

Abstract
Heparanase (HPSE) is the dominant mammalian endoglycosidase and important tumorigenic, angiogenic, and pro-metastatic molecule. Highest levels of HPSE activity have been consistently detected in cells possessing highest propensities to colonize the brain, emphasizing the therapeutic potential for targeting HPSE in brain metastatic breast cancer (BMBC). Lapatinib (Tykerb) is a small-molecule and dual inhibitor of human epidermal growth factor receptor1 and 2 (EGFR and HER2, respectively) which are both high-risk predictors of BMBC. It was approved by the US Food and Drug Administration for treatment of patients with advanced or metastatic breast cancer. However, its role is limited in BMBC whose response rates to lapatinib are significantly lower than those for extracranial metastasis. Because HPSE can affect EGFR phosphorylation, we examined Roneparstat, a non-anticoagulant heparin with potent anti-HPSE activity, to inhibit EGFR signaling pathways and BMBC onset using lapatinib-resistant clones generated from HER2-transfected, EGFR-expressing MDA-MB-231BR cells. Cell growth, EGFR pathways, and HPSE targets were assessed among selected clones in the absence or presence of Roneparstat and/or lapatinib. Roneparstat overcame lapatinib resistance by inhibiting pathways associated with EGFR tyrosine residues that are not targeted by lapatinib. Roneparstat inhibited the growth and BMBC abilities of lapatinib-resistant clones. A molecular mechanism was identified by which HPSE mediates an alternative survival pathway in lapatinib-resistant clones and is modulated by Roneparstat. These results demonstrate that the inhibition of HPSE-mediated signaling plays important roles in lapatinib resistance, and provide mechanistic insights to validate the use of Roneparstat for novel BMBC therapeutic strategies.
Abbreviations: ANOVA, analysis of variance; BR, HER2-transfected MDA-MB-231BR; BMBC, brain metastatic breast cancer; COX-2, cyclooxygenase-2; DME/F-12, Dulbecco’s modified Eagle’s/F-12 medium; ERK, extracellular signal-regulated kinase; EGFR, human epidermal growth factor receptor1; FACS, fluorescence activated cell sorting; FAK, focal adhesion kinase; FBS, fetal bovine serum; HER2, human epidermal growth factor receptor2; HPSE, heparanase; HS, heparan sulfate; Ls/Lr BR clones, lapatinib-sensitive/lapatinib-resistant BR clones; MAPK, mitogen-activated protein kinase; MMP-9, matrix metalloprotease-9; PBS, phosphate-buffered saline; PI3K, phosphoinositide 3-kinase; STR, short tandem repeat.
Introduction
The family of epidermal growth factor receptors (HER/ErbB) plays pivotal roles in the regulation of breast cancer progression and metastasis [1]. One of the four HER family members, epidermal growth factor receptor1 (EGFR, HER1, or ErbB1) is overexpressed in 25% to 80% of breast cancers [2], while another, epidermal growth factor receptor2 (HER2, neu, or ErbB2), is amplified and/or overexpressed in approximately 20% to 30% of primary breast cancers [3]. Lapatinib (Tykerb, GW572016) is a small-molecule and dual inhibitor of EGFR and HER2. Its use was approved by the US Federal and Drug Administration for therapeutic combinations with capecitabine for the treatment of patients overexpressing HER2 that have received prior therapy, including anthracycline and the humanized HER2 antibody trastuzumab (Herceptin) [4]. Brain metastatic breast cancer (BMBC) is frequently detected in patients overexpressing HER2 and EGFR [5]. However, either systemic or recent targeted therapies using lapatinib are only minimally effective with BMBC response rates far lower (approximately 5%) than those with extracranial metastases [6], [7], [8].
Molecular mechanisms countering lapatinib resistance are not fully understood and subject of intense investigation because of their therapeutic implications. Roles of lapatinib in cell survival and proliferation are dependent and selective upon EGFR and HER2 phosphorylation and tyrosine kinase catalytic activity [4], [6], [7], [8]. For example, EGFR and HER2 signal through the phosphoinositide 3-kinase (PI3K) pathway and lapatinib was shown to block downstream signaling via PI-3K/Akt and mitogen-activated protein kinase (MAPK) pathways in breast cancer cells by interrupting baseline and ligand-stimulated activity [6], [7], [9]. However, not all EGFR/HER2-expressing breast cancer cells respond to lapatinib, particularly if cells overexpress EGFR such as the brain-seeking MDA-MB-231BR variant [5], [7].
Heparanase (HPSE) is the dominant mammalian endoglycosidase (endo-β-D-glucuronidase), cleaving heparan sulfate (HS) to fragments which retain biological activity. By this action, HPSE releases important HS/heparin-binding angiogenic and growth factors, affecting their levels and biological potency [10], [11], [12]. HPSE functions are not limited to HS cleavage or the release of HS-sequestered growth factors but also affect clustering, shedding, and mitogenic activity of HS proteoglycans, e.g., cell surface syndecans, which are the main HPSE targets [10], [11], [12], [13]. Heparanase activity correlates with the metastatic potential of cancer cells, a notion that is well-supported experimentally and clinically [10], [11], [12], [13], [14], [15], [16], [17], [18], [19]. Of relevance, highest HPSE levels have been consistently detected in tumor cells selected to possess highest propensities to colonize the brain [14], [15], [16] with the recent evidence for its expression in patient-isolated breast cancer circulating tumor cells competent to generate brain metastasis in xenografts [20]. Apart from its well-characterized enzymatic activity, heparanase was also shown to exert enzymatic-independent functions, e.g., acting as a signal transducer and regulator of cell adhesion [21] and cytoskeletal dynamics [22]. A recent report also showed that HPSE augmented EGFR phosphorylation that correlated with head and neck tumor progression [18]. Heparanase can thus initiate broad effects that dramatically alter the microenvironment and stimulate tumor cell growth and metastasis. Altogether, these notions raised the following questions – Is HPSE implicated in lapatinib resistance of breast cancer cells expressing EGFR and HER2? If so, by which mechanism(s)?
We hypothesized that HPSE is implicated in mechanisms of lapatinib resistance of breast cancer cells expressing EGFR/HER2, and promotes alternative signaling pathways which are not inhibited by lapatinib, with HPSE inhibition suppressing tumor growth and BMBC. To examine this, we used Roneparstat, a chemically modified heparin lacking anti-coagulant activity and a potent inhibitor of heparanase activity [19], [23]. We selected and used lapatinib-resistant clones generated from the human brain-colonizing MDA-MB-231BR breast cancer cell line [24], then studied Roneparstat-mediated actions related to the lapatinib resistance in these clones. We show that the inhibition of HPSE activity by Roneparstat overcomes lapatinib resistance and suppresses cell growth in vitro and BMBC onset in vivo. These findings provide the molecular basis to potentially employ Roneparstat in therapeutic approaches of lapatinib-resistant breast cancers, particularly breast cancer brain metastasis.
