

Abstract
Objective
SIRT1 has been proposed to be a key signaling node linking changes in energy metabolism to transcriptional adaptations. Although SIRT1 overexpression is protective against diverse metabolic complications, especially in response to high-fat diets, studies aiming to understand the etiology of such benefits are scarce. Here, we aimed to identify the key tissues and mechanisms implicated in the beneficial effects of SIRT1 on glucose homeostasis.
Methods
We have used a mouse model of moderate SIRT1 overexpression, under the control of its natural promoter, to evaluate glucose homeostasis and thoroughly characterize how different tissues could influence insulin sensitivity.
Results
Mice with moderate overexpression of SIRT1 exhibit better glucose tolerance and insulin sensitivity even on a low fat diet. Euglycemic-hyperinsulinemic clamps and in-depth tissue analyses revealed that enhanced insulin sensitivity was achieved through a higher brown adipose tissue activity and was fully reversed by housing the mice at thermoneutrality. SIRT1 did not influence brown adipocyte differentiation, but dramatically enhanced the metabolic transcriptional responses to β3-adrenergic stimuli in differentiated adipocytes.
Conclusions
Our work demonstrates that SIRT1 improves glucose homeostasis by enhancing BAT function. This is not consequent to an alteration in the brown adipocyte differentiation process, but as a result of potentiating the response to β3-adrenergic stimuli.
Keywords: SIRT1, Energy homeostasis, Insulin resistance, Brown adipose tissue
1. Introduction
SIRT1 is a NAD+-dependent protein deacetylase and the best studied mammalian homolog of the yeast enzyme Sir2, a protein with an established capacity to influence yeast replicative lifespan [1]. In mammals, SIRT1 targets include a constellation of transcription factors and enzymes with key roles in mitochondrial biogenesis, lipid catabolism, cholesterol homeostasis and gluconeogenesis [1]. Studies using SIRT1 activating compounds (STACs) in mammals have highlighted how SIRT1 might have pleiotropic metabolic benefits. Dietary supplementation with STACs led to increased mitochondrial biogenesis in diverse mouse tissues, including skeletal muscle and brown adipose tissue (BAT), which in turn protected against high-fat diet (HFD)-induced obesity and many of its metabolic comorbidities [2]. The specificity of these compounds, however, has been called into question and is still unclear [3]. To overcome this problem, transgenic mouse models with a moderate SIRT1 overexpression were generated. In them, a single copy of a large genomic construct (174 kb) containing the entire Sirt1 gene in its natural genomic context was integrated [4]. This led to a 2–4-fold overexpression of SIRT1 in tissues from heterozygous mice for the transgene (SIRT1Tg/−) [4]. While similar to wild-type (WT) mice when fed low fat diets (LFD), SIRT1Tg/− mice were protected against HFD-induced insulin resistance, despite similar body weight gain [4]. These observations were confirmed in an independent SIRT1 overexpressing mouse line (SirBACO) generated by the Accili lab [5]. In light of the above results, we reasoned that the generation of a homozygous transgenic mouse (SIRT1Tg/Tg) might lead to a more marked phenotype, likely closer to that observed with STACs. Here, we describe how SIRT1Tg/Tg mice display enhanced energy expenditure (EE), glucose tolerance and insulin sensitivity even when fed an LFD. We demonstrate that this phenotype stems from a higher BAT activity and that SIRT1Tg/Tg mice do not show major muscle or liver functional changes on LFD. Using immortalized brown adipocytes from SIRT1 transgenic mice we further demonstrate that the effects of SIRT1 on BAT biology do not derive from differences in the brown adipocyte differentiation process, but from a higher response to β3-adrenergic stimuli. Altogether, our work illustrates how SIRT1 gain-of-function can improve insulin sensitivity by acting as a gauge for the BAT response to β3-adrenergic stimuli.
2. Material and methods
2.1. Animal care
The SIRT1 transgenic model has already been described in Ref. [4]. In contrast to that publication, we used homozygote transgenic male mice SIRT1Tg/Tg which had been backcrossed to C57Bl/6N background. Unless otherwise specified, mice were kept in a standard temperature- and humidity-controlled environment with a 12:12-h light–dark cycle. Mice had nesting material and ad libitum access to water and commercial LFD or HFD (D12450J and D12492, respectively, from Research Diets Inc.). All animal experiments were carried according to national Swiss and EU ethical guidelines and approved by the local animal experimentation committee under licenses 2519 and 2519.1-3. For thermoneutrality studies, mice were housed in temperature controlled cabinets at 30 °C.
2.2. Animal phenotyping
All clinical tests were carried out according to standard operational procedures (SOPs) established and validated within the Eumorphia program (http://empress.har.mrc.ac.uk/) [6]. Mice were weighed and the food consumption was measured each week on the same day. Body composition was determined by Echo-MRI (Echo Medical Systems, Houston, TX, USA) and oxygen consumption (VO2), respiratory exchange ratios (RER) were monitored by indirect calorimetry using the comprehensive laboratory animal monitoring system (CLAMS; Columbus Instruments, Columbus, OH, USA). EE was estimated using VO2 and VCO2 values from indirect calorimetry, using the following equation: EE (in kJ/h) = (15.818 × VO2) + (5.176 × VCO2) [7]. Food intake and activity (horizontal (XD) and vertical (Z)) was also monitored using the CLAMS during a 24 h period. Daily voluntary activity was measured by providing a running wheel to the mice and monitoring the running distance. Grip tests, treadmill and cold tests were performed as previously described in Ref. [8]. Maximal running speed and VO2 were evaluated using a calorimetric treadmill (Columbus instruments, Columbus, OH, USA) with an incremental speed protocol. During the run, VO2 and VCO2 were measured. The experiment was stopped when mice showed obvious signs of exhaustion. Glucose and insulin tolerance was analyzed by measuring blood glucose following intraperitoneal injection of 2 g/kg glucose or 0.3 U insulin/kg (human insulin actrapid, Lilly), respectively, after a 12 h fast. For HFD-fed mice, the insulin dose was increased to 0.75 U/Kg. Unless otherwise specified, animals were sacrificed at 8 a.m. after a 12 h fast, in order to stabilize systemic parameters and to allow the measurement of blood biochemistry in the fasting state. Blood samples were collected in EDTA-coated tubes and plasma was isolated after centrifugation. Plasma insulin was determined in plasma samples using specific ELISA kits (Merck Millipore, Darmstadt, Germany). All other plasma parameters were measured using Dimension® Xpand Plus (Siemens Healthcare Diagnostics AG, Dudingen, Suisse). Tissues were collected upon sacrifice and flash-frozen in liquid nitrogen.
2.3. Hyperinsulinemic-euglycemic clamp
Hyperinsulinemic-euglycemic clamps were performed at Phyiogenex (Physiogenex SAS, France) according to standardized procedures, following the Guide for the Care and Use of Laboratory animals and French laws. Mice (n = 12 per genotype) underwent a surgery procedure to insert a catheter in the femoral vein. Clamp was performed five days post-surgery on 6 h fasted mice. Following a first blood collection, mice received a bolus of D-[3-3H] glucose and then radiotracer (30 μCi/min/kg) infusion started for up to 210 min. Insulin was simultaneously infused at 4 mU/kg/min for the first 100 min and at 12 mU/kg/min for the last 110 min. Glucose infusion rate was adjusted according to blood glucose levels until the two respective euglycemic steadystates were reached. Moreover, 1 h before the end of the clamp, a bolus injection of 14C-2Deoxy-Glucose was performed. At the end of the 210-minute perfusion, mice were euthanized, several tissues were collected and 14C-radioactivity was measured to determine glucose utilization in epididymal and sub-cutaneous WAT, brown adipose tissue, vastus lateralis skeletal muscle and liver.
2.4. Respirometry studies
Respirometry studies in homogenates from freshly extracted BAT and liver or in permeabilized EDL muscle were performed using high-resolution respirometry (Oroboros Oxygraph-2k; Oroboros Instruments, Innsbruck, Austria), as described previously in Ref. [9] with minor modifications. All respirometry experiments were performed on fresh tissues immediately following dissection. Liver and BAT were homogenized (mechanical permeabilization) in amino acid-depleted respirometry medium (0.5 mM EGTA, 3 mM MgCl2, 60 mM K-lactobionate, 10 mM KH2PO4, 20 mM HEPES, and 110 mM sucrose, pH 7.1) and the equivalent of 2 mg wet tissue was added to the experimental chamber. Glycolytic EDL skeletal muscle was placed in relaxing buffer (in mM: 2.8 Ca2K2EGTA, 7.2 K2EGTA, 5.8 ATP, 6.6 MgCl2, 20 taurine, 15 Na2 phosphocreatine, 20 imidazole, 0.5 dithiothreitol, and 50 MES, pH 7.1) immediately after dissection, and fibers were gently separated. Following sarcolemmal permeabilization in relaxing buffer supplemented with 0.005% (wt/vol) saponin, the tissue was equilibrated in respirometry medium [in mM: 0.5 EGTA, 3 MgCl2, 60 K-lactobionate, 20 taurine, 10 KH2PO4, 20 HEPES, 110 sucrose and 0.1% (wt/vol) bovine serum albumin, pH 7.1]. Thereafter, skeletal muscle fibers were blotted for ∼30 s to remove the excess of medium and 1.0–2.5 mg of tissue was added to each chamber. All the tissues were assessed in respirometry medium. Oxygen flux (denoted as “Leak” in figures) was measured by adding malate (final concentration 2 mM), pyruvate (10 mM) and glutamate (20 mM) in the absence of ADP. Complex I-driven oxidative phosphorylation (denoted as “C I” in figures) was quantified by the addition of ADP (5 mM; 0.5 mM for tests in BAT). This was followed by the addition of succinate (10 mM) for convergent electron flow through both complex I and II (denoted as “C I + II” in figures). Subsequently, carbonylcyanide-4-(trifluoromethoxy)-phenyl-hydrazone (FCCP) was titrated to achieve maximum flux through the electron transfer system (denoted as “ETS” in figures). Finally, electron transport through complex I (denoted as “ETS C II” in figures) and III was inhibited by the sequential addition of rotenone (0.1 μM) and antimycin A (2.5 μM), respectively. The remaining O2 flux after inhibition with Antimycin A (O2 flux independent of the ETS) was subtracted from the values of each of the previous steps. O2 flux values are expressed relative to tissue wet weight per second.
