

Abstract
Switchgrass (Panicum virgatum) is a polyploid, outcrossing grass species native to North America and has recently been recognized as a potential biofuel feedstock crop. Significant phenotypic variation including ploidy is present across the two primary ecotypes of switchgrass, referred to as upland and lowland switchgrass. The tetraploid switchgrass genome is approximately 1400 Mbp, split between two subgenomes, with significant repetitive sequence content limiting the efficiency of re-sequencing approaches for determining genome diversity. To characterize genetic diversity in upland and lowland switchgrass as a first step in linking genotype to phenotype, we designed an exome capture probe set based on transcript assemblies that represent approximately 50 Mb of annotated switchgrass exome sequences. We then evaluated and optimized the probe set using solid phase comparative genome hybridization and liquid phase exome capture followed by next-generation sequencing. Using the optimized probe set, we assessed variation in the exomes of eight switchgrass genotypes representing tetraploid lowland and octoploid upland cultivars to benchmark our exome capture probe set design. We identified ample variation in the switchgrass genome including 1 395 501 single nucleotide polymorphisms (SNPs), 8173 putative copy number variants and 3336 presence/absence variants. While the majority of the SNPs (84%) detected was bi-allelic, a substantial number was tri-allelic with limited occurrence of tetra-allelic polymorphisms consistent with the heterozygous and polyploid nature of the switchgrass genome. Collectively, these data demonstrate the efficacy of exome capture for discovery of genome variation in a polyploid species with a large, repetitive and heterozygous genome.
Keywords: exome, switchgrass, polyploidy, copy number variant, presence/absence variant, Panicum virgatum
Introduction
Switchgrass (Panicum virgatum L.) is a perennial C4 grass native to North America, indigenous to southern Canada through the central USA and into northern Mexico (Moser et al., 2004). Switchgrass has the potential to produce abundant biomass in the context of abiotic stress due to its deep root system and the potential for multiple harvests per year (Schmer et al., 2008). Switchgrass is present as two primary ecotypes, referred to as upland and lowland. Upland ecotypes tend toward a smaller growth habit with narrower leaves and stems, while lowland ecotypes are better suited to longer growing seasons, with broader leaves, later flowering times, and greater biomass (Porter, 1966; Casler et al., 2012). All ecotypes of switchgrass are polyploid with the majority of upland ecotypes octoploid (2n = 8x = 72) and lowland ecotypes typically tetraploid (2n = 4x = 36), although aneuploidy has been observed (Hopkins et al., 1996; Lu et al., 1998; Costich et al., 2010).
While traditionally cultivated for soil conservation, pasture, and forage, switchgrass is also of interest as a biofuel feedstock crop (McLaughlin and Kszos, 2005; Kole et al., 2012). Genetic and genomic approaches such as genome-wide association studies and genomic selection have the potential to identify important loci and alleles that could enhance the rate of improvement of switchgrass cultivars for use as a biofuel feedstock source. To date, switchgrass genomic resources remain limited. Transcript assemblies have been constructed from Sanger-derived expressed sequence tag libraries, 454 pyrosequences, and RNA-Seq libraries (Tobias et al., 2005, 2008; Palmer et al., 2012; Wang et al., 2012; Zhang et al., 2013; Childs et al., 2014). Recently, the genome sequence of the AP13 individual of Alamo, a lowland tetraploid cultivar, was reported by the Joint Genome Institute (http://www.phytozome.net/dataUsagePolicy.php?org=Org_Pvirgatum) (Casler et al., 2011). Since the switchgrass genome is polyploid and enriched in repetitive sequences (Sharma et al., 2012), challenges are presented with respect to genome assembly. Using analysis of bacterial artificial chromosome end sequences, at least 33% of the switchgrass genome is estimated to be composed of known repeat elements with an estimated density of one gene per 16.4 kb, dependent on whether the region is euchromatic or heterochromatic (Sharma et al., 2012). In addition, switchgrass is largely self-incompatible, and as a consequence, developed cultivars maintain high levels of heterozygosity (Talbert et al., 1999; Taliaferro, et al., 1983; Martinez-Reyna and Vogel, 2002; Liu et al., 2012a).
Several methods to obtain data on genome diversity in multiple accessions in highly repetitive and/or polyploid genomes have been used. Comparative genome hybridization (CGH) has been used effectively to identify gene copy number variants (CNV) in maize (Springer et al., 2009; Swanson-Wagner et al., 2010), soybean (McHale et al., 2012), and human (Kallioniemi et al., 1992). Exome capture followed by sequencing of the captured DNA fragments has been effective in highly complex genomes (Winfield et al., 2012) and presents an alternative to CGH for targeted capture of genic sequence and identification of polymorphisms. In brief, a nucleotide probe set is designed to the genic regions of a reference genome or transcriptome, next-generation sequencing genomic DNA libraries are constructed from the target genomes, and then hybridized to the exome capture probes. After discarding unbound fragments, the captured nucleotides are amplified and sequenced using a next-generation sequencing platform, allowing high depth of coverage over the target regions (Bainbridge et al., 2010). While there are variations in the capture method (solid versus liquid phase) and in the sequencing platform, exome capture sequencing has been demonstrated to be effective in maize (Liu et al., 2012b), wheat (Winfield et al., 2012), barley (Mascher et al., 2013), pine (Neves et al., 2013), potato (Uitdewilligen et al., 2013), and human (Walsh et al., 2010), allowing researchers to sequence genic regions and avoid sequencing the highly repetitive regions of the genome.
In this study, we describe the development of an exome capture probe set for switchgrass, allowing cost effective sequencing of non-repetitive, genic regions of a polyploid grass species. The efficacy of this system is demonstrated with eight switchgrass genotypes representing four tetraploid lowland and four octoploid upland cultivars. Single nucleotide polymorphisms (SNPs), CNV, and presence/absence variants (PAVs) were identified by aligning exome capture sequence reads from the surveyed genotypes against the switchgrass AP13 reference genome. We also report on the suitability of exome capture as a method for detection of structural variation in switchgrass, and the challenges involved with variant detection in a polyploid species.
Results and Discussion
Probe design, optimization, and assessment
At the start of this project, a reference genome for switchgrass was not available and thus, probes for use in exome capture were designed using a set of Sanger-derived P. virgatum transcript sequences available from PlantGDB (Duvick et al., 2008) and a custom transcriptome assembly of pyrosequencing-derived P. virgatum transcript sequences available from the National Center for Biotechnology Information (NCBI). In total, 1 393 704 probes of 50–100 nucleotides in length representing 104 324 transcript sequences (approximately 80 Mb) were synthesized on a prototype array to assess probe performance as described previously (Mascher et al., 2013). Using this prototype array, CGH was performed using four tetraploid individual plants representing lowland (Alamo and Kanlow) and upland cultivars (Summer and Dakotah). After assessing probe performance on the CGH array, a prototype exome capture sequencing probe set was constructed by: (i) removing probes that resulted in over- or under-representation of signal intensities in the CGH experiment; and (ii) selecting a subset of probes that represents approximately 50 Mb of target space. Using the same four cultivars as used in the CGH, exome capture sequencing was performed and the results were used to rebalance the probe set as described previously (Mascher et al., 2013). The final probe set used in this study (904 693 probes) is available from Roche-NimbleGen as the SeqCap EZ Developer probe pool ‘120911_Switchgrass_GLBRC_R_EZ_HX1'. In total, 64 496 transcript sequences obtained from PlantGDB (http://plantgdb.org/) and a de novo assembly of pyrosequencing-derived transcript sequences that represents 52.7 Mb of sequence were used to select the final set of probes. Alignment of the probe set back to the original transcript sequences used in the design resulted in 868 851 probes (96%) aligning to the transcript dataset, of which 861 454 met the alignment criteria of 50% coverage and 90% identity (Figure S1a). Alignment of the probe set to the Joint Genome Institute (JGI) Release 0 of the switchgrass genome (http://www.phytozome.net/dataUsagePolicy.php?org=Org_Pvirgatum) resulted in 869 680 (96%) aligned probes, of which 837 359 met the alignment criteria of 50% coverage and 90% identity (Figure S1b). Of these, 693 738 (77%) aligned to annotated genic regions in the Release 0 annotated switchgrass genome, with 143 621 aligning to unannotated regions of the genome assembly. Of the probes aligned to genic regions, approximately 72% aligned to a single gene (Figure S1b) with the remainder aligning to two or more genes (Figure S1b). Of the annotated genes, the probe set represents a total of 50 038 805 bp composed of 168 961 exons in 44 873 genes in Release 0 of the AP13 reference genome.
