

Abstract
Extensive changes in DNA methylation are common in cancer and may contribute to oncogenesis through transcriptional silencing of tumor-suppressor genes1. Genome-scale studies have yielded important insights into these changes2, 3, 4, 5 but have focused on CpG islands or gene promoters. We used whole-genome bisulfite sequencing (bisulfite-seq) to comprehensively profile a primary human colorectal tumor and adjacent normal colon tissue at single-basepair resolution. Regions of focal hypermethylation in the tumor were located primarily at CpG islands and were concentrated within regions of long-range (>100 kb) hypomethylation. These hypomethylated domains covered nearly half of the genome and coincided with late replication and attachment to the nuclear lamina in human cell lines. We confirmed the confluence of hypermethylation and hypomethylation within these domains in 25 diverse colorectal tumors and matched adjacent tissue. We propose that widespread DNA methylation changes in cancer are linked to silencing programs orchestrated by the three-dimensional organization of chromatin within the nucleus.
Main
We performed comprehensive methylome analysis of a CpG island (CGI) methylator phenotype (CIMP)-high6, stage 3 primary colon adenocarcinoma harboring a KRAS mutation resulting in p.Gly12Asp. We estimated the tumor DNA content of the sample at 67% using microarray-based SNP genotyping (Supplementary Figs. 1,2). We used bisulfite-seq7 to generate sequences of 76 billion uniquely alignable bp (28× average genome coverage) for the tumor sample and sequences of 87 billion bp (32× coverage) for a normal adjacent colon mucosa sample from the same individual (Supplementary Note). Approximately 80% of all genomic CpG dinucleotides were covered with five or more uniquely mapped sequencing reads in both samples (Supplementary Tables 1,2). Bisulfite-seq methylation levels showed strong concordance (Pearson correlations (r) of 0.93–0.97) with Illumina Infinium HumanMethylation27k methylation measurements. Regions of copy number alterations (114 Mb with gains and 223 Mb with losses) also yielded similar DNA methylation results for bisulfite-seq and the Infinium array (Supplementary Fig. 3). DNA methylation at non-CpG (CpH) cytosine contexts was almost undetectable, as has been reported for somatic cell lines and which is in contrast to human embryonic stem cells (hESCs) and induced pluripotent stem cells8 (Supplementary Fig. 4). A representative 10-kb region of the genome shows dramatic differences between the tumor and normal colon tissue; DNA methylation in the tumor is higher within the CGI promoter region but is lower outside of the CGI (Fig. 1).
Figure 1. Bisulfite-seq of a colon tumor and adjacent normal mucosa.

Individual sequencing reads and summary methylation levels are shown within a 10-kb region around the STK33 gene promoter for the normal adjacent colon tissue (top) and matched colon tumor (bottom). Reads are shown without respect to strand orientation and are colored to indicate the percentage of CpG dinucleotides methylated within the read (reads with no CpGs are indicated in yellow). The percent methylation tracks summarize the percentage of reads methylated for each CpG dinucleotide (black dots) as well as the average methylation within sliding windows of five CpGs (solid brown graph). The methylation difference track at the bottom shows the average methylation difference between tumor and normal tissue within sliding windows of five CpGs, with red indicating tumor hypermethylation and green indicating tumor hypomethylation.
We investigated global DNA methylation changes by comparing the methylation of tumor to adjacent normal mucosa in genome-wide windows as small as two adjacent CpG dinucleotides and as large as 20 kb (Supplementary Fig. 5). At all window sizes, the vast majority of windows were methylated in both tissues, but two clear clusters of normally unmethylated windows were present at window sizes less than 5 kb (Fig. 2a). Based on these clusters, we identified discrete elements by screening for methylation within windows of five adjacent CpGs, defining those with an average methylation level <5% as unmethylated and those with a level >35% as methylated. This allowed the identification of 5,163 elements that were unmethylated in normal colon cells and methylated in the tumor (methylation prone) and 21,134 elements that were unmethylated in both (methylation resistant). Although less abundant, we identified 662 elements methylated in normal colon tissue and unmethylated in the tumor (methylation loss).
Figure 2. Three distinct methylation classes at focal elements.
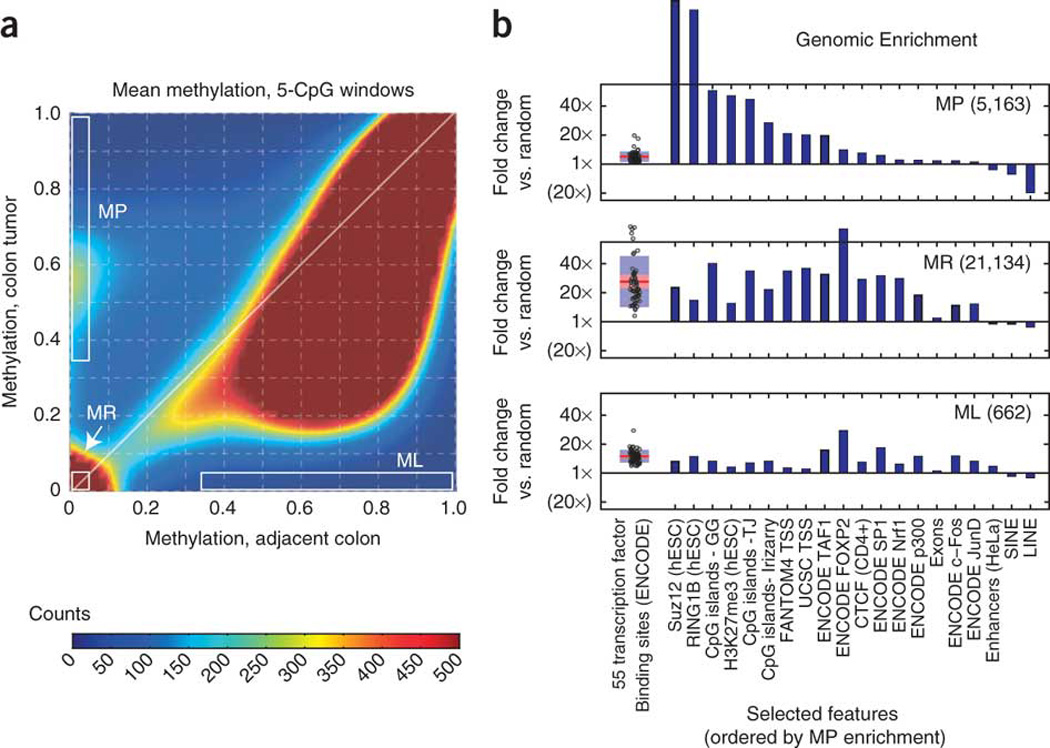
(a) Density plot of the average DNA methylation within all windows of five adjacent CpG dinucleotides on chromosome 4. Distinct subsets of methylation-prone (MP) and methylation-resistant (MR) windows are visible as high-density clusters, whereas the methylation-loss (ML) region is low density. (b) Comparison of each methylation class to ENCODE protein-DNA binding (ChIP-seq) data9 and other genomic features (for the full version, see Supplementary Fig. 6). We determined genomic enrichment by dividing the proportion of overlapping elements within each methylation class by the proportion of overlapping elements within size-matched, randomly generated genomic locations (shown as fold changes). All transcription factors are shown in a boxplot (left), and selected genomic features are shown as individual bars (right).