Materials and Methods
Tissue Culture, Establishment of Lapatinib-Resistant Clones, and Clones Characterization
The human brain metastatic MDA-MB-231BR cell line was obtained from Dr. Toshiyuki Yoneda (The University of Texas Health Science Center-San Antonio, TX). It was derived from MDA-MB-231 parental cells by six sequential cycles of selection followed by cell injection into the internal carotid artery of nude mice, and resulting in augmented abilities to generate brain metastasis over the parental counterpart [24]. The MDA-MB-231BR clone transfected with HER2 (named BR for brevity; original cells possess low HER2 levels) (Figure S3) [5] was provided by Dr. Patricia Steeg (National Cancer Institute, Bethesda, MD). MDA-MB-231 parental, MDA-MB-231BR, and BR variant cell lines were authenticated by short tandem repeat (STR) DNA fingerprinting analyses for 16 loci, and data compared to the database of the Characterized Cell Line Core at MD Anderson Cancer Center (Houston, TX). Lapatinib-resistant cell lines were obtained from surviving BR cells exposed to increasing lapatinib concentrations (0.1, 0.5, 1, 5, and 10 μmol/L) in DMEM/F12 culture medium for 4 to 5 weeks. Resistance at each dose was assessed by comparing the growth of each resistance derivative to parental BR cells. Next, surviving cells were treated chronically in vitro with lapatinib (1 μmol/L) [25]. Medium supplemented with this lapatinib concentration was changed every 2 to 3 days with cells being continuously exposed to lapatinib for a three-months period, and resulting in the generation of BR lapatinib-resistant (BR-Lr) clones. Lapatinib-sensitive (BR-Ls) clones were also obtained from BR cells using the limiting dilution method (single-cell colonies) in 96-well plates [25]. BR-Lr and BR-Ls clones were cultured in Dulbecco’s Modified Eagle Medium plus F12 (DMEM/F12) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Invitrogen), and 1% penicillin and streptomycin (Life Technologies). All BR-Lr clones were cultured in a humidified, 5% CO2 incubator at 37°C, passaged twice weekly along the same schedule, and used only at low passage and if Mycoplasma negative. Experimental metastasis assays were periodically performed to test cell in vivo tumorigenic abilities. These studies were accomplished per protocol approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine, and involved all steps of animals sacrifice and amelioration of suffering. Cell cycle analyses based upon fluorescence activated cell sorting (FACS) were carried out using the FACSCalibur instrument at the flow cytometry core facility of Baylor College of Medicine, and analyzed using a CellQuest software (BD Bioscience).
Antibodies, Reagents, and Inhibitors
Primary antibodies include mouse anti-human heparanase monoclonal antibody which was obtained from Cedarlane Laboratories (Burlington, NC). Rabbit anti-human heparanase was kindly provided by Dr. Israel Vlodavsky (The Rappaport Institute-Technion, Haifa, Israel). The other primary antibodies were purchased from Cell Signaling (Danvers, MA). Secondary antibodies included: goat anti-rabbit IgG [H+L]-HRP and goat anti-mouse IgG [H+L]-HRP that were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), while biotinylated universal anti-rabbit/mouse IgG [H+L] was obtained from Vector Laboratories (Burlingame, CA). Lapatinib (Tykerb) was purchased from Sellek, Inc. (Abilene, TX), and Roneparstat (SST0001 or 100NA, RO-H) was provided by Sigma-tau Research S.A. (Mendrisio, Switzerland) under a material transfer agreement. Roneparstat’s chemical structure and functionalities as potent HPSE inhibitor have been described elsewhere [19], [23]. Based upon its coefficient in lipid phase and the number of hydrogen bonds on the steroid, it is predicted that Roneparstat is able to penetrate the blood-brain barrier or the blood-tumor barrier. Roneparstat was prepared as a stock solution following the manufacturer’s instructions, and diluted in culture medium directly before its use. The focal adhesion kinase (FAK) inhibitor PF 573,228 was purchased from Sigma-Aldrich (St. Louis, MO) and used treating cultured cells at concentrations (30 to 100 nM) reported to significantly decrease FAK phosphorylation without toxicity [26].
Western Blotting and Heparanase Activity Assays
Cells were serum-starved for 24 hours, then indicated proteins were examined for expression levels by Western blotting, as previously described [14], [27]. Heparanase activity in cells and mouse serum was examined using the heparan sulfate degrading enzyme assay kit (TakaRa Mirus.Bio, Madison, WI) following the manufacturer’s instructions, as previously reported [13], [14], [27].
Colony Forming Assays
Assays were performed as previously described [25], [28]. Briefly, 1.5 ml of DMEM containing 10% FBS and 0.5% low-melting agarose were poured into 60-mm Petri dishes, in triplicate. The layer was covered with HER2-transfected BR or BR-Lr clones in 1.5 ml of 0.35% low-melting agarose containing DMEM/F12 supplemented with 10% FBS. Medium was changed every 3 days and dishes were incubated for additional 2 weeks, then stained with crystal violet. Colonies of > 50 μm in size (diameter) were counted under a phase-contrast microscope and colony numbers plotted. At least three independent experiments were performed and data validated for statistical significance.
ELISA Assays
BR and selected lapatinib-resistant and sensitive BR clones (BR-Lr and BR-Ls, respectively) were examined for levels of secreted HPSE using the human heparanase ELISA kit (EIAab Science Ltd., Haifa, Israel), following the manufacturer’s protocol. The kit provides a quantitative HPSE determination in cell-conditioned media and lysates using a biotin-conjugated polyclonal antibody specific for heparanase. HPSE concentration was assessed by color changes induced by the biotin-conjugated antibody and enzyme-conjugated avidin, and by comparing the O.D. of samples to the standards supplied by the kit (for details refer to www.eiaab.com). Syndecan-1 levels were examined in cell conditioned media collected from BR-Lr, BR-Ls or HER2-transfected BR cells with or without drugs treatment, using the human syndecan ELISA kit (Abcam, Inc., Cambridge, MA), per protocol provided by the manufacturer (Figures S1 and S2).
Immunohistochemistry
Proteins of interest were examined using formalin-fixed, paraffin-embedded specimens of primary and BMBC tissues obtained from models of experimental metastasis (xenografts). Sections were fixed with 4% formaldehyde in PBS, permeabilized with 0.1% Triton X-100, and blocked with 10% normal goat serum followed by incubation with indicated primary antibody (1:50 to 1:100 dilution) for 18 hours at 4°C, then by the secondary antibody (1:400 dilution) incubation for 1 hr at room temperature (25°C). Staining was performed using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA). Results were examined by pathologists blinded to study groups. Positivity of staining was distributed throughout a 0 to 3 + intensity scale: 0 corresponded to background staining; 1 +, weak staining; 2 +, moderate staining; 3 +, strongest staining, pathologically assessed.
Experimental BMBC Mouse Model
Athymic nude mice (nu/nu, 5–6 weeks old) were purchased from Harlan Sprague Dawley, Inc. (Indianapolis, IN), and maintained at the accredited animal facility of Baylor College of Medicine (BCM). All studies were conducted according to NIH animal use guidelines and protocol approved by the BCM Animal Care Committee. Parental BR and BR-Lr cells were injected into the third mammary fat pad (0.5 × 106 cells/cell line/mouse; n = 5 mice/treatment group) and drug treatment began seven days following cell injection. Mice were randomly divided into four treatment groups: 1) phosphate-buffered saline (PBS, control); 2) lapatinib (30 mg/kg/day); 3) Roneparstat (30 mg/kg/day); 4) lapatinib plus Roneparstat (30 mg/kg/day each). These reagents were delivered via Alzet osmotic pumps (DURECT Corporation, Cupertino, CA). To examine additive roles of Roneparstat and lapatinib inhibiting tumor growth, primary tumors were monitored weekly, and tumor weight was calculated by the following formula: tumor weight (mg) = d2 × D/2, where d and D are the shortest and the longest diameter, respectively. After 28 days of drug treatment, primary tumors were excised and wet weights recorded. To assess whether the combination of Roneparstat and lapatinib could significantly inhibit BMBC after primary tumors were removed under sterile surgical conditions, mice continued to receive drug treatment for additional two weeks by injecting drugs at same doses. Next, whole brains were excised from the animals and fixed in Bouin’s solution. Serial sections were analyzed by hematoxylin and eosin (H&E) staining to assess presence of brain metastatic lesions. BMBC was analyzed by the Cri Vectra Intelligent system (Cambridge Research & Instrumentation, Inc., Boston, MA) that is capable of visualizing and quantifying tumor cells at a single-cell level (green color) [20].