Exome capture sequence analyses
Exome capture sequencing was performed on a total of eight switchgrass individuals representing seven switchgrass cultivars with representatives from both upland and lowland ecotypes (Table 1). Extracted DNA was subjected to exome capture, followed by paired-end sequencing on the Illumina platform, generating between 145 and 208 million 76-nucleotide reads following quality filtering (Table S1). Filtered reads for each genotype were aligned to both the hard-masked and unmasked version of the AP13 Release 0 switchgrass genome (Joint Genome Institute; http://www.phytozome.com/dataUsagePolicy.php?org=Org_Pvirgatum_v0.0) using bowtie (Langmead et al., 2009). If reads were permitted to align to multiple locations in the unmasked reference genome sequence, between 76 and 92% of the reads could be aligned (Table S2). A small decrease in percentage of reads mapping was observed with each genotype when reads were aligned to the hard-masked genome sequence (Table S2) indicating that the probe design process, as well as the exome capture technique, was efficient at excluding repetitive regions of the genome. For variant detection, only uniquely mapping reads were used and between 42.4 and 53.0% of reads uniquely aligned to the hard-masked reference genome. The AP13 individual, from which the reference genome was constructed, had the greatest percentage of reads that aligned (Table 2). A higher percentage of reads aligned in the lowland cultivars compared with the upland cultivars (Table 2), with the exception of lowland genotype W4. This is consistent with a previous study using simple sequence repeats (SSRs) that showed genetic differentiation between upland octoploids and the lowland tetraploid Alamo from which the reference sequence originated (Zalapa et al., 2011).
Table 1.
Genotype, cultivar, ecotype, and polyploid level for switchgrass samples used in exome capture sequencing in this study
| Genotype | Cultivar | Ecotype | Cultivar origin | Ploidy |
|---|---|---|---|---|
| A5 | Alamo | Lowland | Texas | 4x |
| AP13 | Alamo | Lowland | Texas | 4x |
| SG5-1 | SG5 | Lowland | Unknown | 4x |
| W4 | Wabasso | Lowlanda | Florida | 4x |
| Car1 | Carthage | Upland | North Carolina | 8x |
| P2 | Pathfinder | Upland | Nebraska | 8x |
| She2 | Shelter | Upland | West Virginia | 8x |
| Tr5 | Trailblazer | Upland | Nebraska | 8x |
W4 genotype exhibits lowland phenotype and nuclear chromosome number, but upland chloroplast DNA markers (Zalapa et al., 2011).
Table 2.
Alignment statistics of exome capture sequencing reads to the switchgrass Release 0 hard-masked genome
| Genotype (cultivar, ecotype) | Ploidy | % Reads mapping | Bases covered | Bases covered (>5x) | Exons with coverage | Introns with coverage | Total no. variants detected |
|---|---|---|---|---|---|---|---|
| A5 (Alamo, lowland) | 4x | 49.6 | 274 919 498 | 114 379 992 | 212 723 | 165 173 | 259 949 |
| AP13(Alamo, lowland) | 4x | 53.0 | 316 696 165 | 161 214 328 | 223 107 | 173 130 | 137 698 |
| SG5-1 (SG5, lowland) | 4x | 49.3 | 258 263 021 | 107 238 627 | 209 273 | 162 970 | 222 623 |
| W4 (Wabasso, lowland) | 4x | 44.2 | 235 259 106 | 122 862 995 | 212 101 | 164 618 | 291 484 |
| Car1 (Carthage, upland) | 8x | 46.2 | 225 884 145 | 110 555 957 | 214 679 | 166 790 | 569 434 |
| P2 (Pathfinder, upland) | 8x | 43.0 | 248 703 345 | 132 893 273 | 216 841 | 167 913 | 415 583 |
| She2 (Shelter, upland) | 8x | 45.8 | 227 248 483 | 101 204 570 | 207 331 | 161 402 | 327 980 |
| Tr5 (Trailblazer, upland) | 8x | 42.4 | 240 573 988 | 129 138 338 | 215 064 | 166 872 | 393 503 |
| Total | 517 152 642 | 244 127 862 | 245 219 | 136 819 | 1 395 501 | ||
| Concordant | 105 225 303 | 70 957 952 | 184 883 | 104 913 | 578 078 |
Bases covered indicates the number of bases of the genome with a depth of coverage of at least one uniquely aligned read, while bases covered >5x indicates the number of bases of the genome with a depth of coverage of at least five reads. Exons and introns with coverage indicate the number of exons and introns with a minimum depth of coverage of one. The switchgrass genome (counting only representative gene models) contains 269 451 exons and 203 573 introns. No. of variants indicates the number of sequence variants identified where the position was covered by at least five reads. For an allele to be called a SNP, the alternate allele needed to be represented by at least two reads, or 5% of the total reads, whichever was greater. Concordant refers to bases and variants where there was coverage in all eight genotypes.
While the probe sequences were designed from transcript (exonic) sequences, the captured DNA fragments are genomic and thus the sequenced reads include the targeted exonic sequences as well as non-targeted homologous/homeologous exonic sequences that cross-hybridize with the probes, flanking non-genic sequences, and intronic sequences (Figure 1). Overall, between 226 and 317 Mb (16.6–23.3%) of the switchgrass genome could be aligned with at least one read from the exome capture sequences, with AP13 having the highest coverage of the genome, and this was most likely attributable to the enrichment of AP13 sequences included in the probe design and the construction of the reference genome from AP13 (Table 2). In total, 517 Mb of the genome aligned to a captured read from at least one genotype with 105 Mb having at least one read from all eight genotypes (Table 2). At the exon level, between 63.0 and 83.5% of the predicted exons in the annotated Release 0 switchgrass genome had at least 1× coverage from these alignments with 184 883 exons covered in all genotypes (Table 2). With respect to intronic sequence, between 82.1–86.6% of the annotated introns were represented by at least one read, with 104 913 introns covered in all genotypes (Table 2).
Figure 1.
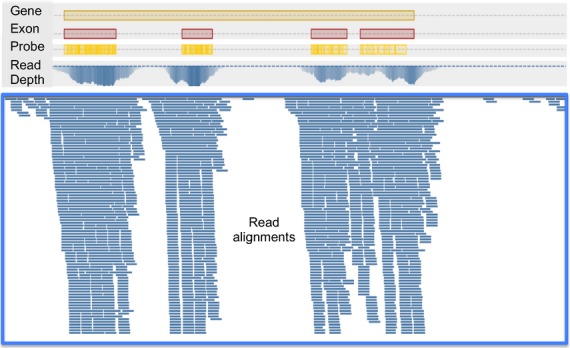
Example of read alignments from exome capture sequencing and alignment to the switchgrass reference genome.
Screenshot from the Tablet visualization program demonstrating the alignment positions of reads obtained from switchgrass exome capture. The top yellow box delineates the locus Pavirv00038283, which encodes a nitrogen metabolic regulation related protein annotated in the AP13 reference genome. The red boxes underneath the gene indicate predicted exon positions while the yellow boxes with embedded tick marks indicate probe positions. Underneath the gene level graphs, the read depth is plotted as an intensity graph with the blue-boxed region showing individual reads from exome capture sequencing of AP13 aligned against the AP13 reference genome.
A concern for exome capture based genome diversity estimations in switchgrass is the variation of genome ploidy levels between samples. To determine how the ploidy of the queried genotypes affected depth of coverage, the average number of bases with a specified minimum depth of coverage across all tetraploids and octoploids was determined (Figure 2). Total coverage between the two ecotypes across the whole genome is consistent at the lower end of the coverage spectrum while at higher depths total coverage varies between upland and lowland genotypes. However, depth of coverage in the probe target regions is highly consistent between the two ecotypes at all observed depths, with the number of covered bases also consistent across varying coverage depths. The process of exome capture involves hybridization to the probe sequence and thus, each probe has the potential to capture allelic, paralogous, homologous and/or homeologous sequences depending on the extent of sequence identity between the probe and the captured sequence. These data suggest that employment of NimbleGen exome capture and sequencing in switchgrass is not appreciably affected by an ascertainment bias in probe design, use of a single reference genome for read alignment, or by ploidy of the sample.
Figure 2.
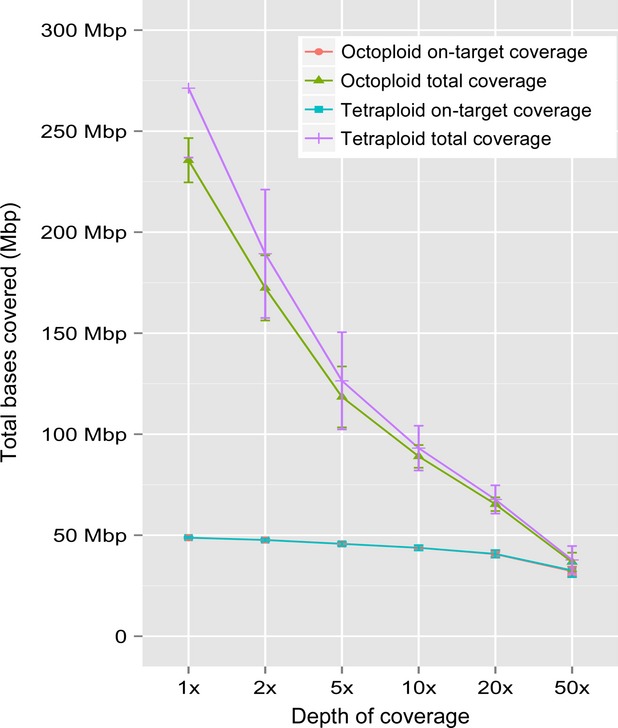
Coverage across switchgrass ecotypes.
