

Abstract
Objective
Glucagon is a hormone with metabolic actions that maintains normoglycemia during the fasting state. Strategies enabling either inhibition or activation of glucagon receptor (Gcgr) signaling are being explored for the treatment of diabetes or obesity. However, the cardiovascular consequences of manipulating glucagon action are poorly understood.
Methods
We assessed infarct size and the following outcomes following left anterior descending (LAD) coronary artery ligation; cardiac gene and protein expression, acylcarnitine profiles, and cardiomyocyte survival in normoglycemic non-obese wildtype mice, and in newly generated mice with selective inactivation of the cardiomyocyte Gcgr. Complementary experiments analyzed Gcgr signaling and cell survival in cardiomyocyte cultures and cell lines, in the presence or absence of exogenous glucagon.
Results
Exogenous glucagon administration directly impaired recovery of ventricular pressure in ischemic mouse hearts ex vivo, and increased mortality from myocardial infarction after LAD coronary artery ligation in mice in a p38 MAPK-dependent manner. In contrast, cardiomyocyte-specific reduction of glucagon action in adult GcgrCM−/− mice significantly improved survival, and reduced hypertrophy and infarct size following myocardial infarction. Metabolic profiling of hearts from GcgrCM−/− mice revealed a marked reduction in long chain acylcarnitines in both aerobic and ischemic hearts, and following high fat feeding, consistent with an essential role for Gcgr signaling in the control of cardiac fatty acid utilization.
Conclusions
Activation or reduction of cardiac Gcgr signaling in the ischemic heart produces substantial cardiac phenotypes, findings with implications for therapeutic strategies designed to augment or inhibit Gcgr signaling for the treatment of metabolic disorders.
Keywords: Glucagon, Myocardial infarction, Glucagon receptor, Cardiomyocytes, Heart, Diabetes, Fatty acid metabolism
1. Introduction
Glucagon is a 29 amino acid peptide hormone secreted from pancreatic islet α-cells that plays a critical role in maintenance of euglycemia, predominantly by increasing hepatic glucose output. Activation of glucagon receptor (Gcgr) signaling promotes glycogenolysis and enhanced gluconeogenesis, and regulates pathways controlling hepatic lipid oxidation and lipid secretion. Although the actions of glucagon are classically viewed as essential for prevention of hypoglycemia in the face of limited nutrient availability or excess insulin action [1], Gcgr signaling also controls cell survival pathways, as genetic interruption of Gcgr signaling increases the susceptibility to hepatic injury [2].
A single Gcgr is expressed not only in liver, but in extrahepatic tissues including the central and peripheral nervous system, pancreatic islets, adipose tissue, kidney, blood vessels and heart [3,4]. In the pancreas, glucagon potentiates glucose-dependent insulin secretion, whereas activation of Gcgr signaling in the brain regulates hepatic glucose production, control of appetite and body weight [5]. Glucagon actions in adipose tissue and kidney are less understood, but have been linked to control of fatty acid and glucose metabolism.
Although glucagon levels normally decrease during a meal, glucagon secretion is inappropriately increased in many subjects with type 2 diabetes (T2D) [1,6]. Over the last several decades, experimental studies attenuating glucagon action using glucagon immunoneutralizing antisera, Gcgr antagonists, antisense Gcgr oligonucleotides and Gcgr−/− mice have demonstrated amelioration of hyperglycemia in experimental models of diabetes [1,7]. Collectively, these findings have raised enthusiasm for glucagon antagonism as a potential therapeutic strategy for T2D. Indeed, GCGR antagonists and antisense oligonucleotides targeting hepatic GCGR expression robustly lower glucose in clinical trials of human subjects with T2D. However, mechanism-based toxicities noted in preclinical studies, including dyslipidemia, and transaminase elevations [2,8], have also been reported in clinical studies. Hence, the risk:benefit proposition for partial attenuation of GCGR signaling in diabetic humans requires further evaluation.
Complementary efforts are exploring whether partial enhancement of glucagon action, together with agonism of the glucagon-like peptide-1 receptor (GLP-1R), may be useful for the treatment of diabetes and/or obesity [9,10]. Oxyntomodulin, a naturally occurring proglucagon-derived peptide, contains the 29 amino acid sequence of glucagon plus a carboxyterminal extension and exerts potent glucoregulatory and anorectic actions in rodents and humans through activation of the GLP-1 and glucagon receptors [11,12]. More recent studies have demonstrated that simultaneous activation of the glucagon and GLP-1 receptors using synthetic balanced co-agonists produces potent glucoregulatory activity and greater weight loss than observed with GLP-1R agonists alone [13]. Hence there is also considerable interest in understanding the metabolic consequences and therapeutic potential arising from partial selective activation of GCGR signaling.
The increasing interest in development of drugs that reduce or activate GCGR signaling for the treatment of metabolic disorders such as diabetes and obesity raises important questions about the cardiovascular actions and safety of such agents. Current understanding of glucagon action in the heart is limited, and activation of Gcgr signaling in this organ has been reported to be either beneficial or harmful, depending on the experimental or clinical context [14–17]. We have now examined the consequences of manipulating Gcgr signaling in the non-diabetic ischemic mouse heart. Our findings reveal that exogenous glucagon impairs survival following ligation of the left anterior descending (LAD) coronary artery, actions requiring p38 MAP kinase. In contrast, GcgrCM−/− mice with cardiac-specific inactivation of the Gcgr display a cardioprotective phenotype, associated with reduced accumulation of incompletely oxidized fatty acid metabolites in the heart. These findings have implications for pharmaceutical efforts directed at manipulating GCGR signaling for the treatment of human disease.
2. Methods
2.1. Mice and reagents
Inducible αMHCCre (stock 005657) [18] and FLPe (stock 005703) transgenic mice in the C57BL/6 background were obtained from the Jackson Laboratory. GcgrCM−/− mice were generated by crossing αMHCCre mice with GcgrFlox mice [19] in the C57BL/6 background. LAD coronary artery ligation was used to induce myocardial infarction (MI) in 12–14-week-old male mice as described in Ref. [20]. All mice were housed (5 per cage) under a light/dark cycle of 12 h in the Toronto Centre for Phenogenomics (TCP) animal facility, with free access to food and water except where noted. All procedures were conducted according to protocols and guidelines approved by the TCP Animal Care Committee. Genotypes were determined through analysis of genomic DNA prepared from tail snips. Tamoxifen (Sigma Aldrich, 50 mg/kg) dissolved in corn oil was administered for 5 consecutive days to 6- or 7-week-old male αMHCCre or GcgrCM−/− mice to induce Cre expression. Before any cardiac assessment or procedure, all mice were allowed 6 weeks to recover after the last tamoxifen injection, as Cre expression in the heart often induces a transient cardiomyopathy that dissipates 5 weeks after tamoxifen-induced Cre expression [21]. Glucagon (Sigma) 30 ng/g body weight or saline in 10% gelatin was administered to C57BL/6 mice as described in Ref. [8], 3 injections daily, with or without 2 injections daily of 1 μmol/g body weight SB203580 (p38 MAPK inhibitor, Sigma) for 7 days. Blood pressure and heart rates were measured using a telemetry system (DSI technology) as described in Ref. [22]. The rat Gcgr adenovirus (AdGcgr) has been described previously in Ref. [2].
2.2. Ischemia/reperfusion
Global no-flow ischemia in Langendorff-perfused hearts was induced as described in Ref. [23]. Hearts underwent a 30 min aerobic perfusion with Krebs-Henseleit buffer, followed by a 30 min global no-flow ischemia, and either a 50 or 60 min reperfusion period during which left ventricular developed pressure (LVDP) was recorded (Biopac Systems Canada Inc.). In a separate set of hearts 1 μg/mL glucagon was administered 20 min prior to ischemia.
2.3. Myocardium metabolic profiling
Mass spectrometry-based metabolic profiling was performed to determine myocardial levels of acylcarnitines and organic acids [24]. Triacylglycerol (TAG) was extracted from frozen myocardial tissue (∼20 mg) with a 2:1 chloroform-methanol solution and quantified with a commercially available enzymatic assay kit (Wako Pure Chemical Industry) as described in Ref. [25].
2.4. Heart histology
Animals were anesthetized using avertin (250 mg/kg body weight ip injection). The chest cavity was opened to expose the heart and 1 M KCl was injected into the apex to arrest the heart in diastole. The heart was perfusion-fixed with 4% buffered formalin at physiological pressure, post-fixed in formalin, embedded in paraffin, and sectioned at 6 μm, and stained with Masson's Trichrome or processed for Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). TUNEL staining was performed using the ApopTag peroxidase kit for apoptosis (EMD Millipore). Cardiac morphometry was performed on mid-ventricular cross-sections using Aperio ImageScope Viewer software (Aperio Technologies). The infarcted area was calculated as a % of total LV area. Cardiac hypertrophy was quantified as the heart weight-to-body weight ratio.
