

Abstract
Human beta defensin-3 (HβD-3) is a host-defense protein exhibiting antibacterial activity towards both Gram-negative and Gram-positive bacteria. There is considerable interest in the function of this protein due to its increased salt tolerance and activity against Gram-positive S. aureus. In this study, analogs of HβD-3 devoid of N and C terminal regions are investigated to determine the influence of specific structural motif on antimicrobial activity and selectivity between Gram-positive and Gram-negative bacteria. Circular dichroism, fluorescence and solid-state NMR experiments have been used to investigate the conformation and mode of action of HβD3 analogs with various model membranes to mimic bacterial inner and outer membranes and also mammalian membranes. Our studies specifically focused on determining four major characteristics: (i) interaction of HβD3 analogs with phospholipid vesicles composed of zwitterionic PC or anionic PE:PG vesicles and LPS; (ii) conformation of HβD3-peptide analogs in the presence of PC or PE:PG vesicles; (iii) ability of HβD3 analogs to permeate phospholipid vesicles composed of PC or PE:PG; and (iv) activities on bacteria cells and erythrocytes. Our results infer that the linear peptide L25P and its cyclic form C25P are more active than L21P and C21P analogs. However, they are less active than the parent peptide, thus pointing towards the importance of the N terminal domain in its biological activity. The variation in the activities of L21P/C21P and L25P/C25P also suggest the importance of the positively charged residues at the C terminus in providing selectivity particularly to Gram-negative bacteria.
Keywords: Antimicrobial peptide, HβD3, solid state NMR, fluorescence, dye leakage
Introduction
Antimicrobial peptides (AMPs), the principal component of the innate immune system, are short (mostly 20–50 amino acids) cationic peptides exhibiting both antimicrobial as well as immune-modulatory activities. In general, mammalian AMPs are classified into two broad families, namely, cathelicidins and defensins. On proteolytic cleavage, the well conserved N-terminal cathelin domain of cathelicidins give rise to an active peptide consisting only the C-terminus fragment. Many of the cathelicidins are stored within the neutrophil secretory granules as inactive pro-peptide in addition to being secreted in the mucosal surfaces [1, 2]. On the other hand, defensins, another prominent member of the mammalian cationic AMPs, ranges from 3–5 kDa in size [3]. In general defensins can be characterized into three sub groups namely, α-, β-defensins, which are distinguished by the presence of disulfide bridges between six conserved cysteine residues and the θ-defensins, which are macrocyclic in nature. Similar to cathelicidins, defensins also requires proteolytic digestion for its activation. Defensins show a broad range of activity against both Gram-positive and Gram-negative bacteria, yeast, and some enveloped viruses [4, 5]. Apart from that, they also have been reported to act as chemokines, activating the adaptive immune response [6, 7].
The α- and β-defensins can be found in various vertebrate species and are seen in the granules of immune cells, epithelial tissue, body fluids and mucosal surfaces. The human neutrophil proteins HNP1–3, the first characterized human defensins are the α-defensins, which are released upon the invasion and phagocytosis of the infecting bacterium [8]. Subsequently, a set of 13 peptides, later discovered in the granules of bovine neutrophils, are referred to as β-defensins [9]. Despite the lack of consensus in sequence similarity among various defensins, β-defensins possess a structurally conserved tertiary scaffold consisting of a triple stranded β sheet core stabilized primarily through disulfide bridges. Thus, the six highly conserved cysteine residues play a critical role in the formation of three disulfide bridges in all of the β defensins [10].
The first β-defensin isolated from human hemofiltrates was HβD1, expressed primarily in the kidney and female urogenital tract. [11]. HβD1 is expressed constitutively, meaning its expression does not depend upon the presence of bacteria or any external inflammatory stimuli [12] and the second β-defensin, HβD2, was isolated from the skin of psoriasis patients [13]. It is active against Gram-negative bacteria and yeast; however, it remains bacteriostatic against Gram-positive bacteria and Staphylococcus aureus [4]. The third human β-defensin, HβD3, was a 45 residue cationic peptide found in epidermal keratinocytes of the psoriasis patients. HβD3 shares low sequence similarity with other β-defensins such as HβD1 and HβD2, and possesses twice the amount of net positive charge density [14–17]. HβD-3 has gained more attention in the recent past for its broad-spectrum of antibacterial activity against both Gram-negative and Gram-positive bacteria [14, 18, 19], even in the presence of high salt concentration. The primary sequence of HβD-3 peptide and its analogues are shown in Table 1.
Table 1.
The amino acid sequences of native HβD-3 peptide and its analogs containing shorter and longer amino acid sequences. The linear and cyclic forms of HβD-3 peptide analogs were used for measuring various biological activities and biophysical studies.
| Peptide | Sequence | Molecular weight | Net charge |
|---|---|---|---|
| HβD-3 | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK | 5161 | 11 |
| L21P | GRCAVLSWLPKEEQIGKCSTR | 2362 | 2 |
| L25P | LSCLPKEEQIGKWSTRGRKSCRRKK | 2975 | 7 |
| C21P | GRCAVLSWLPKEEQIGKCSTR | 2360 | 2 |
| C25P | LSCLPKEEQIGKWSTRGRKSCRRKK | 2973 | 7 |
The solution structure of HβD-3 consists of three anti-parallel β-sheets and a short helical loop in the amino-terminal region of the peptide with most of the charged residues arranged nearby the C-terminal region [20]. As the peptide is small in size, the disulfide bridges between the Cys-Cys play an important role in stabilizing its tertiary structure. Additionally, it has been shown that HβD-3 has the ability to form an amphipathic symmetrical dimer structure through the β2 strand, which upon folding exhibits increasingly positively charged surface [20].
Interestingly, HβD-3 exhibits antimicrobial activity irrespective of its cysteine disulfide framework [18], suggesting that disulfide bond may not be essential for its antimicrobial activity. Therefore, a systematic biophysical approach is necessary to confirm its structure-function relationship in an ad-hoc manner. It is well documented that the cell-lytic activities of antimicrobial peptides are thought to proceed by peptide binding to the cell membrane surface and then permeabilizing the membrane lipid bilayer. Previous studies suggested that the unstructured antimicrobial peptides may fold into a folded conformation upon binding to the membrane and subsequently permeabilize the membrane [21, 22].