2.5. Western blotting
Cells were lysed in lysis buffer (50 mM Tris–HCL pH7.5, 150 mM NaCl, EDTA 5 mM, NP40 1%, sodium butyrate 1 mM, protease inhibitors). Proteins were quantified using BCA assay (Pierce). For western blotting, proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Antibodies are listed in Supplemental Table 1. All spliced together non-contiguous lanes from the same gels are marked using a thin black line and noted in the figure. The images for the full, unedited gels are available upon request.
2.6. Determination of mitochondrial vs. nuclear DNA ratio
Total genomic DNA was extracted from liver, skeletal muscle (gastrocnemius) and brown adipose tissue using a DNeasy Blood and Tissue kit (Qiagen) according to the manufacturer's instructions. Mitochondrial and nuclear DNA (COX2 and HK2, respectively) were measured by quantitative PCR using SYBR Green. Primers are listed in Supplemental Table 2.
2.7. RNA extraction and qPCR
Total mRNA from all studied tissues was extracted according to the instructions provided by the manufacturer, using TRIZOL (Life technologies). The mRNA concentration was measured using a Nanodrop 1000 (Thermo Scientific, Wilmington, MA). Conversion to cDNA was performed using SuperScript II (Life technologies) with oligo (dT2), Random Hexamers primers and RNAsine (Roche) according to the protocol provided by the manufacturer. Quantification of mRNA expression was performed using the SYBR Green real time PCR technology (Roche). Reactions were performed in duplicate in a 384-well plate using the Light Cycler (Roche). Primers are listed in Supplemental Table 2. Gene expression was normalized with β2-microglobulin and cyclophillin as housekeeping genes. Relative gene expression between genotypes was assessed through the ΔΔCt method [10].
2.8. Microarray
Illumina microarrays were used to profile the gene expression levels of approximately 25,000 genes, in the BAT of WT and SIRT1Tg/Tg mice (n = 6 per group). The mRNA samples were hybridized on a single chip and raw microarray data were analyzed using the Illumina genome studio software. The results of the microarray are available in the GEO database with the accession number GSE62324. To test for sets of related genes that might be systematically altered in SIRT1Tg/Tg BAT, we used a Gene Set Enrichment Analysis (GSEA), a method which combines information from the members of previously defined sets of genes (e.g. curated biological pathways) to increase signal relative to noise and improve statistical power to detect subtle changes [11]. Complete details on the method for this analysis are available on the http://www.broad.mit.edu/gsea website. Briefly, genes from the microarray were first ranked according to the expression difference (signal to noise ratio) between genotypes. The extent of association was then measured by a non-parametric running sum statistic termed the enrichment score (ES), and the maximum ES (MES) was recorded over each gene set. Permutation tests were used to assess the statistical significance of the MES, which is calculated as the fraction of the 100 random permutations of the gene list in which the top pathway gave a stronger result than that observed in the actual data. The unadjusted nominal P value estimates the statistical significance of a gene set without adjusting for gene set size or multiple hypothesis testing, whereas the false discovery rate (FDR) statistic adjusts for both. In this analysis, an FDR of approximately 30% was considered acceptable. The nominal P-values for the gene set are indicated in the corresponding panels.
2.9. Citrate synthase activity
Citrate synthase activity was performed on tissue accordingly to the protocol of Sigma's Citrate Synthase Assay Kit (CS0720).
2.10. Triglycerides and glycogen content
Triglycerides and Glycogen content were performed on tissue accordingly to the protocol of Bioassay System Assay Kit (ETGA-200 and E2GN-100, respectively).
2.11. Lipolysis
Lipolysis was determined on freshly extracted tissues using the ZenBio assay kit (LIP-3-NC), incubating the tissues in assay buffer supplemented with a vehicle or with isoproterenol 1 μM during 5 h at 37 °C under agitation and then glycerol release were measured.
2.12. Cell culture
Primary brown adipocytes cells were obtained from the interscapular BAT of WT or SIRT1 transgenic mice and immortalized as previously described in Ref. [12]. Immortalized brown pre-adipocytes were grown in “growth medium” (GM) (DMEM supplemented with 10% FBS, 20 nM Insulin, and 1.5 nM 3,3′,5 Triiodothyronine (T3)). For differentiation, cells were growth until 90% confluence with GM and next stimulated during 36 h with “differentiation medium” (GM supplemented of 0.5 μM dexamethasone, 1 μM rosiglitazone, 0.125 μM indomethacin and 0.5 mM isobutylmethylxanthine (IBMX)). Then, cells were cultivated in GM until final differentiation (day 6 of differentiation). Adipocytes are fully differentiated when they exhibited multilocular lipid droplets in their cytoplasm. For experiments, norepinephrine (A0937, SIGMA) or CL316,243 (C5976, SIGMA) were added to GM at 1 μM concentrations unless otherwise specified.
2.13. Statistical analyses
Statistical analyses were performed with Prism software (GraphPad). Differences between two groups were analyzed using Student's t test (two-tailed), and multiple comparisons were analyzed by ANOVA with a Bonferroni post hoc test. A P-value less than 0.05 was considered significant. Data are expressed as means ± SEM.
3. Results
3.1. SIRT1Tg/Tg mice display higher energy expenditure and glucose tolerance
SIRT1Tg/Tg mice displayed a moderate SIRT1 overexpression in skeletal muscle, liver, white adipose tissue (WAT) and BAT, both at the mRNA and protein levels (Figure 1A,B). In contrast to the heterozygous mice, where changes were reported to be 2–4-fold [4], up to 10-fold increases in SIRT1 expression were observed in the WAT from SIRT1Tg/Tg mice. When fed an LFD, SIRT1Tg/Tg mice displayed a similar body weight (Figure 1C) and composition (Figure 1D) as WT mice. However, SIRT1Tg/Tg showed a marked increase in O2 consumption and EE both during the light and the dark phases (Figure 1E and Figure S1A-B). The respiratory exchange ratio (RER) of SIRT1Tg/Tg mice was lower than in WT mice during the light phase, indicating a higher use of lipid energy substrates (Figure 1F and Figure S1C). The bigger amplitude in RER changes between the light and dark phases (Figure 1F) suggests an enhanced flexibility to shift between carbohydrate and lipid energy sources. While food intake was similar between genotypes (Figure 1G), the CLAMS analysis revealed a clear tendency to decreased activity in SIRT1Tg/Tg mice (Figure S1D). The lower spontaneous activity of SIRT1Tg/Tg mice was more evident when daily voluntary activity was analyzed in regular housing cages (Figure 1H). This is an interesting aspect, as SIRT1Tg/- mice also showed a tendency towards lower activity on LFD [4], and this decrease was largely significant in the SirBACO mice [5]. Also, the treatment with STACs has been reported to decrease spontaneous activity in mice [8,13]. Hence, systemic SIRT1 activation might reduce spontaneous activity, which could explain why SIRT1Tg/Tg mice have a similar body weight despite higher EE.
Figure 1.
