

Abstract
Congenital tyrosine hydroxylase deficiency (THD) is found in autosomal-recessive Dopa-responsive dystonia and related neurological syndromes. The clinical manifestations of THD are variable, ranging from early-onset lethal disease to mild Parkinson disease-like symptoms appearing in adolescence. Until 2014, approximately 70 THD patients with a total of 40 different disease-related missense mutations, five nonsense mutations, and three mutations in the promoter region of the tyrosine hydroxylase (TH) gene have been reported. We collected clinical and biochemical data in the literature for all variants, and also generated mutant forms of TH variants previously not studied (N = 23). We compared the in vitro solubility, thermal stability, and kinetic properties of the TH variants to determine the cause(s) of their impaired enzyme activity, and found great heterogeneity in all these properties among the mutated forms. Some TH variants had specific kinetic anomalies and phenylalanine hydroxylase, and Dopa oxidase activities were measured for variants that showed signs of altered substrate binding. p.Arg233His, p.Gly247Ser, and p.Phe375Leu had shifted substrate specificity from tyrosine to phenylalanine and Dopa, whereas p.Cys359Phe had an impaired activity toward these substrates. The new data about pathogenic mechanisms presented are expected to contribute to develop individualized therapy for THD patients.
Keywords: tyrosine hydroxylase deficiency, THD, Dopa responsive dystonia
Introduction
Tyrosine hydroxylase (TH; MIM #191290) is an aromatic amino acid hydroxylase (AAAH) that catalyzes the rate-limiting step in the synthesis of the catecholamines, dopamine, adrenaline, and noradrenaline. The enzyme is structurally and functionally related to phenylalanine hydroxylase (PAH; MIM #612349) and tryptophan hydroxylase 1 and 2 (TPH1; MIM #191060, TPH2; MIM #607478). All AAAHs are dependent on the cofactor tetrahydrobiopterin (BH4), molecular oxygen, and ferrous iron to hydroxylate their substrate. In addition to the hydroxylation of tyrosine (l-Tyr) to form dihydroxyphenylalanine (l-Dopa, TH can catalyze the hydroxylation of phenylalanine (l-Phe) to l-Tyr [Fitzpatrick, 1999], and the oxidation of l-Dopa [Haavik, 1997] (Fig.1).
Figure 1.
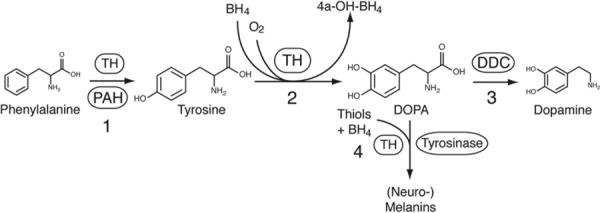
Enzymatic activities of tyrosine hydroxylase. In addition to the hydroxylation of l-Tyr to form l-Dopa, TH can catalyze the hydroxylation of l-Phe to l-Tyr, and the oxidation of l-Dopa.
In the brain, TH is mainly expressed in dopaminergic neurons in the ventral tegmental area and substantia nigra pars compacta, and in the noradrenergic neurons of the locus coeruleus. In the periphery, TH is mainly found in sympathetic neurons and in the adrenal medulla [Nagatsu and Ichinose, 1991]. In humans, a single TH gene encodes four main isoforms of TH protein (hTH), generated by alternative splicing of pre-mRNA [Grima et al., 1987]. Alternative splicing of mRNA results from the use of two donor sites in exon 1 and inclusion/exclusion of exon 2. Isoform 1 (hTH1) has no insertion and isoform 4 (hTH4) has the longest insertion of 31 additional amino acid residues. The nomenclature of missense and nonsense mutations in the TH gene in the literature refers to the corresponding amino acid substitution of the full-length form of the enzyme (hTH4). However, in some previous studies of TH variants, the nomenclature of the mutations has been based on hTH1 [Knappskog et al., 1995; Ludecke et al., 1996; Royo et al., 2005; Calvo et al., 2010]. The alternative splicing affects the enzyme regulation by phosphorylation, but only modest differences in the catalytic properties of the isoforms have been reported [Haavik et al., 1991; Nasrin et al., 1994; Gordon et al., 2009]. hTH1 is most abundant, and is also the isoform studied most commonly in vitro as a recombinant enzyme.
Mutations in the TH gene are found in patients with tyrosine hydroxylase deficiency (THD; MIM #605407), which is associated with autosomal-recessive Dopa-responsive dystonia and related neurological syndromes with predominantly motor symptoms [Willemsen et al., 2010]. The THD patients reported in the literature have symptoms ranging from mild Parkinson´s disease (PD)-like characteristics to severe neurodegenerative encephalopathy. Willemsen et al. (2010) reviewed the clinical and biochemical data on 36 patients and the literature describing phenotypes and genotypes of THD. They proposed to categorize the patients into two subgroups, THD type A and B, based on clinical features. Type A was defined as a progressive extrapyramidal movement disorder with onset in infancy or childhood. Type B is a more severe, complex encephalopathy with onset in the neonatal period or early infancy. The majority of patients belong to the type A category, and these patients generally respond well to treatment with l-Dopa. In contrast, patients who suffer from type B THD are often poor responders to substitution treatment with l-Dopa, and are possibly more prone to l-Dopa-induced dyskinesias [Pons et al., 2013]. Even within these groups, there is great variation in phenotype of patients with mutations in the TH gene. To date, no correlation between genotype and phenotype has been established. Homozygosity of the p.Arg233His mutation has been found in six type A and three type B patients. The p.Leu236Pro mutation occurred homozygously in one type A and two type B patients. However, all patients with at least one promoter mutation had the type A THD [Willemsen et al., 2010]. As all reported missense mutations of the TH gene in THD affect amino acid residues that are present in all isoforms of TH (hTH1-4), their effects on protein function should be evident in all four isoforms.
So far, seven of the reported missense mutations in the TH gene have been characterized at the protein level. Thus, reduced stability and/or activity was found for hTH1 p.Leu205Pro [Ludecke et al., 1996], p.Gln381Lys (corresponding to p.Leu236Pro and p.Gln412Lys in hTH4) [Knappskog et al., 1995], p.Thr245Pro, p.Thr283Met, p.Arg306His, p.Thr463Met (corresponding to p.Thr276Pro, p.Thr314Met, p.Arg337His, p.Thr494Met in TH4) [Royo et al., 2005], and most recently the TH mutant p.Arg202His (corresponding to p.Arg233His in TH4) [Calvo et al., 2010], which is the most common mutation found in THD patients [Willemsen et al., 2010].
The aim of the present study was to perform a molecular characterization and genotype–phenotype analyses of all protein coding variants reported in THD where clinical data are available. In addition to the seven missense mutations characterized biochemically earlier [Knappskog et al., 1995; Ludecke et al., 1996; Royo et al., 2005; Calvo et al., 2010], we characterized the enzymatic and biophysical properties of proteins carrying each of the 21 TH missense mutations and one nonsense mutation that have been reviewed [Willemsen et al., 2010], and a recently described Norwegian missense mutation, that is, p.Arg441Pro [Haugarvoll and Bindoff, 2011].
Here, we report remarkable variable effects of the disease mutations on TH, affecting the solubility, stability, activity, and substrate specificity of the enzyme. This novel knowledge about the molecular mechanism underlying disease progression has potential implications for future diagnosis and management of THD.
Materials and Methods
Materials
6R-tetrahydrobiopterin (BH4) was purchased from Dr. B. Schircks Laboratories (Jona, Switzerland). BL(21)D3pLysS Escherichia coli, and The Amplex® Red Hydrogen Peroxide/Peroxidase Assay kit were purchased from Invitrogen™ (Thermo Fisher Scientific Inc., Waltham, MA, US). Complete Protease inhibitor cocktail, EDTA-free and Lysis-B reagent were obtained from Roche (Mannheim, Germany). Other reagents for the TH activity assay were of analytical grade and purchased from Sigma (St. Louis, MO), unless otherwise stated.
Construction of Plasmids
Mutations were introduced into the wild-type (wt)-hTH1 cDNA on the pET3a-hTH1 vector [Le Bourdelles et al., 1991] by PCR-based mutagenesis using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA). NCBI Reference Sequence: NM_199292.2. Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence. Mutagenesis was carried out using primers from Invitrogen™ (Supp. Table S1). Introduction of the correct mutation and exclusion of other mutations were verified by Sanger sequencing of the whole coding region.
Expression and Purification of Proteins Expressed in E. coli
Recombinant wt human TH, isoform 1 (hTH1), and the mutant TH were expressed in BL(21)D3pLysS E. coli (Invitrogen™). The bacteria were grown at 37°C in LB medium containing 50 μg ml−1 of ampicillin and 34 μg ml−1 of chloramphenicol. The expression of T7 polymerase was induced at OD600 nm = 0.8 by addition of 1 mM isopropyl 6-D-thiogalactopyranoside. Temperature was decreased to 25°C after induction, and the bacteria were harvested after 6-h incubation. Bacteria pellets were kept at −20°C until purification.
Bacteria (from 1 l of culture) were diluted in 20 mM Tris/HCl pH 7.6, containing 5% sucrose (w/v), 1 mM dithiothreitol (DTT), 1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride, and one tablet Complete Protease inhibitor cocktail, EDTA-free (Roche)/25 ml, and disrupted by passage through a French press (type FA-073; SLM Instruments, Urbana, IL) at 69 MPa. The lysate was centrifuged at 12,000g for 20 min, and the supernatant used for purification by heparin sepharose chromatography [Haavik et al., 1991]. The purified enzymes were concentrated and stored in liquid nitrogen until used.
