

Abstract
It was reported that TNF receptor type II signaling, which has the capacity to stimulate CD4+ forkhead box P3+ (Foxp3+) regulatory T cells (Tregs), activated the noncanonical NF-κB pathway in an IKKα-dependent manner. Therefore, we studied the role of IKKα in the homeostasis of Treg population. To this end, we generated a mouse strain with conditional knockout of IKKα in CD4 cells (Ikkαf/f:CD4.Cre) that showed a >60% reduction in the number of Tregs in the thymus and peripheral lymphoid tissues, whereas the number of Foxp3− effector T cells (Teffs) remained at a normal level. The function of Tregs deficient in IKKα was examined using Rag1−/− mice cotransferred with naive CD4 cells (nCD4s). Although wild-type (WT) Tregs inhibited colitis induced by transfer of WT nCD4s, IKKα-deficient Tregs failed to do so, which was associated with their inability to reconstitute Rag1−/− mice. Furthermore, nCD4s deficient in IKKα also failed to reconstitute Rag1−/− mice and were defective in proliferative responses in vitro and in vivo. Thus, our study reveals a novel role of IKKα in the maintenance of a normal Treg population and in the control of expansion of CD4 T cells. These properties of IKKα may be exploited as therapeutic strategies in the treatment of major human diseases.—Chen, X., Willette-Brown, J., Wu, X., Hu, Y., Howard, O. M. Z., Hu, Y., Oppenheim, J. J. IKKα is required for the homeostasis of regulatory T cells and for the expansion of both regulatory and effector CD4 T cells.
Keywords: Foxp3, proliferation, colitis, lymphopenic mouse
CD4 T cells play central roles in orchestrating innate and adaptive immune responses (1). In response to stimulation by antigen-presenting cells (APCs), CD4 T cells proliferate in a process referred to as clonal expansion and differentiate into specialized effector T cells (Teffs), which are essential for the development of adaptive immune responses (2). The magnitude and duration of an immune response are also controlled by a subset of CD4 T cells, namely CD4+ forkhead box P3+ (FoxP3+) regulatory T cells (Tregs), which represent ∼10% of peripheral CD4 T cells and are indispensable for the maintenance of immune homeostasis and for the prevention of autoimmune responses (3). However, Tregs also attenuate immune responses against tumor antigens and therefore represent a major immunosuppressive mechanism by which tumors evade immune surveillance (4). Further understanding of signaling pathways by which the homeostasis of Tregs is regulated can help design novel treatments for autoimmunity and cancer.
NF-κB is the prototypical proinflammatory transcription factor that participates in the regulation of genes important in both innate and adaptive immunity (5). The IKK is required for the activation of NF-κB through phosphorylation and degradation of the endogenous IκBs (6). The IKK complex consists of 3 subunits: IKKα (IKK1; CHUK); IKKβ (IKK2); and a regulatory protein, IKKγ (NEMO or NF-κB essential modulator). IKKβ and IKKγ, but not IKKα, are required for canonical NF-κB activation (7). In contrast, noncanonical NF-κB signaling is independent of IKKβ and IKKγ, whereas IKKα is required for this pathway through the phosphorylation and processing of p100, leading to the generation of p52/RelB heterodimers and activation of target genes (8). In addition, IKKα was also reported to regulate gene expression in an NF-κB-independent manner (9).
Our previous studies showed that TNF activates and expands Tregs through TNF receptor type II (TNFR2) (10), one of the TNF receptors (TNFRs) preferentially expressed by highly suppressive mouse and human Tregs (11, 12). TNFR2 appears to play a key role in maintaining a normal-sized Treg population in the steady state as well as in the expansion of Tregs in response to inflammatory stimulation because the number of Tregs was markedly reduced in mouse strains deficient in TNFR2 or TNFR2 ligands, and Tregs deficient in TNFR2 failed to expand in an inflamed colon (13). It was reported that TNF-TNFR2 interaction results in the activation of noncanonical NF-κB pathway through p100 processing (14) and that IKKα is required for the activation of this pathway (8, 15). In addition to TNFR2, we also reported that a number of costimulatory TNFR superfamily (TNFRSF) members collectively promoted the expansion and function of peripheral Tregs (16). Furthermore, it has been recently reported that costimulation of TNFR2 together with other TNFRSF members is required for thymic generation of Tregs (17). Similar to TNFR2, some other members of the TNFRSF also elicited noncanonical NF-κB signaling through IKKα (18). Therefore, we hypothesized that IKKα may be a major component of signaling pathway of TNFR2 and the other TNFRSF members in the maintenance of a normal population of Tregs.
It has been shown that T cells can develop normally in the absence of IKKα (8, 19, 20); however, mutation of IKKα resulted in greater susceptibility of T cells to apoptosis (20). More recently, Li et al. (15) reported that IKKα can transcriptionally regulate Th17 immune responses. These studies were mainly based on the IkkαAA knockin mice (8) in which the activating phosphorylation sites of IKKα protein, Ser176 and Ser180, are replaced by 2 alanines (AA), thereby abolishing the activation of its kinase activity. Nevertheless, genetic ablation of IKKα in CD4 cells is likely to provide more definitive evidence regarding the role of IKKα in the function of CD4 cells. To this end, we generated a mouse strain with conditional knockout (KO) of IKKα in CD4 cells (Ikkαf/f:CD4.Cre mice). As expected, the number of Tregs was markedly reduced in the thymus and peripheral lymphoid tissues of Ikkαf/f:CD4.Cre mice. However, our studies showed that both Tregs and Teffs isolated from Ikkαf/f:CD4.Cre mice failed to repopulate lymphopenic Rag1−/− mice. The in vitro proliferative responses of IKKα-deficient naive CD4 T cells to T-cell receptor (TCR) stimulation were markedly reduced. The proliferation of total CD4 cells deficient in IKKα was also markedly reduced in the recipient Rag1−/− mice. Thus, IKKα is important for the maintenance of a normal Treg population, but it also contributes to the proliferative expansion of CD4+ Teffs.
MATERIALS AND METHODS
Mice
Rag1−/− and Ly5.2 C57BL/6 mice were provided by the Animal Production Area of the National Cancer Institute (NCI) (Frederick, MD, USA). Ikkαf/f:CD4.Cre mice were generated by crossing homozygous Ikkαf/f mice (21) to CD4.Cre transgenic mice initially obtained from Taconic Biosciences (Derwood, MD, USA) and maintained in the Animal Production Area of NCI. Mice were backcrossed 7 times onto a C57BL/6 background, and genotypes were determined by PCR and Western blot. Frederick National Laboratory for Cancer Research is accredited by AAALAC International and follows the Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the Guide for Care and Use of Laboratory Animals (22). Anti-mouse antibodies (Abs) were purchased from BD Biosciences (San Diego, CA, USA) consisting of anti-mouse CD3 (145-2C11), CD4 (GK1.5), CD8a (53-6.7), CD25 (PC61), CD45 (30F11), CTLA-4 (UC10-4F10-11), TNFR2 (TR75-89), TNF (MP6-XT22), interferon γ (INFγ; XMG1.2), and IL-17A (TC11-18H10). Leukocyte Activation Cocktail was also purchased from BD Biosciences. Functional grade purified anti-mouse CD3e (eBio500A2) and CD28 (37.51) Abs, Foxp3 Staining Set (FJK-16s), and anti-mouse TCRβ Ab (H57-597), CD45.2 (104), and CD45RB (C363.16A) were purchased from eBioscience (San Diego, CA, USA). Human recombinant TGF-β1 was from R&D Systems (Minneapolis, MN, USA). CellTraceth Violet was purchased from Life Technologies (Grand Island, NY, USA).