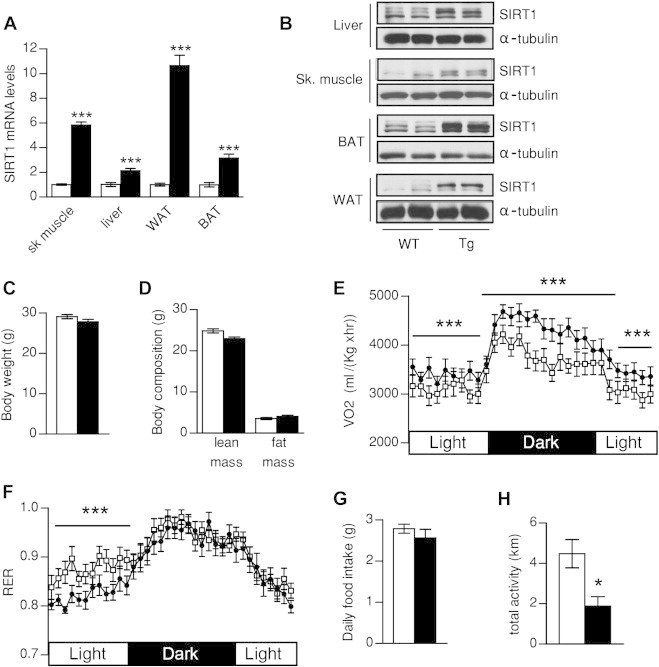
Evaluation of energy homeostasis in SIRT1Tg/Tg mice. (A, B) 20 week-old wild-type (WT) and SIRT1Tg/Tg (Tg) mice were sacrificed to measure (A) the mRNA and (B) protein levels of SIRT1 in the indicated tissues. (C, D) Three months old WT and Tg mice were fed ad libitum with a low fat diet and (C) body weight and (D) body composition were measured through EchoMRI. (E) Oxygen consumption (VO2) and (F) Respiratory exchange ratio (RER) were measured by indirect calorimetry using a comprehensive laboratory animal monitoring system (CLAMS). (G) Food intake a measured during the indirect calorimetry tests. (H) Voluntary wheel running activity was measured in 20-week-old WT and Tg mice. All values are presented as mean ± SEM of n = 10–14 mice for each genotype. *indicates statistical significant difference between WT (white bars and circles) and Tg mice (black bars and circles) at P < 0.05.
We next evaluated if the marked differences of SIRT1Tg/Tg mice on energy homeostasis could have an impact on glucose metabolism. Blood glucose and insulin levels were similar between WT and SIRT1Tg/Tg mice, either in the fed or fasted state (Table 1). SIRT1Tg/Tg mice, however, displayed a higher glucose tolerance (Figure 2A) and response to insulin (Figure 2B), based on intraperitoneal glucose and insulin tolerance tests, respectively. In order to evaluate the key tissues contributing to the higher glucose tolerance and insulin response, we performed a hyperinsulinemic-euglycemic clamp on WT and SIRT1Tg/Tg. Both phenotypes required similar glucose infusion rates (GIR) at low insulin concentrations (4 mU/kg/min) (Figure 2C). However, when insulin levels were raised to asses peripheral glucose uptake (12 mU/kg/min), SIRT1Tg/Tg mice required a significantly higher GIR (Figure 2C). In these conditions, insulin equally decreased hepatic glucose production in both genotypes (Figure 2D), illustrating that differences in hepatic function do not account for the higher GIR rate. Glucose disposal rates were also calculated for individual tissues in response to the insulin challenges. Glucose uptake was similar between genotypes in skeletal muscle, liver, epididymal and sub-cutaneous WAT (Figure 2E). In contrast, BAT glucose uptake was higher in SIRT1Tg/Tg mice (Figure 2E) suggesting that BAT is the main responsible for the higher GIR required to maintain euglycemia in SIRT1Tg/Tg mice.
Table 1.
Blood biochemistry.
| Parameters | WT (n = 6) | Tg (n = 6) |
|---|---|---|
| Glycemia (mg/dL) – Fed | 150.63 ± 3.12 | 155.14 ± 6.01 |
| Glycemia (mg/dL) – Fasted | 75.67 ± 4.8 | 81.14 ± 5.1 |
| Insulinemia (ng/mL) – Fed | 2.73 ± 0.34 | 2.74 ± 0.30 |
| Insulinemia (ng/mL) – Fasted | 1.10 ± 0.21 | 1.05 ± 0.10 |
| Triglycerides (mmol/L) | 1.08 ± 0.13 | 1.50 ± 0.19 |
| Free Fatty Acid (mmol/L) | 0.63 ± 0.11 | 0.75 ± 0.09 |
| Total Cholesterol (mmol/L) | 2.8 ± 0.2 | 2.8 ± 0.2 |
| HDL-Cholesterol (mmol/L) | 2.78 ± 0.15 | 2.74 ± 0.25 |
| LDL-Cholesterol (mmol/L) | 0.11 ± 0.03 | 0.09 ± 0.02 |
Figure 2.
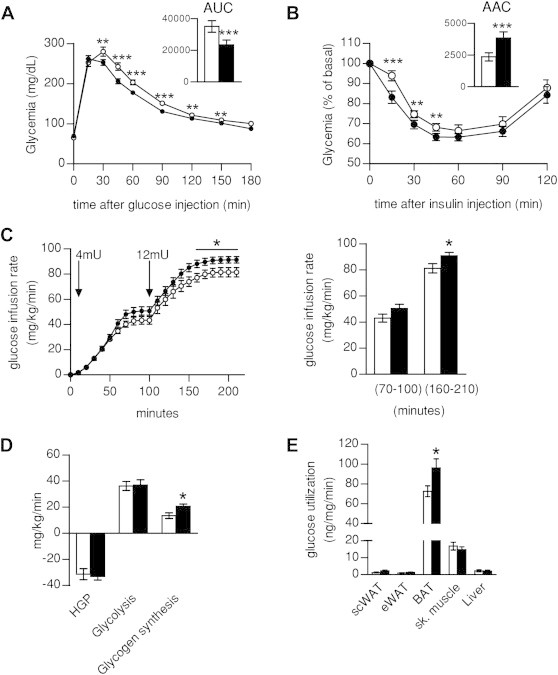
SIRT1 overexpression improves insulin sensitivity through higher BAT glucose uptake. (A) An intraperitoneal glucose tolerance test and (B) an introperitoneal insulin tolerance test were performed on 4-month-old WT and Tg mice. Area under the curve (AUC) or area above the curve (AAC) calculations are present on the top right of each glucose excursion. (C–E) Hyperinsulinemic-euglycemic clamp was performed on WT and SIRT1Tg/Tg mice fed on low fat diet. Glucose Infusion Rate (GIR) measured at two different levels of insulin infusion (4 and 12 mU/kg/min) (C), glucose fluxes (D) and Glucose uptake in different tissues (inguinal sub-cutaneous and epididymal WAT, BAT, vastus lateralis skeletal muscle and liver) (E) are represented. All values are presented as mean ± SEM of n = 12–14 mice for each genotype. * indicates statistical significant difference between WT (white bars and circles) and Tg mice (black bars and circles) at P < 0.05.
These observations contrast with those in SIRT1Tg/− mice, where higher glucose tolerance was only observed on HFD [4,5]. In this sense, SIRT1Tg/Tg mice also displayed a strong protection against glucose intolerance (Figure S2A) and insulin resistance (Figure S2B) on HFD, despite a similar body weight gain as WT mice (Figure S2C).
3.2. Skeletal muscle and liver are not affected in SIRT1Tg/Tg mice on low fat diet
The fact that SIRT1Tg/Tg mice did not display higher insulin sensitivity in skeletal muscle is in line with recent reports using transgenic models with even higher SIRT1 overexpression [14,15]. However, these results do not rule out other possible contributions of SIRT1 transgenesis to muscle metabolic properties. In this sense, mitochondrial biogenesis and higher oxidative capacity are believed to be two key metabolic actions triggered by SIRT1 in skeletal muscle [16]. To further explore these areas, we first, submitted the mice to grip and treadmill tests in order to evaluate muscle force and endurance, respectively. However, none of them were improved by SIRT1 transgenesis (Figure 3A,B). In line with this, no differences in maximal running speed or maximal VO2 could be found between genotypes (Figure S3A-B). No structural or oxidative profile differences were observed between the gastrocnemius muscles of WT and SIRT1Tg/Tg mice, based on succinate dehydrogenase stainings (data not shown). WT and SIRT1Tg/Tg mice also showed no differences when evaluating mitochondrial vs. nuclear DNA ratios (Figure 3C), citrate synthase (CS) activity (Figure 3D) or the levels of mitochondrial-related mRNAs and proteins in gastrocnemius muscle (Figure 3E,F). The protein content of mitochondrial respiratory complex subunits was also unaffected in highly oxidative (soleus) or glycolytic (EDL) muscles (Figure S3C). Interestingly, an upregulation of several transcriptional regulators, such as the peroxisome proliferator-activated receptor-α (PPARα) and the Forkhead O box (FOXO) 1 and 3a, was observed in SIRT1Tg/Tg muscles. However, the above results suggest that these changes were not enough to affect muscle endurance and mitochondrial function. To further substantiate the lack of differences in mitochondrial function between WT and SIRT1Tg/Tg mice, we performed respirometry analyses in permeabilized EDL muscle fibers. Uncoupled respiration (leak) through complex I, stimulated with malate, pyruvate and glutamate, was not affected by SIRT1 transgenesis (Figure 3G). Similarly, maximal respiration in the coupled state, with electron input through complex I alone (C I) or through complex I + II – after the addition of succinate – (C I + II) was comparable across genotypes (Figure 3G). Maximum electron transport system (ETS) capacity was also similar, even after the addition of the complex I inhibitor rotenone (ETS CII) (Figure 3G). Hence, no major differences in mitochondrial function could be observed in the skeletal muscle from WT and SIRT1Tg/Tg mice.
