

Abstract
Mouse models have increased our understanding of the pathogenesis of medulloblastoma (MB), the most common malignant pediatric brain tumor that often forms in the cerebellum. A major goal of ongoing research is to better understand the early stages of tumorigenesis and to establish the genetic and environmental changes that underlie MB initiation and growth. However, studies of MB progression in mouse models are difficult due to the heterogeneity of tumor onset times and growth patterns and the lack of clinical symptoms at early stages. Magnetic resonance imaging (MRI) is critical for noninvasive, longitudinal, three-dimensional (3D) brain tumor imaging in the clinic but is limited in resolution and sensitivity for imaging early MBs in mice. In this study, high-resolution (100 μm in 2 hours) and high-throughput (150 μm in 15 minutes) manganese-enhanced MRI (MEMRI) protocols were optimized for early detection and monitoring of MBs in a Patched-1 (Ptch1) conditional knockout (CKO) model. The high tissue contrast obtained with MEMRI revealed detailed cerebellar morphology and enabled detection of MBs over a wide range of stages including pretumoral lesions as early as 2 to 3 weeks postnatal with volumes close to 0.1 mm3. Furthermore, longitudinal MEMRI allowed noninvasive monitoring of tumors and demonstrated that lesions within and between individuals have different tumorigenic potentials. 3D volumetric studies allowed quantitative analysis of MB tumor morphology and growth rates in individual Ptch1-CKO mice. These results show that MEMRI provides a powerful method for early in vivo detection and longitudinal imaging of MB progression in the mouse brain.
Abbreviations: 2D, two-dimensional; 3D, three-dimensional; Cb, cerebellum; CKO, conditional knockout; GCP, granule cell precursor; IHC, immunohistochemistry; MB, medulloblastoma; MEMRI, Mn-enhanced MRI; Mn, manganese; MRI, magnetic resonance imaging; Ptch1, Patched-1; SHH, Sonic Hedgehog; T1w, T1-weighted; T2w, T2-weighted; Td, tumor doubling time (in weeks)
Introduction
Pediatric central nervous system tumors are the leading cause of cancer-related mortality in childhood. In this age group, medulloblastoma (MB) is the most frequent malignant brain tumor accounting for approximately 20% of all cases. MB is an embryonal tumor found in the cerebellum (Cb) that typically appears between the ages of three and nine but is also found in infants and adults [1], [2]. With advances in aggressive treatment strategies combining surgery, cranio-spinal radiotherapy (in children ≥ 3 years of age), and chemotherapy, overall survival rates for patients with MB approaches 70% to 80% [3], [4], [5]. In spite of this progress in patient outcome, clinical management of MB still remains a challenge since patients who do survive often suffer devastating effects of the multimodal therapy, such as major long-term neurocognitive and neuroendocrine sequelae and a significant risk of developing secondary tumors [6], [7], [8], [9]. For this reason, characterization of MB pathogenesis is the focus of intense research in neuro-oncology, with the hope that a greater understanding of the biologic pathways disrupted in this disease will lead to the development of novel and less harmful therapies specifically targeted to the abnormal molecular signatures of this developmental cerebellar tumor.
Significant progress has been made in our understanding of the pathogenesis of MB as a result of genome-wide profiling of human MB samples [10], [11], [12]. There is now clear evidence that MB is not a single tumor entity but that it comprises at least four subgroups, including those related to mutations in the Wingless and Sonic Hedgehog (SHH) signaling pathways, each associated with distinct gene expression signatures, transcriptomes, histopathologic phenotypes, and prognoses [13], [14]. Furthermore, mouse models have been reported relevant to all four known MB subgroups [3], [15], [16], [17], [18], [19], [20]. From both the clinical and preclinical studies, consensus is forming that identification of the subgroup status and further subclassification [21] of these subgroups will enable treatment strategies tailored to individual tumors, which should translate into improved patient outcomes.
Despite this progress, detailed understanding of the differences between these MB subgroups and the degree of heterogeneity that exists within and between subgroups is still unclear. For example, the SHH-MB subgroup, consisting of approximately 30% of human MBs, is one of the most studied subtypes and has been recapitulated in several genetic mouse models [3], [15] and was recently subdivided into three human subtypes [21]. SHH is an essential pathway that normally regulates the proliferation of one of the major cell populations within the developing Cb, the granule cell precursors (GCPs) [22], [23]. Several studies have demonstrated a connection between increased SHH signaling and MB tumorigenesis, most notably due to loss-of-function mutations in Patched-1 (Ptch1) [24], [25], [26] and activating mutations in Smoothened[27], [28], the two key receptors of the pathway. Likewise, it has been substantially shown that GCPs are susceptible to malignant transformation and subsequent MB formation through oncogenic activation of the SHH pathway, revealing GCPs as the primary cell of origin of SHH-MBs [29], [30]. Interestingly, there is emerging clinical evidence of complex molecular heterogeneity within MBs, including within this well-characterized SHH subgroup [21], [31], [32], [33], [34], [35], which could indicate distinct cellular etiologies, specific altered signaling pathways, or differences in the timing and location of the cells of origin and genetic mutations driving tumor formation and progression. Therefore, it seems likely that additional uncharacterized MB subgroupings could be present and that additional large human and preclinical studies are needed to further dissect their biologic basis and degree of clinical relevance.
Given the importance of mouse models in this research, there is a clear need for efficient, high-throughput methods for analyzing MB phenotypes. Analysis of current mouse MB models has been mostly limited to static, two-dimensional (2D) information acquired with traditional histologic methods. Complementing these data, in vivo imaging modalities should provide a powerful approach for noninvasive tumor detection in mouse models, which are often limited by incomplete penetrance. Additionally, sensitive imaging techniques could allow the characterization and monitoring of tumor progression and the volumetric quantification of changes in response to novel therapies. In particular, detection and analysis of the early stages of tumorigenesis is especially important for brain tumors like MB, given that early stages of disease are asymptomatic and advanced-stage tumors may not accurately reflect the most relevant initiating or driver mutations for tumor progression.
Due to its excellent tissue contrast and high penetration and resolution, clinical magnetic resonance imaging (MRI) has played a major role in the diagnosis and management of human brain tumors. However, there are certain challenges for the use of MRI in preclinical brain tumor research. For instance, the utility of in vivo MRI in mouse tumor systems relies on the development of imaging protocols for high-throughput screening in models that traditionally have been limited by incomplete penetrance, as well as high-resolution in vivo analysis even at the early tumor stages. In addition, the establishment of MRI protocols for longitudinal imaging over extended periods of time is crucial for dynamic qualitative and quantitative analysis of tumor progression, which should provide a better stratification of experimental cohorts based on tumor phenotype and behavior. Previous studies have used conventional MRI techniques for morphologic brain tumor imaging in mouse MB models, but similar to most human studies [36], [37], [38], [39], mouse MB MRI reports have largely focused on detection and monitoring of tumor burden at relatively advanced tumor stages. For example, T2-weighted (T2w) MRI has been used to define the location and extent of advanced MBs in mouse models [40], [41], [42], while gadolinium-enhanced T1-weighted (T1w) imaging has been used to evaluate breakdown of the blood-brain barrier (BBB) and leakage of the contrast agent into MB tumors [40]. Because gadolinium enhancement depends on tumor type and stage and is often observed at later stages of tumor progression, gadolinium-enhanced imaging has not been used extensively for early tumor detection in the clinic, especially for MB.
The overall goal of the current study was to implement an in vivo MRI protocol for early detection and characterization of tumor progression in a mouse model of SHH-MB. Manganese (Mn)–enhanced MRI (MEMRI) has been used extensively for both anatomic and functional neuroimaging studies in mice. MEMRI relies on the properties of paramagnetic Mn2 + ions to produce MR contrast. Besides multiple roles in normal physiology, Mn2 + can function as a calcium (Ca2 +) analog, entering neurons through voltage-gated Ca2 + channels and resulting in positive enhancement on T1w MR images of the tissues where it accumulates. Once in neurons, Mn2 + is also transported along axons and can cross synapses to accumulate in adjacent neurons [43], [44], [45], [46], [47], [48]. We have previously demonstrated the utility of MEMRI for detailed analysis of brain and Cb development in normal and mutant mice, from embryonic to adult stages [49], [50], [51], [52], [53], [54]. Given the increased signal and high degree of anatomic definition in the Cb obtained with MEMRI [53], we reasoned that this would be a good potential method for analyzing abnormalities in cerebellar anatomy caused by the presence of MB lesions.
The specific objectives of this study were three-fold. First, we aimed to apply and optimize an in vivo MEMRI protocol for the detection and high-resolution imaging of early stages of MB tumorigenesis in Ptch1–conditional knockout (CKO) mice, a model of the SHH-MB subgroup that arises from very few Ptch1 mutant GCPs. Second, we sought to implement a time-efficient MRI protocol for high-throughput screening of tumor presence in Ptch1-CKO mice. Third, we aimed to use these protocols for longitudinal imaging and volumetric analysis of MB growth patterns to noninvasively characterize the progression of individual MB lesions from early to advanced stages.
