

Abstract
Transcription factor p53 plays a critical role in the cellular response to stress stimuli. We have seen that p53 dissociates selectively from various promoter sites as a result of oxidation at long-range through DNA-mediated charge transport (CT). Here, we examine this chemical oxidation and determine the residues in p53 that are essential for oxidative dissociation, focusing on the network of cysteine residues adjacent to the DNA-binding site. Of the eight mutants studied, only the C275S mutation shows decreased affinity for the Gadd45 promoter site. However, both mutations C275S and C277S result in substantial attenuation of oxidative dissociation, with C275S causing the most severe attenuation. Differential thiol labeling was used to determine the oxidation states of cysteine residues within p53 after DNA-mediated oxidation. Reduced cysteines were iodoacetamide-labeled, whereas oxidized cysteines participating in disulfide bonds were 13C2D2-iodoacetamide-labeled. Intensities of respective iodoacetamide-modified peptide fragments were analyzed by mass spectrometry. A distinct shift in peptide labeling toward 13C2D2-iodoacetamide-labeled cysteines is observed in oxidized samples, confirming that chemical oxidation of p53 occurs at long range. All observable cysteine residues trend toward the heavy label under conditions of DNA CT, indicating the formation of multiple disulfide bonds among the cysteine network. On the basis of these data, it is proposed that disulfide formation involving C275 is critical for inducing oxidative dissociation of p53 from DNA.
Transcription factor p53 is one of the most heavily studied human proteins due to its marked prevalence of mutation in human cancer. Over half of all human cancers display mutations in the p53 gene, with the majority of these mutations localized to the DNA-binding domain.1−3 Although much research has been conducted on this protein and its many roles within the cell, the precise mechanisms by which p53 senses cellular stresses and influences cellular fate are still largely unknown. We have previously shown that DNA-mediated charge transport (CT) can sequence-selectively promote the oxidative dissociation of p53 bound to DNA.4 Here, we examine the mechanisms by which DNA-mediated oxidation is sensed by p53 and how the resulting dissociation from DNA occurs.
A major focus of our laboratory has been the characterization of long-range CT through DNA.5−9 We have found that oxidative damage to DNA can occur from a distance because of the migration of electron holes through the π-stacked bases. Ground-state CT has been observed to occur over 100 base pairs (34 nm) through DNA.10 However, perturbations in the intervening base pair stack, such as abasic sites and base mismatches, severely attenuate DNA CT. In a cellular environment, oxidative damage can occur by reactive oxygen species attacking DNA, and we have found that oxidative DNA damage can also occur from a distance in vivo.5,6,11 The one-electron oxidation potential of guanine is the lowest of the bases (+1.29 V), therefore making it the most readily oxidized base.12−14 Thus, a known hallmark of DNA CT oxidation is the formation of DNA damage products at 5′ guanines of guanine doublets and triplets.15 However, certain amino acid functional groups, such as the thiol group of cysteine, possess lower one-electron oxidation potentials than guanine and could thermodynamically be oxidized in DNA-bound proteins.
The chemistry of thiols located near the DNA base stack was investigated to determine whether thiol redox chemistry could be modulated via DNA CT. Electrochemistry experiments on a graphite surface have shown that a disulfide incorporated into the backbone of an oligonucleotide can be reduced to the corresponding thiol groups by the application of negative potential.16 Additionally, DNA CT induced by a distally bound anthraquinone (AQ) photooxidant is able to promote oxidation of neighboring thiol groups incorporated into the backbone of an oligonucleotide, resulting in the formation of a disulfide bond.17
An intriguing feature of p53, which binds DNA as a tetramer, is that it contains nine highly conserved cysteine residues within the DNA-binding domain.18 These cysteines are purported to play a variety of roles, including tetramer formation, Zn2+ binding, and sequence-specific interaction with the p53 response element (Figure 1). The response element consensus motif is 5′-RRRCWWGYYY-3′, with R representing a purine, Y representing a pyrimidine, and W representing either an adenine or a thymine.18 The p53 consensus sequence contains two response elements that may be separated up to 13 bases, with one monomer of p53 binding to each quarter site (5′-RRRCW-3′). Within each p53 monomer, three cysteine residues (C176, C238, and C242) and one histidine (H179) coordinate a zinc ion that is believed to be structurally necessary for DNA binding.2,19−21 Located close to the Zn2+, but not participating in metal binding, is C182. Closer to the DNA–p53 interface are the remaining conserved residues of interest: C124, C135, C141, C275, and C277. Nestled into the major groove, C277 is capable of forming a hydrogen bond within the purine region of the p53 response element quarter site.20,21 C275 is located 7.0 Å away from C277, from sulfur atom to sulfur atom. Residues C124, C135, and C141 are found as a cluster situated deeper into the core of the DNA-binding domain, with C275 being 7.0 Å away from C135. Chen and co-workers have reported these residues as being reduced in their structural characterizations of the p53 DNA-binding site; however, disulfide formation is plausible based on the proximity of these residues with respect to one another.20,21
Figure 1.
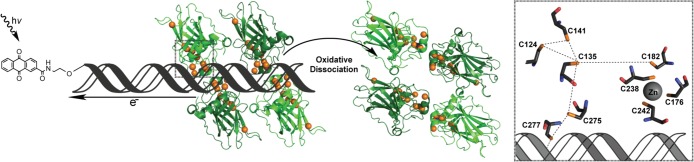
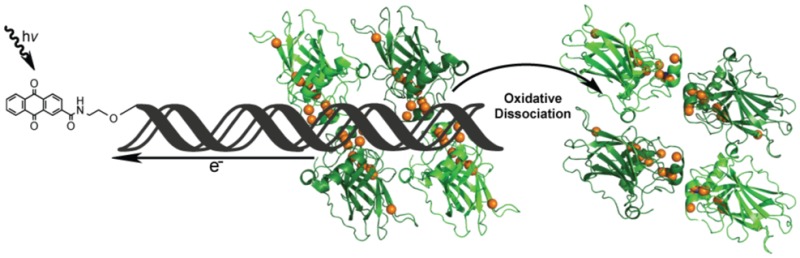
Scheme of p53 oxidation through DNA-mediated charge transport. Oxidation is initiated by AQ excitation, causing it to abstract an electron from DNA. This electron hole equilibrates among the π-stacked bases, ultimately localizing to a low redox potential guanine site. If the trapped electron hole localizes to the DNA–p53 interface, then the bound p53 protein may be oxidized due to amino acids with lower one-electron oxidation potentials than that of guanine. The oxidation of DNA-bound p53 causes the formation of a disulfide bond and leads to the dissociation from DNA. The orange spheres represent the sulfur atoms of each cysteine residue within the p53 DNA-binding domain, making them candidates for oxidation via DNA CT and subsequent disulfide formation. The DNA–p53 interface is examined in greater detail in the corresponding boxed region to the right. This diagram depicts the nine conserved cysteine residues within a DNA-bound p53 monomer in relation to one another and the DNA based on the 3KMD crystal structure.20
One can imagine these conserved cysteine residues electronically coupling to promoter site DNA and playing a role in the redox modulation of p53. A model of p53 oxidation in response to DNA CT is illustrated in Figure 1. Oxidation of p53 is initiated at a distance by the photoexcitation of AQ covalently tethered to DNA, injecting an electron hole into the DNA base stack.4 This oxidizing equivalent is then shuttled through the π-stacked base pairs and localizes to sites of low redox potential. If the electron hole localizes to a site to which protein is bound, such as the p53 promoter site, then the hole can oxidize the lower redox potential amino acid residues within close proximity to the DNA. This oxidation of p53 leads to dissociation from the DNA, ultimately altering gene regulation in response to genomic stress while leaving the DNA undamaged.22 Experiments using electrophoretic mobility shift assays (EMSA) have determined that p53 responds selectively to oxidation via DNA CT, causing the protein to dissociate from various promoter sites. We have seen that the location of guanine residues within a p53 promoter site dictates whether DNA-bound p53 can be oxidized through DNA CT.22 This sequence selectivity in DNA-mediated oxidation of p53 indicates an element of control, causing oxidative dissociation of p53 when bound to certain promoter sites but not to others. This selectivity in response to DNA CT may correlate with the biological regulation of genes controlled by p53 under conditions of oxidative stress.