Figure 3.
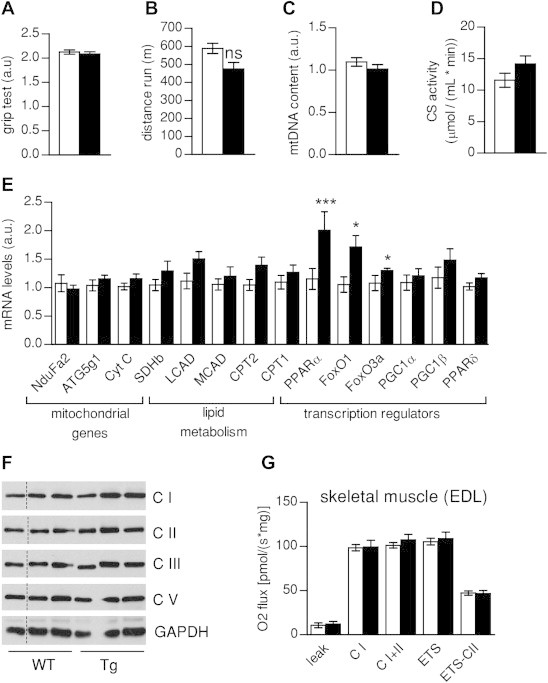
Muscle function is not affected by SIRT1 transgenesis. (A) Muscle force was evaluated in wild type (WT) and SIRT1Tg/Tg (Tg) mice through a grip test. (B) Running distance was evaluated by submitting mice to an endurance treadmill test. (C) Mitochondrial DNA content in gastrocnemius muscle was measured and normalized to nuclear DNA copy number. (D) Citrate synthase activity was measured in quadriceps muscle. (E) Total mRNA was extracted from gastrocnemius muscles and used for qPCR analysis of the markers indicated. (F) Protein analysis of mitochondrial markers in total homogenates of quadriceps muscle (thin black lines on gels are used for lanes that were run on the same gel but were non-contiguous). (G) Oxidative phosphorylation and electron transfer system capacity in permeabilitzed EDL muscle fibers of WT and Tg mice. All values are presented as mean ± SEM of n = 8–10 mice for each genotype. * indicates statistical significant difference between WT (white bars and circles) and Tg mice (black bars and circles) at P < 0.05.
Next, we also examine the consequences of SIRT1 transgenesis in liver tissue on LFD. While the hyperinsulinemic-euglycemic clamp revealed no major effects of SIRT1 transgenesis in liver glucose turnover on LFD (Figure 2D,E), the liver accounts for the benefits of SIRT1 transgenesis on glucose metabolism upon HFD [4,5]. To further evaluate hepatic glucose metabolism in our SIRT1Tg/Tg mice on LFD diet, we challenged the mice with a pyruvate tolerance test. After the injection of pyruvate, a gluconeogenic precursor, glucose excursions were comparable between WT and SIRT1Tg/Tg mice (Figure 4A). We also failed to detect differences between the two genotypes on histological Oil red'O stainings (Figure 4B) or at the level of hepatic triglyceride or glycogen content (Figure 4C,D). As in muscle, no changes could be found on mitochondrial DNA content (Figure 4E), on CS activity (Figure 4F) or at the mRNA and protein level of several mitochondrial components (Figure 4G,H). In line with the comparable response to pyruvate, the expression of key gluconeogenic genes, such as glucose 6-phosphatase (G6Pase) and phopho-enol-pyruvate carboxylase (PEPCK), was similar between genotypes (Figure 4G). As in muscle, some transcriptional regulators of lipid metabolism were upregulated, including PPARα, FOXO1 and FOXO3a (Figure 4G). Interestingly, respirometry analyses revealed a higher complex II activity in SIRT1Tg/Tg livers (Figure 4I). However, based on the absence of differences in hepatic metabolism upon pyruvate and insulin challenges, the physiological relevance of these increases is unclear.
Figure 4.
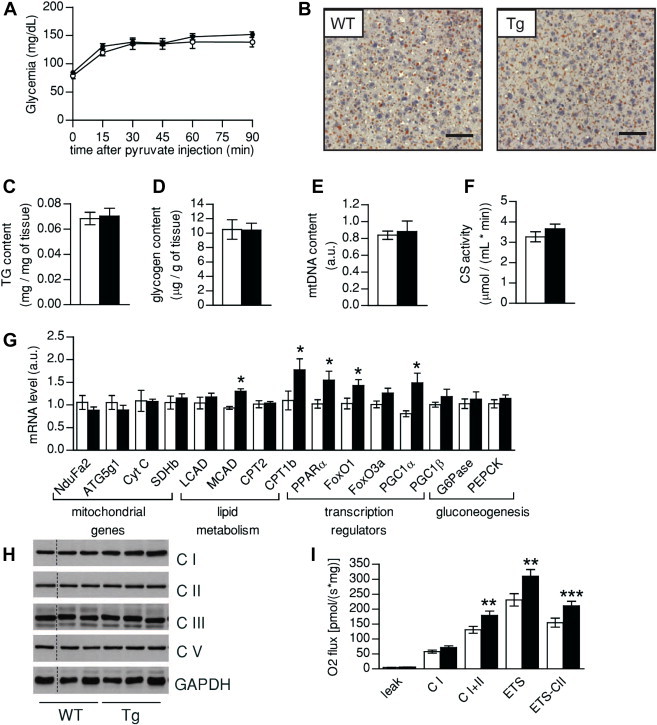
Hepatic function is not critically affected by SIRT1 transgenesis. (A) Blood glucose curves after a pyruvate (2 g/kg) challenge. (B) Oil Red'O staining in WT and Tg livers after an overnight fast (bar = 600 μm) (C, D) Hepatic triglyceride (C) and glycogen (D) content was measured after an overnight fast. (E) Mitochondrial DNA content was measured and normalized to nuclear DNA copy number. (F) Citrate synthase activity in liver. (G) Total liver mRNA was extracted and used for qPCR analysis of the markers indicated. (H) Protein analysis of mitochondrial markers in total liver homogenates (thin black lines on gels are used for lanes that were run on the same gel but were non-contiguous). (I) Oxidative phosphorylation and electron transfer system capacity in liver homogenates of WT and Tg mice. All values are presented as mean ± SEM of n = 8–10 mice for each genotype. * indicates statistical significant difference between WT (white bars and circles) and Tg mice (black bars and circles) at P < 0.05.
3.3. Brown adipose function is improved in SIRT1 transgenic mice
The lack of effects on WAT insulin response in SIRT1Tg/Tg mice on LFD (Figure 2E) was in line with the absence of differences in the weight of different WAT depots (Figure S4A) or their histological aspect (Figure S4B). Accordingly, we found no alterations in mitochondrial function (Figure S4C) or lipolytic capacity (Figure S4D) in the WAT of SIRT1Tg/Tg mice.
Finally, we focused on the BAT function in SIRT1Tg/Tg mice, as it is the tissue most likely explaining the differences in glucose homeostasis between genotype (Figure 2E). Strikingly, the color of the BAT from SIRT1Tg/Tg mice was more intense (Figure 5A), likely due to a lower accumulation of lipids, as reflected in the much smaller lipid droplets observed through hematoxylin/eosin staining (Figure 5B). We next evaluated BAT thermogenic function by placing the mice at 6 °C and monitoring body temperature for 5 h. While no significant differences on the basal state, SIRT1Tg/Tg mice maintained better their body temperature during the cold challenge (Figure 5C), suggesting a higher thermogenic activity. As in other tissues, we found no differences between genotypes in mitochondrial DNA copy number (Figure 5D) or in the content of respiratory complexes subunits at protein and mRNA levels (Figure 5E and Figure S5A, respectively). To infer the possible mechanisms by which SIRT1Tg/Tg mice displayed altered BAT function, we performed microarray analyses (results available in the GEO database with the accession number GSE62324). Geneset enrichment revealed a strongly significant (nominal P-value < 0.01) increase of genes related to fatty acid metabolism and, more in particular, lipid catabolism (Figure 5F and Figure S5B). The microarray results were validated by quantitative real-time PCR (qPCR), which confirmed that genes related to β-oxidation were strongly induced in the BAT of SIRT1Tg/Tg mice (Figure 5G). This way, key genes for lipid oxidation, such as the long-chain acyl-CoA dehydrogenase (LCAD), the medium-chain acyl-CoA dehydrogenase (MCAD) or the carnitine palmitoyltransferase 1b (CPT1b), were significantly elevated in SIRT1Tg/Tg mice (Figure 5G). This was, again, in line with an upregulation of PPARα and FOXOs. Interestingly, the protein and mRNA levels of the uncoupling protein UCP1 were also elevated in SIRT1Tg/Tg BAT (Figure 5E and G, respectively). The key role of UCP1 in energy dissipation could explain the higher EE (Figure 5E,F) and thermogenic potential (Figure 5C) observed in SIRT1Tg/Tg mice. In line with this, respirometry analyses revealed an almost 2-fold increase in uncoupled respiration (leak) in the BAT from SIRT1Tg/Tg mice, and a higher maximal ETS capacity (Figure 5H). Concordantly, CS activity was also increased (Figure 5I). Finally, we examined the lipolytic rates in WT and SIRT1Tg/Tg BATs. The results obtained illustrate how the basal lipolytic rates in the BAT from SIRT1Tg/Tg are ∼20% higher than those of their control WT mice (Figure 5J). This result is in line with the higher basal phosphorylation of the hormone sensitive lipase (HSL) observed in protein homogenates (Figure 5E), and could explain the lower lipid accumulation observed through histology (Figure 5B). Also, the mRNA level of the type II iodothyronine deiodinase (DIO2) was also increased in the BAT of SIRT1Tg/Tg mice, supporting a higher catabolic rate (Figure 5G). Altogether, these data suggest that SIRT1Tg/Tg mice have a higher thermogenic function and lipid oxidation-related gene expression in BAT. While SIRT1 activation has been classically linked to enhanced mitochondrial biogenesis [1] mitochondrial content was not affected in the BAT from SIRT1Tg/Tg mice, despite a markedly higher SIRT1 activity, as testified by the reduced acetylation of NFκB and FoxO1 (Figure 5K).
