

Abstract
Objective
Elevated serum levels of the lymphangiogenic factors VEGF-C and -D have been observed in obese individuals but their relevance for the metabolic syndrome has remained unknown.
Methods
K14–VEGFR-3–Ig (sR3) mice that constitutively express soluble-VEGFR-3–Ig in the skin, scavenging VEGF-C and -D, and wildtype (WT) mice were fed either chow or high-fat diet for 20 weeks. To assess the effect of VEGFR-3 blockage on adipose tissue growth and insulin sensitivity, we evaluated weight gain, adipocyte size and hepatic lipid accumulation. These results were complemented with insulin tolerance tests, FACS analysis of adipose tissue macrophages, in vitro 3T3-L1 differentiation assays and in vivo blocking antibody treatment experiments.
Results
We show here that sR3 mice are protected from obesity-induced insulin resistance and hepatic lipid accumulation. This protection is associated with enhanced subcutaneous adipose tissue hyperplasia and an increased number of alternatively-activated (M2) macrophages in adipose tissue. We also show that VEGF-C and -D are chemotactic for murine macrophages and that this effect is mediated by VEGFR-3, which is upregulated on M1 polarized macrophages. Systemic antibody blockage of VEGFR-3 in db/db mice reduces adipose tissue macrophage infiltration and hepatic lipid accumulation, and improves insulin sensitivity.
Conclusions
These results reveal an unanticipated role of the lymphangiogenic factors VEGF-C and -D in the mediation of metabolic syndrome-associated adipose tissue inflammation. Blockage of these lymphangiogenic factors might constitute a new therapeutic strategy for the prevention of obesity-associated insulin resistance.
Keywords: Vegfc, Obesity, Adipose tissue, Inflammation, Macrophages
Abbreviations: sR3, soluble-VEGFR-3; HFD, high-fat diet; SWAT, subcutaneous white adipose tissue; EWAT, epididymal white adipose tissue; CM, conditioned medium
1. Introduction
Obesity is a growing public health problem and its prevalence has reached epidemic proportions in recent decades [1]. Obesity and associated metabolic syndrome are accompanied by heightened levels of proinflammatory mediators, not only systemically but also locally in metabolically critical tissues such as adipose tissue, liver and skeletal muscle [2]. A plethora of studies has shown that an increase in proinflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-6 and macrophage chemotactic protein (MCP)-1 results in adipose tissue insulin resistance [3–6], adipocyte lipolysis [7] and hepatic steatosis [6], eventually leading to systemic insulin resistance (reviewed in Ref. [8]). Major changes of immune cell populations – especially of macrophages – within adipose tissue play a key role in propagating obesity-induced adipose tissue inflammation [9–11]. Macrophage polarization is generally classified as M1 (proinflammatory) or M2 (anti-inflammatory) phenotype, mirroring the Th1–Th2 lymphocyte subsets [12–14]. In obesity, increased numbers of macrophages have been found in adipose tissue, with a shift towards the M1-polarized state that elicits effector functions by the production of a surplus of proinflammatory cytokines and promotes adipocyte insulin resistance [15]. While the increase in macrophage numbers in obesity has been largely attributed to an enhanced expression of MCP-1 by the hypertrophied adipocytes [16] or lipid sensing of macrophages via CD36 [17], the contribution of other adipose-derived factors in macrophage recruitment is yet to be determined.
Elevated serum levels of VEGF-A, -C [18] and -D [19] have been reported in overweight and in obese subjects, and serum VEGF-C levels appear to be more closely correlated with metabolic and lipid parameters than serum VEGF-A levels in obese patients [20]. VEGF-C and -D are potent activators of lymphatic vessel growth (lymphangiogenesis) via VEGFR-3 signaling [21,22], and impaired lymphatic vessel function is associated with increased adipocyte accumulation [23], whereas lymphatic vessel function is reduced in obese individuals [24] as well as in obese mice [25]. Previously, VEGF-A has been implicated in the pathogenesis of metabolic syndrome since overexpression of VEGF-A in adipose tissue promotes accumulation of an M2-polarized macrophage population and associates with improved insulin sensitivity [26,27]. While no major adipose tissue phenotype was found in VEGF-D deficient mice [28], the biological role of the VEGF-C/VEGF-D/VEGFR-3 axis in obesity has remained largely unknown.
To investigate the potential role of VEGF-C and -D in diet-induced obesity, we subjected K14–sVEGFR-3–Ig transgenic mice (sR3 mice), which express the extracellular domain of human VEGFR-3, fused to the Fc domain of the human Ig gamma chain, under the control of the keratin 14 promoter [29], to high-fat diet (HFD). The sR3 protein scavenges endogenous VEGF-C and -D, resulting in a lack of dermal lymphatic vessels and in lymphedema [29]. We also treated db/db mice with a VEGFR-3 blocking antibody during the development of insulin resistance. Our results identify a key role for the VEGF-C/VEGF-D–VEGFR-3 pathway in the development of adipose tissue insulin resistance by contributing to the recruitment of inflammatory macrophages. Therefore, the blockade of VEGF-C and -D might represent a new therapeutic approach to alleviate diet-induced insulin resistance.
2. Results
2.1. Comparable weight gain and lipid uptake in sR3 and WT mice
In sR3 mice, the sR3 protein secreted by epidermal keratinocytes captures the lymphangiogenic growth factors VEGF-C and -D, thereby inhibiting VEGFR-3 signaling [29]. We found that sR3 mice had a lack of podoplanin-positive lymphatic vessels in the tail and back skin, confirming the transgene activity (Figure 1A and data not shown). The sR3 protein was also detectable by ELISA in the subcutaneous white adipose tissue (SWAT), which is in close proximity to the skin where the transgene is produced (Figure S1). In contrast, it was not detectable in the epididymal white adipose tissue (EWAT) or in the liver of sR3 transgenic mice (Figure S1). An X-gal staining performed on mice expressing the LacZ enzyme under control of the VEGFR-3 promoter (Flt4CreErt2; Rosa26LacZ double knock-in mice [30]) revealed that there were VEGFR-3 positive lymphatic vessels in tissues such as the mesentery and the intestine (Figure S2A and B), but there were no VEGFR-3 expressing cells in the adipose tissue except for collecting lymphatic vessels and lymph nodes (Figure S2C).
Figure 1.
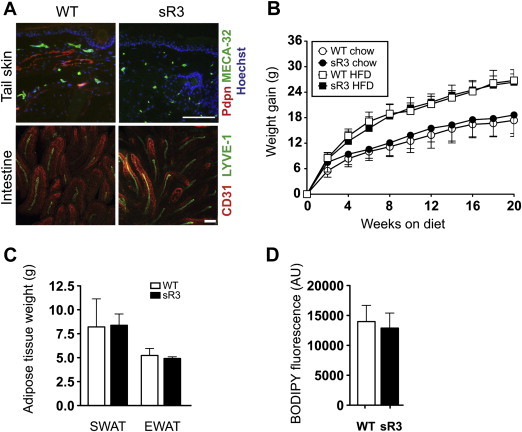
Comparable weight gain, fat distribution and intestinal lipid uptake in sR3 mice. (A) Compared to WT mice, sR3 mice lack podoplanin positive dermal lymphatic vessels in the tail. Whole-mount imaging of intestinal villi of WT and sR3 mice revealed no differences in lacteal or blood vessel structure. Scale bars = 100 μm. (B) sVEGFR-3–Ig expression does not influence weight gain or adipose tissue distribution during chow diet or HFD (n = 8–13, data analyzed with repeated measures ANOVA; genotype effect on weight gain P = 0.30). (C) CT scan analyses and further calculation showed comparable adipose tissue weights in mice on HFD (n = 5–6). (D) Determination of BODIPY fluorescence in the serum 1 h after oral ingestion revealed similar lipid uptake in WT and sR3 mice (n = 3–4, two-tailed Student's t-test). All data are mean ± SD.