Methods
Animals
All mice used in this study were maintained under protocols approved by the Institutional Animal Care and Use Committee at New York University School of Medicine. Ptch1-CKO mice (Ptf1aCre/+;Ptch1fl/fl) were generated by mating Ptf1aCre/+ mice [55], which were also heterozygous for a floxed allele of the Ptch1 gene (Ptch1fll +), with homozygous Ptch1fl/fl mice [56]. The genotype of each mouse was confirmed by polymerase chain reaction of tail biopsy, using primers for Ptch1[56] and Cre [57]. For MEMRI, a 30 mM MnCl2 solution of manganese chloride tetrahydrate (Sigma-Aldrich-221279) in isotonic saline was injected intraperitoneally (IP) 24 hours before imaging, at a dose per weight of 0.4 or 0.5 mmol/kg (50 and 62.5 mg MnCl2 per kg body weight for mice aged 3 weeks or less and 1 month or more, respectively). This dose corresponded to a single IP injection of 0.4 to 0.5 ml of MnCl2 solution per 30-g mouse. This MnCl2 dose was similar to the dose documented in the previous work using MEMRI for brain imaging [49], [53], [54], [58]. At this dose, no obvious adverse effects were noticed as a consequence of MnCl2 administration in this study.
Mouse Brain MRI
MRI was performed in a 7-Tesla, 200-mm diameter horizontal bore magnet (Magnex Scientific, Yarnton, United Kingdom) interfaced to a Bruker Biospec Avance II console (Bruker BioSpin MRI, Ettlingen, Germany) with actively shielded gradients (750 mT/m; BGA9S; Bruker) and using a 25-mm (inner diameter) quadrature Litz coil (Doty Scientific, Columbia, SC). For each imaging session, mice were anesthetized with isoflurane (5% in air for initial induction followed by 1.0-1.5% in air delivered through nosecone during imaging) and placed in mouse holders [59] (Dazai Research Instruments, Toronto, Ontario) that secured the body and head to facilitate reproducible setup and to minimize motion artifacts. Physiological body temperature was maintained using a warm air system, and respiration and heart rates were monitored using MR compatible equipment (SAII; SA Instruments, Stony Brook, NY). In vivo MRI images were obtained using a three-dimensional (3D) T1w gradient echo sequence for high-throughput screening [echo/repetition times (TE/TR) = 4/15 ms; flip angle (FA) = 18°; field of view = 19.2 mm × 19.2 mm × 12 mm; matrix = 128 × 128 × 80, providing 150-μm isotropic resolution in approximately 15 minutes] and/or a higher-resolution 3D T1w gradient echo self-gated sequence [60] to minimize motion artifacts (TE/TR = 3.6/50 ms; FA = 40°; field of view = 25.6 mm × 25.6 mm × 25.6 mm; matrix = 256 × 256 × 256, providing 100-μm isotropic resolution in 1 hour and 49 minutes). Selected mice were also scanned with a 2D multislice T2w fast spin echo “Rapid Acquisition with Refocused Echoes” (RARE) sequence (TE/TR = 8.9/4000 ms; effective TE = 35 ms; number of averages = 4; six 0.5-mm 2D slices; 2D matrix = 256 × 256, providing 80-μm in-plane resolution in approximately 9 minutes).
MRI Analysis
After reconstruction, MRI images were analyzed using AMIRA Imaging software (V5.2; Visage Imaging, San Diego, CA). Data sets were oriented to allow visualization of axial, sagittal and coronal planes through the brain. For quantitative analysis, volumetric measurements and 3D renderings were generated from MEMRI images using a combination of manual and semi-automated segmentation tools as previously reported [51], [53], [61]. Whole brain, Cb, and tumor regions were delineated and a mask was created for each area. Once the masks of the entire regions were obtained and corrected, they were represented three-dimensionally using the Surface Rendering modules in AMIRA. For quantification purposes, the total volume from the segmented and rendered regions (in voxels) was measured using the AMIRA Tissue Statistics function and converted to mm3. As previously described, MEMRI images had excellent contrast and allowed detailed visualization of normal cerebellar anatomy [49], [53], [58]. In our study, advanced tumors and early pretumoral lesions were visualized as non-enhanced areas of negative contrast within the enhanced layers of the cerebellar folia. Consequently, tumor tissue was identified, segmented, and manually refined for every section while comparing to normal mouse brain anatomy at the different imaging planes using MEMRI images of control littermates. Segmentations of selected tumors were correlated with histology at each stage to validate the MEMRI results. To compare the difference in MRI detection and volumetric measurements between the high-resolution 100-μm and high-throughput 150-μm sequences, we selected a group of mice at 3 weeks and scanned them sequentially with both MEMRI protocols. We then independently segmented the early pretumoral lesions visible in each MEMRI sequence and calculated their volumes with AMIRA. Using the statistical analysis software OriginPro 7.5 (OriginLab Corp, Northampton, MA), we performed a two-sample paired t test to investigate the difference between the two MRI protocols for each tumor volume measurement at the P < .05 level of confidence.
Tumor Growth Analysis
To quantify tumor growth characteristics, longitudinal MEMRI images of individual animals were obtained and volumes were calculated according to the protocol described above. Each individual Ptch1-CKO mouse had three to seven longitudinal scans between 3 and 26 weeks of age, and the imaging time points were separated by at least 2 weeks. For each mouse, the tumor volume was fitted to an exponential growth model:
| (1) |
where t is the time in weeks, V0 is the initial volume (extrapolated back to t = 0), and the exponent b is converted to the tumor doubling time (Td) in weeks [62] using the following formula:
| (2) |
The correlation coefficient R2 was computed for each exponential fit. For comparison between individuals, each growth curve was normalized to its initial volume (V0), and the normalized curves were displayed on a semi-logarithmic plot.
Histology and Immunohistochemistry
For histologic analyses, mice were perfused transcardially with 4°C phosphate-buffered saline (PBS) mixed with heparin (5000 heparin units/l), followed by 4°C 4% paraformaldehyde. After perfusion, brains were dissected and post-fixed with 4% paraformaldehyde at room temperature for 2 hours. Tissue was then processed for paraffin embedding, and 10-μm sagittal sections were acquired, covering the entire Cb and tumor of each mouse. Normal Cb and MB tumors were examined using hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC) with heat antigen retrieval in citric acid buffer (pH = 6).
For IHC, the primary antibodies used were rabbit anti-Ki67 (1:500; Abcam ab15580) and mouse anti-p27 (1:1000, BD Biosciences 610241), followed by anti-rabbit Alexa Fluor 555 (1:1000) and anti-mouse Alexa Fluor 488 (1:1000) for secondary antibody staining. Slides were counterstained with Hoechst or 4′6-diamidino-2-phenylindole (DAPI) for nuclear visualization and imaged on an inverted microscope (Zeiss Axio Observer.A1 microscope with Axiovision software). IHC for Ki67 was also visualized in selected slides using streptavidin-HRP conjugate, developed with DAB and counterstained with hematoxylin. After staining, slides were coverslipped and imaged using a bright-field microscope (Nikon Eclipse E400) or a digital slide scanner (Leica SCN400F).
For some slides, Ki67/p27 staining was combined with Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining to assess apoptosis. For this, sections were permeabilized with 0.5% Triton X-100 in PBS for 10 minutes, washed with PBS, and preincubated for 15 minutes with TdT buffer 1 × supplemented with 1 mM CoCl2. Terminal transferase (Roche Product No. 03333566001) and Biotin-16-dUTP (Roche Product No. 11093070910) were added, and the reaction was carried out as recommended by the manufacturer on sections for 1 hour at 37°C and the reaction was stopped with 0.01% Tween 20 in PBS. Signal was revealed by streptavidin-conjugated to Alexa Fluor 647, diluted 1:500 in PBS.
Captured images were analyzed using Adobe Photoshop and SlidePath Digital Image Hub (Leica Biosystems, Buffalo Grove, IL). For analysis of tumor differentiation (using Ki67/p27 staining), two to three slides per tumor (containing both lateral and medial sections) were examined and qualitatively assessed for the presence of differentiated regions in five to ten images per slide at both × 10 and × 20 magnification. For semi-quantitative analysis, we acquired representative 10 × images covering the entire tumor region in each slide (15-35 images per tumor depending on tumor size) and determined the fraction of images that showed isolated regions of differentiation for each tumor: (−) for 0 to 5%, (+) for 5% to 25%, (++) for 25% to 50%, and (+++) for > 50%.
Results
In vivo MEMRI Allowed Noninvasive Detection of MB Tumors in Ptch1-CKO Mice
As demonstrated previously [49], [53], in vivo MEMRI images showed region-specific signal and contrast enhancement in the mouse brain and allowed more detailed visualization of normal Cb morphology when compared to a non-MEMRI image (Figure 1, A and B). Abnormal Cb lesions were detected and easily delineated as hypointense regions within the enhanced Cb (Figure 1, C–E) in 74 of 92 Ptch1-CKO mice imaged with MEMRI between 11 and 28 weeks of age (80%). In selected Ptch1-CKO mice with MBs, gadolinium-enhanced T1w MRI showed heterogeneous patterns of enhancement similar to reports in other mouse MB models [40], which was not useful for delineating the lesion margins (Supplemental Fig. 1). Within the group of 74 lesion-bearing Ptch1-CKO mice imaged with MEMRI between 11 and 28 weeks of age, 47 mice presented symptoms commonly observed in advanced Cb tumors including weight loss, domed head, hunched posture, and ataxia at the time of sacrifice.
Figure 1.