We compared these three methylation classes to genomic annotations and ENCODE9 protein-DNA interactions (chromatin immunoprecipitation sequencing (ChIP-seq)) by examining genomic enrichment relative to randomly selected regions in the genome (Fig. 2b and Supplementary Fig. 6). Although only 29% of methylation-prone elements corresponded to known promoters (transcription start sites (TSS)), they almost universally (95%) coincided with CGIs10 and were highly enriched for marks of polycomb repressive complex 1 and 2 activity in hESCs. Although earlier work has shown enrichment of polycomb sites at methylation-prone promoters6, 11, 12, 13, non-promoter regulatory regions have not been well characterized. We found that non-promoter regions containing the known enhancer marks p300 (ref. 14) and H3K27ac15 were more likely to be methylation resistant than promoters, but those non-promoter regions that were methylation prone were, like promoters, primarily at CGIs and were highly overlapping with polycomb marks. Binding of the transcription factors Sp1, Nrf1 or YY1 can protect CGIs from cancer-specific DNA methylation4, 16, and we found this protective property to extend to most of the 55 transcription factors present in ENCODE; methylation-resistant elements were strongly enriched for almost all factors (median enrichment 22×), whereas methylation-prone elements were only weakly enriched (median enrichment 4×). Similarly, methylation-resistant elements had 29× enrichment for CTCF insulator binding sites17 (51% of methylation-resistant elements overlapped a CTCF site), whereas methylation-prone elements only had 7× enrichment for these sites (13% of all methylation-prone elements overlapped a CTCF site). Consistent with an earlier report18, methylation-prone elements were strongly depleted of Alu repeats and other short and long interspersed elements relative to methylation-resistant and methylation-loss elements (Fig. 2b).
We performed microarray expression analysis and found that methylation-prone promoters, both CGI and non-CGI, were associated with low expression in normal colon tissue and with loss of expression in the tumor (Supplementary Fig. 8). Genes silenced in the tumor gained methylation across an entire CGI promoter (MGMT) (Fig. 3a) or within an isolated portion of a promoter (MAF). We used the program HOMER19 to identify sequence motifs enriched within either methylation-resistant or methylation-prone elements (Fig. 3b and Supplementary Figs. 13–15). In agreement with a recent study4, methylation-prone elements were enriched for CA and GA dinucleotide repeats, and methylation-resistant elements were enriched for numerous sequences matching known transcription factor binding motifs, including Nrf1, Sp1, GABPA, YY1 and NF-Y (Fig. 3b and Supplementary Fig. 14).
Figure 3. Focal methylation classes correspond to distinct epigenomic and sequence signatures.
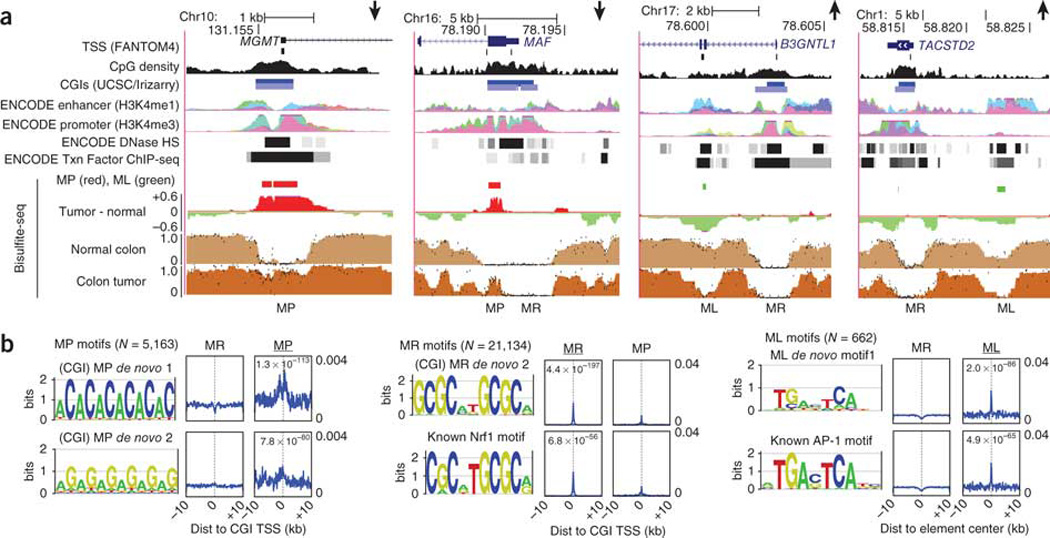
(a) UCSC Genome Browser plots of two downregulated (MGMT and MAF) and two upregulated (B3GNTL1 and TACSTD2) genes reveal that elements of the methylation-prone (MP), methylation-resistant (MR) and methylation-loss (ML) classes often coincide with a combination of promoter or enhancer histone modifications (H3K4 methylation), DNase I hypersensitivity (HS) and transcription-factor binding. In the enhancer and promoter tracks, each color represents an individual ENCODE cell line, and all cell lines are combined in the DNase HS and transcription factor tracks. (b) Significant results from HOMER19 sequence motif searches within each of the three methylation classes (for the full results, see the Supplementary Figs. 13–15). Because methylation-prone and methylation-resistant elements most often corresponded to CGI TSS, alignments for these two classes are relative to the oriented TSS, whereas those for the methylation-loss class (right) show alignments relative to the center of the unoriented methylation-loss element. Matches to known motifs from the HOMER database are shown below the de novo motif they match (Nrf1 and AP-1).