Solubility of TH, wt, and Mutants
hTH1- and THD-associated mutants were expressed as described above and bacteria were harvested by centrifugation at 4,000g for 20 min at 4°C. Cell pellets (5 ml cultures) were resuspended in Lysis-B reagent containing Complete Protease inhibitor cocktail, EDTA-free following the protocol provided by the manufacturer (Roche). Aliquots of the extracts were centrifuged at 10,000g for 20 min. Proteins in the soluble and insoluble fractions were analyzed by SDS-PAGE, with quantification of the bands by Quantity One software [McKinney et al., 2004].
Assays of Enzyme Activity and Data Analysis
TH activity was assayed at 25°C in a standard reaction mixture containing 200 μM BH4, 2 mM DTT, 10 μM Fe(II) ammonium sulfate, 0.1 mg ml−1 catalase, and 50 μM tyrosine (l-Tyr) in 40 mM Na-Hepes pH 7.0. The enzyme was diluted to 10 μM subunit concentration (0.56 mg ml−1) in buffer-containing 10 μM Fe(II) ammonium sulfate and 0.5 mg ml−1 BSA and preincubated on ice. The formation of Dopa was measured by HPLC with fluorimetric detection [Haavik and Flatmark, 1980]. PAH activity of wt and mutant TH was assayed in the same way as TH activity, with phenylalanine (l-Phe) replacing l-Tyr. l-Tyr formation was detected by HPLC. l-Dopa oxidase activity of wt and mutant TH was assayed by a radiochemical method as previously described [Haavik, 1997]. To determine the enzyme kinetic parameters of wt and mutant TH, the concentration of BH4 was varied (0–500 μM), keeping l-Tyr levels constant (50 μM), or l-Tyr concentrations were varied (0–200 μM), and BH4 levels were kept constant (200 μM). The Km values for l-Phe were determined using 0–300 μM l-Phe and 200 μM BH4.
We used nonlinear regression curve fitting in SigmaPlot (version 9.0; Systat Software, Inc., San Jose, CA) to determine the kinetic parameters for TH, wt, and mutants, using either a simple Michaelis–Menten (MM) equation or MM with substrate inhibition (Eq. 1). To obtain a robust comparison between mutant proteins, the number of parameters was kept at a minimum. Simple MM equation was used if no clear evidence for substrate inhibition was found in the substrate range used here (two or more measurements showing decreased activity at higher substrate concentrations). For the cofactor, Vmax and Km values were fitted to a MM equation to get more robust fits, and the Hill coefficients were obtained using nonlinear regression with the Hill equation (Eq. 2).
| (1) |
| (2) |
Thermal Stability of TH Activity
Wild type and mutant forms of TH were diluted in 10 μM Fe(II) ammonium sulfate and 0.5 mg ml−1 BSA to 10 μM enzyme subunit (0.56 mg ml−1), and incubated at 37°C, 45°C, and 55°C. Aliquots were taken at different time points and assayed for remaining TH activity as described above.
Oxygraphic Measurements
Oxygraphic assays of TH activities were carried out in a high-resolution respirometer (Oroboros Oxygraph, Innsbruck, Austria) [Gnaiger et al., 1995], essentially as described [Fossbakk and Haavik, 2005]. To reduce the background oxygen consumption, TH assay was modified as follows. The concentrations of Fe(II), BH4, and DTT was reduced to 4 μM, 125 μM, and 1.25 mM, respectively. Dioxygen concentration was digitally recorded at time intervals of 2 sec. Reaction rates were calculated as the negative time derivatives of dioxygen concentration and corrected for background from instrument and autoxidation of the incubation medium containing substrates and cofactors without enzyme. Typical background oxidation rate was 13.9 ± 2.6 pmol s−1 ml−1 at an initial oxygen concentration of 240 μM.
Detection of Hydrogen Peroxide (H2O2)
An AmplexRed kit (Invitrogen™) was used to quantify the amount of H2O2 produced as a byproduct in the TH reaction. The enzyme was incubated in 40 mM Na-HEPES pH 7.0, 50 μM l-Tyr, 30 μM BH4, and 1 μM Fe(II) ammonium sulfate for 4 min at 25°C before AmplexRed reagent was added. Further, we followed the procedure described in the protocol provided by the manufacturer.
Molecular Modeling/Prediction Analysis
The protein structural model for the localization of the TH variants was prepared based on the human TH X-ray diffraction structure (PDB 2XSN, a truncated form of the enzyme lacking the N-terminal first 192 residues and which was crystalized with a Zn2+ replacing the Fe2+ in the catalytic site). The active site of TH was modeled using the structure of the catalytic domain of human PAH with BH4 and 3-(2-thienyl)-l-alanine (PDB 1KW0) as template. Accelrys Discovery Studio v3 1.1.11157 was used for the preparation of structural models. Predicted severity of the mutations were estimated in MutPred (mutpred.mutdb.org) [Li et al., 2009]. FoldX (v3.0; foldx.crg.es) [Schymkowitz et al., 2005] was used to estimate the difference in protein stability (ΔΔG) of the mutants relative to wt-TH.
Results
Expression Efficiency and Solubility of wt and mutant TH in E. coli
We here report for the first time a detailed characterization of 23 missense mutations in TH found in THD patients. An overview of the nucleotide position of these and other THD mutations and the position of the amino acids altered in the three-dimensional structure of TH are shown in Figure2. In addition, we include a similar detailed characterization of seven THD mutations reported in four previous studies (Fig.2B, blue residues). Three promoter variants (c.-69T>A, c.-70G>A, and c.-71C>T) and protein variants (p.Gly294Arg, p.Gly315Ser, p.Ala385Val, and p.Gly408Arg) where incomplete clinical data have been reported are also shown in Figure2, but they have not been analyzed here. TH mutations reported after the start of the study [Chi et al., 2012; Stamelou et al., 2012; Cai et al., 2013] were not analyzed biochemically.
Figure 2.
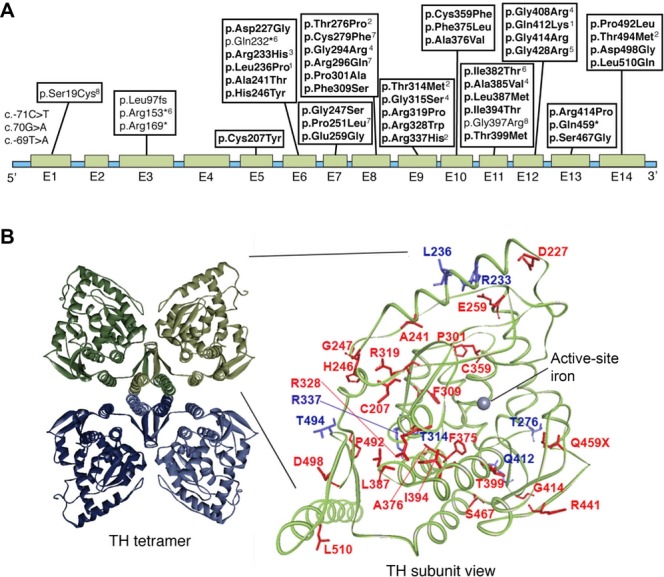
Mutations in the TH gene. A: Schematic drawing of the genomic structure and location of mutations in human TH gene reported by November 2013. Mutations without annotation are from Willemsen et al. (2010), and the mutations in bold are those studied here. Reference sequence: NM_199292.2 (UniGene) transcript variant a (hTH4 with 528 amino acid). 1[Knappskog et al., 1995; Ludecke et al., 1996], 2[Royo et al., 2005], 3[Calvo et al., 2010], 4 [Mak et al., 2010], 5[Stamelou et al., 2012], 6[Chi et al., 2012], 7[Giovanniello et al., 2012], 8[Cai et al., 2013]. B: Tetrameric TH including the catalytic and tetramerization domains (left, PDB 1TOH) with one subunit detailed using human TH crystal structure (right, PDB 2XSN). All THD mutations expressed here for the first time are located in the catalytic and tetramerization domain (right amplification), and are colored red, whereas mutations characterized by others [Knappskog et al., 1995; Ludecke et al., 1996; Royo et al., 2005; Calvo et al., 2010] are shown in blue. The model was prepared using the software Accelrys Discovery Studio.
Wild type and mutated forms of TH were produced without fusion partners in E. coli, and soluble enzyme was purified by Heparin Sepharose chromatography [Haavik et al., 1991]. Bacteria were homogenized, and soluble and insoluble fractions were analyzed with SDS-PAGE with Coomassie-blue staining. For the wt TH, 43% of the protein appeared in the soluble fraction; however, the solubility of the THD mutants varied between 7% and 50% (Fig.3A). The amount of soluble TH protein in the bacterial lysates for the TH mutants p.Cys207Tyr, p.Asp227Gly, p.Ala241Thr, p.His246Tyr, p.Gly247Ser, p.Pro301Ala, p.Phe309Ser, p.Leu387Met, p.Thr399Met, p.Gly414Arg, p.Gln459*, and p.Ser467Gly was not significantly different from wt TH. However, for the missense variants p.Glu259Gly, p.Arg319Pro, p.Arg328Trp, p.Cys359Phe, p.Phe375Leu, p.Ala376Val, p.Ile394Thr, p.Arg441Pro, p.Pro492Leu, p.Asp498Gly, and p.Leu510Gln, the TH protein in the soluble fraction was significantly reduced compared with wt TH (P < 0.05 or <0.005, t-test) (Fig.3A), where the mutants marked in bold showed the largest decrease in solubility (four to sevenfold).
Figure 3.