In this study, we analyzed the structural and functional aspects of both the linear and cyclic fragments of HβD-3 at the membrane interface level using several low-resolution biophysical techniques such as CD, dye leakage, fluorescence etc in conjunction with high resolution techniques like solid-state NMR spectroscopy and theoretical study employing molecular dynamics simulation. The primary objective of this study is to understand the mechanism of interaction between HβD-3 and the model lipid membranes, in order to explain its biological activities against bacteria and blood cells. Therefore, it is mandatory to work on the native sequence to narrow down the minimal required sequence for the antimicrobial activity to subsequently synthesize the active analogues [23, 24].
Materials and Methods
Materials
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) were purchased from Avanti Polar Lipids, Inc. and used without further purification. All of the reagents for peptide synthesis and cleavage were purchased from Applied Biosystems (Foster City, CA) and Aldrich (Milwaukee, WI), respectively. Fmoc-protected amino acids were purchased from Advanced ChemTech (Louisville, KY). Chloroform and methanol were purchased from Aldrich (Milwaukee, WI), and all other reagents were obtained from Fisher (Pittsburgh, PA). Carboxyfluoroscein, and buffer materials were purchased from Sigma Aldrich.
Peptide synthesis and characterization
Peptide analogs of HβD-3 were synthesized on an automated ABI-431A (Applied Biosystems, Foster City, CA) peptide synthesizer using Fmoc chemistry on a 0.1 mmole scale. The first amino acid was attached to the Wang resin (Peptides International, Louisville, KY) using the symmetric anhydride method. The attachment of the first amino acid was monitored by the deprotection of Fmoc group and the resin was further used for the synthesis if the attachment was >90%. All the protected amino acids were purchased from either ABI or Novabiochem. The amino acids were coupled as their esters and double acylation were carried out wherever required. After synthesis, the peptide was cleaved from the resin by acid cleavage containing TFA:anisole:cresol in the ratio 1:0.1:0.1 (v/v) and was precipitated using cold ether. The precipitated peptide was washed with ether, dried and dissolved in water and finally lyophilized to solid. The powder was purified by RP-HPLC. The major fraction was collected, lyophilized and stored in powder form. The oxidation of the peptides was carried out using its purified linear analog in 30% DMSO for 16–18 hrs at room temperature and purified by RP-HPLC. All purified peptides were characterized by MALDI-TOF mass spectrometry using CHCA as a matrix. The primary sequences and their corresponding average masses are given in Table 1.
Antimicrobial activity Assay
Varying concentrations of peptide prepared by doubling serial dilution ranging from 100 μg/mL to 0.1 ng/mL to the sterile 96 well microtiter plate together with 10 μL 108 cells/mL of mid log phase bacterial suspension grown in MH broth and washed twice with phosphate buffer of pH 7.4 and incubated for 3 hours. MH broth was added to each well and incubated at 37 °C for overnight. MICs were set as the lowest concentration of the peptide at which there was no growth above the inoculated level of bacteria as obtained from absorbance measured at 600 nm.
Preparation of Vesicles
Lipid films were prepared by mixing required volumes of different lipids drawn from chloroform stock solution, evaporating under a stream of N2 gas, and drying the residual solvents under high vacuum for overnight. The dried lipid film was then resuspended in 5 mM HEPES buffer containing 120 mM salt for about an hour. The solution was then vortexed continuously for ~2 minutes. The resultant turbid lipid dispersion was later sonicated with a constant power setting of 2 for about 20 minutes in an ice cooled water bath. The turbid lipid suspension slowly turned to clear. Later, the solution was removed and centrifuged twice in two different tubes at 4000 rpm for about 4 minutes. This process generated small unilamellar vesicles (SUVs), which had the least scatter and were stable. The phosphorus content in lipid stock solutions was determined by using a standard spectrophotometric analysis [25].
Lipid Assay
As different lipids have varied absorbance maxima, different calibration curves were prepared. Known concentrations of lipids (measured by phosphate assay) were used for preparing the calibration curves. The calibration curves prepared for synthetic lipids, POPE: POPG (3:1) and POPC have absorbance maximum at 464 nm and 436 nm, respectively. The concentration of lipids calculated by this assay was used for all other experiments in this study.
Peptide Aggregation by Fluorescence Spectroscopy
The ability of peptides to self-aggregate in aqueous solution was monitored by tryptophan fluorescence. To 1 ml of aqueous buffer, different concentrations of a peptide were added and scanned from 300 to 450 nm by exciting the fluorophore at 280 nm and monitoring the shift in the wavelength of the emission maximum. Peptides were titrated in the concentration range of 20–40 μM. Peptides that have the ability to undergo self-aggregation showed an increase in the peptide concentration. All the peptides mentioned in Table 1 displayed a blue shift in a region of 4–7 nm, due to the relocation of the tryptophan to a hydrophobic environment (data not shown). Based on the results obtained, concentrations of peptide samples were chosen such that there is no aggregation of peptide in solution phase.
Peptide-Lipid Interaction by Tryptophan Fluorescence
Lipid-peptide interaction was measured with a Varian Cary Eclipse spectrofluorometer (Walnut Creek, CA) at 25 °C. This was studied by monitoring the changes in the tryptophan fluorescence spectra of the peptide upon the addition of SUVs of different lipids. Tryptophan fluorophore is quite sensitive to the environment and the transfer of fluorophore from a polar to non-polar environment is often accompanied by a blue shift in the fluorescence emission maximum. The excitation wavelength was set at 280 nm and the emission was scanned from 300 to 450 nm with a slit width of 5 nm for both. The blue shift was observed upon the addition of peptide to buffer containing lipids, indicating the binding of the peptide to lipid. The change in the intensity and shift in the wavelength was monitored at different lipid or LPS to peptide concentration ratios. Measured intensity values were corrected from a control experiment (in the absence of peptide) for the light-scattering effects of the SUVs at each lipid concentration. In order to reduce the contributions of high scattering background signals for DLS and CD, small unilamellar vesicles were used [26].