T-cell transfer model of colitis
Naive CD4+CD25−CD45RBhi T cells were isolated from Ikkαf/f:CD4.Cre mice or control mice and injected intraperitoneally into Rag1−/−-immunodeficient recipients (4 × 105 cells/mouse) alone, or cotransferred with Ikkαf/f:CD4.Cre mouse-derived or control mouse-derived CD4+CD25+CD45RBlo Tregs at a ratio of 5:1. Mice were monitored weekly for the clinical symptoms of colitis such as rectal bleeding, loose feces/diarrhea, rough/hunched posture, and loss of body weight by animal facility staff. Any mice losing >20% of their starting body weight or showing severe signs of disease were euthanized. In some experiments, total CD4+ cells were purified with MACS (Miltenyi Biotec Incorporated, Anburn, CA, USA) from spleens of Ly5.2 mice (CD45.1+) or Ikkαf/f:CD4.Cre mice (CD45.2+) and mixed at a 1:1 ratio. The cells were labeled with CellTraceth Violet. The cells (2 × 106 cells/mouse) were transferred into Rag1−/− mice. In some experiments, CD4+ cells were MACS purified from wild-type (WT) control (Ikkαf/f), CD4.Cre, or Ikkαf/f:CD4.Cre mice, and cells were transferred separately into Rag 1−/− mice, at the same number as cells derived from Ikkαf/f:CD4.Cre mice. Two weeks after transfer, mice were sacrificed, and phenotype and number of transferred cells in the recipient mice were analyzed.
Cell purification
Single-cell suspensions from spleen and mesenteric lymph nodes (mLNs) were obtained by filtration through a 70-µm cell strainer (BD Labware, San Jose, CA, USA). Preparation of colon lamina propria (cLP) cells was as previously described (23). In brief, colons were rinsed in PBS and cut into ∼0.3 cm pieces. Intestinal epithelial cells were removed by incubation with calcium- and magnesium-free PBS containing 10% fetal calf serum (FCS) and 5 mM EDTA. Colon tissues then were digested with RPMI 1640 medium containing 10% FCS and 1 mg/ml collagenase type 4 (Worthington Biochemical Corporation, Lakewood, NJ, USA) for 30 min at 37°C. CD4+ T cells were purified using magnetic beads coated with anti-CD4 Ab (clone L3T4) according to the manufacturer’s instructions (Miltenyi Biotec Incorporated). Subsequently, the CD4+ cells were stained with anti-CD4, anti-CD25, and anti-CD45RB Abs and sorted into naive CD4+CD25−CD45RBhi T cells and CD4+CD25+ Tregs (>94% of Foxp3+ cells) using Cytomation MoFlo cytometer (Fort Collins, CO, USA).
In vitro proliferation of T cell
Flow-sorted naive CD4 cells (nCD4s) from spleen and LNs of WT control mice (Ikkαf/f) or Ikkαf/f:CD4.Cre mice were seeded into a 96-well plate at 104–105 per well. The cells were stimulated with plate-bound anti-CD3e Ab alone, plate-bound anti-CD3e Ab and soluble anti-CD28 Ab (2 µg/ml), or soluble anti-CD3e Ab (2 µg/ml) and APCs (2 × 105 cells/wells) isolated from WT control mice or from Ikkαf/f:CD4.Cre mice. The plate-bound anti-CD3 Ab was achieved by incubating a plate containing 10 µg/ml anti-CD3e Ab in 50 µl PBS per well in 37°C for overnight. CD4-depleted, irradiated (3000 rad) splenic cells were used as APCs. The cells were cultured in RPMI 1640 medium (Lonza BioWhittaker, Walkersville, MD, USA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA), 2 mM glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, and 50 μM 2-ME. Cells were pulsed with 1 µCi [3H]thymidine (PerkinElmer Life Sciences, Boston, MA, USA) per well for the last 6 h of the culture period. The proliferation was evaluated by [3H]thymidine incorporation.
Flow cytometry
After blocking Fc receptor, cells were incubated with appropriately diluted Abs. Acquisition was performed using a LSRII (BD Biosciences), and data analysis was conducted using FlowJo software (Tree Star Incorporated, Ashland, OR, USA). For intracellular cytokine staining, cells were restimulated with Leukocyte Activation Cocktail for 4 h. Fluorescence-activated cell sorting (FACS) analysis was gated on the live cells only by using the LIVE/DEAD Fixable Dead Cell Stain Kit (Life Technologies).
Statistical analysis
Comparison of data was analyzed by 2-tailed Student’s t test or 1- or 2-way ANOVA test, as indicated in the figure legends. The statistical analysis was performed with Prism 6.0 (GraphPad Software Incorporated, La Jolla, CA, USA).
RESULTS
The number of Tregs in the thymus and peripheral lymphoid organs of Ikkαf/f:CD4.Cre mice is markedly reduced
CD4 cell-specific conditional IKKα KO mice (designated as Ikkαf/f:CD4.Cre or KO) were generated by crossing homozygous Ikkαf/f mice (21) with mice carrying a specific CD4.Cre transgene. Mice were backcrossed 7 times onto a C57BL/6 background, and genotypes were determined by PCR and Western blot (Supplemental Fig. 1). Ikkαf/f mice were used as controls (designated as Ikkα-WT or WT), unless otherwise specified.
Mice with selective ablation of IKKα in CD4 cells were viable and grew normally. They had grossly normal thymus, spleen, mLNs, inguinal LNs, and axillary LNs. The proportion of CD4 single-positive (SP) and CD8 SP cells in the thymus of the Ikkαf/f:CD4.Cre mice was the same as from IKKα-WT control mice (Supplemental Fig. 2A). The peripheral lymphoid organs of Ikkαf/f:CD4.Cre mice had a normal proportion of total CD3+ cells in their spleens and LNs (Supplemental Fig. 2B). There was no reduction in the proportion of CD4 cells and no increase in the proportion of CD8 cells in the splenic and LN’s CD3+ population of Ikkαf/f:CD4.Cre mice (P > 0.05; Supplemental Fig. 2C). The total cellularity in the lymphoid tissues of Ikkαf/f:CD4.Cre mice was the same as in WT control mice (Supplemental Fig. 2D).