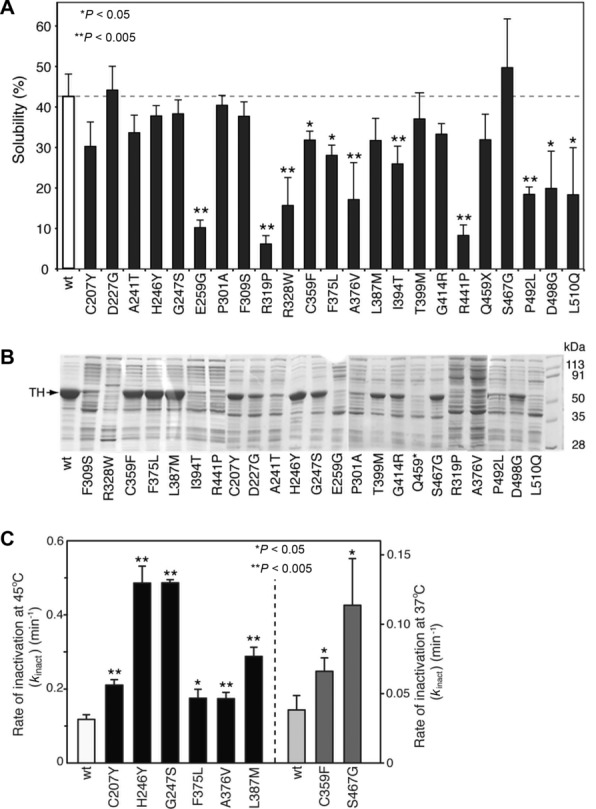
Solubility and stability of the missense variants. A: The solubility of wt and mutant TH was compared. The fraction of soluble TH (mean ± SD for N = 3–6) is given from measurements upon lysis of the bacteria (Materials and Methods). Protein in the soluble fraction of mutant TH p.Cys207Tyr, p.Asp227Gly, p.Ala241Thr, p.His246Tyr, p.Gly247Ser, p.Pro301Ala, p.Phe309Ser, p.Leu387Met, p.Thr399Met, p.Gly414Arg, p.Gln459*, and p.Ser467Gly was not significantly different from wt TH. For mutant TH, p.Glu259Gly, p.Arg319Pro, p.Arg328Trp, p.Cys359Phe, p.Phe375Leu, p.Ala376Val, p.Ile394Thr, p.Arg441Pro, p.Pro492Leu, p.Asp498Gly, and p.Leu510Gln protein in the soluble fraction was significantly reduced compared with wt TH (*P < 0.05 or **P < 0.005, t-test). B: Shows the protein yield of wt and mutant TH. Coomassie-blue SDS-PAGE gel (12% acrylamide) showing total protein from E. coli lysates purified by Heparin Sepharose chromatography. Last lane show molecular weight standards. The position of TH protein is indicated (left) and the lanes are marked with enzyme variant. C: Compares the thermal inactivation rates of wt and mutant forms of TH. The figure shows the rate of thermal inactivation as a measure of stability of wt TH and the mutants p.Cys207Tyr, p.His246Tyr, p.Gly247Ser, p.Phe375Leu, p.Ala376Val, p.Leu387Met, p.Cys359Phe, and p.Ser467Gly. Enzyme activity was assayed as described in Methods after incubation at 45°C or 37°C. Pure protein samples diluted in 10 μM Fe(II) ammonium sulfate and 0.5 mg ml BSA were incubated at 45°C or 37°C for 0–30 min. Aliquotes were assayed for TH activity at different time points and the rate constant of inactivation (kinact) was determined by fitting to an exponential decay curve (Methods). Values are given as means ± SEM and represent data from three independent experiments. The rates of inactivation of mutants TH p.Cys359Phe and p.Ser467Gly were too high at 45°C, and the values given in this figure were obtained at 37°C (right panel). The stability of each of the mutants was significantly different from wt TH (*P < 0.05, or **P < 0.005, t-test).
The solubility of TH in the bacterial extracts also affected the amount of soluble enzyme that was recovered after partial purification by heparin sepharose chromatography. Thus, the yield of p.Cys207Tyr, p.His246Tyr, p.Gly247Ser, p.Cys359Phe, p.Phe375Leu, p.Leu387Met, p.Thr399Met, p.Gly414Arg, p.Ser467Gly, and p.Asp498Gly was similar (>50%) to that of wt TH. The TH p.Asp227Gly, p.Ala241Thr, p.Pro301Ala, p.Phe309Ser, p.Pro492Leu, p.Gln459*, and p.Ile394Thr mutants had intermediate (10%–50%) purification yield, whereas TH protein was hardly detectable for the TH mutants p.Arg328Trp, p.Arg441Pro, p.Glu259Gly, p.Arg319Pro, p.Ala376Val, and p.Leu510Gln (Fig.3B).
Thermal Stability
We next assessed the thermal stability of wt and mutated forms of TH by estimating their first-order inactivation rate constant (kinact) at 45°C, a measure of the kinetic stability of the enzymes (Fig.3C). For the TH mutants, p.Cys359Phe and p.Ser467Gly, we report kinact constants measured at 37°C, as the rate at 45°C was too high to be measured accurately. Interestingly, all mutants tested had significantly higher rates of inactivation compared with wt TH (P < 0.05 or < 0.005, t-test). The most dramatic changes were found for the TH mutants p.His246Tyr, p.Gly247Ser, and p.Ser467Gly where kinact increased >threefold, whereas the p.Leu387Met mutant had a twofold higher kinact.
Enzyme Activity and Kinetic Properties of wt and Mutated Forms of TH
The enzymatic activity of wt and mutant TH was measured using standardized in vitro assay conditions as described above. The TH mutants p.Asp227Gly, p.Glu259Gly, p.Phe309Ser, p.Arg319Pro, p.Arg328Trp, p.Ile394Thr, p.Arg441Pro, p.Gln459*, p.Pro492Leu, and p.Leu510Gln had no measurable enzyme activity (<0.2% compared with wt TH) and were therefore not included in a detailed study of their enzyme kinetic properties. Less than 20% residual activity was found for TH mutants p.Cys207Tyr, p.Ala241Thr, p.Pro301Ala, p.Cys359Phe, p.Phe375Leu, p.Ala376Val, p.Thr399Met, p.Gly414Arg, p.Ser467Gly, and p.Asp498Gly, whereas mutants p.His246Tyr, p.Gly247Ser, and p.Leu387Met had 50%–100% activity compared with wt TH. All mutants with measurable enzyme activity were investigated further to determine their kinetic properties (Table1).
Table 1.
Kinetic Properties of wt and Mutant TH
| Protein | Mutation | E.S.a | Activity (%) | Vmax(Tyr) (nmol min−1 mg−1) | Km(Tyr) (μM) | Ksi(Tyr) (μM) | Activity (%) | Vmax(BH4)b (nmol min−1mg−1) | Km(BH4)b (μM) | Hill coefficientc |
|---|---|---|---|---|---|---|---|---|---|---|
| wt THd | 100 | 2460 ± 420 | 5.1 ± 2.2 | 59 ± 18 | 100 | 591 ± 33 | 39 ± 8.4 | 0.73 ± 0.23 | ||
| wt THe | − | 1100 ± 140 | 17 ± 4.0 | 92 ± 25 | - | 800 ± 100 | 46 ± 1.5 | 0.56 ± 0.07 | ||
| wt THf | 100f | 3570 ± 36i | 46 ± 1.0 | 46 ± 1.0 | − | − | 13 ± 2.0 | − | ||
| wt THg | TNT | 100g | 164 ± 9 | 9.9 ± 0.30 | − | − | − | 53 ± 3.0 | 1.2 | |
| 100g | 259 ± 13 | 6.6 ± 0.20 | n.d. | − | n.d. | n.d. | 1.5 | |||
| p.Cys207Tyrd | c.620G>A | 18 | 454 ± 22 | 83 ± 8.7 | n.a. | 23 | 136 ± 4.7 | 8.2 ± 0.41 | ≈2 | |
| p.Asp227Glyd | c.680A>G | <0.2 | n.d. | n.d. | n.d. | <0.2 | n.d. | n.d. | n.d. | |
| p.Arg233Hise | c.698A>G | 6.0 | 150 ± 30 | 13 ± 4.0 | 131 ± 52 | 15 | 90 ± 10 | 28 ± 5.7 | 0.88 | |
| p.Leu236Proh | c.707T>C | TNT | 16 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| 1.5 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | |||
| HEK293 | 0.3 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | ||
| p.Ala241Thrd | c.721G>A | 1.7 | 42 ± 12 | 23 ± 11 | 108 ± 72 | 4.2 | 25 ± 2.1 | 6.7 ± 3.4 | ||
| p.His246Tyrd | c.736C>T | 59 | 1440 ± 111 | 6.8 ± 1.7 | 59j | 55 | 328 ± 30 | 16 ± 4.7 | 1.2 ± 0.50 | |
| p.Gly247Serd | c.739G>A | 48 | 1189 ± 53 | 11.8 ± 0.87 | 49 ± 4.0 | 43 | 253 ± 9.7 | 6.6 ± 1.1 | 1.3 ± 0.36 | |
| p.Glu259Glyd | c.776A>G | <0.2 | n.d. | n.d. | n.d. | <0.2 | n.d. | n.d. | n.d. | |
| p.Thr276Prof | c.826A>C | 161f | 5730 ± 143i | 66 ± 5.0 | 73 ± 7.0 | − | − | 14 ± 1.0 | n.d. | |
| p.Pro301Alad | c.901C>G | 1.1 | 28 ± 11 | 14 ± 8.6 | 32 ± 21 | 4.7 | 28 ± 1.5 | 9.6 ± 2.0 | 0.48 ± 0.18 | |
| p.Phe309Serd | c.926T>G | <0.2 | n.d. | n.d. | n.d. | <0.2 | n.d. | n.d. | n.d. | |
| p.Thr314Metf | c.941C>T | 24f | 857 ± 18 | 39 ± 2.0 | 37 ± 2.0 | − | 10 ± 1.0 | − | ||
| p.Arg319Prod | c.956G>C | <0.2 | n.d. | n.d. | n.d. | <0.2 | n.d. | n.d. | n.d. | |
| p.Arg328Trpd | c.982C>T | < 0.2 | n.d. | n.d. | n.d. | <0.2 | n.d. | n.d. | n.d. | |
| p.Arg337Hisf | c.1010G>A | 111f | 3950 ± 89 i | 42 ± 3.0 | 48 ± 3.0 | − | − | 10 ± 1.0 | n.d. | |
| p.Cys359Phed | c.1076G>T | 13 | 329 ± 4.2 | 38 ± 1.1 | n.a. | 8.3 | 49 ± 2.7 | 8.2 ± 1.8 | 0.88 ± 0.36 | |
| p.Phe375Leud | c.1125C>G | 9.8 | 241 ± 20 | 56 ± 11 | n.a. | 43 | 256 ± 11 | 36 ± 5.2 | 1.27 ± 0.16 | |
| p.Ala376Vald | c.1127C>T | 8.4 | 325 ± 107 | 13 ± 6.9 | 30 ± 16 | 25 | 146 ± 13 | 15 ± 4.4 | 1.21 ± 0.36 | |