Circular Dichroism (CD) Spectroscopy
Circular dichroism (CD) spectra were recorded from AVIV CDS Model 62Ds spectropolarimeter at room temperature using a 1 cm quartz cuvette. Spectra were recorded with a bandwidth of 1 nm, a scan speed of 50 nm/min, and a step resolution of 0.2 nm. An average of four scans per sample was collected. The scans were recorded in the range of 185–250 nm (for lipid samples from 200–240 nm). Proper blanks were recorded using the peptide free suspension and were subtracted from the corresponding sample spectra. The different media used for recording the spectra were aqueous buffer of HEPES (5 mM, 120 mM NaCl, pH 7.4) and lipid vesicles (SUVs of POPC and 3:1 POPE:POPG). The final spectra were plotted as mean residue molar ellipticity versus wavelength.
Dye Leakage
Carboxyfluorescein dye entrapped SUVs were prepared as described elsewhere (24). The dye-containing vesicles were then purified by gel filtration chromatography, using a Sephadex G-75 column. Aliquots of peptide solution were added to a stirred suspension of SUVs and the fluorescence emission intensity as a function of time was recorded using the excitation wavelength of 490 nm and the emission wavelength of 520 nm. The maximum leakage from each sample was determined by adding 1% TritonX-100.
Hemolytic Assay
Hemoglobin release from sheep erythrocytes upon treatment with peptides was taken as a measure of their hemolytic activity. Amount of released hemoglobin was determined by measuring absorbance at 414 nm. Red blood cells (Colorado Serum, Denver, CO) were centrifuged and washed four times with PBS (0.15 M NaCl, 0.005 M phosphate buffer, pH 7.4). Then 100 μL of red blood cells were added to the wells of a 96-well plate, and 100 μL of the peptide solution (in PBS) were added to each well. The plates were incubated for 1 h at 37°C, and centrifuged at 200 × g for 10 min. Absorbance of the supernatants was measured at 414 nm, and 0% and 100% hemolysis was determined in PBS and 0.1% Triton X-100, respectively.
Solid-State NMR Spectroscopy
POPE and POPG lipids were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Samples were prepared by mixing 3:1 POPE and POPG lipids and 1 mol% of peptide were added. The solution was then dried under a stream of nitrogen gas followed by vacuum pumping overnight. The lipid film was thereafter placed in a humidity chamber at 40 °C for about 36 hours.
Two-dimensional proton-detected local field (PDLF) NMR has made it possible to obtain clear Pake-type C-H dipolar coupling spectra, which help to probe molecular order, structure, and dynamics. We have used MLVs of POPE:POPG (3:1 mole ratio) as a control and 1 mol % of HBD-3 analog C25P in POPE:POPG (3:1 ratio) MLVs to understand the specific interaction of the peptide with lipid vesicles. These experiments help us to identify the individual sites where the peptide interacts with the lipid. In these PDLF experiments, the 1H 90° pulse length was 5.39 μs and a 5 s pulse delay was used.
Solid-state NMR experiments were carried on a Varian Infinity 400 MHz solid-state NMR spectrometer operating with the 13C resonance frequency of 100.6 MHz and proton resonance frequency of 400.1 MHz. A Chemagnetics temperature controller was used to maintain the sample temperature, and each sample was equilibrated at 37 °C for at least 20 minutes before each experiment. A 4 mm double-resonance MAS probe (Chemagnetics) was used (ZrO2 rotors, Teflon spacers and end-caps) for experiments on POPE:POPG MLVs at 5.15 kHz spinning speed. Data processing was accomplished using the Spinsight software (Varian) on a Sun SPARC workstation.
Molecular Dynamics Simulation
Molecular dynamics simulation methodology has been adopted in the present work to study the dynamic property of peptide L25P in the POPC bilayer. A linear sequence of L25P and a helical structure of L25P was prepared using tleap module of Amber [27]. In order to generate the random-coil structure for L25P, a simulation of 5 ns was performed in water box with fully extended linear structure. All the peptide structures were processed for gas-phase energy minimization and simulated annealing as per the protocol reported in the literature [28]. POPC bilayer was prepared with the peptides embedded within the bilayer and solvated with a 10Å water box (SPC water models [29]). The system was charge neutralized with seven chloride (Cl-) counter ions. All the simulations were carried out in Desmond using OPLS_2005 force-field [30] at constant temperature and pressure of 300K and 1atm respectively. All the initial setting of system minimization, equilibrium and temperature control and various remaining simulation parameters were similar to the published protocol [28]. The described systems were continued for a simulation spanning 50 ns time scale. Computation of the electrostatic potential was carried out by solving Adaptive Poisson-Boltzmann solver (APBS) as reported in the literature [31]. All the computations were carried out using GPU workstation enabled with Tesla C2075.
Results and discussion
Designing of peptides
The three dimensional solution structure of HβD3 reveals an unstructured N terminal region (Gly1-Lys8) followed by a short helical segment that precedes a three-stranded antiparallel β sheet, common for all three β defensins. The C terminal region of the peptide is found to be rich in positively charged residues [20]. The antiparallel β sheet region is assumed to be the membrane-binding region, maintaining a perfect amphipathic structure with the positively charged residues distinctly separated from the polar face. These charge separations probably mediates the initial interaction with the negatively charged membrane and assist the hydrophobic face to insert into the membrane (Fig. 1). Previously, it was reported that the removal of the first five N terminal residues, Gly1-Thr5, did not alter the activity of this peptide against the Gram positive bacteria, such as S. aureus, despite a reduction in its activity against a range of Gram negative bacteria [32]. However, recent studies on chimeric peptides constructed of N and C terminal region of HβD1 and HβD3 show that the chimeras containing the N and C terminal regions of HβD3 and middle region of HβD2 or the chimeras containing the N terminal region of HβD3 but with the middle and C terminal region of HβD2 were more potent against a broad range of bacteria when compared to either of the parent defensin peptides [33]. To understand the role of N terminal extended region and the helical segment that follows it in endowing the native peptide with antimicrobial activity and selectivity towards Gram positive and Gram negative bacteria, we chose to investigate the peptide analogues L25P (Leu21-Lys45), L21P (Gly16-Arg36), C25P and C21P (cyclic forms of L25P and L21P, respectively) from the native HβD3 peptide (Table 1).
Fig 1.

The three-dimensional solution structure of HβD3 (pdb acquisition code, 1KJ6) with the highlighted region showing the positive charge density, could be responsible to interact with membrane and hence it’s antimicrobial activity.