Compared to methylation-prone and methylation-resistant elements, those elements losing methylation in the tumor (methylation loss) occurred less frequently at promoters (3%) and CGIs (26%) but were generally enriched in ENCODE transcription factor binding sites including TAF1 (Fig. 2b; median enrichment 11.4×), suggesting that many of these elements act as either unannotated promoters or transcriptional enhancers. Methylation-loss elements, whether at a promoter or an enhancer, were more likely to be associated with genes that gained expression in the tumor (Supplementary Fig. 7), which is in contrast to methylation-prone elements. B3GNTL1 and TACSTD2 are both upregulated in the tumor and contain methylation-loss elements within putative enhancers with sites for the Fos and Jun transcription factors (Fig. 3a). The predominantly over-represented sequence motif within methylation-loss elements corresponded to the AP-1 binding sequence of the Fos-Jun dimer (Fig. 3b), making it tempting to speculate that methylation loss reflects chromatin remodeling initiated by Fos-Jun at these sites, a process known to play a crucial role in intestinal proliferation and oncogenesis20.
Genome-wide methylation changes at varying window sizes showed that the majority of the genome that was methylated could be resolved into two distinct fractions in windows of 20 kb (Fig. 4a). One fraction was markedly hypomethylated in the tumor, resembling the partially methylated domains (PMDs) that occur in somatic cell lines but not hESCs8. Based on this profile, we identified PMDs genome wide by searching for 10-kb windows with an average methylation of 20–60% and then collapsing these into domains longer than 100 kb; in all, 44% of the tumor genome was contained within these PMD domains (Fig. 4b). We found diverse somatic cell lines to share close to 75% of the PMDs8, and we found that about 75% of IMR-90 fibroblast PMDs were contained within colon tumor PMDs. Although tumor PMD regions had slightly reduced methylation in normal colon cells (Fig. 4a), virtually none of them satisfied our PMD criteria (Fig. 4b), indicating a shared property of immortalized cell lines and tumors that is absent from normal somatic tissues.
Figure 4. Hypermethylated CGIs fall within long, tumor-specific PMDs.
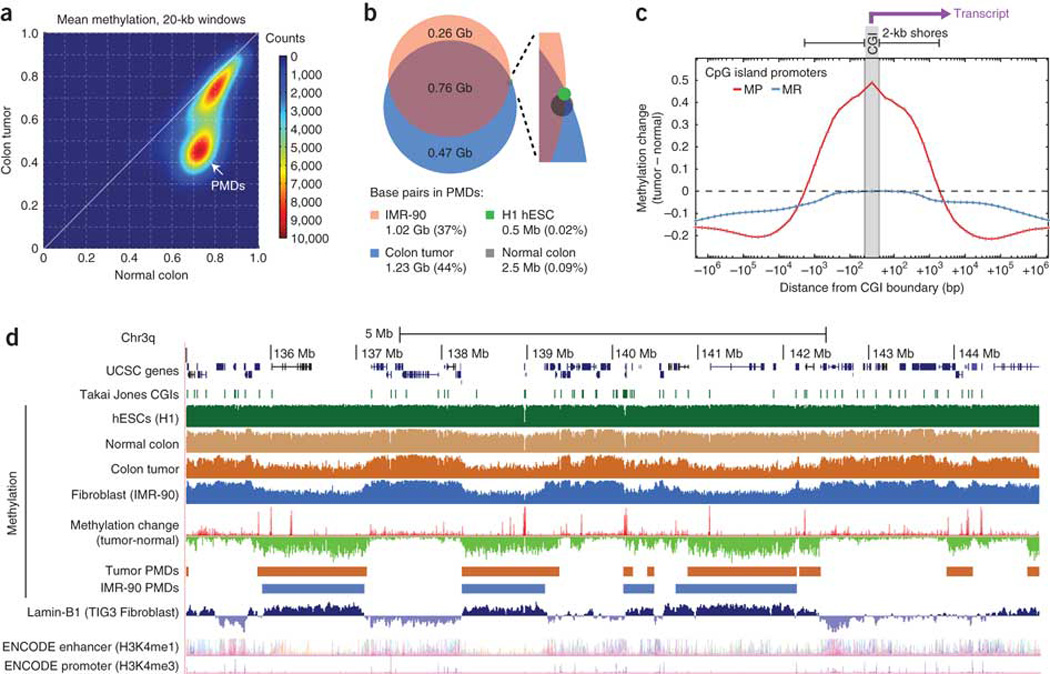
(a) Density plot of average DNA methylation within all 20-kb windows on chromosome 4 showing a distinct subset of windows representing PMDs in the tumor but not normal colon tissue. (b) We identified PMDs for four cell types by searching for 100-kb partially methylated windows (see text), and we compared the percentage of the genome contained within PMDs between the tumor and normal colon tissue along with two other cell types7. (c) The average methylation change is shown as a function of distance from CGI promoters for all promoters that were unmethylated in the normal colon (with mean methylation <0.2). We divided promoters into methylation-prone (MP; with mean tumor methylation >0.3) and methylation-resistant (MR; with mean tumor methylation <0.2), and the plots are oriented to show the transcribed region toward the right side. (d) UCSC Genome Browser plot of a representative 10-Mb region on chromosome 3q showing substantial overlap between colon tumor and IMR-90 PMDs, Lamin-B1 marks and focal hypermethylation (methylation-resistant elements are visible as red spikes in the methylation change track). Lamin-B1 and ENCODE enhancer and promoter tracks are from the UCSC annotation database.