| p.Leu387Metd | c.1159C>A | 115 | 2830 ± 1450 | 24 ± 17 | 24 ± 18 | 147 | 866 ± 18 | 55 ± 4.3 | 0.65 ± 0.24 | |
| p.Ile394Thrd | c.1181T>C | <0.2 | n.d. | n.d. | n.d. | <0.2 | n.d. | n.d. | n.d. | |
| p.Thr399Metd | c.1196C>T | 4.6 | 112 ± 6.4 | 11 ± 2.2 | n.a. | 21 | 126 ± 33j | 19 ± 9.2j | n.d. | |
| p.Gln412Lysg | c.1234C>A | TNT | 18g | 30 ± 4.0 | 34 ± 2.0 | n.a. | − | − | 34 ± 4.0 | 0.8 |
| 41g | 106 ± 5.0 | 40 ± 1.9 | n.a. | − | n.d. | n.d. | 1.2 | |||
| p.Gly414Argd | c.1240G>A | 2.7 | 67 ± 7.5 | 27 ± 8.1 | 59j | 15 | 88 ± 4.4 | 15 ± 2.7 | 1.6 ± 0.3 | |
| p.Arg441Prod | c.1322G>C | <0.2 | n.d. | n.d. | n.d. | <0.2 | n.d. | n.d. | n.d. | |
| p.Gln459*d | c.1375C>G | < 0.2 | n.d. | n.d. | n.d. | <0.2 | n.d. | n.d. | n.d. | |
| p.Ser467Glyd | c.1399A>G | 19 | 595 ± 71 | 27 ± 8.7 | n.a. | 48 | 286 ± 10 | 6.9 ± 1.0 | 1.0 ± 0.3 | |
| p.Pro492Leud | c.1475C>T | <0.2 | n.d. | n.d. | n.d. | <0.2 | n.d. | n.d. | n.d. | |
| p.Thr494Metf | c.1481C>T | 116f | 4140 ± 107i | 43 ± 3.0 | 44 ± 3.0 | − | − | 9.0 ± 1.0 | n.d. | |
| p.Asp498Glyd | c.1493A>G | 5.9 | 179 ± 57 | 9.3 ± 5.4 | 46 ± 28 | 13 | 74 ± 6.0 | 6.4 ± 2.2 | 0.4 ± 0.5 | |
| p.Leu510Glnd | c.1529T>A | <0.2 | n.d. | n.d. | n.d. | <0.2 | n.d. | n.d. | n.d. |
NCBI Reference Sequence: NM_199292.2. Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence, with the initiation codon as codon 1.
Expression system other than E. coli.
Estimated using standard MM equation.
The Hill coefficient for BH4 was estimated from the data using sigmoid kinetics (Eq. 2).
This work.
[Calvo et al., 2010].
[Royo et al., 2005].
[Knappskog et al., 1995].
[Ludecke et al., 1996].
Calculated from reported kcat values using TH subunit molecular weight of 56 kDa.
Ksi(Tyr) for wt during fitting of kinetic parameters.
kFitted with substrate inhibition (Eq(1), Ksi = 187 μM obtained.
n.a., not apparent; n.d., not determined.
We observed differences in all kinetic constants assessed. As shown in Table1, some of the mutants had up to 16- and 1.4-fold increased Km values for l-Tyr and BH4, respectively. However, the majority of the TH mutants had decreased Km values for BH4 and increased Km values for tyrosine compared with wt TH. The mutants p.His246Tyr and p.Gly247Ser had about 50% lower Vmax values, but only moderately changed Km(Tyr), whereas they had increased affinity for BH4 and had lost their negative cooperativity for the cofactor. Together with p.Leu387Met, they therefore had no clear enzyme kinetic aberrancies that could explain their impaired enzyme function. Interestingly, several of the mutants showed no apparent substrate inhibition with tyrosine (n.a. for Ksi[Tyr] in Table1).
THD Mutants Show Altered Substrate Selectivity
In addition to the main physiological substrates tyrosine and BH4, TH can utilize alternative amino acid substrates (Fig.1) and many synthetic BH4 derivatives [Teigen et al., 2004]. The altered Km values could indicate that some mutations affect the substrate binding sites in the enzyme, possibly also leading to altered substrate specificity. To explore this possibility, the PAH and Dopa oxidase activities were measured for enzyme variants that had relatively intact catalytic activity and stability, but altered kinetic properties. The mutant p.Arg233His characterized previously [Calvo et al., 2010] was included in these experiments. As for the wt enzyme, all TH variants tested also had significant PAH activities (Table2). The p.Phe375Leu mutant had in fact increased Vmax for phenylalanine along with a 10-fold decrease in its tyrosine hydroxylase activity. However, this efficiency was moderated by an increase in the Km value for both substrates, making the substrate specificity only twofold different between tyrosine and phenylalanine. All mutants except the p.Cys359Phe mutant had lost much of their substrate selectivity, mainly driven by increased Km for tyrosine and lowered Km for phenylalanine. The p.Gly414Arg mutant was in fact a more efficient hydroxylase of phenylalanine than of tyrosine. The opposite situation was found for the p.Cys359Phe mutant, which had lost more of its efficiency toward phenylalanine than for tyrosine.
Table 2.
Amino Acid Substrate Specificity of wt TH and Selected Mutants
| Protein (mutation) | Substrate | Vmax (nmol min−1mg−1) | Km (μM) |  |
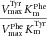 |
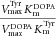 |
|---|---|---|---|---|---|---|
| wt TH | l-Tyr | 2460 ± 417 | 5.1 ± 2.2 | 484 | 88 | 114 |
| l-Phe | 1640 ± 148 | 296 ± 69 | 5.5 | |||
| l-Dopa | 126 ± 11 | 29.8 ± 8.5 | 4.2 | |||
| p.Arg233His | l-Tyr | 150 ± 30 | 13.0 ± 4.0 | 11.5 | 4.8 | 7.7 |
| (c.698A>G) | l-Phe | 352 ± 33 | 149 ± 22 | 2.4 | ||
| l-Dopa | 1.6 | 1.0 | 1.5 | |||
| p.Gly247Ser | l-Tyr | 1189 ± 53 | 11.8 ± 0.9 | 101 | 16 | 36 |
| (c.739G>A) | l-Phe | 734 ± 46 | 119 ± 25 | 6.2 | ||
| l-Dopa | 50 ± 9.4 | 17 ± 12 | 2.8 | |||
| p.Cys359Phe | l-Tyr | 329.4 ± 4.2 | 37.6 ± 1.1 | 8.8 | 80 | |
| (c.1076G>T) | l-Phe | 73.2 ± 5.7 | 663 ± 99 | 0.1 | ||
| l-Dopa | n.m. | n.m. | ||||
| p.Phe375Leu | l-Tyr | 241 ± 20 | 56 ± 11 | 4.3 | 2.2 | 11 |
| (c.1125C>G) | l-Phe | 1960 ± 44 | 993 ± 37 | 2.0 | ||
| l-Dopa | 77 ± 30 | 202 ± 135 | 0.4 | |||
| p.Leu387Met | l-Tyr | 2830 ± 1450 | 24 ± 17 | 118 | 21 | 131 |
| (c.1159C>A) | l-Phe | 1407 ± 78 | 258 ± 38 | 5.5 | ||
| l-Dopa | 62 ± 14 | 68 ± 39 | 0.9 | |||
| p.Gly414Arg | l-Tyr | 66.8 ± 7.5 | 26.6 ± 8.1 | 2.5 | 0.5 | 1.3 |
| (c.1240G>A) | l-Phe | 452 ± 17 | 92 ± 12 | 4.9 | ||
| l-Dopa | 35.1 ± 7.2 | 18 ± 13 | 1.9 |
The table summarizes the different enzyme kinetic parameters obtained for wt TH and some of the mutants, using l-Tyr, l-Phe, or l-Dopa as substrate. We did not observe substrate inhibition kinetics for l-Phe or l-Dopa, which is why only Vmax and Km values are reported. The data for l-Tyr are taken from Table1. Data points were fitted using standard MM kinetics by nonlinear regression (Sigma plot) as described in Materials and Methods section. Values are given as the best estimate ± standard error of estimate. n.m., not measured.
l-Dopa has been shown to be a substrate for TH (Fig.1), with comparable Vmax and Km values as for tyrosine [Haavik, 1997]. The TH-mediated oxidation of Dopa is thought to lead to the formation of neuromelanin and possibly to be involved in Dopa-mediated toxicity [Haavik and Toska, 1998]. The Dopa oxidase activity of wt TH and 12 mutants with >2% residual TH activity was compared. Under experimental conditions that were optimized for tyrosine hydroxylation, the wt enzyme oxidized both substrates, but with a Vmax/Km ratio for tyrosine that was more than 100-fold higher than for l-Dopa. As shown in Table2, p.Cys359Phe and to a lesser extent p.Leu387Met had increased selectivity for tyrosine over l-Dopa compared with the wt, whereas the other mutants (p.Arg233His, p.Gly247Ser, p.Phe375Leu, p.Leu387Met, and p.Gly414Arg) showed lowered substrate selectivity. In particular, the pGly414Arg mutant was a more efficient PAH than tyrosine hydroxylase and was approximately similarly efficient in oxidizing l-Dopa as tyrosine.