Several groups have worked to investigate the intricacies of p53 oxidation at a molecular level, an area of which little information is known after more than 30 years of research. The idea of redox modulation of p53 first arose in work showing that p53 can bind promoter sites selectively under reducing conditions but not under oxidizing conditions.23 More recently, Fersht and co-workers investigated the reactivity of cysteine residues by alkylation in an effort to stabilize mutant p53 observed in cancer.24 Using nanospray ionization (nESI) mass spectrometry, they determined that C141 and C124 react first with alkylating agents and are therefore the most reactive cysteine residues, followed by C135, C182, and C277. Langridge-Smith and co-workers have utilized top-down and middle-down Fourier transform ion cyclotron resonance (FT ICR) mass spectrometry to determine the reactivity of cysteine residues within p53 oxidized by H2O2.25 They determined that C182 and C277 exhibit significant modification with N-ethylmaleimide and were deemed to be the most reactive residues. However, the high reactivity of these residues was determined to be primarily due to their high solvent accessibility, which may not be the dominant factor in DNA-bound p53 oxidation in vivo. Work has also been done to map oxidized cysteine residues in H2O2-treated p53 by nESI FT ICR mass spectrometry.26 This work showed that oxidation of the p53 core domain by H2O2 caused a loss of Zn2+ binding within p53, with corresponding formation of two disulfide bonds among C176, C182, C238, and C242. Our laboratory found, using MALDI-TOF mass spectrometry, that DNA-mediated oxidation of p53 might proceed via formation of a disulfide bond involving C141 and an undetermined second cysteine.4
Here, we continue to investigate p53 cysteine oxidation promoted at a distance through DNA CT. Specifically, we aim to resolve the interplay of cysteine oxidation within the p53 DNA-binding domain through the study of p53 mutants. Using EMSA, we investigate the effect of select p53 mutations on DNA binding affinity as well as the ability to undergo oxidative dissociation from the Gadd45 promoter site. The Gadd45 promoter site was chosen since p53 is known to readily bind this sequence and has been shown to dissociate upon oxidation via DNA CT.4 To determine if oxidative dissociation of p53 occurs concurrently with disulfide bond formation and to probe the specific residues involved, we employed a differential thiol labeling technique targeting cysteine residue oxidation states through the use of isotopically distinct iodoacetamide labels. The sequentially labeled samples were proteolytically digested, and labeled peptide fragment intensities were examined on a QTRAP 6500 LC-MS/MS and directly compared. Through this methodology, we are able to characterize the redox states of individual cysteine residues and observe disulfide formation within p53 oxidized at a distance through DNA CT.
Experimental Procedures
Synthesis and Modification of Oligonucleotides
DNA was synthesized using standard solid-phase automated synthesis, modified with anthraquinone (AQ), and radiolabeled as described previously.22,27,28 The DNA used in the following experiments contains the Gadd45 promoter site (underlined) with a 12 base 5′ linker. Constructs both without photooxidant (light control, LC) and with AQ were made. AQ: 5′-AQ-AAA TCA GCA CTA CAG CAT GCT TAG ACA TGT TC-3′. LC: 5′-AAA TCA GCA CTA CAG CAT GCT TAG ACA TGT TC-3′. Complement: 5′-GAA CAT GTC TAA GCA TGC TGT AGT GCT GAT TT-3′.
Protein Preparation
The p53′ protein is a full-length human p53 containing three stabilizing mutations: M133L, V203A, and N268D.29 All subsequent mutants studied are in addition to the p53′ mutations and incorporated by site-directed mutagenesis (QuikChange II, Agilent), with resulting sequences verified by Laragen (primer sequences are located in Supporting Information Table 1). The p53′ protein and subsequent mutants were purified as previously described.22,30
Electrophoretic Mobility Shift Assay of p53′ and Mutants
For the determination of apparent KD’s for each mutant, varied concentrations of each p53′ mutant were added to 25 nM Gadd45 response element DNA in the presence of 5 μM competitor DNA duplex (5′-GGAAAAAAAAAAAAAAAAAAACC-3′) (IDT), 0.1% NP-40 (Surfact-Amps NP-40, Thermo Scientific), and 0.1 mg/mL BSA in p53 buffer (20 mM Tris-HCl, pH 8.0, 20% glycerol, 100 mM KCl, 0.2 mM EDTA). Samples were prepared at ambient temperature, allowed to incubate for 20 min, and electrophoresed on a 10% TBE polyacrylamide native gel (Bio-Rad) in 0.5× TBE buffer at 4 °C and 50 V for 1.5 h. DNA from the gel was transferred to Amersham Hybond-N nucleotide blotting paper (GE Healthcare) with a semidry electroblotter (Owl HEP-1) for 1 h at 175 mA in transfer buffer (25 mM Tris-HCl, pH 8.5, 200 mM glycine, 10% methanol). The blots were exposed to a phosphorimaging screen (GE Healthcare), imaged with a STORM 820 or Typhoon FLA 9000 scanning system (GE Healthcare), and analyzed using ImageQuant TL and OriginPro.
Samples prepared for p53 oxidation assays contained 25 nM p53 tetramer under the same conditions as those listed above for the majority of the mutants. Two mutants were assayed at higher p53 concentrations due to their higher apparent KD values: Y236F-p53′ at 50 nM tetramer and C275S-p53′ at 125 nM tetramer. Samples were made at 4 °C and irradiated in an ice bath for varying lengths of time (0, 5, 15, 30, 45, and 60 min) by solar simulator (ORIEL Instruments) with a UVB/UVC long-pass filter. These samples were then analyzed by EMSA as described above, and data were normalized to the corresponding unirradiated control. The change in p53 binding was determined by monitoring the free DNA signal over the total DNA signal in each lane. Data are an average of a minimum of three assay replicates, and the error is reported as the standard error of the mean.