Figure 5.
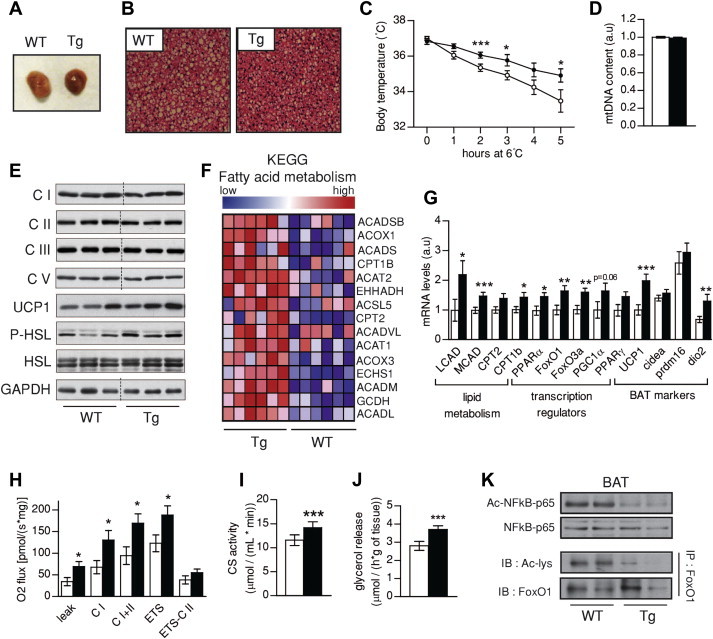
Brown adipose function is improved in SIRT1 transgenic mice. (A) Pictures of brown adipose tissue (BAT) from WT and Tg mice. (B) Hematoxylin and eosin stainings on the BAT of wild type (WT) and SIRT1Tg/Tg (Tg) mice (bar = 600 μm) (C) Brown adipose thermogenic function was evaluated by placing WT and Tg mice at 6 °C for 5 h. (D) Mitochondrial DNA content in BAT from WT and Tg mice, normalized to nuclear DNA copy number. (E) Western Blots were performed to evaluate the protein levels of proteins in BAT from WT and Tg mice (thin black lines on gels are used for lanes that were run on the same gel but were non-contiguous). (F) Gene set enrichment analyses of gene expression profiles of BAT from WT and Tg mice. (G) Total mRNA was extracted from BAT and used for qPCR analysis. (H) Oxidative phosphorylation and electron transfer system capacity in BAT. (I) Citrate synthase activity in BAT from WT and Tg mice. (J) Lipolysis was evaluated by measuring glycerol release in isolated BAT. (K) SIRT1 activity was tested in WT and Tg BAT using the acetylation status of RelA(p65) and FOXO1 as readout. All values are presented as mean ± SEM of n = 8–10 mice for each genotype. * indicates statistical significant difference between WT (white bars and circles) and Tg mice (black bars and circles) at P < 0.05.
3.4. SIRT1 transgenesis affects BAT response to adrenergic stimulation
We next aimed to understand whether the changes in BAT biology elicited by SIRT1 transgenesis were derived from alterations in the BAT differentiation process or on its function. For this purpose, we immortalized primary brown pre-adipocytes from WT and SIRT1 transgenic mice. SIRT1 transgenesis did not affect the cellular morphology (Figure 6A), lipid droplet accumulation (Figure S6A) and TG content (Figure 6B) of the cultured brown adipocytes. Brown adipocyte differentiation markers, such as Prdm16, Cidea and PPARγ, were similarly induced at the mRNA and protein level after differentiation between the two genotypes (Figure 6C,D). In line with this, the mRNA expression of these markers in adult BAT from WT and SIRT1Tg/Tg mice was also similar (Figure 6G). These analyses also confirmed that SIRT1 overexpression levels in undifferentiated and differentiated transgenic immortalized brown adipocytes are comparable to those in the SIRT1 transgenic BAT (Figures 1A,B and 6D). Hence, SIRT1 overexpression does not affect brown adipocyte differentiation. Strikingly, while Ucp1 expression, both at the mRNA and protein level, increased upon differentiation of WT brown adipocytes, it did much less in the brown adipocytes from SIRT1 transgenic mice (Figure 6D,E). A recent publication indicated that SIRT1 can amplify the transcriptional response to PKA signaling [17]. We hence reasoned that the increased UCP1 levels observed in SIRT1Tg/Tg BAT could be consequent to a higher response of SIRT1 transgenic adipocytes to β3-adrenergic stimulation. To test this hypothesis, we analyzed the response of differentiated brown adipocytes to adrenergic stimulation using norepinephrine (NE) or the synthetic agonist CL316,243 (CL). Both agents induced a comparable increase in p-HSL after 5 h of treatment (Figure 6F). This indicates that early signaling responses to β3 stimulation are not altered by SIRT1 transgenesis. However, the transcriptional response of Ucp1 to NE or CL was dramatically enhanced in SIRT1 brown adipocytes (Figure 6F,G and Figure S6B). The enhanced transcriptional response to β3 agonists in SIRT1 transgenic adipocytes was certified using another adrenergic target in the BAT, the PPARγ coactivator 1α (PGC-1α) [17] (Figure S6C). The transcriptional responsiveness of SIRT1 transgenic adipocytes to adrenergic stimulation was higher at concentrations as low as 10 nM, as testified by Ucp1 mRNA analyses (Figure 6G). Similarly, SIRT1 transgenic brown adipocytes achieved maximal Ucp1 transcriptional response to adrenergic stimulation at earlier time-periods than WT adipocytes (Figure 6H). Thus, our results indicate that SIRT1 grants brown adipocytes with a higher transcriptional responsiveness to β3 agonists.
Figure 6.
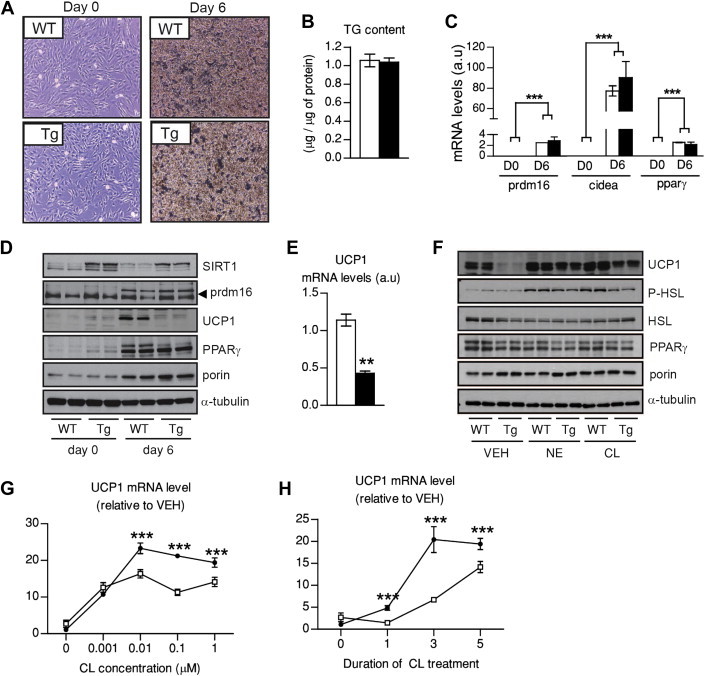
SIRT1 transgenic adipocytes display an exhacerbated response to β3-adrenergic stimulation. Brown pre-adipocytes from wild-type (WT) and SIRT1 transgenic (Tg) mice were isolated and immortalized. (A) The morphology of immortalized brown adipocytes was evaluated before (day 0) and after (day 6) differentiation. (B) Total triglyceride content in differentiated brown adipocytes was evaluated. (C) Total mRNA levels were extracted from pre-adipocytes and differentiated adipocytes and used for qPCR analysis. (D) Total protein extracts were used to evaluate diverse differentiation markers in differentiated and undifferentiated adipocytes. (E) Ucp1 expression was measured in total mRNA extracts of differentiated brown adipocytes. (F) Differentiated WT and Tg adipocytes were stimulated with 1 μM of norepinephrine (NE) or 1 μM of CL316,243 (CL) during 5 h at 37 °C. Then, total proteins were extracted and used for western blot analysis. (G) WT and Tg brown adipocytes were treated with CL in a dose–response fashion for 5 h at 37 °C and then total mRNA was extracted to measure Ucp1 expression. (H) WT and Tg brown adipocytes were treated with 1 μM CL and incubated at 37 °C. Then, total mRNA was extracted at the times indicated to measure Ucp1 expression. All values are presented as mean ± SEM of at least n = 4 independent experiments, each of them run in triplicate. * indicates statistical significant difference between WT (white bars and circles) and Tg mice (black bars and circles) at P < 0.05.