The extent of total and on-target bases with 1-, 2-, 5-, 10-, 20-, or-50 fold sequence coverage in lowland and upland ecotypes. Coverage was averaged at the specified fold of coverage across each ecotype and plotted for both the whole genome (green and purple lines) and for target regions only (red and aqua lines). Note that the two target region lines (aqua and red) are on top of each other and appear as a single line.
At approximately 15 Gbp of sequencing output, 84–88% of the target sequences had a greater than five-fold depth of coverage (Figure 3a). Exome capture sequencing can be readily multiplexed to increase cost efficiency and throughput. To understand how reduced quantities of sequence data would affect read depth coverage of both the target and non-target regions, simulations of higher levels of multiplexing were undertaken by sampling subsets of reads from the initial capture pool and aligning these reads against the reference genome (Figure 3a). With approximately 7.5 Gbp of sequence, 80–85% of the targeted sequence retained five-fold or greater coverage, and at 3.7 Gbp of sequence output, 70–78% of targeted positions still retained a five-fold or greater coverage, corresponding to between 36–41 Mbp of the targeted exome. If the sequence output is decreased to 1.75 Gbp, the coverage decreases more substantially to between 54–68%, and the differences in coverage between samples become much more apparent. As expected, the median depth of coverage for all samples decreases in a linear fashion as sequence output decreases, indicating that analyses that rely on depth of coverage variations may be more affected by multiplexing than analyses relying on a depth threshold (Figure 3b).
Figure 3.

Distribution of on-target coverage across switchgrass genotypes obtained from exome capture sequencing.
The percentage of target bases with a depth of coverage of five or greater (a) and the median coverage of target bases (b) are plotted against total sequencing output. Regression lines were obtained through fitting the model y ˜ a*log(x) + b for (a) and a linear model for (b).
Single nucleotide polymorphism
Using an average of 13.5 Gb of sequence per genotype and a stringent filter of a read depth of five and a minimum of two reads for an alternate allele, we identified 1 395 501 total loci with a SNP in at least one of our eight genotypes (Table 2, Table S3). After filtering for positions with no missing data points, variant detection revealed 578 078 unique loci with SNPs (Table 3, Table S3). Overall, the majority of SNPs were bi-allelic (84%) followed by tri-allelic (15%), and tetra-allelic (1%) when compared across all individuals (Table 3). A total of 231 843 SNPs occurred only within a single ecotype (i.e., upland or lowland), with upland genotypes yielding approximately twice as many ecotype-restricted SNPs than lowland genotypes. Private SNPs, i.e. SNPs restricted to a single genotype, were limited in three of the lowlands (A5, AP13, SG5-1) and more abundant in W4 and the upland octoploids (Table 3). Undoubtedly, an expansion of surveyed genotypes will reduce the number of private SNPs and provide more confident annotations of ecotype-specific SNPs as well.
Table 3.
Single nucleotide polymorphisms detected from exome capture sequencing of eight switchgrass genotypes using stringent coverage criteria across the panel
| Genotype | No. SNPs |
|---|---|
| Total SNPs | 578 078 |
| Upland-restricted | 162 556 |
| Lowland-restricted | 69 287 |
| Private SNPs | |
| A5 | 8930 |
| AP13 | 7404 |
| SG5-1 | 7615 |
| W4 | 21 699 |
| Car1 | 22 207 |
| P2 | 24 007 |
| She2 | 18 491 |
| Tr5 | 21 097 |
| Allelic diversity | |
| Bi-allelic | 487 966 |
| Tri-allelic | 85 806 |
| Tetra-allelic | 4306 |
Exome capture sequencing reads were aligned to the switchgrass Release 0 reference genome and stringent criteria (>5x depth of coverage, no missing data or positions with less than the requisite coverage, and minor allele frequency >0.05. For each position, when alternate alleles were present, at least one of those alleles was required to be supported by at least two reads to call a SNP in that genotype). Private SNPs indicates the number of SNPs restricted to that genotype. Upland and lowland restricted SNPs indicate the number of SNPs that were present only in that ecotype.
As the reference genome to which the reads were aligned is derived from AP13, it is not unexpected that AP13 had the lowest exonic SNP density, suggesting limited sequencing errors in the reference genome and a high degree of separation in the assembly of the subgenomes, including alleles, and in mapping reads to their cognate location (Table 4). While A5 and SG5-1 had higher exonic SNP densities compared with AP13, the density was less than that observed in W4 and all of the upland octoploids, consistent with previous studies using SSRs that showed clear genetic differentiation between cultivars due to geographic origin, ecotype, and ploidy (Zalapa et al., 2011). All of the upland genotypes and W4 had greater SNP density in the intronic regions compared to their exonic regions (Table 4). However, A5, AP13, and SG5-1 had a lower SNP density in intronic regions compared with exonic regions. Furthermore, even for W4 and the upland genotypes, the overall SNP density in introns was not substantially higher than that observed in exons. This does not appear to be an artifact of total Mb of coverage of the annotated AP13 intronic space, as the SNP density was determined based on the number of intronic and exonic bases having at least five-fold coverage in each individual. However, the lower than expected SNP density in introns may be due to the distribution of reads within introns, which is significantly skewed towards the regions flanking the intron/exon splice site (Figure 1). Previous work in humans demonstrated that regions of exons and introns nearest the exon-intron boundaries have lower SNP density compared to regions distal to the boundaries, presumably due to the requirement for sequence conservation in areas involved in mRNA splicing (Majewski and Ott, 2002). Examination of read depth distribution in the introns of the W4 individual (Figure S2) shows that, overall, the intronic regions outside of the intron-exon boundaries (defined here as the 15 bp nearest the boundary) have a substantially lower depth of coverage compared with the 15 bp regions flanking the exon-junction. However, in exons, the read distribution is more comparable between the flanking regions and the remainder of the exon with high depths of coverage for the majority of the exonic space (Figure S3). This indicates that for introns, despite the total Mb of intronic bases with the minimum coverage to call polymorphisms (five reads), the skew in coverage near the conserved intron/exon boundaries results in a lower overall SNP density in introns than anticipated. Another possible explanation for the division in intronic density between these three individuals and W4 and the upland genotypes is that these three lowland individuals are more closely related to the reference cultivar AP13 and highly divergent alleles in these individuals may fail to align to the AP13 reference genome and thus are not called in our pipeline. This is a limitation of all read-based polymorphism detection methods in which reads derived from highly diverged alleles will fail to align to the allele represented in the reference genome sequence thereby resulting in an underestimation of polymorphism.
Table 4.
Density of detected single nucleotide polymorphisms within covered intronic and exonic sequence
| Genotype | SNPs/kbp exon | SNPs/kbp intron | Exon bases covered | Intron bases covered | Exon SNPs | Intron SNPs |
|---|---|---|---|---|---|---|
| Lowland (4x) | ||||||
| A5 | 3.78 | 3.53 | 34 165 964 | 23 300 957 | 129 285 | 82 285 |
| AP13 | 2.30 | 1.58 | 35 560 711 | 24 360 088 | 81 732 | 38 412 |
| SG5-1 | 3.63 | 3.39 | 33 196 576 | 22 148 226 | 120 371 | 75 130 |
| W4 | 4.79 | 5.08 | 33 029 595 | 21 142 397 | 158 115 | 107 449 |
| Upland (8x) | ||||||
| Car1 | 5.01 | 5.23 | 34 388 417 | 22 997 111 | 172 291 | 120 172 |
| P2 | 4.46 | 4.75 | 34 848 309 | 22 662 065 | 155 524 | 107 730 |
| She2 | 4.72 | 5.02 | 32 808 272 | 21 813 549 | 154 854 | 109 445 |
| Tr5 | 4.43 | 4.69 | 34 231 228 | 22 555 598 | 151 535 | 105 853 |
SNPs were identified where a position had at least 5x depth of coverage and at least two reads supported the alternate allele at that position, with a minor allele frequency of >0.05. Where multiple alternate alleles existed at a position, at least one of those alleles was required to be supported by at least two reads. Exon and intron bases covered indicates the number of exon and intron bases in each genotype where the depth of coverage is greater than five-fold. SNP density was determined using the number of intron and exon bases with sufficient coverage to call SNPs (five-fold depth of coverage or greater).