Additionally, to elucidate the significance of the positively charged clusters residing at the C terminus of the native peptide, we specifically designed L21P and C21P excluding the C terminal residues present in L25P and C25P (Table 1). Further, we have retained the native Cys-Cys disulfide linkages in all the analogues, to rule out the possibility of non-native disulfide bond formations. In case of L25P and C25P we have replaced Cys 40 with Ser and Cys33 with Trp maintaining the native Cys23-Cys41 bond. Similarly, in the case of L21P and C21P, we replaced Cys25 with Trp, thereby to retain the native Cys18-Cys33 bonding. Since Trp can be used as a fluorescence probe due to its intrinsic fluorescence property, replacement of Cys with Trp aided us to perform the binding studies using fluorescence spectroscopy techniques.
Antimicrobial activity
The antimicrobial activity of the HβD3 analogs was performed against Gram-negative Salmonella enterica and Gram-positive Staphylococcus aureus. Both L25P and C25P were seen to be equally active against the strains tested, with inhibition of 40 % bacterial growth at a concentration of 50 μg/ml. Interestingly, L21P and C21P were also active against S. aureus, inhibiting 40 % growth at 50 μg/ml concentration, but could only inhibit 18 % and 22 %, respectively, of the growth of S. enterica at the same peptide concentration (Table 2). The increased activity of L25P and C25P could be explained by the presence of more hydrophobic residues and net positive charges. At molecular level, a more hydrophobic amphipathic peptide will interact better with the hydrophobic core present in the microbial membranes. However, none of analogs exhibited as potent activity as the native HβD3 peptide [32] [33], suggesting a distinct functional role for the presence of the N-terminal region in the native peptide.
Table 2.
Antimicrobial activities of HβD3-peptide analogs towards Gram-negative and Gram-positive bacteria. Representative table indicates the inhibition of growth (in %) as a function of peptide concentration (A) in Salmonella enterica (Gram-negative bacteria) and (B) in Staphylococcus aureus (Gram-positive bacteria).
| Peptide name | Percentage inhibition of growth | |||
|---|---|---|---|---|
| S. aureus | S. enterica | |||
| 50 μg/ml | 100 μg/ml | 50 μg/ml | 100 μg/ml | |
| L25P | 37 % | 40 % | 38 % | 50 % |
| C25P | 37 % | 40 % | 37 % | 45 % |
| L21P | 35 % | 38 % | 16 % | 18 % |
| C21P | 37 % | 37 % | 22 % | 24 % |
Membrane binding studies
The interaction of the peptide with either the zwitterionic or negatively charge membrane was studied using Trp as a fluorescent probe. Tryptophan is a unique amino acid capable of both making both hydrophobic and polar interactions. Based on the polarity of the environment in which it is present, its fluorescence property also varies.
Firstly, we investigated the fluorescence binding studies of HβD3 peptide analogs in the presence of LPS. LPS is the chief permeability barricade present in the outer membrane of the Gram-negative bacteria that hinders the entry of various antimicrobial agents, giving Gram-negative bacteria an advantage to survive. For its activity, the antimicrobial peptides need to cross this LPS layer to gain entry into the inner membrane and disrupt it, leading to cell death. The fluorescence of L25P and C25P showed a blue shift of 10 nm and 7 nm, respectively in the presence of LPS, indicating that the linear form of peptide binds slightly better than the cyclic peptide (Fig 2A). This result is in good agreement with the antimicrobial activity that we see for these peptides against Gram-negative S. enterica. Contrarily, L21P and C21P did not show any significant blue shift in the presence of LPS, which correlates well with their low activity against Gram-negative bacteria (Figure 2A). Further, upon addition of anionic POPE:POPG (3:1) vesicles, both C25P and L25P peptides exhibited a significant blue-shift increase of ~ 10 nm. Whereas, the shorter peptides, L21P and C21P, showed a blue shift of only 7 nm, indicating that the long peptides bind relatively stronger than the shorter ones. The subsequent shift in emission wavelength of the HβD3- analogs were plotted as a function of the lipid:peptide molar ratio and is shown in Figure 2B. Overall, in the case of HβD3- analogs, the fluorescence blue-shift effect can be explained by the direct interaction of this residue with the hydrophobic core of POPE:POPG vesicle, suggesting the penetration of tryptophan residues into the phospholipid bilayer. The fluorescence experiment was also performed in the presence of zwitterionic POPC vesicles for the HβD3 peptide analogs. It is found that the analogue peptides experience a very little blue shift of ~3 nm (Figure 2C) which indicates that these analogues possibly do not interact with the mammalian mimic membranes.
Fig 2.
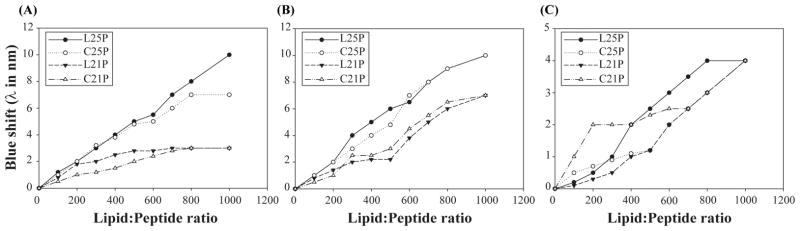
Fluorescence emission maximum of HβD3-peptide analogs in the presence of SUVs (A) POPC, (B) POPE:POPG (3:1) and (C) LPS at room temperature. Shift in emission maximum as a function of lipid:peptide ratio is represented in the graph. Maximum blue shift was observed for L25P and C25P in POPE:POPG vesicles indicating a strong penetration of the hydrophobic parts of the peptides into lipid vesicles.
Secondary structure of human defensin peptides
Circular dichoism (CD) spectroscopy has been used as a standard method to determine the secondary structures of proteins and peptides in aqueous buffer and its interaction with receptor or membrane. The secondary structure of the HβD-3 peptide analogues were characterized by CD measurement in aqueous buffer, POPC and POPE:POPG (3:1) vesicle (Figure 3). The cyclic as well as linear form of HβD-3 peptide analogues (C21P, C25P, L21P and L25P) showed good solubility in aqueous buffers without forming any large insoluble aggregates at the concentrations used herein. The CD spectra of the peptides (C21P, C25P and L21P) in aqueous solution as well as in the vesicle containing anionic or zwitterions, showed minimum polarity around 200 nm, indicating that these peptides failed to adopt any secondary structure in these media (data not shown). In contrast, the linear peptide L25P showed interesting conformational characteristics under the same sample condition. In the presence of HEPES buffer and zwitterionic POPC vesicle suspensions, L25P are least structured (Fig 3). However, in the presence of POPE:POPG (3:1) vesicle, pronounced structural differences were observed for L25P. The CD spectrum of this linear peptide showed double minima around 208 and 222 nm, which is characteristic of an α-helical structure in the presence of anionic vesicle (Fig 3). The adoption of a helical structure upon attachment to the membrane strongly, associates it with its antimicrobial activity. We could not record the CD spectra for HβD3-analogs in liposomes due to the turbidity of the sample caused by aggregation or fusion at a higher ratio of phospholipid.
