

Abstract
The voltage-gated sodium channel (Nav) plays a key role in regulation of neuronal excitability. Aberrant regulation of Nav expression and/or function can result in an imbalance in neuronal activity which can progress to epilepsy. Regulation of Nav activity is achieved by coordination of a multitude of mechanisms including RNA alternative splicing and translational repression. Understanding of these regulatory mechanisms is complicated by extensive genetic redundancy: the mammalian genome encodes ten Navs. By contrast, the genome of the fruitfly, Drosophila melanogaster, contains just one Nav homologue, encoded by paralytic (DmNa v). Analysis of splicing in DmNa v shows variants exhibit distinct gating properties including varying magnitudes of persistent sodium current (INaP). Splicing by Pasilla, an identified RNA splicing factor, alters INaP magnitude as part of an activity-dependent mechanism. Enhanced INaP promotes membrane hyperexcitability that is associated with seizure-like behaviour in Drosophila. Nova-2, a mammalian Pasilla homologue, has also been linked to splicing of Navs and, moreover, mouse gene knockouts display seizure-like behaviour.
Expression level of Navs is also regulated through a mechanism of translational repression in both flies and mammals. The translational repressor Pumilio (Pum) can bind to Na v transcripts and repress the normal process of translation, thus regulating sodium current (INa) density in neurons. Pum2-deficient mice exhibit spontaneous EEG abnormalities. Taken together, aberrant regulation of Nav function and/or expression is often epileptogenic. As such, a better understanding of regulation of membrane excitability through RNA alternative splicing and translational repression of Navs should provide new leads to treat epilepsy.
Keywords: Excitability, Drosophila, Epilepsy, Paralytic, Splicing, Translational repression
Introduction
The regulation of neuronal excitability—primarily the ability to maintain action potential firing within physiological constraints—is an important mechanism for maintenance of neuronal stability [1]. Without such regulation, chronic changes in levels of synaptic excitation have the potential to destabilise neural circuits leading to an imbalance in neuronal activity. One consequence of activity imbalance is seizure, which if recurrent is termed epilepsy [2]. The voltage-gated sodium channel (Nav) plays a key role in the regulation of neuronal excitability because its activation results in action potential firing. It is perhaps, therefore, not surprising that many mechanisms that act to stabilise neuronal activity do so through modifying the activity of this class of ion channel [1, 3–7].
Ten genes (SCN1A-SCN11A), encoding pore-forming α-subunits, are present in mammals [8]. This relatively high number is, however, insufficient to support the wide diversity of Nav kinetics reported in the nervous system. Diversity of signalling is critically reliant on additional mechanisms such as alternative splicing, RNA editing, and protein modification (i.e., phosphorylation) [9–12, 4]. However, whilst the importance of posttranscriptional and posttranslational modifications is appreciated for refining activity of channel subtypes, understanding of the mechanisms that neurons employ to determine which form of Nav to express remains rudimentary. In contrast to mammals, the genome of the fruitfly Drosophila melanogaster contains only one Nav channel homologue: encoded by paralytic (DmNa v) [13, 5]. The lack of redundancy, coupled with a high degree of structural and functional homology, makes DmNav an advantageous model with which to study the role of this ion channel family [14, 15]. In this review, we use DmNav as a model to summarise recent findings relating to how neurons generate diversity in Nav channel activity and to stabilise neuronal circuit function when faced with changing levels of synaptic excitation.
Alternative splicing generates diversity in Nav channel activity
Alternative splicing involves the substitution, removal, and/or inclusion of exonic sequences within a pre-messenger RNA (mRNA) to produce transcripts encoding related protein isoforms [9]. Estimates indicate that ~95 % of human genes are alternatively spliced [16, 17]. Variant transcripts of DmNa v, first reported by Loughney et al., (1989), were among the first evidence for the existence of alternative splicing of this family of gene products. Subsequent studies in Drosophila, Musca, and cockroach have identified 15 alternatively spliced exons [18, 19, 14, 20, 15]. Importantly, alternative splicing of exons is replicated in mammalian Nav channels [21–23]. Spliced exons are conserved across evolutionarily diverged species, strongly indicative of fundamental physiological importance.
A recent structure-function study has described the effects to DmNav channel kinetics of alternative splicing [15]. Of the 15 known splice decisions, two splice events are mutually exclusive incorporating one of either a pair of exons (C/D and K/L). Both exon pairs are membrane spanning, contributing to domains IIS4–5 and IIIS3–4, respectively. The remaining 11 spliced exons (J, 7, 8, I, A, B, E, F, 22, H, 23) are independent and cytoplasmic. Heterologous expression of DmNa v splice variants in Xenopus oocytes shows that such splicing imparts specific attributes to channel kinetics. For example, inclusion of exon F results in a hyperpolarising shift in activation kinetics, indicative of increased excitability for those neurons that express F-containing variants. By contrast, inclusion of exons J and E results in a depolarising shift of activation voltages which are predicted to reduce neuron excitability. On the other hand, channels expressing exon H inactivate at more depolarised voltages, predicted to make neurons more excitable. Finally, the choice to include mutually exclusive exons K or L markedly affects the magnitude of the persistent current (INaP) that arises from incomplete inactivation of the channel [24, 5, 25]. Inclusion of exon K results in a smaller INaP relative to that observed from expression of transcripts that contain exon L, in otherwise identical channels. Increasing INaP leads to an increased frequency of action potential firing [26, 5]. Figure 1 summarises the known splicing events of DmNa v, and the effect on channel kinetics and/or INaP is summarised in Table 1. Of course, the caveat to heterologous expression is that the nature of the cell membrane of the cell type used may influence the kinetics of expressed channels compared to expression, in this instance, in Drosophila neurons [27]. Attempts to express DmNa v variants in Drosophila neurons, using the well-characterised GAL4/UAS system, has repeatedly failed to produce functional channels, for unknown reasons (Lin and Baines, unpublished observations).
Fig. 1.
