastolic Ca2+ ratio is progressively elevated in rank order of Ca2+‐CaM affinity. Relatively low Ca2+‐affinity D96V and D130G‐CaM show a significantly slower decay rate for 1‐Hz stimulation. **P<0.01; ****P<10−4. n=10. C, Representative caffeine‐induced Ca2+ transients. Scale bars: y‐axis: 0.5 F340/F380 units; x‐axis: 5 s. D, Sarcoplasmic reticulum Ca2+ is not significantly different between WT CaM and long QT CaMs. E, Caffeine‐induced Ca2+‐transient decay rate is not significantly different, suggesting no change in NCX function. WT indicates wild type.
Isoproterenol Restores Entrainment for LQTS‐CaM Mutations
The LQTS‐CaM mutants D96V and D130G cause abnormal Ca2+ transients at elevated stimulation frequencies (Figures 4 and 5) and exhibit elevated cytosolic Ca2+ levels (Figure 8). Higher Ca2+ levels could drive arrhythmogenic activity analogous to that seen for CPVT.21 Faster pacing rates protect against catecholamine‐induced triggered activity in CPVT models22; however, Ca2+‐transient abnormalities are exacerbated at even modestly higher rates (≥1 Hz) in FVM expressing relatively low Ca2+‐affinity LQTS‐CaM mutants (Figure 5). Isoproterenol (ISO) enhances the rate of Ca2+ reuptake to the SR in developing cardiomyocytes (Figure 9A and 9Ai), and this effect is preserved for cardiomyocytes expressing LQTS‐CaM mutants (Figure 9A and 9Ai). Given the propensity of defective Ca2+ reuptake observed in D96V and D130G‐CaM expressing cardiomyocytes, we tested the hypothesis that ISO treatment alleviates these LQTS‐CaM mutant‐induced Ca2+‐transient loss of entrainment. Figure 9B shows a representative D96V‐CaM‐expressing cardiomyocyte. Prior to ISO addition, a 2:1 block occurs (Figure 9B, upper left). ISO induces a partial rescue of 1:1 entrainment manifested as a spontaneous transition from 2:1 to 1:1 (stimulus to Ca2+ transient; Figure 9B, upper right). This type of behavior was scored as “partial rescue” because of spontaneous restoration and reversion of pathological Ca2+ transients. The relatively lower Ca2+ affinity D130G‐CaM exhibited alternans at 0.5 Hz pacing frequency (Figure 9C, upper left). ISO rapidly results in full restoration of normal Ca2+ transients in this example (Figure 9C, upper right). Overall, ISO caused some degree of restoration of normal Ca2+ homeostasis in 65% of cells tested. For D96V‐CaM‐expressing cardiomyocytes, 9 and 2 of 17 cells showed partial and full restoration, respectively. For D130G‐CaM‐expressing cardiomyocytes, 4 and 3 of 11 cells showed partial and full restoration, respectively. In sharp contrast, acute ISO led to cell death in 6 of 9 CPVT‐CaM mutant‐expressing cardiomyocytes and had no beneficial effects in the remaining cells tested. These data suggest that slowed Ca2+ reuptake contributes to LQTS‐CaM–associated arrhythmogenesis, and enhancing the rate of reuptake is a plausible mechanism for ameliorating LQTS‐CaM dysfunction.
Discussion
The major new finding of this study is that CaM mutants associated with LQTS cause significantly slowed CDI of ICa,L and alter Ca2+ homeostasis in FVM. We also excluded a major contribution of dysregulated Na current caused by LQTS‐CaM mutations. In contrast, the CPVT‐CaM mutant N54I had no effect on CDI but disrupted Ca2+ homeostasis. We conclude that LQTS‐CaM evokes the novel mechanism of CDI slowing to initiate the sequence of cellular physiological events leading to ventricular arrhythmia.
This study shows a rank‐order relationship between Ca2+‐CaM affinity and the propensity for abnormal Ca2+ homeostasis. This relationship holds for the 3 tested LQTS‐CaM mutants and is distinct for CPVT‐CaM mutant N54I. LQTS is driven by a prolongation of the ventricular cellular AP duration, and AP duration is determined by the sum of ionic conductance, principally net voltage‐gated K+ channels and LTCC. CaM prebound to LTCC confers a direct relationship between CaM alterations of LTCC current and AP duration, whereby increased Ca2+ influx prolongs AP duration. LQTS‐CaM mutations occur de novo in the carboxyl‐terminus (C‐lobe) of CaM.2 The C‐lobe of LTCC‐bound CaM senses Ca2+ entry through single LTCC channel openings, and this occurs in a restricted space where local [Ca2+] can reach levels as high as 60 to 100 μmol/L.23–24 The LQTS‐CaM mutation F142L C‐lobe Ca2+‐affinity Kd is 15 μmol/L, well below the estimated local [Ca2+], and thus may explain the relatively subtle effects observed with F142L. The D130G Ca2+ Kd is 150 μmol/L, which is consistent with a greater propensity to promote pathological Ca2+ transients among LQTS‐CaM mutants tested. Recent studies also quantified the LQTS‐CaM LTCC effects25 based on the conceptual framework of an enzyme‐inhibitor‐like tuning of LTCC‐CaM signaling.26–27 In contrast, the CPVT‐CaM mutation N54I is located on the N‐lobe and shows sensitive, near normal Ca2+ affinity.4 This supports the contention that LQTS‐CaM mutation primarily targets LTCC, whereas CPVT‐CaM leaves LTCC function intact.
