

Abstract
Large hospital-based clinical laboratories must be prepared to rapidly investigate potential infectious disease outbreaks. To challenge the ability of our molecular diagnostics laboratory to use whole-genome sequencing in a potential outbreak scenario and identify impediments to these efforts, we studied 84 invasive serotype emm59 group A streptococcus (GAS) strains collected in the United States. We performed a rapid-response exercise to the mock outbreak scenario using whole-genome sequencing, genome-wide transcript analysis, and mouse virulence studies. The protocol changes installed in response to the lessons learned were tested in a second iteration. The initial investigation was completed in 9 days. Whole-genome sequencing showed that the invasive infections were caused by multiple subclones of epidemic emm59 GAS strains likely spread to the United States from Canada. The phylogenetic tree showed a strong temporal-spatial structure with diversity in mobile genetic element content, features that are useful for identifying closely related strains and possible transmission events. The genome data informed the epidemiology, identifying multiple patients who likely acquired the organisms through direct person-to-person transmission. Transcriptome analysis unexpectedly revealed significantly altered expression of genes encoding a two-component regulator and the hyaluronic acid capsule virulence factor. Mouse infection studies confirmed a high-virulence capacity of these emm59 organisms. Whole-genome sequencing, coupled with transcriptome analysis and animal virulence studies, can be rapidly performed in a clinical environment to effectively contribute to patient care decisions and public health maneuvers.
INTRODUCTION
Infectious disease outbreaks can quickly decimate a population, causing substantial human morbidity, mortality, and economic loss (1–3). Large hospital-based diagnostic laboratories must be highly vigilant, since they likely will be the first to recover the pathogen and perhaps to recognize the potential for an outbreak. That is, a concerted effort is needed for clinical laboratories to rapidly and effectively respond to naturally occurring or nefariously spreading microbial agents. To this end, our molecular diagnostics laboratory recently validated whole-genome sequencing of microbes as a clinical test. This assay is routinely used to identify unknown organisms and investigate pathogens of uncertain taxonomic assignment (4, 5). We have also devised a standardized rapid-response protocol to evaluate strains implicated in a perceived outbreak. The overarching goal is to generate sufficient data in as short of a time as possible to infer relatedness among epidemiologically linked strains and understand the molecular processes contributing to epidemics. These data are useful in informing patient care decisions and guiding public health maneuvers. Importantly, whole-genome sequence data may provide information about the source of the outbreak, identify dissemination routes, and exclude or confirm the possibility of genetic modifications that suggest bioterrorism (1, 2, 5). Genomic findings can also be used to create rapid diagnostic tests, effective therapies, and vaccines (3, 6).
To challenge the rapid-response capability of our clinical laboratory and identify impediments, we constructed a mock outbreak scenario using serotype emm59 group A streptococcus (GAS) as a model pathogen. GAS was used because we have an extensive genome sequence database and expertise with basic, clinical, and translational studies on this organism. Thus, it was a best-case scenario. Serotype emm59 GAS organisms are typically an uncommon cause of human disease (7–9); however, a hypervirulent clone recently arose, and its progeny spread rapidly throughout Canada, causing many hundreds of severe invasive infections (10–12). The surveillance data from the Centers for Disease Control and Prevention now indicate that the frequency of emm59 GAS infections is increasing in some areas of the United States, particularly Minnesota (13). To test the hypothesis that U.S. cases comprise a single-clone group, to determine whether Canadian and U.S. organisms are genetically related, and if so, to identify possible dissemination routes, we sequenced the genomes of all 67 emm59 GAS recovered by the Active Bacterial Core Surveillance (ABCs) program during 2010 to 2012. RNA sequencing and mouse infection studies were also performed. Follow-up epidemiological investigations were conducted by the Minnesota Department of Health. The lessons learned from the rapid-response exercise led to changes in our standard operating procedure, and a second iteration of whole-genome sequencing and phylogenetic analysis was performed on an additional 17 emm59 GAS strains recovered in Minnesota in 2013.
MATERIALS AND METHODS
Bacterial strain samples.
This investigation is based on 84 serotype emm59 GAS strains that were recovered from patients with invasive (sterile-site) infections in the United States (see Table S1 in the supplemental material). All strains are part of the comprehensive population-based surveillance studies that are collected by the Active Bacterial Core Surveillance (ABCs) program administered by the Centers for Disease Control and Prevention (CDC) (13). The ABCs performs active laboratory- and population-based surveillance in 10 geographically disparate sites to collect all GAS isolates recovered from patients with invasive infections.
Whole-genome sequencing and bioinformatic analysis.
Whole-genome sequencing was performed as previously described using a MiSeq instrument (Illumina, Inc., San Diego, CA) (4). The strains were accessioned, and bacteria were collected from Trypticase soy agar containing 5% sheep blood (Becton Dickinson and Company, Franklin Lakes, NJ) using a sterile 1-μl calibrated loop and transferred to a 2-ml tube containing 1 ml of Tris-EDTA and 0.1-mm silica spheres (Lysing matrix B; MP Biomedicals, Santa Ana, CA). The cells were lysed using ballistic disintegration (FastPrep96 automated homogenizer; MP Biomedicals), and genomic DNA was extracted according to the manufacturer's instructions (DNeasy 96 blood and tissue kit; Qiagen, Inc., Valencia, CA). The quality and quantity of genomic DNA were confirmed (double-stranded DNA [dsDNA] BR assay kit and Qubit 2.0 fluorometer; Invitrogen, Life Technologies, Grand Island, NY). Dual-indexed sequencing libraries were prepared according to the manufacturer's instructions (NexteraXT DNA sample preparation kit; Illumina).