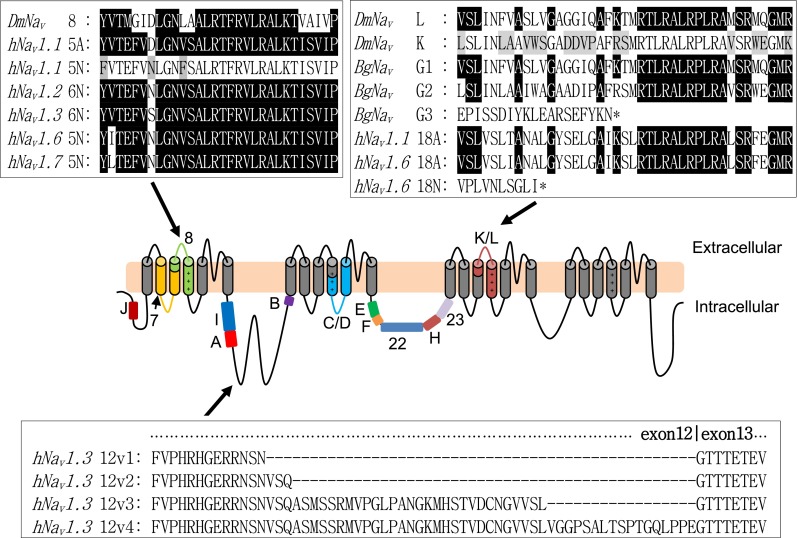
Schematic of the predicted topology of the voltage-gated sodium channel showing approximate locations of Drosophila spliced exons. Cytoplasmic DmNa v exons J, 7, 8, I, A, B, E, F, 22, H, 23 are optional, while exons C/D and K/L are mutually exclusive. DmNa v exon 8 is conserved in human Navs as mutually exclusive spliced exons 5A and 5N (6A and 6N in hNa v 1.2 and hNa v 1.3 due to different exon numbering in the consensus gene sequence), and identical residues are shown in black boxes. Exon 5A and 5N of hNa v 1.1 differ by 3 amino acids, shown in grey boxes in the 5N sequence. Mutually exclusive DmNa v spliced exon L and cockroach BgNa v exon G1 are identical and are conserved in human: exon 18A of hNa v 1.1 and hNa v 1.6. DmNa v exons K and L differ by 16/41 residues (shown in grey boxes in the exon K sequence). Inclusion of BgNa v exon G3 and hNa v 1.6 exon 18N generated a truncated channel. Exon 12 of hNa v 1.3 is located in the intracellular loop between domains I and II. By using different splice donor sites in exon 12, four spliced variants, 12v1, 12v2, 12v3, and 12v4 can be generated. The amino acid sequences are obtained as follows: DmNa v 8, K, and L [15]; hNa v 5A and 5N [61]; hNa v 1.6 18A and 18N [23]; BgNa v G1, G2, and G3 [20]; hNa v 1.3 12v1, 12v2, 12v3, and 12v4 [30]
Table 1.
Summary of spliced Na v exons that are known to affect channel kinetics. Predicted influence on neuron excitability due to splicing are stated, increased (↑), decreased (↓), or complex (?)
| Channel | Exon | Expression system | Predicted effect on cell excitability (by changing) | References |
|---|---|---|---|---|
| DmNa v | J, E | Xenopus oocytes | ↓ (act →) | [15] |
| DmNa v | F | Xenopus oocytes | ↑ (act ←) | [15] |
| DmNa v | H | Xenopus oocytes | ↑ (inact →) | [15] |
| DmNa v | K | Xenopus oocytes | ↓ (INaP amplitude ↓) | [15] |
| DmNa v | L | Xenopus oocytes | ↑ (INaP amplitude ↑) | [15] |
| hNa v 1.1 | 5A | HEK293T | ↑ (INaP amplitude ↑, inact →) | [61] |
| hNa v 1.1 | 5N | HEK293T | ↓ (INaP amplitude ↓, inact ←) | [61] |
| hNa v 1.3 | 12v1 | Xenopus oocytes | ↓ (inact ←) | [30] |
| hNa v 1.3 | 12v2 | Xenopus oocytes | ↓ (act →) | [30] |
| hNa v 1.3 | 12v3 | Xenopus oocytes | ↑ (inact →) | [30] |
| hNa v 1.3 | 12v4 | Xenopus oocytes | ↑ (act ←) | [30] |
| BgNa v | B | Xenopus oocytes | ↑ (INaT amplitude ↑) | [28] |
| BgNa v | G1 | Xenopus oocytes | ? (INaT amplitude ↓, act ←, inact →) | [20] |
| BgNa v | G2 | Xenopus oocytes | ? (INaT amplitude ↑, act →, inact ←) | [20] |
Specific changes observed to channel kinetics are as follows: depolarising (→) or hyperpolarising (←) shifts in activation (act) or inactivation (inact) and/or increased (↑) or decreased (↓) transient (INaT) or persistent sodium current (INaP) amplitude
Both embryonic and adult Drosophila CNS expresses a wide diversity of DmNa v splice forms. However, the profile of splicing differs between these two stages. This is indicative that different Nav properties are required at each stage and that these differences are achieved through splicing. Differences of spliced exons expressed in these two stages include a greater usage of exon J (89 %) but not of exon F (10 %) in adults and vice versa in embryos (10 % exon J and 78 % exon F) [14, 15]. However, the physiological significance of these differences is not clear. It is interesting to note that DmNa v transcripts which lack a majority of common cytoplasmically located spliced exons result in channels with shifted activation and inactivation kinetics towards hyperpolarised and depolarised voltages, respectively, and which also exhibit a much larger INaP. These properties are predicted to make neurons highly excitable [15]. Similarly, analysis of splicing of Na v in other insects shows that it is important for functional properties of the expressed channel. For example, exclusion of optional exon B (located at the linker between the domains I and II, but not equivalent to exon B in Drosophila) in cockroach sodium channels (BgNa v) potentiates the amplitude of the fast-activating and inactivating INa transient current (INaT), which is likely to increase cell excitability (Table 1) [28]. Indeed, an emerging theme is that splicing in of optional exons primarily reduces channel activity and hence, membrane excitability, in order to suit the requirements of neural signalling.
Splicing in intracellular coding regions of mammalian Navs can also result in changes to channel activity. For example, the human Na v 1.3 (SCN3A) alternative spliced exon 12, which encodes an intracellular loop between domains I and II, results in the generation of multiple isoforms. By using multiple splice donor sites in exon 12, four different variants are produced: 12v1, 12v2, 12v3, and 12v4. The variant 12v4, when compared to 12v2, seemingly increases membrane excitability by shifting activation kinetics of the expressed INa. By contrast, inactivation kinetics showed a shift toward hyperpolarising potentials for 12v1 over 12v3, indicative that expressing 12v1 might be expected to decrease membrane excitability (Table 1) [29, 30]. Taken together, Nav gating properties can be determined by the inclusion of exons to alter membrane excitability. However, details of how inclusion of specific spliced exons change gating of the affected channels remains to be determined. A possible mechanism for altering channel kinetics is the phosphorylation state of the channel [30, 31]. Analysis of the amino acid sequence of human Nav1.3 splice variants revealed the presence of two additional phosphorylation sites (protein kinase C on Ser631⁄632 and casein kinase II on Ser646) in 12v3 and 12v4 that are absent from other variants [30]. Changing membrane excitability through phosphorylation in the I-II linker of Nav may influence current amplitude without significantly affecting gating properties [11, 32–36].
Persistent Na current and membrane excitability
The persistent Na current (INaP) has been identified to play critical roles in regulating membrane excitability [37]. Moreover, numerous point mutations in human Na vs, identified in patients with epilepsy, potentiate this component of the voltage-gated Na current (INa) [26]. Interestingly, INaP is also a primary target of some clinically used antiepileptic drugs, including phenytoin, valproic acid, and lamotrigine. [38–40]. It is significant, therefore, that the magnitude of this current can be altered through alternative splicing. However, our understanding of the molecular machinery that regulates splicing of Na vs is poor. This is unfortunate because a fuller understanding may offer new leads for antiepileptic drug design.