The proportion of Foxp3-expressing Tregs in the total thymocyte population or CD4+CD8− SP thymocytes was 0.22 or 2.23%, respectively, in Ikkαf/f:CD4.Cre mice, which was reduced by >60% as compared with the proportion in WT mice (0.61 or 5.63%, respectively; P < 0.01; Fig. 1A). The proportion of Tregs in peripheral LN cells was 2.17% and of the CD4+ cells was 4.5% in Ikkαf/f:CD4.Cre mice. In contrast, WT mouse LN cells contained 4.73%, and CD4+ LN cells contained 11.2% of Tregs (Fig. 1B). Likewise, the proportion of Tregs in total splenic cells and in the CD4+ splenic cells was 1.06 and 5.86%, respectively, in Ikkαf/f:CD4.Cre mice, and 2.72 and 11.2%, respectively, in WT mice (P < 0.01; Fig. 1C). In the WT mice, the average number of Tregs in the spleen, thymus, and mLNs was 2.01 × 106, 0.52 × 106, and 1.06 ×106, which was reduced to 0.71 × 106, 0.15 × 106, and 0.29 × 106, respectively, in the KO mice (P < 0.01–0.001; Fig. 1). Thus, the number of peripheral Tregs in Ikkαf/f:CD4.Cre mice was reduced by >70%. Therefore, IKKα appears to play an important role in the thymic generation and peripheral homeostasis of Tregs. Nevertheless, Tregs present in the Ikkαf/f:CD4.Cre mice were phenotypically similar to their WT counterparts and expressed high levels of CD25 and CTLA4 and a lower level of CD45RB (data not shown).
Figure 1.

The number of Tregs in Ikkαf/f:CD4.Cre mice is markedly reduced. Cells from the thymus, spleen, and LNs of WT mice and KO mice were stained with CD45, CD3, CD4, CD8, and Foxp3 Abs. The proportion of Foxp3+ cells was analyzed by FACS. A) Proportion of Foxp3+ cells in total thymocytes, in CD4+CD8− SP thymocytes, and the number of Foxp3+ cells in the thymus. B) Proportion of Foxp3+ cells in total LN cells, in CD4+ LN cells (pooled from mesenteric, axillary, and inguinal regions), and the number of Foxp3+ cells in the mLNs. C) Proportion of Foxp3+ cells in total splenic cells, in CD4+ splenic cells, and the number of Foxp3+ cells in spleen. Number in the FACS data indicates the proportion of gated cells. The summary of proportion or number of Tregs is shown as mean ± SD. Data shown are representatives of at least 3 separate experiments with similar results. Comparison (n = 3–5 mice per group, 2-tailed Student’s t test) between indicated groups is shown: **P < 0.01; ***P < 0.001.
To examine whether IKKα-deficient nCD4s were intrinsically defective in the expression of Foxp3, flow-sorted nCD4s from Ikkαf/f:CD4.Cre mice were stimulated with plate-bound anti-CD3 and soluble anti-CD28 in the presence of TGF-β. The results show that Foxp3 can normally be induced in IKKα-deficient nCD4s (data not shown), indicating that the reduction in Tregs is not caused by the impaired capacity to express Foxp3 induced by TGF-β.
IKKα-deficient Tregs fail to inhibit colitis in Rag 1−/− mice induced by transfer of nCD4s
Previously, we (13) and Housley et al. (24) showed that Tregs deficient in TNFR2 failed to inhibit colitis in Rag 1−/− mice induced by transfer of WT nCD4s. Therefore, we hypothesized that IKKα deficiency may also result in defective suppressive function of Tregs in the inflammatory milieu. To test this idea, flow-sorted WT CD4+CD25−CD45RBhigh naive cells were transferred alone or cotransferred with WT Tregs or IKKα-deficient Tregs into Rag 1−/− mice. As expected, transfer of WT nCD4s alone induced colitis in Rag1−/− mice, as shown by the reduction of body weight (Fig. 2A) and enhanced thickness of the wall of the colon (Fig. 2C). This pathogenic effect of WT nCD4s was inhibited by the cotransfer of WT Tregs, but not IKKα-deficient Tregs (Fig. 2A, C). Furthermore, transfer of IKKα-deficient nCD4s alone failed to induce colitis (P < 0.01–0.05; Fig. 2B, C). We also noticed that the size of spleens and LNs correlated with the degree of inflammation. Spleens from Rag1−/− mice treated with WT nCD4s (134 mg on average) or cotreated with IKKα-deficient Tregs (123 mg on average) were >2-fold larger than those from mice cotreated with WT nCD4s and WT Tregs (58 mg on average), or treated with IKKα-deficient nCD4s alone (40 mg on average; Fig. 2C, D; P < 0.01–0.001).
Figure 2.

In vivo effect of Tregs and Teffs from Ikkαf/f:CD4.Cre mice on the development of colitis in Rag 1−/− mice. nCD4s (CD4+CD25−CD45RBhi) and Tregs (CD4+CD25+CD45RBlo) were flow sorted from WT mice and Ikkαf/f:CD4.Cre mice. WT nCD4s (4 × 105 cells/mouse) were transferred alone or cotransferred with WT or KO Tregs at a 5:1 ratio into Rag1−/− mice. As a comparison, KO nCD4s (4 × 105 cells/mouse) were also transferred alone into Rag 1−/− mice. A) Body weight change of mice transferred with WT nCD4s alone or cotransferred with WT Tregs or KO Tregs. B) Body weight change of mice transferred with WT nCD4s or KO nCD4s. Comparison (2-tailed Student’s t test) between WT nCD4-transfer group with other groups is shown: * P < 0.05; ** P < 0.01; *** P < 0.001. C) Representative photographs of colon, spleen, and LNs. D) Weight of spleens (n = 7–10). Dashed line indicates the weight of Rag1−/− mouse spleen without transfer of T cells. Data shown are representative of 3 separate experiments with similar results. Comparison (1-way ANOVA test) between indicated groups is shown: **P < 0.01; ***P < 0.001.
WT naive cells, once administered to Rag 1−/− mice, became activated and produced proinflammatory INFγ and IL-17A in the cLP (Fig. 3A). As we reported previously (13), WT Tregs had the capacity to inhibit expression of INFγ but enhanced IL-17A expression by nCD4s (P < 0.05–0.01; Fig. 3A–C). In contrast, IKKα-deficient Tregs failed to do so (Fig. 3A–C). Cytokine expression profile of IKKα-deficient Teffs could not be determined because of the paucity of T cells recovered from Rag1−/− mice administered IKKα-deficient nCD4s. Therefore, IKKα is critical for the in vivo function of both Tregs and Teffs after transfer into Rag1−/− mice.
Figure 3.