To investigate the relationship between promoter hypermethylation and PMD hypomethylation, we calculated methylation levels in variably sized windows surrounding CGI promoters (Fig. 4c). Promoters that were hypermethylated within about 1 kb of the CGI boundary tended to be more hypomethylated starting from about 10 kb to more than a Mb away. This is apparent in a 10-Mb region (Fig. 4d) where focal hypermethylation peaks (red spikes in the methylation change track) are found primarily within hypomethylated PMDs. This relationship held true genome wide even after controlling for expression levels of the associated genes (Supplementary Fig. 9), with 57% of methylation-prone elements and only 19% of methylation-resistant elements being located within PMDs (Supplementary Fig. 6). At gene promoters, associated hypomethylation occurred both upstream and downstream of the TSS (Fig. 4c), indicating that observations about differential methylation at gene bodies21, 22 may be at least partially a consequence of these longer, multi-gene PMDs.
Previously, multi-gene domains of long-range epigenetic silencing (LRES) were identified on the basis of gene expression in prostate cancer cells23, 24. Some prostate LRESs from these studies24 clearly coincided with our colon PMDs, and overall the genes within these two sets overlapped significantly (P < 0.0001; Supplementary Figs. 10,16). However, our PMDs did not significantly overlap with prostate LRESs at the bp level, which is likely to be a consequence of the lower resolution of the LRES study. We did, however, observe a striking correspondence between tumor PMDs and nuclear-lamina–associated domains (LADs25) in TIG3 fibroblast cells (Fig. 4d). Because dynamic association and dissociation with the nuclear lamina has been implicated as a key mechanism in the developmental regulation of long-range gene silencing26, we investigated a large region containing several PMD boundaries specific to either the colon tumor or IMR-90 fibroblasts (Fig. 5a). This region contains two tumor suppressor genes subject to frequent epigenetic silencing in epithelial tumors, NRG1 (ref. 27) and SFRP1 (ref. 28), both of which had hypermethylated promoters and reduced expression in our tumor. Both genes fell within colon-tumor–specific PMDs, with the SFRP1 promoter defining a PMD boundary present in IMR-90 cells but not the tumor (Fig. 5b). Determining whether such cell-type–specific boundaries arise during normal lineage specification or oncogenesis will require additional study, but recent work has shown that loss of key boundary elements such as CTCF sites can cause aberrant spreading of silencing domains in cancer29. Profiling tumor PMDs will allow exploration into whether chromosomal rearrangements can also lead to aberrant silencing boundaries.
Figure 5. Properties of PMD boundaries.
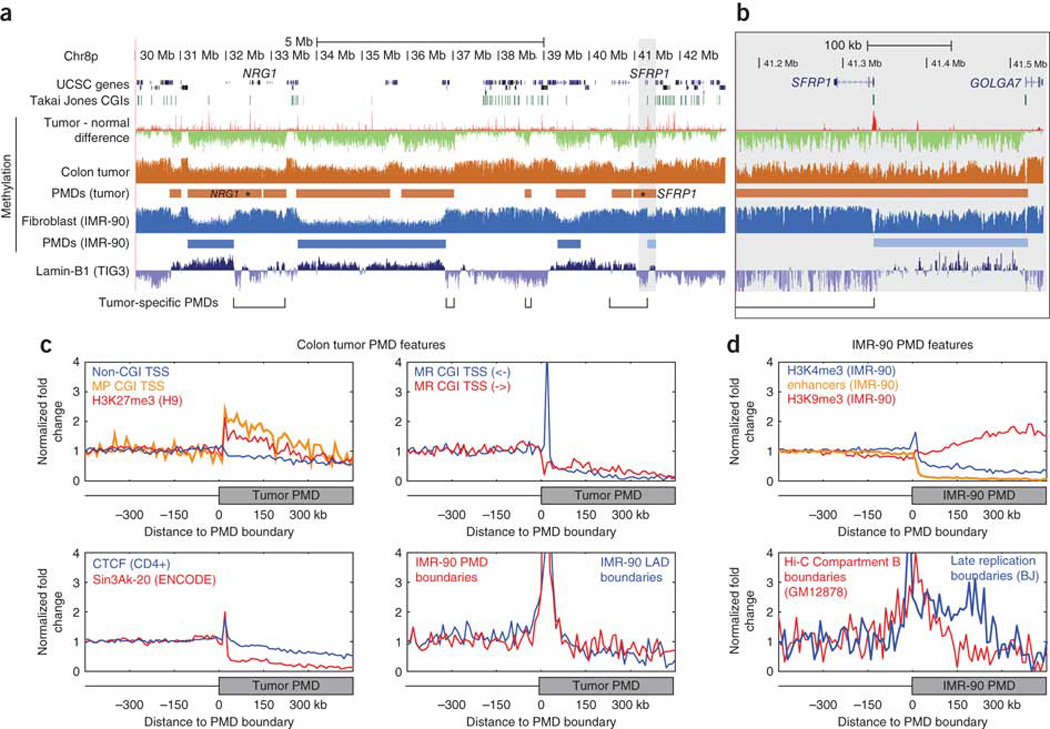
(a) UCSC Genome Browser plot of a 13-Mb region with several PMD boundaries specific to either the colon tumor or IMR-90 fibroblasts7. Tumor-specific PMD regions are annotated, showing that the two epithelial tumor suppressors NRG1 and SFRP1 fall within these regions. (b) A higher resolution view of the highlighted area surrounding SFRP1 showing that the gene promoter is hypermethylated in the tumor and defines a cell-type–specific PMD boundary in IMR-90 cells. (c,d) Average genomic density of a number of annotation features is plotted for 10-kb bins relative to colon tumor (c) and IMR-90 (d) PMD boundaries. Plots are oriented with regions outside the PMD to the left of the midpoint and regions inside the PMD to the right of the midpoint, as shown in the diagrams below each plot. We normalized the genomic density by dividing the value within each bin by the average density within bins lying outside of PMDs. For complete boundary plots, see Supplementary Figure 11.