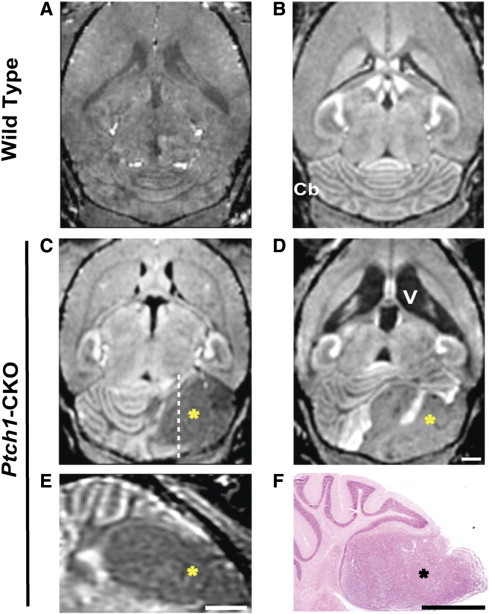
In vivo MEMRI detection of MB tumors in Ptch1-CKO mice. Compared to non–Mn-enhanced T1w MRI (A), post-contrast signal enhancement in axial MEMRI image (B) enabled visualization of normal brain and Cb anatomy in wild-type mice. MEMRI also allowed detection of advanced MB tumors as hypointense areas (*) within the Cb of Ptch1-CKO mice, typically located in the Cb hemispheres (C and D), and demonstrated severely enlarged ventricles (V) in some mice (D). Panel E shows a sagittal section from the mouse shown in C (dashed line indicates the approximate location of the section) and highlights the MB tumor (*). A matched histologic section (F), stained with H&E, showed good correlation of tumor location with MEMRI (E). Scale bars, 1 mm for A to F (scale on D for A–D).
On MEMRI, symptomatic mice typically had either unilateral focal (N = 36/47) or bilateral widespread (N = 11/47) hypointense tumors, primarily located within the anterior regions of the Cb hemispheres. These advanced-stage tumors measured 112.2 ± 92.0 mm3 in volume (N = 47, mean ± SD) and generally showed displacement of midline structures together with variable degrees of hydrocephalus (Figure 1, C and D). Histopathologic analysis confirmed that regions of negative contrast in MEMRI images correlated to MB tumor tissue in matched H&E-stained sections (Figure 1, E and F). In asymptomatic mice beyond 11 weeks of age (N = 27/74), MEMRI also identified mice with intermediate-sized tumors (N = 18/27, 23.5 ± 11.5 mm3) or small focal Cb lesions (N = 9/27, 5.4 ± 2.0 mm3). Quantitative analysis of the volume of tumors and brain ventricles showed large variability in tumor size and degree of hydrocephalus between symptomatic mice in the same age groups (Supplemental Table 1; N = 47). These results suggest variability in tumor onset time and/or growth rates in the Ptch1-CKO mice, similar to other mouse MB models [3]. The obvious challenges of studying tumor biology based on age or clinical symptoms highlight the need for noninvasive imaging methods to detect and monitor tumor progression.
In vivo MEMRI Enabled Characterization of the Early Stages of MB Formation
To investigate early stages of MB formation, high-resolution (100-μm) in vivo MEMRI images were obtained from postnatal day 14 (P14) and P21 wild-type (N = 10) and Ptch1-CKO (N = 16) mice (Figure 2). At these early time points, Ptch1-CKO mice were observed to preserve the overall normal foliation pattern; however, abnormal hypointense areas, 100 to 200 μm or more in thickness, were detected on the surface of the Cb of Ptch1-CKO mice, often between the folia (Figure 2, A–D). Matching histologic sections with MEMRI images from individual mice (N = 12) showed excellent qualitative agreement and confirmed these hypointense areas as regions of hyperplasia and persistence of the P14 to P21 external granule cell layer, the proliferative zone of the postnatal Cb that is normally depleted by P16 (Figure 2, C–F). These regions of persistent external granule cell layer, observed as hypointense areas on MEMRI, are referred to as “pretumoral lesions” in this paper. 3D renderings were generated from MEMRI images to examine the morphology, spatial distribution, and volume of histologically validated early pretumoral lesions at P14 to P21 (Figure 2G). The average lesion volume at P14 to P21 was 6.70 ± 3.31 mm3, and three distinct phenotypes were observed: (1) unilateral lesion (N = 2/12), (2) bilateral lesion that appeared to be connected (N = 6/12), and (3) multiple separated lesions (N = 4/12; Supplemental Video 1). All of the lesions had a predominant anterior component (N = 12), while many also had a smaller posterior component (N = 8/12), using lobule VIb in the vermis and crus I in the hemispheres as the reference structures dividing anterior from posterior.
Figure 2.
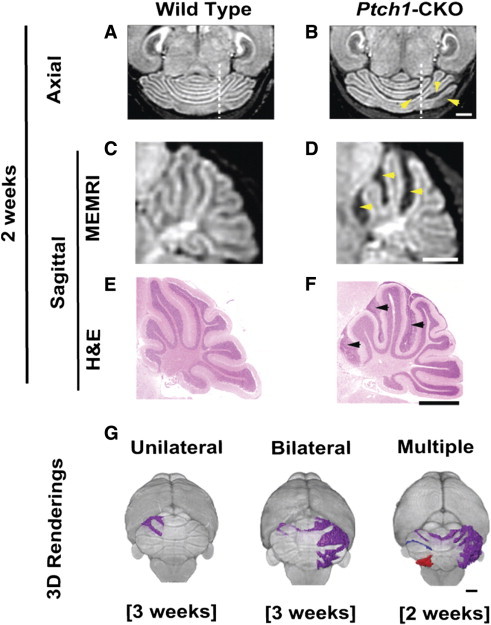
MEMRI allowed detection of early-stage MB lesions in Ptch1-CKO mice. Axial (A and B) and sagittal (C and D) MEMRI images (dashed lines on A and B show the approximate location of the sagittal sections shown in C and D, respectively) revealed multiple hypointense areas (yellow arrowheads) in the cerebella of Ptch1-CKO mice (B and D) compared to wild-type controls (A and C) at 2 to 3 weeks of age. Matched sagittal H&E sections (E and F) demonstrated accurate histologic correlation between areas of negative contrast in MEMRI (D; yellow arrowheads) and early pretumoral lesions (F; black arrowheads). Representative 3D volume renderings from MEMRI data demonstrate distinct patterns of Cb lesions seen in individual Ptch1-CKO mice at 2 to 3 weeks of age. These early pretumoral lesions were detected as unilateral, bilateral, and multiple (separated lesions shown in different colors; see Supplemental Video 1 for more detailed visualization), as demonstrated in G. Scale bars, 1 mm.
MEMRI Is More Sensitive for the Detection of Early Pretumoral MB Lesions than Conventional MRI
Accurate detection of early stages of tumor formation is critical for enhancing the value of MRI in preclinical studies. Conventional MRI of brain tumors typically uses T2w images for assessing tumor location and morphology [40], [41], [63]. Both T1w MEMRI images and T2w RARE images were acquired in the same Ptch1-CKO mice to assess the sensitivity of each protocol to detect MB lesions at distinct stages of tumor formation. As previously described, tumor tissue and areas of hydrocephalus showed low signal intensity on T1w MEMRI, whereas these regions showed high signal intensity on T2w RARE images (Figure 3). At late stages (11-28 weeks old, N = 9), severe brain deformity and large tumors were successfully detected in both types of images; however, the Cb foliation pattern and the tumor margins were better demarcated with MEMRI (Figure 3, A–D). At early stages (P14-P21, N = 9), hypointense pretumoral lesions surrounded by enhanced cerebellar folia were clearly visible on MEMRI, while the majority of those same lesions were undetectable on RARE due to the lack of contrast between tumor and normal Cb (Figure 3, E–H). Specifically, a total of 24 distinct pretumoral lesions was visualized with MEMRI in the group of P14 to P21 Ptch1-CKO mice (N = 9), but only 4 of 24 (17%) of those early pretumoral lesions were also detected with RARE. Volumetric analysis based on MEMRI data revealed that the average size of the early pretumoral lesions detectable with both MEMRI and RARE was 3.10 ± 1.14 mm3 (minimum = 2.29 mm3) compared to 1.21 ± 1.19 mm3 (minimum = 0.12 mm3) for those lesions only detected with MEMRI. These results establish MEMRI as a more sensitive method for detection of early pretumoral MB lesions in the Ptch1-CKO mice.
Figure 3.
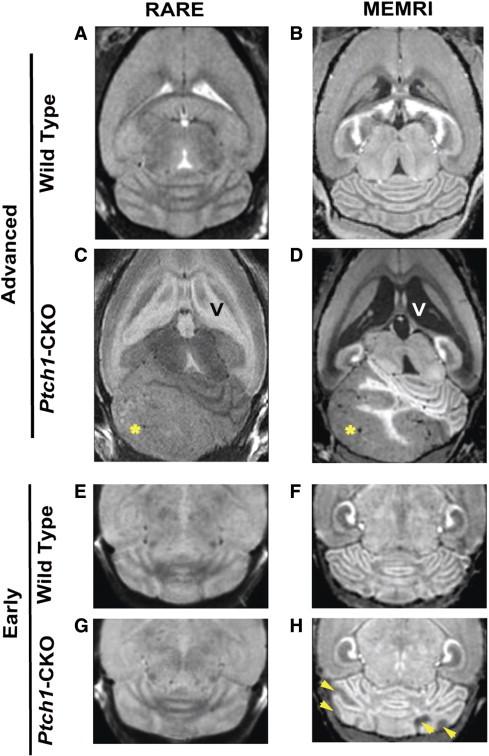
MEMRI was more sensitive for detecting early-stage MB than conventional T2w MRI. Representative examples are shown for wild-type (A, B, E, and F) and Ptch1-CKO mice (C, D, G, and H) scanned with both non-contrast T2w RARE (left panels; A, C, E, and G) and post-contrast T1w MEMRI sequences (right panels; B, D, F, and H). These images show improved tissue contrast in MEMRI compared to RARE images and show higher sensitivity for visualization of tumor tissue (*) using MEMRI (D) compared to RARE (C), especially for detection of early pretumoral lesions with MEMRI (H, arrowheads), which could not be detected with RARE (G). Scale bars, 1 mm.