Fig 3.

Secondary structure of HβD3-peptide analog in different media as revealed by CD spectroscopy. CD spectra of L25P peptide in HEPES (5 mM, 120 mM NaCl, and pH 7.4) buffer (represented by plus), POPC vesicle suspension (represented by open triangle), and in PE:PG vesicle suspensions (represented by closed diamond). The spectra were measured in 1 cm cuvette with 8 μM peptide concentration.
Peptide-induced leakage from carboxyfluorescein trapped lipid vesicles
Four representative peptides were tested for their ability to disrupt small unilamellar vesicles (SUV) prepared from POPC or POPE:POPG (3:1) lipids. Peptides at increasing concentrations were mixed with the SUVs (100 μM), which were pre-treated with the carboxyflouresceine dye. In our study the lipid-peptide molar ratio ranges from 1:l0 to 1:l00.
Only insignificant amount of membrane disruption was observed for all the HβD3 peptide analogs in the presence of dye trapped in POPC vesicle suspension, signifying their inactivity towards mammalian mimic membranes (Figure 4A). However, all the four analogs exhibit substantial membrane disruption activity in the presence of dye trapped in POPE:POPG SUVs (Figure 4B). Further, these peptides have more permeating activity in anionic lipid vesicles, that mimic the bacterial membrane, whereas, less active towards zwitterionic vesicles resembling mammalian membrane (Figure 4A). Additionally, L25P and C25P are the most potent peptides compared to L21P and C21P because of the presence of highly crowded positive charges at the C terminus and hydrophobic residues.
Fig 4.

Changes in the rate of carboxyfluorescein (CF) release upon the addition of HβD3-peptide analogs in zwitterionic and anionic vesicle suspensions at room temperature. Graph represents the percentage of CF-dye leakage as a function of the molar concentration of the HβD3-peptide analogs in vesicles. Peptide-induced CF-dye leakage in (A) POPC vesicle and (B) POPE:POPG (3:1) vesicle suspensions until a total peptide concentration of 10 μM. HβD3-peptide analogs did not induce CF leakage in zwitterionic POPC vesicles, however a significant CF release was observed in the presence of PE:PG vesicles under the conditions tested.
Hemolytic activity
Previous studies have shown that HβD3 has no hemolytic activity at physiological salt concentrations [14]. In this study, we performed hemolytic assay for all the analogs to find out hemolytic activity of the analogs. Interestingly, all the HβD3 analogues showed less than 5% and 10% of hemolysis for a maximum concentration of 100 and 200 μg/ml concentrations, respectively. These results are in agreement with the fact that all of the analogs do not interact with, or cause dye leakage from zwitterionic POPC vesicles that are mammalian membrane mimetic.
Solid-state NMR studies of peptide-lipid interaction
From the CD, fluorescence and dye-leakage assays we clearly see that the HBD-3 analog C25P peptide has the strongest affinity towards the mimics of the bacterial model membranes that are negatively charged compared to the mimics of the mammalian membranes. This result also serves as excellent criteria for any AMPs to discriminate its activity between bacterial and mammalian cell types. Moreover C25P being a cyclic peptide may be more bioavailable when compared to its linear counterpart and it is also found to be less cytotoxic towards human red blood cells. Hence, it is more meaningful to understand the specific nature of the lipid-peptide interaction at atomic-resolution using novel solid-state NMR experiments. Hence, we used solid-state NMR experiments on HBD-3 analogue C25P in the presence of the POPE:POPG MLVs. Phosphorus-31 NMR experiments under stationary conditions were carried out on POPE:POPG MLVs with and without C25P, by varying the concentration of the peptide from 0 to 5 mole %, to understand the effect of C25P interaction with lipid headgroups. The resultant spectra revealed significant peptide-induced distortions in the lamellar phase 31P line-shapes (data not shown). But, within the detection limit of NMR experiments, the spectra revealed the absence of any non-lamellar phases in the sample. The absence of any isotropic peak in the spectra suggests that the peptide does not function via detergent-type mechanism of membrane disruption.[34]
To probe the peptide-induced structural changes in the hydrophobic region of lipid bilayers, 2D R-PDLF [35] experiments were carried out as explained in our previous studies [36, 37]. 2D R-PDLF spectra were obtained with and 1 mole % C25P (data not shown). Comparing the difference in the C-H dipolar coupling values of the control (POPE:POPG MLVs at 3:1 ratio) and C25P (1 mol %) added to POPE:POPG MLVs at a ratio of 3:1, we could differentiate and identify certain peaks in the acyl chain region as well as the head group region. 1D C-H dipolar coupling spectral slices extracted from the 2D R-PDLF spectra near the alpha-carbon region of the lipid are shown in Fig 6. The C-H dipolar couplings spectra indicate that the interaction of the peptide with lipids induce motional disorder that result in the averaging of the observed C-H dipolar couplings. These results support the idea that the HBD-3 analog, C25P peptide, has the ability to fold into a relatively more ordered structure upon binding to negatively charged membranes. The negatively charged vesicles in turn can induce conformational transitions of the peptide in a way to increase the amphipathicity of the peptide. This sequel of conformational inductions enables the peptide to interact with bacterial membranes more effectively.
Fig. 6.

1H-13C dipolar coupling spectral slices extracted from 2D R-PDLF spectra obtained from 3:1 POPE:POPG MLVs without (A and C) and with (B and D) 1 mole % C25P at room temperature.