IKKα-deficient Tregs failed to suppress pathogenic Th1 responses in vivo. nCD4s and Tregs from WT mice and Ikkαf/f:CD4.Cre mice were transferred alone or cotransferred with WT or KO Tregs at a 5:1 ratio into Rag1−/− mice, as described in Fig. 2. After 8 wk, cLPLs were isolated. The intracellular expression of IFNγ and IL-17A by initially transferred naive WT CD4 cells presented in the cLPLs was analyzed by FACS, gating on TCRβ+CD4+Foxp3− cells. Representative FACS plot (A) and summary (B and C, mean ± SEM; n = 3–8) are shown. Data shown are representative of 3 separate experiments with similar results. Comparison (1-way ANOVA test) of indicated groups is shown: *P < 0.05; **P < 0.01.
IKKα-deficient CD4 cells, both Tregs and Teffs, fail to repopulate in the lymphopenic mice
It has been reported that the administered Foxp3+ and Foxp3− T cells replicated similarly in the lymphopenic mouse (25, 26). Thus, the malfunction of IKKα-deficient Tregs and Teffs might therefore be attributed to their inability to expand in lymphopenic Rag1−/− mice. To test this, we first analyzed the proportion of transferred CD4 cells, identified by expression of CD4 and CD3, in total CD45+ leukocytes present in the mLNs, and in the draining LNs of colon. As shown in Fig. 4A, B, roughly half of the leukocytes recovered from mLNs were transferred CD4 T cells (41.9–54.5%) in Rag 1−/− mice inoculated with WT nCD4s alone or together with WT Tregs or KO Tregs. In contrast, only 8.3% of leukocytes in mLNs in the mice injected with IKKα-deficient nCD4s were transferred T cells, which was ∼7-fold lower than that in mice injected with WT nCD4s (P < 0.0001; Fig. 4A, B). The proportion of Foxp3+ cells present in mLNs was also analyzed. Only a minor fraction (<1%) of total transferred cells was Foxp3+ cells in Rag 1−/− mice given either WT nCD4s alone or KO nCD4s alone (Fig. 4C, D). Therefore, none or an extremely low number of nCD4s were converted into Foxp3-expressing Tregs in Rag1−/− mice. In Rag1−/− mice given WT nCD4s and WT Tregs, there was ∼20% of Foxp3+ cells in the total CD4 cell population; the majority of them should represent initially transferred WT Tregs. In contrast, only 2.2% of Foxp3+ cells were recovered when WT nCD4s were transferred with IKKα-deficient Tregs, which was markedly lower than in mice with cotransferred WT Tregs (P < 0.001; Fig. 4C, D). We examined other tissues, including LNs in the axillary and inguinal regions, spleen, and cLP, and observed similar results (data not shown). Thus, IKKα expression is crucial for the repopulation of CD4 T cells, including both Tregs and Teffs, in the lymphopenic environment.
Figure 4.

IKKα is required for the repopulation of nCD4 T cells and Tregs in Rag1−/− mice. nCD4s and Tregs from WT mice and Ikkαf/f:CD4.Cre mice were transferred alone or cotransferred with WT or KO Tregs at a 5:1 ratio into Rag1−/− mice, as described in Fig. 2. After 8 wk, mice were sacrificed. A and B) The proportion of transferred CD4 T cells in the total CD45+ leukocyte population in mLNs of recipient Rag 1−/− mice was analyzed by FACS. (A) shows the representative FACS analysis (gating on CD45+ cells), and (B) shows the summary of data (mean ± SD; n = 7–9). Comparison (1-way ANOVA test) of KO nCD4 transfer alone with other groups is shown: ***P < 0.001, C, D) The proportion of Foxp3+ cells in transferred CD4 T cells present in the mLNs of recipient Rag 1−/− mice was analyzed by FACS, gating on CD45+CD3+CD4+ cells. The typical FACS data are shown in (C), and summary of data is shown in (D) (mean ± SD; n = 7–9). Compared (1-way ANOVA test) of WT nCD4s plus WT Tregs with other groups is shown: ***P < 0.001. The number in the FACS data indicates the proportion of cells in the respective gating. Data shown are representative of 3 separate experiments with similar results.
IKKα-deficient CD4 cells are defective in their proliferative expansion in vivo
It has been reported that unfractionated CD4 cells with mutant IKKα normally repopulated Rag 1−/− mice (15). Therefore, we further compared the capacity of IKKα-deficient total CD4 cells to competitively expand in Rag1−/− mice with WT CD4 cells inoculated in the same mouse. To this end, CD4 cells from spleens of congenic Ly5.2 WT B6 mice (CD45.2−) or from Ikkαf/f:CD4.Cre mice (CD45.2+) were isolated with MACS, mixed at a 1:1 ratio and then stained with CellTraceth Violet (Fig. 5A). The cells were transferred (2 × 106 cells/mouse) into Rag1−/− mice. After 2 wk, recipient Rag1−/− mice were sacrificed. The proportion of WT and KO CD4 T cells was analyzed by FACS, gating on the live CD45+CD4+TCRβ+ population. As shown in Fig. 5B, the majority of transferred T cells present in the spleen of recipient Rag 1−/− mice were WT CD4 cells (>80%), whereas KO cells comprised less than one-quarter of total T cells (P < 0.001), which was markedly reduced from the initial one-half of total transferred cells.
Figure 5.

IKKα is crucial for competitive expansion of CD4 T cells in Rag1−/− mice. CD4 cells from spleens of Ly5.2 WT B6 mice (CD45.2−) or Ikkαf/f:CD4.Cre mice (CD45.2+) were isolated by MACS and mixed at a 1:1 ratio and were labeled with CellTraceth Violet (A). The cells (2 × 106 cells/mouse) were transferred into Rag1−/− mice. After 2 wk, recipient Rag1−/− mice were sacrificed. The proportion of WT and KO T cells in total transferred T cells (CD45+CD4+TCRβ+ cells) present in the spleen was analyzed by FACS (B) (left shows the typical FACS histogram, and right shows the summary; n = 3). Data shown are representatives of 3 separate experiments with similar results. Proliferation of WT CD4 cells (C) or KO CD4 cells (D) was analyzed by FACS, gating on their respective congenic marker. Typical FACS data are shown in (C) and (D). Summary of proportion of spontaneous and homeostatic proliferation cells is shown in (E), and the MFI of FxCycle Violet of both forms of proliferation is shown in (F) (n = 10), pooled from 2 separate experiments. Number in the FACS data indicates the proportion of gated cells or MFI. Comparison (2-tailed Student’s t test) between indicated groups is shown: **P < 0.01; ***P < 0.001.
To determine whether IKKα-deficient CD4 T cells were more susceptible to cell death, WT and IKKα-deficient CD4 cells were stimulated with plate-bound CD3 and soluble CD28. After 24 h stimulation, the proportion of dead cells, as identified by positive propidium iodide (PI) staining, was analyzed by FACS. The results showed that IKK–α-deficient CD4 cells were not more susceptible than WT cells to activation-induced cell death upon in vitro TCR stimulation (Supplemental Fig. 3A). Furthermore, cell viability of WT and KO CD4 T cells in the peripheral blood of Rag 1−/− recipient mice was analyzed by LIVE/DEAD Fixable Near-IR Stain (Life Technologies). The results also clearly showed that IKKα deficiency did not result in greater cell death in vivo (Supplemental Fig. 3B). Thus, the results from both in vitro and in vivo experiments excluded the possibility that the reduction of IKKα-deficient CD4 T cells in Rag 1−/− mice was due to cell death.