Intrigued by the SFRP1 boundary promoter, we investigated the Genome-wide distribution of various genomic annotations with respect to PMD boundaries (Fig. 5c and Supplementary Fig. 11). We confirmed that methylation-prone CGI promoters were enriched within PMDs, but we found that they were most abundant within the first 150 kb of the PMDs, a pattern similar to that of the hESC polycomb mark relative to LAD boundaries25 (Fig. 5c, upper left). Conversely, methylation-resistant CGI promoters were depleted within PMDs but were strongly enriched within 10 kb of the boundary itself, as shown at SFRP1 (Fig. 5c, upper right). Only methylation-resistant promoters facing away from the PMD were enriched at the boundary; those promoters facing into the PMD were depleted, which is suggestive of a mechanistic link between gene transcription and PMD boundary formation. Comparisons to ENCODE ChIP-seq data revealed other factors that were enriched at PMD boundaries, including CTCF and SIN3A, the latter of which was enriched at the PMD boundary but was almost completely absent within the PMD itself (Fig. 5c, lower left). LAD boundaries from IMR-90 cells strongly coincided with colon tumor PMD boundaries (Fig. 5c, lower right).
We used IMR-90 data to investigate the relationship between PMD boundaries and histone-modification profiles in the same cells30 (Fig. 5d, top). The promoter-associated H3K4me3 mark was enriched at PMD boundaries and was somewhat depleted within PMDs, whereas the combinatorial H3K4me1/3 enhancer signature31 was almost completely absent within PMDs. The heterochromatin-associated H3K9me3 mark was enriched within deeply internal portions of PMDs. IMR-90 PMD boundaries also coincided with boundaries of late-replication domains in fibroblasts32 (Fig. 5d, bottom), a feature that may contribute mechanistically to their DNA methylation loss over repeated cell divisions33. In the three-dimensional structure of the nucleus, IMR-90 PMDs corresponded to one of the two major nuclear compartments identified using whole-genome chromatin conformation capture (Hi-C) in lymphoblastoid cells34.
We confirmed the spatial association of hypermethylation and hypomethylation within PMDs using DNA methylation array data from an independent and diverse set of 25 colon and rectal tumors and matched adjacent tissue6 (Fig. 6). We used stringent tumor to normal comparisons to characterize array features in one of four categories for each tumor: methylation resistant, methylation prone, partial methylation loss or constitutively methylated (Fig. 6a and Supplementary Fig. 12). The extent of hypermethylation and hypomethylation were highly correlated within each tumor (Fig. 6b), suggesting the presence of a single cancer cell population that accumulates both alterations simultaneously. In each tumor, methylation-prone and partial–methylation-loss loci were preferentially localized within the tumor PMDs relative to the invariant methylation-resistant and constitutively methylated loci (Fig. 6c). Two conclusions can be drawn from these observations: (i) colorectal tumors in general contain hypomethylated PMDs relative to adjacent normal colon, and (ii) focal hypermethylation and long-range hypomethylation are associated within these PMD domains. These two phenomena appear to be linked through a developmentally regulated26 and evolutionarily conserved35 mechanism involving the higher-order organization of chromatin within the nucleus and DNA replication timing32. How the two are decoupled in immortalized cell lines, which do have clear PMDs but not widespread focal hypermethylation, will give insights into the specific gene-silencing mechanisms used by cancer cells36. These findings show the power of bisulfite-seq to detect both local changes (that is, at promoters, enhancers or insulators) and higher-order chromatin structure in a single assay using clinical DNA samples.
Figure 6. Tumor-specific hypermethylation and hypomethylation are correlated and are strongly enriched within PMDs in a diverse set of 25 colon tumors.