2.5. Glucose tolerance
12–14-week-old male mice were fasted overnight (16–18 h), and glucose (1.5 mg/g body weight) was administered orally (through a gavage tube) or via injection into the peritoneal cavity (intraperitoneal glucose tolerance test). Blood samples were drawn from the tail vein at 0, 15, 30, 60, 90, and 120 min post-glucose administration, and blood glucose and insulin levels were measured as described in Ref. [26].
2.6. Western blotting
Hearts were collected from fasted mice (5 h) 30 min following ischemia or sham surgery, washed in Krebs buffer containing 11 mM glucose and frozen. Frozen hearts were powdered and homogenized in buffer containing 50 mM Tris HCl, pH 8, 1 mM EDTA, 10% glycerol, 0.02% Brij-35. Western blotting was carried out as described in Refs. [27,28] and blots were visualized using an enhanced chemiluminescence Western blot detection kit (Perkin Elmer) and quantified with Carestream Molecular Imaging Software (Kodak).
2.7. RNA analyses
RNA was isolated from cardiac extracts using TRI reagent (Sigma). First-strand cDNA was synthesized from total RNA using the SuperScript III reverse transcriptase synthesis system (Invitrogen). Real-time polymerase chain reaction was performed with the ABI Prism 7900 Sequence Detection System using TaqMan Gene Expression Assays and TaqMan Universal PCR Master Mix (Applied Biosystems). Relative levels of mRNA transcripts were quantified using the 2-ΔCt method and normalized to levels of peptidyl-prolyl isomerase A (Ppia-cyclophilin) RNA.
2.8. Cell culture
Atrial cardiomyocytes were isolated as described in Ref. [22]. HL-1 atrial cardiac myocytes were provided by Dr. William Claycomb (Louisiana State University, New Orleans), and cultured in Claycomb Media (Sigma–Aldrich) with 10% FBS, 1% penicillin/streptomycin, 0.1 mM norepinephrine, and 2 mM l-glutamine. Cells were seeded onto 6-well plates (BD Falcon) coated with 0.02% gelatin/0.5% fibronectin. Confluent cells were serum-starved and supplemented with Claycomb Media without FBS and norepinephrine prior to infection with Adβ-Gal or AdGcgr at 10× multiplicity of infection. 24 h following infection, cells were treated with either PBS or glucagon for 3 h in supplemented Claycomb Media without FBS and norepinephrine. Cellular injury was induced by H2O2 [29].
2.9. Statistical analysis
Results are presented as mean ± SEM. Statistical significance was determined using 1- or 2-way analysis of variance with Bonferroni post hoc tests (as appropriate) using GraphPad Prism 4.0 (GraphPad Software Inc). Statistical significance was noted when p < 0.05.
3. Results
3.1. Glucagon impairs outcomes during experimental MI in a p38 MAPK-dependent manner
As recent studies of the related Glp1r revealed issues with receptor localization and surprising chamber-specific receptor expression [22,30,31], we first assessed Gcgr expression in the mouse heart using both regular PCR and primers corresponding to the full length open reading frame, as well as quantitative real-time PCR (Supplementary Figure 1A,B). Consistent with a previous report in Ref. [32], Gcgr expression was most abundant in RNA from the right atria but easily detectible in RNA from both right and left ventricle. In contrast, Glp1r expression was restricted to the atria as previously described in Refs. [22,32]. The expression of the Gipr was much less atrial-biased, relative to the Glp1r, whereas Glp2r mRNA transcripts were not detected in the mouse heart (Supplementary Figure 1A,B).
To determine the effects of glucagon treatment on cardiovascular outcomes during MI, we treated mice with glucagon (3× daily, via subcutaneous injection, 30 ng/g body weight) prior to and following LAD coronary artery ligation (Figure 1A). No significant changes in random glycemia or body weight were observed with glucagon injections (Supplementary Figure 1C,D), however, glucagon significantly reduced survival following MI and was associated with a significant increase in TUNEL-positive apoptotic cardiac myocytes (48 h post-MI), although no effect on adverse LV remodeling or infarct scar formation was observed (Figure 1B–D). These negative cardiovascular outcomes required p38 MAPK; glucagon increased p38 MAPK phosphorylation in both the aerobic and ischemic heart (Figure 1E), and co-treatment with SB203580 (p38 MAPK inhibitor) prevented the glucagon-mediated reduction in MI survival (Figure 1B), as well as the increase in TUNEL-positive cardiac myocytes (Figure 1D).
Figure 1.
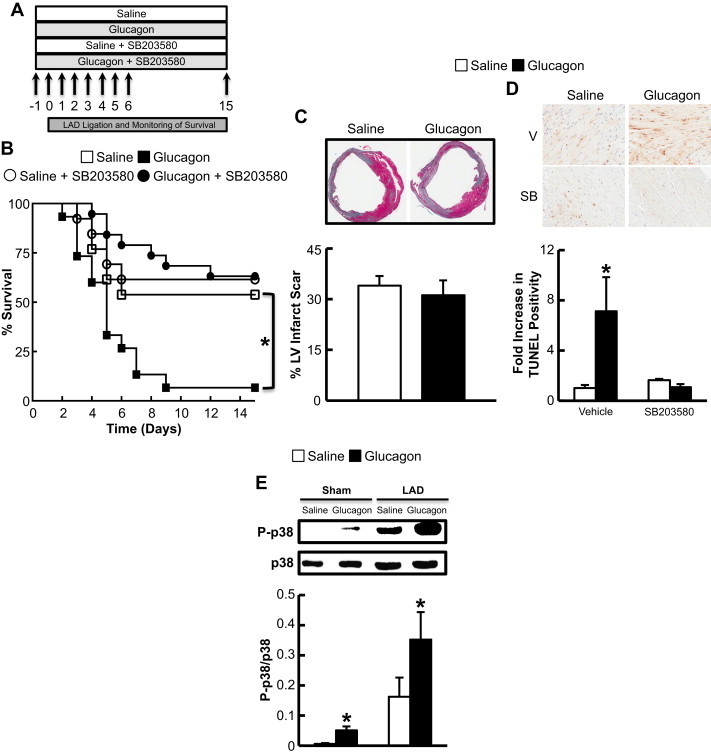
Glucagon impairs survival after MI in a p38 MAPK-dependent manner. (A) Schematic of overall study design. Mice were injected with vehicle/glucagon/SB203580 for 7 days starting 1 day prior to LAD coronary artery ligation and sacrificed 15 days later. (B) Survival following LAD coronary artery ligation in C57BL/6 mice treated with saline or glucagon (30 ng/g) with or without co-administration of the p38 MAPK inhibitor (SB203580 1 μmol/kg) for 1 week. *p < 0.05 saline vs. glucagon. Data are mean ± S.E.M (LAD n = 13–15 per treatment). (C) Infarct size assessed in mice described in (B) at day 15. Data are mean ± S.E.M (LAD n = 5–6 per treatment). (D) C57BL/6 mice treated with saline or glucagon (30 ng/g) with or without co-administration of the p38 MAPK inhibitor (SB203580 1 μmol/kg; SB) were subjected to LAD coronary artery ligation for 48 h to assess TUNEL positive cardiac myocytes. *p < 0.05 saline vs. glucagon. Data are mean ± S.E.M (LAD n = 7–8 mice per treatment). (E) p38 MAPK phosphorylation in C57BL/6 mice treated with saline or glucagon (30 ng/g) with or without SB203580 (1 μmol/kg) 48 h post-LAD coronary artery ligation or sham surgery. *p < 0.05 saline vs. glucagon group. Data are mean ± S.E.M (n = 3–5).
3.2. Glucagon activates PPARα in a p38 MAPK-dependent manner in cardiac myocytes, and increases cardiac myocyte apoptosis in vitro
As glucagon increases hepatocyte PPARα activity in a p38 MAPK-dependent manner [8], we examined whether similar regulation occurs in the heart. Treatment of HL-1 atrial cardiac myocytes (which do not express the endogenous Gcgr) with an adenovirus encoding the rat Gcgr (AdGcgr) followed by treatment with glucagon (20 nM) for 24 h increased the expression of PPARα target genes (Figure 2A). In contrast, no such changes were observed in HL-1 cells treated with a control adenovirus (Adβ-Gal, Supplementary Figure 1E). Furthermore, glucagon (20 nM for 24 h) increased luciferase activity directed by a PPARα response element in AdGcgr-infected HL-1 cells (Figure 2B), but not in Adβ-Gal-infected HL-1 cells (Supplementary Figure 1F). Furthermore, SB203580 abolished the glucagon-mediated increase in PPARα luciferase activity in AdGcgr-infected HL-1 cells, illustrating the dependency on p38 MAPK activity (Figure 2C). We also observed an increase in nuclear PPARα translocation in both glucagon-treated adult cardiac myocytes (Figure 2D) and AdGcgr infected HL-1 cells (Figure 2E), which was abolished by pre-treatment with SB203580 (Figure 2E).
Figure 2.