Based on our above results, we observed that HβD3 peptide analogs distinguish themselves from the native HβD3 antimicrobial peptide in the following characteristics: (i) they are significantly unstructured in the membrane as revealed by CD spectroscopy; (ii) C25P and L25P peptides penetrate more into the hydrophobic core of negatively charged membrane and less into the zwitterionic membrane as revealed by tryptophan fluorescence experiments; (iii) despite their unordered structure, they insert into the hydrophobic core of the lipid vesicles and presumably form pores as revealed by carboxyfluorescein-dye leakage recovered from fluorescence experiments; (iv) L25P peptide is significantly unstructured in aqueous buffer and in zwitterionic vesicles, whereas it adopts an α-helical structure in the presence of anionic vesicle suspensions, which corresponds to its activity in permeating bacterial mimicking negatively charged membranes.
The data presented in this article show a direct correlation between the ability of the peptide to insert itself into phospholipid membranes and its capability to permeate them (Fig 7). Presumably, the large number of positive charged residues at the C terminal region facilitates the initial binding to the negatively charged membrane through electrostatic interaction, whereby they adopt a perfectly amphipathic structure stabilized by hydrophobic interaction with the flexible N terminal arm propelling the disintegration of the membrane. A similar behavior for lactoferrampin was observed upon interaction with LPS [38]. The N-terminal region adopted an alpha helical conformation and oriented itself at an angle of 45° in LPS micelles, while the flexible C-terminal region facilitated the disintegration of the micelle paving way for the lysis of the cell. We are in a process of designing synthetic antimicrobial peptides on the basis of structure-activity relationship established for HβD3 in an attempt to come up with a novel spectrum of molecules having antimicrobial activity against a broad range of pathogens, antiendotoxic activity and negligible cytotoxicity. Such realized molecules would qualify as a promising candidate for peptide-based antibiotics in combating pathogenic diseases.
Fig. 7.

Structural snapshots adopted from molecular dynamics trajectory (0, 25 and 50 ns), revealing the conformation of L25P in POPC bilayer. The phosphate atoms are shown in spheres and the peptide is shown in a cartoon representation. Tryptophan (cyan), arginine (pale blue) and lysine (pink) residues are shown as spheres (A, C and E). Electrostatic potential of L25P peptide was calculated by solving APBS with an -surface potential representation using ±4 kT/e values (B, D and F) for similar snapshots. All figures were prepared using Pymol.
Molecular Dynamics Simulations
A plethora of studies using MD simulations have reported the structure, peptide-peptide interactions, peptide-lipid interactions and modes of action of antimicrobial peptides. Particularly, MD simulation studies utilizing model membranes and experimental constraints are highly valuable to obtain insights into the physiological function of antimicrobial peptides. Such studies can address many valuable biological questions. For example, the roles of lipid charge, lipid bilayer thickness, and lipid bilayer composition can be better understood by suitably combining experimental measurements with MD simulations. In fact, the differences in the membrane composition between bacterial and mammalian membranes are key in differentiating the function of AMPs [39–41]. Studies have shown that the presence of anionic lipids enhances the membrane binding affinity of AMPs to bacterial membranes, while the presence of cholesterol in mammalian membranes enhances the selectivity of AMPs by inhibiting the AMP-induced membrane disruption [42]. In a similar context, we have investigated the dynamicity of peptide L25P in POPC bilayer. Two systems were considered for molecular dynamics: one with helical conformation and another with random-coil conformation as described in the methodology section. RMSD analysis (Supporting Information Fig 1A) and the peptide conformation reveal the trajectory to be much conserved from 12 to 38 ns duration owing to a deviation within 2 Å range. A jump in conformation seems to take place for helical L25P in the later part of trajectory from 40 to 50 ns time scale as evidenced from the plot. A similar RMSD analysis was also carried out for the C terminal region (R22RKK25) and tryptophan region (K12WS14) to mark their relative deviation. Tryptophan region of L25P seems to be stable within the hydrophobic core of the POPC bilayer. It is thus the corresponding plot seems to be stable throughout the time scale window. The RMSD of C terminus region was found to be much fluctuating. It can be presumed that pattern of RMSD plot for the full peptide L25P is mainly driven by the C terminus RMSD. It should be noted that the initial model adopted for the simulation was a perfect helical structure for L25P (Fig 7A). A marked perturbation in the secondary structure was observed for L25P by the end of the simulation time scale, which suggests that the helical conformation was not consistent in the lipid environment. This result correlates well with the CD spectrum indicating a perturbation of the secondary structure in the presence of a lipid environment.
Interestingly the hypothesis in context to the membrane disruption by the positively charged residues (C terminus) is well judged from our theoretical investigation. Figure 7(C and E) corresponds to the snapshot of L25P conformation in a POPC bilayer at 25 and 50 ns, respectively. Positively charged residues (Arg and Lys) present at the C terminus of the peptide were found to interact with the negatively charged phosphate groups of the lipid. This interaction was important as it enforces the membrane disruption with their dynamics. It can be evidenced from the figure that with the course of the simulation the terminal residues were protruding out of the micelle environment. The membrane disintegration is governed by the electrostatic interaction (Figure 7B, D and F), where the electropositive charge of terminal residues disrupts the phosphate head group and accordingly the peptide was distended out. The extended part of our simulations, which includes the random-coil structure of L25P, also shows the similar disintegration of the lipid membrane. The phenomenon is found to be governed by a similar electrostatic interaction between the positively charged C terminal residues and the negatively charged phosphate groups. Notably, it will be difficult to adopt any particular secondary structure in a conventional MD simulation, because the potential energy barrier cannot be supplemented with molecular mechanics. It is hence not be correct to be judgemental regarding the adoption of a helical conformation from a random-coil for L25P.
Supplementary Material
Figure S1. RMSD plot for a helical conformation of L25P (A) and a random-coil (B) in POPC bilayer.
Fig 5.

Hemolytic activities of HβD3-peptide analogs against sheep erythrocytes. Grey colored bars represents hemolytic activity against erythrocytes at 200 μg/ml and blue colored bar represents hemolytic activity against erythrocytes at 100 μg/ml.
Highlights.
Underlining structural motifs of HβD-3 analogs designed for antimicrobial activity.
Elucidation of conformational details with lipid vesicles and lipopolysaccharides.
Cell membrane permeabilizing ability of HβD-3 analogs in phospholipid vesicles.
Activity of HβD-3 analogs against bacterial cell and human erythrocytes.
Role of charged terminal residues found to enhance the activity profile.
Acknowledgments
This study was supported by research funds from NIH (AI054515 to A.R.) and American Heart Association (to A.R.).