Using a 75-bp paired-end protocol, we first sequenced the genomes of all 67 emm59 GAS strains recovered by the ABCs in the 10 collection sites in the United States during 2010 to 2012. Libraries yielding a minimum of 13-fold coverage were analyzed as described below. The libraries with low genomic coverage were sequenced again on a second MiSeq run using a 50-bp paired-end protocol. After the completion of the 9-day mock outbreak investigation, which included whole-genome sequencing, RNA sequencing, and mouse virulence studies (Fig. 1 to 4), a second iteration of whole-genome sequencing and phylogenetic analysis was performed on the 17 emm59 GAS strains recovered in Minnesota in 2013 (Fig. 2). The libraries generated for the second iteration of whole-genome sequencing and phylogenetic analysis were prepared as described above and sequenced using a 250-bp single-end protocol. The three sequencing runs were performed on one MiSeq instrument using the default software settings for the Generate FASTQ on-instrument workflow.
FIG 1.
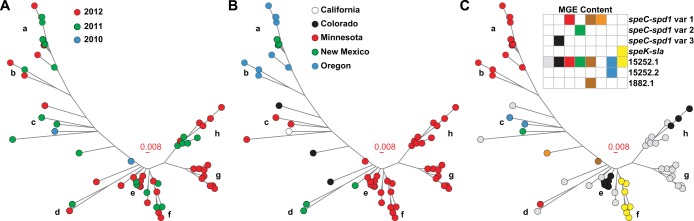
Phylogenetic analysis of 67 serotype emm59 GAS strains recovered in the United States in 2010 to 2012. The inferred genetic relationships are shown based on 165 concatenated nonredundant SNP loci. The branches of the neighbor-joining tree with ≥2 strains per node are labeled a to h. A clear pattern of temporal, spatial, and mobile gene content structures is evident. (A) Temporal structuring is color coded by year of strain isolation. (B) Spatial structuring is color coded by state of strain isolation. (C) Mobile genetic element (MGE) content is color coded by the combination of prophage and prophage-associated genes present in each strain. Three distinct variants of the prophage-encoding streptococcal pyrogenic exotoxin C (speC) and streptococcal phage-encoded DNase1 (spd1) genes were identified. The subclonal lineage on branch f has prophage-encoding streptococcal pyrogenic exotoxin K (speK) and phospholipase A2 (sla) genes. Prophages 15252.1, 15252.2, and 1882.1 were previously described in genome of the reference strains MGAS15252 and MGAS1882, respectively.
FIG 4.
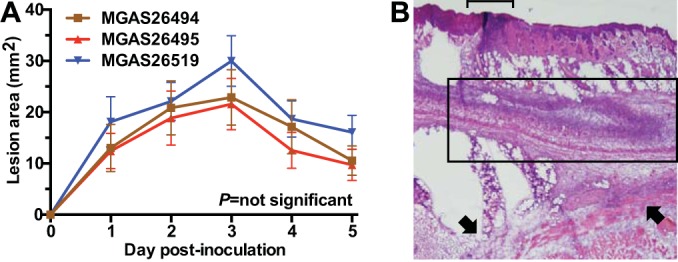
Serotype emm59 GAS strains recovered in the United States are highly virulent in a mouse model of skin and soft tissue infection. (A) Strains MGAS26494, MGAS26495, and MGAS26515 caused large skin lesions in mice. Data are shown as means ± standard errors of the means. (B) Frozen section analysis performed rapidly on specimens collected 72 h postinoculation (day 7 of the study) demonstrates an ulcerated lesion (bracket), severe tissue necrosis (boxed region) and dissemination to the deep and lateral margins (arrows). The images are shown at ×4 original magnification.
FIG 2.
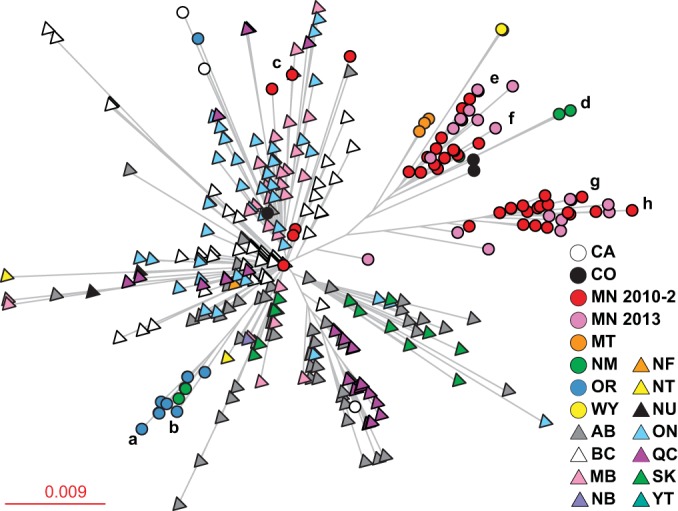
Phylogenetic analysis of all sequenced serotype emm59 GAS strains recovered in Canada and the United States. The inferred genetic relationships are shown based on 956 concatenated nonredundant SNP loci from 353 strains. The branches of strains sequenced in the rapid-response exercise are labeled a to h relative to those in Fig. 1. Spatial structuring is color coded by the province or state of origin. The emm59 strains recovered in Minnesota in 2013 that were analyzed during a second iteration of the rapid-response drill are shown in pink. Canadian provinces (triangles): AB, Alberta; BC, British Columbia; MB, Manitoba; NB, New Brunswick; NF, Newfoundland; NT, Northwest Territory; NU, Nunavut; ON, Ontario; QC, Quebec; SK, Saskatchewan; and YT, Yukon Territory. States (circles): CA, California; CO, Colorado; MN, Minnesota; MT, Montana; NM, New Mexico; OR, Oregon; WY, Wyoming.