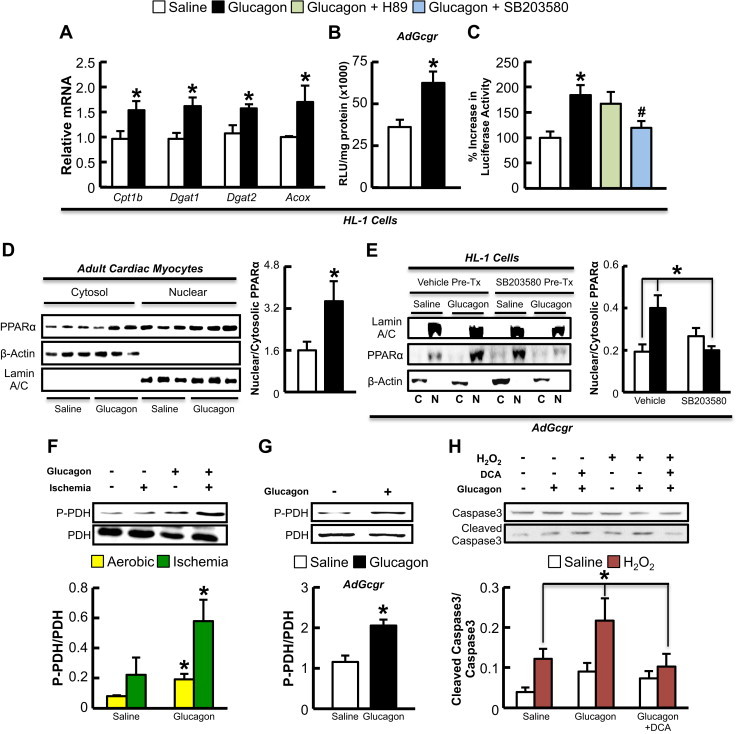
Glucagon increases PPARα-dependent gene expression, PPARα nuclear translocation, cleaved caspase 3 levels, and PDH phosphorylation. (A) HL-1 cells infected with adenovirus expressing the rat Gcgr cDNA were treated with saline or 20 nM glucagon for 3 h for assessment of Cpt1b, Dgat1, Dgat2, and Acox mRNA expression. (B,C) HL-1 cells were infected with Adβ-gal or AdGcgr for 24 h followed by transfection with a PPARα gene promoter-luciferase construct. Cells were treated with saline or 20 nM glucagon for 3 h, with or without the PKA inhibitor (H89) or p38 MAPK inhibitor (SB203580) and luciferase expression was quantified as Relative Light Units (RLU). (D) Primary cultures of murine atrial cardiac myocytes were treated with saline or 20 nM glucagon for 3 h and cells were harvested for Western blot analysis of cytoplasmic and nuclear protein expression. (E) HL-1 cell lines infected with AdGcgr were treated with saline or 20 nM glucagon with and without SB203580 and cells were harvested for nuclear (N) and cytoplasmic (C) protein analysis. (F) C57BL/6 mice were treated with exogenous glucagon (30 ng/g) every 8 h for 24 h to assess PDH phosphorylation in hearts in mice that underwent 30 min occlusion of the LAD coronary artery or sham surgery (n = 6 per treatment). (G) PDH phosphorylation in HL-1 cells infected with AdGcgr for 24 h followed by a 3 h treated with glucagon. (H) AdGcgr infected HL-1 cell lines were treated for 24 h with 100 μM H2O2 and/or 20 nM glucagon during the final 3 h of H2O2 treatment to detect cleaved caspase 3 levels. Separate groups of cells were treated with or without 1.5 mM DCA (PDH activator via inhibiting the PPARα target gene, PDK4) for 24 h concomitantly with H2O2. *p < 0.05, saline vs. glucagon. #p < 0.05, glucagon + SB203580 vs. glucagon alone. All cell culture experiments were repeated at least twice, n-4-6 per condition.
We next determined whether pyruvate dehydrogenase (PDH, the rate-limiting enzyme of glucose oxidation) activity was altered in hearts and HL-1 cells treated with glucagon. Glucagon increased PDH phosphorylation (indicative of reduced PDH activity) in both the aerobic/ischemic heart (Figure 2F), and in HL-1 cells infected with AdGcgr (Figure 2G), but not in cells infected with Adβ-Gal (Supplementary Figure 1G). Glucagon also increased hydrogen peroxide (H2O2)-induced caspase-3 cleavage, an effect negated via pretreatment with the PDH kinase inhibitor, dichloroacetate (DCA) (Figure 2H). Pre-treatment with DCA also reduced expression of the pro-apoptotic protein, Bax, but had no effect on expression of the anti-apoptotic protein, Bcl-2, in AdGcgr-infected HL-1 cells treated with H2O2 and glucagon (Supplementary Figure 2).
3.3. Glucagon regulates acylcarnitine profiles in the heart
Although myocardial ischemia reduces fatty acid oxidation rates due to the reduction in oxygen supply, we observed a robust increase in long chain acylcarnitines in ischemic hearts 24-h post-glucagon and 30 min post-MI (Figure 3). The most likely interpretation of this profile is that glucagon triggered an increase in β-oxidation, but that flux through the TCA cycle, electron transport chain, and oxidative phosphorylation was limited by ischemia, resulting in accumulation of incompletely oxidized fats. Interestingly, Krebs cycle intermediates were not altered in glucagon-treated hearts (Supplementary Figure 3), suggesting that the increase in fatty acid oxidation may have provided acetyl CoA for the Krebs cycle to offset the apparent decrease in PDH activity.
Figure 3.
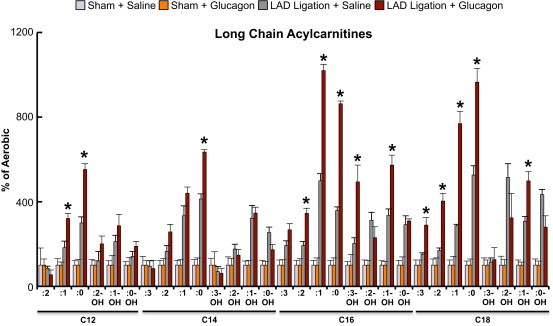
Glucagon increases long chain acylcarnitine levels in the ischemic heart. Glucagon (30 ng/g body weight) or saline injections were administered subcutaneously every 8 h over a 24 h period (4 total injections at 0, 8, 16, and 24 h) and hearts were harvested after a 5 h fast and subjected to metabolomics assessment to determine acylcarnitine content. Ischemic hearts were collected 30 min following LAD coronary artery ligation surgery (surgery took place 4 h into the 5 h fast). Data is expressed as percent of levels in aerobic mouse hearts for each respective group.*p < 0.05 saline vs. glucagon-treated LAD coronary artery ligation group. Data are mean ± S.E.M (n = 5 per group).
3.4. Cardiomyocyte-specific deletion of the Gcgr produces a cardioprotective phenotype in response to ischemic injury
As increased glucagon action produces negative effects on the ischemic heart, we hypothesized that reducing glucagon action should protect against ischemic injury. Mice with global germline deletion of the Gcgr exhibit mild hypoglycemia, and increased levels of proglucagon-derived peptides, Fgf-21 and bile acids [26,33–35], complicating interpretation and mechanistic attribution of cardiac phenotypes arising from whole body loss of the Gcgr. Indeed, secondary increases in GLP-1 and Fgf-21 in Gcgr−/− mice may exert independent cardioprotective actions [36,37]. Accordingly, we generated a tamoxifen-inducible cardiomyocyte-specific Gcgr knock out mouse (GcgrCM−/−; Supplementary Figure 4A). Treatment with tamoxifen (50 mg/kg IP) for 5 consecutive days decreased cardiac Gcgr mRNA expression by ∼85% without affecting Gcgr mRNA expression in the liver and kidney (Supplementary Figure 4B–D). GcgrCM−/− mice appeared phenotypically normal, exhibiting normal weight gain and similar glucose tolerance relative to α myosin heavy chain-Cre (αMHCCre) littermate controls (Supplementary Figure 5). Following LAD coronary artery ligation, GcgrCM−/− mice exhibited a marked increase in survival, reduced cardiac hypertrophy, and substantially less adverse LV remodeling (Figure 4). A significant reduction in mRNA levels of (a) key PPARα target genes and (b) mRNA transcripts encoding regulators of lipid metabolism was detected in GcgrCM−/− aerobic hearts, with similar trends detected in ischemic hearts (Figure 5A). PPARα protein expression was also reduced in sham/aerobic but not in ischemic GcgrCM−/− hearts; ischemia alone reduced cardiac PPARα protein expression (Figure 5B). In contrast, levels of Akt and GSK3β phosphorylation were unaltered in ischemic hearts from GcgrCM−/− mice, but Akt phosphorylation was increased in aerobic/sham hearts from GcgrCM−/− mice (Figure 5B). Furthermore, PDH phosphorylation showed a trend towards reduction in both sham/aerobic and ischemic hearts from GcgrCM−/− mice (Figure 5B), consistent with a role for glucagon action in the control of cardiac PDH activity and glucose oxidation.
Figure 4.