Statistical Analyses
At least three independent experiments were performed and all results were validated by analysis of variance (ANOVA) performed on the data with experiment specified as the random effect. Values are expressed as mean ± SD of at least triplicate samples. P < .01 was considered statistically significant.
Results
Generation of Lapatinib-Resistant Clones from HER2-Transfected MDA-MB-231BR Cells
HER2-transfected MDA-MB-231BR (named BR for brevity) cells were exposed to increasing concentrations of lapatinib (0.1–10 μmol/L in DMEM/F12 containing 10% FBS medium) for 4–5 weeks. Lapatinib-resistant BR clones (BR-Lr) were selected from surviving cells exposed to highest concentration of lapatinib (10 μmol/L). Clones were then selected following a chronic exposure to lapatinib (1 μmol/L) for three months and subsequently characterized. This lapatinib concentration was found to be sufficient to maintain resistance to the drug and did not alter HER2 expression in these cells (Figure S3) [25]. Five lapatinib-resistant single clones were obtained, amplified by long-term culturing, and named BR-Lr1, BR-Lr2, etc (Figure 1A). Lapatinib-resistant (BR-Lr) clones were also obtained from original MDA-MB-231BR cells using the limiting dilution method in 96-well plates (Figure S3) [25]. Single clones were selected in the absence of lapatinib, and the sensitivity of these clones to lapatinib was tested by survival assays (Figure 1A) [25], [28]. We used BR-Lr1 and BR-Lr2 as lapatinib-resistant clones in all subsequent experiments. Further, because BR-Ls cell growth characteristics were highly similar to parental BR cells, we used the latter as experimental control in all experiments.
Figure 1.
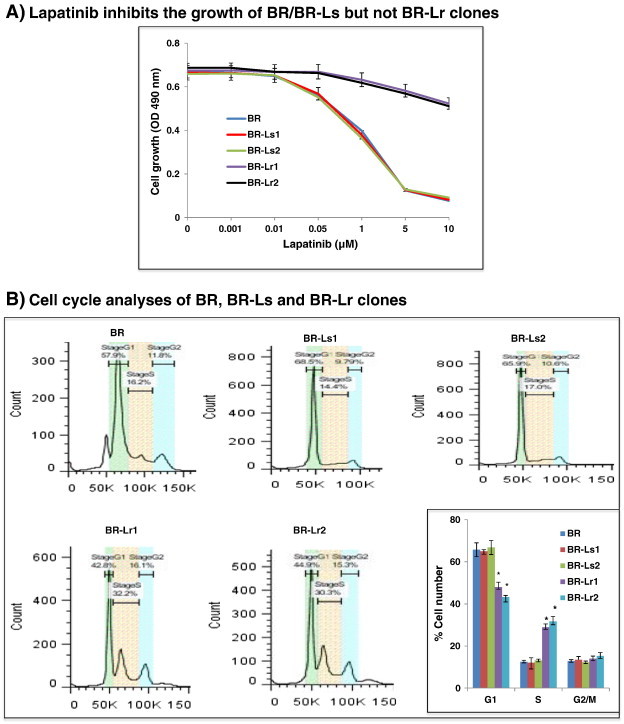
Selection and growth of lapatinib-resistant BR clones. A. Graph displaying the generation of lapatinib-resistant clones from surviving HER2-transfected, EGFR expressing MDA-MB-231BR (named BR for brevity) cells [7]. Resistant clones were developed by first exposing BR cells to increasing lapatinib concentrations (0.1, 0.5, 1, 5 and 10 μmol/L in DMEM/F12 culture medium). This period of selection lasted for 4–5 weeks. Medium supplemented with lapatinib (1 μmol/L) [25] was changed every 2–3 days, and surviving cells were treated chronically in vitro with this lapatinib concentration for 3 months. Afterwards, clones developing resistance to lapatinib were selected for amplification and characterization. B. Representative cell cycle analyses of BR, BR-Ls and BR-Lr clones by fluorescence-activated cell sorting (FACS) and their quantitation. Bars represent the mean ± SD of three independent determinations. *P < .01 was considered statistically significant. C. Colony formation (soft-agar assays) by BR and selected BR-Lr clones in the presence of lapatinib (1 μM). Colonies of > 50 μm in diameter size were counted and colony number statistically quantified. All data were analyzed by ANOVA. Values are expressed as mean ± SD of triplicate samples. P < .01 was considered statistically significant. Refer to Materials and Methods for experimental details.
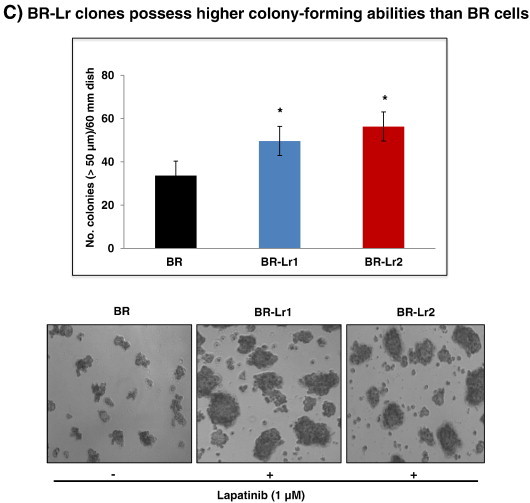
To define the nature of growth inhibition of BR parental and selected BR-Lr/BR-Ls clones, considering that cancer cell proliferation is largely driven by cell cycle alterations, we examined the cell proliferation status by cell-cycle analyses using FACS. BR-Lr clones grew at higher rates and had a lower percentage (15–20%) cells in G1 while higher one (20–25%) in S phase compared to BR or BR-Ls clones (Figure 1B) [28]. Next, we examined BR-Lr growth abilities employing colony formation assays. Lapatinib-resistant cells exhibited a significantly higher colony formation (52% and 56% for BR-Lr1 and BR-Lr2, respectively) compared to BR cells, suggesting that lapatinib-resistant clones possess stronger proliferative capabilities (Figure 1C).