Examination of Ca2+ transients reveals mutant‐CaM effects that are not predicted by examining LTCC function alone. LQTS‐CaM slows CDI, but CaM‐N54I has no effect on CDI; however, both classes of LQTS‐CaM mutations D96V, D130G, and N54I result in abnormal Ca2+ transients. Recent work demonstrated that CPVT‐CaM, but not LQTS‐CaM, induces inappropriate SR Ca2+ release12 driven by aberrant CaM‐RyR2 interactions (cf8). In the present study, we uniquely show that the nature of the Ca2+‐transient phenotype differs in cardiomyocytes expressing LQTS‐CaM–associated mutations D96V and D130G compared with cells expressing CPVT‐CaM mutants. The predominant pattern of pathological Ca2+ transients exhibited by cells expressing these 2 LQTS‐CaM mutants was slowed SR Ca2+ reuptake, whereas cells expressing the CPVT‐CaM mutant N54I showed a combination of alternans and 2:1 blockade. The latter finding is consistent with other models of CPVT.28 Moreover, in mouse models of CPVT, catecholamines trigger activity in the ventricular myocardium,29–31 and the catastrophic effect of ISO on CaM‐N54I FVM is consistent with expectations of CPVT. A dominant contributor to twitch Ca2+‐transient relaxation is phospholamban‐SERCa function. In this vein, ISO is predicted to have 2 major effects: increasing the rate of SR Ca2+ reuptake through phospholamban‐SERCa signaling and increasing Ca2+ influx by LTCC activation. Additional considerations reveal the benefit of assessing Ca2+ transients as an integrative measure of function. For example, an ISO‐induced increase of K+ conductance simultaneously acts to shorten AP duration, blunting the time course of LTCC Ca2+ influx. ISO is well established to speed Ca2+ transient kinetics and increase SR Ca2+ load. If LQTS‐CaM effects were limited only to slowing of CDI, then we would have expected that ISO would not restore normal Ca2+ transients. The finding that ISO restored normal Ca2+ transients in LQTS reveals distinctions between CPVT‐CaM and LQTS‐CaM mutations and highlights the unexpected contribution of augmented Ca2+ reuptake as a future therapeutic direction for LQTS‐CaM patients. Applying therapeutics involving increased SR Ca2+ reuptake has plausible future possibilities considering early successes of the Calcium Up‐regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) trial.32
Study Limitations
For heterologous expression of NaV1.5 we selected 2 prominently expressed products of alternative splicing: the canonical form expressed in adult heart and a developmentally regulated splice variant that is expressed prominently in fetal and infant heart. Evidence exists for protein expression of both splice isoforms. Other SCN5A splice variants have been demonstrated at the mRNA level but not at the protein level, and their developmental regulation is uncertain. The most straightforward interpretation of our NaV1.5 data is that augmentation of late INa is not to the main pathophysiological mechanism responsible for the LQTS‐CaM phenotype; however, we cannot exclude that another unstudied splice variant conspires with mutant CaM to evoke a pathological phenotype. In contrast, our data on Ca2+ channel modulation argues effectively that delayed CDI is a more plausible mechanism for LQTS associated with CaM mutations.
Relatively late development stage FVM served as a cellular model to study the effects of LQTS‐CaM–associated mutations. These late‐stage FVM express a full complement of mature cardiomyocyte physiological responses including Ca2+‐induced Ca2+ release20,33 and β‐adrenergic receptor–mediated responses.34–35 As with any cellular system, there are limitations. First, although the LTCC multiprotein complex is fully functional, the same issues discussed above for expression of various channel protein isoforms applies. Second, this is a cellular model, and behaviors of dispersed cells may not report native physiological responses. Although this system allows us to use a common background to compare the influence of CaM mutations, it will be important to follow up studies in more complex in vivo settings.