Oxygraphic Measurements and Coupling Efficiency
Suboptimal substrate binding could potentially also affect the reaction stoichiometry and reaction coupling of TH. For the related enzyme PAH and other oxidoreductases, some disease-associated mutations show altered reaction stoichiometry with uncoupling of substrate consumption to product formation [Kemsley et al., 2003]. Excess oxygen consumption in the enzyme reaction can lead to the formation of reactive oxygen species (ROS) as side products. To detect possible uncoupling of their hydroxylation reaction, all mutant forms of TH studied here were screened for enzyme activity using an oxygraphic assay [Fossbakk and Haavik, 2005]. For the TH variants p.His246Tyr, p.Gly247Ser, p.Phe375Leu, p.Leu387Met, and p.Ser467Gly that had high catalytic efficiency, we obtained high-quality oxygraphic recordings of oxygen consumption during the enzyme reaction. Samples were taken at fixed time intervals during the reaction, and the formation of l-Dopa was measured by HPLC.
As previously described [Rostrup et al., 2008], the wt TH reaction has a characteristic pattern of oxygen consumption with an initial burst phase and a steady-state reaction rate after 1–2 min [Flatmark et al., 1999]. This reaction pattern and the time courses for oxygen consumption in the oxygraphic assay and l-Dopa production (measured by HPLC) and coupling ratios were similar for wt TH and the mutant TH variants that had measurable activity (Supp. Fig. S1). To further investigate whether any of the mutants had an impaired reaction coupling efficiency, resulting in the production of ROS, we compared the production H2O2 by wt TH and 23 mutant enzymes. However, we found no significant difference in H2O2 production by these enzymes (data not shown).
Discussion
In human samples sequenced so far, p.Val112Met appears to be the only common TH missense variant (frequency 0.418) [Ludecke and Bartholome, 1995]. In contrast to this nonpathogenic amino acid change in the regulatory domain, missense mutations reported in THD are almost exclusively found in the catalytic or tetramerization domains (Fig.2). This also allowed us to purify the mutant proteins using heparin sepharose, which relies on an intact heparin-binding site in the N-terminal [Daubner and Piper, 1995]. Our characterization of 23 of these mutants are in accordance with previous studies of seven other variants of TH found in THD, that all show decreased stability or activity compared with wt TH [Knappskog et al., 1995; Ludecke et al., 1996; Royo et al., 2005; Calvo et al., 2010]. However, various mutated forms of TH show great diversity in stability and activity compared with wt TH (Table3).
Table 3.
Summary of Features of Mutant TH and Phenotypic Classification of Associated THD (Type A and B)
| Mutation | Type THD | Solubility | Residual activity | Thermal stability | Dominant feature | MutPred predictiona | ΔΔGb (kcal mol−1) |
|---|---|---|---|---|---|---|---|
| p.Cys207Tyrc | A | Moderately reduced | <20% | Moderately reduced | Activity | 0.790, −M*, +P** | 2.3:4.0 |
| p.Asp227Glyc | A | Not reduced | <0.2% | n.d. | Activity | 0.960 | 6.6:6.7 |
| p.Arg233Hisd | A/B, homozygous | 14% | Moderately reduced | Activity | 0.980 | 2.6:2.5 | |
| p.Leu236Proe | A/B, homozygous | Reduced | 16%f | Activity | 0.842, −S* | 4.5:4.5 | |
| 1.5% | Activity | ||||||
| 0.3% g | Activity | ||||||
| p.Ala241Thrc | A/B | Moderately reduced | <5% | n.d. | Activity | 0.858 | 0.56:1.0 |
| p.His246Tyrc | A | Not reduced | >50% | Severely reduced | Stability | 0.784 | −0.35:−0.49 |
| p.Gly247Serc | A | Not reduced | <50% | Severely reduced | Stability | 0.797, −C* | 4.3:3.7 |
| p.Glu259Glyc | A | Sign reduced | n.d. | n.d. | Solubility/stability | 0.816 | 5.3:5.0 |
| p.Thr276Proh | A | – | 100% | Sign reduced | Stability | 0.845, −S* | −0.19:−0.19 |
| p.Pro301Alac | B, homozygous | Not reduced | <5% | n.d. | Activity | 0.892 | 2.6:2.6 |
| p.Phe309Serc | B, homozygous | Moderately reduced | <0.2% | n.d. | Activity | 0.869, −S*, +D** | 4.3:4.1 |
| p.Thr314Meth | A | – | 24% | Severely reduced | Stability | 0.956 | 4.5:4.6 |
| p.Arg319Proc | A | Sign reduced | <0.2% | n.d. | Solubility/stability | 0.863, −M* | 4.3:4.4 |
| p.Arg328Trpc | B | Sign reduced | <0.2% | n.d. | Solubility/stability | 0.918, −M* | 1.6:1.4 |
| p.Arg337Hish | A | – | 100% | Severely reduced | Stability | 0.920 | 7.7:5.9 |
| p.Cys359Phec | B, homozygous | Moderately reduced | <10% | Severely reduced | Activity, stability | 0.720 | 2.8:4.5 |
| p.Phe375Leuc | B | Moderately reduced | <10% | Moderately reduced | Activity | 0.962 | 1.1:1.1 |
| p.Ala376Valc | A | Sign reduced | <10% | Moderately reduced | Solubility | 0.972 | 1.9:1.8 |
| p.Leu387Metc | A | Moderately reduced | >50% | Severely reduced | Stability | 0.881 | 0.58:0.65 |
| p.Ile394Thrc | A | Sign reduced | <0.2% | n.d. | Solubility/stability | 0.817, +D*, +P*, +U* | 2.8:2.8 |
| p.Thr399Metc | B | Moderately reduced | <5% | n.d. | Activity | 0.853 | 0.0:0.1 |
| p.Gln412Lyse | A, homozygous | Reduced | 18%f | Activity | 0.808, +M*, +U* | 0.33:0.45 | |
| 65% | Activity | ||||||
| p.Gly414Argc | A | Moderately reduced | <10% | n.d | Activity, stability | 0.844, −U* | 1.8:1.9 |
| p.Arg441Proc | A | Sign reduced | <0.2% | n.d. | Solubility/stability | 0.557 | 3.8:3.8 |
| p.Gln459*c | B | Moderately reduced | <0.2% | n.d. | Activity | - | 0.87:0.82 |
| p.Ser467Glyc | A | Not reduced | <20% | Severely reduced | Thermal stability | 0.901, −S*, −D* | 1.7:1.6 |
| p.Pro492Leuc | B | Sign reduced | <0.2% | n.d. | Solubility/stability | 0.884 | 7.6:1.0 |
| p.Thr494Meth | A | Sign reduced | 100% | Solubility | 0.921 | −0.25:−0.83 | |
| p.Asp498Glyc | A | Sign reduced | <10% | n.d. | Activity, solubility/stability | 0.840, −S* | 2.7:1.3 |
| p.Leu510Glnc | A | Sign reduced | <0.2% | n.d. | Solubility/stability | 0.801, +D* | 2.3:0.2 |
Predicted severity of the mutation was estimated in MutPred [mutpred.mutdb.org) [Li et al., 2009]. The numbers denote probability of deleterious/disease-associated mutation, letters denote gain (+) or loss (-) of disorder (D), stability (S), catalytic activity (C), modification by methylation (M), by phosphorylation (P), or by ubiquitination (U), with confidence P < 0.05 (*) and P < 0.01 (**). Modification need not be at the mutated residue.
FoldX (v3.0; foldx.crg.es) [Schymkowitz et al., 2005] was used to estimate the difference in protein stability (ΔΔG) of the mutants relative to wt TH. The crystal structure of human TH (PDB: 2XSN) was used as template. Predictions were performed using both the tetrameric structure or a monomer of TH (ΔΔG[tetramer]:ΔΔG[monomer]). Values are given pr. mol TH subunit. The greater the positive value, the larger is the predicted destabilization of TH by the mutation.
This work.
[Calvo et al., 2010].
Protein produced using a TNT in vitro translation system.
Protein expressed in HEK293 cells.
[Royo et al., 2005].
n.d., not determined.