Bioinformatics processes were performed using a workstation with two 6-core dual-threaded Intel Xeon processors, 32 GB random access memory (RAM), and an Ubuntu operating system. The data were secured by the institutional firewall. The number and quality of sequence reads for each FASTQ file were confirmed using FastQC (Babraham Bioscience Technologies, Cambridge, United Kingdom). The emm types were identified using emm_pipeline.pl (14). Polymorphisms were identified first using VAAL (15) and later using Musket, Novoalign, and Freebayes (15). Variant call format (VCF) files were filtered using VCFtools, with the parameter QUAL of >20. The reference GAS emm59 strain MGAS15252 (GenBank accession no. CP003116) was selected because its genome was sequenced to closure, and it has been used in multiple animal virulence studies (10). The gene content was determined by mapping reads to an emm59-centric pseudo-pan-genome, as previously described (15). The sequence data were deposited at NCBI as PRJNA194066.
Genome-wide RNA transcript analysis.
The RNA transcripts were measured by RNA sequencing (RNA-Seq) analysis. Triplicate cultures of each strain were grown to mid-exponential (optical density at 600 nm [OD600], 1) and late-exponential (OD600, 1.5) phases. The RNA-Seq libraries were prepared according to the manufacturer's instructions (Epicentre Biotechnologies, Madison, WI) and sequenced using a 50-bp single-end protocol. To generate a sufficient number of sequence reads for transcript analysis (average, 1 million reads per library), two runs were performed on one MiSeq instrument using the default software settings for the Generate FASTQ on-instrument workflow. The two FASTQ files generated for each library were concatenated, and the number and quality of sequence reads were confirmed using FastQC. The RNA transcripts were quantified from the concatenated FASTQ files using the default software settings (CLC bio, Cambridge, MA), with differences of >1.5-fold and a P value of <0.05 after applying Bonferroni's correction for multiple comparisons was considered significant. Hyaluronic acid production was measured using an enzyme-linked immunosorbent assay (ELISA) (Corgenix, Inc., Broomfield, CO).
Mouse model of invasive infection.
Strain virulence was tested in a mouse model of invasive infection, as previously described (10). Cryopreserved stocks of bacteria were prepared, and SKH1-hrBR mice (Charles River BRF, Houston, TX) were inoculated subcutaneously with 1 × 108 CFU. Lesions were measured with a digital caliper, and the mean abscess area was compared using a 2-way analysis of variance (ANOVA), with a P value of <0.05 considered significant. For microscopic examination, the lesions were removed en bloc and processed for frozen section histopathology. Representative micrographs were obtained using a BX5 microscope fitted with a DB70 digital camera (Olympus, Tokyo, Japan). The study protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the Houston Methodist Research Institute.
RESULTS
Whole-genome sequencing and phylogenetic analysis of serotype emm59 GAS strains recovered in the United States.
We first investigated 67 emm59 GAS strains collected by the CDC ABCs during 2010 to 2012 from patients with invasive infections in California (n = 1), Colorado (n = 4), Minnesota (n = 46), New Mexico (n = 6), and Oregon (n = 10) (see Table S1 in the supplemental material). To challenge the ability of our clinical laboratory to expeditiously respond to an outbreak scenario and identify impediments to providing actionable information, the study was designed as a mock rapid-response exercise. Our goal was to acquire a genome-level understanding of patient isolates in a clinically relevant time frame. We used whole-genome sequencing, bioinformatics processes, RNA sequencing, and a mouse model of invasive infection to complete the study in 9 days (Fig. 5).
FIG 5.
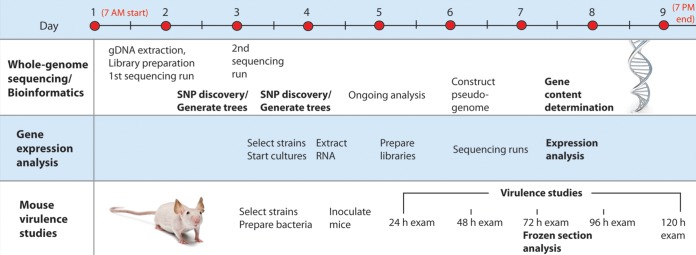
Timeline of the rapid-response exercise. To investigate the molecular underpinnings of a mock outbreak of invasive emm59 GAS recovered in the United States, whole-genome sequencing, bioinformatics processes, RNA sequencing, and mouse virulence studies were completed in 9 days. gDNA, genomic DNA.