Abbreviations
- CD
Circular Dichroism
- DMPC
1,2-Dimyristoyl-sn-Glycero-3-Phosphocholine
- DMPG
1,2-Dimyristoyl-sn-Glycero-3-Phospho-rac-(1-glycerol)
- DMSO
Dimethyl Sulfoxide
- DSC
Differential Scanning Calorimetry
- HβD-3
Human beta defensin-3
- LPS
lipopolysaccharides
- NMR
Nuclear Magnetic Resonance
- PDLF
Proton-Detected Local Field
- POPC
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPG
1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phospho-rac-(1-glycerol)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frohm M, Agerberth B, Ahangari G, Ståhle-Bäckdahl M, Lidén S, Wigzell H, Gudmundsson GH. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. Journal of Biological Chemistry. 1997;272:15258–15263. doi: 10.1074/jbc.272.24.15258. [DOI] [PubMed] [Google Scholar]
- 2.Durr U, Sudheendra U, Ramamoorthy A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochimica et Biophysica Acta (BBA)-Reviews on Biomembranes. 2006;1758:8–25. doi: 10.1016/j.bbamem.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 3.Kaiser V, Diamond G. Expression of mammalian defensin genes. Journal of Leukocyte Biology. 2000;68:779–784. [PubMed] [Google Scholar]
- 4.Schröder JM. Epithelial antimicrobial peptides: innate local host response elements. Cellular and Molecular Life Sciences CMLS. 1999;56:32–46. doi: 10.1007/s000180050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dhople V, Krukemeyer A, Ramamoorthy A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochimica et Biophysica Acta (BBA)-Reviews on Biomembranes. 2006;1758:1499–1512. doi: 10.1016/j.bbamem.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Territo M, Ganz T, Selsted M, Lehrer R. Monocyte-chemotactic activity of defensins from human neutrophils. Journal of Clinical Investigation. 1989;84:2017. doi: 10.1172/JCI114394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chertov O, Michiel DF, Xu L, Wang JM, Tani K, Murphy WJ, Longo DL, Taub DD, Oppenheim JJ. Identification of defensin-1, defensin-2, and CAP37/azurocidin as T-cell chemoattractant proteins released from interleukin-8-stimulated neutrophils. Journal of Biological Chemistry. 1996;271:2935–2940. doi: 10.1074/jbc.271.6.2935. [DOI] [PubMed] [Google Scholar]
- 8.Ganz T, Selsted ME, Szklarek D, Harwig S, Daher K, Bainton DF, Lehrer RI. Defensins. Natural peptide antibiotics of human neutrophils. Journal of Clinical Investigation. 1985;76:1427. doi: 10.1172/JCI112120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selsted ME, Tang YQ, Morris W, McGuire P, Novotny M, Smith W, Henschen A, Cullor J. Purification, primary structures, and antibacterial activities of beta-defensins, a new family of antimicrobial peptides from bovine neutrophils. Journal of Biological Chemistry. 1993;268:6641–6648. [PubMed] [Google Scholar]
- 10.Bauer F, Schweimer K, Klüver E, Conejo-Garcia JR, Forssmann WG, Rösch P, Adermann K, Sticht H. Structure determination of human and murine β-defensins reveals structural conservation in the absence of significant sequence similarity. Protein Science. 2001;10:2470–2479. doi: 10.1110/ps.24401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valore EV, Park CH, Quayle AJ, Wiles KR, McCray PB, Jr, Ganz T. Human beta-defensin-1: an antimicrobial peptide of urogenital tissues. Journal of Clinical Investigation. 1998;101:1633. doi: 10.1172/JCI1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao C, Wang I, Lehrer RI. Widespread expression of beta-defensin hBD-1 in human secretory glands and epithelial cells. FEBS letters. 1996;396:319–322. doi: 10.1016/0014-5793(96)01123-4. [DOI] [PubMed] [Google Scholar]
- 13.Harder J, Bartels J, Christophers E, Schroder J. A peptide antibiotic from human skin. Nature. 1997;387:861–861. doi: 10.1038/43088. [DOI] [PubMed] [Google Scholar]
- 14.Harder J, Bartels J, Christophers E, Schroder J. Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. Journal of Biological Chemistry. 2001;276:7–13. doi: 10.1074/jbc.M008557200. [DOI] [PubMed] [Google Scholar]
- 15.Sawai M, Jia H, Liu L, Aseyev V, Wiencek J, McCray PB, Jr, Ganz T, Kearney W, Tack B. The NMR structure of human beta-defensin-2 reveals a novel alpha-helical segment. Biochemistry. 2001;40:10–16. doi: 10.1021/bi002519d. [DOI] [PubMed] [Google Scholar]
- 16.García JR, Jaumann F, Schulz S, Krause A, Rodríguez-Jiménez J, Forssmann U, Adermann K, Klüver E, Vogelmeier C, Becker D. Identification of a novel, multifunctional β-defensin (human β-defensin 3) with specific antimicrobial activity. Cell and tissue research. 2001;306:257–264. doi: 10.1007/s004410100433. [DOI] [PubMed] [Google Scholar]
- 17.Schutte B, Mitros J, McCray PB., Jr Discovery of five conserved beta -defensin gene clusters using a computational search strategy. Proceedings of National Academy of Sciences of the United States of America. 2002;99:29–33. doi: 10.1073/pnas.042692699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Z, Hoover DM, Yang D, Boulègue C, Santamaria F, Oppenheim JJ, Lubkowski J, Lu W. Engineering disulfide bridges to dissect antimicrobial and chemotactic activities of human β-defensin 3. Proceedings of the National Academy of Sciences. 2003;100:8880–8885. doi: 10.1073/pnas.1533186100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoover DM, Wu Z, Tucker K, Lu W, Lubkowski J. Antimicrobial characterization of human β-defensin 3 derivatives. Antimicrobial agents and chemotherapy. 2003;47:2804–2809. doi: 10.1128/AAC.47.9.2804-2809.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schibli D, Hunter H, Aseyev V, Starner T, Wiencek J, McCray PB, Jr, Tack B, Vogel H. The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. Journal of Biological Chemistry. 2002;277:79–89. doi: 10.1074/jbc.M108830200. [DOI] [PubMed] [Google Scholar]
- 21.Domadia PN, Bhunia A, Ramamoorthy A, Bhattacharjya S. Structure, interactions, and antibacterial activities of MSI-594 derived mutant peptide MSI-594F5A in lipopolysaccharide micelles: role of the helical hairpin conformation in outer-membrane permeabilization. Journal of the American Chemical Society. 2010;132:18417–18428. doi: 10.1021/ja1083255. [DOI] [PubMed] [Google Scholar]