Heparanase Expression, Activity, and Secretion in BR/BR-Ls/BR-Lr Cells
To test the hypothesis that heparanase plays an important role in mechanisms of lapatinib resistance, BR/BR-Ls/BR-Lr cells were examined for HPSE expression (Western blotting/ELISA assays), HPSE secretion (ELISA), and activity (HS-degrading enzyme assays). Higher HPSE expression was detected in BR-Lr1/2 clones, notably the 50 kDa active HPSE subunit (Figure 2A) [11]. Similarly, HPSE was approximately 1.2 to 1.5 fold higher in BR-Lr than BR or BR-Ls cell lysates, however increased (1.5-fold) heparanase activity was observed in the cell conditioned medium of all lapatinib-resistant BR clones compared to BR counterparts (Figure 2, B and C, respectively). Third, to determine whether elevated HPSE activity enhances shedding of target HS proteoglycans, e.g., cell surface syndecan-1 [13], [29] in lapatinib-resistant BR cells, syndecan-1 levels were analyzed in the cell conditioned medium by ELISA. Higher (~ 50%) syndecan-1 expression was detected in BR-Lr clones compared to parental BR cells (Figure S1), providing further indication that HPSE activity may be relevant in lapatinib resistance mechanisms driving an aggressive tumor phenotype [30].
Figure 2.
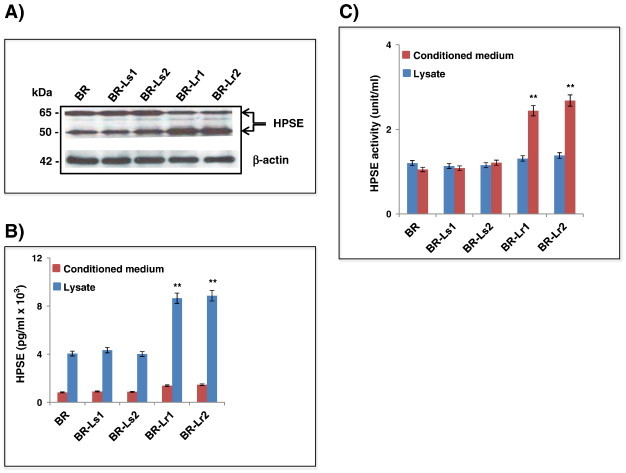
Heparanase (HPSE) expression and activity in HER2-transfected BR, lapatinib-sensitive and lapatinib-resistant selected clones (BR-Ls and BR-Lr, respectively). A. Western blotting analyses showing expression of HPSE in its latent (65 KDa) and active (50 kDa) forms [11], [27]. They were performed as previously described [14], [27]. B. Heparanase expression in cell lysates and conditioned medium of BR/BR-Ls/BR-Lr cells by ELISA. C. Heparanase activity in cell lysates and conditioned medium of BR/BR-Ls/BR-Lr cells. HPSE activity was assesses as previously described [13], [14]. All data were analyzed for statistical significance. Values are expressed as mean ± SD of three independent determinations. **P < .001, ANOVA. Refer to Materials and Methods for experimental details.
Inhibition of HPSE-Mediated Signaling by Roneparstat in BR-Lr Cells
To evaluate roles of heparanase conferring lapatinib resistance, we blocked heparanase using Roneparstat, then investigated the extent of HPSE-mediated signaling inhibition in BR-Lr clones. Abilities of HPSE to affect ERK/FAK and EGFR-associated signaling have been previously reported [12], [18]. Phosphorylated FAK (pFAK) levels were greatly decreased in BR-Lr clones, treated either with Roneparstat or Roneparstat additive to lapatinib, and compatible with decreased EGFR (Y845) and ERK phosphorylation (Figure 3A). Importantly, while Roneparstat affected the phosphorylation of EGFR at tyrosine position 1173 (pEGFR, Y1173), it also reduced the phosphorylation of pEGFR at 992 position (Y992) which is not a target of lapatinib (Figure 3A) [7]. Second, we examined effects of Roneparstat treatment on the growth of BR-Lr cells. Cell growth was profoundly inhibited by Roneparstat, either alone or in the presence of lapatinib (Figure 3B). This suggests that Roneparstat can inhibit the phosphorylation of HPSE downstream signaling, specifically pEGFR (Y845), pFAK and pMAPK, thus decreasing BR-Lr cell growth abilities [26]. Third, to determine whether the inhibition of phosphorylated FAK (pFAK) modulates cell growth, we treated cells with the FAK inhibitor PF 573,228. The phosphorylation of EGFR (Y845), Src, and ERK1/2 expression was reduced by PF 573,228 treatment (Figure 3C). Of note, BR-Lr clones were significantly more sensitive to PF 573,228, exhibiting a strong reduction (70% to 75%) in colony formation (Figure 3D). Fourth, we examined the expression of MMP-9 and COX2 which are known to be regulated by HPSE [27]. The expression of MMP-9 and Cox-2 was affected by Roneparstat with reduced MMP-9 and COX-2 protein levels following Roneparstat treatment. This reduction was rescued by the addition of recombinant human HPSE (rHPSE) (Figure 3E; see also Figure S2). Lastly, we validated the effect of HPSE inhibition on lapatinib resistance employing siRNA approaches. HPSE-specific siRNA (SMART pool siRNA) [14] inhibited HPSE expression in both BR and BR-Lr clones (Figure 4A). Cell colony formation abilities also depended upon siRNA inhibition and correlated with HPSE siRNA concentration-dependent effects (Figure 4B).