Detected SNPs, both on-target and total SNPs, appear to increase in a linear relation to sequencing output, with no plateau detected even at 16 Gbp of sequencing output (Figure 4a). Also, the number of SNPs detected in genotypes more diverged from the reference genotype remained higher than that of more closely related genotypes even as read number decreased (Figure 4b). This can be attributed to several factors, first being that deeper read coverage of intronic regions will permit increased SNP discovery as more intron sequence reaches the requisite read coverage for SNP calling. Second, deeper read depth will result in the discovery of more minor alleles in each genotype, especially octoploids as seen in Figure 4(a). Third, any bias in the library preparation, capture process, or sequencing method will be amplified with deeper coverage such that systematic errors will be called as SNPs.
Figure 4.

Numbers of single nucleotide polymorphisms identified from eight switchgrass genotypes.
The numbers of single nucleotide polymorphisms (SNPs) detected between the eight sampled genotypes and the AP13 reference sequence across the whole genome (a) or only in targeted regions (b) are shown. Regression lines were determined by fitting a linear plot to the average of the number of SNPs detected.
To demonstrate the efficacy of exome sequencing as a technique for characterizing switchgrass germplasm, we conducted phylogenetic analysis on all eight genotypes using 487 965 bi-allelic SNP loci containing no missing data (Figure 5). Upland and lowland genotypes clustered with other members of their ecotype, demonstrating the ability of exome-derived SNPs to accurately categorize switchgrass germplasm. Both AP13 and A5 are individuals from the Alamo cultivar and surprisingly, AP13 did not cluster closely with A5. These two individuals originated from different seed lots of Alamo, potentially originating from different locations and/or generations of seed multiplication (Casler et al., 2012; M. D. Casler, unpublished data) and thus may be more genetically distinct than individuals from Alamo that originate from a single seed lot (Gunter et al., 1996). Alternatively, variation among individuals within cultivars could be quite high. Genotype W4 is an individual from the lowland tetraploid cultivar Wabasso and was previously shown to be genetically distinct from the other lowland tetraploid Alamo and SG5 cultivars, based on SSR markers (Zalapa et al., 2011). Interestingly, W4 exhibits an insertion in its chloroplast genome previously believed to be diagnostic for the upland ecotype, yet clearly maintains a lowland-type tetraploid nuclear genome and phenotypic habitat, suggestive of ancient upland-lowland hybridization (Zalapa et al., 2011; Jakubowski et al., 2012).
Figure 5.
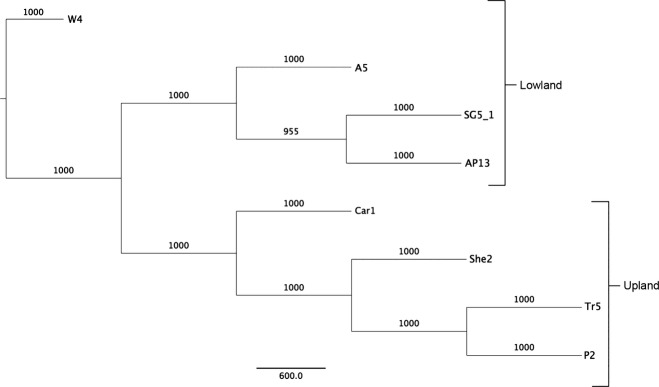
Genetic distance tree of switchgrass cultivars generated using single nucleotide polymorphism data generated from exome capture sequencing.
Neighbor-joining tree illustrating the genetic distance between the eight switchgrass genotypes sampled in this study. Cultivars included among the genotypes include the lowlands Alamo [A5 (4x) and AP13 (4x)], SG5 (SG5-1, 4x), and Wabasso (W4, 4x) and the uplands Carthage (Car1, 8x), Pathfinder (P2, 8x), Shelter (She2, 8x), and Trailblazer (Tr5, 8x). In total, 487 965 bi-allelic single nucleotide polymorphisms with no missing data were used to construct the tree.
Structural variation in switchgrass ecotypes
Copy number variation is a form of structural variation which, relative to a single reference genome, is manifested as a reduction or increase in the number of loci. These variations can result from deletions, insertions, or duplications and have been shown to be associated with disease in humans (Lupski et al., 1991; Chartier-Harlin et al., 2004) as well as in grass species such as maize (Springer et al., 2009; Swanson-Wagner et al., 2010), sorghum (Zheng et al., 2011), rice (Hurwitz et al., 2010), and barley (Munoz-Amatriain et al., 2013). Historically, CNV is detected through array hybridization, with the strength of the hybridization signal given as the indicator of amplification or deletion of the query locus. Detection of CNVs using next-generation sequencing technologies utilizes variation in read depth as well as read pair information to identify putative CNVs (Bolon et al., 2011; Chia et al., 2012; Duan et al., 2013). Using the exome capture sequencing dataset, we evaluated CNV in the surveyed genotypes using normalized depth of read coverage relative to AP13 and uniquely aligning reads. Using the approach described in maize (Chia et al., 2012), edger was used to generate fold change in read depth, and using the cutoff criteria described in Swanson-Wagner et al. (2010), ‘up-CNV’ and ‘down-CNV’ were identified. In total, 946 up-CNVs and 7227 down-CNVs were identified in the seven genotypes (Tables 5, S4 and S5). Presence–absence variants (PAVs) represent an extreme class of down-CNVs and were identified in this study using a criterion of a minimum of five reads detected in AP13 and no reads detected in the query genotype. A total of 3336 PAVs was identified in the seven query genotypes, indicating extreme levels of CNV in switchgrass (Tables 5, S6).
Table 5.
Exome capture copy number variation and presence/absence variation detected in upland (octoploid) and lowland (tetraploid) individuals
| Genotype | Ploidy | Up-CNV | Down-CNV | PAV |
|---|---|---|---|---|
| A5 (Alamo, lowland) | 4x | 109 | 2432 | 914 |
| AP13(Alamo, lowland) | 4x | NA | NA | NA |
| SG5 (SG5, lowland) | 4x | 101 | 2236 | 1047 |
| W4 (Wabasso, lowland) | 4x | 342 | 1806 | 1113 |
| Car1 (Carthage, upland) | 8x | 186 | 2501 | 1152 |
| P2 (Pathfinder, upland) | 8x | 325 | 1179 | 827 |
| She2 (Shelter, upland) | 8x | 116 | 2398 | 1249 |
| Tr5 (Trailblazer, upland) | 8x | 346 | 1307 | 961 |
| Tetraploid and lowland restricted | 4x | 267 | 2560 | 963 |
| Octoploid and upland restricted | 8x | 469 | 2265 | 1117 |
| Total, unique | 946 | 7227 | 3336 |
Tetraploid restricted indicates copy number variants and presence/absence variants detected only in tetraploid samples, and octoploid restricted indicates copy number variants detected only in octoploid individuals. Total, unique indicates the total number of unique copy number variants and presence/absence variants detected in all seven individuals.
Copy number variants and PAVs from each genotype were further examined to identify ecotype and ploidy-restricted CNVs and PAVs. For up-CNVs, 267 CNVs were restricted to tetraploid lowland ecotypes while 469 up-CNVs were restricted to octoploid upland ecotypes (Table 5). Examination of Pfam domain enrichment in the JGI AP13 annotated reference genes with up-CNV (Tables S7, S8) showed enrichment in the protein kinase domain (PF00069) in both the upland and lowland genotypes, which is consistent with the high copy number known for protein kinases in plant genomes (International Rice Genome Sequencing Project, 2005). Various domains typically found in transposable elements (PF03108, PF10551, PF13456) were also enriched in the up-CNVs in both lowland and upland genotypes. This is not surprising based on the abundance of repetitive transposons in switchgrass (Sharma et al., 2012) and suggestive of incomplete repeat masking during the annotation process. Domains found in other genes that are members of large gene families were enriched in up-CNVs, but different domains were enriched in the two ecotypes. In lowland ecotypes, the zein seed storage protein domain (Song et al., 2001) and the prolamin-like family domain (Xu and Messing, 2009), both known to be found in large gene families in grass species, were enriched in up-CNVs. In upland ecotypes the pentatricopeptide (PPR) repeat (Geddy and Brown, 2007) was enriched in up-CNVs. Interestingly, transcription factors were also enriched in up-CNVs in uplands including the KNOX domain (PF03790, PF03791) known to be involved in plant development by regulating meristem maintenance and organ patterning (Hake et al., 2004).
For the down-CNVs, 2560 were restricted to lowland tetraploid ecotypes, while 2265 were restricted to upland octoploid genotypes. For PAVs, 963 were restricted to lowland tetraploid ecotypes while 1117 were restricted to upland octoploid ecotypes (Table 5). Highly enriched Pfam domains observed in ecotype-restricted down-CNVs and PAVs included PPR domains, leucine-rich repeat (LRR) domains (Michelmore and Meyers, 1998), and transposable element-related domains, domains which are found frequently in large gene families (Tables S9, S10, S11 and S12). In soybean, genes encoding a nucleotide binding domain which is typically found in LRR disease resistance genes were significantly enriched in CNVs (McHale et al., 2012) while in Arabidopsis, PAVs were predicted for one-third of the nucleotide binding LRR genes in a survey of 18 accessions (Guo et al., 2011). As this class of genes is hypothesized to be rapidly evolving to adapt to pathogen divergence, detection of down-CNVs in switchgrass may be reflective of reduced hybridization of the probe sequences and/or reduced alignment of highly diverged sequence reads to the AP13 reference genome. Interestingly, several domains of unknown function were enriched in both upland and lowland down-CNVs and PAVs, suggesting that these are highly variable regions of the switchgrass genome.