In early behavioural screens of Drosophila, different single-gene mutations were identified that induce a seizure-like phenotype when flies are exposed to strong sensory stimuli. Following a mechanical shock, such as vortexing or harsh-tapping of the culture vial, bang-sensitive (bs) mutant flies exhibit a stereotyped sequence of seizure-like spasms, followed by a period of paralysis, and then a second recovery seizure-like phase that precedes a more complete recovery (Fig. 2) [41, 42]. Despite the evolutionary distance, the resemblance in epileptiform activity between fly and humans and the response to clinical antiepileptic drugs make bs mutants an accepted model for studying epilepsy [42–49]. One such bs mutant—slamdance (sda)—which has a deficiency of aminopeptidase N, exhibits increased seizure-like activity in response to electrical stimulation in the larval stage. Detailed electrophysiology shows that INaP is significantly increased in central motoneurons in this mutant [48]. A molecular analysis reveals that splicing of DmNa v is similarly altered in the sda mutation to favour inclusion of exon L at the expense of exon K [25]. As previously described (see above), inclusion of exon L results in channels that, when expressed in Xenopus oocytes, exhibit a larger INaP [15]. Seizure-like behaviour, in response to electric shock, along with the increased INaP and increased inclusion of L isoform are all reversed by feeding larvae with antiepileptic drugs (AEDs) including phenytoin (Phy) and gabapentin (Gbp) [25, 48]. Thus, a better understanding of how INaP is regulated, particularly through splicing, may be beneficial for epilepsy therapy.
Fig. 2.

Drosophila bang-sensitive mutant behaviour. Brief vortexing (~10 s) of the culture vial, containing bang-sensitive mutant flies, induces a stereotyped sequence of seizure-like spasms, followed by a period of paralysis, and then a recovery seizure-like phase that precedes a normal but refractory phase followed ultimately by a complete recovery
Activity-dependent alternative splicing regulation of INaP expression
Seizures can be induced in both mammals and flies through ingestion of proconvulsants such as picrotoxin (PTx) [50, 46]. PTx elicits seizure through antagonism of the GABAA receptor-suppressing synaptic inhibition [51, 52]. Remarkably, we showed that enhancement of synaptic activity in wild-type larvae, through ingestion of PTx, is sufficient to increase inclusion of exon L in DmNa v, increasing INaP as a consequence and inducing a bang-sensitive phenotype. Conversely, seizure activity can be rescued via enhancing synaptic inhibition in sda through ingestion of GABA [25]. Both manipulations suggest that the ‘decision’ to splice either exon L or K is dictated by neuronal activity: i.e. activity-dependent. Increasing synaptic excitatory input results in greater inclusion of exon L, which in turn increases INaP and membrane excitability [25]. Increased excitability would be predicted to further increase inclusion of exon L up to a maximum of 100 % (which is observed in sda and other bs mutants). Such a self-reinforcing cycle provides a plausible, although untested, explanation of the clinical observation in which untreated seizures beget seizures, i.e. paroxysmal activity has the potential to promote susceptibility to further seizures [53, 54].
Splicing of Na v transcripts in response to activity provides an important mechanism for inducing changes in excitability. Because of the complexity of the mammalian CNS, with its larger number of expressed Navs, the extent of splicing and its functional consequences are not well understood. However, the high degree of homology between DmNav and its mammalian counterparts allows us to use the former to guide future studies in mammals. Drosophila exon K/L (located in homology domain III S3-4) is conserved in the homologous domain III from insect to mammal [20, 18, 23], although the outcome of splicing differs. Splicing at this location in cockroach produces three mutually exclusive transcripts that contain spliced exons G1, G2, or G3. G3 contains a stop codon and generates a nonfunctional channel, whereas G1 and G2 result in channels that differ in peak INaT amplitude, gating properties (Table 1) and sensitivity to deltamethrin, a pyrethroid insecticide [20]. In mammals, this same region is also spliced in Na v 1.1 and Na v 1.6—resulting in the inclusion of exons 18A or 18N [21–23]. Exon 18A predominates in adult brain and 18N in embryo and nonneuronal tissues. Similar to the cockroach exon G3, mammalian exon 18N contains a stop codon and generates a truncated channel. These truncated Nav channels that contain only the first two domains, express mainly in nonneuronal tissues, and are hypothesised to be a ‘fail-safe’ mechanism to prevent the expression of functional Navs in nonexcitable cells [55, 23, 21, 22].
A second splicing event in mammalian Navs is noteworthy because it occurs at the equivalent S3-4 region of homologous domain I. Similar to DmNa v, splicing at exon 5 in Na v 1.1 is mutually exclusive with the choice of either exons 5A or 5N (again for adult and neonatal). Alternative splicing in this region is also observed in Na v 1.2, 1.3, 1.6 and Na v 1.7 in both human and mouse [56–60]. In human Nav1.1, three amino acids differ between exon 5A and 5N; however, the channels exhibit distinct gating properties. Heterologous expression of human Nav1.1-5N, in HEK293T cells, produces channels which exhibit more rapid inactivation and reduced INaP compared to Nav1.1-5A. Whilst much needs to be learnt about this splice event, these results suggest that splicing at this location is sufficient to confer changes in neuronal excitability (Table 1) [61]. Intriguingly, inclusion of neonatal exon 6N is increased in both Na v 1.2 and Na v 1.3 following electrical or kainate-induced seizure in adult rat hippocampus [62, 63], perhaps indicative that splicing may similarly be activity-regulated in mammals, as it is in the fly.
Pasilla/Nova, critical factors involved in activity-dependent alternative splicing
A screen of RNA-binding proteins in Drosophila first identified Pasilla (Ps) to be sufficient to regulate splicing of mutually exclusive exons K and L in DmNa v [64]. The inclusion of exon K is significantly increased to 50 % in a ps loss-of-function mutant indicating that the presence of Ps is necessary for the inclusion of exon L [15]. Loss of one copy of ps is also sufficient to rescue the bs-associated seizure behaviour of sda mutants and, moreover, to also prevent PTx-induced seizure in WT background (Fig. 3) [25]. These data suggest that Ps is required for the underlying activity-dependent splicing mechanism. Ps, which contains a K-homology (KH) RNA-binding domain [65, 66], encodes the Drosophila homologue of the human neuro-oncological ventral antigen 1 and 2 (Nova-1 and Nova-2, respectively) proteins [67, 68]. Nova-1 and Nova-2 are expressed to high levels in brain, however, in largely nonoverlapping patterns [69–71]. By recognising YCAY-motifs, located either in introns or 3’UTRs of target transcripts, Nova1/2 regulate neuronal alternative splicing and also mediate transportation of some target transcripts between the nucleus and cytoplasm [68, 72, 73]. Splicing in at least 17 ion channel genes, including Na v s, is predicated to be regulated by Nova-2 [74, 75]. Significantly, overexpression of Nova-2 in HEK293 cells results in an increase in the Na v 1.1-5N splice variant [75]. In support of this, Nova-2 and Na v 1.1-5N transcript abundance are upregulated in temporal neocortical tissue of mesial temporal lobe epilepsy patients. [75]. The relationship between Nova expression and epilepsy has been further examined by EEG recordings in Nova-2 +/− heterozygous mice (Nova-2 −/− mice die within 2–3 weeks of birth). Perturbing Nova steady-state levels in Nova-2 +/− heterozygous mice gives rise to cortical hyperexcitability and also to spontaneous generalised seizure discharge [73]. Moreover, Nova localization shifts from primarily nuclear to cytoplasmic within 2–4 h after pilocarpine-induced seizure [73]. Taken together, these findings strongly implicate perturbation of Nova-2 function contributes to epileptogenesis. The corollary would be that manipulation of Nova activity might be antiepileptic. The conservation of function between Ps and Nova offers the exciting opportunity to utilise Drosophila to rapidly identify molecules that might influence Nova function.
