

Abstract
Cutaneous atrophy is the major adverse effect of topical glucocorticoids; however, its molecular mechanisms are poorly understood. Here, we identify stress-inducible mTOR inhibitor REDD1 (regulated in development and DNA damage response 1) as a major molecular target of glucocorticoids, which mediates cutaneous atrophy. In REDD1 knockout (KO) mice, all skin compartments (epidermis, dermis, subcutaneous fat), epidermal stem, and progenitor cells were protected from atrophic effects of glucocorticoids. Moreover, REDD1 knockdown resulted in similar consequences in organotypic raft cultures of primary human keratinocytes. Expression profiling revealed that gene activation by glucocorticoids was strongly altered in REDD1 KO epidermis. In contrast, the down-regulation of genes involved in anti-inflammatory glucocorticoid response was strikingly similar in wild-type and REDD1 KO mice. Integrative bioinformatics analysis of our and published gene array data revealed similar changes of gene expression in epidermis and in muscle undergoing glucocorticoid-dependent and glucocorticoid-independent atrophy. Importantly, the lack of REDD1 did not diminish the anti-inflammatory effects of glucocorticoids in preclinical model. Our findings suggest that combining steroids with REDD1 inhibitors may yield a novel, safer glucocorticoid-based therapies.
Keywords: glucocorticoid, glucocorticoid receptor, mTOR, REDD1, skin atrophy
Introduction
Glucocorticoid hormones are essential regulators of proliferation, differentiation, and metabolism in skin. They are also effective anti-inflammatory drugs widely used to treat the hyperproliferative and inflammatory skin diseases such as atopic dermatitis and psoriasis (Schäcke et al, 2006; Schoepe et al, 2006). Unfortunately, their beneficial therapeutic effects are often accompanied by numerous adverse effects including skin atrophy, characterized by a profound loss in skin thickness and elasticity combined with decreased barrier function. Skin atrophy involves all skin compartments, that is, epidermis, dermis, sebaceous glands, and subcutaneous (s.c.) fat. Typical epidermal changes include a reduction in thickness, decreased number and size of keratinocytes, diminished stratum corneum and intercellular lipid lamella (Jablonska et al, 1979; Lehmann et al, 1983; Zheng et al, 1984; Lubach & Kietzmann, 1988; Schoepe et al, 2006). These changes are combined with an altered orientation and packing of collagen and elastin fibers, and decreased cellularity in the dermis (Lehmann et al, 1983; Schoepe et al, 2006). In addition, in mice topical glucocorticoids and in patients intradermally injected glucocorticoids induce drastic atrophy/lypolysis of s.c. fat (Woodbury & Kligman, 1992; Imagawa & Ohkuma, 2010). Although steroid-induced skin atrophy is well known and characterized morphologically, the underlying molecular mechanisms are poorly understood.
Glucocorticoids act via a specific receptor (the glucocorticoid receptor, GR), which is a ligand-dependent transcription factor (Adcock, 2001; Necela & Cidlowski, 2004; Vandevyver et al, 2012; Ratman et al, 2013). In the absence of glucocorticoids, GR resides in the cytoplasm in a complex with molecular chaperones that inhibit GR nuclear import. Upon ligand binding, GR undergoes phosphorylation, dimerization, and nuclear translocation. Gene transactivation (TA) by glucocorticoids requires binding of GR homodimers to palindromic glucocorticoid-responsive elements (GRE) in gene promoters. Transrepression (TR) by glucocorticoids is mediated by diverse mechanisms including interaction between GR and other transcription factors, such as major pro-inflammatory factors NF-κB and AP-1 (De Bosscher et al, 2003; Necela & Cidlowski, 2004; Schäcke et al, 2006; Chebotaev et al, 2007a; Ratman et al, 2013). TR by GR is critical for anti-inflammatory effects of glucocorticoids. In contrast, many metabolic side effects of oral steroids related to the maintenance of the hypothalamic–pituitary–adrenal axis, glucose metabolism, and osteoporosis are largely dependent on TA (Schäcke et al, 2006, 2009; De Bosscher et al, 2010; Nixon et al, 2013; Ratman et al, 2013).
We discovered recently that glucocorticoids induced a robust activation of REDD1 (regulated in development and DNA damage response) in mouse and human skin (Baida et al, 2013). REDD1 is a stress response gene induced by hypoxia, DNA damage, nutrient or energy deprivation, or by endoplasmic reticulum stress (Shoshani et al, 2002; Ellisen et al, 2002; Brugarolas et al, 2004; Lin et al, 2005; Sofer et al, 2005; Wang et al, 2006; DeYoung et al, 2008). REDD1 is also activated by glucocorticoids and is a direct GR target (Wang et al, 2006; Shimizu et al, 2011). It inhibits mammalian target of rapamycin (mTOR) by stabilizing the tuberous sclerosis protein 1 (TSC1)–TSC2 inhibitory complex (Brugarolas et al, 2004; Ellisen, 2005; Wang et al, 2006; DeYoung et al, 2008; Mata et al, 2011). mTOR regulation by REDD1 contributes to the control of cell growth and size in Drosophila and in mammals. These findings suggest that abnormalities of REDD1 signaling may disrupt energy homeostasis (Ellisen et al, 2002; Shoshani et al, 2002; Sofer et al, 2005; Katiyar et al, 2009).
REDD1 is involved in another atrophogenic effect of glucocorticoids, muscle waste (Wang et al, 2006; Shimizu et al, 2011). It is also well understood that cross-talk between anabolic mTOR and catabolic GR is important to maintain the mass of skeletal muscle (Shimizu et al, 2011). Such cross-talk, as well as the role of REDD1 in steroid-induced atrophy in the skin, has not been considered.
REDD1 KO mice (Brafman et al, 2004; Sofer et al, 2005) are resistant to a variety of pathological conditions caused by stress, such as oxidative stress in the retina, emphysema induced by tobacco smoke, and ceramide-induced apoptosis of the lung epithelial cells (Brafman et al, 2004; Yoshida et al, 2010; Kamocki et al, 2013). However, neither adverse nor therapeutic effects of glucocorticoids have been studied on REDD1 KO background.
Here, we used REDD1 KO mice to examine the role of REDD1 in skin. We report that REDD1 KO animals are resistant to glucocorticoid-induced skin atrophy compared to wild-type isogenic animals. Similarly, organotypic raft cultures of primary human keratinocytes after REDD1 knockdown were completely protected from the hypoplastic effects of glucocorticoids. In contrast, REDD1 was dispensable for the anti-inflammatory action of glucocorticoids. In agreement, the comparison of transcriptional response to glucocorticoids in the epidermis of wild-type and REDD1 KO mice revealed that REDD1 is critically important for the TA by the glucocorticoids, especially the genes related to lipid and protein metabolism/catabolism, but not for the TR of the genes related to glucocorticoid anti-inflammatory effects.
Results
Topical glucocorticoids induce skin atrophy and activate REDD1 expression
We used clinically relevant glucocorticoid regiments known to induce significant skin atrophy in mice and in patients (Schoepe et al, 2006, 2010; Chebotaev et al, 2007b). B6D2 mice (F1 C57Bl/6 × DBA) that we used previously to study the side effects of glucocorticoids (Chebotaev et al, 2007b) were treated with medium potency glucocorticoid fluocinolone acetonide (FA, 2 μg/animal). For human volunteers, we used one of the most potent steroids, clobetasol propionate (CBP, 0.05% cream). In mice, a 2-week treatment caused a marked 50% epidermal thinning and 50% depletion of the interfollicular basal keratinocytes (Fig1A and B). A similar degree of epidermal hypoplasia was observed in humans after 2-week treatment; however, the negative effect on basal keratinocytes was less pronounced (Fig1E and F).
Figure 1. Topical glucocorticoids induce epidermal atrophy and REDD1 mRNA expression.

- A–G B6D2 mice were treated topically with acetone (vehicle control) or glucocorticoid FA (2 μg/animal), every 72 h for 2 weeks. Human volunteers were treated with 0.05% CPB cream applied to the right arm skin once or daily for 2 weeks. Untreated skin from the left arm was used as a control. H&E staining of mouse skin (A) and human skin (E). Scale bars are 20 μm (A) and 40 μm (E). Morphometric analysis of epidermal thickness and number of basal keratinocytes in mouse skin treated with FA 1, 2, and 4 times (B), and human skin treated daily for 2 weeks (F). REDD1 mRNA expression in mouse epidermis 8 h after 1st, 2nd, and 4th applications of FA (C, Q-PCR), in mouse epidermis and s.c. adipose, 24 h after FA (D, Q-PCR), and in human skin 24 h after single (G, volunteers V4 and V5) and 2-week (G, volunteers V1, V2, V3, RT–PCR) treatment with CBP. RPL27 was used as a cDNA normalization control. In human skin, the means ± SD were calculated in each individual sample compared to the untreated skin from the same individual (30 measurements/condition). In mouse skin, the means ± SD were calculated for three individual skin samples/condition in one representative experiment (30 measurements/condition) out of three experiments. Q-PCR results are the means ± SD calculated for three individual RNA samples/condition. Statistical analysis for differences between treatment and control was done by the unpaired two-tailed t-test.