Selective Cysteine Labeling with Iodoacetamide Tags
Proteins p53′, C275S-p53′, and C141S-p53′ were studied to observe changes in cysteine oxidation state in DNA-bound p53 upon long-range DNA CT. Each sample consisted of 100 μL of 1.0 μM Gadd45 DNA (LC or AQ), 2.0 μM p53′ monomer, 0.1% NP-40, and 5.0 μM competitor DNA, in p53 buffer. Samples were prepared at 4 °C and allowed to incubate for 20 min prior to aliquoting. Samples for irradiation were aliquoted into a low-profile 96-well PCR plate (Bio-Rad) at 10 μL each, placed in an ice-water bath, and irradiated for 1 h by solar simulator with a UVB/UVC long-pass filter. Unirradiated samples remained in the dark at 4 °C for the duration of the other irradiations. Samples were adjusted to 6 M guanidine hydrochloride (GdmCl), by the addition of 8 M GdmCl in 20 mM Tris, 100 mM KCl, 0.2 mM EDTA, at pH 7.8. The samples were transferred to Amicon Ultra 0.5 mL 30 kDa cutoff centrifugal filter units (Millipore) and centrifuged at 13 000g for 15 min. The concentrated samples, ∼30 μL, were then treated with a 100-fold molar excess of iodoacetamide (single-use, Thermo Scientific) with respect to the number of cysteine residues present. The reaction was allowed to continue for 1 h in the dark, shaking at 250 rpm. Samples were diluted with 6 M GdmCl and centrifuged, repeatedly, until the concentration of remaining iodoacetamide was at least 100-fold below the number of cysteine residues and concentrated to ∼30 μL. Dithiothreitol (DTT) was added at a 10-fold molar excess of the reactive species present in the sample, cysteine, and remaining iodoacetamide, to reduce disulfides. This reduction was allowed to incubate for 20 min at ambient temperature in the dark, shaking at 250 rpm. The same molar concentration of Tris(2-carboxyethyl)phosphine (TCEP-Neutral, Calbiochem) as that of DTT was then added to further ensure disulfide reduction and allowed to incubate, as above, for another 20 min. Samples were diluted with 6 M GdmCl and centrifuged, repeatedly, until the concentration of remaining DTT and TCEP were at a molar concentration 1000-fold below the number of cysteine residues present and the total volume was concentrated to ∼30 μL. To each sample was added 13C2D2-iodoacetamide (Aldrich) in H2O at a 100-fold molar excess with respect to the cysteine residues and remaining reducing agents present. This reaction was allowed to continue for 4 h at ambient temperature, shaking at 250 rpm, in the dark. The samples were diluted using 100 mM Tris, pH 8.5, to decrease the GdmCl concentration. The sample was repeatedly diluted and centrifuged until the final GdmCl concentration was below 0.1 M GdmCl in a final sample volume of ∼30 μL and dried in vacuo. The dry sample pellet was dissolved in 40 μL of 8 M urea in 100 mM Tris-HCl, pH 8.5. One microliter of 0.1 μg/μL lysyl endopeptidase (WAKO) dissolved in 100 mM Tris-HCl, pH 8.5, was added to each sample and allowed to incubate for 4 h at ambient temperature in the dark. The samples were subsequently diluted with 100 mM Tris-HCl, pH 8.5, to a final concentration of 2 M urea and adjusted to 1 mM CaCl2. Trypsin (1 μL of 0.5 μg/μL) (Promega) in water was added to each sample and allowed to incubate in the dark overnight at ambient temperature. The following morning, each sample was adjusted to 5% formic acid to simultaneously inhibit protease activity and protonate tryptic peptides; samples were then dried in vacuo. Dry samples were suspended into 50 μL of 0.1% TFA and sonicated for 5 min. Stagetips were made in-house with Empore Extraction disk C-18 membranes (3M) for desalting the peptide samples.31 The stagetip was washed once with 100 μL of 80% acetonitrile in 0.1% TFA and twice with 100 μL 0.1% TFA prior to sample loading, centrifuging for 3 min at 3000 rpm between each round. Samples were loaded to the stagetip by centrifugation and then washed twice with 100 μL of 0.1% TFA. The sample was eluted with 100 μL of 80% acetonitrile in 0.1% TFA into a fresh collection tube. The eluent was dried in vacuo and stored at −20 °C until analysis.
Multiple Reaction Monitoring (MRM) Mass Spectrometry
Each protein sample, 500 fmol per injection, was dissolved in 2% acetonitrile with 0.2% formic acid (FA). To ensure consistency among sample sets and to help validate proper peak assignment by retention time, iRT peptide standards (BIOGNOSYS) were added. Samples were examined on the ABSciex QTRAP 6500 LC-MS/MS system, equipped with an Eksigent ekspert nanoLC 425 pump, ekspert nanoLC400 autosampler, ekspert cHiPLC, and Analyst software. Samples were separated on a cHiPLC Chrom XP C18-CL 3 μm trap column, 120 Å (200 μm × 0.5 mm), inline with a cHiPLC Chrom XP C18-CL 3 μm column, 120 Å (75 μm × 150 mm) using a 45 min linear gradient of acetonitrile in 0.2% FA at a flow rate of 300 nL/min. An unscheduled transition list of cysteine-containing peptides with both respective iodoacetamide labels, as well as iRT peptide standards, was generated by Skyline and exported to the QTRAP for quantitation (Supporting Information Table 1).32 Raw data files generated by the QTRAP were imported back into Skyline, where peak areas were then integrated and exported for further processing. Observable and quantifiable peptide fragments include the following: C124, [121, 132] SVTCTYSPALNK; C135, [133, 138] LFCQLAK; C141, [140, 156] TCPVQLWVDSTPPPGTR; C182, [182, 196] CSDSDGLAPPQHLIR; and C275 and C277, [274, 280] VCACPGR. Two cysteine-containing peptide fragments were unobservable in our methods due to unfavorable mass/charge of the fragments: C176, [174, 180] RCPHHER; C229, C238, and C242, [213, 248] HSVVVPYEPPEVGSDCTTIHYNYMCNSSCMGGMNRR. Various proteases were evaluated; however, this large peptide fragment could not be further cleaved given the inherent amino acid sequence of p53′.
Results
Mutant p53′ Affinity for the Gadd45 Promoter Site
To understand the chemistry of p53 oxidation from a distance through DNA CT, individual residues within the DNA-binding domain were selectively mutated. We used a pseudo-wild-type p53, termed p53′, that incorporates three stabilizing mutations (M133L, V203A, and N268D) while remaining redox active.29 All other mutants studied were created by site-directed mutagenesis of the p53′ plasmid. The following cysteine residues were mutated to similarly sized but redox-inactive serine: C124, C135, C141, C182, C275, and C277. Two other mutations studied include Y236F and N239Y. These mutations were chosen since they are within close proximity to the cysteine residues in question and involve the addition or deletion of a similarly redox-active tyrosine (+0.9 V).11 This cohort of p53 mutants was studied by EMSA to determine if any changes in binding affinity to the Gadd45 promoter site were evident without photooxidation.