To determine the genetic relationships among the strains comprising the mock outbreak, whole-genome sequencing was performed. A 75-bp paired-end protocol was selected to balance the generation of sufficient data for bioinformatics analysis with a short instrument run time. The first phylogenetic tree was created 49 h after beginning the exercise (Fig. 5). The results from the strains having sufficient sequence read coverage for evaluation confirmed that all were closely related genetically to the Canadian emm59 epidemic clone. That is, within 2 days, we unambiguously confirmed the spread of the Canadian epidemic organisms to multiple disparate sites in the United States. Libraries with low genomic coverage were sequenced again using a 50-bp paired-end protocol. A shorter read length was used to generate data more quickly (Fig. 5). The results demonstrated that these strains were also closely related genetically to the Canadian emm59 epidemic clone.
Phylogenetic analysis revealed that all GAS strains were closely related. The 67 emm59 strains differed, on average, by 17 polymorphisms (mean, 15 single-nucleotide polymorphisms [SNPs] and 2 indels; range, 0 to 25 SNPs and 0 to 6 indels). This very low level of sequence diversity is similar to the results from previous investigations of the Canadian emm59 epidemic clone (10–12, 16). Five sets of genomically indistinguishable strains were identified (see Table S1 in the supplemental material), indicating that those infections likely arose from direct human-to-human transmission. A follow-up epidemiological investigation discovered that a common community group, Native Americans, had a higher proportion of invasive emm59 GAS infections than other groups (P < 0.001, chi-square test), including strains with identical genome sequences. For example, among one set of genetically identical strains, 3 were recovered from Native American patients who lived in the same metropolitan area, and a fourth was recovered from another member of the Native American community (see Table S1).
A review of all publically available emm59 GAS genome sequence data demonstrated that, albeit small, sufficient genetic diversity exists among the strains comprising the epidemic clone used to define distinct subclonal lineages (10–12). To test the hypothesis that strains recovered in the United States have a nonrandom temporal-spatial distribution, a neighbor-joining tree was constructed. This analysis revealed that most strains were assigned to 8 major branches, with a few singletons falling in intervening regions (Fig. 1A). A strong temporal structure was identified, with strains at the outermost tips of each branch being recovered more recently than the strains located near the node (Fig. 1A). A strong spatial structure was also identified, with all branches except one being composed of strains recovered from a single state (Fig. 1B). Furthermore, strains recovered from the same county were also closely clustered (see Table S1 in the supplemental material). Taken together, these data are consistent with a model of local transmission and subclonal diversification following the introduction of a progenitor strain to each discrete region. To determine whether the U.S. strains were derived from a single source or multiple sources, a neighbor-joining tree was created using polymorphism data generated from the U.S. strains sequenced in the first and second iterations of this rapid-response exercise and all other available sequenced emm59 GAS genomes. Phylogenetic analysis demonstrated that the strains recovered in Minnesota likely had two distinct origins, British Columbia and Manitoba (Fig. 2). Similarly, the strains recovered in New Mexico were genetically allied with the strains originating from British Columbia or Alberta. These data suggest that the Canadian emm59 epidemic clone was imported to the United States via multiple independent events.
Gene content analysis of serotype emm59 GAS strains recovered in the United States.
The genomes of most bacterial species can be divided into two portions. The core genome contains the genes present in all strains of that species. In contrast, the accessory genome contains genes that are unique to some strains and represents an important source of gene content variation. In GAS, mobile genetic elements (MGEs) commonly carry genes encoding secreted toxins or resistance to antimicrobial agents (17). To evaluate the gene contents of the U.S. strains, we mapped sequence reads to an emm59-centric GAS pseudo-pan-genome. The core genome was 100% conserved. However, we detected unexpected diversity in the MGEs. Overall, 8 distinct combinations were identified (Fig. 1C). The superimposition of MGE content onto the phylogenetic tree revealed that strains located within each branch tend to have the same prophage collection, implying identity by descent.
Genome-wide RNA transcript analysis of serotype emm59 GAS strains recovered in the United States.