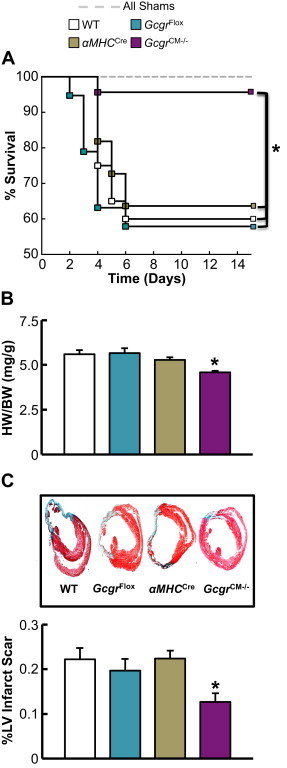
Loss of cardiac Gcgr signaling enhances survival following MI and attenuates adverse LV remodeling. (A) LAD coronary artery ligation was performed in 11–14-week-old GcgrCM−/− and littermate control mice and survival was monitored for 15 days following surgery. *p < 0.05 GcgrCM−/− vs. each control group. Data are mean ± S.E.M (SHAM; n = 10 per genotype and LAD; n = 11–23 per genotype). (B) Heart weight:body weight (HW:BW) ratio 15 days following LAD coronary artery ligation. *p < 0.05 GcgrCM−/− vs. GcgrFlox and wild-type (WT) mice. Data are mean ± S.E.M (n = 8–11 per genotype). (C) Masson's trichome staining of infarcted heart and quantification of % left ventricular (LV) infarct scar area 15 days following LAD coronary artery ligation. *p < 0.05 GcgrCM−/− vs. littermate control mice. Data are mean ± S.E.M (n = 9–11 per genotype).
Figure 5.
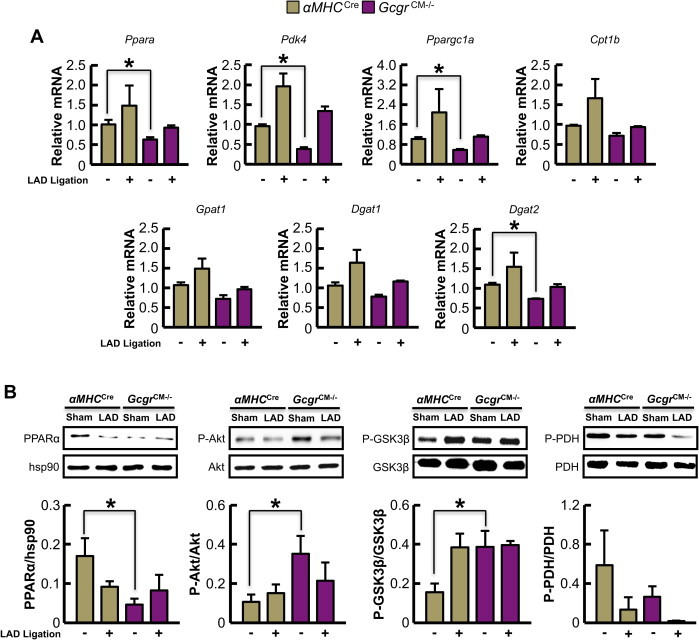
Selective loss of Gcgr signaling in cardiomyocytes leads to reduced expression of genes and proteins regulating fatty acid metabolism. (A) Quantification of mRNA transcript levels from sham/aerobic and ischemic hearts (30 min post-LAD coronary artery ligation) from 12-week-old αMHCCre and GcgrCM−/− mice fasted for 5 h (n = 4). *p < 0.05 GcgrCM−/− vs. αMHCCre. Data are mean ± S.E.M. (B) Protein expression/phosphorylation in fasted (5 h) hearts from 12-week-old αMHCCre and GcgrCM−/− mice subjected to sham surgery or LAD coronary artery occlusion for 30 min *p < 0.05 for αMHCCre vs. GcgrCM−/− mice. Data are mean ± S.E.M (n = 4 mice in each group).
3.5. Acylcarnitine profiling suggests reduced myocardial fatty acid oxidation in GcgrCM−/− mice
We observed a significant reduction in several long chain acylcarnitine species in sham/aerobic hearts from GcgrCM−/− mice (Figure 6A), suggesting that eliminating glucagon action in heart reduces substrate burden on the fatty acid oxidation pathway. Consistent with the likelihood that this metabolic effect was specific to fatty acid oxidation and not an overall decrease in oxidative metabolism, we observed a decrease in medium chain acylcarnitines in sham/aerobic hearts from GcgrCM−/− mice, whereas myocardial C2 (acetylcarnitine) levels were similar between GcgrCM−/− and αMHCCre mice (Figure 6B). In addition, TAG content and the majority of Krebs cycle intermediates were similar in GcgrCM−/− and αMHCCre sham/aerobic hearts (Figure 6C–I). Similar results were observed in GcgrCM−/− and αMHCCre ischemic hearts 30 min post-MI, with the exception of an increase in myocardial TAG content in GcgrCM−/− hearts (Figure 6J–R). Ischemia itself reduces fatty acid oxidation and results in the mobilization of myocardial TAG stores (∼2.2 vs. 1.1 μmol/g wet weight in αMHCCre aerobic and ischemic hearts, respectively), but the additional reduction in fatty acid oxidation in GcgrCM−/− ischemic hearts likely explains their increase in myocardial TAG content relative to ischemic hearts from their αMHCCre littermates. Comparable results were observed in the hearts of GcgrCM−/− and αMHCCre mice fed a high fat diet for 6 months, with the majority of long chain acylcarnitines trending lower in GcgrCM−/− hearts (Supplementary Figure 6).
Figure 6.
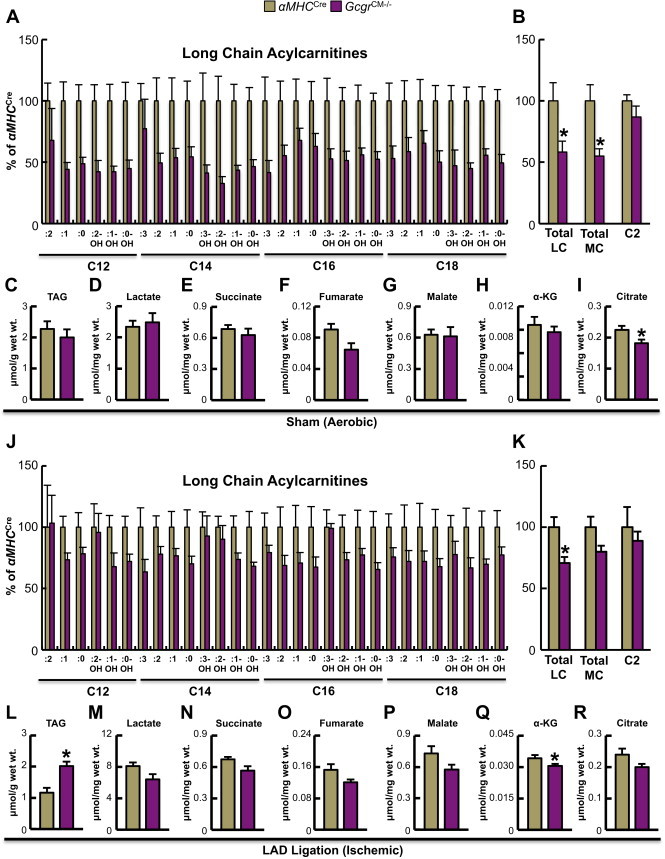
Targeted metabolomics reveals reduced fatty acid oxidation in GcgrCM−/− hearts. (A) Acylcarnitine levels in aerobic hearts harvested from fasted (5 h) αMHCCre and GcgrCM−/− mice (n = 5 per genotype). Values are expressed as percent of levels in hearts from αMHCCre mice. (B) Aerobic myocardial total long chain (LC), medium chain (MC) and acetyl (C2) acylcarnitines.*p < 0.05 αMHCCre vs. GcgrCM−/−. Levels of triacylglycerol (TAG) (C), lactate (D) and Krebs cycle intermediates (E–I) in 5 h fasted sham/aerobic hearts. *p < 0.05 αMHCCre vs. GcgrCM−/−. (J) Acylcarnitine levels in hearts harvested 30 min following cardiac ischemia from 5 h fasted αMHCCre and GcgrCM−/− mice (n = 5 per genotype). Values are expressed as percent of αMHCCre mice values. (K) Levels of total LC, MC and acetyl (C2) acylcarnitines in ischemic hearts.*p < 0.05 αMHCCre vs. GcgrCM−/−. TAG (L) and lactate (M) content in ischemic hearts. *p < 0.05 αMHCCre vs. GcgrCM−/−. (N–R) Levels of Krebs cycle intermediates in hearts harvested 30 min following cardiac ischemia from 5 h fasted αMHCCre and GcgrCM−/− mice. *p < 0.05 αMHCCre vs. GcgrCM−/−.
3.6. Glucagon attenuates recovery of left ventricular developed pressure during ischemia/reperfusion injury
We next determined whether reduced or enhanced cardiac Gcgr signaling modulates outcomes in response to ex vivo ischemia/reperfusion (I/R) injury. Isolated hearts from Gcgr−/− mice exhibited enhanced recovery of LV developed pressure (LVDP) following I/R injury, whereas glucagon treatment impaired recovery of LVDP in isolated hearts from WT mice (Figure 7A–F).