Each mutant protein was evaluated by EMSA, and the apparent KD values were determined using varied concentrations of the p53′ mutants in the presence of 25 nM Gadd45 DNA (LC or AQ) in p53 buffer with 5 μM competitor DNA, 0.1% NP-40, and 0.1 mg/mL BSA. The determined apparent KD values are listed in Table 1. The majority of the chosen mutations did not significantly change the binding affinity of these proteins to the Gadd45 promoter site as compared to that of p53′, with or without AQ. The baseline of binding affinity is shown by p53′ with KD values of 1.6 ± 0.6 nM and 2.4 ± 1.1 nM of p53 tetramer for LC and AQ, respectively. C124S-p53′, C135S-p53′, C141S-p53′, and C277S-p53′ all share similar values as that of p53′, with apparent KD values below 5 nM p53′ tetramer. Two mutants exhibited a slight decrease in affinity, at 9.7 ± 4.3 nM (LC) and 8.2 ± 4.7 nM (AQ) tetramer for Y236F-p53′ and 15.1 ± 1.8 nM (LC) and 13.7 ± 4.4 nM (AQ) tetramer for C182S-p53′. Notably, the C275S-p53′ mutant displays severely attenuated affinity for the Gadd45 promoter site with apparent KD values of 56 ± 13 nM (LC) and 54 ± 8 nM (AQ).
Table 1. Relative Dissociation Constants of Mutant p53 Bound to the Gadd45 Promoter Site.
| mutant of p53a | KD LC DNA (nM tetramer)b | KD AQ DNA (nM tetramer)b |
|---|---|---|
| p53′ | 1.6 ± 0.6 | 2.4 ± 1.1 |
| C124S | 3.7 ± 0.5 | 4.29 ± 0.04 |
| C135S | 4.4 ± 2.8 | 3.1 ± 1.2 |
| C141S | 4.6 ± 1.2 | 3.7 ± 0.3 |
| C182S | 15.1 ± 1.8 | 13.7 ± 4.4 |
| Y236F | 9.7 ± 4.3 | 8.2 ± 4.7 |
| N239Y | 1.2 ± 0.4 | 1.0 ± 0.1 |
| C275S | 56 ± 13 | 54 ± 8 |
| C277S | 3.0 ± 0.8 | 2.3 ± 0.5 |
All mutants contain the stabilizing mutations M133L, V203A, and N268D.
The apparent KD of p53′ (in tetramer units) was determined at 25 nM duplex, 5 μM dAdT, 0.1% NP-40, 0.1 mg/mL BSA in 20 mM Tris-HCl (pH 8.0), 20% glycerol, 100 mM KCl, and 0.2 mM EDTA at ambient temperature, and the sample was electrophoresed at 50 V on a 10% polyacrylamide gel in 0.5× TBE at 4 °C.
Oxidative Dissociation of p53′ Mutants through DNA CT
Additional EMSAs were employed to determine if any of these mutations altered the ability of p53′ to oxidatively dissociate from the Gadd45 promoter site. Changes in p53′ binding to the Gadd45 promoter site with respect to irradiation time for each mutant were quantified, and the results are shown in Figure 2. A representative EMSA autoradiogram is provided in Supporting Information Figure 1. Most samples were composed of 25 nM p53′ tetramer and 25 nM Gadd45 DNA in the presence of 5 μM competitor DNA, 0.1% NP-40, and 0.1 mg/mL BSA in p53 buffer. Y236F-p53′ and C275S-p53′ were assayed at higher protein concentrations, 50 nM tetramer and 125 nM tetramer, respectively, to ensure protein–DNA binding due to their higher apparent KD values. The fraction change in p53′ binding is determined as the free DNA signal divided by the sum of the free DNA and p53-bound DNA signals, normalized to the unirradiated control. Each mutant was analyzed over a minimum of three replicates, with the error bars reflecting the standard error of the mean. Previous experiments with the same construct, although with an intervening mismatch, showed an inhibition of oxidative dissociation, demonstrating that oxidation of p53 is DNA-mediated, as opposed to involving a direct AQ–protein interaction.4
Figure 2.

EMSA analysis to determine the activity of mutant p53 bound to the Gadd45 promoter site upon distally induced DNA-mediated oxidation. Solid markers represent AQ samples, and hollow markers represent LC samples. The data are representative of the average of a minimum of three replicates, with the error given as the standard error of the mean. Samples contained 25 nM mutant p53′ tetramer and 25 nM Gadd45 DNA in the presence of 5 μM competitor DNA, 0.1% NP-40, and 0.1 mg/mL BSA in p53 buffer (20 mM Tris-HCl, pH 8.0, 20% glycerol, 100 mM KCl, and 0.2 mM EDTA). Two mutants were assayed at higher protein concentrations due to their higher apparent KD values: Y236F-p53′ at 50 nM tetramer and C275S-p53′ at 125 nM tetramer.
The behavior of p53′ is the standard to which each mutant is compared in Figure 2. The EMSAs of p53′ oxidation reveal minimal oxidative dissociation from the LC-Gadd45 DNA (white), lacking the pendant AQ photooxidant. However, the p53′ protein readily dissociated from the AQ-Gadd45 DNA (black), with 31.0 ± 1.2% total p53′ dissociation upon 60 min of irradiation. The LC-Gadd45 DNA samples across all of the mutants behave similarly, with minimal dissociation upon irradiation irrespective of additional mutations. As compared to the p53′ protein, several mutants displayed a slight increase in the amount of dissociation from the AQ-Gadd45 DNA upon irradiation: C141S-p53′ (37.9 ± 2.7%), Y236F-p53′ (37.2 ± 2.3%), C135S-p53′ (34.0 ± 5.0%), and C124S-p53′ (33.4 ± 8.6%). Conversely, several mutants displayed a slight attenuation in the oxidative dissociation of p53 upon irradiation: C182S-p53′ (27.2 ± 3.0%), N239Y-p53′ (25.5 ± 0.9%), and C277S-p53′ (22.6 ± 2.9%). The most notable difference is observed with C275S-p53′, which reaches a maximum of only 13.3 ± 2.5% protein dissociation upon irradiation and is not within error of any other mutant.
Analysis of Cysteine Oxidation in p53′ by Mass Spectrometry
Using multiple reaction monitoring (MRM) through sensitive analytical mass spectrometry, we directly examined the formation of disulfide bonds within p53′ and mutants from a distance through DNA CT. An overview of the cysteine labeling protocol used to differentially label cysteine residues within p53 respective to oxidation state is shown in Figure 3. Using this methodology, one can distinguish whether individual cysteine residues in the protein are participating in a disulfide bond. After protein oxidation is induced from a distance by irradiation of the AQ-DNA, the protein is denatured in 6 M GdmCl and treated with iodoacetamide. Reduced cysteine residues in p53′ will react with iodoacetamide (red), whereas oxidized cysteine residues participating in disulfide bonds remain chemically unavailable. Removal of excess iodoacetamide and subsequent reduction of all disulfide bonds allow for accessibility of the newly reduced cysteine residue thiol groups to react with the isotopically heavy 13C2D2-iodoacetamide (blue). The protein is then proteolytically digested, desalted by C18 stagetip, and analyzed on a QTRAP 6500 LC-MS/MS. Representative chromatograms of the acquired data for the peptide fragment containing C124 from a p53′ sample set are shown at the bottom of Figure 3. The peak areas for both the iodoacetamide (red) and 13C2D2-iodoacetamide (blue) labeled fragments were analyzed in Skyline and then directly compared.32 These data clearly show the trend toward 13C2D2-iodoacetamide label with the AQL sample, whereas controls (LCD, LCL, and AQD, see Figure 3) were predominated by the isotopically light iodoacetamide label.