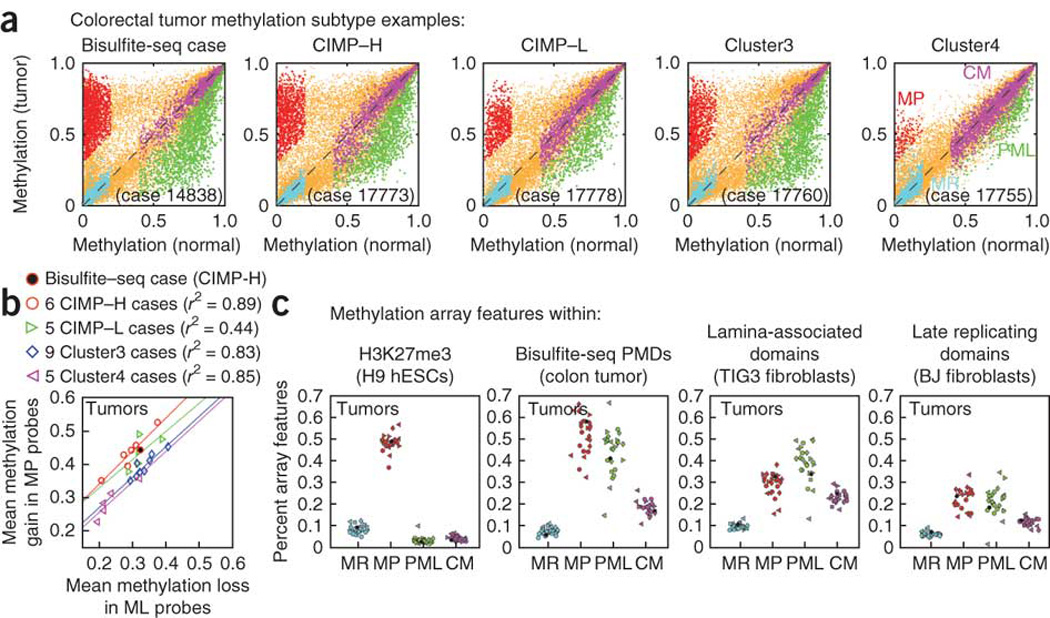
(a) Infinium HumanMethylation27k array values (β values) for five representative tumors, each compared to adjacent normal colon mucosa from the same individual. The tumor sequenced using bisulfite-seq (from individual 14838) is shown alongside one tumor of each methylation subtype from ref. 6, and colored points indicate probes identified as one of four methylation classes: methylation prone (MP, red), methylation resistant (MR, cyan), partial methylation loss (PML, green) and constitutively methylated (CM, purple). Probes not clearly falling into one of these categories are shown in orange. (b) The mean hypermethylation of methylation-prone probes (tumor β minus normal β) and the mean hypomethylation of methylation-loss probes (normal β minus tumor β) show a strong linear correlation (Pearson r = 0.80) across all samples. Colored lines indicate the best robust linear regression fit for each methylation subtype. (c) For each tumor-normal comparison, the fraction of microarray features falling within different genomic regions (H3K27me3, bisulfite-seq PMDs, and so on) is shown, with features separated by methylation class (methylation resistant, methylation prone, methylation loss and constitutively methylated). Shapes indicate tumor subtype as in panel b, with the bisulfite-seq data colored solid black.
URLs
Bisulfite-seq maps, http://epigenome.usc.edu/; University of Southern California High Performance Computing Center, http://www.usc.edu/hpcc/; MAQ, http://maq.sourceforge.net/; in-house Java library, http://sourceforge.net/projects/ngsgenomelibs/; Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/; Salk Epigenomics, http://neomorph.salk.edu/human_methylome/; UCSC ENCODE portal, http://genome.ucsc.edu/ENCODE/.
Methods
Tissue samples
Sequencing samples were picked from a recent study profiling promoter DNA methylation of 125 colorectal tumors and 25 normal adjacent tissues6. BRAF mutation analyses was performed on all samples using pyrosequencing, and we excluded those with the BRAF mutation resulting in p.Val600Glu and those with microsatellite instability from selection. We then ranked tumors by average gain of methylation among a set of cancer-specific promoters and picked a strongly hypermethylated tumor from the CIMP-high class. The tumor harbored a KRAS mutation resulting in p.Gly12Asp and was from a 60-year-old male with stage 3 primary colon adenocarcinoma. Tumor and adjacent normal mucosa DNA were obtained from the Ontario Tumor Bank (with the accessions OTB14838T for tumor and OTB14838N for adjacent tissue). Informed consent was obtained for all subjects and was approved for the described analyses by the University of Southern California institutional review board.
Bisulfite-seq library construction and sequencing
DNA libraries from each sample were prepared using a method similar to a previous study7 but with a number of customizations. Briefly, sequencing adapters with fully methylated cytosines (Integrated DNA Technologies) were used to create Illumina Genome Analyzer IIx sequencing libraries followed by bisulfite conversion (Zymo EZ DNA Methylation Kit, Zymo Research) and PCR amplification. Two libraries were made for each sample, and each library was PCR amplified by dividing it into nine independent PCR reactions and then pooling the PCR products.
Attachment of the library DNA to the Genome Analyzer flow cell was performed on an Illumina Cluster Station fluidics device. Single-end DNA sequencing (76-bp reads) was performed using the Illumina Genome Analyzer IIx as previously described7. A total of 63 and 67 lanes were sequenced for the normal colon and tumor samples, respectively. Reads passing the Illumina chastity quality filter were retained, resulting in 1,694,273,737 reads for normal samples and 1,658,970,379 reads for tumor samples (Supplementary Table 1).
Alignment and extraction of methyl-cytosine levels
Genome build hg18 (NCBI v.36) was used for all analyses. MAQ (see URLs) was used for sequence alignment, using the '-c' option to match any C or T in the sequencing read to a C in the reference genome. SAMtools37 was used to perform processing and merging of BAM files, and duplicate reads starting at the same genomic position were removed for each of the two sequencing libraries per sample. Reads were also filtered out if they had a MAQ mapping quality score of less than 30, which removed alignments with many mismatches as well as those reads aligning equally well to multiple locations in the genome. An in-house Java library (see URLs) was used to transform BAM alignments to percent methylation for each cytosine in the genome. Each cytosine in the reference genome was included for analysis if it was covered by three or more C or T reads on the bisulfite-converted strand, greater than 90% of all reads on the strand were either C or T and greater than 90% of reads on the opposite strand (that is, the 'G' strand, which is not affected by bisulfite conversion) were G. Cytosines were considered high-confidence CpGs if greater than 90% of reads at the following position were G or high-confidence CpHs if greater than 90% of reads at the next position were H (A,C,T). For the colon tumor sample, of 49,057,680 reference genome CpGs covered by sequence reads, 96.2% (47,179,160) were classified as high-confidence CpGs. In the normal colon mucosa, 47,886,080 of 49,101,921 CpGs (97.5%) were classified as high-confidence CpGs.