Figure 3.
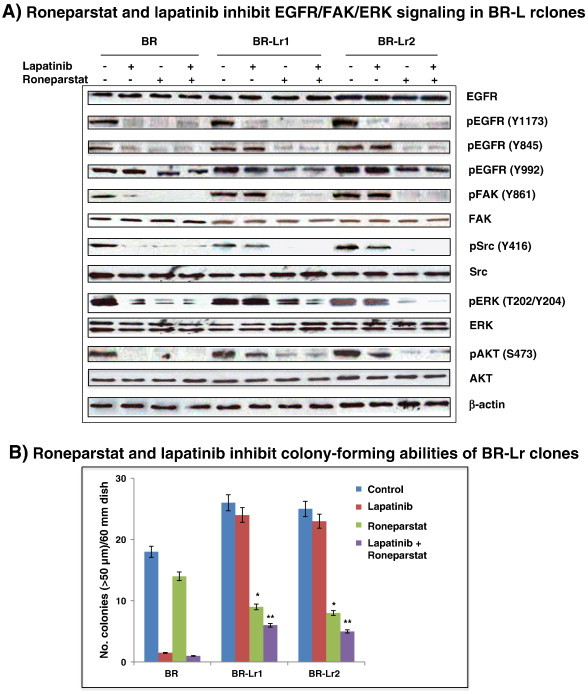
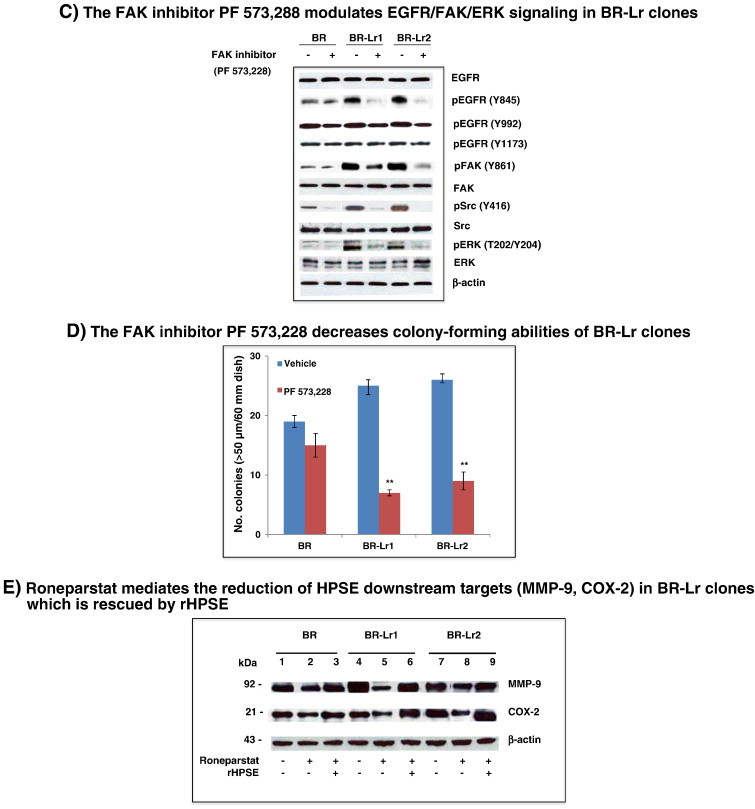
Roneparstat and lapatinib inhibit EGFR and FAK-associated signaling in HER2-transfected BR and BR-Lr clones (BR, BR-Lr1 and BR-Lr2, respectively). A. Western blotting analysis of BR, BR-Lr1 and BR-Lr2 cells. Cells were seeded in six-well plates and serum-starved for 24 hours. Cells were then treated as indicated [PBS (control), Roneparstat (30 mg/kg/day), lapatinib (1 μM) without or with Roneparstat at same dose (30 mg/kg/day)]. Following 24 hours incubation at 37°C, cells were collected and cell lysates prepared for Western blot analyses. Representative results are shown. β-actin indicates equal loading (control). B. Roneparstat and lapatinib inhibited the colony formation of BR-Lr clones. Bars represent the mean ± SD of three independent determinations. *P < 0.01 was considered statistically significant. C. Western blotting analyses showing the inhibition of focal adhesion kinase (FAK) to modulate EGFR/FAK/ERK signaling in BR-Lr clones. The FAK inhibitor PF 573,228 (Sigma-Aldrich, St. Louis, MO) was used in all experiments at concentrations (30-100 nM) that avoid off-targets effects [26]. Representative results are shown with BR cells analyzed in parallel as control. β-actin indicates equal loading (control). D. FAK inhibition by PF 573,228 decreases colony-forming abilities of BR-Lr clones. Values are expressed as mean ± SD of three independent determinations. **P < 0.001, ANOVA. E. Roneparstat-mediated reduction of HPSE downstream effects is rescued by addition of recombinant human HPSE (rHPSE). Western blotting analyses expression of HPSE-mediated targets MMP-9 and COX-2 [14] in BR/BR-Lr1/BR-Lr2 following treatment with Roneparstat (30 mg/kg/day) in the absence or presence of rHPSE (500 ng/ml) [12]. β-actin indicates equal loading (control). All data were analyzed for statistical significance. Values are expressed as mean ± SD of three independent determinations. **P < 0.001, ANOVA. Refer to Materials and Methods for experimental details.
Figure 4.
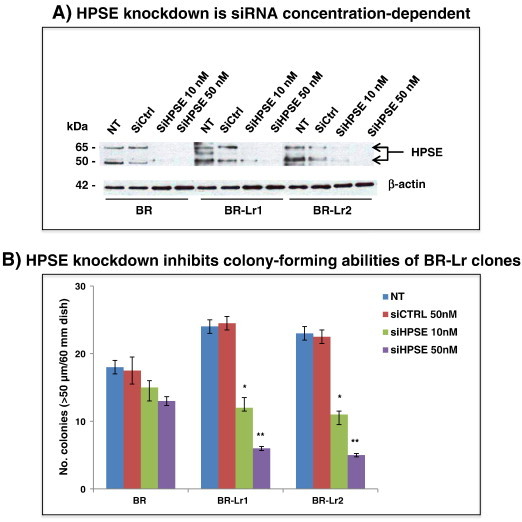
Heparanase knockdown decreases colony formation of BR-Lr clones. Cells (BR, BR-Lr1, BR-Lr2) were transfected with HPSE SMARTpool siRNA (siHPSE 10 nM, 30 nM) or non-target control (siCtrl) for 48 hours [14], then treated with lapatinib (1 μM) for 24 hours. Next, cell lysates were prepared and HPSE protein levels were determined by Western blot analyses (A) while cell growth abilities were examined by colony formation assays (B). Representative results are shown. β-actin was used for equal loading (control). Data were analyzed for statistical significance. Values are expressed as mean ± SD of triplicate samples. *P < 0.01; **P < 0.001, ANOVA. Refer to Materials and Methods for experimental details.
Roneparstat Sensitizes Lapatinib-Mediated Inhibition of Cell Growth and BMBC Onset
Heparanase roles in tumor invasion and metastasis are long-known [10], [11] with a recent report implicating Roneparstat to alter tumor growth by heparanase activity inhibition [23]. Because of the presence of high HPSE activity in selected BR-Lr clones, we hypothesized that Roneparstat could overcome lapatinib resistance by inhibiting HPSE actions in these cells. Accordingly, we examined effects of Roneparstat on primary tumor growth and metastasis by injecting BR-Lr clones into nude mice followed by various drug treatments [PBS control, lapatinib (1 μM) without or with Roneparstat (30 mg/kg per day), and Roneparstat alone (30 mg/kg per day)]. These reagents were administered to animals by Alzet osmotic pumps after 7 days of tumor cell injection into mammary fat pads. They were delivered for 28 days with weekly monitoring for tumor development. Once observed, tumors were removed and their extent, number, size, and wet-weight were assessed among treatment modalities. Tumor size was approximately 40% smaller in Roneparstat/lapatinib-treated animals than lapatinib alone with the tumor weight also reduced (62% reduction; P < .001) in Roneparstat/lapatinib-treated group compared to lapatinib only-treated animals (Figure 5A).
Figure 5.