Therefore, we further studied the proliferation of IKKα-deficient CD4 T cells in Rag 1−/− mice, which were inoculated with FxCycle Violet (Life Technologies)-stained WT and IKKα-deficient CD4 T cells at a 1:1 ratio. After 2 wk of transfer, both WT and IKKα-deficient CD4 T cells exhibited a rapid, burst-like proliferation that leads to the loss of detectable FxCycle Violet Stain, and a slow steady proliferation with detectable gradient dilution of FxCycle Violet Stain (Fig. 5C, D), which was consistent with a previous report (27). However, >99% of WT CD4 T cells were found in the population undergoing rapid proliferation, and <1% of cells were in the slow proliferative population. In contrast, a markedly reduced population of rapid proliferative cells (82.1%) was found in IKKα-deficient CD4 cells (Fig. 5D, E; P < 0.01). More importantly, IKKα-deficient CD4 cells in both rapid and slow proliferative subsets showed a markedly higher intensity of FxCycle Violet Stain, compared with their WT counterparts (Fig. 5C, D, F; P < 0.01–0.001), indicating that IKKα-deficient CD4 cells as a whole were less proliferative than WT CD4 cells. The same results were also found in T cells recovered from LNs in the mesenteric, axillary, and inguinal regions and in the peripheral blood (data not shown), mitigating the contribution of a potential different trafficking pattern of IKKα-deficient CD4 cells to the results. Thus, IKKα is critically important for the competitive expansion of CD4 cells in the lymphopenic Rag 1−/− mice.
To exclude the confounding effect of a potential difference in genetic background between WT B6 Ly5.2 mice and KO (Ikkαf/f:CD4.Cre) mice, we further compared the proliferative capacity of CD4 cells isolated from WT (Ikkαf/f) mice or CD4.Cre mice with CD4 cells isolated from KO mice (Ikkαf/f:CD4.Cre) by inoculating the same number of cells separately into Rag 1−/− mice. As shown in Fig. 6A, B, 17 d after inoculation of 1.6 × 106 of WT CD4 cells, 12.3% of splenic CD45+ cells were derived from transferred CD4 cells. In contrast, only 1.19% of splenic CD45+ cells were derived from transferred CD4 cells when donor cells were from KO mice (P < 0.01), consistent with data shown in Fig. 4A. Consequently, the absolute number of transferred cells in the spleen of recipient mice was 1.13 × 106 cells on average when donor cells were from WT mice, which was markedly reduced to 0.03 × 106 cells on average when donor cells were from KO mice (P < 0.01; Fig. 6C). Apparently, the difference in transferred cell number was more profound than the difference in their proportion in CD45+ cells, which was due to the reduced number of total leukocytes in lymphoid organs from Rag 1−/− mice receiving KO CD4 cells compared with those in the recipient mice administered WT CD4 cells (data not shown).
Figure 6.
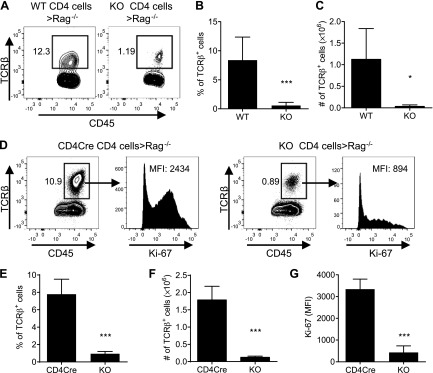
Impaired capacity of unfractionated CD4 T cells to repopulate in Rag 1−/− mice. A–C) CD4 cells from WT (Ikkαf/f mice) and Ikkαf/f:CD4.Cre mice were isolated by MACS. The cells (1.6 × 106 cells/mouse) were transferred into Rag1−/− mice. After 17 d, recipient Rag1−/− mice were sacrificed. The proportion of WT and KO T cells (TCRβ+ cells) in total CD45+ leukocytes present in the spleen was analyzed by FACS. A) Representative FACS plots. The analysis of TCRβ+ cells was gated on CD45+ splenic cells. B) Summary of the proportion of TCRβ+ cells in CD45+ splenic cells (mean ± SD; n = 4–6). C) The number of transferred cells in the recipient mouse spleen (mean ± SD; n = 4–6). D–G) CD4 cells from CD4.Cre mice or KO (Ikkαf/f:CD4.Cre) mice were isolated by MACS. The cells (4 × 106 cells/mouse) were transferred into Rag1−/− mice. After 14 d, recipient Rag1−/− mice were sacrificed. The proportion and Ki-67 expression by CD4.Cre or KO T cells (TCRβ+ cells) present in the spleen were analyzed by FACS. D) Representative FACS plots. The analysis of TCRβ+ cells was gated on CD45+ splenic cells, and analysis of Ki-67 was gated on TCRβ+ cells. E) Summary of the proportion of TCRβ+ cells in CD45+ splenic cells (mean ± SD; n = 4). F) The number of transferred cells in the recipient mouse spleen (mean ± SD; n = 4). G) Summary of Ki-67 expression (MFI, mean fluorescence intensity) by transferred cells. Data shown are representative of 2 separate experiments with similar results. Number in the FACS data indicates the proportion of gated cells or MFI. Comparison (2-tailed Student’s t test) between indicated groups is shown: *P < 0.05; ***P < 0.001.
We next examined the expansion of unfractionated CD4 cells from CD4.Cre mice or from KO mice (Ikkαf/f:CD4.Cre) transferred into Rag 1−/− mice from 2 wk previously. The proportion of transferred CD4.Cre T cells recovered from Rag 1−/− mouse spleen was 10.9% of the total CD45+ leukocytes. In contrast, only 0.89% of IKKα-deficient T cells were found in the Rag 1−/− mouse spleen (Fig. 6D, E; P < 0.001). Consequently, there were >14-fold more T cells present in the spleen of mice administered CD4.Cre cells (1.79 × 106 cells/spleen) compared with mice given IKKα-deficient T cells (0.13 × 106 cells/spleen; Fig. 6F; P < 0.001). The reduced number of IKKα-deficient cells was attributed to their impaired proliferation because their Ki-67 expression was markedly lower than the expression on the CD4.Cre cells (Fig. 6D, G; P < 0.001). In addition to analyzing the spleen, we also examined the proportion, number, and Ki-67 expression by transferred CD4.Cre cells and KO cells in the axillary/inguinal LNs, mLNs, colon lamina propria lymphocyte (cLPL), and blood and obtained similar results (data not shown). Therefore, the impaired proliferative expansion of IKKα-deficient CD4 cells in the lymphopenic mice was not due to the CD4.Cre expression but attributable to the lack of IKKα.