To investigate if the neutralization of VEGF-C and -D might affect diet-induced obesity, four-week-old male sR3 mice and their wildtype (WT) littermates on the FVB background were fed with either chow or high-fat diet (HFD) for 20 weeks. WT and sR3 mice gained a similar amount of weight under HFD (WT: 26.8 ± 2.5 g, n = 8; sR3: 26.4 ± 1.9 g, n = 13; P = 0.708). Similarly, the weight gain under chow diet was comparable for both genotypes (WT: 17.4 ± 3.8 g, n = 9; sR3: 18.4 ± 4.3 g, n = 13; P = 0.477; Figure 1B). Computed-tomography scan analyses and further calculation showed comparable weights of SWAT (WT: 8.2 ± 1.3 g, n = 5; sR3: 8.3 ± 0.6 g, n = 6; P = 0.92) and of EWAT (WT: 5.2 ± 0.3 g; sR3: 4.9 ± 0.1 g; P = 0.42) between WT and sR3 mice (Figure 1C). Since the sR3 protein is detectable in the systemic circulation of sR3 mice [29,31], we next investigated whether intestinal lipid uptake by lacteal lymphatic vessels might be impaired. Whole-mount stainings of intestines revealed normal lacteal vessels in sR3 mice (Figure 1A). Moreover, similar amounts of the lipophilic dye BODIPY were found in sera of sR3 and WT mice at 1 h after intra-gastric administration (WT: 14,000 ± 1347 arbitrary units (AU), n = 4; sR3: 12,920 ± 1442 AU, n = 3; P = 0.61; Figure 1D), indicating that the circulating sR3 did not interfere with the lipid absorption of intestinal lacteal vessels.
2.2. sR3 mice on HFD have smaller SWAT adipocytes
Since insulin resistance is usually associated with hypertrophic obesity, we investigated potential systemic effects of the sR3 protein and quantified the size of adipocytes under HFD and chow diet. We found that the SWAT, but not the EWAT adipocytes were markedly smaller in sR3 mice than in WT mice under chow diet (Figure 2A and B). After 20 weeks of HFD, the adipocyte size was significantly increased in both WT and sR3 mice but the SWAT adipocytes were still smaller in sR3 mice than in WT mice (Figure 2A). The size distribution of SWAT adipocytes was then plotted in a cumulative frequency graph (Figure 2C). There was a significant leftward shift in the cumulative frequency of adipocyte sizes in sR3 mice under both diets (chow WT n = 5, sR3 n = 10; P < 0.001; HFD WT n = 5, sR3 n = 9; P < 0.001; two-sample Kolmogorov–Smirnov test).
Figure 2.
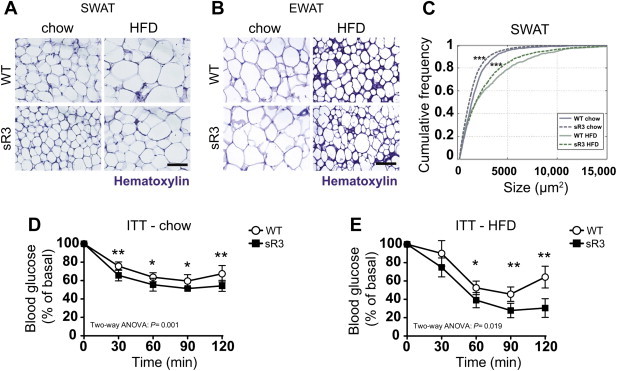
sR3 mice have smaller subcutaneous adipocytes and are more insulin sensitive. (A) Compared to WT controls sR3 mice have smaller SWAT adipocytes under chow diet and HFD. (B) EWAT adipocyte size is not altered. Scale bars = 100 μm. (C) Cumulative distribution plot of SWAT adipocyte sizes between the groups shows a leftward shift in sR3 mice, both on chow and on HFD (***P < 0.001; two-sample Kolmogorov–Smirnov test). (D, E) Insulin tolerance test (ITT) showing increased insulin sensitivity of sR3 mice under chow (n = 7 each) and HFD (n = 5 each) in response to insulin stimulation. Blood glucose levels of sR3 mice and WT controls before i.p. administration of insulin were set to 100% (curves are compared using a two-way ANOVA, effect of genotype is shown; *P < 0.05 and **P < 0.01 compared to WT control at the same time point with two-tailed Student's t-test). All data are mean ± SD.
2.3. Enhanced insulin sensitivity of sR3 mice
Since adipocyte size is inversely correlated with insulin sensitivity [32], we next investigated if sR3 mice have enhanced insulin sensitivity. We found that fasting insulin levels were significantly lower in sR3 mice than in their WT littermates under chow diet (Table S1). Fasting insulin levels were also lower in the sR3 mice than in WT mice after HFD. The fasting glucose levels were similar in both genotypes (Table S1). The homeostatic model assessment of insulin resistance (HOMA-IR) index is frequently used as an indicator of insulin resistance, and metabolic syndrome development is associated with higher HOMA-IR values [33]. We found lower HOMA-IR indices in sR3 mice than in WT mice under both chow and HFD (Table S1). Insulin tolerance tests revealed a significant improvement in the response to insulin after 24 weeks of chow or HFD in sR3 mice compared to WT mice (Figure 2D and E). Fasting–refeeding experiments showed enhanced clearance of FFAs from the serum of sR3 mice under chow diet, indicating better functionality of the adipocytes in these mice (Table S1).
2.4. sR3 mice have reduced hepatic lipid accumulation under HFD
We next investigated ectopic lipid accumulation in the liver. The livers of WT mice were enlarged and appeared yellow after 20 weeks of HFD, whereas the livers of sR3 mice seemed to be protected from hepatic steatosis and looked markedly different (Figure 3A). Oil Red O stainings of frozen liver sections confirmed reduced lipid accumulation in sR3 livers, especially under HFD, as compared to WT mice (Figure 3B). Consistent with this finding, the liver triglyceride content was significantly reduced in sR3 mice under HFD (WT: 307.4 ± 6.8, n = 8; sR3: 223.4 ± 7.7 mg triglyceride/g liver; n = 13; P < 0.001; Figure 3C), while the liver triglyceride content was only slightly reduced in chow fed sR3 mice (WT: 43.1 ± 3.5, n = 8; sR3: 32.3 ± 4.3 mg triglyceride/g liver, n = 13; P = 0.098). The sinusoidal and vascular morphology of the livers appeared similar in WT and sR3 mice, as well as the coverage by blood vessels, determined by measurement of the CD31 positive area (Figure 3D and E). Consistent with the lipid accumulation phenotype, WT mice under HFD showed a significant reduction of AKT phosphorylation in the liver upon insulin stimulation (Figure 3F and G), whereas the livers of sR3 mice under HFD maintained high levels of AKT phosphorylation upon insulin stimulation, indicating that they were protected from steatosis-induced insulin resistance (Figure 3F and G).
Figure 3.