Figure 3.
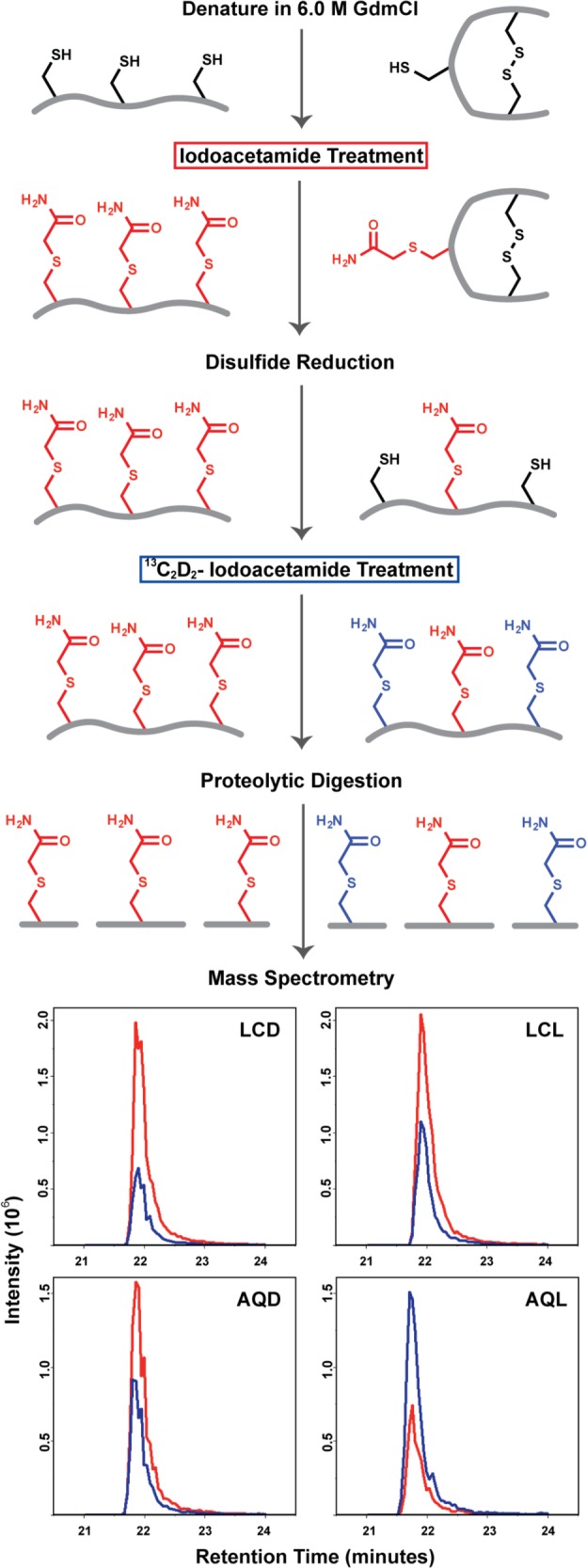
Procedure for differential thiol labeling of cysteine residues. Examples of the labeling procedure are depicted for a fully reduced protein (left) and its corresponding oxidized, disulfide containing counterpart (right). After oxidation from a distance through DNA CT, the protein sample is denatured in 6 M GdmCl and treated with iodoacetamide. Cysteine residues in a reduced state will react with iodoacetamide (red), whereas cysteine residues participating in disulfide bonds remain chemically unavailable. Removal of excess iodoacetamide followed by reduction of all disulfide bonds allow for accessibility of newly reduced thiol groups to react with the second 13C2D2-iodoacetamide label (blue). The protein is then proteolytically digested, peptide fragments are analyzed on a QTRAP 6500 LC-MS/MS, and peak areas are integrated in Skyline. Representative chromatograms of the C124 containing SVTCTYSPALNK peptide fragment from a p53′ sample set are shown as relative intensities of iodoacetamide (red) and 13C2D2IAA (blue) peptides detected. The four traces represent the following: LCD, light control dark; LCL, light control light; AQD, anthraquinone dark; and AQL, anthraquinone light.
Proteins p53′, C275S-p53′, and C141S-p53′ were studied by mass spectrometry to observe changes in cysteine oxidation in DNA-bound p53′ promoted at a distance through DNA CT. We monitored the changes of cysteine residues in p53′ as our standard of comparison. We also examined C275S-p53′ since it displayed the least oxidative dissociation by EMSA as well as C141S-p53′ since C141 was previously implicated in potential disulfide formation through DNA CT.4 The floating-bar plots for each peptide fragment depict the fraction of the total signal of heavy and light modified species, totaling 1.0 (Figure 4). The fraction of 13C2D2-iodoacetamide-labeled species is represented in positive values (black), and the fraction of iodoacetamide-labeled species is represented in negative values (white). These cumulative data sets are represented with individual protein mutants located in rows and corresponding cysteine-containing peptide fragments in columns. Each sample set per mutant is composed of 4 variants, corresponding to DNA used (LC or AQ) and irradiation (D-dark, L-light). The data represent the average of three replicates for the C124, C135, C141, and C182 peptide fragments. The data for C275 and C277 represent the average of two replicates. The error is represented as the standard error of the mean. Peptide fragments corresponding to C176, C229, C238, and C242 could not be observed due to an unfavorable mass/charge ratio.
Figure 4.

Determination of cysteine oxidation states by MRM mass spectrometry. Proteins p53′, C275S-p53′, and C141S-p53′ were studied to observe changes in cysteine oxidation induced through DNA CT. Cumulative data are depicted with individual mutant proteins localized in rows and the corresponding cysteine-containing peptide fragments in columns. The floating-bar plots for each peptide fragment are depicted as the fraction of the total signal of both heavy and light modified species, totaling 1.0. The fraction of 13C2D2-iodoacetamide-labeled species is represented in positive values (black), and the fraction of iodoacetamide-labeled species is represented in negative values (white). Each plot is composed of four samples: LCD, light control dark; LCL, light control light; AQD, anthraquinone dark; and AQL, anthraquinone light. The value (white) located within the AQL floating bar represents the percent change in heavy labeling of AQL sample with respect to the average of the corresponding LCD, LCL, and AQD controls.