The proportion and number of Tregs present in CD4 cells from CD4.Cre and KO mice before and after transfer were also studied. First, we compared the number of Tregs in the spleen, mLNs, and thymus of CD4.Cre and KO mice. Similar to the presence of Tregs in the WT and KO mice (Fig. 1), the proportion of Foxp3+ cells in the CD45+ cells, or in the CD4+ T cells and the absolute number of Foxp3+ Tregs in these organs, was markedly higher in the CD4.Cre mice than in the KO mice (Supplemental Fig. 4A, C; P < 0.05–0.001). Before transfer, 11.9% of MACS-purified CD4 cells from CD4.Cre mice were Foxp3+ Tregs, whereas only 5.01% were Foxp3+ Tregs in CD4 cells derived from KO mice (Supplemental Fig. 4D). Two weeks after transferring into Rag 1−/− mice, the proportion of Foxp3+ Tregs in the CD4 cells recovered from recipient mice was the same as before transfer, i.e., 2- to 3-fold more Tregs in T cells derived from CD4.Cre mice than cells from KO mice (P < 0.001; Supplemental Fig. 4E). These data further confirmed that IKKα deficiency is the cause of the reduction of Tregs.
Proliferative responses of IKKα-deficient nCD4s to in vitro TCR stimulation are impaired
It has been proposed that IKKα may play a role in T-cell proliferative response to a weak stimulant but has no impact when T cells are strongly stimulated (15). Therefore, we further examined the role of IKKα in the proliferative response of CD4 cells to TCR stimulation with different potencies in vitro. Because CD4 cells from WT and Ikkαf/f:CD4.Cre mice contain a different number of Tregs that may interfere with the results, flow-sorted CD4+CD25−CD445RBhi cells were used in this study. WT or IKKα-deficient nCD4s (104–105 cells/well) were stimulated with soluble CD3 Ab plus APCs derived from WT mice (Fig. 7A), from Ikkαf/f:CD4.Cre mice (Fig. 7B), or stimulated with plate-bound anti-CD3 Ab alone (Fig. 7C) or plate-bound anti-CD3 plus soluble anti-CD28 Ab (Fig. 7D). Under all stimulation conditions, the nCD4s from WT mice were markedly more proliferative than nCD4s from Ikkαf/f:CD4.Cre mice (Fig. 7; P < 0.05–0.001). These data clearly show that IKKα is required for maximal proliferation of CD4 cells to TCR stimulation in vitro regardless of the strength of TCR stimulation.
Figure 7.

Proliferative responses of nCD4s from Ikkαf/f:CD4.Cre mice to TCR stimulation. CD4+CD25− cells were flow sorted from spleens and LNs pooled from 3 WT or 3 Ikkαf/f:CD4.Cre mice. The cells were seeded to the round-bottom 96-well plate at 104–105 cells/well in triplicate. The cells were stimulated with soluble anti-CD3 Ab and APCs from WT mice (A) or from Ikkαf/f:CD4.Cre mice (B), or cells were stimulated with plate-bound anti-CD3 Ab alone (C) or plate-bound anti-CD3 Ab and soluble anti-CD28 Ab (D) for 72 h. Proliferation was measured by [3H]thymidine incorporation assay. Data (mean ± SD; n = 3) shown are representatives of 3 separate experiments with similar results. Data were analyzed with 2-way ANOVA test: *P < 0.05; **P < 0.01; ***P < 0.001.
DISCUSSION
NF-κB has been shown to play an important role in the homeostasis and function of Tregs. For example, it was reported that c-Rel regulates the differentiation of Tregs (28–31). The Ubc13-IKKβ signaling axis was shown to promote the maintenance of suppressive function and phenotypic stability of Tregs (32), and IKKβ was also shown to be involved in the generation of Tregs (33). In this study, we found that specific ablation of IKKα in CD4 cells resulted in a marked reduction of Tregs in the thymus as well as in the peripheral lymphoid tissues. Because the signaling of TNFR2 has been shown to activate the noncanonical NF-κB pathway (14), this phenotype of IKKα correlates well with mouse strains deficient in receptor or ligands of TNFR2 (13). Therefore, in addition to the well-known effect of canonical NF-κB pathway on Tregs, the alternative activation of NF-κB pathways is also required for the homeostasis of Tregs.
Noncanonical NF-κB activation relies on accumulation of NF-κB-inducing kinase (NIK) and subsequent phosphorylation of IKKα; thus, both NIK and IKKα are key noncanonical NF-κB signaling components (18). Mouse strain with mutant NIK manifested impaired the capacity to generate Tregs (34). More recently, it was shown that NIK plays an essential cell-intrinsic role in peripheral Treg maintenance (35). The correlation in the reduction of Tregs in the thymus and periphery in mouse strains deficient in NIK and IKKα recapitulated the crucial role of noncanonical NF-κB pathways in the thymic generation and peripheral homeostasis of Tregs. Unlike the canonical pathway, activation of noncanonical NF-κB is quite restricted to a subset of TNFR family members, including TNFR2 (18). Therefore, costimulatory TNFRSF members may synergistically activate the noncanonical NF-κB pathway through NIK and IKKα, which may explain why they can collectively promote thymic generation of Tregs (17) and expansion of peripheral Tregs (36). Indeed, OX40 has been recently shown to trigger induction of NIK and activation of noncanonical NF-κB in CD4 T cells (37). This effect of OX40 may synergize with signaling of TNFR2 and result in the maximal activation of Tregs (36). In addition to costimulatory TNFRSF members, activation of the noncanonical NF-κB pathway may also function as an underlying mechanism of a Treg-promoting effect of other molecules. For example, CD28 is required for both thymic generation of Tregs (35) as well as survival and expansion of Tregs in the periphery (34). This action of CD28 is at least partially dependent on its activating effect of the noncanonical NF-κB pathway (37). Therefore, the possibility that the noncanonical NF-κB pathway may be a therapeutic target for up- or down-regulation of Treg activity should be investigated.