In summary, LQTS‐ and CPVT‐associated CaM mutations both disrupt Ca2+ homeostasis in murine cardiomyocytes, but there is a sharp dichotomy in pathways leading to pathological phenotypes. LQTS‐CaM mutations occurring on the C‐lobe of CaM reduce Ca2+ affinity and, consequently, cause slowing of LTCC kinetics by attenuating CDI. In contrast, CPVT‐CaM mutant N54I has no effect on LTCC kinetics. As expected for CPVT, β‐adrenergic stimulation irreversibly overloaded cytosolic Ca2+ but also partially restored entrainment. This suggests a potential proof of principle for genotype‐specific treatment of “calmodulinopathies.”
Sources of Funding
This work was supported by NIH grants HL083374 (George) and HL074091 (Satin).
Disclosures
None.
Acknowledgments
Gail Sievert provided technical support.
References
- 1.Napolitano C, Bloise R, Monteforte N, Priori SG. Sudden cardiac death and genetic ion channelopathies: long qt, brugada, short qt, catecholaminergic polymorphic ventricular tachycardia, and idiopathic ventricular fibrillation. Circulation. 2012; 125:2027-2034. [DOI] [PubMed] [Google Scholar]
- 2.Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, Papagiannis J, Feldkamp MD, Rathi SG, Kunic JD, Pedrazzini M, Wieland T, Lichtner P, Beckmann BM, Clark T, Shaffer C, Benson DW, Kaab S, Meitinger T, Strom TM, Chazin WJ, Schwartz PJ, George AL., Jr Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013; 127:1009-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makita N, Yagihara N, Crotti L, Johnson CN, Beckerman B, Shigemizu D, Watanabe H, Ishikawa T, Aiba T, Mastantuono E, Tsunoda T, Nakagawa H, Tsuji Y, Tsuchiya T, Yamamoto H, Miyamoto Y, Endo N, Kimura A, Ozaki K, Motomura H, Suda K, Tanaka T, Schwartz PJ, Meitinger T, Kaab S, Shimizu W, Chazin WJ, George AL. Calm2 mutations associated with atypical juvenile long qt syndrome. Circulation. 2013; 128:A13371 [Google Scholar]
- 4.Nyegaard M, Overgaard MT, Sondergaard MT, Vranas M, Behr ER, Hildebrandt LL, Lund J, Hedley PL, Camm AJ, Wettrell G, Fosdal I, Christiansen M, Borglum AD. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012; 91:703-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams J. The complexity of gene expression, protein interaction, and cell differentiation. Nat Educ. 2008; 1:110 [Google Scholar]
- 6.Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage‐gated ca2+ channels. Neuron. 2004; 41:745-754. [DOI] [PubMed] [Google Scholar]
- 7.Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of ca2+ channel inactivation in controlling heart excitation. PNAS. 2002; 99:17185-17190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, Guo T, Oda T, Chakraborty A, Chen L, Uchinoumi H, Knowlton AA, Fruen BR, Cornea RL, Meissner G, Bers DM. Cardiac myocyte z‐line calmodulin is mainly ryr2‐bound and reduction is arrhythmogenic and occurs in heart failure. Circ Res. 2014; 114:295-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi N, Takahashi N, Xu L, Smithies O, Meissner G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle ca release channel. J Clin Invest. 2007; 117:1344-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamaguchi N, Xu L, Pasek DA, Evans KE, Meissner G. Molecular basis of calmodulin binding to cardiac muscle ca(2+) release channel (ryanodine receptor). J Biol Chem. 2003; 278:23480-23486. [DOI] [PubMed] [Google Scholar]
- 11.Arnaiz‐Cot JJ, Damon BJ, Zhang XH, Cleemann L, Yamaguchi N, Meissner GW, Morad M. Cardiac calcium signaling pathologies associated with defective calmodulin regulation of type 2 ryanodine receptor. J Physiol. 2013; 591:4287-4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang HS, Nitu FR, Yang Y, Walweel K, Pereira L, Johnson CN, Faggioni M, Chazin WJ, Laver D, George AL, Jr, Cornea RL, Bers DM, Knollmann BC. Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res. 2014; 114:1114-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cribbs LL, Martin BL, Schroder EA, Keller BB, Delisle BP, Satin J. Identification of the t‐type calcium channel (ca(v)3.1d) in developing mouse heart. Circ Res. 2001; 88:403-407. [DOI] [PubMed] [Google Scholar]