We recently reported that REDD1 was at the top of the list of genes up-regulated in mouse epidermis upon FA treatment (Baida et al, 2013). Using Q-PCR, we showed that REDD1 mRNA was increased by ∼4.5-fold in the epidermis of B6D2 mice, 4–24 h after the first FA application (Fig1C and D), and stayed significantly above the control level for the duration of treatment. REDD1 mRNA was also strongly up-regulated in human skin treated with CBP for 24 h–2 weeks (Fig1G).
Because the atrophic changes in mouse epidermis due to FA treatment were paralleled by the complete depletion of s.c. adipose tissue (Fig1A), we assessed REDD1 in adipose. The purity of s.c. adipose isolation was verified using specific adipocyte and keratinocyte markers (Supplementary Fig S1). We found that REDD1 expression was equally activated in s.c. adipose and epidermis (FigD and Supplementary Fig S1).
REDD1 protein level is tightly regulated in epidermis and correlates with mTOR activity and autophagy
Both immunostaining and Western blot analysis revealed that REDD1 protein was barely detectable in the skin of adult wild-type mice (Fig2A and B). FA induced REDD1 expression as early as 4–8 h after first application and following subsequent applications during chronic FA treatment, albeit to a lesser extent (Fig2B). The multiband pattern of REDD1 signal on Western blots was reported previously and may reflect REDD1 phosphorylation (Katiyar et al, 2009; Li et al, 2012; Regazzetti et al, 2012). The increase in REDD1 protein in the skin in response to FA was confirmed by immunostaining, which showed that REDD1 was mostly expressed in the cytoplasm of mouse keratinocytes throughout the epidermis (Fig2A).
Figure 2. REDD1 protein expression in epidermis is tightly regulated and correlates with inhibition of mTOR and autophagy.
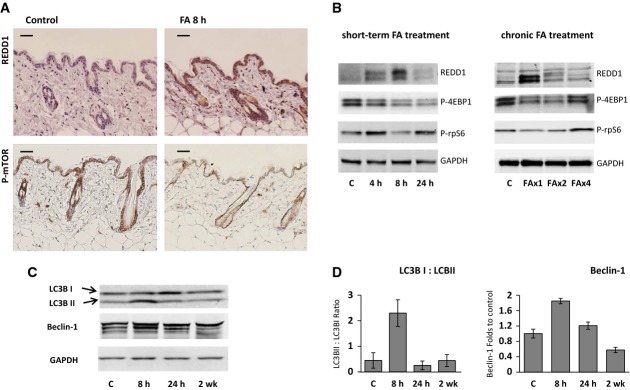
- A Immunohistochemical staining of mouse skin treated with acetone or FA for 8 h for REDD1 (upper panels) and phosphorylated mTOR-Ser2448 (lower panels). Scale bars are 20 μm.
- B Western blot analysis of REDD1 protein and phosphorylation of down-stream mTOR target proteins 4E-BP1 and rpS6 in murine epidermis. GAPDH is used as a normalization control.
- C, D Glucocorticoids induce autophagy in epidermis. Western blot analysis of Beclin-1 and conversion of light chain 3 (LC3) from LC3-I to LC3-II (C). Quantification of LC3-I to LC3-II conversion and Beclin-1 expression (D). The means ± SD were calculated using Western blots from two independent experiments (each lane is whole-cell protein from three pulled individual samples of epidermis).
The observed differences between REDD1 induction dynamics at mRNA and protein levels (especially at the 24-h time point and at the end of chronic FA treatment, compare Fig1C and D, and Fig2B) are likely due its short half-life (Katiyar et al, 2009). REDD1 expression is regulated via multiple mechanisms including translational repression by miR-221 which was reported to regulate REDD1 in murine hepatic progenitor cells (Pineau et al, 2010). In agreement, pri-miR-221 was significantly increased at the time points when REDD1 protein levels started to decline, for example, 24 h after single FA application, and after the first week of the chronic treatment (Supplementary Fig S5). This inverse correlation suggests that mature miR-221 is possibly involved in control of REDD1 induction by steroids in skin.
Because REDD1 is a known inhibitor of mTOR signaling (Sofer et al, 2005; Wang et al, 2006; DeYoung et al, 2008; Shimizu et al, 2011), we tested whether FA inhibits mTOR activity in the skin. In previous studies, mTOR activity has been monitored by phosphorylation of its major substrates, 4E-BP1 (eukaryotic initiation factor 4E binding protein 1) and S6K1 (ribosomal p70/S6 kinase 1) as well as ribosomal protein S6 (rpS6), a substrate of S6K1 (Checkley et al, 2011). As basal level of S6K1 phosphorylation was low in skin of B6D2 mice, we used 4E-BP1 and rpS6 as preferred markers. We showed that FA blocked mTOR activity in the skin. Moreover, there was an inverse correlation between phosphorylation of mTOR targets and REDD1 expression in the epidermis. By the time when REDD1 expression reached its peak (8 h after 1st and 2nd FA applications), the repression of mTOR activity by FA was most pronounced (Fig2A and B). We further confirmed this inverse correlation using immunostaining for REDD1 and active, phosphorylated at Ser2448 mTOR (Fig2A).
Previous studies showed that mTOR inhibition by glucocorticoids in other tissues causes autophagy (Molitoris et al, 2011; Shimizu et al, 2011). In accordance, we observed in FA-treated skin the conversion of light chain 3 (LC3) from its free form (LC3-I) to a membrane-bound state (LC3-II), a key step that initiates autophagy in mammalian cells (Klionsky et al, 2012). LC3-I to LC3-II conversion reached its maximum 8 h after FA treatment and paralleled with the increase of another autophagy marker Beclin-1, highest REDD1 expression and strongest repression of mTOR activity (Fig2C and D).
REDD1 KO mice are resistant to the depletion of stem cells and skin atrophy by glucocorticoids
To assess the causative role of REDD1 in therapeutic and side effects of glucocorticoids in the skin, we compared REDD1 KO mice (Brafman et al, 2004) with isogenic controls (B6x129). REDD1 KO mice displayed mild epidermal hyperplasia and slightly increased keratinocyte proliferation (Fig3C and data not shown). There were no significant changes in early and medium/late differentiation markers including keratins K5, K10, loricrin, and involucrin (Supplementary Fig S2).
Figure 3. REDD1 KO mice are resistant to glucocorticoid-induced skin atrophy.

- A, B H&E staining (A) and Masson's trichrome staining: Dermis/collagen fibers are blue, muscle is red, nuclei are dark red, and cytoplasm is red/pink (B). Arrows point to epidermis and brackets indicate subcutaneous adipose (A) and dermis (D). Scale bars are 20 μm.
- C, D Morphometric analysis of epidermal thickness and dermal cellularity as described in Materials and Methods. Changes in epidermal thickness (C) are presented as % to wild-type control epidermis. Changes in dermal cellularity (D) are presented as % to corresponding control skin. The means ± SD were calculated for three individual skin samples in one representative experiment (30 measurements/condition) out of two experiments. Statistical analysis for differences between treatment and control and between control wild-type and REDD1 KO epidermal thickness was done by the unpaired two-tailed t-test.
In contrast, the lack of REDD1 strongly attenuated the effects of glucocorticoids in the skin. REDD1 KO animals showed considerable resistance to FA-induced epidermal atrophy compared to wild-type animals. In wild-type mice, chronic FA treatment reduced epidermal thickness by 50%, compared to < 20% in REDD1 KO mice (Fig3A and C). Similarly, s.c, adipose tissue was significantly protected from the atrophy upon REDD1 knockout (Fig3A and B). Glucocorticoids induce severe thinning of collagen and elastin fibrous network and decrease cellularity of the dermis (Woodbury & Kligman, 1992; Schoepe et al, 2006). Using Masson's trichrome staining, we showed that REDD1 KO minimized FA effect on dermal fibers and caused significant protection of the dermal cells (Fig3B and D, and higher magnification images in Supplementary Fig S6).
Our previous studies showed that GR activation has a profound negative effect on epidermal stem cells (SCs) in the bulge of hair follicles (Chebotaev et al, 2007b,c). In agreement, chronic FA treatment of wild-type mice completely eliminated the CD34+ follicular SCs (only 5% of hair follicles were CD34 positive) and diminished by ∼40% the numbers of p63-positive progenitors in the basal layer of epidermis (Fig4A and C). Importantly, in REDD1 KO animals treated with FA, CD34+ SCs and p63+ keratinocytes were largely preserved: 40% of hair follicles remained CD34 positive, and the number of p63+ basal keratinocytes decreased only by 20% (Fig4A and C).
Figure 4. Protective effect of REDD1 KO on p63+ progenitors and CD34+ follicular epithelial stem cells.