A shift toward increased 13C2D2-iodoacetamide labeling indicates that the cysteine of interest has become oxidized and is participating in a disulfide bond. For p53′ and C141S-p53′ sample sets, the AQL samples show a marked increase in 13C2D2- iodoacetamide labeling over that of the LCD, LCL, and AQD control samples. The protein p53′ does indeed undergo chemical oxidation through DNA-mediated DNA CT. Interestingly, the C275S-p53′ sample set depicts a different interplay of oxidation states than that observed for p53′ and C141S-p53′. The overall baseline of 13C2D2-iodoacetamide corresponding to the C135 and the C182 peptides is significantly higher across all four samples. The C124, C141, and C277 peptides in C275S-p53′ behave more similarly to the other sample sets with a distinct, albeit a less intense, increase in the AQL samples as compared to the controls.
Discussion
Although much work has been done to elucidate the redox-dependent binding of p53 to different promoter sites, relatively little is known about the chemistry of p53 oxidation at a molecular level. We are particularly interested in how the protein may be coupled into a charge transport pathway with DNA and how DNA-mediated oxidation of p53 may affect the affinity of p53 for individual promoter sites. The conserved cysteine residues not involved in Zn2+ binding are of particular interest due to their biologically accessible oxidation potential, close proximity to DNA, and ability to form disulfide bonds. In our studies, we sought to determine the role of various cysteine residues (C124, C135, C141, C182, C275, and C277) within the DNA-binding domain of p53 through mutagenesis. The cysteine-to-serine mutation was chosen since serine is structurally similar to cysteine but does not contain the redox-active sulfur atom. Two other mutations involving redox-active tyrosine residues (Y236F and N239Y) were investigated as well, as tyrosine has the same one-electron oxidation potential as that of cysteine (+0.9 V), also making it accessible to photooxidation by DNA-bound AQ.11
Effect of Select Mutations on p53′ Binding Affinity
Each mutant of p53′ was first evaluated by determining changes in affinity for the Gadd45 promoter site. All comparisons were made against the observed affinity of p53′ tetramer for Gadd45 DNA, which was determined to be 1.6 ± 0.6 nM and 2.4 ± 1.1 nM of tetramer for LC and AQ, respectively. The majority of our chosen mutations did not significantly alter the binding affinity of these proteins to the Gadd45 promoter site. C124S-p53′, C135S-p53′, C141S-p53′, N239Y-p53′, and C277S-p53′ all share similar affinities as that of p53′, with apparent KD values below 5 nM p53′ tetramer, indicating that C124, C135, C141, N239, and C277 do not play a significant role in modulating p53 binding affinity to DNA. Y236F-p53′ and C182S-p53′ both exhibited a slight decrease in affinity, with corresponding apparent KD values between 8 and 15 nM p53 tetramer. This indicates that the integrity of Y236 and C182 within the protein may contribute to binding affinity through necessary DNA–protein contacts or protein–protein interactions in tetramer formation. Notably, the C275S-p53′ mutant displays severely attenuated affinity for the Gadd45 promoter site with KD values of 56 ± 13 (LC) and 54 ± 8 nM (AQ). This finding demonstrates that the integrity and likely positioning of C275 are necessary for the high-affinity binding of p53 to promoter site DNA.
Effect of Select Mutations on Oxidative Dissociation
How do these mutations affect the oxidative dissociation of DNA-bound p53? The behavior of p53′ is the standard to which each mutant was compared. For p53′, 31% p53′ dissociation is seen relative to controls after 60 min of irradiation of DNA-tethered AQ. Oxidative dissociation from the AQ-Gadd45 DNA is equal to or slightly increased for C124S-p53′, C135S-p53′, C141S-p53′, and Y236F-p53′ upon irradiation. Slightly increased dissociation suggests that the integrity of these residues is not essential. In contrast, several mutants did cause attenuation in oxidative dissociation. The C182S-p53′ mutation appears to slightly decrease oxidative dissociation. The N239Y-p53′ mutation also shows a slight decrease in dissociation; since tyrosine has the same redox potential as cysteine and is within close proximity of the DNA, the added tyrosine residue may become oxidized, preventing electron hole migration to other cysteine residues.11 Interestingly, while known to be a stabilizing mutation within p53, N239Y has been observed in colorectal cancer somatic cell mutations.29,33,34 It is noteworthy that the C277-p53′ mutant binds Gadd45 DNA with comparable affinity as that of p53′ but does not appear to dissociate as readily, at 22% and not within error of p53′. This result indicates that C277 may be a necessary element for the oxidative dissociation of p53, perhaps through coupling into the DNA CT pathway and initiating disulfide formation with the nearby C275. Indeed, the most significant difference observed with the mutants is the severe attenuation of oxidative dissociation of C275S-p53′, with a maximum of only 13% dissociation. Thus, it is evident that C275 plays a critical role in the affinity of p53 for its promoter site as well as enables oxidative dissociation. Interestingly, the mutation of C275 has been observed in lung cancer.35 The attenuation of oxidative dissociation in both C275S and C277S suggests the possibility that these residues form a key disulfide bond upon oxidation. The formation of a disulfide between C275 and C277 would also remove DNA contacts, lowering DNA affinity overall, and enabling p53 dissociation. The observed amounts of oxidative dissociation of C275S-p53′ and C277S-p53′ are, however, not equal; this variation is likely due to the differing location of the cysteine residues with respect to the DNA bases conveying the electron hole.
Mass Spectrometry Results to Characterize Cysteine Oxidation States
Mass spectrometry studies were carried out to understand the chemistry of DNA-mediated p53 oxidation. A differential thiol labeling method was devised to determine the oxidation state of specific cysteine residues within p53. The sequential use of iodoacetamide, reducing agents, and isotopically distinct 13C2D2-iodoacetamide enable us to label cysteine residues depending on their respective oxidation state. A shift toward greater 13C2D2-iodoacetamide labeling in comparison to that of controls, as monitored through MRM mass spectrometry, indicates oxidation of that residue and its disulfide participation. We were able to study six of the ten cysteine residues present within the DNA-binding domain through this technique. We were unable to detect C176 since it is located in a very small and highly charged peptide fragment [RCPHHER], resulting in an unfavorable mass/charge ratio. Three cysteine residues (C229, C238, and C242), all residing within one extraordinarily large peptide fragment [HSVVVPYEPPEVGSDCTTIHYNYMCNSSCMGGMNRR], could not be further digested proteolytically and could therefore not be detected within the limits of our instrumentation. The remaining six cysteine residues are readily detected and quantifiable. However, C275 and C277 reside within the same peptide fragment, so secondary ion intensities were utilized to deconvolute mixed species containing both iodoacetamide and 13C2D2-iodoacetamide.
It is important to note that these mass spectrometry data indicate directly that the DNA-bound p53′ protein can be oxidized from a distance through DNA-mediated CT. Residues bound to the DNA, and not those most accessible to solution, are oxidized, funneling oxidative damage from the DNA helix and into the protein. This DNA-mediated process promotes p53′ dissociation from the Gadd45 promoter site.