In the event of a large outbreak, it is important to rapidly obtain as much data as possible that have bearing on the molecular basis of the pathogen-host interaction. Such information can be used to create new diagnostic tests, effective therapies, and vaccines. This type of data can be obtained in many ways, including genome-wide RNA transcript analyses and animal model virulence studies. For example, a key discovery of the European “sprout outbreak” investigation was that some antibiotics induced Shiga toxin expression by the epidemic Escherichia coli clone (3). To gain additional data about the molecular pathogenesis of U.S. emm59 strains, a genome-wide transcript analysis was performed. Three strains were selected using the following criteria: (i) the strains were recovered from patients in Minnesota, since a single state was used to emulate a regional outbreak scenario, and emm59 strains were the most common GAS serotype recovered in Minnesota during the collection dates; (ii) the strains were located on different branches of the phylogenetic tree; and (iii) the strains had the most frequent (wild-type) alleles for all major transcription regulatory genes. A nonrandom overrepresentation of polymorphisms occurring in the regulatory genes CovR-CovS and Mga has been reported in Canadian emm59 strains (12). Among these 67 U.S. strains, 29 had a polymorphism in CovR-CovS or Mga (P < 0.001 for each gene, chi-square test). Although the implication of this finding remains uncertain, a mutation of CovR-CovS or Mga in other GAS serotype strains significantly alters virulence (18, 19). The GAS strains MGAS26494, MGAS26495, and MGAS26519 met our criteria. The cultures were started on day 3 (immediately after the genome data were analyzed), and transcript analyses were completed on day 7 (Fig. 5). Pairwise comparisons of the RNA sequence data revealed a highly similar genome-wide expression profile, which is compatible with the clonal background of epidemic emm59 GAS strains (Fig. 3A). The expression of only 16 to 32 and 25 to 40 genes was significantly altered at the mid- and late-exponential phases of growth, respectively. Five differentially expressed genes encoding virulence factors or transcription regulators were of particular interest (Fig. 3B). The hyaluronic acid synthase (has) operon was significantly upregulated in strain MGAS26494. The hyaluronic acid capsule is a key GAS virulence factor (20). To test the hypothesis that the overexpression of has operon genes alters the capsular phenotype, colonies grown on solid medium were visually inspected, and hyaluronic acid was measured. Consistent with our hypothesis, the colonies of strain MGAS26494 were strikingly larger and more mucoid (Fig. 3C) and expressed significantly more hyaluronic acid (Fig. 3D). To identify the genetic basis for an altered capsule phenotype, the genome sequence data were reexamined. We discovered a 38-bp deletion located in the has operon promoter of strain MGAS26494. This polymorphism, which occurs in a noncoding region of the genome, was not recognized as being possibly important to strain virulence during our initial analysis because we focused on nonsynonymous (amino acid changing) polymorphisms and nonsense mutations. The deleted nucleotides contain a consensus-binding site for CovR, a well-studied regulator of capsule biosynthesis and GAS virulence (19). The deletion of the CovR binding site likely derepresses the has operon, resulting in increased gene expression and enhanced capsule production. An additional unexpected finding was the significantly altered expression of genes encoding the YesM-YesN two-component system in strain MGAS26494 (Fig. 3B). The YesM-YesN two-component system directly or indirectly regulates ∼40% of the transcriptome (mostly during the stationary phase of growth) in serotype emm1 strains but has not been studied in emm59 GAS strains (21). Since a genomic explanation for the altered expression of yesM and yesN was not immediately apparent, the genome sequence data were reanalyzed using an alternative bioinformatics process. The new pipeline discovered that strain MGAS26494 represents a mixed population, with approximately half of the sequence reads having a polymorphism in the promoter region of yesM and yesN. We postulate that this promoter mutation, located adjacent to the −10 box, results in the altered expression of yesM and yesN and may explain the altered expression of 8 other genes under its regulatory control. Taken together, the RNA sequence data add to the list of regulatory genes identified by whole-genome sequencing that are undergoing diversifying selection in epidemic emm59 GAS.
FIG 3.
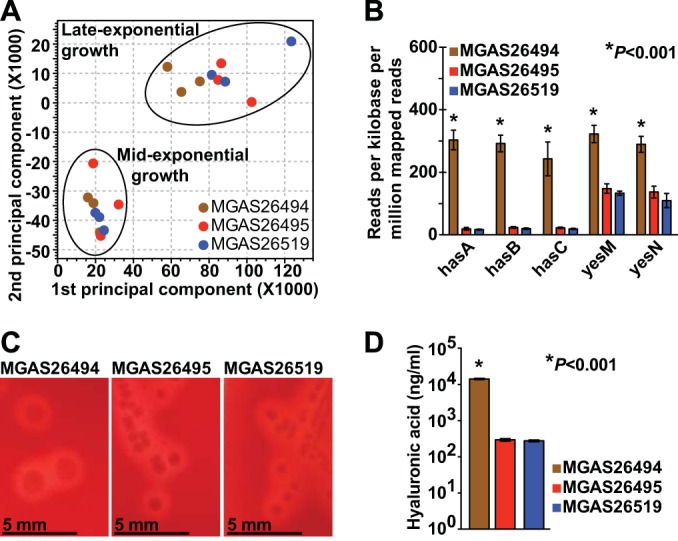
Gene expression analysis of serotype emm59 GAS strains recovered in the United States. Three strains collected from patients in Minnesota that were assigned to different branches of the phylogenetic tree (MGAS26494, branch f; MGAS26495, branch c; MGAS26515, branch b) and have wild-type alleles for all major transcriptional regulators were selected for RNA sequencing. (A) Principal component analysis is shown for gene expression profiles of strains grown to mid- and late-exponential phase (lower and upper circles, respectively). (B) Genes encoding the hyaluronic acid capsule operon (hasA, hasB, and hasC) and the two-component transcriptional regulator yesM and yesN were significantly overexpressed in strain MGAS26494. Data are shown as means ± standard errors of the means. (C and D) Strains MGAS26494, MGAS26495, and MGAS26515 were grown on Trypticase soy agar containing 5% sheep blood. Consistent with capsule gene upregulation, strain MGAS26494 demonstrated a markedly larger and hypermucoid colony phenotype (×1 original magnification) and produced significantly more hyaluronic acid. Data are shown as means ± standard errors of the means.
Serotype emm59 GAS strains recovered in the United States are highly virulent in a mouse model of invasive infection.