Fig. 3.

Pasilla is required for activity-dependent inclusion of exon L of DmNa v. Prolonged mean recovery time to electroshock of third instar larvae (i.e. increased severity of seizure) is observed in both slamdance (sda) mutants and picrotoxin (PTx)-fed WT flies. Analysis of splicing of DmNa v in whole CNS of such larvae shows that inclusion of exon L increased to ~100 %. In sda, loss of one copy of pasilla (sda +/−, ps +/−) is sufficient to decrease the inclusion of exon L and to rescue seizure-like behaviour. Similarly, removal of one copy of ps in WT larvae (ps +/−) diminishes PTx-induced seizure, as well as inclusion of exon L. Data are taken from [25]
Sodium channel expression and homeostasis
Control of neuron excitability is known to be achieved at the genomic level through transcriptional regulation of Na v channel genes [1, 3, 76, 6]. In Drosophila CNS, the regulation of the voltage-gated sodium current (INa) can be achieved through activity-dependent alteration of DmNa v mRNA level [77, 4, 5, 78]. Removal of excitatory synaptic inputs to motoneurons, achieved by expressing tetanus toxin light chain in all central neurons, significantly increased DmNa v transcript abundance and also the magnitude of INa in motoneurons. On the other hand, enhanced excitatory synaptic release, achieved by increasing cAMP level in the CNS, decreased both mRNA level and INa [77, 5]. This homeostatic mechanism is ideally suited to allow membrane excitability to track the degree of synaptic excitation to which a neuron is exposed (i.e. neuronal homeostasis).
Mammalian neurons (e.g. rat) exhibit the same type of activity-dependent homeostasis of membrane excitability [3]. Deprivation of synaptic excitation in cortical neuron cultures, achieved by chronically blocking glutamatergic signalling, resulted in increased Na v 1.6 mRNA expression, INa, and membrane excitability [79]. The underling mechanism of this homeostatic regulation, in both flies and mammals, requires the protein Pumilio (Pum) [79, 80, 5, 78]. Pumilio is a member of the Pum and FBF (PuF) RNA-binding protein family [81, 82] and is evolutionarily conserved in many species including yeast (Saccharomyces cerevisiae), C. elegans, Drosophila, Anopheles, zebrafish, Xenopus, mouse, and human [82, 83].
In the fly CNS, activity-dependent increase in Pum level results in the translational repression of DmNa v transcripts, reducing INa and membrane excitability [5, 78]. This mechanism is dynamic, such that decreasing levels of synaptic excitation results in decreased Pum level, increased DmNa v transcript abundance, and potentiation of membrane excitability. In rat cortical neurons, the level of Pum was similarly observed to be activity-dependent, mirroring the mechanism observed in the fly [79, 84]. Pum is able to repress translation through binding a specific motif—termed Nanos response element (NRE) [85]—present in both DmNa v and rat Na v 1.6 transcripts [79, 78]. Once Pum is bound to a transcript, cofactors Nanos [86] and brain tumour [86] are recruited to form a quaternary RNA-protein complex that causes transcript deadenylation [87] and consequently repression of translation. An 8-nucleotide core motif UGUA(A/U/C)AUA [88] of the NRE is sufficient for the binding of Pum to DmNa v transcripts [78], and this motif exists in about 10 % of all Drosophila transcripts [88]. Notably, those 10 % of transcripts were only interrogated for NREs present in the 3’UTR region; however, Pum binds to the NRE located in the 3’ end of the open reading frame (ORF) in both DmNa v and rat Na v 1.6 [79, 78]. Therefore, there might be many more Pum targets yet to be identified.
In a genome-wide screening of transcripts associated with the RNA-binding region of Pum, more than 1,000 distinct mRNAs were identified [88]. This suggests that Pum is broadly involved in posttranscriptional regulation of many genes. Indeed, in addition to regulating translation of Na vs, Pum has also been implicated to regulate dendritogenesis [89, 90], expression of glutamate receptors [91], and aspects of memory and learning in higher brain centres [92]. Behaviour training of long-term memory (LTM) produced by spaced training (ten training sessions with a 15-min rest interval between each session), compared to anaesthesia-resistant memory (ARM) produced by massed training (ten training sessions without rest intervals), resulted in pum mRNA upregulation. Pum mutant flies also showed defects in LTM formation. [92]. Pum regulates NMJ morphology via negative regulation of the translational factor eIF-4E expression by directly binding to an NRE in the 3’UTR of the eIF-4E transcript [90]. Pum loss-of-function mutants show enhanced expression of eIF-4E and upregulated GluRIIA expression and increased frequency of spontaneous neurotransmitter release [91]. Thus, Pum is seemingly central to many aspects of CNS function, not least of which is homeostatic control of neuronal excitability. In this regard, it is significant that in mouse, Pum2 deficiency leads to spontaneous EEG abnormalities and lower seizure thresholds to the proconvulsant pentylenetetrazole [93]. Similar to Pum, the Na v 1.6 transcript is upregulated in CELF4 (CUGBP, ELAV-like family member 4) deficient mice [94]. CELF4 is similarly a brain-specific neuronal RNA-binding protein and binds to the 3’UTR of Na v 1.6. Because mammalian Nav1.6 is the primary determinant of action potential initiation and main contributor of INaP in excitatory neurons, upregulated Na v 1.6 mRNA results in increasing neuronal excitability [95]. Consequently, CELF4 deficient mice exhibit both convulsive and nonconvulsive (absence-like) seizures and also have a lower seizure threshold [94, 96]. These findings demonstrate that understanding the regulation of INa or INaP via RNA-binding proteins is a potentially important approach for epilepsy therapy.
Summary and outlook
Voltage-gated sodium channels are important determinants for controlling membrane excitability. Regulation of Nav activity is achieved, at least in part, by coordination of RNA alternative splicing and translational repression of Na v transcripts (Fig. 4). When one considers additional mechanisms of regulation of Nav channel activity, including RNA editing [28], phosphorylation [33, 32, 11, 4], trafficking [97, 98], and degradation [98–100], it becomes clear that these channels are subject to both considerable and diverse regulation consistent with the high level of channel diversity observable in the multitude of neuron types in the human brain. The utilisation of model systems, including Drosophila, offers the significant opportunity to rapidly progress understanding in these and related areas.
Fig. 4.