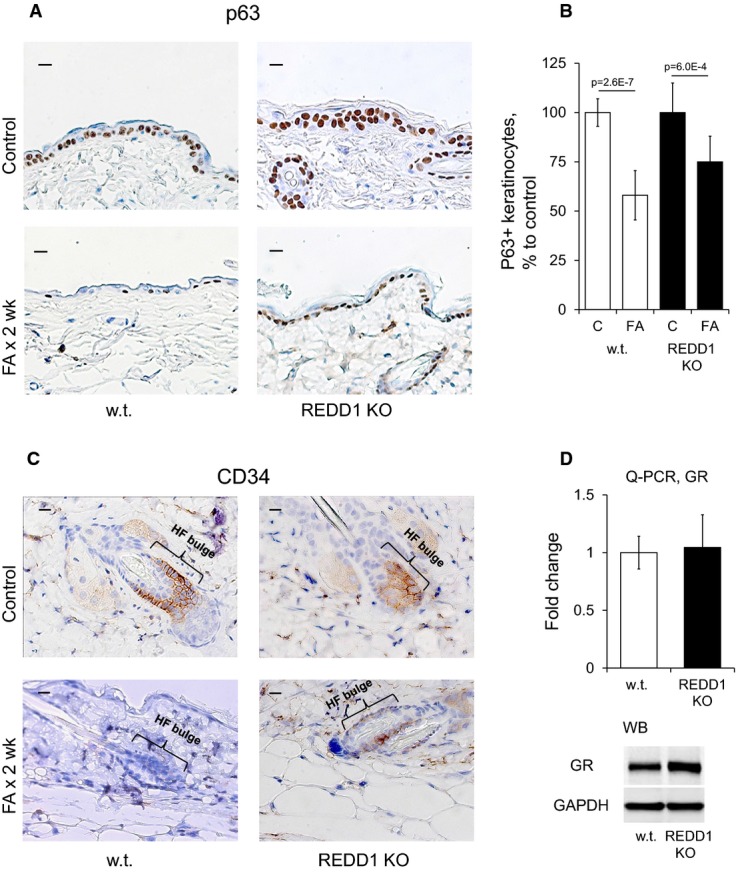
- A–C Expression of p63 (A) and CD34 (C). Scale bars are 10 μm. Analysis of p63 staining (B). The number of p63+ basal keratinocytes/total number of basal keratinocytes is presented as % to the corresponding control epidermis.
- D Similar GR expression in epidermis of wild-type and REDD1 KO mice determined by Q-PCR and Western blotting. Rpl27 and GAPDH used as a normalization controls, respectively.
REDD1 KO mice retain sensitivity to the anti-inflammatory effect of glucocorticoids
To further assess the consequences of REDD1 knockout for glucocorticoid therapy, we measured their responses to FA in a model where inflammation/edema is induced by topical irritant croton oil (CO) in the mouse ear. This assay is typically employed to test anti-inflammatory effects of GR ligands (Schäcke et al, 2004; Park et al, 2006; Schoepe et al, 2010). REDD1 KO animals and isogenic controls were equally responsive to inflammation (Fig5). Remarkably, FA relieved this inflammatory response with equal potency in wild-type and REDD1 KO mice as assessed by changes in ear morphology and the weight of ear punch (Fig5).
Figure 5. Similar sensitivity of REDD1 KO and wild-type animals to the anti-inflammatory effect of glucocorticoids.
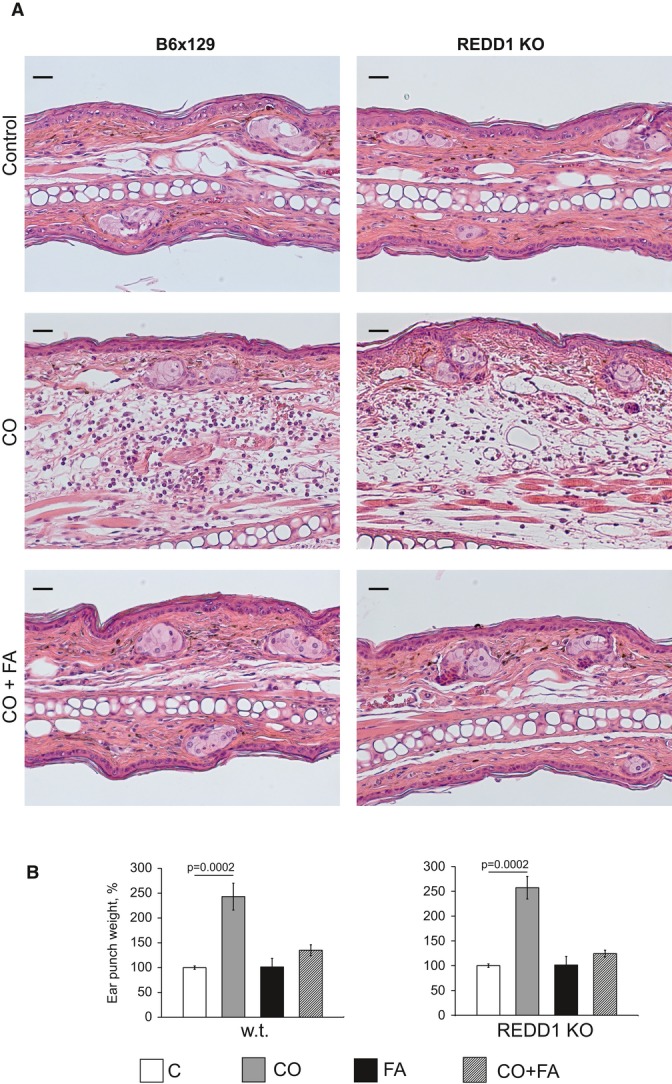
- A H&E staining. Scale bars are 20 μm.
- B Ear punch weight. Results are presented as % to corresponding (wild-type or REDD1 KO) control ear weight. The means ± SD were calculated for six individual ear punches/condition in one representative experiment (out of three experiments). Statistical analysis for differences between treatment and corresponding control was done by the unpaired two-tailed t-test.
REDD1 knockdown reduces hypoplastic effect of glucocorticoids in organotypic raft cultures
To evaluate possible contribution of REDD1 to glucocorticoids' action in human skin, we used 3-dimensional organotypic raft cultures (ORC) made of primary human epidermal keratinocytes (NHEK, Getsios et al, 2009) as described previously (Schoepe et al, 2010). We prepared ORCs from NHEK infected with control pGIPZ- or shREDD1-expressing lentiviruses (Fig6A). Both short-term (6 h) and chronic (7 days) treatment with CBP induced REDD1 mRNA and protein in control ORCs; this induction was completely abolished in shREDD1 ORCs (Fig5C and D). In agreement, mTOR activity was increased in shREDD1-expressing rafts compared to pGIPZ controls, with or without CBP treatment, as was evidenced by increased 4E-BP1 phosphorylation (Fig6C).
Figure 6. Down-regulation of REDD1 protects organotypic raft cultures from the hypoplastic effect of glucocorticoids.

- A, B H&E and BrdU staining of raft cultures (BrdU+ cells are indicated by arrowheads). Scale bars are 10 μm.
- C Western blot analysis of REDD1, GR, and mTOR substrate 4E-BP1 phosphorylation. GAPDH was used as a loading control.
- D Q-PCR analysis of REDD1 expression in rafts.
- E Analysis of CBP effect on basal keratinocyte number (left) and keratinocyte proliferation (number of BrdU+ basal keratinocytes/total number of basal keratinocytes, right) is presented as % to corresponding control rafts.
In pGIPZ-infected ORCs, the number and especially proliferation (BrdU incorporation) of basal keratinocytes were decreased by 25–40% after 7-day treatment with CBP (Fig6A, B and E). In contrast, shREDD1-expressing ORCs were completely protected from the hypoplastic/anti-proliferative effect of CBP (Fig6A, B, and E).
REDD1 plays an important role in GR signaling: asymmetrical effect on GR TA and TR
The reduced sensitivity of REDD1 KO mice and shREDD1 ORCs to steroid-induced atrophy suggested that lack of REDD1 could affect either GR expression or GR function. Q-PCR and Western blotting showed that GR expression was not significantly changed/slightly increased either in the epidermis of REDD1 KO mice (Fig4D) or in ORCs after shREDD1 knockdown (Fig6C).
To investigate the global role of REDD1 in GR signaling, we performed genome-wide expression profiling of the epidermis of REDD1 and wild-type mice treated with vehicle (control) or FA using Illumina mouse whole-genome gene array (GEO Submission GSE59151). Gene expression was measured 24 h post-FA application, when both TA and TR are usually fully developed (Wu et al, 2004). Differential expression was identified based on the adjusted P-value threshold of 0.05.
REDD1 KO had only minor effect on baseline gene expression with only three genes were up-regulated and 23 were down-regulated in REDD1 KO compared to wild-type controls (Supplementary Table S1, and heatmaps Fig7A and Supplementary Fig S4).
Figure 7. Global effect of REDD1 KO on the expression of glucocorticoid-responsive genes in murine epidermis.

- A A heatmap of gene expression as analyzed by the Mouse Whole-Genome Gene Expression BeadChips (Illumina). Columns show normalized gene expression for individual animals (red: increased, green: decreased expression).
- B Venn diagrams show overlap of differentially expressed genes between REDD1 KO and wild-type mice (adjusted P < 0.05). Arrows connect sections of the heatmap with corresponding Venn diagrams.
- C Most enriched gene ontology (GO) categories for biological processes and molecular functions (fold-change enrichment ≥ 3, P < 0.01), associated with transrepression and transactivation of the glucocorticoid-responsive genes in REDD1 KO and wild-type mice.
- D Array validation by Q-PCR for genes from the different GO categories. Note: lack of overlap in GO categories between KO and wild-type mice associated with up-regulated genes and significant overlap in the function of down-regulated genes.
In contrast, REDD1 appeared central for gene regulation by glucocorticoids. In REDD1 KO animals, ∼20% fewer genes responded to FA than in wild-type mice: (338 versus 397 up-regulated and 337 versus 441 down-regulated, Fig7B and Supplementary Table S2). More importantly, the lack of REDD1 specifically altered TA branch of GR signaling. Only 30% FA-induced genes were also activated in REDD1 KO mice. In contrast, there was a 50% overlap between genes inhibited by FA in REDD1 KO and wild-type animals (Fig7B). The detailed analysis of differentially regulated individual genes in two genotypes is presented in Supplementary Table S2.