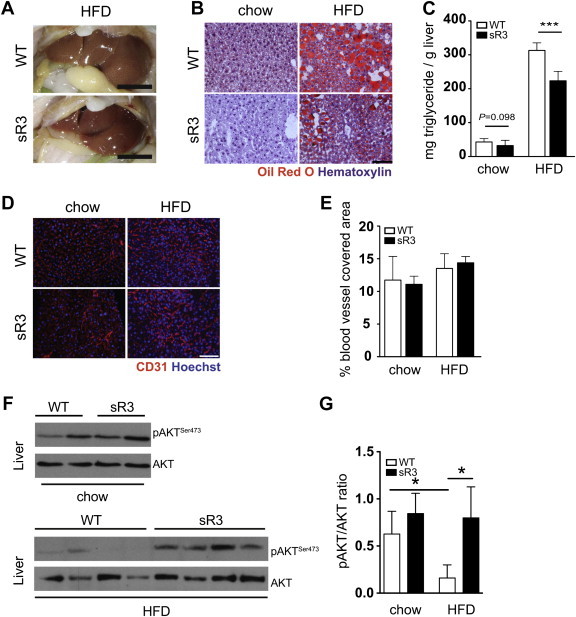
sR3 mice exhibit reduced ectopic lipid accumulation after HFD. (A) Representative appearance of steatotic livers of WT and sR3 mice after 20 weeks on HFD, note the reduction in liver steatosis in the sR3 mice. Scale bars = 1 cm. (B) Oil red O staining of liver sections confirmed a reduction in lipid accumulation in the livers of sR3 mice under both chow and HFD for 20 weeks, compared to WT mice. Scale bar = 100 μm. (C) Triglyceride content measurements of the livers showed a slight reduction in the livers of sR3 mice on chow (P = 0.098), and a highly significant reduction under HFD (n = 8–13, two-tailed Student's t-test, ***P < 0.001). (D) Immunofluorescence stains for CD31 (red) and (E) subsequent image analysis showed similar coverage of liver sections with blood vessels and unaltered sinusoidal morphology. Scale bar = 100 μm. (F) Representative western blot analysis and (G) quantification of insulin-stimulated AKT phosphorylation of liver samples, showing increased AKT phosphorylation in the sR3 mice as compared to WT controls under HFD. Phospho-AKT levels were normalized to respective total AKT (n = 4–6, two-tailed Student's t-test, *P < 0.05). All data are mean ± SD.
2.5. sR3 mice on the C57BL/6 background show a similar phenotype
To investigate whether the genetic background might affect the phenotype, we next crossed sR3 mice into the C57BL/6 background (sR3 B6 mice). After 10 weeks of HFD, the sR3 B6 mice had a similar body weight compared to their WT littermates (Figure S3A) but had lower fasting glucose (WT: 11.3 ± 1.5; sR3 B6: 7.4 ± 1.6 mmol/L, n = 3–4 per group; P = 0.034) and fasting insulin (WT: 2.4 ± 0.7; sR3 B6: 1.4 ± 0.4 ng/mL, n = 3–4 per group; P = 0.052) levels, resulting in significantly lower HOMA-IR values (WT: 32.6 ± 12.6; sR3 B6: 13.9 ± 2.1, n = 3–4 per group; P = 0.030; Figure S3B). The sR3 B6 mice had smaller adipocytes in both SWAT and EWAT (median adipocyte size SWAT, WT: 3,642 ± 511.2; sR3 B6: 2,025 ± 332.8 μm2, n = 3–4 per group; P = 0.004; EWAT WT: 7,001 ± 1,164; sR3 B6: 5,179 ± 557.5 μm2, n = 3–4 per group; P = 0.038; Figure S3C). This was reflected by a significant leftward shift in the cumulative frequency of SWAT and EWAT adipocyte sizes in sR3 mice (SWAT: P < 0.001; EWAT: P = 0.002; two-sample Kolmogorov–Smirnov test; Figure S3C). The sR3 B6 mice also had reduced hepatic lipid accumulation (WT: 214.5 ± 36.1; sR3: 156.9 ± 19.7 mg triglyceride/g liver; n = 3–4 per group; P = 0.041; Figure S3D). SWAT macrophages of sR3 B6 mice under HFD showed an approximately 50% reduced expression of CD11c (P = 0.049) and Tnf (P = 0.027), indicating a less inflammatory phenotype (Figure S3E).
2.6. Increased M2/M1 macrophage ratio in the SWAT of sR3 mice on HFD
Recent studies have reported an altered balance of M1 and M2 macrophage subsets in obese adipose tissue [15,34], with a shift towards M1-polarization of macrophages. Since sR3 mice showed enhanced insulin sensitivity under both chow and HFD, we next investigated adipose tissue macrophages and their polarization status. We analyzed the CD11b+/F4/80+ populations in the adipose stromal-vascular fraction for expression levels (mean fluorescence intensity, MFI) of MHC class II antigen (M1 marker) and CD206 (M2 marker) by FACS analyses (Figure 4A and Figure S4A). We found that sR3 mice on HFD had a higher percentage of CD11b+/F4/80+ macrophages in the stromal-vascular fraction of the SWAT than WT mice (WT: 8.6 ± 1.6%, n = 3; sR3: 24.1 ± 6.0%, n = 7; P = 0.003; Figure 4B) with an increased M2-subset in this population (Figure 4B). Importantly, sR3 mice had significantly increased CD206/MHC II ratios in the SWAT after 20 weeks of chow (WT: 3.3 ± 1.8, n = 3; sR3: 10.7 ± 4.2, n = 3; P = 0.049; Figure 4B) and HFD (WT: 1.3 ± 0.6, n = 3; sR3: 6.7 ± 4.5, n = 7; P = 0.007; Figure 4B), indicating a less inflammatory microenvironment. In the EWAT, this difference was more subtle (Figure S4B). We also found that expression of CD68 (a common macrophage marker) in total SWAT was comparably increased under HFD in sR3 mice and WT mice, whereas expression of CD11c (an M1 marker) was significantly lower in sR3 mice than in WT mice under HFD, further supporting a shift of macrophage polarization towards M2 in the sR3 mice (Figure S5).
Figure 4.
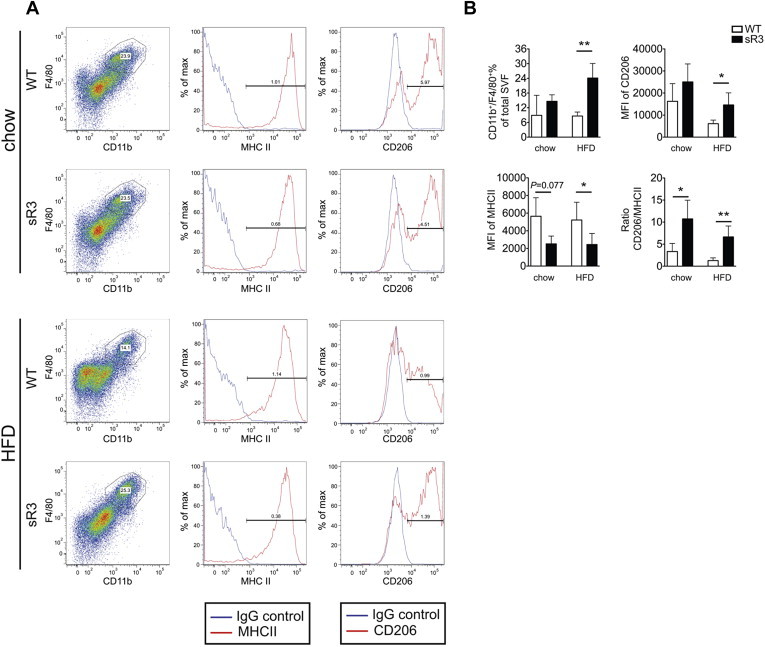
sR3 mice have an increased ratio of M2/M1 polarized macrophages in subcutaneous adipose tissue. (A) Representative FACS plots. (B) FACS analyses showed that sR3 mice had an increase in CD11b+/F4/80+ macrophages, as well as higher ratios of M2/M1 polarized macrophages. This difference was due to decreased expression of MHCII and increased expression of CD206 in the CD11b+/F4/80+ macrophages within the stromal-vascular fraction (n = 3–7, two-tailed Student's t-test, *P < 0.05, **P < 0.01). All data are mean ± SD.