Figure 7.
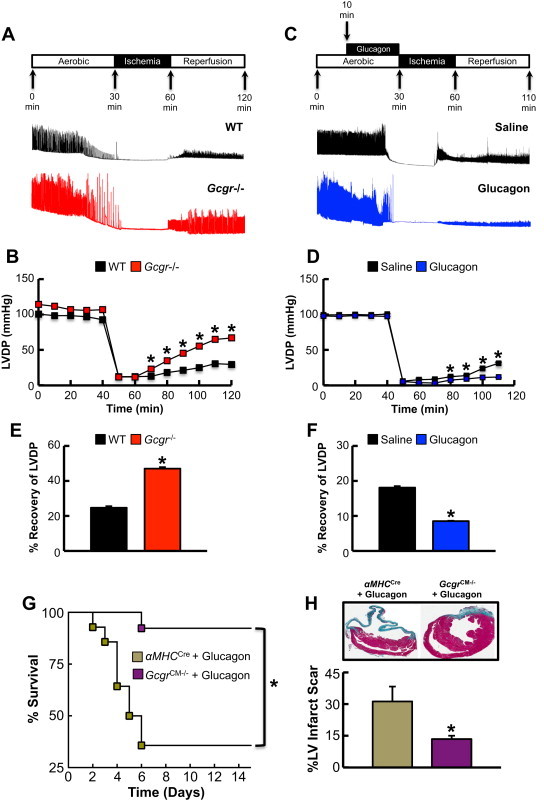
Loss of Gcgr signaling protects whereas glucagon impairs recovery of LV developed pressure (LVDP) after I/R injury in the isolated heart ex vivo. (A–F) Schematic depiction of peptide infusions, ischemia and reperfusion times, and representative LVDP recordings and data from isolated perfused hearts. LVDP measurements in isolated perfused Gcgr−/− (B,E) and WT (D,F) hearts subjected to I/R injury with and without glucagon (1 μg/mL for 20 min) (D,F). For E&F, graph depicts the percentage recovery rate of LVDP following ischemia. Data shown are means ± S.E.M. (n = 3–6 per genotype). *p < 0.05 compared with the control group. (G,H) Glucagon impairs survival after myocardial infarction in a cardiac Gcgr-dependent manner. (G) Left anterior descending coronary artery ligation (LAD) surgeries were performed in 11–14-week-old αMHCCre and GcgrCM−/− mice treated with 30 ng/g body weight glucagon given by subcutaneous injection (every 8 h for 7 days). Survival was monitored for 15 days following surgery. (H) Infarct size was assessed 15 days following LAD coronary artery ligation. *p < 0.05 αMHCCre WT vs. GcgrCM−/− mice. Data are mean ± S.E.M (n = 12–13 per genotype).
3.7. Glucagon fails to increase mortality or impair LV remodeling in ischemic GcgrCM−/− mice
To determine whether the glucagon-induced increase in mortality and adverse LV remodeling following MI (Figure 1) was due to direct activation of the cardiac glucagon receptor, we treated GcgrCM−/− mice and their αMHCCre littermates with glucagon (30 ng/g body weight; 3× daily, via subcutaneous injection) prior to and following LAD coronary artery ligation. Although glucagon increased mortality and infarct size in αMHCCre control mice, it had no effect on these parameters in GcgrCM−/− mice (Figure 7G,H). Hence the deleterious effects of glucagon on the cardiac response to ischemic injury are not indirect and require a functional cardiomyocyte Gcgr.
4. Discussion
Our findings demonstrate that exogenous glucagon administration has negative actions on the ischemic heart, whereas reduction in cardiac glucagon action in GcgrCM−/− mice results in marked cardioprotection. In both situations, changes in fatty acid oxidation correlate with cardiac outcomes, as multiple long-chain acylcarnitines are increased by glucagon in the ischemic heart, whereas GcgrCM−/− mice have reduced levels of these metabolites under ischemic conditions. Taken together these findings strongly and independently support the importance of both pharmacological and physiological Gcgr signaling in the cardiac response to ischemic injury.
Previous studies have provided conflicting data on the role of glucagon on the myocardium. Treatment with glucagon worsened the recovery of cardiac power in the isolated working rat heart during global no-flow ischemia/reperfusion [16] and cardiomyopathy developed in a patient with a glucagonoma that was completely reversed upon tumor resection [38]. In contrast, glucagon improved LV minute work and contractility in dogs with acute MI [39]. Furthermore, glucagon administration to 6 patients with AMI produced positive inotropic effects and temporarily improved cardiogenic shock [40], whereas glucagon increased cardiac performance and reduced LV failure without altering myocardial oxygen consumption in dogs subjected to MI [41]. Although heart rate and systolic blood pressure trended lower in non-ischemic GcgrCM−/− mice (data not shown), these differences were not statistically significant perhaps due to the small number of animals analyzed. Our results support the contention that pharmacological glucagon agonism is deleterious to the ischemic heart, as glucagon significantly enhanced mortality following MI, whereas GcgrCM−/− mice exhibited a cardioprotective phenotype following LAD coronary artery ligation-induced myocardial injury. Thus, reduction of glucagon receptor signaling in the heart should be explored as a novel approach for the attenuation of ischemic myocardial injury. However, whether cardiac GCGR signaling can be safely, selectively and effectively targeted pharmacologically in humans without incurring additional systemic liabilities is currently unknown.
Although the mechanism(s) via which glucagon receptor signaling modulates cardiovascular outcomes following MI remain incompletely identified, changes in myocardial fatty acid oxidation appear to be implicated. Acylcarnitine profiling indicated that glucagon caused accumulation of incompletely oxidized lipids in the heart, as has also been seen in experimental models of heart failure [42]. In skeletal muscle, accumulation of acylcarnitines has been described in obesity and type 2 diabetes. Relief of acylcarnitine accumulation by reducing entry of fatty acids into the mitochondria relieves substrate overload and enhances insulin action [24] and conversely, increasing acyl CoA/acylcarnitine accumulation by transgenic knockout of carnitine acyl transferase (CrAT) impairs insulin sensitivity [43]. Here, we observed increased acylcarnitines in response to glucagon administration in the ischemic heart, consistent with activation of the early, but not later phases of fatty acid oxidation, whereas heart-specific deletion of Gcgr appeared to reduce substrate pressure on the fatty acid oxidation pathway. Consistent with the metabolic profile, glucagon treatment of cardiac myocytes increased PPARα activity, a known activator of the β-oxidative machinery, whereas PPARα downstream target gene mRNA expression was reduced in hearts from GcgrCM−/− mice.
PPARα is a key regulator of fatty acid oxidation in the heart [44,45] and cardiac-specific PPARα overexpression (a) worsens the recovery of cardiac function during ex vivo I/R injury in the isolated working heart [46] and (b) induces a diabetic-like cardiomyopathy [45]. Furthermore, genetic elimination of PPARα in dominant-negative NADPH oxidase transgenic mice reversed their increased infarct size and cardiac myocyte apoptosis in response to in vivo I/R injury [47] and elimination of PPARα in cardiac-specific aryl hydrocarbon nuclear translocator deficient mice reversed their associated cardiomyopathy and lipotoxicity [48]. Likewise, PPARα deficient mice are protected against both ex vivo I/R injury and streptozotocin-induced diabetic cardiomyopathy [46,49]. In contrast, treatment with the PPARα agonist, GW7647, reduced infarct size in CD1 mice following temporary occlusion of the LAD coronary artery [50], whereas the PPARα agonist, fenofibrate, improved recovery following ex vivo I/R injury in hearts from mice with diet-induced obesity [51]. While direct PPARα agonism increases fatty acid oxidation, chronic peripheral PPARα agonism actually increases hepatic fatty acid oxidation rates, which decreases circulating lipids without changes in cardiac PPARα activity [51]. Acknowledging the ongoing controversy regarding PPARα agonism, fatty acid oxidation and ventricular function, our findings with glucagon are consistent with a negative role for Gcgr-dependent PPARα activity in the ischemic heart.
Consistent with previous studies of glucagon action in the liver [8] we demonstrated that glucagon activates PPARα in the heart in a p38 MAPK dependent manner. However, p38 MAPK has also been demonstrated to increase cardiac injury through mechanisms independent of changes in fatty acid oxidation, such as an increase in intracellular acidosis which decreases the efficiency of contractile function [52], or via interaction with TAK1-binding protein 1 to enhance cardiac myocyte apoptosis [53]. Thus, other mechanisms may also contribute to our observed phenotypes via activation of p38 MAPK activity.