The Pfam domain enrichment data suggested that up-CNVs, down-CNVs, and PAVs were associated with genes belonging to large gene families, consistent with observations seen in maize using CGH (Swanson-Wagner et al., 2010). Thus, we determined the representation of these structure variants relative to paralogous gene family membership. Clustering of the entire predicted switchgrass proteome (Release 0, 65 878 genes encoding 70 071 proteins) with the predicted proteomes of five other grass species (rice, maize, sorghum, B. distachyon, S. italica) yielded 23 811 (36.1%) lineage-specific switchgrass genes while 29 759 (45.2%) switchgrass genes were ‘core orthologs’ and present in an orthologous cluster with all six grass species including switchgrass (Table 6). Interestingly, a higher percentage of CNV and PAVs were lineage-specific, consistent with studies in maize (Swanson-Wagner et al., 2010) that revealed CNVs were associated with lineage-specific genes. In contrast, CNVs and PAVs were under-represented in the core ortholome (Table 6). Using a less stringent definition of the ‘core ortholome’ to address annotation issues with any one of the six grass genomes, a similar pattern of under-representation of CNVs and PAVs were observed. If the ortholome was defined as having a switchgrass protein and at least two other grass species (i.e., a three species cluster), 63.7% of the switchgrass proteome was present in a cluster with 26.9–36.0% of the up-CNVs, 36.6–44.2% of the down-CNVs, and 30.8–35.8% PAVs present in an orthologous cluster depending on the genotype. Thus, as with maize, these CNVs and PAVs may not serve essential functions in switchgrass or may be annotation artifacts. Indeed, the switchgrass reference genome and annotation are early drafts and the discovery of enrichment of domains associated with transposable elements in genes with up-CNV suggests incomplete repeat masking.
Table 6.
Distribution of all switchgrass genes and genes with copy number and presence/absence variation in switchgrass-specific genes and core ortholome in grass species
| Genotype | Total up-CNVs | Total down-CNVs | Total PAVs | Switchgrass-specific genes |
Core ortholome in grass species |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total number of switchgrass-specific genes (%) | Up-CNVs in switchgrass-specific gene family (%) | Down-CNVs in switchgrass-specific gene family (%) | PAVs in switchgrass-specific gene family (%) | Total number of switchgrass genes conserved among grass species (%) | Up-CNVs in conserved genes (%) | Down-CNVs in conserved genes (%) | PAVs in conserved genes (%) | ||||
| A5 | 109 | 2432 | 914 | 69 (63.3) | 1113 (45.8) | 463 (50.7) | 12 (11.0) | 427 (17.6) | 85 (9.3) | ||
| SG5 | 101 | 2236 | 1047 | 60 (59.4) | 1007 (45.0) | 522 (49.9) | 10 (9.9) | 408 (18.2) | 99 (9.5) | ||
| W4 | 342 | 1806 | 1113 | 178 (52.0) | 966 (53.5) | 558 (50.1) | 37 (10.8) | 358 (19.8) | 121 (10.9) | ||
| Car1 | 186 | 2501 | 1152 | 23 811 (36.1) | 113 (60.8) | 1132 (45.3) | 559 (48.5) | 29 759 (45.2) | 27 (14.5) | 392 (15.7) | 87 (7.6) |
| P2 | 325 | 1179 | 827 | 160 (49.2) | 644 (54.6) | 430 (52.0) | 31 (9.5) | 211 (17.9) | 57 (6.9) | ||
| She2 | 116 | 2398 | 1249 | 70 (60.3) | 1068 (44.5) | 610 (48.8) | 15 (12.9) | 407 (17.0) | 102 (8.2) | ||
| Tr5 | 346 | 1307 | 961 | 187 (54.0) | 685 (52.4) | 510 (53.1) | 36 (10.4) | 248 (19.0) | 79 (8.2) | ||
In total, 65 878 switchgrass genes were used in these analyses. To determine switchgrass-specific genes and core ortholome among grass species, predicted switchgrass proteins were used along with the predicted proteomes of rice, maize, sorghum, Setaria, and Brachypodium in OrthoMCL analyses as described in the Experimental Procedures. When genes are conserved in the all six Poaceae species, they are defined as core ortholome.
Copy number and presence/absence variation is not limited to single genes interspersed along the chromosome and larger tracts of CNV and PAV have been reported in other species (Springer et al., 2009; Cook et al., 2012; Iovene et al., 2013). To assess the genome distribution of structural variation in switchgrass, we mapped the Release 0 genes to the pseudomolecules of Release 1, which has 636 Mb of the total 1230 Mb assembled genome anchored to 18 pseudomolecules (http://www.phytozome.net/panicumvirgatum_er.php). As shown in Figure S4, only a few up-CNVs were found in clusters while many down-CNVs including PAVs were found in clusters; however, the majority of these are composed of two genes and may represent minor structural variation between the switchgrass genotypes. A small subset of these structural variants spanned multiple genes and may represent more substantial structural variation in the switchgrass genome. The biggest cluster was observed in a region of chromosome 8b (Figure 6). Genes across this region encode an organelle RNA recognition domain protein, protein kinase domain protein, cathepsin propeptide inhibitor domain protein, triose-phosphate transporter family protein as well as conserved genes of no known function and a hypothetical gene. Currently, a substantial portion of the switchgrass genome remains unanchored to a linkage group and is not represented in the Release 1 pseudomolecules, thereby limiting our ability to accurately detect larger-scale CNV and introgressions. When a fully anchored and complete switchgrass genome sequence is available, we can determine the extent of larger-scale structural variation at the ecotype, population, and individual level including potential introgressions.
Figure 6.

Log2 ratios between the number of reads from each query genotype and that of the AP13 reference are shown for genes at approximately 53 Mb on chromosome 8b.
(a) Plot of log2 ratios of average read depth in the query genotype relative to the AP13 reference genotype on a per gene basis.
(b) Heatmap of log2 ratios for genes that span the region in (a) denoted with a black bar plus flanking genes of this cluster.
Some previous studies have shown that certain classes of genes have high rates of polymorphism and thus are fast evolving (Clark et al., 2007; McNally et al., 2009). As copy number variation could confound determination of SNP density, we removed all genes with CNV and then classified the genes into a high-SNP density class if they had SNP densities >1 standard deviation from the mean (Table S13). Using Pfam domain annotations, enrichment tests were performed to identify gene ontology (GO) terms associated with high-SNP density genes (Fisher's exact test, P ≤ 0.05, Table S14). Pfam domains involved in various levels of protein interaction were enriched in the high-SNP density genes consistent with previous studies examining intra-species diversity (Clark et al., 2007; McNally et al., 2009). The most highly enriched domains in those genes are protein kinase domains, LRR domains, and PPR domains (Table S14). As discussed above, LRR receptor kinases are found in a large gene family that play a critical role in plant pathogen response (Michelmore and Meyers, 1998), and possess hyper-variable regions as well as high rates of diversification. PPR domain containing genes are a very large gene family in plants whose functions have not been fully elucidated but have been shown to be extremely variable between species and are not strongly conserved (Geddy and Brown, 2007).
Conclusions
We have generated a robust probe set to interrogate genetic diversity in exonic regions of switchgrass. We were able to demonstrate limited ascertainment bias in the probe design, exome capture process, and bioinformatics analyses using lowland tetraploid as well as upland octoploid genotypes. We were able to identify ample genetic variation between genotypes of lowland and upland ecotypes as well as within the two ecotypes, which is consistent with previous studies that showed genetic differentiation of switchgrass based on geographic origin, ploidy and ecotype. Furthermore, we identified structural variation in the switchgrass genome that may be a contributor to phenotypic variation.