2.7. CM of sR3 SWAT macrophages promote adipogenic differentiation
It has been previously found that adipocytes of lean mice secrete Th2 cytokines to maintain an M2-promoting milieu [35], whereas diet-induced obesity and hypertrophy of adipocytes lead to adipose tissue inflammation and an M1 conversion of resident macrophages [15]. M1 type macrophages produce proinflammatory factors that diminish insulin sensitivity and inhibit adipocyte differentiation [36–38]. We next studied if macrophages from WT and sR3 adipose tissue have different effects on the adipogenic differentiation of 3T3-L1 cells, a widely used model of adipocyte differentiation [39]. CD11b+ cells, which represent a mixture of M1 and M2 macrophages, were isolated from the SWAT of chow fed sR3 mice and WT mice by magnetic-activated cell sorting (MACS). We incubated 3T3-L1 cells with 10% conditioned media (CM) collected from cultures of these cells (after 24 h) and assessed adipogenic differentiation after 10 days. CM from WT macrophages reduced the adipogenic differentiation of 3T3-L1 cells significantly, compared to the effects of CM from sR3 macrophages (Figure 5A and B). The inhibitory effect was stronger when higher macrophage numbers were used (Figure 5A and B).
Figure 5.
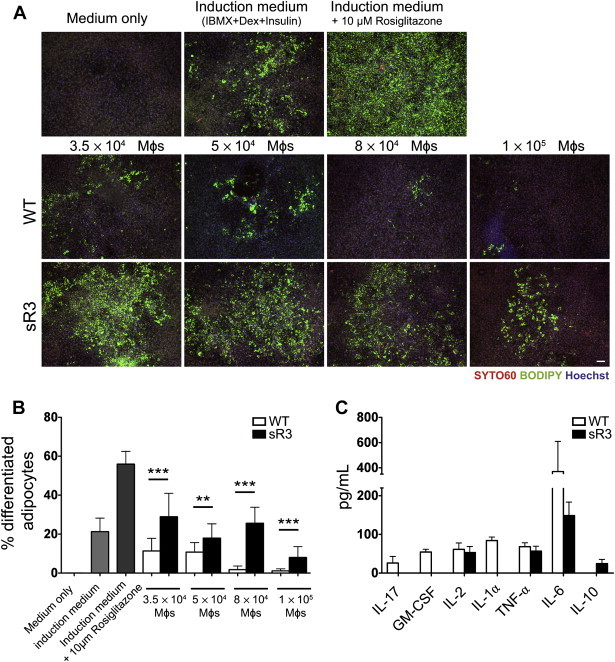
Conditioned media (CM) from WT, but not sR3 SWAT macrophages inhibit adipogenic differentiation of 3T3-L1 cells. (A) Images of terminally differentiated 3T3-L1 adipocytes stained with BODIPY 493/503 (green, lipid droplets), Syto60 (red, cytosol) and Hoechst (blue, nuclei, scale bar = 100 μm). (B) Quantification showed a significant decrease in adipogenic differentiation of the adipocytes treated with CM from WT macrophages, compared to adipocytes treated with sR3 macrophage CM (n = 4 each, two-tailed Student's t-test, **P < 0.01, ***P < 0.001). (C) Decreased (IL-6) or absent (IL-17, GM-CSF, IL-1α) proinflammatory cytokines in CM of sR3 SWAT macrophages. IL-10 (a Th2 cytokine) was only detectable in sR3 macrophage CM (n = 4 each). All data are mean ± SD.
These data suggested that macrophages from sR3 adipose tissue might secrete less proinflammatory (Th1) cytokines that inhibit adipocyte differentiation. Indeed, cytokine analyses revealed that CM from sR3 macrophages lacked several proinflammatory cytokines (GM-CSF, IL-1α and IL-17) that were detected in CM from WT macrophages, and that they tended to have lower levels of IL-6 (Figure 5C). Conversely, the anti-inflammatory cytokine IL-10 was detectable only in the CM of sR3 macrophages (Figure 5C). These results are consistent with our findings that macrophages in the SWAT of WT mice were more of the M1-type, and they indicate that these macrophages might contribute to insulin resistance by reducing the differentiation of preadipocytes into mature adipocytes.
2.8. VEGF-C and -D promote chemotactic migration of murine macrophages
Since sR3 protein neutralizes VEGF-C and -D, we next investigated if VEGF-C and -D might exert direct effects on macrophage recruitment and/or polarization. In WT mice, gene expression of both Vegfc and Vegfd was significantly upregulated in subcutaneous adipocytes after 20 weeks of HFD, as compared to chow diet (relative gene expression normalized to levels in chow: Vegfc: chow 1.0 ± 0.4, HFD 3.9 ± 1.3; n = 4; P < 0.001; Vegfd: chow 1.0 ± 0.8, HFD 3.8 ± 1.4; n = 4; P < 0.001; Figure 6A). Analyses of Vegfc and Vegfd expression, together with CD11b, in total SWAT and EWAT of lean (C57BL/6) and obese (db/db) mice revealed that CD11b, Vegfc and Vegfd were also increased in EWAT in obesity (Figure S6). To investigate if VEGF-C or -D might have direct effects on murine macrophage polarization, CD11b+ peritoneal macrophages were isolated by MACS and incubated with IFN-γ for M1 polarization, with a combination of IL-4/IL-10/IL-13/M-CSF for M2 polarization, or with VEGF-C or -D. While IFN-γ upregulated the expression of MHC class II (an M1 marker) and induced a “fried-egg” like cell morphology in agreement with classical activation, the combination of IL-4/IL-10/IL-13/M-CSF increased the expression of CD206, an M2 marker, and induced an elongated cellular morphology (alternative activation) in these macrophages. None of the two cellular polarization features were observed after incubation with VEGF-C or -D (Data not shown). Both VEGF-C and -D, however, significantly induced chemotactic migration of macrophages in a transwell migration assay (BSA control: 100 ± 20.1%; VEGF-C: 173.5 ± 32.8%, P = 0.014; VEGF-D: 229.8 ± 50.9%, P = 0.003, n = 4 per group; Figure 6B).
Figure 6.