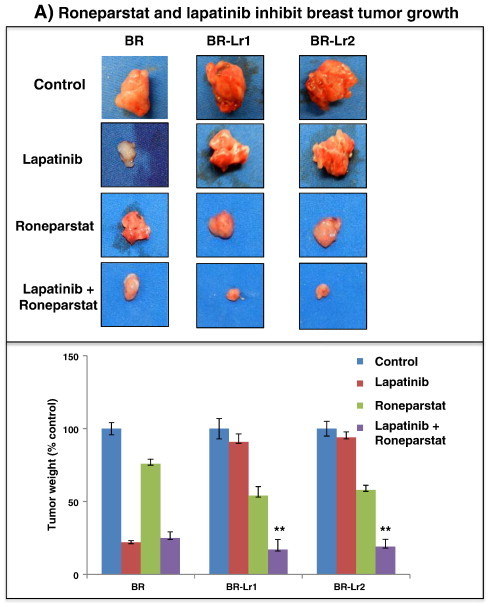
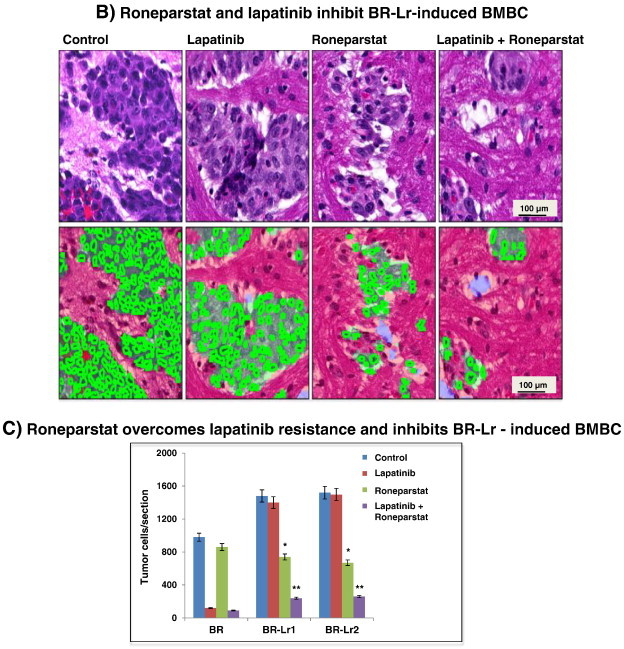
Roneparstat and lapatinib inhibit breast tumor growth and BMBC onset in xenografts. A. HER2-transfected parental BR cells (BR) and selected BR-Lr clones (BR-Lr1, BR-Lr2) were injected into the third mammary fat pad (0.5 × 106 cells/cell line/mouse; n = 5 mice/treatment group). Tumor growth was monitored weekly by measuring length and width with a digital caliper. Tumors were subsequently harvested and photographed. Representative images are shown (top). Tumor weight was calculated and compared among treatment groups (bottom). Values are expressed as mean ± SD of triplicate samples. **P < .001, ANOVA. B. Hematoxylin and eosin (H&E) sections from animals with BMBC following BR-Lr cell injection. Sections were randomly selected and analyzed by the Cri-Vectra-inForm system (Cambridge Research & Instrumentation, Inc.) which highlights and enumerates single tumor cells (in green) but not adjacent stromal cells [20]. Representative images of BR-Lr-induced BMBC are shown for the various treatments. C. Quantification of BMBC tumor burden represented by the number of single tumor cells detected and analyzed by the Cri-Vectra-inForm system [20]. Primary tumors from different treatment groups were removed when they reached the same size, then the extent of BMBC tumor burden was measured by Cri-Vectra-inForm. Data were analyzed for statistical significance. Values are expressed as mean ± SD of triplicate samples. *P < .01; **P < .001, ANOVA. Refer to Materials and Methods for experimental details.
To analyze whether combinatorial Roneparstat and lapatinib treatment inhibited BR-Lr cell-induced BMBC, delivery of drugs continued for additional two weeks within animal sub-groups. Mice were then sacrificed, brains surgically removed, and examined for BMBC presence. A distinct reduction (> 60%; P < .001; primary tumors within treatment groups were removed when reaching the same size) of the number of BMBC cells in Roneparstat-treated mice was observed compared to lapatinib alone or control groups (Figure 5, B and C). These results suggest that Roneparstat is able to significantly inhibit BR-Lr cell-mediated tumor growth and brain metastasis. Second, to evaluate whether the inhibition effect on tumor growth and metastasis resulted from HPSE inhibition, we analyzed HPSE activity in the serum of experimental mice according to drug treatment. We detected HPSE activity to be significantly lower (60% to 75% reduction; P < .001) in Roneparstat and Roneparstat/lapatinib-treated animal groups compared to lapatinib alone (Figure S4). Third, to translate signaling results (Figure 3) to in vivo settings, we investigated phosphorylation levels of EGFR (Y845), FAK and ERK1/2 in BR-Lr-induced BMBC tumors. We observed levels to be reduced following Roneparstat and Roneparstat/lapatinib treatments of experimental enimals (Figure 6, A–C). Altogether, findings indicate that Roneparstat sensitizes lapatinib inhibition of tumor growth in BR-Lr clones by a regulation of heparanase activity and heparanase downstream signaling and targets.
Figure 6.
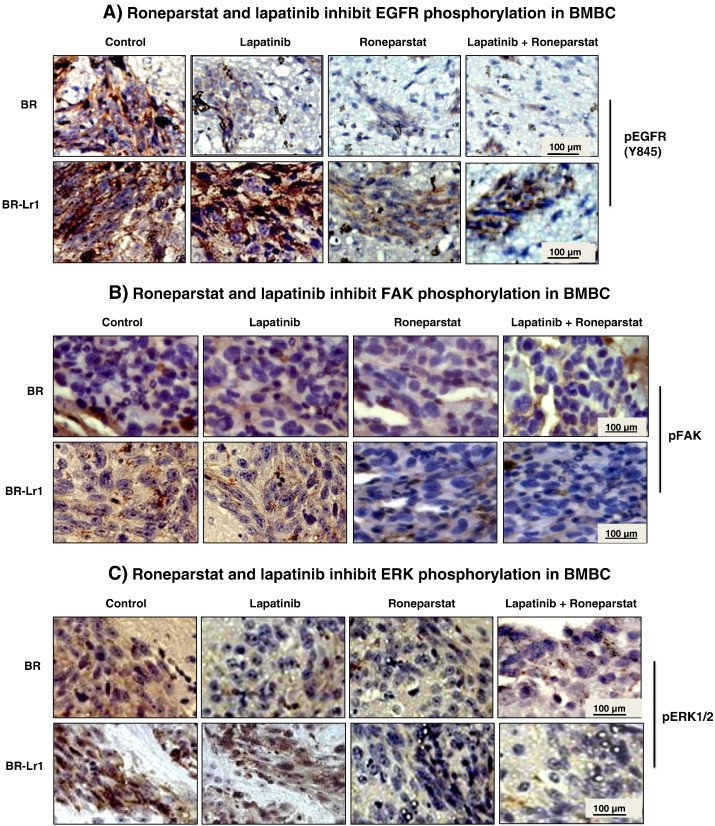
Roneparstat and lapatinib inhibit EGFR/FAK/ERK phosphorylation of BR-Lr cell-induced BMBC. A–C. Immunohistochemical BMBC analyses of treated HER2-transfected BR and BR-Lr clones displaying reduced EGFR (Y845) (A), FAK (B), and ERK1/2 (C) phosphorylation in response to Roneparstat and lapatinib treatment. Data obtained using the BR-Lr1 clone are shown and representative of all BR-Lr clones investigated. Immunohistochemistry was performed as described in Materials and Methods (refer also to reference [7]).
Discussion
The molecular mechanisms of resistance to lapatinib in breast cancer remain poorly understood, particularly in brain metastatic breast cancer (BMBC). The present study was designed to identify mechanisms by which cells expressing EGFR/HER2, both high-risk predictors for BMBC onset, develop resistance to lapatinib. We hypothesized that compensatory signaling originating independently of the EGFR/HER2 pathways may exist and be responsible to impart added resistance to the drug. We demonstrated the relevance of HPSE in lapatinib resistance with Roneparstat inhibitor regulating downstream pathways that lapatinib fails to inhibit, and reducing breast tumor growth and brain metastasis.
Several breast cancer model cell systems have known capabilities of generating BMBC (e.g., MDA-MB-231P/BR, MDA-MB-435, MDA-MB-361 and MDA-MB-468) [5], [7], [31]. However, reasons by which these cells acquire resistance to lapatinib treatment are still unclear and understudied. Among these model cell systems, MDA-MB-231BR is most studied because of its high propensities to generate BMBC in experimental metastasis assays. Further, a report showed that lapatinib inhibited the phosphorylation of HER2/EGFR downstream signaling proteins modulating cell proliferation/migration abilities and BMBC propensities of MDA-MB-231BR cells, either with or without HER2 presence [7].