Identification of partially methylated domains
Partially methylated domains were identified by scanning all windows of at least 10 kb and having at least ten individual cytosines contained within CpG dinucleotides (each CpG dinucleotide contains two cytosines, one on each strand). All overlapping partially methylated windows were collapsed into a single PMD. Only those PMDs longer than 100 kb were used in subsequent analysis. Because of the preponderance of methylation-resistant promoters found within 10 kb of PMD boundaries (Fig. 5c), we shortened each PMD by 10 kb on either end for the analysis of validation tumors (Fig. 6b).
Identification of focal methylation changes
Unmethylated regions were identified by scanning all windows of at least ten individual CpG cytosines within five CpG dinucleotides (each CpG dinucleotide contains two cytosines, one on each strand). Only those cytosines covered by at least three cytosine or thymine reads were counted, and each CpG dinucleotide was assigned a weighting factor defined as the span (in bp) between the next CpG dinucleotide upstream and the next CpG dinucleotide downstream. A weighted average was calculated for each window, and those windows with an average DNA methylation of less than 5% in both tumor and adjacent normal tissue were categorized as methylation resistant. Those windows with methylation of less than 5% in the adjacent normal tissue and greater than 35% in the tumor were characterized as methylation prone, and those windows with methylation of greater than 35% in the adjacent normal tissue and less than 5% in the tumor were characterized as methylation loss. Two or more overlapping regions from a single methylation class were merged into one. For enrichment of functional annotations within these regions (Fig. 3b,c), elements of each methylation class within 500 bp were merged into a single locus. Elements were not merged for motif analysis (Fig. 3d).
Copy-number analysis using Illumina 1M SNP arrays
We performed SNP analyses using the Illumina 1M SNP array platform (Illumina) in order to assess tumor purity and copy-number variation in the colon tumor and normal adjacent samples. Tumor purity was assessed using the large deleted segment on chromosome 1p at position chr1:1–37,740,361. We selected all SNPs with a B-allele frequency between 0.1 and 0.9 (that is, SNPs that are heterozygous AB in the diploid cell fraction and have a deleted A- or B-in the haploid tumor cell fraction). The mean haploid allele frequency μaf of these 2,194 probes was 0.762 (s.d. of 0.050 between probes yielded a 95% confidence interval of 0.760–0.764). The percentage of haploid cells is calculated as 2 − (1/μaf) or 0.688 (95% confidence interval of 0.684–0.692). To identify copy-number alterations genome wide, we used genoCNV38 in matched tumor/normal mode (genoCNA) and output domains of copy number 1, 2 and 3 or more in the tumor (Supplementary Figure 2). A total of 223 Mb in 196 domains were determined to be copy number 1 (deletion), 114 Mb in 598 domains were determined to be copy number 3 or more (increase) and the remainder of the data (2,566 Mb) were determined to be diploid.
Gene expression data
The expression data processing is described in reference 6 and is available at the NCBI Gene Expression Omnibus (GEO) (see URLs) under accession number GSE25070.
Twenty-five paired colon tumor and normal validation samples
Infinium HumanMethylation27k array data were downloaded for 25 tumor and normal colon samples from reference 6 (GEO GSE25062). Methylation subtype labels (CIMP-H, CIMP-L, Cluster 3 and Cluster 4) were taken from the GEO record and represented an unsupervised clustering of 125 colon tumors from two cohorts: (i) 100 samples from the Ontario Tumor Bank ('OTB' collection), which included the sample from individual 14838 described in detail in this study, and (ii) 26 paired tumor and adjacent tissue samples from the Groene Hart Hospital in The Netherlands ('CL' collection). For our validation analysis in Figure 6, we used only the CL collection, as it was completely independent from the OTB tumor studied and it allowed us to compare differences between tumor and non-tumor methylation levels. We removed one individual (17768) because of a potential sample swap, yielding 25 total pairs (Supplementary Fig. 12).
We identified methylation classes for the CL pairs as follows. Methylation-resistant probes for an individual tumor sample were defined as those with a normal tissue β (methylation) value of less than 0.2 and a tumor β within 0.5 s.d. of the probe mean among the 25 normal tissue samples. Methylation-prone probes were those with a normal tissue β of less than 0.2 and a tumor β more than 5 s.d. above the probe mean among the 25 normal tissue samples. Constitutively-methylated probes were those with a normal tissue β of greater than 0.5 and a tumor β within 0.5 s.d. of the mean of the probe among the 25 adjacent normal tissue samples. Partial–methylation-loss probes were those with a normal tissue β of greater than 0.5 and a tumor β more than 5 s.d. below the mean of the probe among the 25 normal tissue samples.
Other external data sets
Bisulfite-seq data for the H1 and IMR-90 cell lines7 were downloaded from the Salk Epigenomics website (see URLs). FANTOM4 5′ TSS annotations39 were downloaded from the Genome Biology website. ChIP-seq data from the UCSC ENCODE portal (see URLs), HiC data34 from GEO GSE18199 and replication timing data are from reference 32.
Supplementary Material
Acknowledgments
We acknowledge generous support of the University of Southern California Epigenome Center by the Kenneth T. and Eileen L. Norris Foundation. We are grateful to S. Hansen for providing DNA replication data. High performance computing support was provided by the University of Southern California High Performance Computing Center (see URLs). We are greatly indebted to Denise Culhane for her superb proofreading skills.
The work described in this manuscript was not supported by nor will it benefit Epigenomics, AG.
Footnotes
Accession codes.
Sequence data and alignments from this study are available under the dbGaP accession code PHS000385.
Accession codes
Referenced accessions
Gene Expression Omnibus
Contributions
The project was conceived and the experiments were designed by P.W.L., D.J.W., B.P.B., D.V.D.B. and T.H. The Bisulfite-seq library construction and Genome Analyzer sequencing were performed by D.J.W., J.F.A. and D.V.D.B. The Infinium genotyping and data analysis was performed by B.P.B., motif analysis by H.N. and pipeline automation by B.P.B. and Z.R. Bisulfite-seq data processing and analysis were performed by B.P.B., Z.R. and H.N. Validation samples were collected and analyzed by C.P.E.L., C.M.v.D., R.A.E.M.T., B.P.B., D.J.W. and T.H. The manuscript was prepared by B.P.B. and P.W.L., and the study was supervised by P.W.L.