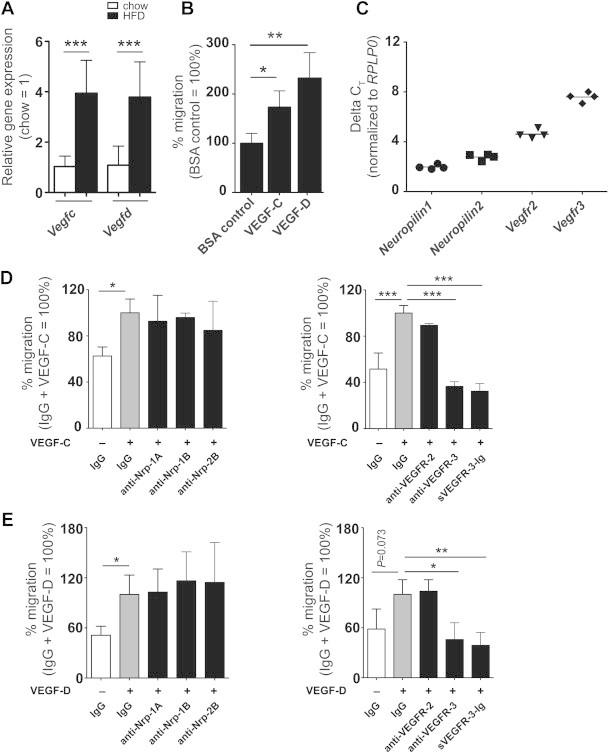
VEGF-C and VEGF-D are chemotactic for macrophages. (A) The mRNA expression of Vegfc and Vegfd was increased in the adipocytes of WT mice after 20 weeks on HFD (n = 4 each). (B) Increased migration of peritoneal macrophages towards VEGF-C and VEGF-D (200 ng/mL, n = 3 mice per group, representative results from three independent experiments). (C) Real-time PCR demonstrated expression of VEGF-C/-D receptors and co-receptors in adipose tissue macrophages (average Ct value for Rplp0 is 20.1). (D, E) Blockage of neuropilin-1 or -2 did not influence macrophage migration towards VEGF-C or VEGF-D. Blockage of VEGFR-2 had no effect on macrophage migration towards VEGF-C and VEGF-D but migration was strongly inhibited by an anti-VEGFR-3 antibody and by sVEGFR-3–Ig (sR3 protein). *P < 0.05, **P < 0.01, ***P < 0.001. All data are mean ± SD.
2.9. Macrophage migration towards VEGF-C and -D is mediated by VEGFR-3
Since both VEGF-C and -D promoted macrophage migration, we next explored whether adipose tissue macrophages express the receptors that bind VEGF-C and -D, namely VEGFR-2, VEGFR-3 and their co-receptors neuropilin-1 and neuropilin-2. CD11b+ adipose tissue macrophages were isolated from WT mice by MACS and gene expression was studied by quantitative real-time PCR. We found that adipose tissue macrophages expressed Nrp1 and Nrp2, as well as Vegfr2 and Vegfr3 (Figure 6C).
To elucidate which of these receptors might mediate the migration of macrophages towards VEGF-C and -D, we performed transwell migration assays in the presence of receptor blocking monoclonal antibodies against neuropilin-1, neuropilin-2, VEGFR-2 or VEGFR-3. Blockage of neuropilin-1 or neuropilin-2 did not inhibit macrophage migration towards VEGF-C (VEGF-C + IgG: 100 ± 11.9%; anti-Nrp-1A: 92.7 ± 22.5%; anti-Nrp-1B: 95.9 ± 3.8%; anti-Nrp-2B: 84.8 ± 25.4%; one-way ANOVA: NS; n = 3; Figure 6D) or VEGF-D (VEGF-D + IgG: 100 ± 23.2%; anti-Nrp-1A: 102.8 ± 27.7%; anti-Nrp-1B: 116.3 ± 34.7%; anti-Nrp-2B: 114.4 ± 47.8%; one-way ANOVA: NS; n = 3; Figure 6E). Similarly, blockage of VEGFR-2 had no major effect (Figure 6D and E). In contrast, incubation with a blocking antibody against VEGFR-3, or neutralization with sR3 protein both strongly inhibited macrophage migration towards VEGF-C (VEGF-C + IgG: 100.0 ± 6.5%; anti-VEGFR-2: 89.4 ± 1.9%, one-way ANOVA: NS; anti-VEGFR-3: 36.5 ± 4.1%, P < 0.001; sVEGFR-3–Ig: 32.3 ± 6.7%, P < 0.001; n = 3, Figure 6D) and VEGF-D (VEGF-D + IgG: 100.0 ± 17.6%; anti-VEGFR-2: 103.8 ± 13.8%, one-way ANOVA: NS; anti-VEGFR-3: 45.7 ± 20.2%, P < 0.05; sVEGFR-3–Ig: 38.8 ± 15.5%, P < 0.01; n = 3, Figure 6E).
2.10. Vegfr3 mRNA is expressed by M1 but not M2 macrophages
Based on the findings that VEGF-C and -D are chemotactic for macrophages, and that the sR3 mice have an increased ratio of M2/M1 macrophages in their adipose tissue, we next investigated whether VEGFR-3 is differentially expressed by the M1 versus M2 macrophages. Mouse bone marrow-derived monocytes were differentiated into macrophages by M-CSF treatment for 6 days and were then polarized to M1 with IFN-γ treatment, or to M2 with a combination of IL-4/IL-10/IL-13/M-CSF treatment for 24 h. The M1 polarization was confirmed by the upregulation of the M1-related genes Inos and Tnf (Figure 7A), and the M2 polarization was confirmed by an increased mRNA expression of the M2-related genes Mrc1, Arg1 and Ym1 (Figure 7B). M1-polarized macrophages showed a strong upregulation of Vegfr3 mRNA expression, in contrast to M2-polarized macrophages where Vegfr3 mRNA was downregulated (relative gene expression of Vegfr3 compared to untreated controls: M1 polarized macrophages: 9.4 ± 5.6 fold; P = 0.029; M2 macrophages: 0.14 ± 0.03 fold P = 0.029; and P = 0.045 M1 compared to M2; n = 4 per group; Figure 7A).
Figure 7.
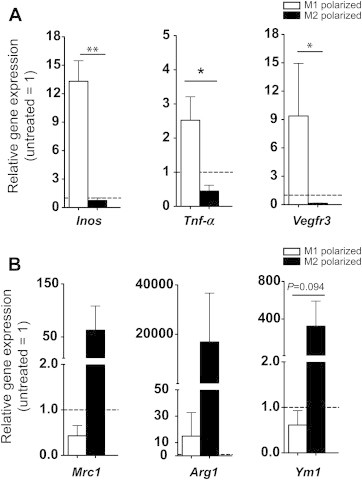
Induction of polarization related markers upon M1 or M2 polarization. (A) M1 polarization associated increase in the expression of Inos and Tnf mRNA. (B) M2 polarization increases the expression of the mRNA levels of Mrc1, Arg1 and Ym1. Vegfr3 mRNA expression is upregulated in M1, and downregulated in M2 polarized macrophages. Representative data from 5 independent experiments performed with 4 biological replicates per experimental group. Dashed line represents expression level in unstimulated macrophages. *P < 0.05, **P < 0.01. All data are mean ± SD.