Animal infection studies are important for testing hypotheses that have bearing on genetic alterations and strain virulence. For example, nonhuman primate experiments were crucial to understanding virulence differences among the isolates of the pandemic H1N1 influenza A virus (22). Similarly, compared to a historic emm59 strain, the Canadian epidemic clone was shown to be significantly more virulent in mouse and monkey infection models (10). To further investigate the pathogen-host interactions of the U.S. strains, we used a mouse model of invasive infection. The mice were inoculated on day 4 (after the genome analyses were complete) and observed for the duration of the exercise (Fig. 5). Consistent with the existing data for the Canadian clone, the U.S. strains rapidly caused large necrotic lesions (Fig. 4A). Frozen section (rapid) histopathology analysis, completed in approximately 4 h, confirmed that each strain was highly virulent, causing marked tissue destruction and dissemination (Fig. 4B).
DISCUSSION
Whole-genome sequencing is an invaluable tool for microbial pathogenesis research (23). It has been used to identify unknown organisms, discover new mechanisms of antimicrobial resistance, and understand the virulence of isolates of uncertain taxonomic status (5, 24). Whole-genome sequencing of microbes was previously validated in our molecular diagnostics laboratory, and it is run once per week as a routine clinical test. Additional runs are performed as needed for scenarios, such as outbreak investigations. Inasmuch as whole-genome sequencing has been used to study infectious disease outbreaks (1, 2), we challenged the ability of our clinical laboratory to rapidly respond to a mock outbreak scenario. Our team of 7 pathologists and infectious disease investigators, 3 public health professionals, and 2 laboratory technologists completed the investigation in 9 days. The work was performed concurrently with normal patient care activities. We provide a model strategy for large hospital-based clinical laboratories to rapidly investigate potential infectious disease outbreaks. All instruments, reagents, and software used are commercially available, so the protocol can be readily implemented in other settings.
Each step of the mock outbreak investigation, including whole-genome sequencing, RNA sequencing, and mouse virulence studies, generated data that might be crucial to answering questions bearing on patient care and public health activities. The results demonstrated that multiple subclones of highly virulent emm59 GAS strains, likely imported from Canada, caused these infections in the United States in 2010 to 2012. The genome sequence data revealed a disproportionate number of polymorphisms altering major transcriptional regulators and unexpected diversity in mobile genetic elements (prophage). The RNA sequence data also helped us recognize polymorphisms leading to increased hyaluronic acid capsule expression and altered regulation by a two-component system. In principle, these findings could be used to develop rapid molecular tests similar to those proposed for classifying epidemic E. coli O157:H7 (25). This sort of assay could be readily used for point-of-care testing in emergency departments, small hospitals, and public health laboratories. Likewise, animal virulence data, including rapid frozen section histopathology, provided additional information bearing on pathogen-host interactions and might support an evaluation of new therapies or vaccines. Finally, informed by the discovery of strains having indistinguishable genome sequences, we uncovered an unexpected association of emm59 GAS and Native Americans.
The lessons learned from this exercise prompted a review of the disaster preparedness plan for our laboratory. We now have a standard operating procedure for using whole-genome sequencing and virulence analyses as part of the response of our clinical laboratory to potential outbreaks, including naturally occurring or possibly illegitimately released pathogens. The exercise also identified important opportunities for process improvement. After protocol changes were implemented, a second iteration of the rapid-response drill was performed using emm59 strains recovered in Minnesota in 2013 (Fig. 2). First, a new bioinformatics process was constructed to take advantage of innovations occurring since the initial pipeline was validated. The new bioinformatics process includes read correction algorithms, multithreaded read mappers, and variant callers that can handle mixed-read populations (13). The alternative bioinformatic process, as noted previously, was crucial for discovering an unexplained alteration of yesM and yesN gene expression in one strain. We also installed secure file transfer protocol (FTP) and network-attached storage to facilitate rapid data transfer between the sequencing instrument, bioinformatics workstations, and local and international collaborators. Second, we are prospectively creating tools for analyzing important pathogens, such as Staphylococcus aureus and Klebsiella pneumoniae (1, 26), because compared to our initial study in which 2 days were spent evaluating gene content, the analysis was completed in ∼4 h during the second iteration, since the pseudo-pan-genome had already been constructed. Third, animal protocols and reagent inventories must be actively managed. The mouse experiments were quickly initiated using a preexisting protocol for studying GAS pathogenesis and in-house animals. Communication with the IACUC is now included in our rapid-response protocol, including contingencies to quickly approve new studies and expedite animal purchases in an emergency situation. Conversely, our RNA sequencing experiment was slowed due to inclement weather that caused delayed shipment of the reagents. Given the important discovery of capsule gene overexpression uncovered by the RNA sequence data, this delay might have detrimentally impacted patient care. As a result, we are evaluating electronic solutions for inventory management. Clinical laboratory directors must also effectively communicate needs to private sector companies. Although many companies have knowledgeable technical support staff, most are available to troubleshoot problems only during standard business hours. This arrangement is not suitable for large clinical laboratories that operate continuously. We experienced a 1-h delay in the first sequencing run that was caused by a typographical error in the sample sheet, which would have been prolonged if sequencing were performed at a time when technical support was unavailable.