Current understanding of the role of IKKα in the proliferative responses of CD4 T cells to TCR stimulation is also largely based on the studies of IKKαAA mice. It was reported that the proliferative responses of T cells (20) or CD4 cells (15) with a mutant IKKα to anti-CD3 plus anti-CD28 stimulation were not reduced. However, our data clearly show that both Tregs and Teffs isolated from Ikkαf/f:CD4.Cre mice failed to expand upon transfer into lymphopenic Rag1−/− mice. Furthermore, the in vitro proliferative responses of CD4 cells to TCR stimulation were markedly reduced. The noncanonical NF-κB pathway is largely responsible for the activation of RelB, and RelB activation is known to be required for the maximal T-cell activation (38). In IKKα mutant CD4 cells, RelB was inhibited, whereas proliferative responses were normal, which is likely caused by the reduction in IκBα resynthesis and consequently compensates for the inhibition of RelB activation in IKKα-mutated CD4 cells (15). Furthermore, the ratio of p100:p52 was increased by up to 4-fold in CD4 cells isolated from Ikkαf/f:CD4.Cre mice compared with CD4 cells from WT mice (Supplemental Fig. 1), consistent with a previous finding that IKKα was required for the processing of p100 and the generation of p52 (8). p100 is an inhibitor of the canonical NF-κB pathway (39), which is crucial for the biologic function of CD4 T cells (40), and p100 can down-regulate TCR signaling (41). It has been shown that the balance between p100 and p52 had a major impact on the activation of T cells (42). Therefore, the increased ratio of p100:p52 was at least partially attributable to the impaired proliferative responses of IKKα-deficient CD4 cells. Nevertheless, IKKα was shown to inhibit the proliferation of other cell types (43–45). Thus, the effect of IKKα on cellular proliferation appears to be cell-type specific.
Li et al. (15) reported that adoptive transfer of CD4+ T cells expressing mutant IKKα (IKKαAA) into Rag 1−/− mice resulted in a marked reduction in the development of experimental autoimmune encephalomyelitis (EAE) induced by myelin oligodendrocyte glycoprotein (MOG) peptide compared with transfer of WT CD4 cells. However, there was no difference in the number of splenic T cells between Rag1−/− recipient mice that received WT and IKKαAA cells (15). In the study by Li et al. (15), large numbers of total CD4 T cells (107 cells/mouse) were transferred into Rag1−/− mice, which was >35-fold than the number of naive CD4 T cells (4 × 105 cells/mouse) used in our transfer studies. Presumably, in contrast with the transfer of a smaller number of nCD4s, repopulation by a large number of unfractionated splenic CD4 cells used in the study by Li et al. (15) may depend less on the expansion in Rag1−/− mice. In addition, genetic ablation of IKKα is also likely to have a different effect on CD4 cells than a mutation of IKKα. Nevertheless, IKKαAA CD4 T cells recovered from EAE-inducing Rag1−/− mice were also defective in their proliferative responses to MOG peptide in vitro as shown in the study by Li et al. (15), which probably reflects the effect of IKKα on the proliferative response of CD4 cells as revealed by our study.
Taken together, our study reveals a novel role of IKKα in the maintenance of the population of Treg cells and in the control of expansion of CD4 T cells. These properties of IKKα may be exploited for therapeutic strategies in the treatment of major human diseases. For example, inhibition of IKKα may eliminate Treg activity by reducing the numbers of Tregs. Because activation of macrophages is inhibited by both IKKα (43–45) and Tregs (46), inhibition of IKKα may also markedly enhance macrophage-mediated innate defense against invading pathogens and cancer. On the other hand, an inhibitor of IKKα may prove useful in the treatment of T-cell proliferative disorders, such as autoimmune inflammation and hematopoietic malignancies. These potential beneficial effects of targeting IKKα should be addressed in future studies.
Supplementary Material
Acknowledgments
The authors thank Dr. Na-Young Song, Trivett L. Anna, and Czarra T. Kelli for their help in this study. The authors thank the U.S. National Cancer Institute (NCI)-Frederick Cancer Inflammation Program Fluorescence Cytometry Core (Kathleen B. Noer, Roberta M. Matthai, and Guity Mohammadi) for expert technical assistance with flow cytometry. This project has been funded in whole or in part with federal funds from the U.S. National Institutes of Health (NIH) NCI, under contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH NCI, Center for Cancer Research.
Glossary
- Ab
antibody
- APC
antigen-presenting cell
- cLP
colon lamina propria
- cLPL
colon lamina propria lymphocyte
- EAE
experimental autoimmune encephalomyelitis
- FACS
fluorescence-activated cell sorting
- FCS
fetal calf serum
- FoxP3
forkhead box P3
- IFN
interferon
- KO
knockout
- LN
lymph node
- mLN
mesenteric lymph node
- MOG
myelin oligodendrocyte glycoprotein
- nCD4
naive CD4 cell
- NIK
NF-κB-inducing kinase
- PI
propidium iodide
- RPMI
Roswell Park Memorial Institute medium
- SP
single positive
- TCR
T-cell receptor
- Teff
effector T cell
- TNFR
TNF receptor
- TNFR2
TNF receptor type II
- TNFRSF
TNF receptor superfamily
- Treg
regulatory T cell
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Zhu J., Yamane H., Paul W. E. (2010) Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 28, 445–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaech S. M., Wherry E. J., Ahmed R. (2002) Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2, 251–262 [DOI] [PubMed] [Google Scholar]
- 3.Sakaguchi S., Yamaguchi T., Nomura T., Ono M. (2008) Regulatory T cells and immune tolerance. Cell 133, 775–787 [DOI] [PubMed] [Google Scholar]
- 4.Zou W. (2006) Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 6, 295–307 [DOI] [PubMed] [Google Scholar]
- 5.Dev A., Iyer S., Razani B., Cheng G. (2011) NF-κB and innate immunity. Curr. Top. Microbiol. Immunol. 349, 115–143 [DOI] [PubMed] [Google Scholar]
- 6.Zandi E., Rothwarf D. M., Delhase M., Hayakawa M., Karin M. (1997) The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91, 243–252 [DOI] [PubMed] [Google Scholar]
- 7.Häcker H., Karin M. (2006) Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13. [DOI] [PubMed] [Google Scholar]
- 8.Senftleben U., Cao Y., Xiao G., Greten F. R., Krähn G., Bonizzi G., Chen Y., Hu Y., Fong A., Sun S. C., Karin M. (2001) Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 293, 1495–1499 [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto Y., Verma U. N., Prajapati S., Kwak Y. T., Gaynor R. B. (2003) Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature 423, 655–659 [DOI] [PubMed] [Google Scholar]
- 10.Chen X., Bäumel M., Männel D. N., Howard O. M., Oppenheim J. J. (2007) Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. 179, 154–161 [DOI] [PubMed] [Google Scholar]