Our current studies have several important limitations. First, the majority of our experiments were performed in relatively young, healthy, non-diabetic, non-obese mice. Perturbations of the normal metabolic environment may greatly influence myocardial energy uptake, patterns of fuel utilization, and potentially, cardiovascular outcomes. Moreover, the role(s) of and pathways activated by enhanced or disrupted cardiac Gcgr signaling in the ischemic myocardium of older mice with established cardiovascular disease may be different. Furthermore, it cannot be assumed that gain and loss of Gcgr signaling in the normal, diabetic, or obese human heart will produce similar deleterious or beneficial outcomes. Notably, we have not ascertained whether the acute negative actions of glucagon on the mouse heart reflect achievement of a critical Cmax or total exposure to glucagon. Moreover, whether sustained exposure to glucagon would result in partial desensitization of the cardiac Gcgr signaling pathway has not been examined. Of potential relevance to future studies of the Gcgr and related cyclic AMP-linked cardiac receptors, a recent report described cardioprotective actions of secreted cyclic AMP metabolized to adenosine, which differs substantially from the classical cardiotoxic effects of intracellular cyclic AMP [54]. Hence understanding the effects of glucagon and GLP-1 on intracellular vs. secreted cyclic AMP in the heart may be important. Finally, our experiments focused on the consequences of ischemic cardiac injury, and the importance of Gcgr signaling in experimental models of heart failure under normoglycemic and diabetic conditions requires further elucidation.
There is currently great interest in the therapeutic potential of glucagon/GLP-1 co-agonists and glucagon-containing tri-agonists for the treatment of obesity and/or diabetes [7,10,55]. Our findings demonstrate that pure unopposed acute glucagon agonism has negative effects on the myocardium during ischemic injury, whereas cardiomyocyte-specific elimination of glucagon receptor activity results in robust cardioprotection against MI-induced mortality and adverse LV remodeling. Nevertheless, we did not assess a full range of glucagon doses in our gain of function studies, hence it may be possible to administer glucagon at doses that prevent hypoglycemia (type 1 diabetes), or achieve some degree of weight loss (diabetes and/or obesity), without cardiotoxicity. Whether the combination of glucagon with GLP-1 agonism in the same molecule or as a mixture of two different agonists, will similarly mitigate the potential adverse consequences of unopposed Gcgr activation on the ischemic diabetic heart is an important question [13]. Indeed, observations using the isolated rat perfused heart model suggest that glucagon alone compromised the energetic state of ischemic hearts, whereas a glucagon-GLP-1 dual agonist exerted preferential actions on cardiac energetics without increasing levels of cyclic AMP accumulation, thereby mitigating the adverse effects of glucagon [56]. Furthermore, it cannot be assumed that administration of GCGR antagonists to diabetic subjects, at doses resulting in partial attenuation of GCGR signaling in multiple tissues, will produce a cardiovascular phenotype that mirrors or overlaps our findings in non-diabetic mice with marked selective genetic reduction of Gcgr signaling in cardiomyocytes.
Similar questions surround the dose–response relationships for the cardiovascular actions of glucagon in subjects with type 1 diabetes and pre-existing coronary artery disease. Indeed, the investigational use of combined glucagon-insulin delivery systems for the optimized treatment of type 1 diabetes [57] emphasizes the need to explore the safety of a range of glucagon concentrations in humans at risk for cardiovascular events. Our data highlights the importance of understanding the cardiovascular actions of novel peptide therapies being evaluated for the treatment of patients with metabolic disorders associated with a substantial concomitant risk of developing ischemic heart disease.
Acknowledgments
These studies were funded by an operating grant from the Canadian Institutes of Health Research (136942 and 93749) and by the Canada Research Chairs Program and the Banting and Best Diabetes Centre-Novo Nordisk Chair in Incretin Biology (DJD). We thank Dr. William Claycomb (Louisiana State University, New Orleans) for kindly providing us with the HL-1 atrial cardiac myocytes. We also thank Minsuk Kim for collaborative assistance with ischemia/reperfusion experiments and culturing of adult mouse atrial cardiac myocytes. JRU was supported by fellowships from the Canadian Institutes of Health Research (CIHR), the Alberta Innovates-Health Solutions, and Venture-Sinai.
Conflict of interest
Dr. Drucker has served as an advisor or consultant within the past 12 months to Arisaph Pharmaceuticals Inc., Intarcia Therapeutics, Merck Research Laboratories, MedImmune, Novo Nordisk Inc., NPS Pharmaceuticals Inc., Receptos, Sanofi, and Transition Pharmaceuticals Inc.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
Supplementary Figure 1. G protein-coupled receptor expression in the heart. Glycemia and body weight in mice following glucagon treatment, and the effect of glucagon on PPARα target gene expression and PPARα-directed luciferase activity in Adβ-gal transduced HL-1 cells. (A) Mouse G-protein-coupled receptor transcripts were identified in RNA isolated from individual right atrium (RA), left atrium (LA), right ventricle (RV), and left ventricle (LV) from C57BL/6 mice following reverse transcription-PCR with gene-specific primers that amplify products encompassing the majority of the open reading frame. Southern blotting with an internal 32P-labeled oligonucleotide was used to detect each product. Expected size of full-length PCR product is indicated by the arrow on the left of each panel. Lung and islet were used as positive controls for glucagon-like peptide-1 receptor (Glp1r) expression. White adipose tissue (WAT) and islet were used as positive controls for glucose-dependent insulinotropic polypeptide receptor (Gipr) expression. Liver (Liv) was used as a positive control for glucagon receptor (Gcgr) expression. Jejunum (Jej) was used as a positive control for glucagon-like peptide-2 receptor (Glp2r) expression. Ec1 and Ec2 are from mouse primary endothelial cell preparations. The following primers were used for RT-PCR: Glp1r: Forward: GTACCACGGTGTCCCTCTCA: Reverse: CCTGTGTCCTTCACCTTCCCTA Internal: GGATGGGCTCCTCTCCTAAT: Gipr: Forward: CTGCTTCTGCTGCTGTGGT Reverse: CACATGCAGCATCCCAGA Internal: GTCTGCAGGCTTTGTCTTCC: Gcgr: Forward: CTGCTGCTGCTGCTGTTGG Reverse: ACCTTGGGAGACTACTGGC Internal: GGATTCTGGTGGATCCTGCG: Glp2r: Forward: CTGCTGGTTTCCATCAAGCAA Reverse: TAGATCTCACTCTCTTCCAGA Internal: GCACACGCAATTACATCCAC. (B) G-protein-couple receptor gene expression was examined in RNA isolated from combined left and right atria or combined left and right ventricle samples from a single C57BL/6 mouse using real-time PCR. mRNA levels of the indicated genes are expressed relative to cyclophilin mRNA levels. Glp2r, glucagon-like peptide-2 receptor; Glp1r, glucagon-like peptide-1 receptor, Gcgr, glucagon receptor; Gipr, glucose-dependent insulinotropic polypeptide receptor. List of real-time primers (Applied Biosystems Inc.) are as follows: Glp2r-Mm00558835_m1: Glp1r-Mm0135008_m1: Gcgr-Mm00433546_m1: Gipr-Mm01316344_m1; PPIA-Mm02342430_m1. (C) Random morning non-fasted glucose and (D) body weight in mice repeatedly injected with glucagon as per the schedule shown in Figure 1A. (E) HL-1 cells transduced with Adβ-gal were treated with saline or 20 nM glucagon for 3 h following which RNA was isolated for mRNA expression analysis. Data are mean ± S.E.M (n = 5). (F) HL-1 cells were transduced with Adβ-gal for 24 h followed by transfection with a PPAR-α reporter gene-luciferase construct for another 24 h and luciferase expression was assessed. Data are mean ± S.E.M (n = 5). (G) HL-1 cells transduced with Adβ-gal for 24 h followed by a 3 h treatment with 20 nM glucagon. Cell lysates were analyzed by Western blotting for expression of P-PDH and total PDH. Data are mean ± S.E.M (n = 5).
Supplementary Figure 2. Glucagon increases Bax protein expression in H2O2 treated AdGcgr transduced HL-1 cells. (A, B) Bax and Bcl-2 protein expression in HL-1 cells infected with AdGcgr for 24 h followed by 24 h treatment with 100 μM H2O2 and 20 nM glucagon during the final 3 h of H2O2 treatment. Another group was also treated concomitantly with 1.5 mM DCA (PDH activator via inhibition of the PPARα target gene, PDK4) during H2O2 treatment.
Supplementary Figure 3. Krebs cycle intermediates in aerobic and ischemic hearts. (A–E) Levels of Krebs cycle intermediates in aerobic or ischemic hearts harvested 30 min following LAD coronary artery ligation or sham surgery in mice injected with glucagon (30 ng/g) or saline (every 8 h for 24 h) (n = 5 per group).
Supplementary Figure 4. Generation and characterization of GcgrCM−/− mice. (A) GcgrFlox mice were mated with FLPe mice to remove the neomycin resistance cassette [9]. The resulting GcgrFlox mice were then mated with αMHCCre mice to generate GcgrCM−/− mice. Gcgr mRNA expression in GcgrCM−/− and control mice in liver (B), kidney (C) and heart (D). *p < 0.05 GcgrCM−/− vs. littermate control mice. Data are mean ± S.E.M (n = 5–6 per genotype).