We benefited from this exercise being a best-case scenario. The ABCs previously collected the strains, and our international collaborative team has established expertise in GAS pathogenesis, epidemiology, and genetics. If sequencing and analysis were outsourced, the causative organism was unfamiliar or initially unknown, or the pathogen grew slowly or required biosafety level 3 containment, the pace of our response would have been slowed. Also, the existence of multiple high-quality, well-annotated, closed genomes makes GAS an ideal model. A review of publicly available databases reveals that the depth and breadth of deposited genomes for many medically important pathogens is lacking (4). International efforts, such as a 100K Genome Project (http://100kgenome.vetmed.ucdavis.edu), Global Microbial Identifier (http://www.globalmicrobialidentifier.org), and a collaboration between the Sanger Institute, Pacific Biosystems, and Public Health England will begin to close this gap. Another key barrier is that whole-genome sequencing of microbes remains a relatively slow and costly process. In the event of a catastrophic outbreak or release of a bioterrorism agent, we should ideally generate clinically actionable results within hours rather than days. Finally, gene expression and animal virulence studies, which may be crucial to developing new diagnostics and therapeutics, are inherently slow and cannot be initiated until after interpreting the genome sequence data. If the outbreak were widespread, included subclones with greater genomic diversity, or involved multiple dissemination routes, more isolates, possibly hundreds per day, would need to be evaluated. The magnitude of such a study would overwhelm our surge capacity and likely that of most large laboratories.
In summary, our clinical laboratory was able to rapidly perform whole-genome sequencing and virulence analyses in a mock outbreak scenario. The data generated from each step of the exercise would effectively inform patient care decisions and public health maneuvers. Importantly, building on lessons learned, we are now far better prepared for a bona fide outbreak.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kathryn Stockbauer for editorial assistance and the CDC's Streptococcus laboratory. The images used in Fig. 5 were acquired from Thinkstock by Getty Images.
R.J.O., N.F., and J.M.M. designed the study. R.J.O., N.F., P.K., M.A.S., S.B.B., S.W.L., C.V.B., and C.C. performed the research. K.J.C.-S., R.L., and C.V.B. performed the epidemiology investigation. R.J.O. and J.M.M. wrote the first complete draft of the report, and all the authors provided contributions and suggestions.
This work was supported by the Fondren Foundation and Houston Methodist Hospital, Houston, TX, and the Minnesota Emerging Infections Program and the CDC's Emerging Infections Program Network.
Footnotes
Published ahead of print 24 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.02164-14.
REFERENCES
- 1.Harris SR, Cartwright EJ, Török ME, Holden MT, Brown NM, Ogilvy-Stuart AL, Ellington MJ, Quail MA, Bentley SD, Parkhill J, Peacock SJ. 2013. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect. Dis. 13:130–136. 10.1016/S1473-3099(12)70268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, Eyre DW, Wilson DJ, Hawkey PM, Crook DW, Parkhill J, Harris D, Walker AS, Bowden R, Monk P, Smith EG, Peto TE. 2013. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect. Dis. 13:137–146. 10.1016/S1473-3099(12)70277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D, Klammer A, Peluso P, Lee L, Kislyuk AO, Bullard J, Kasarskis A, Wang S, Eid J, Rank D, Redman JC, Steyert SR, Frimodt-Møller J, Struve C, Petersen AM, Krogfelt KA, Nataro JP, Schadt EE, Waldor MK. 2011. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 365:709–717. 10.1056/NEJMoa1106920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long SW, Williams D, Valson C, Cantu CC, Cernoch P, Musser JM, Olsen RJ. 2013. A genomic day in the life of a clinical microbiology laboratory. J. Clin. Microbiol. 51:1272–1277. 10.1128/JCM.03237-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright AM, Beres SB, Consamus EN, Long SW, Flores AR, Barrios R, Richter GS, Oh SY, Garufi G, Maier H, Drews AL, Stockbauer KE, Cernoch P, Schneewind O, Olsen RJ, Musser JM. 2011. Rapidly progressive, fatal, inhalation anthrax-like infection in a human: case report, pathogen genome sequencing, pathology, and coordinated response. Arch. Pathol. Lab. Med. 135:1447–1459. 10.5858/2011-0362-SAIR.1. [DOI] [PubMed] [Google Scholar]
- 6.Köser CU, Bryant JM, Becq J, Török ME, Ellington MJ, Marti-Renom MA, Carmichael AJ, Parkhill J, Smith GP, Peacock SJ. 2013. Whole-genome sequencing for rapid susceptibility testing of M. tuberculosis. N. Engl. J. Med. 369:290–292. 10.1056/NEJMc1215305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tyrrell GJ, Lovgren M, Ibrahim Q, Garg S, Chui L, Boone TJ, Mangan C, Patrick DM, Hoang L, Horsman GB, Van Caeseele P, Marrie TJ. 2012. Epidemic of invasive pneumococcal disease, western Canada, 2005–2009. Emerg. Infect. Dis. 18:733–740. 10.3201/eid1805.110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. 2009. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect. Dis. 9:611–616. 10.1016/S1473-3099(09)70178-1. [DOI] [PubMed] [Google Scholar]