2.11. VEGFR-3 blockade reduces SWAT macrophages in db/db mice
We next investigated if obesity-associated inflammation and metabolic syndrome in db/db mice might be reduced by inhibiting VEGFR-3 signaling during the development of insulin resistance, using systemic treatment with the VEGFR-3 blocking antibody MF4-31C1 (MF4). After two weeks of treatment, MF4 treated and control IgG treated mice had comparable body weights (Figure 8A) and adipose tissue mass (Figure 8B). Importantly, however, MF4 treated db/db mice were more insulin sensitive in an insulin tolerance test (n = 5 per group; two-way ANOVA P = 0.0122; Figure 8C). The MF4 treated mice also had smaller SWAT adipocytes (median adipocyte size IgG: 9456 ± 822.7; MF4: 7744 ± 535.2 μm2, n = 4 per group; P = 0.013; Figure 8D); EWAT adipocyte size was comparable (median adipocyte size IgG: 11,050 ± 874.1; MF4: 11,874 ± 888.4 μm2; Figure 8D). MF4 treated mice had a significantly reduced percentage of CD11b+/F4/80+ cells in the SWAT (IgG: 29.5 ± 3.7; MF4: 10.0 ± 6.8%; n = 2–3 per group; P = 0.038) and a trend towards reduction in the EWAT (IgG: 30.6 ± 6.6; MF4: 20.3 ± 8.4%; n = 2–3 per group; Figure 8E). This reduction was in line with a subtle reduction in the mRNA levels of CD11b, Tnf and Il6 (Figure 8F). MF4 treated mice had less hepatic lipid accumulation than control mice, as evidenced by reduced Oil Red O staining and lower levels of liver triglycerides (IgG: 153.8 ± 19.5; MF4: 121.1 ± 21.3 mg triglyceride/g liver; n = 3–5 per group; P = 0.074; Figure 8G). Collectively, these findings indicate that blockage of VEGFR-3 signaling during the development of insulin resistance has beneficial effects on the insulin sensitivity and macrophage accumulation in the adipose tissue.
Figure 8.
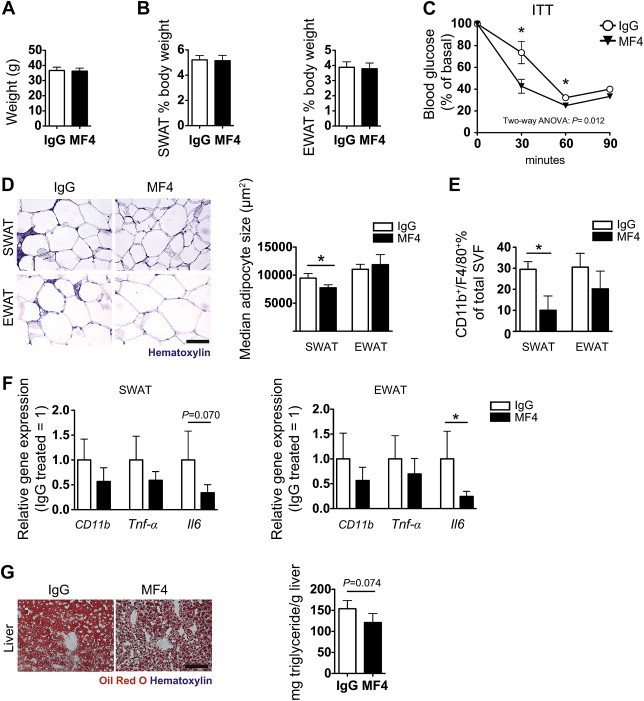
VEGFR-3 blocking antibody treatment impedes obesity-associated adipose tissue inflammation in db/db mice. (A) No difference in weight gain and (B) %SWAT and %EWAT after 2 weeks of treatment. (C) Insulin tolerance test, (curves are compared with two-way ANOVA (effect of treatment is shown); *P < 0.05 compared to IgG control at the same time point with two-tailed Student's t-test). (D) Hematoxylin staining showing smaller SWAT adipocytes in MF4 treated db/db mice, whereas EWAT adipocyte size was not changed. (E) FACS results of CD11b+/F4/80+ cells in stromal-vascular fraction of SWAT and EWAT. (F) Relative gene expression levels of CD11b, Tnf and Il6 in SWAT and EWAT. (G) Reduced ectopic lipid accumulation in the livers of MF4 treated mice as compared to IgG treated mice as shown by Oil Red O staining and triglyceride measurements. Scale bars = 100 μm *P < 0.05 (Two-tailed Student's t-test compared to IgG control). All data are mean ± SD.
3. Discussion
In this study, we show that blockage of the VEGFR-3 ligands VEGF-C and -D improves systemic insulin sensitivity and protects the liver against high-fat diet induced steatosis, associated with reduced adipocyte size and adipose tissue inflammation, and an increased M2/M1 macrophage ratio in adipose tissue.
Even though there were no differences in HFD-induced weight gain and adipose tissue mass between sR3 mice and WT mice on both the FVB and C57BL/6 genetic background, sR3 mice had significantly smaller subcutaneous adipocytes under chow and HFD, and the decrease in subcutaneous adipocyte size was consistent with increased insulin sensitivity as measured by insulin tolerance tests. Enhanced adipose tissue insulin sensitivity leads to a reduction of adipose tissue lipolysis and a reduced spillover to other tissues. Thus, the markedly lower hepatic steatosis in sR3 mice after HFD, together with the enhanced AKT phosphorylation in response to insulin, indicate that sR3 mice are more insulin-sensitive and are protected against HFD-induced insulin resistance. Our finding that the sR3 protein was detectable in the SWAT but not in the EWAT of sR3 mice is in line with the greater effect seen in the SWAT in this model. Since EWAT represents the metabolically more relevant adipose tissue depot, further studies involving the targeted delivery of the sR3 protein to all adipose depots should investigate whether this might result in an even more pronounced effect and in a further reduction of overall macrophage infiltration and inflammation.
Immune cells have emerged as major players in inflammation-mediated metabolic dysfunction [2,8], and a shift in macrophage polarization from the anti-inflammatory M2 phenotype to the proinflammatory M1 phenotype has been reported in obesity [15]. While the related angiogenesis factor VEGF-A was found to promote the recruitment of an M2 polarized population in combination with IL-4 in tumors [40], the potential effects of VEGF-C and -D on adipose tissue macrophage recruitment and polarization have remained unknown. Our finding that Vegfc and Vegfd expression is upregulated in SWAT adipocytes of obese mice is in line with increased VEGF-C and -D serum levels detected in obese patients and suggests that adipose tissue is one of the sources of VEGF-C and -D in obesity [18,19]. While we did not find any direct effects of VEGF-C or -D on adipogenic differentiation of 3T3-L1 cells in vitro (data not shown), the increased VEGF-C and -D expression by adipocytes likely promotes macrophage recruitment into the adipose tissue, since our FACS analyses of adipose tissue macrophages revealed an increased M2/M1 marker expression ratio in the SWAT of sR3 mice where the signaling of both VEGF-C and VEGF-D was inhibited. A likely explanation for this finding is provided by the results of the transwell migration experiments where both VEGF-C and -D induced chemotactic migration of murine macrophages in a VEGFR-3-dependent manner. This finding is in agreement with the reported VEGFR-3 expression by subsets of macrophages [41–44], including our own previous study in human macrophages [45] in which VEGF-C overexpressing tumors had an increased number of peritumoral macrophages. We found that the expression of VEGFR-3 is strongly upregulated during M1 polarization of macrophages but not during M2 polarization, which is in line with the LPS-induction of Vegfr3 expression in macrophages [44]. This differential expression likely results in preferential migration of M1 polarized macrophages towards tissues with elevated levels of VEGF-C and -D. These results indicate that VEGF-C and -D are involved in the specific recruitment rather than the polarization of adipose tissue macrophages. Importantly, treatment of db/db mice (a genetic mouse model of obesity) with a VEGFR-3 blocking antibody for 2 weeks resulted in a significant reduction of adipose tissue infiltrating macrophages and of Tnf and Il6 expression, together with reduced adipocyte size and hepatic lipid accumulation, and an improvement in insulin sensitivity. These findings, together with the reported absence of adipose tissue changes in VEGF-D deficient mice [28] indicate that VEGF-C, rather than VEGF-D, is responsible for the metabolic changes observed in obesity.