- 22.Bhunia A, Domadia PN, Torres J, Hallock KJ, Ramamoorthy A, Bhattacharjya S. NMR structure of pardaxin, a pore-forming antimicrobial peptide, in lipopolysaccharide micelles mechanism of outer membrane permeabilization. Journal of biological chemistry. 2010;285:3883–3895. doi: 10.1074/jbc.M109.065672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sitaram N, Subbalakshmi C, Nagaraj R. Structural and charge requirements for antimicrobial and hemolytic activity in the peptide PKLLETFLSKWIG, corresponding to the hydrophobic region of the antimicrobial protein bovine seminalplasmin. International journal of peptide and protein research. 1995;46:166–173. doi: 10.1111/j.1399-3011.1995.tb01332.x. [DOI] [PubMed] [Google Scholar]
- 24.Saberwal G, Nagaraj R. Cell-lytic and antibacterial peptides that act by perturbing the barrier function of membranes: facets of their conformational features, structure-function correlations and membrane-perturbing abilities. Biochimica et Biophysica Acta (BBA)-Reviews on Biomembranes. 1994;1197:109–131. doi: 10.1016/0304-4157(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 25.Stewart JCM. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Analytical biochemistry. 1980;104:10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 26.Mao D, Wallace B. Differential light scattering and absorption flattening optical effects are minimal in the circular dichroism spectra of small unilamellar vesicles. Biochemistry. 1984;23:2667–2673. doi: 10.1021/bi00307a020. [DOI] [PubMed] [Google Scholar]
- 27.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. Journal of Computational Chemistry. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banerjee V, Kar RK, Datta A, Parthasarathi K, Chatterjee S, Das KP, Bhunia A. Use of a small peptide fragment as an inhibitor of insulin fibrillation process: a study by high and low resolution spectroscopy. Plos One. 2013;8:e72318. doi: 10.1371/journal.pone.0072318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dyer KM, Perkyns JS, Stell G, Pettitt BM. Site-renormalised molecular fluid theory: on the utility of a two-site model of water. Molecular Physics. 2009;107:423–431. doi: 10.1080/00268970902845313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R, Philipp DM, Repasky MP, Zhang LY, Berne BJ, Friesner RA, Gallicchio E, Levy RM. Integrated Modeling Program, Applied Chemical Theory (IMPACT) Journal of Computational Chemistry. 2005;26:1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kar RK, Suryadevara P, Jana J, Bhunia A, Chatterjee S. Novel G-quadruplex stabilizing agents: in-silico approach and dynamics. 2013;31:1497–1518. doi: 10.1080/07391102.2012.742246. [DOI] [PubMed] [Google Scholar]
- 32.Klüver E, Schulz-Maronde S, Scheid S, Meyer B, Forssmann WG, Adermann K. Structure-activity relation of human β-defensin 3: influence of disulfide bonds and cysteine substitution on antimicrobial activity and cytotoxicity. Biochemistry. 2005;44:9804–9816. doi: 10.1021/bi050272k. [DOI] [PubMed] [Google Scholar]
- 33.Jung S, Mysliwy J, Spudy B, Lorenzen I, Reiss K, Gelhaus C, Podschun R, Leippe M, Grötzinger J. Human β-defensin 2 and β-defensin 3 chimeric peptides reveal the structural basis of the pathogen specificity of their parent molecules. Antimicrobial agents and chemotherapy. 2011;55:954–960. doi: 10.1128/AAC.00872-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramamoorthy A. Beyond NMR spectra of antimicrobial peptides: dynamical images at atomic resolution and functional insights. Solid state nuclear magnetic resonance. 2009;35:201–207. doi: 10.1016/j.ssnmr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao X, Edén M, Levitt MH. Recoupling of heteronuclear dipolar interactions in solid-state NMR using symmetry-based pulse sequences. Chemical physics letters. 2001;342:353–361. [Google Scholar]
- 36.Smith PE, Brender JR, Ramamoorthy A. Induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides: the case of islet amyloid polypeptide. Journal of the American Chemical Society. 2009;131:4470–4478. doi: 10.1021/ja809002a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith PE, Brender JR, Dürr UH, Xu J, Mullen DG, Banaszak Holl MM, Ramamoorthy A. Solid-state NMR reveals the hydrophobic-core location of poly (amidoamine) dendrimers in biomembranes. Journal of the American Chemical Society. 2010;132:8087–8097. doi: 10.1021/ja101524z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghosh A, Datta A, Jana J, Kar RK, Chatterjee C, Chatterjee S, Bhunia A. Sequence context induced antimicrobial activity: insight into lipopolysaccharide permeabilization. Molecular bioSystems. 2014;10:1596–1612. doi: 10.1039/c4mb00111g. [DOI] [PubMed] [Google Scholar]
- 39.Hallock KJ, Lee DK, Omnaas J, Mosberg HI, Ramamoorthy A. Membrane composition determines pardaxin’s mechanism of lipid bilayer disruption. Biophys J. 2002;83:1004–1013. doi: 10.1016/S0006-3495(02)75226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Epand RF, Ramamoorthy A, Epand RM. Membrane lipid composition and the interaction of pardaxin: The role of cholesterol. Prot Pept Lett. 2006;13:1–5. [PubMed] [Google Scholar]
- 41.Lee DK, Brender JR, Sciacca MF, Krishnamoorthy J, Yu C, Ramamoorthy A. Lipid composition dependent membrane fragmentation and pore-forming mechanisms of membrane disruption by pexiganan (MSI-78) Biochemistry. 2013;52:3254–3263. doi: 10.1021/bi400087n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramamoorthy A, Lee DK, Narasimhaswamy T, Nanga RPR. Cholesterol reduces pardaxin’s dynamics - a barrel-stave mechanism of membrane disruption investigated by solid-state NMR. Biochim Biophys Acta, Biomembr. 2010;1798:223–227. doi: 10.1016/j.bbamem.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. RMSD plot for a helical conformation of L25P (A) and a random-coil (B) in POPC bilayer.