To quantify the relationship between TA and TR and REDD1 gene status, we assessed Pearson correlation between the fold changes of the differentially expressed (397 activated and 441 down-regulated) genes in wild-type and in REDD1 KO epidermis. We found only a weak positive correlation between FA-induced genes (r = 0.16, P = 0.003), and a robust positive correlation between FA-inhibited genes (r = 0.72, P = 10e-10), indicating much stronger involvement of REDD1 in GR TA, but not TR. This conclusion is also illustrated by the hierarchical clustering of microarray data on the heatmap with high level of similarity between genes repressed by FA in wild-type and REDD1 KO animals (Fig7A, Supplementary Fig S4).
Next, we performed Gene Ontology (GO) enrichment analysis of the differentially expressed genes for each comparison (Supplementary Table S3). The most enriched GO terms associated with the up-regulated genes in wild-type epidermis included metabolic processes (lipids, sphingolipids, and fatty acids), catabolism (lipids, sphingolipids, proteins), and proteolysis. These metabolic and catabolic gene categories were absent among genes up-regulated in REDD1 KO epidermis (Fig7C). Instead, in REDD1 KO keratinocytes, FA up-regulated genes related to autophagy, endosomes, and PI3K signaling. Remarkably, the GO analysis of gene repression showed 70% overlap between REDD1 KO and wild-type animal response, with predominant categories related to the anti-inflammatory effect of glucocorticoids (antigen processing/presentation, and MHC/major histocompatibility complex protein binding) (Fig7C). In addition, FA inhibited genes related to DNA replication, reflective of the anti-proliferative effect of glucocorticoids in wild-type but not in REDD1 KO skin.
Array validation was performed by Q-PCR for six genes from different GO categories, inhibited (Cd74, H2-Ab1) or up-regulated (Fkbp5, Elovl3, Rptn, Socs2) by FA in wild-type epidermis (Fig7D). Remarkably, Cd74 and H2-Ab1 associated with class II MHC and important for antigen presentation and immune response (Pan et al, 2001; Beswick & Reyes, 2009) were similarly down-regulated in wild-type and in REDD1 KO epidermis (Fig7D). The examples of genes differentially activated in wild-type and REDD1 KO epidermis include Elovl3 involved in lipid metabolism (Guillou et al, 2010), Rptn, a multifunctional matrix protein (De Guzman Strong et al, 2010), Socs2, which regulates cell signaling and protein degradation (Larsen & Röpke, 2002), and Fkbp5, a molecular chaperone involved in steroid receptor activation, and also an inhibitor of mTOR and Akt (Li et al, 2011; Vandevyver et al, 2012) (Fig7D). Q-PCR data for all these genes were in perfect agreement with the DNA array analyses (Pierson correlation > 0.9, Supplementary Fig S3).
Overall, the analysis of gene expression at single gene and GO category levels, for the first time, revealed a feed-forward loop whereby GR target REDD1 is necessary for its TA action.
REDD1 does not alter GR phosphorylation and nuclear localization
The major steps required for GR-dependent TA are phosphorylation and nuclear translocation. ORC are notoriously difficult for biochemical analyses using multiple time points. Thus, we used immortalized human keratinocytes (HaCaT), stably infected with control pGIPZ or shREDD1 lentiviruses, in which shREDD1 prevented REDD1 activation by glucocorticoid FA (Fig8A).
Figure 8. REDD1 inhibition did not affect GR phosphorylation and nuclear translocation in HaCaT human keratinocytes.
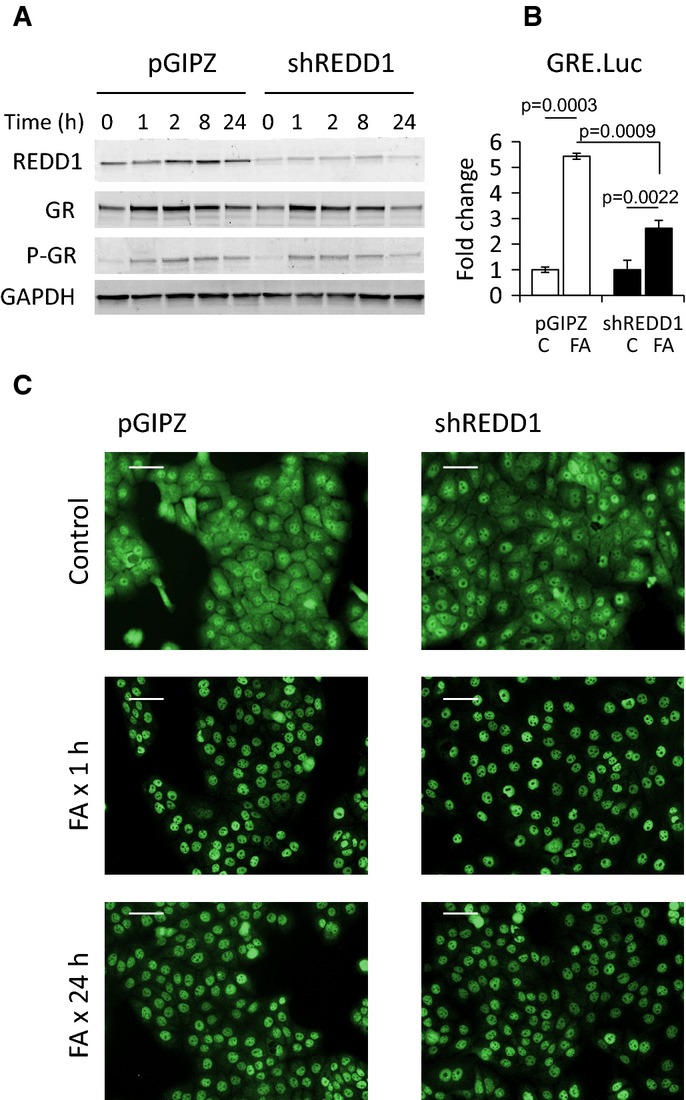
- Western blot analysis of REDD1, GR, and phosphorylated GR-Ser211. GAPDH used as a loading control.
- Reduced induction of Luciferase reporter in shREDD1-HaCaT cells. shREDD1- and pGIPZ-HaCaT cells were infected with GRE.Luc lentivirus and treated with FA (10−6 M) for 24 h. The Luciferase induction is presented as a fold change to corresponding vehicle-treated control. The means ± SD were calculated for three individual wells/group in one representative experiment (out of three experiments). Statistical analysis for differences between groups was done by ANOVA.
- Immunofluorescence analysis of GR nuclear translocation and retention in shREDD1- and pGIPZ-HaCaT cells treated with FA (10−6 M). Scale bars are 10 μm.
In agreement with the impaired activation of GR target genes in REDD1 KO animals, GR activation was strongly decreased by REDD1 knockdown in HaCaT keratinocytes as assessed by Luciferase assay with GRE.Luciferase reporter (Fig8B). Upon ligand binding, GR is typically degraded; however, in some cells, this degradation is minimal and may be preceded by temporary GR accumulation (Yemelyanov et al, 2008). We observed this temporary GR accumulation in HaCaT cells (Fig8A). Despite the difference in GR activity in Luciferase assay, basal GR levels, the dynamics of GR protein changes, GR phosphorylation, and nuclear translocation in response to FA were identical in shREDD1- and pGIPZ-HaCaT keratinocytes (Fig8A and C).
Similar mechanisms control skin and muscle atrophy
Glucocorticoids can induce catabolic/atrophic changes in multiple tissues including muscle, subcutaneous fat, and bone (Wang et al, 2006; Shimizu et al, 2011). We used ProfileChaser software to compare transcriptional responses to glucocorticoids in epidermis and other tissues, using our array data and DNA arrays deposited to NCBI (http://profilechaser.stanford.edu, Engreitz et al, 2010; Dudley et al, 2011). The comparison revealed remarkably high degree of similarity between gene expression patterns in steroid responses of the epidermis and muscle and, unexpectedly, steroid-independent changes in Duchene muscular dystrophy (similarity scores 0.66 and 0.64 accordingly, Table1).
Table 1.
Similar molecular signature of GR in epidermis, s.c. adipose and in muscle
| Skin compartment | GEO | Study title | Organism | Similarity score | Q-value |
|---|---|---|---|---|---|
| Epidermis | GDS2688 | Skeletal muscle response to chronic glucocorticoids | Rattus norvegicus | 0.657 | 0.003 |
| Epidermis | GDS3027 | Duchenne muscular dystrophy | Homo sapiens | 0.64 | 0.004 |
Gene expression in epidermis of wild-type mice treated with FA was analyzed by Illumina oligo arrays (as in Fig6). The gene expression changes in epidermis were compared to the gene expression profiles from publicly available experiments obtained from NCBI Gene Expression Omnibus (GEO) by ProfileChaser software (Stanford University, http://profilechaser.stanford.edu/). The Similarity Score is a weighted Pearson correlation coefficient between arrays. Q-value = FDR, false discovery rate.
Discussion
This work represents comprehensive investigation of the mechanisms of adverse and therapeutic glucocorticoid effects in skin, which combines focused and global, genome-wide methods of analysis.