3.5. Thermoneutrality blunts the differences in glucose homeostasis of SIRT1Tg/Tg mice
The results above illustrate that, upon adrenergic stimulation, SIRT1 transgenic adipocytes have an exacerbated transcriptional response (Figure 6G,H) that allows them to recover UCP1 protein levels to comparable levels of that of WT mice even within a few hours (Figure 6F). We reasoned that if this stimulation had a chronic character, this could lead to even further increases and explain the higher activity of BAT in SIRT1Tg/Tg mice. In this sense, it must be taken into account that at regular housing conditions (∼20–22 °C), mice are below thermoneutrality, which leads to a basal stimulation of the BAT thermogenic function. Considering the above results, this chronic basal adrenergic tone could be responsible for the higher BAT function, Ucp1 expression and EE observed in SIRT1Tg/Tg mice. To test this hypothesis, we housed WT and SIRT1Tg/Tg mice at thermoneutrality (30 °C), which blunts thermogenic activity [18]. In turn, this would allow us to evaluate whether the enhancement of BAT function is responsible for the higher glucose tolerance and insulin sensitivity of SIRT1Tg/Tg mice on LFD. After one month at thermoneutrality, SIRT1Tg/Tg and WT mice were undistinguishable at the level of body weight (Figure 7A), body composition (Figure 7B) and food intake (Figure S7A). As in normal housing conditions the total activity of the SIRT1Tg/Tg mice was also reduced at thermoneutrality (Figure S7C). Importantly, however, thermoneutrality blunted the effect of SIRT1 transgenesis on VO2 (Figure 7C), EE (Figure S7B) and RER (Figure S7D) that we previously observed in normal housing conditions (Figure 1E,F). In addition, thermoneutrality also abrogated the higher glucose tolerance and insulin sensitivity of SIRT1Tg/Tg mice (Figure 7D,E). Interestingly, a higher glycemia was clearly observed in SIRT1Tg/Tg mice 15 min after a glucose load (Figure 7D), even though the physiological relevance of this difference is unclear. These evidences, together with the results obtained during hyperinsuilnemic-euglycemic clamp (Figure 2E), clearly demonstrate that enhanced BAT function is the primordial event by which SIRT1Tg/Tg mice are granted better glucose homeostasis in regular housing conditions. Thermoneutrality increased the lipid droplet size in the BAT of both WT and SIRT1Tg/Tg mice (Figures 5B and 7F), and rendered both genotypes histologically undistinguishable. Similarly, the higher basal expression of lipid oxidation-related genes observed in SIRT1Tg/Tg mice was blunted on thermoneutral conditions (Figure 7G). Interestingly, we observed a dramatic reduction in Ucp1 expression in the BAT of SIRT1Tg/Tg compared to that of WT mice (Figure 7G), which is very reminiscent of the lower Ucp1 mRNA levels observed in cultured primary brown adipocytes. This substantiates that the higher Ucp1 levels in mice housed at normal conditions are consequent to an exacerbated response to a basal adrenergic tone. As expected, thermoneutrality lowered uncoupled respiration (leak) in both WT and SIRT1Tg/Tg mice (Figures 5H and 7H). While similar respiratory profiles were observed in the BAT from WT and SIRT1Tg/Tg mice on thermoneutrality, we noted a tendency to a lower respiratory leak in SIRT1Tg/Tg mice (Figure 7H), in line with the decrease in Ucp1 expression. Interestingly, thermoneutrality did not affect the higher Complex II activity in the livers of SIRT1Tg/Tg mice (Figures 4H and 7H). On one side, this testifies for the BAT-specific impact of the intervention. On the other, it argues that the increase in Complex II activity in the liver is not responsible for the better glucose tolerance of SIRT1Tg/Tg mice on regular housing conditions. Finally, we evaluated whether thermoneutrality also blunted the higher lipolytic rates we observed in SIRT1Tg/Tg mice on non-thermoneutral conditions (Figure 5J). As expected, thermoneutral conditions decreased lipolytic rates in WT mice, as noticed when comparing the values obtained in Figure 5J (white bar; 2.80 ± 0.25 mmol/(mg of tissue*h)) and at thermoneutral conditions (Figure 7I, white bar, 1.51 ± 0.22 mmol/(mg of tissue*h)). Thermoneutrality also blunted the higher lipolytic rates and HSL phosphorylation in the BAT from SIRT1Tg/Tg mice and made them undistinguishable from WT mice (Figure 7I).
Figure 7.
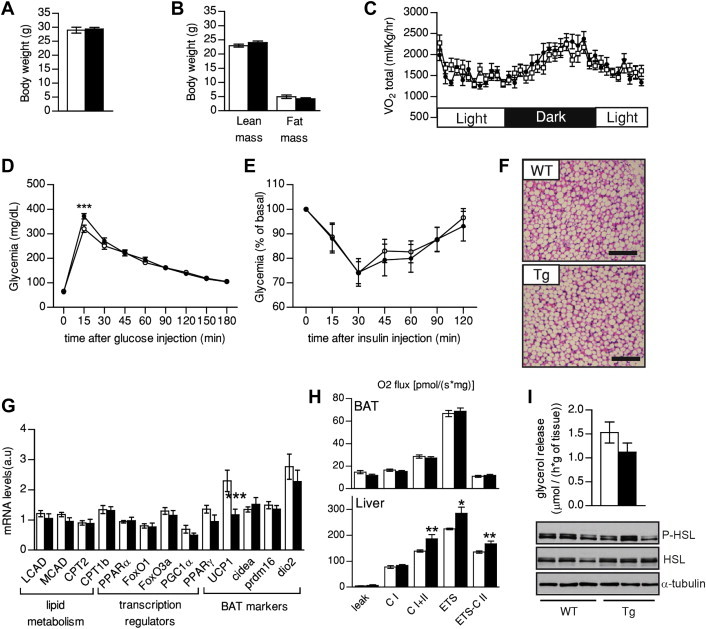
Thermoneutrality blunts the insulin-sensitizing effect of SIRT1 transgenesis. Three months old WT and Tg mice were fed ad libitum with low fat diet and placed at thermoneutrality (30 °C) for one month before phenotyping. Then, (A) Body weight and (B) body composition were measured using Echo-MRI. (C) O2 consumption was evaluated using a comprehensive animal laboratory monitoring system; (D) An intraperitoneal glucose tolerance test and (E) an insulin tolerance test were performed on 4 months old mice. (F) Hematoxylin and eosin stainings of BAT from WT and Tg mice (bar = 600 μm). (G) Total mRNA was extracted from BAT and used for qPCR analysis. (H) Oxidative phosphorylation and electron transfer system capacity in BAT (top) and liver (bottom) of WT and Tg mice. (I) Lipolysis rates were evaluated by measuring glycerol release from BAT. At the bottom HSL and p-HSL levels were evaluated in total BAT protein extracts. All values are presented as mean ± SEM of n = 12 mice for each genotype. * indicates statistical significant difference between WT (white bars and circles) and Tg mice (black bars and circles) at P < 0.05.
4. Discussion
While SIRT1 activation has been linked to diverse metabolic health benefits, the key tissue(s) responsible for such actions remain elusive. Mouse models with a moderate SIRT1 overexpression have shown a key protective role for SIRT1 on liver function upon HFD [4,5]. Here we describe how the expression of just one additional copy of SIRT1 to that of previous works [4,19], is sufficient to enhance BAT activity, leading to increased EE and improved glucose homeostasis even on LFD.