Supplementary Figure 5. Body weight and glucose tolerance in GcgrCM−/− and control mice. (A) Body weight was monitored from 7 to 14 weeks of age before and after tamoxifen injections in GcgrCM−/− and littermate control mice. (B) Blood glucose levels during an oral glucose tolerance test in 10- to 14-week-old mice (n = 11–22 mice per group). Plasma insulin levels at 0 and 15 min following oral glucose challenge (n = 4–9 mice per group). (C) Blood glucose levels during an intraperitoneal glucose tolerance test in 10- to 14-week-old mice (n = 11–22 mice per group). Plasma insulin levels at 0 and 15 min following intraperitoneal glucose challenge (n = 4–9 mice per group). Data are mean ± S.E.M.
. Targeted metabolomics reveals reduced fatty acid oxidation in hearts from high fat fed GcgrCM−/− mice. Acylcarnitine levels in aerobic hearts harvested from 5 h fasted αMHCCre and GcgrCM−/− mice that had been maintained on a high fat diet for 6 months (n = 5 per genotype). Values are expressed as percent of αMHCCre mice values.
References
- 1.Unger R.H., Cherrington A.D. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. Journal of Clinical Investigation. 2012;122(1):4–12. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sinclair E.M., Yusta B., Streutker C., Baggio L.L., Koehler J., Charron M.J. Glucagon receptor signaling is essential for control of murine hepatocyte survival. Gastroenterology. 2008;135(6):2096–2106. doi: 10.1053/j.gastro.2008.07.075. [DOI] [PubMed] [Google Scholar]
- 3.Dunphy J.L., Taylor R.G., Fuller P.J. Tissue distribution of rat glucagon receptor and GLP-1 receptor gene expression. Molecular and Cellular Endocrinology. 1998;141(1–2):179–186. doi: 10.1016/s0303-7207(98)00096-3. [DOI] [PubMed] [Google Scholar]
- 4.Campos R.V., Lee Y.C., Drucker D.J. Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology. 1994;134:2156–2164. doi: 10.1210/endo.134.5.8156917. [DOI] [PubMed] [Google Scholar]
- 5.Ali S., Drucker D.J. Benefits and limitations of reducing glucagon action for the treatment of type 2 diabetes. American Journal of Physiology. Endocrinology and Metabolism. 2009;296(3):E415–E421. doi: 10.1152/ajpendo.90887.2008. [DOI] [PubMed] [Google Scholar]
- 6.Muller W.A., Faloona G.R., Aguilar-Parada E., Unger R.H. Abnormal alpha-cell function in diabetes. Response to carbohydrate and protein ingestion. New England Journal of Medicine. 1970;283(3):109–115. doi: 10.1056/NEJM197007162830301. [DOI] [PubMed] [Google Scholar]
- 7.Habegger K.M., Heppner K.M., Geary N., Bartness T.J., DiMarchi R., Tschop M.H. The metabolic actions of glucagon revisited. Nature Reviews Endocrinology. 2010;6(12):689–697. doi: 10.1038/nrendo.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Longuet C., Sinclair E.M., Maida A., Baggio L.L., Maziarz M., Charron M.J. The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metabolism. 2008;8(5):359–371. doi: 10.1016/j.cmet.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Day J.W., Gelfanov V., Smiley D., Carrington P.E., Chicchi G., Erion M.D. Optimization of co-agonism at GLP-1 and glucagon receptors to safely maximize weight reduction in DIO-rodents. Biopolymers. 2012;98(5):443–450. doi: 10.1002/bip.22072. [DOI] [PubMed] [Google Scholar]
- 10.Sadry S.A., Drucker D.J. Emerging combinatorial hormone therapies for the treatment of obesity and T2DM. Nature Reviews Endocrinology. 2013;9(7):425–433. doi: 10.1038/nrendo.2013.47. [DOI] [PubMed] [Google Scholar]
- 11.Dakin C.L., Small C.J., Batterham R.L., Neary N.M., Cohen M.A., Patterson M. Peripheral oxyntomodulin reduces food intake and body weight gain in rats. Endocrinology. 2004;145(6):2687–2695. doi: 10.1210/en.2003-1338. [DOI] [PubMed] [Google Scholar]
- 12.Wynne K., Park A.J., Small C.J., Patterson M., Ellis S.M., Murphy K.G. Subcutaneous oxyntomodulin reduces body weight in overweight and obese subjects: a double-blind, randomized, controlled trial. Diabetes. 2005;54(8):2390–2395. doi: 10.2337/diabetes.54.8.2390. [DOI] [PubMed] [Google Scholar]
- 13.Day J.W., Ottaway N., Patterson J.T., Gelfanov V., Smiley D., Gidda J. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nature Chemical Biology. 2009;5(10):749–757. doi: 10.1038/nchembio.209. [DOI] [PubMed] [Google Scholar]
- 14.Glick G., Parmley W.W., Wechsler A.S., Sonnenblick E.H. Glucagon. Its enhancement of cardiac performance in the cat and dog and persistence of its inotropic action despite beta-receptor blockade with propranolol. Circulation Research. 1968;22(6):789–799. doi: 10.1161/01.res.22.6.789. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Munoz C., Nieto-Ceron S., Cabezas-Herrera J., Hernandez-Cascales J. Glucagon increases contractility in ventricle but not in atrium of the rat heart. European Journal of Pharmacology. 2008;587(1–3):243–247. doi: 10.1016/j.ejphar.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Goodwin G.W., Taegtmeyer H. Metabolic recovery of isolated working rat heart after brief global ischemia. American Journal of Physiology. 1994;267(2 Pt 2):H462–H470. doi: 10.1152/ajpheart.1994.267.2.H462. [DOI] [PubMed] [Google Scholar]
- 17.Maroko P.R., Kjekshus J.K., Sobel B.E., Watanabe T., Covell J.W., Ross J., Jr. Factors influencing infarct size following experimental coronary artery occlusions. Circulation. 1971;43(1):67–82. doi: 10.1161/01.cir.43.1.67. [DOI] [PubMed] [Google Scholar]
- 18.Sohal D.S., Nghiem M., Crackower M.A., Witt S.A., Kimball T.R., Tymitz K.M. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circulation Research. 2001;89(1):20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 19.Longuet C., Robledo A.M., Dean E.D., Dai C., Ali S., McGuinness I. Liver-specific disruption of the murine glucagon receptor produces alpha-cell hyperplasia: evidence for a circulating alpha-cell growth factor. Diabetes. 2013;62(4):1196–1205. doi: 10.2337/db11-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ussher J.R., Baggio L.L., Campbell J.E., Mulvihill E.E., Kim M., Kabir M.G. Inactivation of the cardiomyocyte Glucagon-Like Peptide-1 Receptor (GLP-1R) unmasks cardiomyocyte-independent GLP-1R-mediated cardioprotection. Molecular Metabolism. 2014;3(5):507–517. doi: 10.1016/j.molmet.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koitabashi N., Bedja D., Zaiman A.L., Pinto Y.M., Zhang M., Gabrielson K.L. Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models. Circulation Research. 2009;105(1):12–15. doi: 10.1161/CIRCRESAHA.109.198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim M., Platt M., Shibasaki T., Quaggin S., Backx P.H., Seino S. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nature Medicine. 2013;19(5):567–575. doi: 10.1038/nm.3128. [DOI] [PubMed] [Google Scholar]
- 23.Ban K., Noyan-Ashraf M.H., Hoefer J., Bolz S.S., Drucker D.J., Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation. 2008;117(18):2340–2350. doi: 10.1161/CIRCULATIONAHA.107.739938. [DOI] [PubMed] [Google Scholar]
- 24.Koves T.R., Ussher J.R., Noland R.C., Slentz D., Mosedale M., Ilkayeva O. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metabolism. 2008;7(1):45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 25.Ussher J.R., Koves T.R., Jaswal J.S., Zhang L., Ilkayeva O., Dyck J.R. Insulin-stimulated cardiac glucose oxidation is increased in high-fat diet-induced obese mice lacking malonyl CoA decarboxylase. Diabetes. 2009;58(8):1766–1775. doi: 10.2337/db09-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali S., Lamont B.J., Charron M.J., Drucker D.J. Dual elimination of the glucagon and GLP-1 receptors in mice reveals plasticity in the incretin axis. Journal of Clinical Investigation. 2011;121(5):1917–1929. doi: 10.1172/JCI43615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koehler J.A., Drucker D.J. Activation of GLP-1 receptor signaling does not modify the growth or apoptosis of human pancreatic cancer cells. Diabetes. 2006;55:1369–1379. doi: 10.2337/db05-1145. [DOI] [PubMed] [Google Scholar]
- 28.Yusta B., Holland D., Koehler J.A., Maziarz M., Estall J.L., Higgins R. ErbB signaling is required for the proliferative actions of GLP-2 in the murine gut. Gastroenterology. 2009;173(3):986–996. doi: 10.1053/j.gastro.2009.05.057. [DOI] [PubMed] [Google Scholar]
- 29.Ban K., Kim H., Cho J., Diamandis E., Backx P.H., Drucker D.J. GLP-1(9-36) protects cardiomyocytes and endothelial cells from ischemia-reperfusion injury via cytoprotective pathways independent of the GLP-1 receptor. Endocrinology. 2010;151(4):1520–1531. doi: 10.1210/en.2009-1197. [DOI] [PubMed] [Google Scholar]