First, we shed new light on the dynamics of molecular responses to topical glucocorticoids in the skin. We have identified REDD1 as an early glucocorticoid-responsive gene in mammalian skin. REDD1 activation in mice occurs 4–8 h after the treatment when it inhibits mTOR and promotes autophagy in the epidermis. Later, at the end of 2-week glucocorticoid treatment, both REDD1 and mTOR activity return to basal levels (Fig2B). This is the time point when mice develop resistance to topical glucocorticoids (tachyphylaxis) (Chebotaev et al, 2007c). In contrast to mice, tachyphylaxis does not occur in patients (Taheri et al, 2013); this could be possibly caused by persistence of REDD1 up-regulation.
This is the first study in which REDD1 KO mice were used to determine the contribution of REDD1 to skin maintenance and response to glucocorticoids. In general, REDD1 role in skin and keratinocytes has not been studied; a single report suggests increased keratinocyte differentiation in vitro due to REDD1 (Ellisen et al, 2002). In our study, REDD1 had minimal effect on the gene expression in adult epidermis, and this is corroborated by the fact that adult REDD1 KO animals have no overt skin phenotype, likely due to low basal REDD1 levels.
At the same time, REDD1 induction by glucocorticoids was critically important for skin atrophy. In REDD1 KO mice, all skin compartments were protected from the atrophogenic effect of steroids. Likewise, ORCs of human epidermis, which are currently the most advanced model of human skin (Getsios et al, 2009; Schoepe et al, 2010), were also protected from hypoplasia by REDD1 knockdown. In contrast to its central role in adverse cutaneous effects of steroids, REDD1 is dispensable for the anti-inflammatory effect of glucocorticoids as FA alleviated inflammation and edema due to croton oil in both REDD1 KO and control mice.
Interestingly, REDD1 knockout safeguarded at least two populations of stem cells: CD34+ follicular epithelial stem cells and p63+ keratinocyte progenitors from the detrimental effects of glucocorticoids. These data along with recent observations that REDD1 is decreased during reprogramming of somatic cells to induced pluripotent stem cells (Corominas-Faja et al, 2013) suggest an important role for REDD1 in stem cells maintenance. CD34 is also a marker of white adipose stem cells (Park et al, 2008), and thus, protection of CD34+ adipocytes could explain the observed protection of s.c. fat in REDD1 KOs.
Recent report documents the inhibitory effect of mTOR on GR function in the muscle (Shimizu et al, 2011); however, the effect of REDD1 and other mTOR inhibitors on GR signaling has not been well studied. Our bioinformatics analysis of transcriptional response to glucocorticoids in the skin identified a novel intriguing role of REDD1 in control of the repertoire of glucocorticoid-regulated genes and the integral functional response to glucocorticoids. Furthermore, the lack of REDD1 predominantly affected gene activation (TA) and not inhibition (TR) by glucocorticoids.
While TA involves binding of GR homodimers to palindromic GREs in the promoters and enhancers of the glucocorticoid-inducible genes, TR is largely independent of GR dimerization and in many cases stems from the direct interaction (tethering) between GR monomer and another transcription factors including pro-inflammatory NF-kB and AP-1. Another TR mechanism is binding of GR monomers to ‘negative GREs’ (Nixon et al, 2013; Ratman et al, 2013). The TA versus TR outcome also depends on the recruitment of steroid hormone receptor coactivators (such as SRC-1, SRC2) and corepressors (such as NCoR, SMRT) (Ratman et al, 2013). As GR phosphorylation and nuclear import/retention were similar in control and shREDD1-HaCaT keratinocytes, it is conceivable that REDD1 modifies conditions for GR dimerization and DNA binding, or impinges on the specter of coregulators recruited by GR.
TR by GR is critical for its anti-inflammatory function, even though the induction of select GR-dependent genes is also needed (Schäcke et al, 2006, 2009; Chebotaev et al, 2007a; Clark et al, 2008; Ratman et al, 2013). In contrast, many side effects of GR are caused by its TA action (Schäcke et al, 2006, 2009; Ratman et al, 2013). Thus, selective GR activators (SEGRA) that shift GR activity toward TR are expected to have a better therapeutic profile than classical glucocorticoids. Indeed, several SEGRA including ZK245186/mapracorat preserve the anti-inflammatory effect of glucocorticoids but do not induce skin atrophy (Schäcke et al, 2009). We demonstrate that REDD1 deletion ‘dissociates’ TA and TR functions of glucocorticoids.
Chronic glucocorticoid therapy induces atrophy in many organs besides skin (Wang et al, 2006; Shimizu et al, 2011; Henneicke et al, 2014). Meta-analysis of ours and published gene arrays revealed strong similarity between changes in transcriptome in glucocorticoid-treated epidermis (our results) and muscle undergoing steroid-dependent and steroid-independent (Duchene atrophy) waste. Thus, molecular mechanisms underlying the atrophy in epidermis and muscle are likely similar and involve REDD1. We also speculate that similar REDD1-dependent mechanisms underlie the steroid atrophy of s.c. adipose as REDD1 was highly induced in s.c. adipocytes, and s.c. fat was protected in REDD1 KO mice from glucocorticoids.
In conclusion, we discovered that REDD1 acts as GR modulator and atrophogen in the skin. Further, our results suggest the clinical relevance of REDD1 as molecular target for safer combination GR-targeted therapies in the skin. We expect that blocking REDD1 by pharmacological inhibitors or RNAi could reduce and possibly alleviate skin atrophy in response to steroids; this is a new strategy for safer glucocorticoid treatments of chronic inflammatory diseases in the skin and other tissues.
Materials and Methods
Chemicals
Fluocinolone acetonide (FA), croton oil (CO), and all other chemicals unless stated otherwise were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). Clobetasol propionate (CBP) was purchased at the pharmacy as 0.05% cream.
Animals and treatments
B6D2 (F1 C57Bl ×DBA) mice used previously to study steroid skin atrophy (Chebotaev et al, 2007c) were obtained from Jackson Laboratory (Bar Harbor, ME, USA). REDD1 KO mice in F1 C57BL/6 × 129SvEv genetic background (B6x129) were generated by Lexicon Genetics Inc. for Quark Pharmaceuticals Inc. REDD1 exon 2 was replaced with LacZ/neo expression cassette (Brafman et al, 2004). Wild-type B6x129 isogenic control females were obtained from Taconic (Germantown, NY, USA).
Seven-week-old females in the telogen stage of the hair cycle were shaved and treated 3 days later. Glucocorticoid FA was applied topically (2 μg/animal) in 200 μl acetone to the back skin once or up to four times every third day as described (Chebotaev et al, 2007c). Control animals were treated with acetone only. Skin was harvested 4–24 h after FA application as indicated in Figure legends. Animals were injected i.p. with bromodeoxyuridine (BrdU, Sigma-Aldrich, 50 μg/g of animal weight) 1 h before skin was harvested. Epidermis and s.c. fat were isolated from the murine dorsal skin mechanically by scraping (Chebotaev et al, 2007c).
In our work with animals, we adhered to ACUC protocols approved by the Northwestern University Animal Care and Use Committee. The protocols specify experimental procedures (topical treatment with glucocorticoids, ear edema test, BrdU injections), mouse strains (C57Blx129; C57BlxDBA, REDD1 KO), animal sex (males and females), age (from 5 weeks to 1 year), number of animals allowed to use, housing details (five animals/cage, regular chow diet, food and water ad libitum, 12 h on/12 h off light exposure), and animal euthanasia. All animals were maintained at the Northwestern University barrier animal facility.
Ear edema test
To evaluate the anti-inflammatory effect of glucocorticoid FA, we used ear edema test (Schäcke et al, 2004; Park et al, 2006; Schoepe et al, 2010). Seven-week-old female animals were pretreated with FA (2 μg in 20 μl of acetone) or vehicle (20 μl of acetone) applied to the back of the ear lobe 1 h before application of nonspecific contact irritant croton oil (CO, 10% solution in 20 μl of acetone). Mice were sacrificed and ears were harvested 9 h after CO application, the time point at which we observed maximum ear edema in B6x129 animals (data not shown). Four-millimeter ear punch biopsies were immediately weighted to assess ear edema. In all experiments, we used 3–4 animals/group; experiments were repeated 2–3 times.
Treatment of human volunteers
Glucocorticoid CBP was applied topically as a 0.05% cream to the skin of right arm of healthy human volunteers (age 32–65) once or every 24 h for 2 weeks. Untreated left arm skin was used as control. Four-millimeter-full-thickness punch skin biopsies were taken 24 h after the last CBP application as indicated in Figure legends. Whole human skin samples were used for the molecular biological studies.
Keratinocyte cell lines and 3-D organotypic raft cultures
Three-dimensional (3-D) organotypic raft cultures (ORC) of human epidermis were made as described in Getsios et al (2009). Briefly, neonatal human epidermal keratinocytes (NHEK) were infected with shRNA- and pGIPZ-expressing lentiviruses followed, 48 h later, by selection for puromycin (2 μg/ml) resistance. Selected keratinocyte cultures were reseeded onto collagen gels with embedded J2-3T3 fibroblasts and cultured at the air–liquid interface as previously described for 3 days to allow the initial phase of epidermis formation. The standard ORC medium contains high 5 × 10−7 M hydrocortisone. In our experiments, ORC were cultured in the medium with 10−8 hydrocortisone during first 3 days and then were treated with glucocorticoid CBP (5 × 10−6 M) or vehicle control (0.05% DMSO) for 7 days. Rafts were treated with BrdU (10−7 M) 1 h before harvesting and fixed in formalin. The epithelial sheets were peeled off the collagen lattice, snap-frozen, protein and RNA extracted, and processed for Western blot analysis and Q-PCR.