Experimental Procedures
Probe design and alignment to the reference genome
Two sets of transcripts were used for probe design. The PlantGDB (Duvick et al., 2008) assembly of P. virgatum Sanger-derived expressed sequence tags (Release 181a; 120 524 unique transcripts) and a custom assembly of P. virgatum pyrosequencing-derived transcript sequences downloaded from NCBI (SRR064785-SRR064802). The pyrosequencing-derived RNA-Seq reads were trimmed of low-quality sequences using the fastq_quality_trimmer tool from the fastx toolkit (version 0.0.13; http://hannonlab.cshl.edu/fastx_toolkit/). Reads <200 nucleotides were discarded, and the quality threshold parameter was set to 20. Fasta and quality files were prepared from the fastq files using the fastq_to_fasta and fastq_quality_converter tools from the fastx toolkit. The runAssembly program from the newbler package (Roche, http://www.454.com/products/analysis-software/) was used to assemble the sequences. A minimum read overlap of 25% and a minimum overlap identity of 90% was required during assembly. In total, 31 871 pyrosequencing-derived transcripts were generated. Both the PlantGDB and pyrosequencing-derived transcript assemblies were filtered by removing sequences <250 nucleotides in length, sequences with more than 10 Ns, and any sequence with more than 50% low complexity sequence. Highly redundant sequences and transposable element-related sequences were removed from the switchgrass transcript assembly collection leaving 104 621 sequences that were used for probe design for CGH testing. Known grass genes important for cell wall synthesis were identified by surveying literature and keyword searches, and the transcript assemblies were aligned to these genes of interest. After initial CGH testing, a final set of probes designed from PlantGDB and pyrosequencing-derived transcript assemblies were selected to provide a total length of probe sequences of approximately 50 Mbp. First, we aligned all switchgrass transcript assemblies to the S. italica predicted proteome (Bennetzen et al., 2012). For each S. italica protein, a single switchgrass sequence was chosen based on highest alignment scores with those switchgrass sequences matching cell-wall-related genes of interest given priority; any switchgrass sequences that had matched any cell-wall-related genes of interest but that did not have alignments with S. italica proteins were also kept. Second, other switchgrass sequences that did not align to the S. italica proteome were randomly chosen until the length of all probes from retained switchgrass sequences totaled 50 Mbp. Sequences with probes with weak CGH signals were not considered. In total, 64 496 sequences (PlantGDB and pyrosequencing-derived) representing 52.7 Mb of transcript sequence were used to select the final set of probes.
Representation of annotated genic sequences by the probes in the Roche-NimbleGen SeqCap EZ Developer probe pool design 120911_Switchgrass_GLBRC_R_EZ_HX1 (904 693 probes) was determined by alignment of the probe sequences to the AP13 reference genome Release 0 (http://www.phytozome.net/dataUsagePolicy.php?org=Org_Pvirgatum) using gmap (version 2013-03-31) (Wu and Watanabe, 2005) with default parameters. After alignment, only probe sequences with alignment of at least 50% of their length and with 90% identity were used for further analyses. To determine exonic coverage in the AP13 reference genome, any exon with at least 1 bp of probe alignment was considered to have coverage. We considered this reasonable given the nature of exome capture reads to trail far beyond the end of the capture probes themselves (Figure 1). Any gene with coverage of at least one exon was considered covered. Total covered exon sequence was the sum of all covered exons, with exon lengths as determined by the switchgrass Release 0 gene annotations. Probes were also checked using these alignment methods and criteria by mapping back to the set of transcripts used in the probe design.
Plant material and DNA isolation
In total, 11 switchgrass individuals, representing 10 cultivars that included upland and lowland ecotypes as well as tetraploid and octoploids were used in this study. For the CGH and pilot exome capture sequencing experiments, the tetraploid lowland cultivars (Alamo and Kanlow) and tetraploid upland cultivars (Summer, Dacotah) genotypes were used. For the exome capture sequencing datasets described in this study (Table 1), the lowland individuals represented four cultivars: Alamo [A5 (4x) and AP13 (4x)], SG5-1 (SG5, 4x), and Wabasso (W4, 4x) while the upland individuals represented four cultivars: Carthage (Car1, 8x), Pathfinder (P2, 8x), Shelter (She2, 8x), and Trailblazer (Tr5, 8x). Total genomic DNA was extracted from freeze-dried leaves using the CTAB extraction protocol (Saghaimaroof et al., 1984).
Exome capture sequencing, read alignment and analysis
Using the eight genotypes listed in Table 1, exome capture sequencing was performed using the protocol established by Roche-NimbleGen for SeqCap EZ Developer library preparation, as detailed in Mascher et al. (2013). Eight separate libraries were constructed and each sample was captured twice and sequenced on the Illumina HiSeq 2000 platform generating 76 nucleotide paired-end reads. Initial read quality assessment was performed using the fastqc program (v0.10.0; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Cutadapt (v1.1; https://code.google.com/p/cutadapt/) was used to remove adapter sequences, PCR primers, and to trim bases with a quality score below 20. Reads shorter than 35 bases in length after trimming were discarded.
Filtered exome capture sequence reads were aligned to the hard-masked P. virgatum Release 0 reference genome (http://www.phytozome.net/dataUsagePolicy.php?org=Org_Pvirgatum) using bowtie version 0.12.7 (Langmead et al., 2009), requiring unique alignments with a maximum of a single mis-matched nucleotide in the first 35 bases of the read. Initial analysis was performed on approximately 16 Gbp of sequencing output from each individual (Table S1). To simulate lower quantities of sequencing output, random sampling of initial reads were used to simulate multiplexing of four, 10, and 20 samples per lane, with the assumption of approximately 32 Gbp of sequence per lane of unplexed data. Initial sequencing data comprised roughly one-half of an Illumina HiSeq lane for each individual, so to simulate four samples per lane, approximately one-half of the reads from the initial read set were randomly selected to make up a simulated multiplexed sample. Approximately one-fifth of the reads were randomly selected to simulate 10 samples per lane, and one-tenth of the reads were randomly sampled to simulate 20 samples per lane. Sampling was performed using the Perl rand() function and each sampling simulation was performed 100 times to ensure accuracy. Alignment criteria were identical to those of the initial analysis. Read depth calculation was performed using the genomeCoverageBed function of the bedtools program (Quinlan and Hall, 2010) with the −d flag set. For simulated multiplexing experiments, the average depth was calculated across all simulations at that multiplexing level (e.g., all 10-plexed experimental coverage was averaged to determine the coverage for a 10-plexed sample). Depth plots were generated using the ggplot2 package for the r statistical package.
Single nucleotide polymorphism analysis
Read alignments that met the mapping criteria were processed with the index, sort, merge, and pileup functions of the samtools package (Li et al., 2009) version 0.1.12a. The −Bcf options were used for the pileup command, and index sort and merge were run with default parameters (Li et al., 2009). Resultant pileup files were filtered with custom Perl scripts. To call a position as polymorphic, at least one sample required a minimum of five reads at the position, and at least two (or at least 5%, whichever is greater) of those reads must represent an alternate allele. In cases where more than one alternate allele is present at a location, at least one of those alleles must be represented by at least two reads or 5% of all reads at that position, whichever is greater. Additional filtering was performed to generate the SNP set used for phylogenetic analysis. In order for a position to be used for this analysis, all individuals must meet the above criteria for this position, and no positions were allowed to have missing data, insufficient depth of coverage to call an allele, or to be considered a minor allele (number of non-reference reads <5% of all reads). Phylogenetic analysis was performed using phylip v. 3.695 (Felsenstein, 1989). Bootstrapping was performed using the seqboot function (1000 replicates). Genetic distances were calculated using the gendist function with default parameters, neighbor-joining trees were generated using neighbor function with default parameters, and a consensus tree was generated using consense with default parameters. Resulting trees were visualized with figtree v. 1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/).
High-SNP density genes were defined as genes with a SNP density at least one standard deviation greater than the mean SNP density for all genes. When determining high-SNP density genes, genes that were found to be CNVs or PAVs were removed from the dataset, with the rationale that high or low levels of polymorphism density may instead be detection of duplicated genes or reduced gene copy number.
Structural variation analyses
To assess CNV and PAV, the numbers of reads that mapped to each gene were counted and normalized by library size, and the depth of coverage was computed for each gene using edger (Robinson et al., 2010). A ratio for gene coverage depth was established by dividing the normalized depth for each gene by the normalized depth of the same gene in the reference AP13 and then taking the log2 of the ratio. P-values were computed using a biological coefficient of variation of 0.4 using edger following the authors' instructions (http://www.bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf). For each genotype, genes with a positive log2 ratio above the 99th percentile value and a P-value <0.05 were identified as up-CNVs, while genes with a log2 ratio lower than the negative value of the up-CNV cutoff and P-value <0.05 were identified as down-CNVs. The initial set of down-CNVs were further examined to identify PAVs that met the criteria of no reads mapped to the gene in the query genotype, where at least 5x coverage for the same gene was observed in the AP13 reference genotype.
To examine structural variation at the genome level, putative CNVs were examined for their distribution along the 18 switchgrass chromosomes. Annotated genes (65 878) from the JGI Release 0 annotation (http://www.phytozome.net/panicumvirgatum.php) were aligned to the Release 1 pseudomolecules using gmap (version 2013-03-31) (Wu and Watanabe, 2005) with default parameters. In total, 65 167 genes aligned at the 50% coverage and 90% identity cutoff criteria, of which, 40 047 genes were assigned a chromosomal location based on the best match. No chromosomal information was available for 25 120 genes as they mapped to the contigs that were not assigned to chromosomes in Release 1. For 40 047 genes with a chromosomal location, assessment of localized clustering of structural variation was conducted with a cluster defined as two or more putative CNVs adjacent on a Release 1 pseudomolecule, and the results were visualized using r (version 3.0.1) (http://www.r-project.org) and MeV Multiple Experiment Viewer (http://www.tm4.org/).