The long (2-hour) acquisition time required for the high-resolution (100-μm) MEMRI protocol could become a limitation for future imaging studies with large cohorts of mice. To address this issue, a high-throughput T1w MEMRI protocol was implemented, with lower spatial resolution (150 μm) but a significantly reduced acquisition time of 15 minutes (Figure 4). Comparing images from the same Ptch1-CKO mice, both the high-resolution and the high-throughput MEMRI protocols allowed visualization of advanced-stage tumors (Figure 4A, N = 12) and early pretumoral lesions (Figure 4B, N = 16). Although images acquired with the high-throughput (150-μm) protocol were pixelated due to the lower resolution, this effect did not have a significant impact on lesion detectability and could be corrected by post-processing. To further investigate the differences between the two MEMRI protocols, quantitative volumetric analysis of MB lesions was performed in a group of P21 mice (N = 7). All pretumoral lesions that were detected at 100 μm were also detected at 150 μm. The calculated total average lesion volume was 6.7 ± 3.8 mm3 for the 100-μm images, compared to 7.8 ± 4.1 mm3 for the 150-μm images. Analysis using a two-sample paired t test revealed that there were no significant differences in the lesion volumes measured with the two protocols (P = .13). Taken together, these results demonstrate that the high-throughput (150-μm) MEMRI protocol could be used for sensitive detection and quantitative analysis of both early pretumoral lesions and advanced-stage MB tumors in a time-efficient manner.
Figure 4.
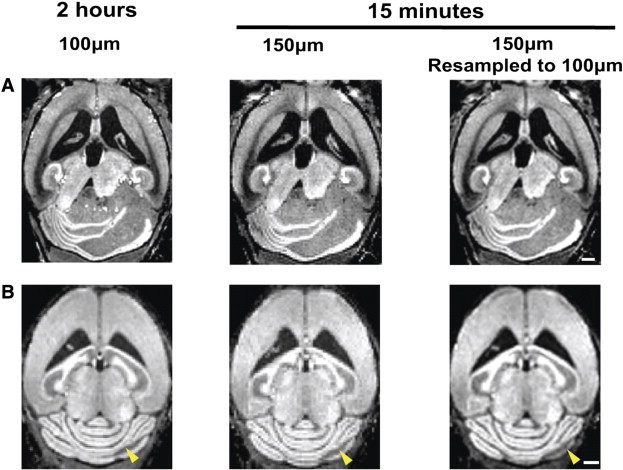
MEMRI detection of advanced and early MB lesions with both high-throughput and high-resolution protocols. MEMRI images of Ptch1-CKO mice demonstrated that both advanced-stage tumors (A) and early pretumoral lesions (B, arrowheads) were detected with either the high-resolution (100 μm; 2 hours) or high-throughput (150 μm; 15 minutes) sequences. High-throughput images were improved by resampling the 150-μm images to 100 μm in post-processing (right panels). Scale bars, 1 mm.
Longitudinal MEMRI Enabled In Vivo Analysis of MB Progression
Longitudinal in vivo imaging of mouse models is critical to analyze growth patterns of developing tumors and for preclinical therapeutic studies. Previous studies of MB formation in Ptch1+/− heterozygous mutant mice have generally defined early stages of MB as up to 8 weeks of age [64], [65], [66]. In this study, in vivo longitudinal MEMRI and volumetric analyses were used to analyze MB progression in Ptch1-CKO mice starting from pretumoral lesions at 3 to 7 weeks through advanced tumor stages at 13 to 20 weeks (Figure 5, N = 21). Specifically, one group of mice was imaged starting at 3 weeks (N = 14), while a second group was begun at a somewhat later stage between 5 and 7 weeks (N = 7). In both groups, pretumoral lesions were detected in each mouse at the initial time point. The average size of detected lesions at 3 weeks (N = 14) was 6.24 ± 5.44 mm3 (min = 0.57, max = 15.8) and that at 5 to 7 weeks (N = 7) was 8.56 ± 6.05 mm3 (min = 0.50, max = 16.3). There was no statistically significant difference in the lesion size between the early imaging time points at 3 and 5 to 7 weeks (P = .4). Over time, the pretumoral lesions progressed and formed MBs in most mice (Figure 5, Figure 6, A and B; N = 16/21, 76%), while in some mice the lesions actually regressed (Figure 6C; N = 5/21, 24%).
Figure 5.
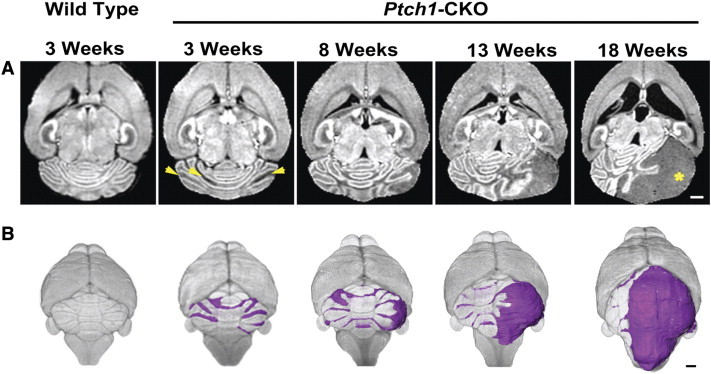
In vivo longitudinal MEMRI enabled imaging tumor progression from early to advanced stages. Axial MEMRI images (A) and 3D volume renderings (B) of a representative individual Ptch1-CKO mouse at sequential time points (3-18 weeks) demonstrate the feasibility of longitudinal imaging using MEMRI. Note also that bilateral pretumoral lesions were observed at early time points (arrowheads), but at later times, only one area exponentially progressed to an advanced MB (*). Representative examples of an axial MEMRI and 3D rendering of a control mouse are shown at the first time point as a reference. Scale bars, 1 mm.
Figure 6.
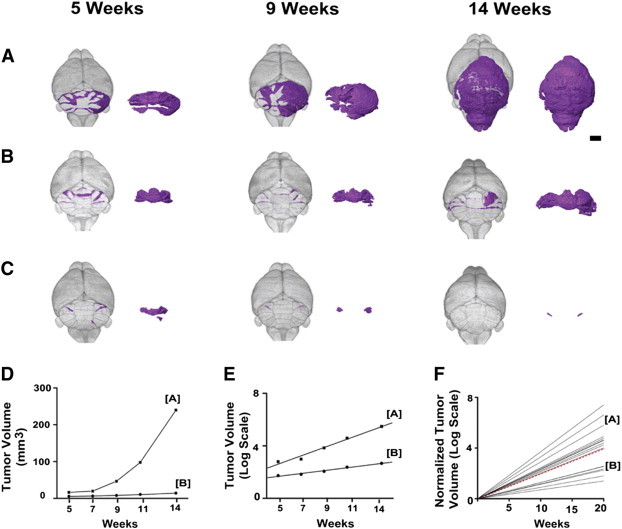
Longitudinal MEMRI revealed variability in MB tumor growth patterns. 3D renderings from longitudinal MEMRI images of representative Ptch1-CKO mice showed a range of MB progression rates from fast (A) to slow (B). In each panel, the segmented MB lesions are shown in color within the whole brain (left) and separately (right). Several examples of early lesions that regressed below detectability with MEMRI were also observed (C; N = 5/21). Quantitative analysis of MB lesion volume (D) demonstrated the distinct growth rate patterns of the mice shown in A and B and also showed excellent fit to an exponential growth model, plotted in semi-log format (E). To compare MB growth rates between mice, lesion volumes were normalized to their initial volumes and again plotted in semi-log format (F; N = 16), demonstrating the variability observed in MB growth rates in the Ptch1-CKO mouse model. Dashed red line represents the mean Td = 4.3 weeks. Scale bar in A, 2 mm.
Both tumor growth and regression were variable between individual Ptch1-CKO mice. Among the mice in which all lesions regressed (N = 5), the average lesion volume at the initial time point was 4.33 ± 4.63 mm3 (min = 1.11, max = 12.5) and most had no detectable lesion by 20 weeks (N = 4/5), while one took 26 weeks to regress below detectability. In the mice with tumors that progressed (N = 16), the average lesion size was 7.9 ± 5.8 mm3 (min = 0.50, max = 16.3), and similarly, there was significant variability in the growth patterns of MBs in the progressing mice (N = 16). At the early stages (3-7 weeks), most Ptch1-CKO had pretumoral lesions that were distributed across a relatively wide region of the Cb (Figure 2, Figure 5, Figure 6), while at more advanced stages tumor growth was predominantly in one region, showing that many of the pretumoral lesions did not develop or were delayed in their progression (Figure 5). Together with the data on the regressing lesions, these results strongly suggest that although pretumoral lesions have the capacity to develop into tumors, they are not predetermined to do so, consistent with earlier studies [64], [66], [67].