HaCaT human keratinocyte cell line is an in vitro spontaneously transformed keratinocytes from histologically normal skin. Line was established by Dr. Fusenig (Boukamp et al, 1988). We have obtained HaCaT cells from Dr. M Denning (Loyola University, Chicago, IL) who received them directly from Dr. Fusenig. It is known that HaCaTs express basal keratin K14 and can express keratins K1/K10 when induced to differentiate by high Ca2+ (Boukamp et al, 1988). We confirmed that our subline of HaCaT cells express keratinocyte differentiation markers using Q-PCR (data not shown). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Cellgro, Manassas, VA, USA) containing 10% FBS (Cellgro) and antibiotics.
Lentiviruses
shREDD1-lentiviral construct
To knockdown REDD1 expression in keratinocytes, we used lentiviral construct expressing shREDD1 targeting REDD1 3′ UTR sequences homologous in human and mouse (clone V2LHS_176476, Thermo Scientific GIPZ Lentiviral shRNA Library). Empty pGIPZ vector (Open Biosystems, GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) was used as a control.
GRE.Luciferase Reporter
GRE.Luc reporter lentiviral construct encoding Firefly Luciferase under transcriptional regulation of glucocorticoid-responsive elements and a control lentiviral construct with Firefly Luciferase under minimal CMV promoter (used as control) were obtained from DNA/RNA delivery Core, SDRC Northwestern University. Lentiviral stocks, packaging, and transduction procedures were performed as described in Zufferey et al (1998) and Yemelyanov et al (2007).
Luciferase assay
HaCat cells expressing Firefly Luciferase under minimal CMV promoter or promoter containing GRE were plated in 12-well plates (three wells/experimental group), grown to ∼60% and treated with FA or vehicle (0.01% DMSO) for 24 h. Luciferase activity was measured using commercial Luciferase Assay (Promega Corp., Madison, WI, USA) and Luminometer TD 20/20 (Turner Designs, Sunnyvale, CA, USA). Luciferase activity of GRE.Luc construct was normalized to Luciferase activity from minimal CMV promoter under the same experimental condition.
Histological analysis and immunostaining
Sections of formalin-fixed, paraffin-embedded skin and ORCs were stained with hematoxylin and eosin (H&E), Masson's trichrome to evaluate the effect on dermis and collagen fibers (Sheehan & Hrapchack, 1980), and with antibodies against BrdU (BD Biosciences, San Jose, CA, USA), keratins 1, 5, and 10, loricrin (Covance, Princeton, NJ), phospho-mTORSer2448 (Cell Signaling Technology, Inc., Danvers, MA, USA), REDD1 (Proteintech Group, Inc, Chicago, IL), p63 (eBioscience, San Diego, CA, USA), and CD34 (Abcam, Cambridge, MA, USA).
GR nuclear translocation was determined by immunofluorescence in HaCaT cells. Cells infected with pGIPZ and shREDD1 lentiviruses were selected with puromycin, seeded on coverslips. After the treatments, cells were fixed with 2% formaldehyde and permeabilized with acetone:methanol (1:1 v/v). After blocking, cells were incubated with primary rabbit anti-GR antibody (H-300, Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by secondary anti-rabbit FITC-conjugated antibody (Jackson Immuno Research). Cell nuclei were counterstained with DAPI (Invitrogen, Life Technologies, Grand Island, NY, USA). Cell and tissue images were taken with AxioCaM HRC camera linked with Zeiss Axioplan2 microscope.
Morphometric analysis
Quantification of the epidermal width and number of basal keratinocytes (as the readouts for skin atrophy and hypoplasia of raft epidermis) was performed in dorsal skin and ORC sections stained with H&E. The number of dermal cells was determined on sections stained with Masson's trichrome in the upper (papillary) dermis that is distinguishable from the lower (reticular) dermis (Driskell et al, 2013). At least 10 individual fields per slide with at least three samples in each experimental group were counted using Axioplan2 microscope software (Carl Zeiss). All measurements are presented as % to corresponding control.
The numbers of BrdU+, p63+, and total number of basal keratinocytes were evaluated in 10 fields of view in each skin/ORC sample under the microscope. Number of BrdU+ and p63+ cells is presented as percent of total number of basal keratinocytes. The number of CD34+ hair follicles among 10 randomly selected hair follicles per skin sample was evaluated under the microscope.
Western blot analysis
The whole-cell protein extracts were prepared using RIPA buffer with protease and phosphatase inhibitor cocktails (Thermo Scientific, Thermo Fisher Scientific Inc., Waltham, MA, USA), resolved by SDS–PAGE on 4–20% gels and transferred to Odyssey nitrocellulose membranes (LI-COR Biosciences, Lincoln, NE, USA). Membranes were blocked with Odyssey Blocking Buffer and incubated with primary antibodies overnight at 4°C, followed by IRDye® secondary antibodies (LI-COR Biosciences). LI-COR Odyssey Imager was used for the band visualization. Equal loading and adequate transfer to the membranes were verified by staining with Ponceau S (Sigma-Aldrich) and with anti-GAPDH (Sigma-Aldrich) antibody. We used Abs against: REDD1 (Proteintech Group, Inc., Chicago, IL), LC3B, phospho-rpS6Ser240/244, phospho-4E-BP1Thr37/46, phospho-GRSer211 (Cell Signaling Technology, Inc.), and GR (H-300 or M-20, Santa Cruz Biotechnology, Inc.).
Abs against REDD1 and Beclin-1 (Fig2B and C) were reported to recognize multiband pattern on Western blots (Katiyar et al, 2009; Li et al, 2012; Regazzetti et al, 2012, and http://www.cellsignal.com/products/primary-antibodies/3738?Ntt=beclin&fromPage=plp), which may reflect phosphorylation status of these proteins.
RNA isolation and quantitative and semi-quantitative RT–PCR
Total RNA from murine epidermis, whole human skin, and cell cultures was isolated with RiboPure kit (Ambion, Life Technologies, Grand Island, NY, USA). Total RNA from murine s.c. adipose was isolated with RNeasy Lipid Tissue Kit (Qiagen, Valencia, CA, USA). The RNA samples were treated with TURBO™ DNase (Ambion). The gene expression was assessed using semi-quantitative two-step RT–PCR and quantitative Q-PCR. Reverse transcription was performed using 1 μg RNA, random hexamers, and M-MLV reverse transcriptase (Invitrogen, Life Technologies), according to manufacturer instructions. The gene-specific primers were designed with NCBI Primer-BLAST (Supplementary Table S4). Q-PCR with SYBR Green detection was performed on the Applied Biosystems® 7000 Real-Time PCR instrument (Life Technologies). Each sample was tested in triplicate, and results were normalized to the expression of the housekeeping Rpl27 gene (de Jonge et al, 2007). For semi-quantitative PCRs, Taq DNA polymerase (Promega) was used, and PCR products were separated on 1.5% agarose gel and visualized using automated imaging system (Bio-Rad, Hercules, CA, USA). RPL27 was used as a normalization control.
Microarray analysis of gene expression
RNA samples were checked for quality and integrity with the Agilent 2100 bioanalyzer and used for microarray analysis. RNA amplification, labeling, and hybridization with the Mouse Whole-Genome Gene Expression BeadChips MouseRef-8 v2.0 (Illumina) were performed at the Genomics Core Facility at the Center for Genetic Medicine at Northwestern University according to Illumina protocols.
Microarray processing was performed using the Limma package (Smyth, 2005), using the neqc function to perform background subtraction using the negative control probes, and quantile normalization using both positive and negative control probes. Differentially expressed probes were identified using the linear model implemented in Limma, applying an adjusted P-value threshold of 0.05. Q-PCR and microarray based gene expression values were compared using linear Pearson correlation.
FA-induced transactivation and transrepression
To quantify the relationship between FA-induced transactivation and transrepression across both phenotypes, we correlated the fold change of the differentially expressed probesets in wild-type, with their corresponding values in REDD1 KO epidermis.
Gene Ontology and pathway analysis
Gene ontology analysis was performed using the DAVID Bioinformatics resource tool (Huang et al, 2009a,b), submitting differentially expressed probes from each experiment, against the probe background of MouseRef 8. DAVID performs a hypergeometric test to identify overrepresented GO terms in a list of differentially expressed probes. Results are presented for categories with greater than or equal to threefold enrichment and P ≤ 0,01.
Gene expression heatmaps
Heatmaps were generated using differentially expressed probesets (adjusted P ≤ 0.05) induced by the application of FA, for the REDD1 KO and wild-type isogenic mice. Samples and genes were hierarchically clustered according to the Euclidean distance of the normalized gene expression values, and clusters were merged using complete linkage. Heatmaps were visualized using the Pheatmap package (http://CRAN.R-project.org/package=pheatmap).
Statistical analysis
Mean and standard deviation values were calculated using Microsoft Excel software. The treatment effects in each experiment were compared by one-way ANOVA or t-test. Differences between groups were considered significant at P < 0.05.
All experiments were repeated two to three times. In animal experiments, we used three to four animals/experimental group. In all figures, the results of one representative experiment are shown as mean values ± SD.