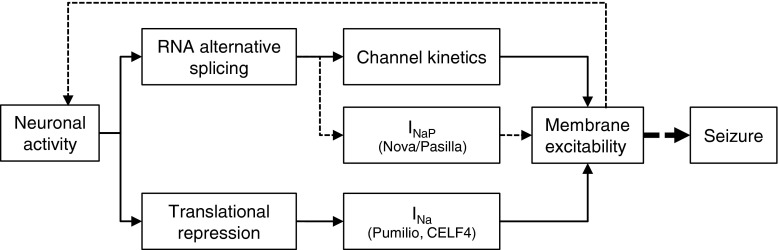
Membrane excitability is regulated by activity-dependent RNA alternative splicing and translational repression of voltage-gated sodium channel transcripts. Control of membrane excitability through Nav activity is achieved by regulation of channel kinetics, current density (INa), and magnitude of persistent Na current (INaP). RNA alternative splicing results in splice variants which exhibit different channel gating properties including activation and inactivation kinetics and INaP. Splicing is regulated, in part, by Pasilla in Drosophila and in humans by its homologue, Nova. In Drosophila, increased synaptic excitation results in increased INaP, which in turn feeds back to further increase synaptic excitation. This self-reinforcing cycle likely further increases INaP (dashed line) leading to seizure. Current density of Nav can be regulated through a mechanism of translational repression of Na v transcripts via Pumilio and possibly CELF4
A particular area where Drosophila is already making a contribution to understanding epilepsy is through modelling human Na v point mutations. A variety of techniques now exist to allow such mutations to be ‘knocked-in’ to DmNa v. Sun et al. [101] recently reported a Na v 1.1 (K1270T) knock-in that recapitulates a mutation associated with genetic epilepsy with febrile seizures plus (GEFS+). Electrophysiological analysis shows this to be a gain-of-function mutation that results in a hyperpolarizing shift in the deactivation potential for INaP. This approach not only serves to validate the genetic basis of human disease, but also provides a sensitised genetic background for high-throughput, low cost, screens to identify novel compounds that have antiepileptic properties. Identification of novel targets, such as splicing regulators, can also be quickly developed as the basis of screens with the potential advantage of identifying antiepileptic compounds which interact with nontraditional targets. By far the most common targets of currently used AEDs are ion channels and, whilst these offer effective therapeutic targets, there might be much to be gained from identifying additional targets which would facilitate combinatorial therapy. Combinations of AEDs are showing promise for the treatment of intractable epilepsy [102].
Use of Drosophila (and other simple model systems) also offers the prospect of exploring the mechanistic basis of epileptogenesis from understanding how small seizures may lead to larger seizures to providing novel approaches to prevent epilepsy from progressing, even when an epilepsy-associated mutation is present. For example, we recently reported that the presence of phenytoin, during embryogenesis when the CNS first forms neural circuits, prevents the normal seizure phenotype characteristic of the Drosophila sda mutant [48]. The inference from this study is that early intervention may be beneficial in blocking epileptogenesis by preventing activity-dependent feedback mechanisms that we spotlight in this review. The finding of conservation of regulatory mechanisms between insects such as Drosophila and mammals validates the use of simpler model organisms to provide better understanding of Na v regulation in humans with an obvious benefit of novel therapies for epilepsy.
Acknowledgments
We thank Dr. Stephanie Schorge (UCL, UK) for commenting on the manuscript. We also thank members of the Baines group for helpful comments and advice. Research in the Baines lab is supported by both the BBSRC (BB/J005002/1) and MRC (MR/J009180/1).
References
- 1.Turrigiano GG, Nelson SB. Thinking globally, acting locally: AMPA receptor turnover and synaptic strength. Neuron. 1998;21(5):933–935. doi: 10.1016/s0896-6273(00)80607-8. [DOI] [PubMed] [Google Scholar]
- 2.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(1):13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 3.Desai NS, Rutherford LC, Turrigiano GG. Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nature neuroscience. 1999;2(6):515–520. doi: 10.1038/9165. [DOI] [PubMed] [Google Scholar]
- 4.Baines RA. Postsynaptic protein kinase A reduces neuronal excitability in response to increased synaptic excitation in the Drosophila CNS. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23(25):8664–8672. doi: 10.1523/JNEUROSCI.23-25-08664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mee CJ, Pym EC, Moffat KG, Baines RA. Regulation of neuronal excitability through pumilio-dependent control of a sodium channel gene. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24(40):8695–8703. doi: 10.1523/JNEUROSCI.2282-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nature reviews Neuroscience. 2006;7(7):563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- 7.Schulz DJ, Baines RA, Hempel CM, Li L, Liss B, Misonou H. Cellular excitability and the regulation of functional neuronal identity: from gene expression to neuromodulation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26(41):10362–10367. doi: 10.1523/JNEUROSCI.3194-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldin AL. Resurgence of sodium channel research. Annual review of physiology. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 9.Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nature reviews Neuroscience. 2007;8(11):819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- 10.Dong K. Insect sodium channels and insecticide resistance. Invertebrate neuroscience : IN. 2007;7(1):17–30. doi: 10.1007/s10158-006-0036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith RD, Goldin AL. Phosphorylation of brain sodium channels in the I–II linker modulates channel function in Xenopus oocytes. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16(6):1965–1974. doi: 10.1523/JNEUROSCI.16-06-01965.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith RD, Goldin AL. Potentiation of rat brain sodium channel currents by PKA in Xenopus oocytes involves the I-II linker. American journal of physiology Cell physiology. 2000;278(4):C638–C645. doi: 10.1152/ajpcell.2000.278.4.C638. [DOI] [PubMed] [Google Scholar]
- 13.Feng G, Deak P, Chopra M, Hall LM. Cloning and functional analysis of TipE, a novel membrane protein that enhances Drosophila para sodium channel function. Cell. 1995;82(6):1001–1011. doi: 10.1016/0092-8674(95)90279-1. [DOI] [PubMed] [Google Scholar]
- 14.Olson RO, Liu Z, Nomura Y, Song W, Dong K. Molecular and functional characterization of voltage-gated sodium channel variants from Drosophila melanogaster. Insect biochemistry and molecular biology. 2008;38(5):604–610. doi: 10.1016/j.ibmb.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin WH, Wright DE, Muraro NI, Baines RA. Alternative splicing in the voltage-gated sodium channel DmNav regulates activation, inactivation, and persistent current. Journal of neurophysiology. 2009;102(3):1994–2006. doi: 10.1152/jn.00613.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature genetics. 2008;40(12):1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 17.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies TG, Field LM, Usherwood PN, Williamson MS. A comparative study of voltage-gated sodium channels in the Insecta: implications for pyrethroid resistance in Anopheline and other Neopteran species. Insect molecular biology. 2007;16(3):361–375. doi: 10.1111/j.1365-2583.2007.00733.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee SH, Ingles PJ, Knipple DC, Soderlund DM. Developmental regulation of alternative exon usage in the house fly Vssc1 sodium channel gene. Invertebrate neuroscience : IN. 2002;4(3):125–133. doi: 10.1007/s10158-001-0014-1. [DOI] [PubMed] [Google Scholar]