Interestingly, we noticed significant differences in growth rates between MBs that progressed (Figure 6, A, B, and D; N = 16). To quantify tumor growth patterns, the lesion/tumor volumes were measured at each time point and fitted to an exponential growth model, given in Eqs. (1), (2), for each mouse (Figure 6, D and E). Regression analysis showed an excellent fit of the volumetric data to the model (R2 > 0.9 in all cases; mean ± SD = 0.96 ± 0.04). Td was highly variable, ranging between 1.8 and 9.9 weeks, with a mean value of 4.29 ± 2.33 weeks. No obvious correlation between Td and tumor volume was observed (Table 1). The variability in MB growth rates between individual Ptch1-CKO mice may potentially be related to molecular differences, such as distinct secondary mutations and epigenetic alterations. Although molecular profiling (e.g., RNA-seq) is beyond the scope of this study, MEMRI will be a useful tool to segregate tumors for such analyses based on their growth rates in future studies.
Table 1.
There Is No Clear Relationship between Tumor Growth Rate and Degree of Differentiation in Ptch1-CKO MBs.
| Mouse ID | Td (Weeks) | Initial Volume (mm3)/[Weeks] | End Volume (mm3)/[Weeks] | Differentiation Index⁎ |
|---|---|---|---|---|
| 1 | 9.9 | 14.7 [3] | 36.4 [15] | +++ |
| 2 | 6.1 | 0.5 [5] | 3.3 [20] | − |
| 3 | 5.9 | 6.2 [3] | 49.8 [20] | +++ |
| 4 | 5.3 | 0.6 [3] | 4.7 [15] | +++ |
| 5 | 5.3 | 4.2 [3] | 14.4 [13] | +++ |
| 6 | 3.5 | 12.9 [3] | 171.5 [20] | – |
| 7 | 3.2 | 0.6 [3] | 11.9 [13] | +++ |
| 8 | 2.9 | 15.8 [3] | 75.5 [15] | + |
| 9 | 2.9 | 3.9 [3] | 184.6 [20] | – |
| 10 | 2.4 | 6.7 [7] | 87.6 [17] | +++ |
| 11 | 2.1 | 16.3 [5] | 239.6 [14] | – |
| 12 | 1.8 | 11.3 [7] | 98.9 [17] | +++ |
(−) 0 to 5%; (+) 5% to 25%; (++) 25% to 50%; (+++) > 50% (see Methods section).
Histopathologic Analysis Showed Variable Levels of Proliferation, Differentiation, and Apoptosis in Ptch1-CKO Mice
MBs are known to show a range of histologic features, which have been partly correlated with age of tumor onset and clinical outcomes in patients. For most SHH-MBs, the presence of either a more classic (little differentiation) or nodular/desmoplastic (extensive differentiation) histologic phenotype has been described [68], [69]. To investigate SHH-MB tumor cytoarchitecture and its possible relationship to tumor growth rates in Ptch1-CKO mice, histologic analyses were performed on a subset of MBs from the longitudinal MEMRI analysis. Tumors were harvested to obtain a range of advanced stages between 13 and 20 weeks and analyzed using IHC for Ki67 (red) and p27 (green) as markers of proliferation and neuronal differentiation, respectively (Figure 7; N = 12). MB tumors studied showed two distinct patterns: one in which expression of Ki67 was dominant, interspersed with scattered differentiated p27-expressing cells, consistent with classic SHH-MB (Figure 7, A, C, E, G, and I), and a second biphasic pattern in which p27 expression was dominant and clustered in multiple isolated regions surrounded by proliferating Ki67-expressing cells (Figure 7, B, D, F, H, and J). Qualitative and semi-quantitative analyses of the histologic phenotypes of the Ptch1-CKO MBs showed that there was variability in the presence and degree of the differentiated regions within individual tumors and that there was no clear correlation between the profiles of differentiation and the measured Td (Table 1).
Figure 7.
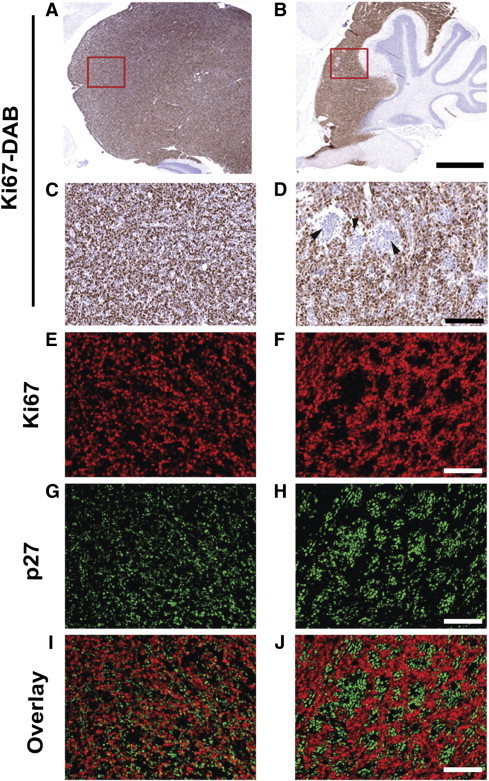
IHC analysis of MB tumors in Ptch1-CKO mice. MB tumors were analyzed using IHC for Ki67 and p27, as markers of proliferation and differentiation, respectively (N = 12). DAB staining for Ki67 at low (× 3; A and B) and high (× 20; C and D) magnification showed the presence of multiple Ki67-negative areas in some tumors (B and D) but in not others (A and C). (Note: the red boxes show the approximate location of C and D on A and B, respectively.) Within the same two tumors, Ki67 (red; E and F)/p27 (green; G and H) double fluorescent staining revealed that the Ki67-negative areas (F) were regions of differentiation with high p27-expression (H and J). Conversely, tumors with uniform Ki67 expression (E) had scattered p27-expressing cells (G) mixed among the Ki67-expressing cells (I). Scale bars, 1 mm in A and B; 50 μm in C to J.
Since the longitudinal MEMRI results indicated distinct tumorigenic potential among lesions in the Ptch1-CKO mice, IHC was used to analyze pretumoral lesions at 3 weeks of age (N = 8 lesions in four mice) and lesions that were not progressing in 13- to 15-week-old tumor-bearing mice (N = 7 lesions in three mice). Sections were stained with H&E for morphology, Ki67 for proliferation, p27 for differentiation, and TUNEL for apoptosis (Figure 8). At 3 weeks, the pretumoral lesions all showed areas of differentiation based on p27-expressing cells, with a majority of Ki67 cells on the outer surface of the lesions (Figure 8, A–D). Most of the early pretumoral lesions showed low levels of TUNEL staining (N = 5/8 in three mice; Figure 8B), but some showed higher TUNEL staining (N = 3/8 in one mouse; Figure 8D). In individual mice at 13 to 15 weeks of age, tumors and smaller lesions that were not progressing (confirmed by MEMRI) were examined. The tumors (N = 3) had doubling times that ranged from 3.2 to 5.3 weeks, and all had the more differentiated p27/Ki67-expression patterns, as well as a uniform distribution of TUNEL-positive cells throughout the tumors (Figure 8, E and I). The smaller lesions that were not progressing (N = 7) showed dominant Ki67 expression with scattered differentiated p27-expressing cells and lower levels of TUNEL, compared to the tumors (Figure 8, F–H and J). Interestingly, in both the early pretumoral and later non-progressing lesions, p27-expressing cells were also observed in the molecular layer, suggesting that some cells within these lesions may still be able to undergo differentiation and migration but at a later stage than normal developing granule cells. Taken together, these results indicate that cells within the early pretumoral and later non-progressing lesions maintain a proliferative potential as well as an ability to differentiate and undergo apoptosis but have not yet undergone the complete malignant transformation necessary to become tumors [66].
Figure 8.
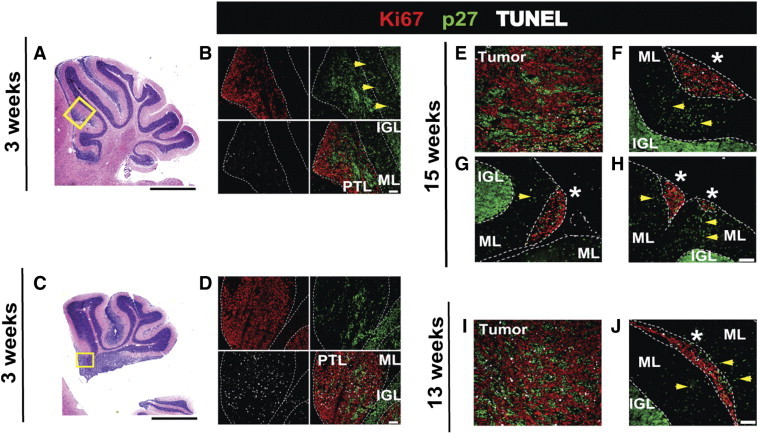
IHC analysis of early pretumoral and later non-progressing lesions. Cerebellar sections show examples of pretumoral lesions (PTL) in two different 3-week-old Ptch1-CKO mice (A and B and C and D). H&E-stained sections (A and C) show the cerebellar morphology and the approximate locations (yellow boxes) of the immunofluorescent sections (B and D) stained for Ki67 (red), p27 (green), and TUNEL (white) and the overlay of all three stains. Examples are also provided for tumors and non-progressing lesions (*) in 15-week-old (E–H) and 13-week-old (I and J) Ptch1-CKO mice. Dashed lines demarcate the PTLs and non-progressing lesions, as well as the molecular layer (ML) and internal granule layer (IGL). Early PTLs (B and D), tumors (E and I), and lesions that were not progressing (F–H and J) all showed high levels of Ki67, with variable p27 and TUNEL staining. p27 expression was more widespread and clustered in the PTLs and tumors when compared to non-progressing lesions. Notably, sections of both PTLs and non-progressed lesions showed some p27-positive cells in the ML between the lesion and the internal granule layer (yellow arrows). Magnification: × 1.5 (A and C) and × 20 (B, D, and E–J). Scale bars, 1 mm in A and C; 50 μm in B, D, and E–J.