Approval of animal and human studies
All animal experiments were performed in compliance with ACUC protocol approved by the Northwestern University Animal Care and Use Committee.
All human studies conformed to the principles set out in the WMA Declaration of Helsinki (http://www.wma.net/en/30publications/10policies/b3/) and the NIH Belmont Report (http://www.hhs.gov/ohrp/humansubjects/guidance/belmont.html). Studies were approved by Northwestern University Institutional Review Board. Written informed consent was received from the participants before the study.
The paper explained.
Problem
Millions of patients worldwide are affected by chronic inflammatory skin diseases, including atopic dermatitis and psoriasis. The glucocorticoids are among the most effective and frequently used anti-inflammatory drugs. Unfortunately, patients chronically treated with topical glucocorticoids develop side effects, including cutaneous atrophy which affects all skin compartments and compromises the barrier function of the skin. Skin atrophy after the long-term use of steroids may be irreversible. The molecular mechanisms of steroid-induced skin atrophy are poorly understood, which prevents the development of novel safer approaches to the glucocorticoid-based therapies.
Results
In this study, we identified REDD1 (regulated in development and DNA damage response 1), a stress-inducible inhibitor of mTOR, as a major molecular target of glucocorticoids which mediates skin atrophy.
In REDD1 KO mice, all skin compartments (epidermis, dermis, and subcutaneous fat) and skin stem cells were significantly protected from the atrophogenic effect of steroids. Likewise, 3-D organotypic raft cultures made from shREDD1-infected primary human keratinocytes were also protected from glucocorticoid hypoplastic effects. In strong contrast to its central role in cutaneous adverse effects of steroids, REDD1 appeared to be dispensable for their anti-inflammatory effects.
Glucocorticoid effects are mediated by their receptor GR, a well-characterized transcription factor. We identified a novel intriguing role of REDD1 as an important GR modulator. REDD1 controls both the repertoire of glucocorticoid-regulated genes and the integral functional response to glucocorticoids. The lack of REDD1 strongly affected activation of genes (including genes involved in metabolism/catabolism of lipids and proteins) by glucocorticoids. At the same time, the inhibition of pro-inflammatory genes related to the anti-inflammatory effects of glucocorticoids remained remarkably similar in wild-type and REDD1 KO mice.
Impact
Our novel observations that in context of REDD1 KO cells GR changes its function could explain the improved therapeutic index of glucocorticoids (preserved therapeutic potential combined with reduced atrophogenic effects) in REDD1 KO animals. Overall, our studies support the development of innovative safer therapies with topical glucocorticoids using REDD1 inhibitors (such as pharmacological inhibitors or siRNA) to reduce/alleviate glucocorticoid-induced skin atrophy. As glucocorticoids are among the most frequently prescribed drugs for the patients with chronic inflammatory diseases and induce atrophogenic effects in different tissues, our findings have important clinical applications beyond the field of dermatology.
Acknowledgments
Work is supported by RO1CA118890 (to IB and RML), Northwestern Memorial Foundation and NUCATS Dixon Translational Research Grants innovation award (to IB), Foglia family foundation grant (to IB), R01DK098242 and U54CA189201 (to JTD), PhRMA Foundation Research Starter Grant (to JTD). Russian Foundation for Basic Research grant 13-04-01707 (to MY) and UICC YY2-2013-001 (to EL). REDD1 KO animals were kindly provided by Quark Pharmaceuticals Inc with the help of Dr. T. Finkel (Nemours Children's Clinic, Orlando, FL). We are thankful to Dr. A. Yemelyanov (Northwestern University, Chicago, IL) for technical consultations. We are grateful to Dr. Volpert (Northwestern University, Chicago, IL) for the discussion of our results. We would like to acknowledge NU genomics, flow cytometry, HTP screening, and mouse phenotyping Core facilities, and NU SDRC pathology, skin tissue engineering and DNA/RNA delivery Cores for technical support.
Author contributions
IB, MY, and JTD designed the experiments; GB, PB, EL, KK, SG, and KY performed the experiments; JTD, BR, and GB performed bioinformatics analysis; and IB and RML wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Review Process File
References
- Adcock IM. Glucocorticoid-regulated transcription factors. Pulm Pharmacol Ther. 2001;14:211–219. doi: 10.1006/pupt.2001.0283. [DOI] [PubMed] [Google Scholar]
- Baida G, Bhalla P, Yuen K, Guo S, Lavker RM, Budunova I. mTOR inhibitor REDD1 protects CD34+ follicular epithelial stem cells and prevents development of steroid-induced cutaneous atrophy. J Invest Dermatol. 2013;133:S243–S246. [Google Scholar]
- Beswick EJ, Reyes VE. CD74 in antigen presentation, inflammation, and cancer. World J Gastroenterol. 2009;15:2855–2861. doi: 10.3748/wjg.15.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brafman A, Mett I, Shafir M, Gottlieb H, Damari G, Gozlan-Kelner S, Vishnevskia-Dai V, Skaliter R, Einat P, Faerman A, et al. Inhibition of oxygen-induced retinopathy in RTP801-deficient mice. Invest Ophthalmol Vis Sci. 2004;45:3796–3805. doi: 10.1167/iovs.04-0052. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebotaev D, Yemelyanov A, Budunova I. The mechanisms of tumor suppressor effect of glucocorticoid receptor in skin. Mol Carcinog. 2007a;46:732–740. doi: 10.1002/mc.20349. [DOI] [PubMed] [Google Scholar]
- Chebotaev D, Yemelyanov A, Zhu L, Lavker RM, Budunova I. The tumor suppressor effect of the glucocorticoid receptor in skin is mediated via its effect on follicular epithelial stem cells. Oncogene. 2007b;26:3060–3068. doi: 10.1038/sj.onc.1210108. [DOI] [PubMed] [Google Scholar]
- Chebotaev DV, Yemelyanov A, Lavker RM, Budunova I. Epithelial cells in the hair follicle bulge do not contribute to epidermal regeneration after glucocorticoid-induced cutaneous atrophy. J Invest Dermatol. 2007c;127:2749–2758. doi: 10.1038/sj.jid.5700992. [DOI] [PubMed] [Google Scholar]
- Checkley LA, Rho O, Moore T, Hursting S, DiGiovanni J. Rapamycin is a potent inhibitor of skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Prev Res. 2011;4:1011–1020. doi: 10.1158/1940-6207.CAPR-10-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AR, Martin JR, Tchen CR. Role of dual specificity phosphatases in biological responses to glucocorticoids. J Biol Chem. 2008;283:25765–25769. doi: 10.1074/jbc.R700053200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corominas-Faja B, Cufí S, Oliveras-Ferraros C, Cuyàs E, López-Bonet E, Lupu R, Alarcón T, Vellon L, Iglesias JM, Leis O, et al. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle. 2013;12:3109–3124. doi: 10.4161/cc.26173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Haegeman G, Elewaut D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr Opin Pharmacol. 2010;10:497–504. doi: 10.1016/j.coph.2010.04.007. [DOI] [PubMed] [Google Scholar]
- De Guzman Strong C, Conlan S, Deming CB, Cheng J, Sears KE, Segre JA. A milieu of regulatory elements in the epidermal differentiation complex syntenic block: implications for atopic dermatitis and psoriasis. Hum Mol Genet. 2010;19:1453–1460. doi: 10.1093/hmg/ddq019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, Ferron SR, Herault Y, Pavlovic G, Ferguson-Smith AC, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. 2013;504:277–281. doi: 10.1038/nature12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, Morgan AA, Sarwal MM, Pasricha PJ, Butte AJ. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med. 2011;3:96–103. doi: 10.1126/scitranslmed.3002648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, Oliner JD, McKeon F, Haber DA. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell. 2002;10:995–1005. doi: 10.1016/s1097-2765(02)00706-2. [DOI] [PubMed] [Google Scholar]
- Ellisen LW. Growth control under stress: mTOR regulation through the REDD1-TSC pathway. Cell Cycle. 2005;4:1500–1502. doi: 10.4161/cc.4.11.2139. [DOI] [PubMed] [Google Scholar]
- Engreitz JM, Morgan AA, Dudley JT, Chen R, Thathoo R, Altman RB, Butte AJ. Content-based microarray search using differential expression profiles. BMC Bioinformatics. 2010;11:603–614. doi: 10.1186/1471-2105-11-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, Dusek RL, Cornwell M, Green KJ. Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J Cell Biol. 2009;185:1243–1258. doi: 10.1083/jcb.200809044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillou H, Zadravec D, Martin PG, Jacobsson A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: insights from transgenic mice. Prog Lipid Res. 2010;49:186–199. doi: 10.1016/j.plipres.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Henneicke H, Gasparini SJ, Brennan-Speranza TC, Zhou H, Seibel MJ. Glucocorticoids and bone: local effects and systemic implications. Trends Endocrinol Metab. 2014;25:197–211. doi: 10.1016/j.tem.2013.12.006. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc. 2009a;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009b;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imagawa K, Ohkuma S. A case of fat injection for treating subcutaneous atrophy caused by local administration of corticosteroid. Tokai J Exp Clin Med. 2010;35:66–69. [PubMed] [Google Scholar]