- 20.Tan J, Liu Z, Nomura Y, Goldin AL, Dong K. Alternative splicing of an insect sodium channel gene generates pharmacologically distinct sodium channels. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22(13):5300–5309. doi: 10.1523/JNEUROSCI.22-13-05300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diss JK, Archer SN, Hirano J, Fraser SP, Djamgoz MB. Expression profiles of voltage-gated Na(+) channel alpha-subunit genes in rat and human prostate cancer cell lines. Prostate. 2001;48(3):165–178. doi: 10.1002/pros.1095. [DOI] [PubMed] [Google Scholar]
- 22.Oh Y, Waxman SG. Novel splice variants of the voltage-sensitive sodium channel alpha subunit. Neuroreport. 1998;9(7):1267–1272. doi: 10.1097/00001756-199805110-00002. [DOI] [PubMed] [Google Scholar]
- 23.Plummer NW, McBurney MW, Meisler MH. Alternative splicing of the sodium channel SCN8A predicts a truncated two-domain protein in fetal brain and non-neuronal cells. The Journal of biological chemistry. 1997;272(38):24008–24015. doi: 10.1074/jbc.272.38.24008. [DOI] [PubMed] [Google Scholar]
- 24.Kiss T. Persistent Na-channels: origin and function. A review. Acta biologica Hungarica. 2008;59(Suppl):1–12. doi: 10.1556/ABiol.59.2008.Suppl.1. [DOI] [PubMed] [Google Scholar]
- 25.Lin WH, Gunay C, Marley R, Prinz AA, Baines RA. Activity-dependent alternative splicing increases persistent sodium current and promotes seizure. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32(21):7267–7277. doi: 10.1523/JNEUROSCI.6042-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stafstrom CE. Persistent sodium current and its role in epilepsy. Epilepsy currents / American Epilepsy Society. 2007;7(1):15–22. doi: 10.1111/j.1535-7511.2007.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia. 2010;51(9):1650–1658. doi: 10.1111/j.1528-1167.2010.02640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song W, Liu Z, Tan J, Nomura Y, Dong K. RNA editing generates tissue-specific sodium channels with distinct gating properties. The Journal of biological chemistry. 2004;279(31):32554–32561. doi: 10.1074/jbc.M402392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dietrich PS, McGivern JG, Delgado SG, Koch BD, Eglen RM, Hunter JC, Sangameswaran L. Functional analysis of a voltage-gated sodium channel and its splice variant from rat dorsal root ganglia. Journal of neurochemistry. 1998;70(6):2262–2272. doi: 10.1046/j.1471-4159.1998.70062262.x. [DOI] [PubMed] [Google Scholar]
- 30.Thimmapaya R, Neelands T, Niforatos W, Davis-Taber RA, Choi W, Putman CB, Kroeger PE, Packer J, Gopalakrishnan M, Faltynek CR, Surowy CS, Scott VE. Distribution and functional characterization of human Nav1.3 splice variants. The European journal of neuroscience. 2005;22(1):1–9. doi: 10.1111/j.1460-9568.2005.04155.x. [DOI] [PubMed] [Google Scholar]
- 31.Chatelier A, Dahllund L, Eriksson A, Krupp J, Chahine M. Biophysical properties of human Na v1.7 splice variants and their regulation by protein kinase A. Journal of neurophysiology. 2008;99(5):2241–2250. doi: 10.1152/jn.01350.2007. [DOI] [PubMed] [Google Scholar]
- 32.Gershon E, Weigl L, Lotan I, Schreibmayer W, Dascal N. Protein kinase A reduces voltage-dependent Na + current in Xenopus oocytes. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1992;12(10):3743–3752. doi: 10.1523/JNEUROSCI.12-10-03743.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M, West JW, Lai Y, Scheuer T, Catterall WA. Functional modulation of brain sodium channels by cAMP-dependent phosphorylation. Neuron. 1992;8(6):1151–1159. doi: 10.1016/0896-6273(92)90135-z. [DOI] [PubMed] [Google Scholar]
- 34.Murphy BJ, Rossie S, De Jongh KS, Catterall WA. Identification of the sites of selective phosphorylation and dephosphorylation of the rat brain Na + channel alpha subunit by cAMP-dependent protein kinase and phosphoprotein phosphatases. The Journal of biological chemistry. 1993;268(36):27355–27362. [PubMed] [Google Scholar]
- 35.Smith RD, Goldin AL. Phosphorylation at a single site in the rat brain sodium channel is necessary and sufficient for current reduction by protein kinase A. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17(16):6086–6093. doi: 10.1523/JNEUROSCI.17-16-06086.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cantrell AR, Smith RD, Goldin AL, Scheuer T, Catterall WA. Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel alpha subunit. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17(19):7330–7338. doi: 10.1523/JNEUROSCI.17-19-07330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yue C, Remy S, Su H, Beck H, Yaari Y. Proximal persistent Na + channels drive spike afterdepolarizations and associated bursting in adult CA1 pyramidal cells. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25(42):9704–9720. doi: 10.1523/JNEUROSCI.1621-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chao TI, Alzheimer C. Effects of phenytoin on the persistent Na + current of mammalian CNS neurones. Neuroreport. 1995;6(13):1778–1780. doi: 10.1097/00001756-199509000-00017. [DOI] [PubMed] [Google Scholar]
- 39.Taverna S, Mantegazza M, Franceschetti S, Avanzini G. Valproate selectively reduces the persistent fraction of Na + current in neocortical neurons. Epilepsy research. 1998;32(1–2):304–308. doi: 10.1016/s0920-1211(98)00060-6. [DOI] [PubMed] [Google Scholar]
- 40.Spadoni F, Hainsworth AH, Mercuri NB, Caputi L, Martella G, Lavaroni F, Bernardi G, Stefani A. Lamotrigine derivatives and riluzole inhibit INa, P in cortical neurons. Neuroreport. 2002;13(9):1167–1170. doi: 10.1097/00001756-200207020-00019. [DOI] [PubMed] [Google Scholar]
- 41.Ganetzky B, Wu CF. Drosophila mutants with opposing effects on nerve excitability: genetic and spatial interactions in repetitive firing. Journal of neurophysiology. 1982;47(3):501–514. doi: 10.1152/jn.1982.47.3.501. [DOI] [PubMed] [Google Scholar]
- 42.Lee J, Wu CF. Electroconvulsive seizure behavior in Drosophila: analysis of the physiological repertoire underlying a stereotyped action pattern in bang-sensitive mutants. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22(24):11065–11079. doi: 10.1523/JNEUROSCI.22-24-11065.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuebler D, Tanouye M. Anticonvulsant valproate reduces seizure-susceptibility in mutant Drosophila. Brain research. 2002;958(1):36–42. doi: 10.1016/s0006-8993(02)03431-5. [DOI] [PubMed] [Google Scholar]
- 44.Reynolds ER, Stauffer EA, Feeney L, Rojahn E, Jacobs B, McKeever C. Treatment with the antiepileptic drugs phenytoin and gabapentin ameliorates seizure and paralysis of Drosophila bang-sensitive mutants. Journal of neurobiology. 2004;58(4):503–513. doi: 10.1002/neu.10297. [DOI] [PubMed] [Google Scholar]
- 45.Tan JS, Lin F, Tanouye MA. Potassium bromide, an anticonvulsant, is effective at alleviating seizures in the Drosophila bang-sensitive mutant bang senseless. Brain research. 2004;1020(1–2):45–52. doi: 10.1016/j.brainres.2004.05.111. [DOI] [PubMed] [Google Scholar]