To investigate the distribution of CNVs or PAVs in paralogous and orthologous gene families, OrthoMCL (Li et al., 2003) with default parameters was used to search for ortholog clusters in the predicted proteomes of six grass species including switchgrass (Release 0), B. distachyon ((The International Brachypodium Initiative, 2010) http://www.phytozome.net/brachy.php), Oryza sativa ((International Rice Genome Sequencing Project, 2005), http://rice.plantbiology.msu.edu/), S. italica [(Bennetzen et al., 2012),http://www.phytozome.net/foxtailmillet.php], Zea mays [maize, (Schnable et al., 2009), http://www.phytozome.net/maize.php], and Sorghum bicolor [sorghum, (Paterson et al., 2009) http://www.phytozome.net/sorghum_er.php]. Switchgrass genes that failed to cluster with any other species or remained as singletons were defined as switchgrass-specific.
Pfam enrichment tests
InterProScan (Zdobnov and Apweiler, 2001) was used to annotate the Release 0 predicted proteins with Pfam domains (Punta et al., 2012). Pfam domain enrichment was performed using a Fisher's exact test with a significance threshold of P < 0.05. Pfam terms meeting this criterion were considered to be enriched.
Data access
Exome capture reads have been deposited in the NCBI SRA under accession number (BioProject ID PRJNA244250). Single nucleotide polymorphisms identified in this study (Table S3) are available for download from the Dryad Digital Repository (http://datadryad.org/) at this DOI (http://doi.org/10.5061/dryad.b52p5).
Acknowledgments
This work was funded by the Department of Energy Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We are grateful to Jeremy Schmutz of the Department of Energy JGI for his work on the switchgrass genome.
Conflict of Interest
The authors from Roche-NimbleGen, Inc. recognize a competing interest in this publication as employees of the company.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Summary of mapping of exome capture probes to the transcriptome design sequences and the switchgrass reference genome Release 0.
Coverage distribution of introns.
Coverage distribution of exons.
Clusters of copy number variants in switchgrass.
Number of raw Illumina reads and reads that passed quality control.
Table S2. Exome capture read alignment statistics.
Table S3. Single nucleotide polymorphisms identified in eight switchgrass genotypes.
Table S4. Genes identified with up-copy number variants in seven switchgrass genotypes using exome capture sequencing.
Table S5. Genes identified with down-copy number variants in seven switchgrass genotypes using exome capture sequencing.
Table S6. Genes with presence/absence variants identified in seven switchgrass genotypes identified by exome capture sequencing.
Table S7. Enrichment of Pfam domains in lowland ecotype up-CNVs.
Table S8. Enrichment of Pfam domains in upland ecotype up-CNVs.
Table S9. Enrichment of Pfam domains in lowland ecotype down-CNVs.
Table S10. Enrichment of Pfam domains in upland ecotype down-CNVs.
Table S11. Enrichment of Pfam domains in lowland ecotype PAVs.
Table S12. Enrichment of Pfam domains in upland ecotype PAVs.
Table S13. Genes with higher than expected SNP density (>1 standard deviation higher than mean) identified in eight switchgrass genotypes by exome capture sequencing.
Table S14. Pfam domains and P-values for genes with high-density single nucleotide polymorphism density.
References
- Bainbridge MN, Wang M, Burgess DL, et al. Whole exome capture in solution with 3 Gbp of data. Genome Biol. 2010;11:R62. doi: 10.1186/gb-2010-11-6-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Schmutz J, Wang H, et al. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012;30:555–561. doi: 10.1038/nbt.2196. [DOI] [PubMed] [Google Scholar]
- Bolon YT, Haun WJ, Xu WW, et al. Phenotypic and genomic analyses of a fast neutron mutant population resource in soybean. Plant Physiol. 2011;156:240–253. doi: 10.1104/pp.110.170811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casler MD, Tobias CM, Kaeppler SM, Buell CR, Wang ZY, Cao PJ, Schmutz J, Ronald P. The switchgrass genome: tools and strategies. Plant Genome. 2011;4:273–282. [Google Scholar]
- Casler MD, Mitchell RB, Vogel KP. Switchgrass. In: Kole C, Joshi CP, Shonnard D, editors. Handbook of Bioenergy Crop Plants. New York: Taylor & Francis; 2012. pp. 563–590. [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Chia JM, Song C, Bradbury PJ, et al. Maize HapMap2 identifies extant variation from a genome in flux. Nat. Genet. 2012;44:803–807. doi: 10.1038/ng.2313. [DOI] [PubMed] [Google Scholar]
- Childs KL, Nandety A, Hirsch C, Gongora-Castillo E, Schmutz J, Kaeppler SM, Casler MD, Buell CR. Generation of transcript assemblies and identification of single nucleotide polymorphisms from seven lowland and upland cultivars of switchgrass. Plant Genome. 2014;7 Doi: 10.3835/plantgenome2013.12.0041. [Google Scholar]
- Clark RM, Schweikert G, Toomajian C, et al. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science. 2007;317:338–342. doi: 10.1126/science.1138632. [DOI] [PubMed] [Google Scholar]
- Cook DE, Lee TG, Guo X, et al. Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science. 2012;338:1206–1209. doi: 10.1126/science.1228746. [DOI] [PubMed] [Google Scholar]
- Costich DE, Friebe B, Sheehan MJ, Casler MD, Buckler ES. Genome-size variation in switchgrass (Panicum virgatum): flow cytometry and cytology reveal rampant aneuploidy. Plant Genome. 2010;3:130–141. [Google Scholar]
- Duan J, Zhang JG, Deng HW, Wang YP. Comparative studies of copy number variation detection methods for next-generation sequencing technologies. PLoS One. 2013;8:e59128. doi: 10.1371/journal.pone.0059128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvick J, Fu A, Muppirala U, Sabharwal M, Wilkerson MD, Lawrence CJ, Lushbough C, Brendel V. PlantGDB: a resource for comparative plant genomics. Nucleic Acids Res. 2008;36:D959–D965. doi: 10.1093/nar/gkm1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP – phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Geddy R, Brown GG. Genes encoding pentatricopeptide repeat (PPR) proteins are not conserved in location in plant genomes and may be subject to diversifying selection. BMC Genomics. 2007;8:130. doi: 10.1186/1471-2164-8-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter LE, Tuskan GA, Wullschleger SD. Diversity among populations of switchgrass based on RAPD markers. Crop Sci. 1996;36:1017–1022. [Google Scholar]
- Guo YL, Fitz J, Schneeberger K, Ossowski S, Cao J, Weigel D. Genome-wide comparison of nucleotide-binding site-leucine-rich repeat-encoding genes in Arabidopsis. Plant Physiol. 2011;157:757–769. doi: 10.1104/pp.111.181990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hake S, Smith HM, Holtan H, Magnani E, Mele G, Ramirez J. The role of knox genes in plant development. Annu. Rev. Cell Dev. Biol. 2004;20:125–151. doi: 10.1146/annurev.cellbio.20.031803.093824. [DOI] [PubMed] [Google Scholar]
- Hopkins AA, Taliaferro CM, Murphy CD, Christian D. Chromosome number and nuclear DNA content of several switchgrass populations. Crop Sci. 1996;36:1192–1195. [Google Scholar]
- Hurwitz BL, Kudrna D, Yu Y, Sebastian A, Zuccolo A, Jackson SA, Ware D, Wing RA, Stein L. Rice structural variation: a comparative analysis of structural variation between rice and three of its closest relatives in the genus Oryza. Plant J. 2010;63:990–1003. doi: 10.1111/j.1365-313X.2010.04293.x. [DOI] [PubMed] [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- Iovene M, Zhang T, Lou Q, Buell CR, Jiang J. Copy number variation in potato – an asexually propagated autotetraploid species. Plant J. 2013;75:80–89. doi: 10.1111/tpj.12200. [DOI] [PubMed] [Google Scholar]
- Jakubowski AR, Price DL, Acharya A, Wei Y, Brummer EC, Kaeppler SM, Casler MD. Natural hybrids and gene flow between upland and lowland switchgrass. Crop Sci. 2012;51:2626–2641. [Google Scholar]
- Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- Kole C, Joshi CP, Shonnard D. Handbook of Bioenergy Crop Plants. Boca Raton, FL: CRC Press; 2012. [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LL, Wu YQ, Wang YW, Samuels T. A high-density simple sequence repeat-based genetic linkage map of switchgrass. G3 (Bethesda) 2012a;2:357–370. doi: 10.1534/g3.111.001503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Ying K, Yeh CT, et al. Changes in genome content generated via segregation of non-allelic homologs. Plant J. 2012b;72:390–399. doi: 10.1111/j.1365-313X.2012.05087.x. [DOI] [PubMed] [Google Scholar]
- Lu K, Kaeppler SM, Vogel KP, Arumuganathan K, Lee D. Nuclear DNA content and chromosome numbers in switchgrass. Great Plains Res. 1998;8:269–280. [Google Scholar]