Discussion
Tumorigenesis is a multistep process in which initiating mutations may lead to pretumoral stages that could further advance to malignancy once exposed to additional oncogenic stimuli. Comparing these early stages to advanced tumors can help determine which mutations are required for early cellular transformation and which mutations drive tumor progression. However, such studies can be challenging in mouse models of brain tumors such as MB since the early tumor stages are asymptomatic and tumor onset and progression can be highly variable within individual mice and among distinct preclinical models. In this study, we demonstrated the utility and optimization of both high-resolution and high-throughput in vivo MEMRI protocols to image MB progression noninvasively over a wide range of tumor stages. Furthermore, our results demonstrated that MEMRI provides an effective and sensitive method for the early detection and growth rate characterization of MBs in mouse models.
SHH-induced MBs are thought to arise primarily from the aberrant proliferation of GCPs stimulated by excess SHH signaling. PTCH1 is a key receptor and antagonist of the SHH signaling pathway and some Ptch1+/− heterozygous mutant mice develop MB [24]. As a result, Ptch1+/− mice have been used extensively to analyze the effects of additional mutations in tumor suppressor/enhancer genes on MB progression [65], [66], [70], [71]. However, these studies have been hindered by low tumor incidence (15-20%) and an unpredictable time of tumor onset in these mice (between 6 and 12 months). To overcome these limitations, several lines of conditional mouse models have also been generated using different Cre-driver lines to inactivate both alleles of the Ptch1 gene, often in the majority of GCPs or cerebellar ventricular zone cells, and to thereby induce early global MB-like growth [25], [29], [30]. In the current study, we used a Ptch1-CKO mouse model using the Ptf1a-Cre driver [55] that deletes Ptch1 in relatively few isolated Ptf1a-expressing GCPs mainly in the anterior embryonic Cb [72] (Raju, in preparation). Our results demonstrate that these Ptch1-CKO mice constitute an excellent model system to study early stages of sporadic SHH-MB tumorigenesis.
Lacking in previous studies of mouse MB models has been an effective noninvasive means of detecting and monitoring early stages of MB progression in vivo. Such a method would enable investigations of growth patterns from early stages of tumor formation, allowing comparisons of different factors that mediate MB tumor progression and/or regression. Importantly, we demonstrated that MEMRI is a highly sensitive approach for imaging MB progression, from early pretumoral lesions to advanced tumor stages. This is a significant advance over conventional MRI methods, allowing noninvasive imaging and in vivo analysis of very early stages of tumorigenesis. Furthermore, our results showed the feasibility of longitudinal MEMRI and the ability to noninvasively follow the progression of individual MBs from early to advanced symptomatic stages. Previous studies using histology have described the presence of early pretumoral lesions in Ptch1+/− mice [25], [64], [65], [73] and in other mouse MB models [74], [75]. Additionally, a number of studies have suggested that some of these pretumoral lesions can give rise to MB and that specific oncogenic events can increase their tumorigenic capacity [66], [67]. However, to our knowledge, this is the first report that demonstrates the in vivo detection and longitudinal imaging of individual pretumoral MB lesions as they progress to advanced tumors.
3D renderings of the longitudinal MEMRI data sets enabled a comprehensive visualization of the morphology of early to advanced lesions. They also provided insight into their dynamic development and showed that they can have different fates. In addition, 3D volumetric studies allowed quantitative analysis of tumor growth rates. In the current study, we found that a simple exponential growth model provided an excellent fit to the data on tumor progression, which was dominated by the larger volumes at advanced stages. A more complex model would be required to describe early MB progression and regression to account for the balance between cell proliferation, differentiation, and apoptosis at the early stages. Our analysis of tumor progression highlighted the presence of different tumor growth patterns in individual Ptch1-CKO mice. This variation of growth rates seen within the population of MBs likely explains differences in the onset time of symptoms in Ptch1-CKO mice and could potentially correlate to underlying molecular signatures among tumors. The variability in tumor progression seen in this and other mouse tumor models also highlights the need for in vivo imaging methods to be included in preclinical studies of therapeutic agents, allowing a more accurate stratification of treatment cohorts based on tumor stage and growth characteristics rather than age or the presence of symptoms. The ability of MRI to quantify changes in growth rates in other types of brain tumors has previously been demonstrated to be useful for preclinical assessment of therapeutic response [41], [62], [76], [77].
Although no obvious neurotoxicity was observed in the current study, it is relevant to mention that at high doses, Mn is known to produce acute and chronic toxic effects [78], [79], [80]. However, MEMRI has been extensively used as an imaging tool in multiple neuroimaging studies at similar or even higher doses than the ones in this study without significant toxicity [46]. In this study, repeated MRI sessions and Mn injections were conducted over a period of weeks to months to track tumor progression. This was well tolerated by the mice at different ages, even as tumors were developing and mice became symptomatic. It should be also noted that studies assessing acute toxic effects of Mn are mostly performed with repeated (20-30) injections in shorter intervals of 24 to 48 hours [79], while in this study injections and subsequent MRI sessions were separated by intervals of at least 2 to 3 weeks. The lengthened period between injections allows the Mn to clear from neural tissues and likely further limits its acute toxicity. Several studies have reported the implementation of fractionated Mn schedules to maximize contrast in long-term MEMRI studies while reducing toxicity [80], [81], an approach that might be worth investigating for future implementation of MEMRI in studies of MB progression.
Taken together, the in vivo MEMRI method described in this paper provides a unique noninvasive approach for the early detection and characterization of SHH-MBs in Ptch1-CKO mice that can be applied to other MB models and other types of Cb tumors. Similar MEMRI methods may also be useful for other brain regions and brain tumor models such as gliomas, relying on negative contrast between enhancing normal brain and non-enhancing tumors for detection. In the future, MEMRI could be combined with other MRI protocols, such as gadolinium-enhanced imaging to assess BBB permeability, dynamic perfusion MRI to study tumor vasculature, or diffusion MRI to examine tissue microstructural changes. These protocols have been used in several clinical reports on MBs, but so far the imaging has been limited to advanced heterogeneous tumors where the results have been variable and still have no clear correlation with pathologic subtype or prognosis [37], [38], [39], [82], [83].
MEMRI could be used in future studies to monitor pretumoral MB lesions and to guide molecular and genetic expression analyses in a stage-dependent manner. Such studies could aid in the temporal and spatial characterization of initiating events for MB formation and lead to greater insights into the relevant changes that distinguish the different tumor stages. Ultimately, as the preclinical models of the four distinct MB subgroups become further refined and additional subtype-specific models emerge, the imaging methods presented in this study could serve as the basis for their characterization. This should enable a better stratification of cohorts in future preclinical therapeutic studies targeted to molecular alterations specific to each MB subgroup.
Acknowledgments
We thank Youssef Zaim Wadghiri for help with pulse sequence optimization, Andrea Mathieux for help with tumor segmentations, and Eva Hernando-Monge for critical review of this manuscript.
Footnotes
This article refers to supplementary materials, which is designated by Supplemental Table 1, Supplemental Figure 1, and Supplemental Video 1 and are available online at www.neoplasia.com.
This research was supported by National Institutes of Health grants R01NS038461 (D.H.T.), R01HL078665 (D.H.T.), R01CA128158 (A.L.J.), and K08NS066083 (G.P.R.), a grant from The Hartwell Foundation (G.P.R.), and Geoffrey Beene Cancer Research Center grant 21680 (A.L.J.). MRI was partially supported by the Small Animal Imaging Core at NYU School of Medicine and by the NYU Cancer Institute through the NIH Cancer Center Support grant (P30CA016087). We thank the Histopathology Core at NYU School of Medicine (also supported by NIH grant P30CA016087) for help with the histologic and IHC analyses.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.10.001.
Appendix A. Supplementary data
Supplemental Fig. 1. Gd enhancement in MB tumors in Ptch1-CKO mice. MEMRI images were acquired using the 15-minute 3D T1w sequence (see Methods section), 24 hours after IP injection of a standard dose of MnCl2. The resulting pre-gadolinium (Gd) images demonstrated advanced MB tumors (*) in each Ptch1-CKO mouse (N = 6). Post-Gd images (right panels) were then acquired from each mouse during the same imaging session, immediately after intravenous injection of Gd-DTPA (Magnevist; 0.1 mM/kg dose) using a tail vein catheter. The post-Gd images showed heterogeneous increase in signal intensity (yellow arrows) within each of the tumors, similar to previous reports (e.g., [40]). Brain tumor uptake of Gd-DTPA has generally been related to contrast leakage due to disruption of the BBB or abnormal tumor vessels, which can differ between individual tumors and tumor type. In the Ptch1-CKO MB tumors, Gd-enhanced MRI was clearly less useful than MEMRI for determining the tumor margins and to assess tumor volume longitudinally and was therefore not pursued further.
Supplemental Table 1. Age Is Not a Good Predictor of Tumor and Brain Ventricle Volume in Ptch1-CKO mice.