Mutations in the TH gene are expected to be relatively mild because severely reduced TH activity is not compatible with life. Mice carrying TH null mutations (Th −/−), die in utero due to the failure in the development of the cardiovascular system, but heterozygous TH deficient mice have a normal life span [Zhou et al., 1995]. Recently, a patient with early-onset PD was reported to have a rare deletion of one entire TH allele and no mutation in the other allele. This patient had no symptoms of disease from the catecholaminergic system before PD was diagnosed at the age of 54, illustrating that neurological symptoms of THD probably require >50% loss of TH activity [Bademci et al., 2010]. THD is a phenotypically heterogeneous disease, and should be a differential diagnosis to consider in patients with symptoms of PD. Furthermore, the authors associated the rare deletion to the PD pathogenesis and the early onset of disease [Bademci et al., 2010]. However, additional studies are necessary to further investigate this matter (see also below).
The catalytic domain of the AAAHs is highly conserved among species spanning from bacteria to mammals [Flydal and Martinez, 2013]. A model of the active site of human TH with bound l-Tyr and BH4 was prepared based on the crystal structure of the catalytic domain of human PAH complexed with BH4 and the substrate analogue 3-(2-thienyl)-l-alanine (PDB 1KW0) [Andersen et al., 2002], which aid to recognize the resides involved in the binding of the pterin cofactor and of tyrosine in TH (Fig.4). The catalytic cleft of TH is about 17 Å deep with a ferrous iron 10 Å from the surface. The iron is coordinated by two histidines and a glutamate (His-361, His-366, and Glu-406; numeration in hTH4) (Fig.4) [Goodwill et al., 1997]. Asp-425 in rat TH (corresponding to Asp-455 in hTH4; Fig.4) has recently been shown to be a determinant for the substrate specificity in TH, by critically avoiding hydrophophic interactions that decrease the preference for l-Tyr without affecting the hydroxylation of l-Phe [Daubner et al., 2013]. Although the ability of TH to hydroxylate l-Phe in addition to l-Tyr could be considered an advantage in phenylketonuria (PKU; MIM #261600) where there is an excess of l-Phe and low availability of l-Tyr, the competitive nature of these substrates could also compromise tyrosine hydroxylase activity and catecholamine synthesis in this condition.
Figure 4.
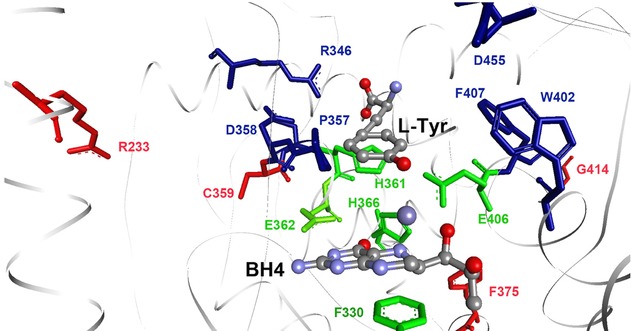
A closer view of the active site of human TH. In green are the iron coordinating triade (His361, His366, and Glu406) and the BH4 binding Glu363 and Phe330. In blue are the substrate interacting residues Arg346, Asp358, Pro357, Trp402, Phe330, and Asp455 (based on the structure of the catalytic domain of human PAH with BH4 and 3-(2-thienyl)-l-alanine (PDB 1KW0)). Residues Arg233, Cys359, Phe375, and Gly414 are shown in red.
Of the mutants where we investigated the amino acid substrate specificity (Table2), p.Arg233His, p.Cys359Phe, p.Phe375Leu, and p.Gly414Arg are mutations structurally positioned to affect active-site residues (Fig.4), and they only showed a moderately reduced solubility and thermal stability (Table3). We found a deleterious kinetic effect for p.Arg233His, as previously reported [Calvo et al., 2010], and in addition, a decrease in the relative preference for l-Tyr as substrate compared with l-Phe and l-Dopa. Cys-359 is one of the six conserved cysteines in TH. The mutation C359F is found homozygous in an Italian boy with type B THD [Brautigam et al., 1999]. As could be expected from the location of this mutation in the substrate-binding site, p.Cys359Phe had a severely reduced activity compared with wt TH (∼10%), and significantly altered affinities for both tyrosine and the cofactor BH4. Interestingly, this mutation led to a higher selectivity for l-Tyr. We also report large kinetic aberrancies for p.Phe375Leu (∼10 or 43% Vmax compared with wt TH), with decreased affinity for l-Tyr (Table1). In a patient with type B THD, this mutation was found in combination with the C-terminal-located p.Ser467Gly mutation that had severely decreased stability. Interestingly, this patient had CSF HVA levels in the low normal range prior to diagnosis, and still responded well to treatment with low doses of l-Dopa [Clot et al., 2009; Doummar et al., 2009].
Mutations closer to the C-terminal had severe effects on the activity and in addition many of the mutations had decreased solubility (as seen for TH p.Gly414Arg, p.Arg441Pro, p.Gln459*, p.Pro492Leu, p.Asp498Gly, and p.Leu510Gln). An exception was TH p.Ser467Gly that had comparable solubility to wt TH, only moderately reduced activity, but severely reduced thermal stability. Together with p.His246Tyr and p.Gly247Ser, these are good candidates for protein stabilization therapy as much of their catalytic activities remained intact. Mutants that showed significant alterations in solubility often also showed severely reduced activity. The TH p.Phe375Leu and p.Ala376Val had significantly decreased solubility, but it was not dramatically reduced and the mutants had considerable remaining activity and only slightly altered thermal inactivation rates. Together with TH p.Leu387Met and p.Cys207Tyr, a combination of several negative effects, including cellular mechanisms not addressed here, could be contributing to the pathogenic phenotype.
Active-site mutations in the AAAHs may interfere with normal reaction mechanism, producing toxic by-products [Kemsley et al., 2003]. Excess oxygen consumption in the TH reaction can lead to the formation of ROS [Haavik et al., 1997]. Although we found no evidence for this among the TH mutants studied here, it might still be considered an additional pathogenic mechanism in vivo, as proposed for PD [Haavik and Toska, 1998; Nakashima et al., 2013].
Within the family of the AAAHs, PAH is the most characterized enzyme with respect to the conformational and catalytic effects of disease-associated mutations and genotype–phenotype relationships [Muntau and Gersting, 2010; Blau et al., 2011]. The majority (⋍60%) of the approximately 850 mutations reported to date in PAH found in hyperphenylalaninemia and PKU are missense mutations (www.biopku.org/pah, accessed march 2014). As observed for many other recessive genetic disorders, the predominant pathogenic mechanism reduced stability or misfolding of mutated enzyme, leading to loss of function [Pey et al., 2007; Scriver, 2007; Gersting et al., 2008]. Studies of both PAH and TPH1 and TPH2 have shown that missense mutations in their catalytic and tetramerization domains have a strong effect on the stability and activity of the enzymes, whereas mutations in the regulatory domain have less impact on these properties [Pey et al., 2007; McKinney et al., 2009]. N-terminal mutations in PAH are often associated with an increased rate of aggregation for recombinantly expressed PAH variants [Pey et al., 2004], but this is less obvious for TH. Different properties of a partially unfolded intermediate may explain these differences within the protein family [Kleppe et al., 1999; Kleppe and Haavik, 2004], and why TH is not as sensitive to mutations in its N-terminal domain [Kleppe and Haavik, 2004]. Although THD is a rare disorder, with less than 70 patients being reported so far, it can be considered a model disease for the group of neurogenetic disorders affecting monoaminergic neurotransmission. Thus, the residual TH biosynthetic capacity is related to symptom severity (Fig.5). It has also been proposed that TH contributes to the progressive degeneration of dopaminergic neurons [Haavik et al., 1997; Haavik and Toska, 1998]. Although THD in general does not seem to be associated with neurodegeneration, the recent discovery of THD precludes a clear assumption about a possible dopaminergic degeneration with progressive neurological deficits in these patients and whether this may be related to certain TH genotypes. The biochemical characterization performed here could help identifying TH variants that have a higher risk of causing cellular damage.
Figure 5.

Mutations found in THD type B patients showed lower activity. The average remaining activity in mutations found in patients classified in THD subtypes A (N = 19 patients) and B (N = 10 patients) [Willemsen et al., 2010] were significantly different (*P < 0.05, t-test).
Whether it is caused by primary THD or by degeneration of catecholaminergic neurons, as found in sporadic or familial PD, dopamine deficiency is usually treated with administration of Dopa or dopamine agonists. High levels of Dopa can even be neurotoxic and be oxidized to reactive intermediates, including quinones, semiquinones, and hydrogen peroxide [Kostrzewa et al., 2002]. Thus, it has been speculated whether oxidation of Dopa by TH can contribute to the progressive degeneration of dopaminergic neurons found in PD [Haavik et al., 1997; Haavik and Toska, 1998]. Possibly, the relatively increased efficacy of Dopa as substrate for p.Arg233His, p.Gly247Ser, p.Phe375Leu, and p.Gly414Arg may be a risk factor for a progressive course of neurological symptoms in patients harboring this mutation. As the role of TH in the generation of neuromelanin and Dopa oxidation in vivo is not settled, the role of the mutants in the production of this pigment is not clear. The in vitro TH assays were performed with 200 μM BH4, which probably is much higher than the physiological levels. Thus, Dopa may be a relatively better substrate for TH in vivo, as the enzyme has a higher cofactor affinity for the l-Dopa oxidase reaction than for tyrosine hydroxylation [Haavik et al., 1997].
In vitro studies of mutant proteins give important information about structural and mechanistic changes caused by a mutation. However, these studies do not provide a complete picture of how mutations affect proteins in vivo. Thus, mutations could also interfere with TH regulation, interaction with other proteins, chaperone interaction, or proteolytic degradation in cells. Mutations can also interfere with mRNA synthesis, stability, localization, or splicing [Chen and Miller, 2008]. Thus, further information could have been obtained if the mutants had been stably expressed in postmitotic human dopaminergic neurons. Despite these limitations, in vitro studies reported here are essential for understanding the molecular mechanisms of disease-causing mutations.