SIRT1 transgenesis did not affect BAT differentiation, yet it dramatically enhanced BAT transcriptional responses to β3-adrenergic stimulation. Hence, the chronic adrenergic tone taking place in mice housed below thermoneutrality drives a higher basal BAT function in SIRT1Tg/Tg mice. In agreement with our results, the SirBACO model has an exacerbated transcriptional response of thermogenic genes after an acute cold exposure [17]. In this case the authors proposed a mechanism in which PKA activation led to the phosphorylation of a residue in the catalytic domain of SIRT1, Ser434, which led to an increase in intrinsic SIRT1 enzymatic activity [17] in the absence of changes in NAD+ levels. In line with this, we did not observe changes in the BAT NAD+ levels between WT and SIRT1Tg/Tg mice (Figure S5C). Further efforts from the Accili lab have demonstrated how SIRT1 amplifies the browning response of WAT in response to cold stimuli [20]. Altogether, these works and ours certify the ability of SIRT1 to amplify the response to β3-adrenergic stimuli. Strikingly, the SirBACO model did not display a basal BAT phenotype [5,17]. Our results on thermoneutrality, however, indicate that small differences in the adrenergic tone can even drive to opposite outcomes of SIRT1 on thermogenic gene expression. This way, SIRT1 transgenesis led to a lower Ucp1 expression in primary brown adipocytes and in BAT from animals at thermoneutral conditions. The repressive action of SIRT1 on Ucp1 is in line with the role of SIRT1 as an energy sensor triggering adaptations aimed to optimize energy production [1]. Accordingly, SIRT1 represses Ucp2 expression in pancreatic β-cells [21,22]. The ability of SIRT1 to shift from a corepressing to a coactivating function on thermogenic gene sets might be determined by environmental cues enhancing, in this case, PKA signaling. In this sense, SIRT1 has also been described as both a positive and negative regulator of PGC-1α and, hence, mitochondrial biogenesis [23,24]. In fact, the simultaneous activation of AMPK signaling is key to determine the outcomes of SIRT1 action on PGC-1α activity and mitochondrial gene expression [25]. In this sense, we did not observe changes in AMPK activity in our SIRT1Tg/Tg model, which might explain the lack of impact on mitochondrial gene expression (Figure S5D). This highlights how small differences in environmental conditions can critically influence SIRT1 actions, which might have strong implications not only for the evaluation of transgenic mouse models, but also for genetic association studies in humans.
Interestingly, higher EE and glucose tolerance were also observed in the first generated model of SIRT1 moderate overexpression [26]. A particularity of that model was a high expression of the transgene in WAT and BAT, but not in the liver or skeletal muscle [26]. More recently, it has been shown that the deletion of SIRT1 in adipose tissues is enough to drive body weight gain and metabolic dysfunction in mice [27]. Conversely, the selective overexpression of SIRT1 in the adipose tissues enhances EE and prevents age-related insulin resistance [28]. Our results refine all these studies by establishing SIRT1 action in the BAT as the key determinant of whole body thermogenic function, EE and glucose homeostasis. Importantly, the higher SIRT1 content did not manifest into an overt phenotype in liver or muscle. Likely, the activity of SIRT1 needs to be triggered by either hormonal or nutritional inputs. In liver, this might only take place upon HFD, when liver phenotypes are clearly observed in SIRT1 transgenic mice [4,5]. The lack of phenotype in skeletal muscle is not entirely surprising, as mice with much higher SIRT1 overexpression in muscle also fail to display any overt alteration [14,15]. Thus, endogenous SIRT1 levels might be able to account for all the necessary SIRT1 activity required to trigger metabolic adaptations in this tissue. Our results also contrast with the largely accepted notion that SIRT1 is a master regulator of mitochondrial biogenesis. In agreement with previous observations [4], the protein or mRNA levels of mitochondrial respiratory complexes were unaffected in SIRT1Tg/Tg tissues. This, does not rule out, however, that SIRT1 overexpression at a higher (>100-fold), non-physiological, level can impact mitochondrial gene expression [14,29].
While SIRT1 can potentially impact on multiple transcriptional programs, lipid oxidation-related gene sets were the most significantly upregulated in the BAT from SIRT1Tg/Tg mice. In fact, SIRT1 transgenesis increased PPARα, FOXO1 and FOXO3a expression in all tissues examined, certifying an intimate link between SIRT1 and these transcriptional regulators in the control of lipid catabolism. Previous reports have demonstrated the ability of SIRT1 to interact with PPARα and enhance its transcriptional activity on lipid catabolism-related genes [30,31]. In this model, SIRT1 would enhance PPARα activity by deacetylating PGC-1α in the PPARα transcriptional complex [30,31]. Conversely, SIRT1 deletion leads to a dramatic decrease in the expression of PPARα target genes, at least in liver [30]. FOXOs are directly deacetylated by SIRT1, an action that targets FOXO activity onto specific gene sets, including oxidative stress genes [32]. Knock-in mice expressing a FOXO1 form mimicking constitutive deacetylation displayed a marked use of lipid as energy source and enhanced lipolytic response to isoproterenol [33], in line with our findings. However, the potent effect of FOXO1 deacetylation on gluconeogenic gene expression [33] is not manifested in our SIRT1Tg/Tg mice. This would confirm that, despite similar overexpression levels in both tissues, SIRT1 transgenesis has a deeper impact in the BAT than in liver, at least on LFD. PPARα and FOXOs, in addition, might also constitute key mediators of the action of SIRT1 on Ucp1 expression, as both directly regulate the Ucp1 promoter in brown fat cells [12,34].
One key question derived from our work is: how does SIRT1 enhance insulin-stimulated glucose uptake in the BAT? One possibility is that higher lipolytic rates and prevention of fat deposition (Figure 5J and B, respectively) might spare the BAT from the accumulation of lipid species that could potentially interfere with insulin signaling, such as ceramides or diacylglycerols (DAGs) [35]. While excessive lipolysis in the WAT can be detrimental for whole body insulin sensitivity [36], this is generally associated with a lipid overflow onto other tissues. In contrast, the higher lipolytic flux in the BAT of SIRT1Tg/Tg mice is fully met by an increase in mitochondrial fatty acid oxidation capacity and uncoupling activity. In skeletal muscle, HSL activity seems to be a critical determinant of insulin sensitivity, as it can prevent ATGL-driven accumulation of DAGs and the consequent disruption of insulin signaling [37]. In this sense, the higher HSL activity in the BAT of SIRT1Tg/Tg mice might act in a similar fashion. In HFD, lower inflammation was a key mechanism by which SIRT1Tg/- mice retained hepatic insulin sensitivity [9]. Interestingly, in our microarrays, cytokine and inflammatory pathways were two of the most significantly downregulated gene-sets in the BAT of SIRT1Tg/Tg mice (Figure S5B), suggesting that a lower baseline inflammation state might also contribute to its higher insulin sensitivity. Recently, using a BAT transplantation model, it has been shown that increasing BAT function promotes an increase in FGF21 and IL-6 expression, which could act in an endocrine fashion to alter insulin sensitivity in other peripheral tissues [38]. This, however, is not the case in our model, as enhanced insulin sensitivity is not manifested in muscle or eWAT (Figure 2E). Accordingly, we failed to detect differences in BAT FGF21 or IL-6 expression between genotypes (data not shown).
A final aspect for discussion is whether SIRT1 activation via transgenesis is comparable to that achieved via improving NAD+ availability. In theory, at similar NAD+ levels, SIRT1 activity will increase directly in line with increases in expression, unless SIRT1 overexpression decreases NAD+ content via excessive consumption. This is not the case in the BAT of SIRT1Tg/Tg mice, where SIRT1 activity is higher, based on the evaluation of NFκB and FOXO1 acetylation (Figure 5K), without affecting NAD+ levels (Figure S5C). Hence, SIRT1 transgenesis should ensure a linear increase in SIRT1 activity with the level of overexpression, at least up to a certain threshold. The efficacy of NAD+ boosting strategies on SIRT1 activity, however, depends on whether intracellular NAD+ levels are rate-limiting for SIRT1 activity in the basal state. Dietary supplementation with the NAD+ precursor nicotinamide riboside (NR) leads to SIRT1 activation in the BAT [39]. This suggests that basal NAD+ levels are rate-limiting for SIRT1 activity in the BAT. However, a NAD+ precursor will fail to activate SIRT1 if NAD+ is not rate-limiting or in tissues with limited capacity to metabolize the precursor into NAD+. In the case of NR, additionally, it also leads to the activation of other sirtuins, such as SIRT3, by increasing NAD+ content in the mitochondrial compartment [39,40]. Therefore the comparison between transgenic and NAD+-related SIRT1 gain-of-function models should be taken carefully. In the case of STACs, resveratrol and SRT1720 have major effects on BAT activity [8,13]. However, STACs have also marked effects in skeletal muscle oxidative capacity and prevent body weight gain on HFD [8,13]. None of these effects have been observed in this or previous studies using models of moderate SIRT1 overexpression [4,5]. Therefore, either STACs increase SIRT1 activity in muscle in a much more stronger fashion than the above mentioned SIRT1 gain-of-function mouse models, or some effects might be influenced by off-target actions on other molecular paths regulating energy metabolism, as discussed recently in Ref. [1].
Altogether, this manuscript describes a key physiological role of SIRT1 in BAT biology and establishes SIRT1 as a sensitive gauge for the adrenergic response of the BAT, a property that could be exploited for therapeutic approaches aimed to ameliorate glycemic control.
Author contributions
M.B., S.S.K., P.J.F.M. and C.C. have taken care of animal management, phenotyping and tissue collection. M.B., M.J., J.R. and C.C. have performed molecular biology analyses. M.B., P.M.G.R. and C.C. have performed respirometry analyses. E.G.C. and A.M.V. have immortalized primary brown adipocytes. M.B., P.M.G.R., M.S. and C.C. have experimentally conceived the project. M.B. and C.C have written the manuscript and all authors have contributed to editing duties.