- 11.Chen X., Subleski J. J., Hamano R., Howard O. M., Wiltrout R. H., Oppenheim J. J. (2010) Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur. J. Immunol. 40, 1099–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X., Subleski J. J., Kopf H., Howard O. M., Männel D. N., Oppenheim J. J. (2008) Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J. Immunol. 180, 6467–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X., Wu X., Zhou Q., Howard O. M., Netea M. G., Oppenheim J. J. (2013) TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J. Immunol. 190, 1076–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rauert H., Wicovsky A., Müller N., Siegmund D., Spindler V., Waschke J., Kneitz C., Wajant H. (2010) Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J. Biol. Chem. 285, 7394–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L., Ruan Q., Hilliard B., Devirgiliis J., Karin M., Chen Y. H. (2011) Transcriptional regulation of the Th17 immune response by IKK(alpha). J. Exp. Med. 208, 787–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X., Hamano R., Subleski J. J., Hurwitz A. A., Howard O. M., Oppenheim J. J. (2010) Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3- conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J. Immunol. 185, 174–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahmud S. A., Manlove L. S., Schmitz H. M., Xing Y., Wang Y., Owen D. L., Schenkel J. M., Boomer J. S., Green J. M., Yagita H., Chi H., Hogquist K. A., Farrar M. A. (2014) Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat. Immunol. 15, 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun S. C. (2011) Non-canonical NF-κB signaling pathway. Cell Res. 21, 71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaisho T., Takeda K., Tsujimura T., Kawai T., Nomura F., Terada N., Akira S. (2001) IkappaB kinase alpha is essential for mature B cell development and function. J. Exp. Med. 193, 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren H., Schmalstieg A., van Oers N. S., Gaynor R. B. (2002) I-kappa B kinases alpha and beta have distinct roles in regulating murine T cell function. J. Immunol. 168, 3721–3731 [DOI] [PubMed] [Google Scholar]
- 21.Liu B., Xia X., Zhu F., Park E., Carbajal S., Kiguchi K., DiGiovanni J., Fischer S. M., Hu Y. (2008) IKKalpha is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell 14, 212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Institute of Laboratory Animal Research, Commission on Life Sciences, National Research Council (1996) Guide for Care and Use of Laboratory Animals, National Academy Press, Washington, D.C. [Google Scholar]
- 23.Griseri T., Asquith M., Thompson C., Powrie F. (2010) OX40 is required for regulatory T cell-mediated control of colitis. J. Exp. Med. 207, 699–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Housley W. J., Adams C. O., Nichols F. C., Puddington L., Lingenheld E. G., Zhu L., Rajan T. V., Clark R. B. (2011) Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J. Immunol. 186, 6779–6787 [DOI] [PubMed] [Google Scholar]
- 25.Yurchenko E., Shio M. T., Huang T. C., Da Silva Martins M., Szyf M., Levings M. K., Olivier M., Piccirillo C. A. (2012) Inflammation-driven reprogramming of CD4+ Foxp3+ regulatory T cells into pathogenic Th1/Th17 T effectors is abrogated by mTOR inhibition in vivo. PLoS One 7, e35572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duarte J. H., Zelenay S., Bergman M. L., Martins A. C., Demengeot J. (2009) Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur. J. Immunol. 39, 948–955 [DOI] [PubMed] [Google Scholar]
- 27.Min B., Yamane H., Hu-Li J., Paul W. E. (2005) Spontaneous and homeostatic proliferation of CD4 T cells are regulated by different mechanisms. J. Immunol. 174, 6039–6044 [DOI] [PubMed] [Google Scholar]
- 28.Isomura I., Palmer S., Grumont R. J., Bunting K., Hoyne G., Wilkinson N., Banerjee A., Proietto A., Gugasyan R., Wu L., McNally A., Steptoe R. J., Thomas R., Shannon M. F., Gerondakis S. (2009) c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J. Exp. Med. 206, 3001–3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long M., Park S. G., Strickland I., Hayden M. S., Ghosh S. (2009) Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 31, 921–931 [DOI] [PubMed] [Google Scholar]
- 30.Ruan Q., Kameswaran V., Tone Y., Li L., Liou H. C., Greene M. I., Tone M., Chen Y. H. (2009) Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 31, 932–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Y., Josefowicz S., Chaudhry A., Peng X. P., Forbush K., Rudensky A. Y. (2010) Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463, 808–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang J. H., Xiao Y., Hu H., Jin J., Yu J., Zhou X., Wu X., Johnson H. M., Akira S., Pasparakis M., Cheng X., Sun S. C. (2012) Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat. Immunol. 13, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gückel E., Frey S., Zaiss M. M., Schett G., Ghosh S., Voll R. E. (2011) Cell-intrinsic NF-κB activation is critical for the development of natural regulatory T cells in mice. PLoS One 6, e20003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang R., Huynh A., Whitcher G., Chang J., Maltzman J. S., Turka L. A. (2013) An obligate cell-intrinsic function for CD28 in Tregs. J. Clin. Invest. 123, 580–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tai X., Cowan M., Feigenbaum L., Singer A. (2005) CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat. Immunol. 6, 152–162 [DOI] [PubMed] [Google Scholar]
- 36.Hamano R., Huang J., Yoshimura T., Oppenheim J. J., Chen X. (2011) TNF optimally activatives regulatory T cells by inducing TNF receptor superfamily members TNFR2, 4-1BB and OX40. Eur. J. Immunol. 41, 2010–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanchez-Valdepenas C., Martin A. G., Ramakrishnan P., Wallach D., Fresno M. (2006) NF-kappaB-inducing kinase is involved in the activation of the CD28 responsive element through phosphorylation of c-Rel and regulation of its transactivating activity. J. Immunol. 176, 4666–4674 [DOI] [PubMed] [Google Scholar]
- 38.Corn R. A., Hunter C., Liou H. C., Siebenlist U., Boothby M. R. (2005) Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J. Immunol. 175, 2102–2110 [DOI] [PubMed] [Google Scholar]
- 39.Basak S., Kim H., Kearns J. D., Tergaonkar V., O’Dea E., Werner S. L., Benedict C. A., Ware C. F., Ghosh G., Verma I. M., Hoffmann A. (2007) A fourth IkappaB protein within the NF-kappaB signaling module. Cell 128, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oh H., Ghosh S. (2013) NF-κB: roles and regulation in different CD4(+) T-cell subsets. Immunol. Rev. 252, 41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Legarda-Addison D., Ting A. T. (2007) Negative regulation of TCR signaling by NF-kappaB2/p100. J. Immunol. 178, 7767–7778 [DOI] [PubMed] [Google Scholar]
- 42.Giardino Torchia M. L., Conze D. B., Jankovic D., Ashwell J. D. (2013) Balance between NF-kappaB p100 and p52 regulates T cell costimulation dependence. J. Immunol. 190, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lawrence T., Bebien M., Liu G. Y., Nizet V., Karin M. (2005) IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature 434, 1138–1143 [DOI] [PubMed] [Google Scholar]
- 44.Li Q., Lu Q., Bottero V., Estepa G., Morrison L., Mercurio F., Verma I. M. (2005) Enhanced NF-kappaB activation and cellular function in macrophages lacking IkappaB kinase 1 (IKK1). Proc. Natl. Acad. Sci. USA 102, 12425–12430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu B., Yang Y., Chernishof V., Loo R. R., Jang H., Tahk S., Yang R., Mink S., Shultz D., Bellone C. J., Loo J. A., Shuai K. (2007) Proinflammatory stimuli induce IKKalpha-mediated phosphorylation of PIAS1 to restrict inflammation and immunity. Cell 129, 903–914 [DOI] [PubMed] [Google Scholar]
- 46.Tiemessen M. M., Jagger A. L., Evans H. G., van Herwijnen M. J., John S., Taams L. S. (2007) CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. USA 104, 19446–19451 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.