- 46.Stilwell GE, Saraswati S, Littleton JT, Chouinard SW. Development of a Drosophila seizure model for in vivo high-throughput drug screening. The European journal of neuroscience. 2006;24(8):2211–2222. doi: 10.1111/j.1460-9568.2006.05075.x. [DOI] [PubMed] [Google Scholar]
- 47.Song J, Tanouye MA. From bench to drug: human seizure modeling using Drosophila. Progress in neurobiology. 2008;84(2):182–191. doi: 10.1016/j.pneurobio.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marley R, Baines RA. Increased persistent Na + current contributes to seizure in the slamdance bang-sensitive Drosophila mutant. Journal of neurophysiology. 2011;106(1):18–29. doi: 10.1152/jn.00808.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parker L, Padilla M, Du Y, Dong K, Tanouye MA. Drosophila as a model for epilepsy: bss is a gain-of-function mutation in the para sodium channel gene that leads to seizures. Genetics. 2011;187(2):523–534. doi: 10.1534/genetics.110.123299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.White HS. Preclinical development of antiepileptic drugs: past, present, and future directions. Epilepsia. 2003;44(Suppl 7):2–8. doi: 10.1046/j.1528-1157.44.s7.10.x. [DOI] [PubMed] [Google Scholar]
- 51.Constanti A. The “mixed” effect of picrotoxin on the GABA dose/conductance relation recorded from lobster muscle. Neuropharmacology. 1978;17(3):159–167. doi: 10.1016/0028-3908(78)90095-3. [DOI] [PubMed] [Google Scholar]
- 52.Jones-Davis DM, Macdonald RL. GABA(A) receptor function and pharmacology in epilepsy and status epilepticus. Current opinion in pharmacology. 2003;3(1):12–18. doi: 10.1016/s1471-4892(02)00015-2. [DOI] [PubMed] [Google Scholar]
- 53.Reynolds EH. Early treatment and prognosis of epilepsy. Epilepsia. 1987;28(2):97–106. doi: 10.1111/j.1528-1157.1987.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 54.Chadwick DW. The treatment of the first seizure: the benefits. Epilepsia. 2008;49(Suppl 1):26–28. doi: 10.1111/j.1528-1167.2008.01446.x. [DOI] [PubMed] [Google Scholar]
- 55.Plummer NW, Galt J, Jones JM, Burgess DL, Sprunger LK, Kohrman DC, Meisler MH. Exon organization, coding sequence, physical mapping, and polymorphic intragenic markers for the human neuronal sodium channel gene SCN8A. Genomics. 1998;54(2):287–296. doi: 10.1006/geno.1998.5550. [DOI] [PubMed] [Google Scholar]
- 56.Sarao R, Gupta SK, Auld VJ, Dunn RJ. Developmentally regulated alternative RNA splicing of rat brain sodium channel mRNAs. Nucleic acids research. 1991;19(20):5673–5679. doi: 10.1093/nar/19.20.5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yarowsky PJ, Krueger BK, Olson CE, Clevinger EC, Koos RD. Brain and heart sodium channel subtype mRNA expression in rat cerebral cortex. Proc Natl Acad Sci U S A. 1991;88(21):9453–9457. doi: 10.1073/pnas.88.21.9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gustafson TA, Clevinger EC, O’Neill TJ, Yarowsky PJ, Krueger BK. Mutually exclusive exon splicing of type III brain sodium channel alpha subunit RNA generates developmentally regulated isoforms in rat brain. The Journal of biological chemistry. 1993;268(25):18648–18653. [PubMed] [Google Scholar]
- 59.Raymond CK, Castle J, Garrett-Engele P, Armour CD, Kan Z, Tsinoremas N, Johnson JM. Expression of alternatively spliced sodium channel alpha-subunit genes. Unique splicing patterns are observed in dorsal root ganglia. The Journal of biological chemistry. 2004;279(44):46234–46241. doi: 10.1074/jbc.M406387200. [DOI] [PubMed] [Google Scholar]
- 60.Kasai N, Fukushima K, Ueki Y, Prasad S, Nosakowski J, Sugata K, Sugata A, Nishizaki K, Meyer NC, Smith RJ. Genomic structures of SCN2A and SCN3A—candidate genes for deafness at the DFNA16 locus. Gene. 2001;264(1):113–122. doi: 10.1016/s0378-1119(00)00594-1. [DOI] [PubMed] [Google Scholar]
- 61.Fletcher EV, Kullmann DM, Schorge S. Alternative splicing modulates inactivation of type 1 voltage-gated sodium channels by toggling an amino acid in the first S3-S4 linker. The Journal of biological chemistry. 2011;286(42):36700–36708. doi: 10.1074/jbc.M111.250225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aronica E, Yankaya B, Troost D, van Vliet EA, Lopes da Silva FH, Gorter JA. Induction of neonatal sodium channel II and III alpha-isoform mRNAs in neurons and microglia after status epilepticus in the rat hippocampus. The European journal of neuroscience. 2001;13(6):1261–1266. doi: 10.1046/j.0953-816x.2001.01502.x. [DOI] [PubMed] [Google Scholar]
- 63.Gastaldi M, Bartolomei F, Massacrier A, Planells R, Robaglia-Schlupp A, Cau P. Increase in mRNAs encoding neonatal II and III sodium channel alpha-isoforms during kainate-induced seizures in adult rat hippocampus. Brain research Molecular brain research. 1997;44(2):179–190. doi: 10.1016/s0169-328x(96)00199-4. [DOI] [PubMed] [Google Scholar]
- 64.Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci U S A. 2004;101(45):15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Siomi H, Matunis MJ, Michael WM, Dreyfuss G. The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic acids research. 1993;21(5):1193–1198. doi: 10.1093/nar/21.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grishin NV. KH domain: one motif, two folds. Nucleic acids research. 2001;29(3):638–643. doi: 10.1093/nar/29.3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302(5648):1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 68.Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444(7119):580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 69.Buckanovich RJ, Posner JB, Darnell RB. Nova, the paraneoplastic Ri antigen, is homologous to an RNA-binding protein and is specifically expressed in the developing motor system. Neuron. 1993;11(4):657–672. doi: 10.1016/0896-6273(93)90077-5. [DOI] [PubMed] [Google Scholar]
- 70.Yang YY, Yin GL, Darnell RB. The neuronal RNA-binding protein Nova-2 is implicated as the autoantigen targeted in POMA patients with dementia. Proc Natl Acad Sci U S A. 1998;95(22):13254–13259. doi: 10.1073/pnas.95.22.13254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seshaiah P, Miller B, Myat MM, Andrew DJ. pasilla, the Drosophila homologue of the human Nova-1 and Nova-2 proteins, is required for normal secretion in the salivary gland. Developmental biology. 2001;239(2):309–322. doi: 10.1006/dbio.2001.0429. [DOI] [PubMed] [Google Scholar]
- 72.Racca C, Gardiol A, Eom T, Ule J, Triller A, Darnell RB. The neuronal splicing factor Nova co-localizes with target RNAs in the dendrite. Frontiers in neural circuits. 2010;4:5. doi: 10.3389/neuro.04.005.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eom T, Zhang C, Wang H, Lay K, Fak J, Noebels JL, Darnell RB. NOVA-dependent regulation of cryptic NMD exons controls synaptic protein levels after seizure. eLife. 2013;2:e00178. doi: 10.7554/eLife.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, Zeeberg BR, Kane D, Weinstein JN, Blume J, Darnell RB. Nova regulates brain-specific splicing to shape the synapse. Nature genetics. 2005;37(8):844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 75.Heinzen EL, Yoon W, Tate SK, Sen A, Wood NW, Sisodiya SM, Goldstein DB. Nova2 interacts with a cis-acting polymorphism to influence the proportions of drug-responsive splice variants of SCN1A. American journal of human genetics. 2007;80(5):876–883. doi: 10.1086/516650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nature reviews Neuroscience. 2003;4(11):885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