The mass spectrometry data furthermore establish which cysteine residues are being oxidized from a distance through DNA CT. In most cysteine residues observed for both the p53′ and the C141S-p53′ sample sets, the AQL samples show a marked increase in 13C2D2-iodoacetamide labeling samples as compared to that of the LCD, LCL, and AQD controls. Thus, cysteine oxidation resulting in disulfide bond formation is occurring among all observable cysteine residues within p53′ and C141S-p53′. However, we are unable to determine whether the disulfide formation is occurring intra- or intermolecularly through our methodologies. Both p53′ and C141S-p53′ show very similar profiles of oxidation with a significant AQL-13C2D2-iodoacetamide increase in all observable cysteine residues: C124, C135, C141, C182, C275, and C277. It should be noted that across all of the samples there is a baseline level of oxidation, indicating some disulfide presence in the protein prior to DNA CT. Nonetheless, it appears that the majority of the cysteines are in the reduced state. Importantly, the fraction of 13C2D2-iodoacetamide labeling is greatly increased upon oxidation resulting from DNA CT. Removal of C141 through the C141S mutation does not appear to alter the DNA binding affinity, oxidative dissociation, or the ability to oxidize any other cysteine residues. This suggests that oxidation of C141 may occur but that its presence is not necessary for modulation of p53′ binding affinity through DNA-mediated oxidation.
The C275S-p53′ sample set depicts a different interplay of oxidation states than that observed in either p53′ or C141S-p53′, however. The overall baseline of 13C2D2-iodoacetamide labeling for C135 and C182 peptide controls are high across all four samples, greater than 60%, and show only a slight increase in the AQL samples over the controls. The C124, C141, and C277 peptides in C275S-p53′ behave more similarly to the other sample sets with a distinct, albeit less intense, increase in the AQL samples with respect to the controls. The smaller shift toward 13C2D2-iodoacetamide labeling in the AQL samples relative to the controls suggests that the absence of C275 disrupts the ability of oxidation to be transferred to the more internal residues.
Oxidative Dissociation of p53′ by Disulfide Formation
By applying the observed data to the network of cysteine residues within p53, we can consider how DNA-mediated oxidation of p53 may occur and how it may lead to changes in protein conformation that decrease affinity for DNA. Reduced p53 binds as a tetramer to the Gadd45 promoter site. Upon DNA oxidation, an electron hole will migrate through the π-stacked bases and localize to DNA sites of low redox potential, such as guanine. This CT occurs on a time scale that is fast compared to that of the irreversible reaction of guanine radicals.36 In the case of the Gadd45 promoter site, the low oxidation potential guanine sites are located within the purine region of the consensus sequence in close proximity to p53 residue C277. Since the redox potential of cysteine (+0.9 V) is lower than that of guanine (+1.29 V), the C277 residue tucked in the major groove near guanine can accept the electron hole, become oxidized, and lose its hydrogen bond to the major groove of DNA.11−14 Due to the solvent accessibility of C277 and its close proximity to C275, further oxidation of C277 by molecular oxygen would allow for loss of a second electron and result in disulfide formation between C277 and C275, located 7.0 Å away. Disulfide formation between these two residues would result in the loss of essential p53–DNA binding contacts, leading to a significant decrease in affinity and causing the dissociation of the oxidized p53 monomer.
Disulfide bonds are known to rearrange among other cysteine residues within close proximity of one another within proteins.37,38 Upon formation of the C277–C275 disulfide bond, a subsequent rearrangement could occur given the presence of many other reduced cysteine residues within close proximity. If this were to occur, then C275 would most likely form a disulfide with C135 (7 Å away). This bond rearrangement would funnel the disulfide bond deeper into the protein and enable C277 to become reduced and possibly reestablish its H-bond to DNA. The disulfide bond could then rearrange once more, resulting in one disulfide bond potentially residing among the inner triad of cysteine residues: C124, C135, and C141.
Thus, well-conserved cysteine residues of p53 provide a chemical platform through which genomic oxidative stress can be directly sensed. Since p53 is a transcription factor presiding over the regulation of hundreds of human genes, the oxidative dissociation of p53 allows for a direct response in p53 gene regulation during times of genomic stress. The extent of oxidative dissociation of p53 depends on the DNA sequence of the promoter site to which it is bound.22 Low redox potential guanine bases located in the purine region of the p53 promoter site allow for electron holes to localize at the DNA–protein interface and to concomitantly oxidize p53. The variability of bases within the promoter site, while fully conforming to the consensus sequence constraints, allows for a tuning of the redox potential at the DNA–protein interface. The DNA sequence of the promoter site determines whether DNA-bound p53 will be able to accept an electron hole and respond to genomic stress. The cysteine residues in the protein create a network, which is coupled to DNA, capable of accepting electron holes via DNA CT. It is through p53 oxidation and disulfide formation that the affinity of p53 for DNA is decreased, leading to the observable oxidative dissociation of DNA-bound p53.
These results thus indicate that DNA-mediated oxidation of p53 is a chemically distinct mechanism for the cell to respond specifically to oxidative damage to the genome. The oxidation of p53 through DNA CT resulting in disulfide formation within a protein is an exciting new chapter in the study of cellular signaling of oxidative stress and the response of p53.
Acknowledgments
We thank Lisa Beckmann for assistance with molecular cloning and protein purification.
Glossary
Abbreviations
- p53′
full-length human p53 with stabilizing mutations M133L, V203A, and N268D
- CT
charge transport
- AQ
anthraquinone
- LC
light control
- EMSA
electrophoretic mobility shift assay
- GdmCl
guanidine hydrochloride
- MRM
multiple reaction monitoring
- nESI
nanospray ionization
- FT ICR
Fourier transform ion cyclotron resonance
Supporting Information Available
Primer sequences used for site-directed mutagenesis, representative p53 oxidation EMSA autoradiogram, and transition list for the peptides monitored by the QTRAP 6500 LC-MS/MS. This material is available free of charge via the Internet at http://pubs.acs.org.