A possible connection between the effects of the blockade of VEGF-C and -D on specific macrophage recruitment and reduction of adipocyte size is provided by the finding that CM from WT, but not from sR3 adipose tissue macrophages, inhibited the adipogenic differentiation of 3T3-L1 cells. There were higher levels of proinflammatory cytokines in CM from WT macrophages than in CM from sR3 macrophages. In particular, IL-17, IL-1α and GM-CSF were detected only in WT macrophage CM. In this context, it is of interest that IL-17 was reported to negatively regulate adipogenesis, glucose homeostasis and obesity [46], and that GM-CSF deficient mice have improved insulin sensitivity despite significantly increased body weight [47]. Together, these results might explain how M2 macrophages in adipose tissue of sR3 mice facilitate adipogenic differentiation of new adipocytes, resulting in smaller and more numerous adipocytes and in enhanced insulin sensitivity.
Collectively, our study reveals that VEGF-C and -D play unanticipated roles in the recruitment of M1 polarized macrophages into obese adipose tissue and in the development of obesity-related insulin resistance. The results of the VEGFR-3 blocking studies in db/db mice indicate potential therapeutic strategies to reduce insulin resistance by blockage of the VEGF-C/VEGF-D–VEGFR-3 axis, for example with an anti-VEGFR-3 antibody or with the sR3 protein that are already available [29,48].
4. Materials and methods
4.1. Mice
Male FvB-TgN(K14-VEGFR-3d1-3-Fc) (sR3) transgenic mice on FVB or C57BL/6 backgrounds [29] and db/db mice (BKS.Cg-Dock7m +/+ Leprdb/J, purchased from The Jackson Lab) were maintained under specific pathogen free (SPF) conditions. Flt4-Cre-ERT2; ROSA26-LSL-LacZ double knock-in mice on a mixed (129/C57BL/6) background [30] were a kind gift from S. Ortega, Spanish National Cancer Research Centre, Madrid, Spain. Starting at the age of 4 weeks, sR3 mice and their WT littermates were provided ad libitum with either chow diet (11% kcal from fat, 31% kcal from protein, and 58% kcal from carbohydrate; Provimi-Kliba, Kaiseraugst, Switzerland) or a high-fat diet (HFD, 60% kcal from fat, 20% kcal from protein, and 20% kcal from carbohydrate; Research Diets Inc., NJ, USA) for 10, 20, 24 or 40 weeks. All experiments were performed in accordance with animal protocols approved by the Kantonales Veterinäramt Zürich.
4.2. Histology
PFA-fixed paraffin-embedded SWAT and EWAT samples were sectioned (10 μm), deparaffinized, rehydrated and stained with hematoxylin. An average of 15–20 images at 20× magnification were acquired per mouse. Adipocyte size was measured using the CellProfiler software (http://www.cellprofiler.org). For lipid staining, liver cryosections (7 μm) were fixed with PFA and stained with Oil Red O as described [49]. For immunofluorescence staining, tail, back skin and liver cryosections (7 μm) were fixed with acetone (−20 °C) and 80% methanol (4 °C), washed in PBS and incubated overnight with a hamster anti-podoplanin antibody (Clone 8.1.1, Developmental Studies Hybridoma Bank, University of Iowa, 1:200) and a rat anti-MECA-32 antibody (BD Pharmingen, 1:200) or a rat anti-CD31 antibody (BD Pharmingen, 1:200) for visualizing lymphatic and blood vessels, respectively. Alexa488-and Alexa594-conjugated secondary antibodies and Hoechst 33342 were added (Invitrogen, Basel, Switzerland), and images were acquired at 20× magnification. For bright field and fluorescence imaging, an Axioskop 2 mot plus microscope (Carl Zeiss, Inc.), equipped with a Plan-APOCHROMAT 10×/0.45 NA objective, an AxioCam MRc camera and a Plan-NEOFLUAR 20×/0.50 objective (Carl Zeiss, Inc.) was used. For bright-field and fluorescence imaging of cell cultures, a Zeiss Axiovert 200 M microscope equipped with a Zeiss AxioCam MRm camera with maximum contrast, a LD-Plan NEOFLUAR 20×/0.4 PhD2 Korr objective was used. Images were acquired with Axiovision software (version 4.7.1).
4.3. Whole-mount immunofluorescence stains
PFA-fixed intestine samples were stained using the following primary antibodies: rabbit anti-mouse LYVE-1 (AngioBio, 1:600), and rat anti-mouse CD31 (BD Pharmingen, 1:250). Further details of the procedures are available in Supplemental Experimental Procedures.
4.4. Intestinal lipid uptake
Eight-week-old male mice were fasted for 3 h before administration of 250 μM BODIPY 493/503 (Invitrogen, Carlsbad, CA) in 50 μL olive oil by oral gavage. After 1 h, mice were sacrificed and the blood was collected via cardiac puncture. Serial dilutions of sera in PBS were made, and the BODIPY fluorescence in the sera was measured at 503 nm with an Infinite M1000 plate reader (Tecan) and the values were normalized to sera of mice that were gavaged with only olive oil.
4.5. Insulin tolerance test
For the insulin tolerance test, mice that were on chow or HFD for 10 or 22 weeks were fasted for 10 h during the light-cycle. The mice were then injected i.p. with 0.75 U/kg (chow) or 2 U/kg (HFD or db/db) human insulin (Sigma). Blood glucose was monitored before the injection and after every 30 min with the Contour glucometer (Bayer HealthCare).
4.6. Insulin-stimulated AKT phosphorylation and western blot
Mice that were on chow or HFD for 24 weeks were fasted for 10 h during the light-cycle and injected i.p. with 0.75 U/kg (chow) or 2 U/kg (HFD) human insulin (Sigma). After 20 min, liver samples were harvested and snap-frozen in liquid nitrogen. The proteins were isolated by homogenizing the liver tissues in ice-cold lysis buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, 5 mM EDTA, 25 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 1% Triton X-100 and 10% glycerol. Western blotting was performed according to standard methods using antibodies against AKT and phospho-AKTSer473 (both from Cell Signaling, 1:1000) and a corresponding horseradish peroxidase-linked secondary antibody (GE Healthcare, 1:6000), followed by a densitometry analysis using ImageJ software (NIH).
4.7. Blocking antibody treatment
Five-week-old male BKS db/db mice were treated with 30 mg/kg of rat anti-mouse VEGFR-3 blocking antibody (mF4-31C1, ImClone; n = 5) or rat IgG control (Sigma; n = 5) every three days for 15 days (five administrations). ITT for db/db mice was performed one day after the final injection as described above.
4.8. Quantification of liver triglycerides
Liver triglycerides were extracted as described previously [50] with minor modifications. Briefly, 50 mg of liver tissue were homogenized in 1 mL hexane:isopropanol (3:2), using a Tissue-lyzer (Qiagen) with steel beads and were centrifuged (24,000 × g, 3 min). 0.5 mL Na2SO4 (470 mM) was added to the supernatant and the mixture was centrifuged (24,000 × g, 3 min). The upper organic phase was collected and left to dry overnight. The lipids were then dissolved in 1 mL Triton-X 100:methanol:buthanol (1:1:3), and the triglyceride levels were measured with the Triglyceride E kit (Wako) following the manufacturer's instructions.