Acknowledgments
We thank the members of the Canto lab for exciting discussions. We thank the members of the EPFL and CNIO animal houses for technical support. We also thank the Genomic platform (P. Descombes, F. Raymond and S. Metairon) at NIHS for technical help and advice on microarray analyses. We would also like to express our gratitude to Physiogenex S.A.S. for their help with the hyperinsulinemic-euglycemic clamp. M.S. is funded by the CNIO and by grants from the MICINN (SAF), the Regional Government of Madrid, the European Research Council (CANCER&AGING; LS1, ERC-2008-AdG), the Botín Foundation, the Ramón Areces Foundation, and the AXA Foundation. P.J.F.M. is funded by the AECC. A.M.V. is funded by the following grant support: SAF2012-33283 (MINECO, Spain), Comunidad de Madrid S2010/BMD-2423, EFSD and Amylin Paul Langerhans Grant and Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM, ISCIII, Barcelona, Spain). P.M.G.R. is funded by the following grant support: BFU2011-24679 (MINECO, Spain) and he is a recipient of a Ramon y Cajal contract: RYC-2009-05158(MINECO, Spain).
Conflict of interest
M.B., M.J., S.S.K., J.R. and C.C. are employees of the Nestlé Institute of Health Sciences S.A.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Canto C., Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacological Reviews. 2012;64(1):166–187. doi: 10.1124/pr.110.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baur J.A., Ungvari Z., Minor R.K., Le Couteur D.G., de Cabo R. Are sirtuins viable targets for improving healthspan and lifespan? Nature Review Drug Discovery. 2012;11(6):443–461. doi: 10.1038/nrd3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pacholec M., Bleasdale J.E., Chrunyk B., Cunningham D., Flynn D., Garofalo R.S. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. Journal of Biological Chemistry. 2010;285(11):8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfluger P.T., Herranz D., Velasco-Miguel S., Serrano M., Tschop M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(28):9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banks A.S., Kon N., Knight C., Matsumoto M., Gutierrez-Juarez R., Rossetti L. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metabolism. 2008;8(4):333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Champy M.F., Selloum M., Zeitler V., Caradec C., Jung B., Rousseau S. Genetic background determines metabolic phenotypes in the mouse. Mammalian Genome. 2008;19(5):318–331. doi: 10.1007/s00335-008-9107-z. [DOI] [PubMed] [Google Scholar]
- 7.Virtue S., Vidal-Puig A. Assessment of brown adipose tissue function. Frontiers in Physiology. 2013:4128. doi: 10.3389/fphys.2013.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagouge M., Argmann C., Gerhart-Hines Z., Meziane H., Lerin C., Daussin F. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127(6):1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 9.Holmstrom M.H., Iglesias-Gutierrez E., Zierath J.R., Garcia-Roves P.M. Tissue-specific control of mitochondrial respiration in obesity-related insulin resistance and diabetes. American Journal of Physiology. Endocrinology and Metabolism. 2012;302(6):E731–E739. doi: 10.1152/ajpendo.00159.2011. [DOI] [PubMed] [Google Scholar]
- 10.Schmittgen T.D., Livak K.J. Analyzing real-time PCR data by the comparative C(T) method. Nature Protocols. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 11.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 12.Ortega-Molina A., Efeyan A., Lopez-Guadamillas E., Munoz-Martin M., Gomez-Lopez G., Canamero M. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metabolism. 2012;15(3):382–394. doi: 10.1016/j.cmet.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Feige J.N., Lagouge M., Canto C., Strehle A., Houten S.M., Milne J.C. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metabolism. 2008;8(5):347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 14.White A.T., Philp A., Fridolfsson H.N., Schilling J.M., Murphy A.N., Hamilton D.L. High-fat diet-induced impairment of skeletal muscle insulin sensitivity is not prevented by SIRT1 overexpression. American Journal of Physiology. Endocrinology and Metabolism. 2014;9(307):E764–E772. doi: 10.1152/ajpendo.00001.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White A.T., McCurdy C.E., Philp A., Hamilton D.L., Johnson C.D., Schenk S. Skeletal muscle-specific overexpression of SIRT1 does not enhance whole-body energy expenditure or insulin sensitivity in young mice. Diabetologia. 2013;56(7):1629–1637. doi: 10.1007/s00125-013-2912-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerhart-Hines Z., Rodgers J.T., Bare O., Lerin C., Kim S.H., Mostoslavsky R. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO Journal. 2007;26(7):1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerhart-Hines Z., Dominy J.E., Jr., Blattler S.M., Jedrychowski M.P., Banks A.S., Lim J.H. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+) Molecular Cell. 2011;44(6):851–863. doi: 10.1016/j.molcel.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cannon B., Nedergaard J. Nonshivering thermogenesis and its adequate measurement in metabolic studies. Journal of Experimental Biology. 2011;214(Pt 2):242–253. doi: 10.1242/jeb.050989. [DOI] [PubMed] [Google Scholar]
- 19.Herranz D., Munoz-Martin M., Canamero M., Mulero F., Martinez-Pastor B., Fernandez-Capetillo O. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nature Communications. 2010;(1) doi: 10.1038/ncomms1001. http://www.nature.com/ncomms/journal/v1/n1/full/ncomms1001.html (Article number 3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiang L., Wang L., Kon N., Zhao W., Lee S., Zhang Y. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell. 2012;150(3):620–632. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moynihan K.A., Grimm A.A., Plueger M.M., Bernal-Mizrachi E., Ford E., Cras-Meneur C. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metabolism. 2005;2(2):105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Bordone L., Motta M.C., Picard F., Robinson A., Jhala U.S., Apfeld J. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biology. 2006;4(2):e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodgers J.T., Lerin C., Haas W., Gygi S.P., Spiegelman B.M., Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434(7029):113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 24.Nemoto S., Fergusson M.M., Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} Journal of Biological Chemistry. 2005;280(16):16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 25.Cantó C., Gerhart-Hines Z., Feige J.N., Lagouge M., Noriega L., Milne J.C. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bordone L., Cohen D., Robinson A., Motta M.C., van Veen E., Czopik A. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell. 2007;6(6):759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- 27.Chalkiadaki A., Guarente L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metabolism. 2012;16(2):180–188. doi: 10.1016/j.cmet.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu C., Bai B., Fan P., Cai Y., Huang B., Law I.K. Selective overexpression of human SIRT1 in adipose tissue enhances energy homeostasis and prevents the deterioration of insulin sensitivity with ageing in mice. American Journal of Translational Research. 2013;5(4):412–426. [PMC free article] [PubMed] [Google Scholar]
- 29.Price N.L., Gomes A.P., Ling A.J.Y., Duarte F.V., Martin-Montalvo A., North B.J. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metabolism. 2012;15(5):675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purushotham A., Schug T.T., Xu Q., Surapureddi S., Guo X., Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metabolism. 2009;9(4):327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Planavila A., Iglesias R., Giralt M., Villarroya F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovascular Research. 2011;90(2):276–284. doi: 10.1093/cvr/cvq376. [DOI] [PubMed] [Google Scholar]
- 32.Brunet A., Sweeney L.B., Sturgill J.F., Chua K.F., Greer P.L., Lin Y. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303(5666):2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 33.Banks A.S., Kim-Muller J.Y., Mastracci T.L., Kofler N.M., Qiang L., Haeusler R.A. Dissociation of the glucose and lipid regulatory functions of FoxO1 by targeted knockin of acetylation-defective alleles in mice. Cell Metabolism. 2011;14(5):587–597. doi: 10.1016/j.cmet.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbera M.J., Schluter A., Pedraza N., Iglesias R., Villarroya F., Giralt M. Peroxisome proliferator-activated receptor alpha activates transcription of the brown fat uncoupling protein-1 gene. A link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. Journal of Biological Chemistry. 2001;276(2):1486–1493. doi: 10.1074/jbc.M006246200. [DOI] [PubMed] [Google Scholar]
- 35.Petersen K.F., Shulman G.I. Etiology of insulin resistance. American Journal of Medicine. 2006;119(5 Suppl. 1):S10–S16. doi: 10.1016/j.amjmed.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Girousse A., Tavernier G., Valle C., Moro C., Mejhert N., Dinel A.L. Partial inhibition of adipose tissue lipolysis improves glucose metabolism and insulin sensitivity without alteration of fat mass. PLoS Biology. 2013;11(2):e1001485. doi: 10.1371/journal.pbio.1001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Badin P.M., Louche K., Mairal A., Liebisch G., Schmitz G., Rustan A.C. Altered skeletal muscle lipase expression and activity contribute to insulin resistance in humans. Diabetes. 2011;60(6):1734–1742. doi: 10.2337/db10-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanford K.I., Middelbeek R.J., Townsend K.L., An D., Nygaard E.B., Hitchcox K.M. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. Journal of Clinical Investigation. 2013;123(1):215–223. doi: 10.1172/JCI62308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canto C., Houtkooper R.H., Pirinen E., Youn D.Y., Oosterveer M.H., Cen Y. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metabolism. 2012;15(6):838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown K.D., Maqsood S., Huang J.Y., Pan Y., Harkcom W., Li W. Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metabolism. 2014;20(6):1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.