Heparanase is well-known to be a potent tumorigenic, angiogenic and pro-metastatic molecule with highest activity levels found in cells selected to possess highest propensities to colonize the brain [14], [16], [27]. For example, we have previously demonstrated that HPSE translocation to nucleolus enhances MDA-MB-231BR cell proliferation through the activation of Topoisomerase I [14] and that HPSE inhibition by siRNA or microRNA can inhibit breast cancer cell growth, invasion and brain metastasis [27]. Our laboratory has previously implicated HPSE in mechanisms of cytoskeletal dynamics and in the cross-talk between tumor cells and vascular brain endothelium within the initial steps leading to BMBC onset [22]. Here, we examined roles of heparanase in newly-developed lapatinib resistant clones derived from HER2-transfected, EGFR-expressing MDA-MB-231BR cells (named BR for brevity) following their exposure to increasing and sequential lapatinib doses. The rationale was to extend the previous investigations using the MDA-MB-231BR model [7] with the knowledge that HPSE can augment EGFR phosphorylation reported in other cancer settings [18]. Our findings indicate that HPSE activity is involved in mechanisms of lapatinib resistance in the BR model system: the potent HPSE inhibitor Roneparstat can significantly sensitize lapatinib inhibition to decrease breast cancer cell growth (colony-forming abilities in soft-agar assays) and brain metastasis in xenografts. We provide evidence that HPSE activity is elevated in lapatinib-resistant BR clones, and that the activity in these clones promoted an enhanced phosphorylation of EGFR (Y845), FAK, ERK1/2, and increased cell growth (Figure 3). Results also indicate that Roneparstat is a potent HPSE inhibitor that can effectively block these signaling pathways to diminish the formation of BMBC in an experimental animal model (Figure 5).
A potential pathway by which HPSE modulate lapatinib resistance is the heparanase/syndecan-1 axis. A recent publication has reported that this axis can be blocked in myeloma cells by Roneparstat [23]. Further, syndecan-1 is increased in several breast cancer types, including HER2 overexpressors [32]. Our data show that levels of shed syndecan-1 was significantly increased in BR-Lr clones compared to parental BR cells (Figure S1). Roneparstat significantly decreased shed syndecan-1 levels in a concentration-dependent manner. Of note, Roneparstat-mediated reduction of shed syndecan-1 was rescued by recombinant human heparanase (rHPSE) in these cells (Figure S2). This can partially explain Roneparstat effectiveness in mechanisms of lapatinib resistance. A possible role of syndecan-1 is to act as a molecular “bridge” and link to FAK/Src signaling since FAK/Src can bind to the cytoplasmic domain of syndecan-1 [29]. Heparanase can enhance syndecan-1 shedding resulting in the activation of FAK/Src signaling and downstream effects at specific EGFR phosphorylation sites, e.g., Y845 (Figures 3, A and C). Other studies have also demonstrated that the activation of ERK requires HPSE activity [30] and that exogeneous HPSE up-regulated FAK/ERK phosphorylation through enhanced fibroblast growth factor receptor binding and signaling [12]. Our findings are consistent with these reports.
Our study has limitations as we did not examine whether the selected BR-Lr clones are also resistant to other HER inhibitors such as trastuzumab targeting HER2 [28] or whether our findings also apply to estrogen receptor-positive human breast cancer cells that develop resistance to lapatinib [25]. Further, lapatinib may be mostly ineffective in BMBC because it has a mediocre brain permeability and may fail to penetrate the brain at sufficient and consistent doses, at least for reliable ones having anti-tumor (cytotoxic) effects. Tumor cells isolated from brain of mice treated with lapatinib can retain lapatinib sensitivity in in vitro conditions [8]. Similarly, the increase of HPSE expression and activity in BR-Lr clones correlated with lapatinib resistance but did not confer resistance to the drug; and how HPSE regulates and/or works in conjunction with other growth/regulatory factors to promote BMBC will have to be elucidated. Lastly, the combinatorial lapatinib-Roneparstat treatment can directly occur at the primary tumor affecting its growth, and that the extent of BMBC (metastasis) be a secondary effect influenced by the primary tumor burden. Although the extent of BMBC regulation following treatment was determined after removing primary tumors with the same size, this remains a possibility to be investigated in future studies (see also Ref. [33]). Regardless, our data put forward the notion that Roneparstat, a small-molecule non-anticoagulant heparin and potent HPSE inhibitor, is effective against BMBC modalities without any evident toxicities or side effects either in cell cultures or animals. Roneparstat inhibition of HPSE and EGFR signaling demonstrate its dramatic effects altering the BMBC microenvironment and the cross-talk between neoplastic and normal brain cells to diminish brain metastasis [15].
In conclusion, we have identified a molecular mechanism of lapatinib resistance in brain metastatic breast cancer, and demonstrated that Roneparstat can overcome lapatinib resistance and inhibit tumor growth and metastasis. Specifically, the combination of Roneparstat and lapatinib inhibited tumor cell growth in vitro and brain metastasis in vivo, presumably through dual targeting of HPSE-mediated pathways within cells of the BMBC microenvironment. Roneparstat outcomes in ongoing clinical trials (Phase I in multiple myeloma) will provide useful information for its potential as a novel therapeutic agent for treatment of BMBC.
Acknowledgments
We would like to thank Dr. Israel Vlodavsky (The Bruce Rappaport Faculty of Medicine, Technion, Israel) for the gift of recombinant human active and latent HPSE. Roneparstat (SST0001 or 100NA, RO-H) is property of Sigma-tau Research Switzerland S.A., and provided to us under a material transfer agreement. This study was supported by NIH grant 1R01 CA160335 and the IDEA Award from the Department of Defense-Congressional Directed Medical Research Programs (W81XWH-11-1-0315) to D.M. Support for STR DNA fingerprinting Core was provided by the Cancer Center Support Grant-funded Characterized Cell Line Core (NCI CA016672). The project was also supported by the Human Tissue Acquisition and Pathology Core at BCM with NIH funding (NCI P30 CA125123), by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574), and by the expert assistance of Joel M. Sederstrom of this Core.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Author contributions: L.Z. performed experiments, data analysis, and wrote the manuscript; M.W. carried out colony formation and ELISA assays, and J.N. completed ELISA and HPSE assays and cell culture. D.M. designed and supervised experiments, and edited the manuscript.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.11.007.