Genotype–Phenotype Correlations
A clear correlation between genotype and phenotype in THD has not been established. The establishment of such a correlation is complicated by the small number of patients reported, and that the majority of patients are compound heterozygous for mutations in the TH gene. Homozygosity of the p.Arg233His mutation is found in six type A and three type B patients. The p.Leu236Pro mutation occurred homozygously in one type A and two type B patients. However, all patients with at least one promoter mutation had the type A THD [Willemsen et al., 2010], indicating that some TH protein was produced despite the presence of the promoter mutation. In THD patients born of related parents, there is also an increased risk of other autosomal-recessive traits/diseases that can complicate the clinical symptoms. There is nevertheless a correlation between some biochemical markers, such as CSF HVA levels, and the severity of their THD symptoms [Willemsen et al., 2010]. Here, we observed a poor correlation between the remaining activity of the TH mutants and CSF HVA content of the patients for which this parameter was available (data not shown). This is not unexpected, as CSF HVA values are highly dependent on the age of the subjects, and the assay protocols that have been used. Interestingly, the clinical subtype (type A or B) of the THD patients could be predicted based on measurements of the in vitro TH activity (Fig.5).
Future Perspectives
As treatment of THD and Parkinson's disease with l-Dopa or dopamine agonists often is inadequate and may only provide temporary symptom relief, searches for treatments that directly target the malfunctioning TH are being conducted. Pharmacological chaperones are compounds that can stabilize the natural protein conformation and protect against denaturation. The cofactor BH4 can actually act as a chaperone when supplemented pharmacologically, stabilizing TH in vivo [Thony et al., 2008]. It has been reported that synthetic pharmacological chaperones can stabilize the human TH mutant p.Arg202His (corresponding to p.Arg233His in TH4) in vitro [Calvo et al., 2010]. Based on the characterization performed here, p.His246Tyr, p.Gly247Ser, p.Leu387Met, p.Ser467Gly, and possibly p.Cys359Phe are good candidates for targeting by pharmacological chaperones as they retained activity and solubility, but had increased thermal inactivation rates. In conclusion, this study provides new insights to the mechanism of pathogenesis of mutations in TH that can be used in the search for new, targeted treatment strategies, and to find stabilizing compounds that may reverse the negative effects of the mutations on the enzyme activity.
Acknowledgments
Jarl Underhaug, Sidsel E. Riise, Guri Matre, and Ali S. Muñoz are thanked for expert technical assistance.
Additional Supporting Information may be found in the online version of this article.
References
- Andersen OA, Flatmark T, Hough E. Crystal structure of the ternary complex of the catalytic domain of human phenylalanine hydroxylase with tetrahydrobiopterin and 3-(2-thienyl)-l-alanine, and its implications for the mechanism of catalysis and substrate activation. J Mol Biol. 2002;320:1095–1108. doi: 10.1016/s0022-2836(02)00560-0. [DOI] [PubMed] [Google Scholar]
- Bademci G, Edwards TL, Torres AL, Scott WK, Zuchner S, Martin ER, Vance JM, Wang L. A rare novel deletion of the tyrosine hydroxylase gene in Parkinson disease. Hum Mutat. 2010;31:E1767–E771. doi: 10.1002/humu.21351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104(Suppl):S2–S9. doi: 10.1016/j.ymgme.2011.08.017. [DOI] [PubMed] [Google Scholar]
- Brautigam C, Steenbergen-Spanjers GC, Hoffmann GF, Dionisi-Vici C, van den Heuvel LP, Smeitink JA, Wevers RA. Biochemical and molecular genetic characteristics of the severe form of tyrosine hydroxylase deficiency. Clin Chem. 1999;45:2073–2078. [PubMed] [Google Scholar]
- Cai C, Shi W, Zeng Z, Zhang M, Ling C, Chen L, Cai C, Zhang B, Li WD. GTP cyclohydrolase I and tyrosine hydroxylase gene mutations in familial and sporadic dopa-responsive dystonia patients. PLoS One. 2013;8:e65215. doi: 10.1371/journal.pone.0065215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo AC, Scherer T, Pey AL, Ying M, Winge I, McKinney J, Haavik J, Thony B, Martinez A. Effect of pharmacological chaperones on brain tyrosine hydroxylase and tryptophan hydroxylase 2. J Neurochem. 2010;114:853–863. doi: 10.1111/j.1471-4159.2010.06821.x. [DOI] [PubMed] [Google Scholar]
- Chen GL, Miller GM. Rhesus monkey tryptophan hydroxylase-2 coding region haplotypes affect mRNA stability. Neuroscience. 2008;155:485–491. doi: 10.1016/j.neuroscience.2008.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi CS, Lee HF, Tsai CR. Tyrosine hydroxylase deficiency in Taiwanese infants. Pediatr Neurol. 2012;46:77–82. doi: 10.1016/j.pediatrneurol.2011.11.012. [DOI] [PubMed] [Google Scholar]
- Clot F, Grabli D, Cazeneuve C, Roze E, Castelnau P, Chabrol B, Landrieu P, Nguyen K, Ponsot G, Abada M, Doummar D, Damier P, et al. Exhaustive analysis of BH4 and dopamine biosynthesis genes in patients with Dopa-responsive dystonia. Brain. 2009;132(Pt 7):1753–1763. doi: 10.1093/brain/awp084. [DOI] [PubMed] [Google Scholar]
- Daubner SC, Avila A, Bailey JO, Barrera D, Bermudez JY, Giles DH, Khan CA, Shaheen N, Thompson JW, Vasquez J, Oxley SP, Fitzpatrick PF. Mutagenesis of a specificity-determining residue in tyrosine hydroxylase establishes that the enzyme is a robust phenylalanine hydroxylase but a fragile tyrosine hydroxylase. Biochemistry. 2013;52:1446–1455. doi: 10.1021/bi400031n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubner SC, Piper MM. Deletion mutants of tyrosine hydroxylase identify a region critical for heparin binding. Protein Sci. 1995;4:538–541. doi: 10.1002/pro.5560040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doummar D, Clot F, Vidailhet M, Afenjar A, Durr A, Brice A, Mignot C, Guet A, de Villemeur TB, Rodriguez D. Infantile hypokinetic-hypotonic syndrome due to two novel mutations of the tyrosine hydroxylase gene. Mov Disord. 2009;24:943–945. doi: 10.1002/mds.22455. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick PF. Tetrahydropterin-dependent amino acid hydroxylases. Annu Rev Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- Flatmark T, Almas B, Knappskog PM, Berge SV, Svebak RM, Chehin R, Muga A, Martinez A. Tyrosine hydroxylase binds tetrahydrobiopterin cofactor with negative cooperativity, as shown by kinetic analyses and surface plasmon resonance detection. Eur J Biochem. 1999;262:840–849. doi: 10.1046/j.1432-1327.1999.00445.x. [DOI] [PubMed] [Google Scholar]
- Flydal MI, Martinez A. Phenylalanine hydroxylase: function, structure, and regulation. IUBMB Life. 2013;65:341–349. doi: 10.1002/iub.1150. [DOI] [PubMed] [Google Scholar]
- Fossbakk A, Haavik J. An oxygraphic method for determining kinetic properties and catalytic mechanism of aromatic amino acid hydroxylases. Anal Biochem. 2005;343:100–105. doi: 10.1016/j.ab.2005.04.043. [DOI] [PubMed] [Google Scholar]
- Gersting SW, Kemter KF, Staudigl M, Messing DD, Danecka MK, Lagler FB, Sommerhoff CP, Roscher AA, Muntau AC. Loss of function in phenylketonuria is caused by impaired molecular motions and conformational instability. Am J Hum Genet. 2008;83:5–17. doi: 10.1016/j.ajhg.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanniello T, Claps D, Carducci C, Carducci C, Blau N, Vigevano F, Antonozzi I, Leuzzi V. A new tyrosine hydroxylase genotype associated with early-onset severe encephalopathy. J Child Neurol. 2012;27:523–525. doi: 10.1177/0883073811420717. [DOI] [PubMed] [Google Scholar]
- Gnaiger E, Steinlechner-Maran R, Mendez G, Eberl T, Margreiter R. Control of mitochondrial and cellular respiration by oxygen. J Bioenerg Biomembr. 1995;27:583–596. doi: 10.1007/BF02111656. [DOI] [PubMed] [Google Scholar]
- Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC. Crystal structure of tyrosine hydroxylase at 2.3 A and its implications for inherited neurodegenerative diseases. Nat Struct Biol. 1997;4:578–585. doi: 10.1038/nsb0797-578. [DOI] [PubMed] [Google Scholar]