- 77.Baines RA, Uhler JP, Thompson A, Sweeney ST, Bate M. Altered electrical properties in Drosophila neurons developing without synaptic transmission. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21(5):1523–1531. doi: 10.1523/JNEUROSCI.21-05-01523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muraro NI, Weston AJ, Gerber AP, Luschnig S, Moffat KG, Baines RA. Pumilio binds para mRNA and requires Nanos and Brat to regulate sodium current in Drosophila motoneurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28(9):2099–2109. doi: 10.1523/JNEUROSCI.5092-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Driscoll HE, Muraro NI, He M, Baines RA. Pumilio-2 regulates translation of Nav1.6 to mediate homeostasis of membrane excitability. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33(23):9644–9654. doi: 10.1523/JNEUROSCI.0921-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schweers BA, Walters KJ, Stern M. The Drosophila melanogaster translational repressor pumilio regulates neuronal excitability. Genetics. 2002;161(3):1177–1185. doi: 10.1093/genetics/161.3.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zamore PD, Williamson JR, Lehmann R. The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. Rna. 1997;3(12):1421–1433. [PMC free article] [PubMed] [Google Scholar]
- 82.Wickens M, Bernstein DS, Kimble J, Parker R. A PUF family portrait: 3’UTR regulation as a way of life. Trends in genetics : TIG. 2002;18(3):150–157. doi: 10.1016/s0168-9525(01)02616-6. [DOI] [PubMed] [Google Scholar]
- 83.Spassov DS, Jurecic R. The PUF family of RNA-binding proteins: does evolutionarily conserved structure equal conserved function? IUBMB life. 2003;55(7):359–366. doi: 10.1080/15216540310001603093. [DOI] [PubMed] [Google Scholar]
- 84.Vessey JP, Vaccani A, Xie Y, Dahm R, Karra D, Kiebler MA, Macchi P. Dendritic localization of the translational repressor pumilio 2 and its contribution to dendritic stress granules. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26(24):6496–6508. doi: 10.1523/JNEUROSCI.0649-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Murata Y, Wharton RP. Binding of pumilio to maternal hunchback mRNA is required for posterior patterning in Drosophila embryos. Cell. 1995;80(5):747–756. doi: 10.1016/0092-8674(95)90353-4. [DOI] [PubMed] [Google Scholar]
- 86.Sonoda J, Wharton RP. Drosophila brain tumor is a translational repressor. Genes & development. 2001;15(6):762–773. doi: 10.1101/gad.870801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chagnovich D, Lehmann R. Poly(A)-independent regulation of maternal hunchback translation in the Drosophila embryo. Proc Natl Acad Sci U S A. 2001;98(20):11359–11364. doi: 10.1073/pnas.201284398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gerber AP, Luschnig S, Krasnow MA, Brown PO, Herschlag D. Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2006;103(12):4487–4492. doi: 10.1073/pnas.0509260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ye B, Petritsch C, Clark IE, Gavis ER, Jan LY, Jan YN. Nanos and pumilio are essential for dendrite morphogenesis in Drosophila peripheral neurons. Current biology : CB. 2004;14(4):314–321. doi: 10.1016/j.cub.2004.01.052. [DOI] [PubMed] [Google Scholar]
- 90.Menon KP, Sanyal S, Habara Y, Sanchez R, Wharton RP, Ramaswami M, Zinn K. The translational repressor pumilio regulates presynaptic morphology and controls postsynaptic accumulation of translation factor eIF-4E. Neuron. 2004;44(4):663–676. doi: 10.1016/j.neuron.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 91.Menon KP, Andrews S, Murthy M, Gavis ER, Zinn K. The translational repressors Nanos and pumilio have divergent effects on presynaptic terminal growth and postsynaptic glutamate receptor subunit composition. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29(17):5558–5572. doi: 10.1523/JNEUROSCI.0520-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dubnau J, Chiang AS, Grady L, Barditch J, Gossweiler S, McNeil J, Smith P, Buldoc F, Scott R, Certa U, Broger C, Tully T. The staufen/pumilio pathway is involved in Drosophila long-term memory. Current biology : CB. 2003;13(4):286–296. doi: 10.1016/s0960-9822(03)00064-2. [DOI] [PubMed] [Google Scholar]
- 93.Siemen H, Colas D, Heller HC, Brustle O, Pera RA. Pumilio-2 function in the mouse nervous system. PloS one. 2011;6(10):e25932. doi: 10.1371/journal.pone.0025932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sun W, Wagnon JL, Mahaffey CL, Briese M, Ule J, Frankel WN. Aberrant sodium channel activity in the complex seizure disorder of Celf4 mutant mice. The Journal of physiology. 2013;591(Pt 1):241–255. doi: 10.1113/jphysiol.2012.240168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chatelier A, Zhao J, Bois P, Chahine M. Biophysical characterisation of the persistent sodium current of the Nav1.6 neuronal sodium channel: a single-channel analysis. Pflugers Archiv: European journal of physiology. 2010;460(1):77–86. doi: 10.1007/s00424-010-0801-9. [DOI] [PubMed] [Google Scholar]
- 96.Wagnon JL, Mahaffey CL, Sun W, Yang Y, Chao HT, Frankel WN. Etiology of a genetically complex seizure disorder in Celf4 mutant mice. Genes brain and behavior. 2011;10(7):765–777. doi: 10.1111/j.1601-183X.2011.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vacher H, Trimmer JS. Trafficking mechanisms underlying neuronal voltage-gated ion channel localization at the axon initial segment. Epilepsia. 2012;53(Suppl 9):21–31. doi: 10.1111/epi.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shao D, Okuse K, Djamgoz MB. Protein-protein interactions involving voltage-gated sodium channels: post-translational regulation, intracellular trafficking and functional expression. The international journal of biochemistry & cell biology. 2009;41(7):1471–1481. doi: 10.1016/j.biocel.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 99.Ekberg JA, Boase NA, Rychkov G, Manning J, Poronnik P, Kumar S. Nedd4-2 (NEDD4L) controls intracellular Na + −mediated activity of voltage-gated sodium channels in primary cortical neurons. The Biochemical journal. 2014;457(1):27–31. doi: 10.1042/BJ20131275. [DOI] [PubMed] [Google Scholar]
- 100.Laedermann CJ, Cachemaille M, Kirschmann G, Pertin M, Gosselin RD, Chang I, Albesa M, Towne C, Schneider BL, Kellenberger S, Abriel H, Decosterd I. Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. The Journal of clinical investigation. 2013;123(7):3002–3013. doi: 10.1172/JCI68996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sun L, Gilligan J, Staber C, Schutte RJ, Nguyen V, O’Dowd DK, Reenan R. A knock-in model of human epilepsy in Drosophila reveals a novel cellular mechanism associated with heat-induced seizure. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32(41):14145–14155. doi: 10.1523/JNEUROSCI.2932-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brigo F, Ausserer H, Tezzon F, Nardone R. When one plus one makes three: the quest for rational antiepileptic polytherapy with supraadditive anticonvulsant efficacy. Epilepsy & behavior : E&B. 2013;27(3):439–442. doi: 10.1016/j.yebeh.2013.03.010. [DOI] [PubMed] [Google Scholar]