Competing financial interests
P.W.L. is scientific advisory board member and consultant for Epigenomics, AG, which has a commercial interest in DNA methylation biomarkers.
Supplementary information
PDF files
1. Supplementary Text and Figures (18M)
Supplementary Note, Supplementary Figures 1–16
Excel files
1. Supplementary Tables 1 and 2 (94K)
Bisulfite-seq summary statistics and Bisulfite-seq detailed statistics by chromosome
Contributor Information
Benjamin P Berman, University of Southern California Epigenome Center, University of Southern California, Keck School of Medicine, Los Angeles, California, USA.
Daniel J Weisenberger, University of Southern California Epigenome Center, University of Southern California, Keck School of Medicine, Los Angeles, California, USA.
Joseph F Aman, University of Southern California Epigenome Center, University of Southern California, Keck School of Medicine, Los Angeles, California, USA.
Toshinori Hinoue, University of Southern California Epigenome Center, University of Southern California, Keck School of Medicine, Los Angeles, California, USA.
Zachary Ramjan, University of Southern California Epigenome Center, University of Southern California, Keck School of Medicine, Los Angeles, California, USA.
Yaping Liu, University of Southern California Epigenome Center, University of Southern California, Keck School of Medicine, Los Angeles, California, USA.
Houtan Noushmehr, University of Southern California Epigenome Center, University of Southern California, Keck School of Medicine, Los Angeles, California, USA.
Christopher P E Lange, Department of Surgery, Groene Hart Hospital, Gouda, The Netherlands; Department of Surgery, Leiden University Medical Center, Leiden, The Netherlands.
Cornelis M van Dijk, Department of Pathology, Groene Hart Hospital, Gouda, The Netherlands.
Rob A E M Tollenaar, Department of Surgery, Leiden University Medical Center, Leiden, The Netherlands.
David Van Den Berg, University of Southern California Epigenome Center, University of Southern California, Keck School of Medicine, Los Angeles, California, USA.
Peter W Laird, University of Southern California Epigenome Center, University of Southern California, Keck School of Medicine, Los Angeles, California, USA.
References
- 1.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 2.Gal-Yam EN, et al. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. USA. 2008;105:12979–12984. doi: 10.1073/pnas.0806437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irizarry RA, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gebhard C, et al. General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res. 2010;70:1398–1407. doi: 10.1158/0008-5472.CAN-09-3406. [DOI] [PubMed] [Google Scholar]
- 5.Noushmehr H, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinoue T, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2011 Jun 9; doi: 10.1101/gr.117523.110. published online, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lister R, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ENCODE Project Consortium et al. User's guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irizarry RA, Wu H, Feinberg AP. A species-generalized probabilistic model-based definition of CpG islands. Mamm. Genome. 2009;20:674–680. doi: 10.1007/s00335-009-9222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Widschwendter M, et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 12.Schlesinger Y, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 13.Ohm JE, et al. A stem cell–like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rada-Iglesias A, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boumber YA, et al. An Sp1/Sp3 binding polymorphism confers methylation protection. PLoS Genet. 2008;4:e1000162. doi: 10.1371/journal.pgen.1000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuddapah S, et al. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Estécio MR, et al. Genome architecture marked by retrotransposons modulates predisposition to DNA methylation in cancer. Genome Res. 2010;20:1369–1382. doi: 10.1101/gr.107318.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aguilera C, et al. c-Jun N-terminal phosphorylation antagonises recruitment of the Mbd3/NuRD repressor complex. Nature. 2011;469:231–235. doi: 10.1038/nature09607. [DOI] [PubMed] [Google Scholar]
- 21.Jones PA. The DNA methylation paradox. Trends Genet. 1999;15:34–37. doi: 10.1016/s0168-9525(98)01636-9. [DOI] [PubMed] [Google Scholar]
- 22.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 23.Frigola J, et al. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat. Genet. 2006;38:540–549. doi: 10.1038/ng1781. [DOI] [PubMed] [Google Scholar]
- 24.Coolen MW, et al. Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity. Nat. Cell Biol. 2010;12:235–246. doi: 10.1038/ncb2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guelen L, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 26.Peric-Hupkes D, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chua YL, et al. The NRG1 gene is frequently silenced by methylation in breast cancers and is a strong candidate for the 8p tumour suppressor gene. Oncogene. 2009;28:4041–4052. doi: 10.1038/onc.2009.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dahl E, et al. Frequent loss of SFRP1 expression in multiple human solid tumours: association with aberrant promoter methylation in renal cell carcinoma. Oncogene. 2007;26:5680–5691. doi: 10.1038/sj.onc.1210345. [DOI] [PubMed] [Google Scholar]
- 29.Witcher M, Emerson BM. Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol. Cell. 2009;34:271–284. doi: 10.1016/j.molcel.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hawkins RD, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen RS, et al. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aran D, Toperoff G, Rosenberg M, Hellman A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011;20:670–680. doi: 10.1093/hmg/ddq513. [DOI] [PubMed] [Google Scholar]
- 34.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pickersgill H, et al. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet. 2006;38:1005–1014. doi: 10.1038/ng1852. [DOI] [PubMed] [Google Scholar]
- 36.Xiang Y, et al. JMJD3 is a histone H3K27 demethylase. Cell Res. 2007;17:850–857. doi: 10.1038/cr.2007.83. [DOI] [PubMed] [Google Scholar]
- 37.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun W, et al. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic Acids Res. 2009;37:5365–5377. doi: 10.1093/nar/gkp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balwierz PJ, et al. Methods for analyzing deep sequencing expression data: constructing the human and mouse promoterome with deepCAGE data. Genome Biol. 2009;10:R79. doi: 10.1186/gb-2009-10-7-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.