- 30.Panjwani N., Mulvihill E.E., Longuet C., Yusta B., Campbell J.E., Brown T.J. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE-/- mice. Endocrinology. 2013;154(1):127–139. doi: 10.1210/en.2012-1937. [DOI] [PubMed] [Google Scholar]
- 31.Pyke C., Heller R.S., Kirk R.K., Orskov C., Reedtz-Runge S., Kaastrup P. GLP-1 receptor localization in monkey and human tissue; novel distribution revealed with extensively validated monoclonal antibody. Endocrinology. 2014;155(4):1280–1290. doi: 10.1210/en.2013-1934. [DOI] [PubMed] [Google Scholar]
- 32.Moore-Morris T., Varrault A., Mangoni M.E., Le Digarcher A., Negre V., Dantec C. Identification of potential pharmacological targets by analysis of the comprehensive G protein-coupled receptor repertoire in the four cardiac chambers. Molecular Pharmacology. 2009;75(5):1108–1116. doi: 10.1124/mol.108.054155. [DOI] [PubMed] [Google Scholar]
- 33.Gelling R.W., Du X.Q., Dichmann D.S., Romer J., Huang H., Cui L. Lower blood glucose, hyperglucagonemia, and pancreatic {alpha} cell hyperplasia in glucagon receptor knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1438–1443. doi: 10.1073/pnas.0237106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J., MacDougall M.L., McDowell M.T., Xi L., Wei R., Zavadoski W.J. Polyomic profiling reveals significant hepatic metabolic alterations in glucagon-receptor (GCGR) knockout mice: implications on anti-glucagon therapies for diabetes. BMC Genomics. 2011;12:281. doi: 10.1186/1471-2164-12-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Omar B.A., Andersen B., Hald J., Raun K., Nishimura E., Ahren B. Fibroblast growth factor 21 (FGF21) and glucagon-like peptide 1 contribute to diabetes resistance in glucagon receptor-deficient mice. Diabetes. 2014;63(1):101–110. doi: 10.2337/db13-0710. [DOI] [PubMed] [Google Scholar]
- 36.Ussher J.R., Drucker D.J. Cardiovascular actions of incretin-based therapies. Circulation Research. 2014;114(11):1788–1803. doi: 10.1161/CIRCRESAHA.114.301958. [DOI] [PubMed] [Google Scholar]
- 37.Planavila A., Redondo I., Hondares E., Vinciguerra M., Munts C., Iglesias R. Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nature Communications. 2013;420:19. doi: 10.1038/ncomms3019. [DOI] [PubMed] [Google Scholar]
- 38.Chang-Chretien K., Chew J.T., Judge D.P. Reversible dilated cardiomyopathy associated with glucagonoma. Heart. 2004;90(7):e44. doi: 10.1136/hrt.2004.036905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puri P.S., Bing R.J. Effects of glucagon on myocardial contractility and hemodynamics in acute experimental myocardial infarction. Basis for its possible use in cardiogenic shock. American Heart Journal. 1969;78(5):660–668. doi: 10.1016/0002-8703(69)90518-3. [DOI] [PubMed] [Google Scholar]
- 40.Eddy J.D., O'Brien E.T., Singh S.P. Glucagon and haemodynamics of acute myocardial infarction. British Medical Journal. 1969;4(5684):663–665. doi: 10.1136/bmj.4.5684.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar R., Molokhia F.A., Norman J.C., Inamdar A.N., Messer J.V., Abelmann W.H. Experimental myocardial infarction. X. Efficacy of glucagon in acute and healing phase in intact conscious dogs: effects on hemodynamics and myocardial oxygen consumption. Circulation. 1972;45(1):55–64. doi: 10.1161/01.cir.45.1.55. [DOI] [PubMed] [Google Scholar]
- 42.Lai L., Leone T.C., Keller M.P., Martin O.J., Broman A.T., Nigro J. Energy metabolic re-programming in the hypertrophied and early stage failing heart: a multi-systems approach. Circulation Heart Failure. 2014 Nov;7(6):1022–1031. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muoio D.M., Noland R.C., Kovalik J.P., Seiler S.E., Davies M.N., DeBalsi K.L. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metabolism. 2012;15(5):764–777. doi: 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Campbell F.M., Kozak R., Wagner A., Altarejos J.Y., Dyck J.R., Belke D.D. A role for peroxisome proliferator-activated receptor alpha (PPARalpha ) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. Journal of Biological Chemistry. 2002;277(6):4098–4103. doi: 10.1074/jbc.M106054200. [DOI] [PubMed] [Google Scholar]
- 45.Finck B.N., Lehman J.J., Leone T.C., Welch M.J., Bennett M.J., Kovacs A. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. Journal of Clinical Investigation. 2002;109(1):121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sambandam N., Morabito D., Wagg C., Finck B.N., Kelly D.P., Lopaschuk G.D. Chronic activation of PPARalpha is detrimental to cardiac recovery after ischemia. American Journal of Physiology. Heart and Circulatory Physiology. 2006;290(1):H87–H95. doi: 10.1152/ajpheart.00285.2005. [DOI] [PubMed] [Google Scholar]
- 47.Matsushima S., Kuroda J., Ago T., Zhai P., Ikeda Y., Oka S. Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1alpha and upregulation of peroxisome proliferator-activated receptor-alpha. Circulation Research. 2013;112(8):1135–1149. doi: 10.1161/CIRCRESAHA.111.300171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu R., Chang H.C., Khechaduri A., Chawla K., Tran M., Chai X. Cardiac-specific ablation of ARNT leads to lipotoxicity and cardiomyopathy. Journal of Clinical Investigation. 2014;124(11):4795–4806. doi: 10.1172/JCI76737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finck B.N., Han X., Courtois M., Aimond F., Nerbonne J.M., Kovacs A. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(3):1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yue T.L., Bao W., Jucker B.M., Gu J.L., Romanic A.M., Brown P.J. Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation. 2003;108(19):2393–2399. doi: 10.1161/01.CIR.0000093187.42015.6C. [DOI] [PubMed] [Google Scholar]
- 51.Aasum E., Khalid A.M., Gudbrandsen O.A., How O.J., Berge R.K., Larsen T.S. Fenofibrate modulates cardiac and hepatic metabolism and increases ischemic tolerance in diet-induced obese mice. Journal of Molecular and Cellular Cardiology. 2008;44(1):201–209. doi: 10.1016/j.yjmcc.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 52.Omar M.A., Verma S., Clanachan A.S. Adenosine-mediated inhibition of 5'-AMP-activated protein kinase and p38 mitogen-activated protein kinase during reperfusion enhances recovery of left ventricular mechanical function. Journal of Molecular and Cellular Cardiology. 2012;52(6):1308–1318. doi: 10.1016/j.yjmcc.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 53.Fiedler B., Feil R., Hofmann F., Willenbockel C., Drexler H., Smolenski A. cGMP-dependent protein kinase type I inhibits TAB1-p38 mitogen-activated protein kinase apoptosis signaling in cardiac myocytes. Journal of Biological Chemistry. 2006;281(43):32831–32840. doi: 10.1074/jbc.M603416200. [DOI] [PubMed] [Google Scholar]
- 54.Sassi Y., Ahles A., Truong D.J., Baqi Y., Lee S.Y., Husse B. Cardiac myocyte-secreted cAMP exerts paracrine action via adenosine receptor activation. Journal of Clinical Investigation. 2014 Dec 1;124(12):5385–5397. doi: 10.1172/JCI74349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tschop M.H., DiMarchi R.D. Outstanding Scientific Achievement Award Lecture 2011: defeating diabesity: the case for personalized combinatorial therapies. Diabetes. 2012;61(6):1309–1314. doi: 10.2337/db12-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Axelsen L.N., Keung W., Pedersen H.D., Meier E., Riber D., Kjolbye A.L. Glucagon and a glucagon-GLP-1 dual-agonist increases cardiac performance with different metabolic effects in insulin-resistant hearts. British Journal of Pharmacology. 2012;165(8):2736–2748. doi: 10.1111/j.1476-5381.2011.01714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Russell S.J., El-Khatib F.H., Sinha M., Magyar K.L., McKeon K., Goergen L.G. Outpatient glycemic control with a bionic pancreas in type 1 diabetes. New England Journal of Medicine. 2014;371(4):313–325. doi: 10.1056/NEJMoa1314474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
. Targeted metabolomics reveals reduced fatty acid oxidation in hearts from high fat fed GcgrCM−/− mice. Acylcarnitine levels in aerobic hearts harvested from 5 h fasted αMHCCre and GcgrCM−/− mice that had been maintained on a high fat diet for 6 months (n = 5 per genotype). Values are expressed as percent of αMHCCre mice values.