- 14.Murphy LL, Moon‐Grady AJ, Cuneo BF, Wakai RT, Yu S, Kunic JD, Benson DW, George AL., Jr Developmentally regulated scn5a splice variant potentiates dysfunction of a novel mutation associated with severe fetal arrhythmia. Heart Rhythm. 2012; 9:590-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crump SM, Andres DA, Sievert G, Satin J. The cardiac l‐type calcium channel distal carboxy terminus autoinhibition is regulated by calcium. Am J Physiol Heart Circ Physiol. 2013; 304:H455-H464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kahlig KM, Lepist I, Leung K, Rajamani S, George AL. Ranolazine selectively blocks persistent current evoked by epilepsy‐associated nanu1.1 mutations. Br J Pharmacol. 2010; 161:1414-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potet F, Chagot B, Anghelescu M, Viswanathan PC, Stepanovic SZ, Kupershmidt S, Chazin WJ, Balser JR. Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J Biol Chem. 2009; 284:8846-8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magyar J, Kiper CE, Sievert G, Cai W, Shi GX, Crump SM, Li L, Niederer SA, Smith NP, Andres DA, Satin J. Rem‐gtpase regulates cardiac myocyte l‐type calcium current. Channels (Austin). 2012; 6:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.George AL., Jr Inherited disorders of voltage‐gated sodium channels. J Clin Invest. 2005; 115:1990-1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korhonen T, Rapila R, Ronkainen VP, Koivumaki JT, Tavi P. Local ca2+ releases enable rapid heart rates in developing cardiomyocytes. J Physiol. 2010; 588:1407-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiss JN, Nivala M, Garfinkel A, Qu Z. Alternans and arrhythmias: from cell to heart. Circ Res. 2011; 108:98-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faggioni M, Hwang HS, van der Werf C, Nederend I, Kannankeril PJ, Wilde AA, Knollmann BC. Accelerated sinus rhythm prevents catecholaminergic polymorphic ventricular tachycardia in mice and in patients. Circ Res. 2013; 112:689-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neher E. Vesicle pools and ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998; 20:389-399. [DOI] [PubMed] [Google Scholar]
- 24.Tay LH, Dick IE, Yang W, Mank M, Griesbeck O, Yue DT. Nanodomain ca(2)(+) of ca(2)(+) channels detected by a tethered genetically encoded ca(2)(+) sensor. Nat Commun. 2012; 3:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Limpitikul W, Joshi‐Mukherjee R, Dick IE, George AL, Yue DT. Calmodulin mutants associated with long qt syndrome suppress inactivation of cardiac l‐type ca2+ currents and prolong action potentials in guinea‐pig ventricular myocytes. Circulation. 2013; 128:A17783 [Google Scholar]
- 26.Liu X, Yang PS, Yang W, Yue DT. Enzyme‐inhibitor‐like tuning of ca(2+) channel connectivity with calmodulin. Nature. 2010; 463:968-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johny MB, Yang PS, Bazzazi H, Yue DT. Dynamic switching of calmodulin interactions underlies ca2+ regulation of cav1.3 channels. Nat Commun. 2013; 4:1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang G, Giovannone SF, Liu N, Liu FY, Zhang J, Priori SG, Fishman GI. Purkinje cells from ryr2 mutant mice are highly arrhythmogenic but responsive to targeted therapy. Circ Res. 2010; 107:512-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rizzi N, Liu N, Napolitano C, Nori A, Turcato F, Colombi B, Bicciato S, Arcelli D, Spedito A, Scelsi M, Villani L, Esposito G, Boncompagni S, Protasi F, Volpe P, Priori SG. Unexpected structural and functional consequences of the r33q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ Res. 2008; 103:298-306. [DOI] [PubMed] [Google Scholar]
- 30.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini‐Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006; 116:2510-2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kashimura T, Briston SJ, Trafford AW, Napolitano C, Priori SG, Eisner DA, Venetucci LA. In the ryr2(r4496c) mouse model of cpvt, beta‐adrenergic stimulation induces ca waves by increasing sr ca content and not by decreasing the threshold for ca waves. Circ Res. 2010; 107:1483-1489. [DOI] [PubMed] [Google Scholar]
- 32.Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, Hajjar RJ. Long‐term effects of aav1/serca2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res. 2014; 114:101-108. [DOI] [PubMed] [Google Scholar]
- 33.Rapila R, Korhonen T, Tavi P. Excitation‐contraction coupling of the mouse embryonic cardiomyocyte. J Gen Physiol. 2008; 132:397-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.An RH, Davies MP, Doevendans PA, Kubalak SW, Bangalore R, Chien KR, Kass RS. Developmental changes in beta‐adrenergic modulation of l‐type ca2+ channels in embryonic mouse heart. Circ Res. 1996; 78:371-378. [DOI] [PubMed] [Google Scholar]
- 35.Nguemo F, Sasse P, Fleischmann BK, Kamanyi A, Schunkert H, Hescheler J, Reppel M. Modulation of l‐type ca(2+) channel current density and inactivation by beta‐adrenergic stimulation during murine cardiac embryogenesis. Basic Res Cardiol. 2009; 104:295-306. [DOI] [PubMed] [Google Scholar]