Movie of early pretumoral MB lesions in a Ptch1-CKO mouse. Representative 3D volume rendering from MEMRI data, demonstrating the morphology and distribution of multiple pretumoral lesions (distinct lesions shown in different colors) in a 2-week old Ptch1-CKO mouse.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Pui C-H, Gajjar AJ, Kane JR, Qaddoumi IA, Pappo AS. Challenging issues in pediatric oncology. Nat Rev Clin Oncol. 2011;8:540–549. doi: 10.1038/nrclinonc.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, Pomeroy SL, Korshunov A, Lichter P, Taylor MD. Medulloblastomics: the end of the beginning. Nat Rev Cancer. 2012;12:818–834. doi: 10.1038/nrc3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rutkowski S, von Hoff K, Emser A, Zwiener I, Pietsch T, Figarella-Branger D, Giangaspero F, Ellison DW, Garre ML, Biassoni V. Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol. 2010;28:4961–4968. doi: 10.1200/JCO.2010.30.2299. [DOI] [PubMed] [Google Scholar]
- 5.Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813–820. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 6.Packer RJ. Childhood brain tumors: accomplishments and ongoing challenges. J Child Neurol. 2008;23:1122–1127. doi: 10.1177/0883073808320758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mabbott DJ, Penkman L, Witol A, Strother D, Bouffet E. Core neurocognitive functions in children treated for posterior fossa tumors. Neuropsychology. 2008;22:159–168. doi: 10.1037/0894-4105.22.2.159. [DOI] [PubMed] [Google Scholar]
- 8.Mabbott DJ, Spiegler BJ, Greenberg ML, Rutka JT, Hyder DJ, Bouffet E. Serial evaluation of academic and behavioral outcome after treatment with cranial radiation in childhood. J Clin Oncol. 2005;23:2256–2263. doi: 10.1200/JCO.2005.01.158. [DOI] [PubMed] [Google Scholar]
- 9.Spiegler BJ, Bouffet E, Greenberg ML, Rutka JT, Mabbott DJ. Change in neurocognitive functioning after treatment with cranial radiation in childhood. J Clin Oncol. 2004;22:706–713. doi: 10.1200/JCO.2004.05.186. [DOI] [PubMed] [Google Scholar]
- 10.Remke M, Ramaswamy V, Taylor MD. Medulloblastoma molecular dissection: the way toward targeted therapy. Curr Opin Oncol. 2013;25:674–681. doi: 10.1097/CCO.0000000000000008. [DOI] [PubMed] [Google Scholar]
- 11.Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Chintagumpala M, Adesina A, Ashley DM, Kellie SJ. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 12.Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, Troost D, Meeteren NS, Caron HN, Cloos J. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3:e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Northcott PA, Korshunov A, Pfister SM, Taylor MD. The clinical implications of medulloblastoma subgroups. Nat Rev Neurol. 2012;8:340–351. doi: 10.1038/nrneurol.2012.78. [DOI] [PubMed] [Google Scholar]
- 15.Lau J, Schmidt C, Markant SL, Taylor MD, Wechsler-Reya RJ, Weiss WA. Matching mice to malignancy: molecular subgroups and models of medulloblastoma. Childs Nerv Syst. 2012;28:521–532. doi: 10.1007/s00381-012-1704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, Finkelstein D, Qu C, Pounds S, Ellison DW. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell. 2012;21:168–180. doi: 10.1016/j.ccr.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, Witt H, Korshunov A, Read TA, Sun JL. An animal model of MYC-driven medulloblastoma. Cancer Cell. 2012;21:155–167. doi: 10.1016/j.ccr.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, Lau J, Masic S, Nguyen K, Yakovenko S. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010;24:1059–1072. doi: 10.1101/gad.1907510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, Grimmer MR, Lau J, Chesler L, Perry A. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell. 2012;21:601–613. doi: 10.1016/j.ccr.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilbertson RJ, Ellison DW. The origins of medulloblastoma subtypes. Annu Rev Pathol. 2008;3:341–365. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- 23.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 24.Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH., Jr. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 25.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 26.Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, Sorensen N, Berthold F, Henk B, Schmandt N. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997;57:2085–2088. [PubMed] [Google Scholar]
- 27.Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, McMahon AP. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66:10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, Hansen S, Knoblaugh SE, Lee D, Eberhart CG. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008;68:1768–1776. doi: 10.1158/0008-5472.CAN-07-5092. [DOI] [PubMed] [Google Scholar]
- 29.Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schuller U, Machold R, Fishell G, Rowitch DH. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Northcott PA, Hielscher T, Dubuc A, Mack S, Shih D, Remke M, Al-Halabi H, Albrecht S, Jabado N, Eberhart CG. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011;122:231–240. doi: 10.1007/s00401-011-0846-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stutz AM. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remke M, Ramaswamy V, Peacock J, Shih DJ, Koelsche C, Northcott PA, Hill N, Cavalli FM, Kool M, Wang X. TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol. 2013;126:917–929. doi: 10.1007/s00401-013-1198-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hervey-Jumper SL, Garton HJ, Lau D, Altshuler D, Quint DJ, Robertson PL, Muraszko KM, Maher CO. Differences in vascular endothelial growth factor receptor expression and correlation with the degree of enhancement in medulloblastoma. J Neurosurg Pediatr. 2014;14:121–128. doi: 10.3171/2014.4.PEDS13244. [DOI] [PubMed] [Google Scholar]
- 37.Eran A, Ozturk A, Aygun N, Izbudak I. Medulloblastoma: atypical CT and MRI findings in children. Pediatr Radiol. 2010;40:1254–1262. doi: 10.1007/s00247-009-1429-9. [DOI] [PubMed] [Google Scholar]
- 38.Chawla A, Emmanuel JV, Seow WT, Lou J, Teo HE, Lim CC. Paediatric PNET: pre-surgical MRI features. Clin Radiol. 2007;62:43–52. doi: 10.1016/j.crad.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 39.Fruehwald-Pallamar J, Puchner SB, Rossi A, Garre ML, Cama A, Koelblinger C, Osborn AG, Thurnher MM. Magnetic resonance imaging spectrum of medulloblastoma. Neuroradiology. 2011;53:387–396. doi: 10.1007/s00234-010-0829-8. [DOI] [PubMed] [Google Scholar]
- 40.Nelson AL, Algon SA, Munasinghe J, Graves O, Goumnerova L, Burstein D, Pomeroy SL, Kim JY. Magnetic resonance imaging of patched heterozygous and xenografted mouse brain tumors. J Neurooncol. 2003;62:259–267. doi: 10.1023/a:1023339902812. [DOI] [PubMed] [Google Scholar]
- 41.Samano AK, Ohshima-Hosoyama S, Whitney TG, Prajapati SI, Kilcoyne A, Taniguchi E, Morgan WW, Nelon LD, Lin AL, Togao O. Functional evaluation of therapeutic response for a mouse model of medulloblastoma. Transgenic Res. 2010;19:829–840. doi: 10.1007/s11248-010-9361-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hekmatyar SK, Wilson M, Jerome N, Salek RM, Griffin JL, Peet A, Kauppinen RA. 1H nuclear magnetic resonance spectroscopy characterisation of metabolic phenotypes in the medulloblastoma of the SMO transgenic mice. Br J Cancer. 2010;103:1297–1304. doi: 10.1038/sj.bjc.6605890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cory DA, Schwartzentruber DJ, Mock BH. Ingested manganese chloride as a contrast agent for magnetic resonance imaging. Magn Reson Imaging. 1987;5:65–70. doi: 10.1016/0730-725x(87)90485-1. [DOI] [PubMed] [Google Scholar]
- 44.Lin YJ, Koretsky AP. Manganese ion enhances T1-weighted MRI during brain activation: an approach to direct imaging of brain function. Magn Reson Med. 1997;38:378–388. doi: 10.1002/mrm.1910380305. [DOI] [PubMed] [Google Scholar]
- 45.Pautler RG, Silva AC, Koretsky AP. In vivo neuronal tract tracing using manganese-enhanced magnetic resonance imaging. Magn Reson Med. 1998;40:740–748. doi: 10.1002/mrm.1910400515. [DOI] [PubMed] [Google Scholar]
- 46.Silva AC, Lee JH, Aoki I, Koretsky AP. Manganese-enhanced magnetic resonance imaging (MEMRI): methodological and practical considerations. NMR Biomed. 2004;17:532–543. doi: 10.1002/nbm.945. [DOI] [PubMed] [Google Scholar]
- 47.Pautler RG. Biological applications of manganese-enhanced magnetic resonance imaging. Methods Mol Med. 2006;124:365–386. doi: 10.1385/1-59745-010-3:365. [DOI] [PubMed] [Google Scholar]
- 48.Silva AC, Bock NA. Manganese-enhanced MRI: an exceptional tool in translational neuroimaging. Schizophr Bull. 2008;34:595–604. doi: 10.1093/schbul/sbn056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wadghiri YZ, Blind JA, Duan X, Moreno C, Yu X, Joyner AL, Turnbull DH. Manganese-enhanced magnetic resonance imaging (MEMRI) of mouse brain development. NMR Biomed. 2004;17:613–619. doi: 10.1002/nbm.932. [DOI] [PubMed] [Google Scholar]