- Jablonska S, Groniowska M, Dabroswki J. Comparative evaluation of skin atrophy in man induced by topical corticoids. Br J Dermatol. 1979;100:193–206. doi: 10.1111/j.1365-2133.1979.tb05561.x. [DOI] [PubMed] [Google Scholar]
- de Jonge HJ, Fehrmann RS, de Bont ES, Hofstra RM, Gerbens F, Kamps WA, de Vries EG, van der Zee AG, te Meerman GJ, ter Elst A. Evidence based selection of housekeeping genes. PLoS One. 2007;2:e898. doi: 10.1371/journal.pone.0000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamocki K, Van Demark M, Fisher A, Rush NI, Presson RG, Hubbard W, Berdyshev EV, Adamsky S, Feinstein E, Gandjeva A, et al. RTP801 is required for ceramide-induced cell-specific death in the murine lung. Am J Respir Cell Mol Biol. 2013;48:87–93. doi: 10.1165/rcmb.2012-0254OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katiyar S, Liu E, Knutzen CA, Lang ES, Lombardo CR, Sankar S, Toth JI, Petroski MD, Ronai Z, Chiang GG. REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A-DDB1 ubiquitin ligase. EMBO Rep. 2009;10:866–872. doi: 10.1038/embor.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen L, Röpke C. Suppressors of cytokine signalling: SOCS. APMIS. 2002;110:833–844. doi: 10.1034/j.1600-0463.2002.1101201.x. [DOI] [PubMed] [Google Scholar]
- Lehmann P, Zheng P, Lavker RM, Kligman AM. Corticosteroid atrophy in human skin. A study by light, scanning, and transmission electron microscopy. J Invest Dermatol. 1983;81:169–176. doi: 10.1111/1523-1747.ep12543603. [DOI] [PubMed] [Google Scholar]
- Li L, Lou Z, Wang L. The role of FKBP5 in cancer aetiology and chemoresistance. Br J Cancer. 2011;104:19–23. doi: 10.1038/sj.bjc.6606014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XH, Ha CT, Fu D, Xiao M. REDD1 protects osteoblast cells from gamma radiation-induced premature senescence. PLoS ONE. 2012;7:e36604. doi: 10.1371/journal.pone.0036604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Qian Y, Shi X, Chen Y. Induction of a cell stress response gene RTP801 by DNA damaging agent methyl methanesulfonate through CCAAT/enhancer binding protein. Biochemistry. 2005;44:3909–3914. doi: 10.1021/bi047574r. [DOI] [PubMed] [Google Scholar]
- Lubach D, Kietzmann M. Investigation of the skin thinning effect of prednicarbate and other corticoids in mouse skin. Skin Pharmacol. 1988;1:200–206. doi: 10.1159/000210774. [DOI] [PubMed] [Google Scholar]
- Mata MA, Satterly N, Versteeg GA, Frantz D, Wei S, Williams N, Schmolke M, Peña-Llopis S, Brugarolas J, Forst CV, et al. Chemical inhibition of RNA viruses reveals REDD1 as a host defense factor. Nat Chem Biol. 2011;7:712–719. doi: 10.1038/nchembio.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitoris JK, McColl KS, Swerdlow S, Matsuyama M, Lam M, Finkel TH, Matsuyama S, Distelhorst CW. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J Biol Chem. 2011;286:30181–30189. doi: 10.1074/jbc.M111.245423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necela BM, Cidlowski JA. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc Am Thorac Soc. 2004;1:239–246. doi: 10.1513/pats.200402-005MS. [DOI] [PubMed] [Google Scholar]
- Nixon M, Andrew R, Chapman KE. It takes two to tango: dimerisation of glucocorticoid receptor and its anti-inflammatory functions. Steroids. 2013;78:59–68. doi: 10.1016/j.steroids.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Pan J, Ju D, Wang Q, Zhang M, Xia D, Zhang L, Yu H, Cao X. Dexamethasone inhibits the antigen presentation of dendritic cells in MHC class II pathway. Immunol Lett. 2001;76:153–161. doi: 10.1016/s0165-2478(01)00183-3. [DOI] [PubMed] [Google Scholar]
- Park KK, Ko DH, You Z, Heiman AS, Lee HJ. Synthesis and pharmacological evaluations of new steroidal anti-inflammatory antedrugs: 9alpha-Fluoro-11beta, 17alpha, 21-trihydroxy-3,20-dioxopregna-1,4-diene-16alpha-carboxylate (FP16CM) and its derivatives. Steroids. 2006;71:83–89. doi: 10.1016/j.steroids.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Park KW, Halperin DS, Tontonoz P. Before they were fat: adipocyte progenitors. Cell Metab. 2008;8:454–457. doi: 10.1016/j.cmet.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Pineau P, Volinia S, McJunkin K, Marchio A, Battiston Terris CB, Mazzaferro V, Lowe SW, Croce CM, Dejean A. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci USA. 2010;107:264–269. doi: 10.1073/pnas.0907904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, De Bosscher K. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol. 2013;380:41–54. doi: 10.1016/j.mce.2012.12.014. [DOI] [PubMed] [Google Scholar]
- Regazzetti C, Dumas K, Le Y, Marchand-Brustel F, Peraldi P, Tanti JF, Giorgetti-Peraldi S. Regulated in Development and DNA Damage Responses-1 (REDD1) protein contributes to insulin signaling pathway in adipocytes. PLoS ONE. 2012;7:e52154. doi: 10.1371/journal.pone.0052154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäcke H, Schottelius A, Döcke WD, Strehlke P, Jaroch S, Schmees N, Rehwinkel H, Hennekes H, Asadullah K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA. 2004;101:227–232. doi: 10.1073/pnas.0300372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäcke H, Rehwinkel H, Asadullah K, Cato AC. Insight into the molecular mechanisms of glucocorticoid receptor action promotes identification of novel ligands with an improved therapeutic index. Exp Dermatol. 2006;15:565–573. doi: 10.1111/j.1600-0625.2006.00453.x. [DOI] [PubMed] [Google Scholar]
- Schäcke H, Zollner TM, Döcke WD, Rehwinkel H, Jaroch S, Skuballa W, Neuhaus R, May E, Zügel U, Asadullah K. Characterization of ZK 245186, a novel, selective glucocorticoid receptor agonist for the topical treatment of inflammatory skin diseases. Br J Pharmacol. 2009;158:1088–1103. doi: 10.1111/j.1476-5381.2009.00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepe S, Schäcke H, May E, Asadullah K. Glucocorticoid therapy-induced skin atrophy. Exp Dermatol. 2006;15:406–420. doi: 10.1111/j.0906-6705.2006.00435.x. [DOI] [PubMed] [Google Scholar]
- Schoepe S, Schäcke H, Bernd A, Zöller N, Asadullah K. Identification of novel in vitro test systems for the determination of glucocorticoid receptor ligand-induced skin atrophy. Skin Pharmacol Physiol. 2010;23:139–151. doi: 10.1159/000270386. [DOI] [PubMed] [Google Scholar]
- Sheehan DC, Hrapchak B. Theory and Practice of Histotechnology. St Louis, MO, USA: The C.V. Mosby Company; 1980. [Google Scholar]
- Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda S, Nakae J, Tagata Y, Nishitani S, Takehana K, et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 2011;13:170–182. doi: 10.1016/j.cmet.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol. 2002;22:2283–2293. doi: 10.1128/MCB.22.7.2283-2293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, et al., editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005;25:5834–5845. doi: 10.1128/MCB.25.14.5834-5845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri A, Cantrell J, Feldman SR. Tachyphylaxis to topical glucocorticoids; what is the evidence? Dermatol Online J. 2013;9:18954–18961. [PubMed] [Google Scholar]
- Vandevyver S, Dejager L, Libert C. On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic. 2012;13:364–374. doi: 10.1111/j.1600-0854.2011.01288.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Kubica N, Ellisen LW, Jefferson LS, Kimball SR. Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J Biol Chem. 2006;281:39128–39134. doi: 10.1074/jbc.M610023200. [DOI] [PubMed] [Google Scholar]
- Woodbury R, Kligman AM. The hairless mouse model for assaying the atrophogenicity of topical corticosteroids. Acta Derm Venereol. 1992;72:403–406. [PubMed] [Google Scholar]
- Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64:1757–1764. doi: 10.1158/0008-5472.can-03-2546. [DOI] [PubMed] [Google Scholar]
- Yemelyanov A, Czwornog J, Chebotaev D, Karseladze A, Kulevitch E, Yang X, Budunova I. Tumor suppressor activity of glucocorticoid receptor in the prostate. Oncogene. 2007;26:1885–1896. doi: 10.1038/sj.onc.1209991. [DOI] [PubMed] [Google Scholar]
- Yemelyanov A, Czwornog J, Gera L, Joshi S, Chatterton RT, Jr, Budunova I. Novel steroid receptor phyto-modulator compound A inhibits growth and survival of prostate cancer cells. Cancer Res. 2008;68:4763–4773. doi: 10.1158/0008-5472.CAN-07-6104. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Mett I, Bhunia AK, Bowman J, Perez M, Zhang L, Gandjeva A, Zhen L, Chukwuek U, Mao T, et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nat Med. 2010;16:767–773. doi: 10.1038/nm.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng PS, Lavker RM, Lehmann P, Kligma AM. Morphologic investigations on the rebound phenomenon after corticosteroid-induced atrophy in human skin. J Invest Dermatol. 1984;82:345–352. doi: 10.1111/1523-1747.ep12260665. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, Trono D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Review Process File