- Gordon SL, Bobrovskaya L, Dunkley PR, Dickson PW. Differential regulation of human tyrosine hydroxylase isoforms 1 and 2 in situ: isoform 2 is not phosphorylated at Ser35. Biochim Biophys Acta. 2009;1793:1860–1867. doi: 10.1016/j.bbamcr.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Grima B, Lamouroux A, Boni C, Julien JF, Javoy-Agid F, Mallet J. A single human gene encoding multiple tyrosine hydroxylases with different predicted functional characteristics. Nature. 1987;326:707–711. doi: 10.1038/326707a0. [DOI] [PubMed] [Google Scholar]
- Haavik J. lDOPA is a substrate for tyrosine hydroxylase. J Neurochem. 1997;69:1720–1728. doi: 10.1046/j.1471-4159.1997.69041720.x. [DOI] [PubMed] [Google Scholar]
- Haavik J, Almas B, Flatmark T. Generation of reactive oxygen species by tyrosine hydroxylase: a possible contribution to the degeneration of dopaminergic neurons. J Neurochem. 1997;68:328–332. doi: 10.1046/j.1471-4159.1997.68010328.x. [DOI] [PubMed] [Google Scholar]
- Haavik J, Flatmark T. Rapid and sensitive assay of tyrosine 3-monooxygenase activity by high-performance liquid chromatography using the native fluorescence of DOPA. J Chromatogr. 1980;198:511–515. doi: 10.1016/s0021-9673(00)80522-1. [DOI] [PubMed] [Google Scholar]
- Haavik J, Le Bourdelles B, Martinez A, Flatmark T, Mallet J. Recombinant human tyrosine hydroxylase isozymes. Reconstitution with iron and inhibitory effect of other metal ions. Eur J Biochem. 1991;199:371–378. doi: 10.1111/j.1432-1033.1991.tb16133.x. [DOI] [PubMed] [Google Scholar]
- Haavik J, Toska K. Tyrosine hydroxylase and Parkinson's disease. Mol Neurobiol. 1998;16:285–309. doi: 10.1007/BF02741387. [DOI] [PubMed] [Google Scholar]
- Haugarvoll K, Bindoff L. A novel compound heterozygous tyrosine hydroxylase mutation (p.R441P) with complex phenotype. J Parkinsons Dis. 2011;1:119–122. doi: 10.3233/JPD-2011-11006. [DOI] [PubMed] [Google Scholar]
- Kemsley JN, Wasinger EC, Datta S, Mitic N, Acharya T, Hedman B, Caradonna JP, Hodgson KO, Solomon EI. Spectroscopic and kinetic studies of PKU-inducing mutants of phenylalanine hydroxylase: Arg158Gln and Glu280Lys. J Am Chem Soc. 2003;125:5677–5686. doi: 10.1021/ja029106f. [DOI] [PubMed] [Google Scholar]
- Kleppe R, Haavik J. Different stabilities and denaturation pathways for structurally related aromatic amino acid hydroxylases. FEBS Lett. 2004;565(1–3):155–159. doi: 10.1016/j.febslet.2004.03.092. [DOI] [PubMed] [Google Scholar]
- Kleppe R, Uhlemann K, Knappskog PM, Haavik J. Urea-induced denaturation of human phenylalanine hydroxylase. J Biol Chem. 1999;274:33251–33258. doi: 10.1074/jbc.274.47.33251. [DOI] [PubMed] [Google Scholar]
- Knappskog PM, Flatmark T, Mallet J, Ludecke B, Bartholome K. Recessively inherited lDOPA-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Hum Mol Genet. 1995;4:1209–1212. doi: 10.1093/hmg/4.7.1209. [DOI] [PubMed] [Google Scholar]
- Kostrzewa RM, Kostrzewa JP, Brus R. Neuroprotective and neurotoxic roles of levodopa (lDOPA) in neurodegenerative disorders relating to Parkinson's disease. Amino Acids. 2002;23(1–3):57–63. doi: 10.1007/s00726-001-0110-x. [DOI] [PubMed] [Google Scholar]
- Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25:2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludecke B, Bartholome K. Frequent sequence variant in the human tyrosine hydroxylase gene. Hum Genet. 1995;95:716. doi: 10.1007/BF00209496. [DOI] [PubMed] [Google Scholar]
- Ludecke B, Knappskog PM, Clayton PT, Surtees RA, Clelland JD, Heales SJ, Brand MP, Bartholome K, Flatmark T. Recessively inherited lDOPA-responsive parkinsonism in infancy caused by a point mutation (L205P) in the tyrosine hydroxylase gene. Hum Mol Genet. 1996;5:1023–1028. doi: 10.1093/hmg/5.7.1023. [DOI] [PubMed] [Google Scholar]
- Mak CM, Lam CW, Siu TS, Chan KY, Siu WK, Yeung WL, Hui J, Wong VC, Low LC, Ko CH, Fung CW, Chen SP, et al. Biochemical and molecular characterization of tyrosine hydroxylase deficiency in Hong Kong Chinese. Mol Genet Metab. 2010;99:431–433. doi: 10.1016/j.ymgme.2009.12.011. [DOI] [PubMed] [Google Scholar]
- McKinney J, Knappskog PM, Pereira J, Ekern T, Toska K, Kuitert BB, Levine D, Gronenborn AM, Martinez A, Haavik J. Expression and purification of human tryptophan hydroxylase from Escherichia coli and Pichia pastoris. Protein Expr Purif. 2004;33:185–194. doi: 10.1016/j.pep.2003.09.014. [DOI] [PubMed] [Google Scholar]
- McKinney JA, Turel B, Winge I, Knappskog PM, Haavik J. Functional properties of missense variants of human tryptophan hydroxylase 2. Hum Mutat. 2009;30:787–794. doi: 10.1002/humu.20956. [DOI] [PubMed] [Google Scholar]
- Muntau AC, Gersting SW. Phenylketonuria as a model for protein misfolding diseases and for the development of next generation orphan drugs for patients with inborn errors of metabolism. J Inherit Metab Dis. 2010;33:649–658. doi: 10.1007/s10545-010-9185-4. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Ichinose H. Comparative studies on the structure of human tyrosine hydroxylase with those of the enzyme of various mammals. Comp Biochem Physiol C. 1991;98:203–210. [PubMed] [Google Scholar]
- Nakashima A, Ota A, Kaneko YS, Mori K, Nagasaki H, Nagatsu T. A possible pathophysiological role of tyrosine hydroxylase in Parkinson's disease suggested by postmortem brain biochemistry: a contribution for the special 70th birthday symposium in honor of Prof. Peter Riederer. J Neural Transm. 2013;120:49–54. doi: 10.1007/s00702-012-0828-5. [DOI] [PubMed] [Google Scholar]
- Nasrin S, Ichinose H, Hidaka H, Nagatsu T. Recombinant human tyrosine hydroxylase types 1–4 show regulatory kinetic properties for the natural (6R)-tetrahydrobiopterin cofactor. J Biochem. 1994;116:393–398. doi: 10.1093/oxfordjournals.jbchem.a124537. [DOI] [PubMed] [Google Scholar]
- Pey AL, Perez B, Desviat LR, Martinez MA, Aguado C, Erlandsen H, Gamez A, Stevens RC, Thorolfsson M, Ugarte M, Martinez A. Mechanisms underlying responsiveness to tetrahydrobiopterin in mild phenylketonuria mutations. Hum Mutat. 2004;24:388–399. doi: 10.1002/humu.20097. [DOI] [PubMed] [Google Scholar]
- Pey AL, Stricher F, Serrano L, Martinez A. Predicted effects of missense mutations on native-state stability account for phenotypic outcome in phenylketonuria, a paradigm of misfolding diseases. Am J Hum Genet. 2007;81:1006–1024. doi: 10.1086/521879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons R, Syrengelas D, Youroukos S, Orfanou I, Dinopoulos A, Cormand B, Ormazabal A, Garzia-Cazorla A, Serrano M, Artuch R. Levodopa-induced dyskinesias in tyrosine hydroxylase deficiency. Mov Disord. 2013;28:1058–1063. doi: 10.1002/mds.25382. [DOI] [PubMed] [Google Scholar]
- Rostrup M, Fossbakk A, Hauge A, Kleppe R, Gnaiger E, Haavik J. Oxygen dependence of tyrosine hydroxylase. Amino Acids. 2008;34:455–464. doi: 10.1007/s00726-007-0547-7. [DOI] [PubMed] [Google Scholar]
- Royo M, Daubner SC, Fitzpatrick PF. Effects of mutations in tyrosine hydroxylase associated with progressive dystonia on the activity and stability of the protein. Proteins. 2005;58:14–21. doi: 10.1002/prot.20293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33(Web Server issue):W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat. 2007;28:831–845. doi: 10.1002/humu.20526. [DOI] [PubMed] [Google Scholar]
- Stamelou M, Mencacci NE, Cordivari C, Batla A, Wood NW, Houlden H, Hardy J, Bhatia KP. Myoclonus-dystonia syndrome due to tyrosine hydroxylase deficiency. Neurology. 2012;79:435–441. doi: 10.1212/WNL.0b013e318261714a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teigen K, Dao KK, McKinney JA, Gorren AC, Mayer B, Froystein NA, Haavik J, Martinez A. Tetrahydrobiopterin binding to aromatic amino acid hydroxylases. Ligand recognition and specificity. J Med Chem. 2004;47:5962–5971. doi: 10.1021/jm0497646. [DOI] [PubMed] [Google Scholar]
- Thony B, Calvo AC, Scherer T, Svebak RM, Haavik J, Blau N, Martinez A. Tetrahydrobiopterin shows chaperone activity for tyrosine hydroxylase. J Neurochem. 2008;106:672–681. doi: 10.1111/j.1471-4159.2008.05423.x. [DOI] [PubMed] [Google Scholar]
- Willemsen MA, Verbeek MM, Kamsteeg EJ, de Rijk-van Andel JF, Aeby A, Blau N, Burlina A, Donati MA, Geurtz B, Grattan-Smith PJ, Haeussler M, Hoffmann GF, et al. Tyrosine hydroxylase deficiency: a treatable disorder of brain catecholamine biosynthesis. Brain. 2010;133(Pt 6):1810–1822. doi: 10.1093/brain/awq087. [DOI] [PubMed] [Google Scholar]
- Zhou QY, Quaife CJ, Palmiter RD. Targeted disruption of the tyrosine hydroxylase gene reveals that catecholamines are required for mouse fetal development. Nature. 1995;374:640–643. doi: 10.1038/374640a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.