- 50.Yu X, Sanes DH, Aristizabal O, Wadghiri YZ, Turnbull DH. Large-scale reorganization of the tonotopic map in mouse auditory midbrain revealed by MRI. Proc Natl Acad Sci U S A. 2007;104:12193–12198. doi: 10.1073/pnas.0700960104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deans AE, Wadghiri YZ, Berrios-Otero CA, Turnbull DH. Mn enhancement and respiratory gating for in utero MRI of the embryonic mouse central nervous system. Magn Reson Med. 2008;59:1320–1328. doi: 10.1002/mrm.21609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu X, Zou J, Babb JS, Johnson G, Sanes DH, Turnbull DH. Statistical mapping of sound-evoked activity in the mouse auditory midbrain using Mn-enhanced MRI. Neuroimage. 2008;39:223–230. doi: 10.1016/j.neuroimage.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szulc KU, Nieman BJ, Houston EJ, Bartelle BB, Lerch JP, Joyner AL, Turnbull DH. MRI analysis of cerebellar and vestibular developmental phenotypes in Gbx2 conditional knockout mice. Magn Reson Med. 2013;70:1707–1717. doi: 10.1002/mrm.24597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu X, Wadghiri YZ, Sanes DH, Turnbull DH. In vivo auditory brain mapping in mice with Mn-enhanced MRI. Nat Neurosci. 2005;8:961–968. doi: 10.1038/nn1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 56.Ellis T, Smyth I, Riley E, Graham S, Elliot K, Narang M, Kay GF, Wicking C, Wainwright B. Patched 1 conditional null allele in mice. Genesis. 2003;36:158–161. doi: 10.1002/gene.10208. [DOI] [PubMed] [Google Scholar]
- 57.Li JY, Lao Z, Joyner AL. Changing requirements for Gbx2 in development of the cerebellum and maintenance of the mid/hindbrain organizer. Neuron. 2002;36:31–43. doi: 10.1016/s0896-6273(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 58.Lee JH, Silva AC, Merkle H, Koretsky AP. Manganese-enhanced magnetic resonance imaging of mouse brain after systemic administration of MnCl2: dose-dependent and temporal evolution of T1 contrast. Magn Reson Med. 2005;53:640–648. doi: 10.1002/mrm.20368. [DOI] [PubMed] [Google Scholar]
- 59.Dazai J, Bock NA, Nieman BJ, Davidson LM, Henkelman RM, Chen XJ. Multiple mouse biological loading and monitoring system for MRI. Magn Reson Med. 2004;52:709–715. doi: 10.1002/mrm.20215. [DOI] [PubMed] [Google Scholar]
- 60.Nieman BJ, Szulc KU, Turnbull DH. Three-dimensional, in vivo MRI with self-gating and image coregistration in the mouse. Magn Reson Med. 2009;61:1148–1157. doi: 10.1002/mrm.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dhenain M, Ruffins SW, Jacobs RE. Three-dimensional digital mouse atlas using high-resolution MRI. Dev Biol. 2001;232:458–470. doi: 10.1006/dbio.2001.0189. [DOI] [PubMed] [Google Scholar]
- 62.Ross BD, Zhao YJ, Neal ER, Stegman LD, Ercolani M, Ben-Yoseph O, Chenevert TL. Contributions of cell kill and posttreatment tumor growth rates to the repopulation of intracerebral 9L tumors after chemotherapy: an MRI study. Proc Natl Acad Sci U S A. 1998;95:7012–7017. doi: 10.1073/pnas.95.12.7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koutcher JA, Hu X, Xu S, Gade TP, Leeds N, Zhou XJ, Zagzag D, Holland EC. MRI of mouse models for gliomas shows similarities to humans and can be used to identify mice for preclinical trials. Neoplasia. 2002;4:480–485. doi: 10.1038/sj.neo.7900269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TT, Lin SM, Wechsler-Reya RJ. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- 65.Corcoran RB, Bachar Raveh T, Barakat MT, Lee EY, Scott MP. Insulin-like growth factor 2 is required for progression to advanced medulloblastoma in patched1 heterozygous mice. Cancer Res. 2008;68:8788–8795. doi: 10.1158/0008-5472.CAN-08-2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kessler JD, Hasegawa H, Brun SN, Emmenegger BA, Yang ZJ, Dutton JW, Wang F, Wechsler-Reya RJ. N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes Dev. 2009;23:157–170. doi: 10.1101/gad.1759909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomas WD, Chen J, Gao YR, Cheung B, Koach J, Sekyere E, Norris MD, Haber M, Ellis T, Wainwright B. Patched1 deletion increases N-Myc protein stability as a mechanism of medulloblastoma initiation and progression. Oncogene. 2009;28:1605–1615. doi: 10.1038/onc.2009.3. [DOI] [PubMed] [Google Scholar]
- 68.McManamy CS, Pears J, Weston CL, Hanzely Z, Ironside JW, Taylor RE, Grundy RG, Clifford SC, Ellison DW. Nodule formation and desmoplasia in medulloblastomas—defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007;17:151–164. doi: 10.1111/j.1750-3639.2007.00058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eberhart CG, Kaufman WE, Tihan T, Burger PC. Apoptosis, neuronal maturation, and neurotrophin expression within medulloblastoma nodules. J Neuropathol Exp Neurol. 2001;60:462–469. doi: 10.1093/jnen/60.5.462. [DOI] [PubMed] [Google Scholar]
- 70.Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, Connelly M, Stewart CF, Gould S, Rubin LL. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1+/−p53−/− mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 71.Ayrault O, Zindy F, Rehg J, Sherr CJ, Roussel MF. Two tumor suppressors, p27Kip1 and patched-1, collaborate to prevent medulloblastoma. Mol Cancer Res. 2009;7:33–40. doi: 10.1158/1541-7786.MCR-08-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sudarov A, Turnbull RK, Kim EJ, Lebel-Potter M, Guillemot F, Joyner AL. Ascl1 genetics reveals insights into cerebellum local circuit assembly. J Neurosci. 2011;31:11055–11069. doi: 10.1523/JNEUROSCI.0479-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim JYH, Nelson AL, Algon SA, Graves O, Sturla LM, Goumnerova LC, Rowitch DH, Segal RA, Pomeroy SL. Medulloblastoma tumorigenesis diverges from cerebellar granule cell differentiation in patched heterozygous mice. Dev Biol. 2003;263:50–66. doi: 10.1016/s0012-1606(03)00434-2. [DOI] [PubMed] [Google Scholar]
- 74.Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 75.Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, Rehg JE, Calabrese C, Solecki D, Eberhart CG. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 2005;19:2656–2667. doi: 10.1101/gad.1368605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee MJ, Hatton BA, Villavicencio EH, Khanna PC, Friedman SD, Ditzler S, Pullar B, Robison K, White KF, Tunkey C. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc Natl Acad Sci U S A. 2012;109:7859–7864. doi: 10.1073/pnas.1114718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dodd CA, Ward DL, Klein BG. Basal Ganglia accumulation and motor assessment following manganese chloride exposure in the C57BL/6 mouse. Int J Toxicol. 2005;24:389–397. doi: 10.1080/10915810500366500. [DOI] [PubMed] [Google Scholar]
- 79.Shukakidze A, Lazriev I, Mitagvariya N. Behavioral impairments in acute and chronic manganese poisoning in white rats. Neurosci Behav Physiol. 2003;33:263–267. doi: 10.1023/a:1022103415424. [DOI] [PubMed] [Google Scholar]
- 80.Grunecker B, Kaltwasser SF, Peterse Y, Samann PG, Schmidt MV, Wotjak CT, Czisch M. Fractionated manganese injections: effects on MRI contrast enhancement and physiological measures in C57BL/6 mice. NMR Biomed. 2010;23:913–921. doi: 10.1002/nbm.1508. [DOI] [PubMed] [Google Scholar]
- 81.Bock NA, Paiva FF, Silva AC. Fractionated manganese-enhanced MRI. NMR Biomed. 2008;21:473–478. doi: 10.1002/nbm.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yeom KW, Mobley BC, Lober RM, Andre JB, Partap S, Vogel H, Barnes PD. Distinctive MRI features of pediatric medulloblastoma subtypes. AJR Am J Roentgenol. 2013;200:895–903. doi: 10.2214/AJR.12.9249. [DOI] [PubMed] [Google Scholar]
- 83.Liu HQ, Yin X, Li Y, Zhang J, Wang Y, Tchoyoson Lim CC, Feng X. MRI features in children with desmoplastic medulloblastoma. J Clin Neurosci. 2012;19:281–285. doi: 10.1016/j.jocn.2011.04.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. Gd enhancement in MB tumors in Ptch1-CKO mice. MEMRI images were acquired using the 15-minute 3D T1w sequence (see Methods section), 24 hours after IP injection of a standard dose of MnCl2. The resulting pre-gadolinium (Gd) images demonstrated advanced MB tumors (*) in each Ptch1-CKO mouse (N = 6). Post-Gd images (right panels) were then acquired from each mouse during the same imaging session, immediately after intravenous injection of Gd-DTPA (Magnevist; 0.1 mM/kg dose) using a tail vein catheter. The post-Gd images showed heterogeneous increase in signal intensity (yellow arrows) within each of the tumors, similar to previous reports (e.g., [40]). Brain tumor uptake of Gd-DTPA has generally been related to contrast leakage due to disruption of the BBB or abnormal tumor vessels, which can differ between individual tumors and tumor type. In the Ptch1-CKO MB tumors, Gd-enhanced MRI was clearly less useful than MEMRI for determining the tumor margins and to assess tumor volume longitudinally and was therefore not pursued further.
Supplemental Table 1. Age Is Not a Good Predictor of Tumor and Brain Ventricle Volume in Ptch1-CKO mice.
Movie of early pretumoral MB lesions in a Ptch1-CKO mouse. Representative 3D volume rendering from MEMRI data, demonstrating the morphology and distribution of multiple pretumoral lesions (distinct lesions shown in different colors) in a 2-week old Ptch1-CKO mouse.