- Lupski JR, Deocaluna RM, Slaugenhaupt S, et al. DNA Duplication associated with Charcot-Marie-Tooth disease type-1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- Majewski J, Ott J. Distribution and characterization of regulatory elements in the human genome. Genome Res. 2002;12:1827–1836. doi: 10.1101/gr.606402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Reyna JM, Vogel KP. Incompatibility systems in switchgrass. Crop Sci. 2002;42:1800–1805. [Google Scholar]
- Mascher M, Richmond TA, Gerhardt DJ, et al. Barley whole exome capture: a tool for genomic research in the genus Hordeum and beyond. Plant J. 2013;76:494–505. doi: 10.1111/tpj.12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale LK, Haun WJ, Xu WW, Bhaskar PB, Anderson JE, Hyten DL, Gerhardt DJ, Jeddeloh JA, Stupar RM. Structural variants in the soybean genome localize to clusters of biotic stress-response genes. Plant Physiol. 2012;159:1295–1308. doi: 10.1104/pp.112.194605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin SB, Kszos LA. Development of switchgrass (Panicum virgatum) as a bioenergy feedstock in the United States. Biomass Bioenergy. 2005;28:515–535. [Google Scholar]
- McNally KL, Childs KL, Bohnert R, et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl Acad. Sci. USA. 2009;106:12273–12278. doi: 10.1073/pnas.0900992106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelmore RW, Meyers BC. Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res. 1998;8:1113–1130. doi: 10.1101/gr.8.11.1113. [DOI] [PubMed] [Google Scholar]
- Moser LE, Burson BL, Sollenberger LE. Warm-Season (C4. Madison, WI: American Society of Agronomy, Crop Science Society of America, Soil Science Society of America; 2004. Grasses. [Google Scholar]
- Munoz-Amatriain M, Eichten SR, Wicker T, et al. Distribution, functional impact, and origin mechanisms of copy number variation in the barley genome. Genome Biol. 2013;14:R58. doi: 10.1186/gb-2013-14-6-r58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves LG, Davis JM, Barbazuk WB, Kirst M. Whole-exome targeted sequencing of the uncharacterized pine genome. Plant J. 2013;75:146–156. doi: 10.1111/tpj.12193. [DOI] [PubMed] [Google Scholar]
- Palmer NA, Saathoff AJ, Kim J, Benson A, Tobias CM, Twigg P, Vogel KP, Madhavan S, Sarath G. Next-generation sequencing of crown and rhizome transcriptome from an upland, tetraploid switchgrass. Bioenergy Res. 2012;5:649–661. [Google Scholar]
- Paterson AH, Bowers JE, Bruggmann R, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- Porter CL. An analysis of variation between upland and lowland switchgrass Panicum virgatum L. in central Oklahoma. Ecology. 1966;47:980–992. [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghaimaroof MA, Soliman KM, Jorgensen RA, Allard RW. Ribosomal DNA spacer-length polymorphisms in barley – Mendelian inheritance, chromosomal location, and population-dynamics. Proc. Natl Acad. Sci. USA. 1984;81:8014–8018. doi: 10.1073/pnas.81.24.8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmer MR, Vogel KP, Mitchell RB, Perrin RK. Net energy of cellulosic ethanol from switchgrass. Proc. Natl Acad. Sci. USA. 2008;105:464–469. doi: 10.1073/pnas.0704767105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, et al. The B73 Maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- Sharma MK, Sharma R, Cao PJ, Jenkins J, Bartley LE, Qualls M, Grimwood J, Schmutz J, Rokhsar D, Ronald PC. A genome-wide survey of switchgrass genome structure and organization. PLoS One. 2012;7:e33892. doi: 10.1371/journal.pone.0033892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R, Llaca V, Linton E, Messing J. Sequence, regulation, and evolution of the maize 22-kD alpha zein gene family. Genome Res. 2001;11:1817–1825. doi: 10.1101/gr.197301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer NM, Ying K, Fu Y, et al. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet. 2009;5:e1000734. doi: 10.1371/journal.pgen.1000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson-Wagner RA, Eichten SR, Kumari S, Tiffin P, Stein JC, Ware D, Springer NM. Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Res. 2010;20:1689–1699. doi: 10.1101/gr.109165.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert LE, Timothy DH, Burns JC, Rawlings JO, Moll RH. Estimates of genetic-parameters in switchgrass. Crop Sci. 1983;23:725–728. [Google Scholar]
- Taliaferro CM, Vogel KP, Bouton JH, McLaughlin SB, Tuskan GA. Reproductive characteristics and breeding improvement potential of switchgrass. In: Overend R, Chornet E, editors. Proceedings of the 4th Biomass Conference of the Americas Biomass, A Growth Opportunity in Green Energy and Value-Added Products. Oxford: Elsevier Sciences; 1999. pp. 147–153. [Google Scholar]
- The International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- Tobias CM, Twigg P, Hayden DM, Vogel KP, Mitchell RM, Lazo GR, Chow EK, Sarath G. Analysis of expressed sequence tags and the identification of associated short tandem repeats in switchgrass. Theor. Appl. Genet. 2005;111:956–964. doi: 10.1007/s00122-005-0030-3. [DOI] [PubMed] [Google Scholar]
- Tobias CM, Sarath G, Twigg P, Lindquist E, Pangilinan J, Penning BW, Barry K, McCann MC, Carpita NC, Lazo GR. Comparative genomics in switchgrass using 61,585 high-quality expressed sequence tags. Plant Genome. 2008;1:111–124. [Google Scholar]
- Uitdewilligen JG, Wolters AM, D'Hoop BB, Borm TJ, Visser RG, van Eck HJ. A next-generation sequencing method for genotyping-by-sequencing of highly heterozygous autotetraploid potato. PLoS One. 2013;8:e62355. doi: 10.1371/journal.pone.0062355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, Pennil C, Nord AS, Mandell JB, Swisher EM, King MC. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc. Natl Acad. Sci. USA. 2010;107:12629–12633. doi: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Zeng X, Iyer NJ, Bryant DW, Mockler TC, Mahalingam R. Exploring the switchgrass transcriptome using second-generation sequencing technology. PLoS One. 2012;7:e34225. doi: 10.1371/journal.pone.0034225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winfield MO, Wilkinson PA, Allen AM, et al. Targeted re-sequencing of the allohexaploid wheat exome. Plant Biotechnol. J. 2012;10:733–742. doi: 10.1111/j.1467-7652.2012.00713.x. [DOI] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- Xu JH, Messing J. Amplification of prolamin storage protein genes in different subfamilies of the Poaceae. Theor. Appl. Genet. 2009;119:1397–1412. doi: 10.1007/s00122-009-1143-x. [DOI] [PubMed] [Google Scholar]
- Zalapa JE, Price DL, Kaeppler SM, Tobias CM, Okada M, Casler MD. Hierarchical classification of switchgrass genotypes using SSR and chloroplast sequences: ecotypes, ploidies, gene pools, and cultivars. Theor. Appl. Genet. 2011;122:805–817. doi: 10.1007/s00122-010-1488-1. [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- Zhang JY, Lee YC, Torres-Jerez I, et al. Development of an integrated transcript sequence database and a gene expression atlas for gene discovery and analysis in switchgrass (Panicum virgatum L.) Plant J. 2013;74:160–173. doi: 10.1111/tpj.12104. [DOI] [PubMed] [Google Scholar]
- Zheng LY, Guo XS, He B, et al. Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor) Genome Biol. 2011;12:R114. doi: 10.1186/gb-2011-12-11-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of mapping of exome capture probes to the transcriptome design sequences and the switchgrass reference genome Release 0.
Coverage distribution of introns.
Coverage distribution of exons.
Clusters of copy number variants in switchgrass.
Number of raw Illumina reads and reads that passed quality control.
Table S2. Exome capture read alignment statistics.
Table S3. Single nucleotide polymorphisms identified in eight switchgrass genotypes.
Table S4. Genes identified with up-copy number variants in seven switchgrass genotypes using exome capture sequencing.
Table S5. Genes identified with down-copy number variants in seven switchgrass genotypes using exome capture sequencing.
Table S6. Genes with presence/absence variants identified in seven switchgrass genotypes identified by exome capture sequencing.
Table S7. Enrichment of Pfam domains in lowland ecotype up-CNVs.
Table S8. Enrichment of Pfam domains in upland ecotype up-CNVs.
Table S9. Enrichment of Pfam domains in lowland ecotype down-CNVs.
Table S10. Enrichment of Pfam domains in upland ecotype down-CNVs.
Table S11. Enrichment of Pfam domains in lowland ecotype PAVs.
Table S12. Enrichment of Pfam domains in upland ecotype PAVs.
Table S13. Genes with higher than expected SNP density (>1 standard deviation higher than mean) identified in eight switchgrass genotypes by exome capture sequencing.
Table S14. Pfam domains and P-values for genes with high-density single nucleotide polymorphism density.