Appendix A. Supplementary data
Figures S1–S4
References
- 1.Olayioye M.A., Neve R.M., Lane H.A., Hynes N.E. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cerra M., Cecco L., Montella M., Tuccillo F., Bonelli P., Botti G. Epidermal growth factor receptor in human breast cancer: comparison with steroid receptors and other prognostic factors. Int J Biol Markers. 1995;10:136–142. doi: 10.1177/172460089501000302. [DOI] [PubMed] [Google Scholar]
- 3.Slamon D.J., Godolphin W., Jones L.A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 4.Rusnak D.W., Alligood K.J., Mullins R.J., Spehar G.M., Arenas-Elliott C., Martin A.M. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW57206) in an expanded panel of human normal and tumor cell lines. Cell Prolif. 2007;40:580–594. doi: 10.1111/j.1365-2184.2007.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palmieri D., Bronder J.L., Herring J.M., Yoneda T., Weil R.J., Stark A.M., Kurek R., Vega-Valle E., Feigenbaum L., Vortmeyer A.O. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007;67(9):4190–4198. doi: 10.1158/0008-5472.CAN-06-3316. [DOI] [PubMed] [Google Scholar]
- 6.Rimawi M.F., Wiechmann L.S., Wang Y.C., Huang C., Migliaccio I., Wu M.F., Gutierrez C., Hilsenbeck S.G., Arpino G., Massarweh S. Reduced dose and intermittent treatment with lapatinib and tratuzumab for potent blockade of the HER pathway in HER2/neu-overexpressing breast tumor xenografts. Clin Cancer Res. 2011;17:1351–1361. doi: 10.1158/1078-0432.CCR-10-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gril B., Palmieri D., Bronder J.L., Herring J.M., Vega-Valle E., Feigenbaum Leiwehr D., Steinberg S.M., Merino M.J., Gilmer T.M., Rubin S.D. Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J Natl Cancer Inst. 2008;100(15):1092–1103. doi: 10.1093/jnci/djn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taskar K.S., Rudraraju V., Mittapalli R.K., Samala R., Thorshelm H.R., Lockman J. Lapatinib distribution in HER2 overexpressing experimental brain metastasis of breast cancer. Pharm Res. 2012;29:770–781. doi: 10.1007/s11095-011-0601-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esteva F.J., Yu D., Hung M.C., Hortobagyi G.N. Molecular predictor of response to trastuzumab and lapatinib in breast cancer. Nat Rev Clin Oncol. 2010;7:98–107. doi: 10.1038/nrclinonc.2009.216. [DOI] [PubMed] [Google Scholar]
- 10.Fux L., Ilan N., Sanderson R.D., Vlodavsky I. Heparanase: busy at the cell surface. Trends Biochem Sci. 2009;34(10):511–519. doi: 10.1016/j.tibs.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ilan N., Elkin M., Vlodavsky I. Regulation, function, and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol. 2006;38(12):2018–2039. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Reiland J., Kempf D., Roy M., Denkins Y., Marchetti D. FGF2 binding, signaling, and angiogenesis are modulated by heparanase in metastatic melanoma cells. Neoplasia. 2006;8(7):596–606. doi: 10.1593/neo.06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reiland J., Sanderson R.D., Waguespack M., Barker S.A., Long R., Carson D.D., Marchetti D. Heparanase degrades syndecan-1 and perlecan heparan sulfate: functional implications for tumor cell invasion. J Biol Chem. 2004;279(9):8047–8055. doi: 10.1074/jbc.M304872200. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L., Sullivan P., Suyama J., Marchetti D. EGF - induced heparanase nucleolar localization augments DNA topoisomerase I activity in brain metastatic breast cancer. Mol Cancer Res. 2010;8(2):278–290. doi: 10.1158/1541-7786.MCR-09-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchetti D., Li J., Shen R. Astrocytes contribute to the brain-metastatic specificity of melanoma cell by producing heparanase. Cancer Res. 2000;60:4767–4770. [PubMed] [Google Scholar]
- 16.Murry B.P., Blust B.E., Singh A., Foster T.P., Marchetti D. Heparanase mechanisms of melanoma metastatic to the brain: development and use of a brain slice model. J Cell Biochem. 2006;97:217–225. doi: 10.1002/jcb.20714. [DOI] [PubMed] [Google Scholar]
- 17.Cohen I., Pappo O., Elkin M., San T., Bar-Shavit R., Hazan R. Heparanase prompts growth, angiogenesis, and survival of primary breast tumors. Int J Cancer. 2006;118:1609–1617. doi: 10.1002/ijc.21552. [DOI] [PubMed] [Google Scholar]
- 18.Cohen-Kaplan V., Doweck I., Naroditsky I., Vlodavsky I., IIan N. Heparanase augments epidermal growth factor receptor phosphorylation: correlation with head and neck tumor progression. Cancer Res. 2008;68(24):10077–10085. doi: 10.1158/0008-5472.CAN-08-2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naggi A., Casu B., Perez M., Cassinelli G., Penco S., Pisano C. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol-splitting. J Biol Chem. 2005;280:12103–12113. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L., Ridgway L., Wetzel M., Ngo J., Yin W., Kumar D., Goodman J.C., Groves M., Marchetti D. The identification and isolation of breast cancer CTCs with brain metastatic competence. Sci Transl Med. 2013;5:84–93. doi: 10.1126/scitranslmed.3005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zetser A., Bashenko Y., Miao H.-Q., Vlodavsky I., Ilan N. Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003;63:7733–7741. [PubMed] [Google Scholar]
- 22.Ridgway L., Wetzel M.A., Epstein A., Marchetti D. Heparanase-induced GEF-H1 signaling regulates the cytoskeletal dynamics of brain metastatic breast cancer cells. Mol Cancer Res. 2012;10(6):689–702. doi: 10.1158/1541-7786.MCR-11-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ritchie J.P., Ramani V.C., Ren Y., Naggi A., Torri G., Casu B. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin Cancer Res. 2011;17(6):1382–1393. doi: 10.1158/1078-0432.CCR-10-2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoneda T., Williams P., Hiraga T., Niewolna M., Nishimura R. A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and brain-seeking clone in vivo and in vitro. J Bone Miner Res. 2001;16(8):1486–1495. doi: 10.1359/jbmr.2001.16.8.1486. [DOI] [PubMed] [Google Scholar]
- 25.Liu L., Greger J., Shi H., Liu Y., Greshock J., Annan R. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009;69(17):6871–6878. doi: 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- 26.Slack-Davis J.K., Martin K.H., Tilghman R.W., Iwanicki M., Ung E.J., Autry C. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–14852. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 27.Zhang L., Sullivan P., Goodman J.C., Gunaratne P.H., Marchetti D. Cancer Res - Priority Report. Vol. 71. 2011. MicroRNA-1258 inhibits breast cancer invasion and metastasis by directly targeting heparanase; p. 645-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang C., Park C.C., Hilsenbeck S.G., Ward R., Rimawi M.F., Wang Y.C., Shou J., Bissell M.J., Osborne K.C., Schiff R. β1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res. 2011;13(4):R84. doi: 10.1186/bcr2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beauvais D.M., Rapraeger A.C. Syndecans in tumor cell adhesion and signaling. Reprod Biol Endocrinol. 2004;2(3):1–12. doi: 10.1186/1477-7827-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purushothaman A., Chen L., Yang Y., Sanderson R. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283(47):32628–32636. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lorger M., Felding-Habermann B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am J Pathol. 2010;176(6):2958–2971. doi: 10.2353/ajpath.2010.090838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen T.L., Grizzle W.E., Zhang K., Hameed O., Siegal G.P., Wei S. Syndecan-1 overexpression is associated with nonluminal subtypes and poor prognosis in advanced breast cancer. Am J Clin Pathol. 2013;140:468–474. doi: 10.1309/AJCPZ1D8CALHDXCJ. [DOI] [PubMed] [Google Scholar]
- 33.Boyango I., Barash U., Naroditsky I., Li J.P., Hammond E., Ilan N. Heparanase cooperates with Ras to drive breast and skin tumorigenesis. Cancer Res. 2014;74(16):4504–4514. doi: 10.1158/0008-5472.CAN-13-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S4