This research was funded by the Ellison Foundation (AG-SS-2079-08), the Moore Foundation for support of the Center for Chemical Signaling at Caltech, the Gordon and Betty Moore Foundation (GBMF775), The Beckman Institute, and National Institutes of Health (1S10OD010788-01A1).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Paveletich N. P.; Chambers K. A.; Pabo C. O. (1993) The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 7, 2256–2564. [DOI] [PubMed] [Google Scholar]
- Joerger A. C.; Fersht A. R. (2007) Structure–function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 26, 2226–2242. [DOI] [PubMed] [Google Scholar]
- Petitjean A.; Mathe E.; Kato S.; Ishioka C.; Tavtigian S. V.; Hainaut P.; Olivier M. (2007) Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 28, 622–629. [DOI] [PubMed] [Google Scholar]
- Augustyn K. E.; Merino E.; Barton J. K. (2007) A role for DNA-mediated charge transport in regulating p53: oxidation of the DNA-bound protein from a distance. Proc. Natl. Acad. Sci. U.S.A. 104, 18907–18912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D.; Holmlin R.; Barton J. K. (1996) Oxidative DNA damage through long-range electron transfer. Nature 382, 731–735. [DOI] [PubMed] [Google Scholar]
- Delaney S.; Barton J. K. (2003) Long-range DNA charge transport. J. Org. Chem. 68, 6475–6483. [DOI] [PubMed] [Google Scholar]
- Muren N. B.; Olmon E. D.; Barton J. K. (2012) Solution, surface, and single molecule platforms for the study of DNA-mediated charge transport. Phys. Chem. Chem. Phys. 14, 13754–13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontz P. A.; Muren N. B.; Barton J. K. (2012) DNA charge transport for sensing and signaling. Acc. Chem. Res. 45, 1792–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereux J.; Boal A.; Barton J. K. (2010) DNA-mediated charge transport in redox sensing and signaling. J. Am. Chem. Soc. 132, 891–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slinker J.; Muren N.; Renfew S. E.; Barton J. K. (2011) DNA charge transport over 34 nm. Nat. Chem. 3, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan J. R.; Tran N. Q.; Ly A.; Ward J. F. (2004) Peptide repair of oxidative DNA damage. Biochemistry 43, 5102–5108. [DOI] [PubMed] [Google Scholar]
- Steenken S.; Jovanovic S. V.; Bietti M.; Bernhard K. (2000) The trap depth (in DNA) of 8-oxo-7,8-dihydro-2′deoxyguanosine as derived from electron-transfer equilibria in aqueous solution. J. Am. Chem. Soc. 122, 2373–2374. [Google Scholar]
- Milligan J. R.; Aguilera J. A.; Nguyen J. V.; Ward J. F. (2001) Redox reactivity of guanyl radicals in plasmid DNA. Int. J. Radiat. Biol. 77, 281–293. [DOI] [PubMed] [Google Scholar]
- Steenken S.; Jovanovic S. V. (1997) How easily oxidizable is DNA? One electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 119, 617–618. [Google Scholar]
- Sugiyama H.; Saito I. (1996) Theoretical studies of GG specific photocleavage of DNA via electron transfer: significant lowering of ionization potential and 5′-localization of HOMO of stacked GG bases in B-form DNA. J. Am. Chem. Soc. 118, 7063–7068. [Google Scholar]
- Gorodetsky A. A.; Barton J. K. (2007) DNA-mediated electrochemistry of disulfides on graphite. J. Am. Chem. Soc. 129, 6074–6075. [DOI] [PubMed] [Google Scholar]
- Takada T.; Barton J. K. (2005) DNA charge transport leading to disulfide bond formation. J. Am. Chem. Soc. 127, 12204–12205. [DOI] [PubMed] [Google Scholar]
- El-Deiry W. S.; Kern S. E.; Pietenpol J. A.; Kinzler K. W.; Vogelstein B. (1992) Definition of a consensus binding-site for p53. Nat. Genet. 1, 45–49. [DOI] [PubMed] [Google Scholar]
- Cho Y.; Gorina S.; Jeffrey P. D.; Pavletich N. P. (1994) Crystal structure of a p53 tumor suppressor–DNA complex: understanding tumorigenic mutations. Science 265, 346–354. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Dey R.; Chen L. (2010) Crystal structure of the p53 core domain bound to a full consensus site as a self-assembled tetramer. Structure 18, 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Zhang X.; Dantas Machado A. C.; Ding Y.; Chen Z.; Qin P. Z.; Rohs R.; Chen L. (2013) Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 41, 8368–8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer K. N.; Barton J. K. (2014) DNA-mediated oxidation of p53. Biochemistry 53, 3467–3475. [DOI] [PubMed] [Google Scholar]
- Parks D.; Bolinger R.; Mann K. (1997) Redox state regulates binding of p53 to sequence-specific DNA, but not to non-specific or mismatched DNA. Nucleic Acids Res. 25, 1289–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaar J. L.; Basse N.; Joerger A. C.; Stephens E.; Rutherford T. J.; Fersht A. R. (2010) Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 19, 2267–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotcher J.; Clarke D. J.; Weidt S. K.; Mackay C. L.; Hupp T. R.; Sadler P. J.; Langridge-Smith P. R. R. (2011) Identification of two reactive cysteine residues in the tumor suppressor protein p53 using top-down FT ICR mass spectrometry. J. Am. Soc. Mass Spectrom. 22, 888–897. [DOI] [PubMed] [Google Scholar]
- Scotcher J.; Clarke D. J.; Mackay C. L.; Hupp T.; Sadler P. J.; Langridge-Smith P. R. (2013) Redox regulation of tumour suppressor protein p53: identification of the sites of hydrogen peroxide oxidation and glutathionylation. Chem. Sci. 4, 1257–1269. [Google Scholar]
- Armitage B.; Yu C.; Devadoss C.; Schuster G. B. (1994) Cationic anthraquinone derivatives as catalytic DNA photonucleases: mechanisms for DNA-damage and quinone recycling. J. Am. Chem. Soc. 116, 9847–9859. [Google Scholar]
- Gasper S. M.; Schuster G. B. (1997) Intramolecular photoinduced electron transfer to anthraquinones linked to duplex DNA: the effect of gaps and traps on long-range radical cation migration. J. Am. Chem. Soc. 119, 12762–12771. [Google Scholar]
- Nikolova P. V.; Henckel J.; Lane D. P.; Fersht A. R. (1998) Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. U.S.A. 95, 14675–14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veprintsev D. B.; Freund S. M.; Andreeva A.; Rutledge S. E.; Tidow H.; Cañadillas J. M.; Blair C. M.; Fersht A. R. (006) Core domain interactions in full-length p53 in solution. Proc. Natl. Acad. Sci. U.S.A. 103, 2115–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R. L. J.; Kalli A.; Smith G. T.; Sweredoski M. J.; Hess S. (2011) Avoiding pitfalls in proteomics sample preparation. Biomacromol. Mass Spectrom. 2, 273–284. [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebischung C.; Gerard J. P.; Gayet J.; Thomas G.; Hamelin R.; Laurent-Puig P. (2002) Prognostic value of P53 mutations in rectal carcinoma. Int. J. Cancer 100, 131–135. [DOI] [PubMed] [Google Scholar]
- Webley K. M.; Shorthouse A. J.; Royds J. A. (2000) Effect of mutation and conformation on the function of p53 in colorectal cancer. J. Pathol. 191, 361–367. [DOI] [PubMed] [Google Scholar]
- Gao H. G.; Chen J. K.; Stewart J.; Song B.; Rayappa C.; Whong W. Z.; Ong T. (1997) Distribution of p53 and K-ras mutations in human lung cancer tissues. Carcinogenesis 18, 473–478. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Stemp E. D. A.; Pulver S. C.; Barton J. K. (1997) Long-range oxidation of guanine by Ru(III) in duplex DNA. Chem. Biol. 4, 389–400. [DOI] [PubMed] [Google Scholar]
- Gilbert H. F. (1995) Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 215, 8–28. [DOI] [PubMed] [Google Scholar]
- Kadokura H.; Katzen F.; Beckwith J. (2003) Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 72, 111–135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.