- 9.Shea PR, Ewbank AL, Gonzalez-Lugo JH, Martagon-Rosado AJ, Martinez-Gutierrez JC, Rehman HA, Serrano-Gonzalez M, Fittipaldi N, Beres SB, Flores AR, Low DE, Willey BM, Musser JM. 2011. Group A streptococcus emm gene types in pharyngeal isolates, Ontario, Canada, 2002–2010. Emerg. Infect. Dis. 17:2010–2017. 10.3201/eid1711.110159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fittipaldi N, Beres SB, Olsen RJ, Kapur V, Shea PR, Watkins ME, Cantu CC, Laucirica DR, Jenkins L, Flores AR, Lovgren M, Ardanuy C, Liñares J, Low DE, Tyrrell GJ, Musser JM. 2012. Full-genome dissection of an epidemic of severe invasive disease caused by a hypervirulent, recently emerged clone of group A streptococcus. Am. J. Pathol. 180:1522–1534. 10.1016/j.ajpath.2011.12.037. [DOI] [PubMed] [Google Scholar]
- 11.Fittipaldi N, Olsen RJ, Beres SB, Van Beneden C, Musser JM. 2012. Genomic analysis of emm59 group A streptococcus invasive strains, United States. Emerg. Infect. Dis. 18:650–652. 10.3201/eid1804.111803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fittipaldi N, Tyrrell GJ, Low DE, Martin I, Lin D, Hari KL, Musser JM. 2013. Integrated whole-genome sequencing and temporospatial analysis of a continuing group A streptococcus epidemic. Emerg. Microbes Infect. 2:e13. 10.1038/emi.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Centers for Disease Control and Prevention. 2013. Active Bacterial Core Surveillance (ABCs) report. Emerging Infections Program Network:group A Streptococcus, 2012. Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/abcs/reports-findings/survreports/gas12.pdf. [Google Scholar]
- 14.Athey TB, Teatero S, Li A, Marchand-Austin A, Beall BW, Fittipaldi N. 2014. Deriving group A streptococcus typing information from short-read whole-genome sequencing data. J. Clin. Microbiol. 52:1871–1876. 10.1128/JCM.00029-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nasser W, Beres SB, Olsen RJ, Dean MA, Rice KA, Long SW, Kristinsson KG, Gottfredsson M, Vuopio J, Raisanen K, Caugant DA, Steinbakk M, Low DE, McGeer A, Darenberg J, Henriques-Normark B, Van Beneden CA, Hoffmann S, Musser JM. 2014. Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc. Natl. Acad. Sci. U. S. A. 111:E1768–E1776. 10.1073/pnas.1403138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown CC, Olsen RJ, Fittipaldi N, Mormon ML, Fort PL, Neuwirth R, Majeed M, Woodward WB, Musser JM. 2014. Spread of virulent group A Streptococcus type emm59 from Montana to Wyoming, USA. Emerg. Infect. Dis. 20:658–660. 10.3201/eid2004.130564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beres SB, Musser JM. 2007. Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS One 2:e800. 10.1371/journal.pone.0000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hause LL, McIver KS. 2012. Nucleotides critical for the interaction of the Streptococcus pyogenes Mga virulence regulator with Mga-regulated promoter sequences. J. Bacteriol. 194:4904–4919. 10.1128/JB.00809-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sumby P, Zhang S, Whitney AR, Falugi F, Grandi G, Graviss EA, Deleo FR, Musser JM. 2008. A chemokine-degrading extracellular protease made by group A streptococcus alters pathogenesis by enhancing evasion of the innate immune response. Infect. Immun. 76:978–985. 10.1128/IAI.01354-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wessels MR, Moses AE, Goldberg JB, DiCesare TJ. 1991. Hyaluronic acid capsule is a virulence factor for mucoid group A streptococci. Proc. Natl. Acad. Sci. U. S. A. 88:8317–8321. 10.1073/pnas.88.19.8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sitkiewicz I, Musser JM. 2006. Expression microarray and mouse virulence analysis of four conserved two-component gene regulatory systems in group a streptococcus. Infect. Immun. 74:1339–1351. 10.1128/IAI.74.2.1339-1351.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Safronetz D, Rockx B, Feldmann F, Belisle SE, Palermo RE, Brining D, Gardner D, Proll SC, Marzi A, Tsuda Y, Lacasse RA, Kercher L, York A, Korth MJ, Long D, Rosenke R, Shupert WL, Aranda CA, Mattoon JS, Kobasa D, Kobinger G, Li Y, Taubenberger JK, Richt JA, Parnell M, Ebihara H, Kawaoka Y, Katze MG, Feldmann H. 2011. Pandemic swine-origin H1N1 influenza A virus isolates show heterogeneous virulence in macaques. J. Virol. 85:1214–1223. 10.1128/JVI.01848-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olsen RJ, Long SW, Musser JM. 2012. Bacterial genomics in infectious disease and the clinical pathology laboratory. Arch. Pathol. Lab. Med. 136:1414–1422. 10.5858/arpa.2012-0025-RA. [DOI] [PubMed] [Google Scholar]
- 24.Long SW, Olsen RJ, Mehta SC, Palzkill TG, Cernoch PL, Perez KK, Musick WL, Rosato AE, Musser JM. 25 August 2014. PBP2a mutations causing high-level ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 10.1128/AAC.03622-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eppinger M, Mammel MK, Leclerc JE, Ravel J, Cebula TA. 2011. Genomic anatomy of Escherichia coli O157:H7 outbreaks. Proc. Natl. Acad. Sci. U. S. A. 108:20142–20147. 10.1073/pnas.1107176108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deleo FR, Chen L, Porcella SF, Martens CA, Kobayashi SD, Porter AR, Chavda KD, Jacobs MR, Mathema B, Olsen RJ, Bonomo RA, Musser JM, Kreiswirth BN. 2014. Molecular dissection of the evolution of carbapenem-resistant multilocus sequence type 258 Klebsiella pneumoniae. Proc. Natl. Acad. Sci. U. S. A. 111:4988–4993. 10.1073/pnas.1321364111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.