4.9. Flow cytometry
Dissected SWAT and EWAT were minced in 2 mg/mL collagenase II (Sigma), buffered with warm Ringer-lactate buffer, and incubated for 1 h at 37 °C. After tissue digestion, floating mature adipocytes were separated from the stromal-vascular fraction (SVF) via centrifugation (200 × g, 5 min at 4 °C). The SVF was either used for flow cytometry or for MACS of macrophages. The macrophage population in the SVF from dissected SWAT (n = 16) and EWAT (n = 20) was stained with the following antibodies: F4/80-APC, CD11b-FITC (both from eBioscience, 1:100) to gate double-positive cells, and CD206-PE and MHCII-PerCP (both from Biolegend, 1:100) to assess distinct macrophage subsets. Each sample was stained in parallel with the corresponding IgG control for CD206 and MHCII. Samples were analyzed using a BD FACSCanto and the FACSDiva software (BD Biosciences). Data were processed with FlowJo software Version 8.7.1 (Tree Star).
4.10. 3T3-L1 adipogenic differentiation assays
3T3-L1 mouse embryonic fibroblasts (preadipocytes) were maintained in high-glucose (4.5 g/L) Dulbecco's modified Eagle's medium (DMEM; Gibco) with 10% bovine serum (Sigma) and penicillin/streptomycin for expansion (maintenance medium). Thereafter, 3T3-L1 cells were plated onto collagen type I-coated plates and were incubated in high-glucose DMEM, supplemented with 10% fetal bovine serum and penicillin/streptomycin (experimental medium, all from Gibco) in a humidified incubator at 5% CO2. Two days post-confluence, cells were incubated in “induction medium”, containing 172 nM insulin, 1 μM dexamethasone and 115 μg/mL isobutyl-methylxanthine (all from Sigma) and 10% macrophage-conditioned media. After 2 days, the medium was changed to insulin medium, which contained 10% macrophage-conditioned media and 172 nM insulin, and this medium was applied every two days. At day 10, cells were fixed with 4% PFA and stained with BODIPY 493/503, Syto60 and Hoechst (all from Invitrogen) to acquire images of lipid droplets, cytosol and nuclei, respectively. The percentage of differentiated adipocytes was analyzed with CellProfiler software as described [49].
4.11. Analysis of gene expression
RNA was extracted from isolated adipocytes or adipose tissue with the RNeasy Mini Kit (Qiagen), and from polarized macrophages and isolated adipose tissue macrophages using the RNeasy Plus Micro Kit (Qiagen). cDNA was transcribed from 500 ng RNA template, using the High Capacity Reverse Transcription kit (Applied Biosystems). The PCR reactions were performed using FastStart SYBR green master mix (Roche) and a 7900HT Fast Real-Time PCR System (Applied Biosystems). Rplp0 (36B4) was used as internal control and fold changes of gene expression were calculated using the ΔΔCT method. The primer sequences are provided in Table S2.
4.12. Transwell migration assay
CD11b+ cells were isolated from the peritoneal lavages of 20-week-old WT mice (n = 3) by MACS (Miltenyi Biotech), and 2 × 104 cells were seeded onto 24-well transwell inserts (pore size 8 μm, Corning). The medium in the bottom chamber consisted of RPMI medium (Gibco) supplemented with 1% FBS and either 200 ng/mL human VEGF-C (kind gift from M. Jeltsch, University of Helsinki, Finland [51]), 200 ng/mL murine VEGF-D (R&D Systems) or 200 ng/mL BSA. For receptor blocking studies, 10 μg/mL of rat anti-mouse VEGFR-2 antibody DC101, rat anti-mouse VEGFR-3 antibody mF4-31C1 (both from Imclone Systems Inc.), sVEGFR-3–Ig (R&D), control rat IgG (Sigma–Aldrich), mouse anti-mouse Nrp-1A, mouse anti-mouse Nrp-1B, mouse anti-mouse Nrp-2B mAb or control mouse IgG (all from Genentech) were added to the bottom chamber, either with or without human VEGF-C (200 ng/mL). After 3 days, the cells on the top of the membrane were removed, and the migrated cells on the bottom side were fixed and stained with Hoechst nuclear stain. Migrated cells were counted manually and their migratory capacity was normalized against the level of migration towards IgG + VEGF-C or IgG + VEGF-D.
4.13. Polarization of bone marrow derived macrophages
Bone marrow derived cells were harvested by flushing the femurs and tibiae of 24-week-old WT mice with sterile PBS. Monocytes (4 × 106) were plated into 6-well plates and differentiated to macrophages by incubation in RPMI medium supplemented with 1% FBS and 100 ng/mL M-CSF (BioLegend) for six days. Cells were washed extensively and polarized towards either M1 macrophages with IFN-γ, or towards M2 macrophages, using a combination of M-CSF, IL-4, IL-13 and IL-10 (all from BioLegend; 20 ng/mL final concentration). Controls were incubated in RPMI medium with 1% FBS. Cell lysates were collected after 24 h for RNA isolation.
4.14. Statistics
Data are presented as mean ± SD, and sample sizes and statistical tests used are indicated in the figure legends. Weight-gain and insulin tolerance test data were analyzed with two-way ANOVA for curve comparison and two-tailed Student's t-test for time-point comparisons. Adipocyte sizes were analyzed using the two-sample Kolmogorov–Smirnov test. Means of two groups were compared with the two-tailed Student's t-test. Means of three or more groups were analyzed with one-way ANOVA with Tukey's post-hoc test. Analyses were performed using GraphPad Prism V6.0 for MacOSX (GraphPad Software, San Diego California, USA), PASWStatistics 18.0 (SPSS Inc, Chicago, IL) or Matlab version 7.12.0.635 R2011a (Mathworks, Inc.). Statistical significance was set at P < 0.05.
Acknowledgments
We thank Jeannette Scholl and Tamara Eicher for technical assistance, Carlos Ochoa and Dr. Sun-Young Yoon for help with animal experiments, Dr. Michael Jeltsch for recombinant VEGF-C, Dr. Bronek Pytowski (ImClone Systems) for DC101 and MF4-31C1 antibodies and the Scientific Center for Optical and Electron Microscopy ScopeM of the Swiss Federal Institute of Technology ETHZ. This work was supported by Swiss National Science Foundation grant 310030B_147087, European Research Council grant LYVICAM, Oncosuisse (KLS-2821-08-2), Krebsliga Zurich (all to M.D.) and Leducq Foundation Transatlantic Network of Excellence grant Lymph Vessels in Obesity and Cardiovascular Disease (11CVD03, to M.D. and K.A.). M.H. was supported by the Sigrid Jusélius Foundation and the Instrumentarium Foundation. Studies in the laboratory of K.A. were also supported by the Academy of Finland and the European Research Council (ERC-2010-AdG-268804).
Conflict of interest
Authors declare no conflicts of interests.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Wang Y., Beydoun M.A., Liang L., Caballero B., Kumanyika S.K. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity (Silver Spring) 2008;16:2323–2330. doi: 10.1038/oby.2008.351. [DOI] [PubMed] [Google Scholar]
- 2.Olefsky J.M., Glass C.K. Macrophages, inflammation, and insulin resistance. Annual Review of Physiology. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 3.Hotamisligil G.S., Shargill N.S., Spiegelman B.M. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 4.Bastard J.P., Jardel C., Bruckert E., Blondy P., Capeau J., Laville M. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. Journal of Clinical Endocrinology and Metabolism. 2000;85:3338–3342. doi: 10.1210/jcem.85.9.6839. [DOI] [PubMed] [Google Scholar]
- 5.Sartipy P., Loskutoff D. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7265–7270. doi: 10.1073/pnas.1133870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanda H., Tateya S., Tamori Y., Kotani K., Hiasa K., Kitazawa R. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. Journal of Clinical Investigation. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laurencikiene J., van Harmelen V., Arvidsson Nordstrom E., Dicker A., Blomqvist L., Naslund E. NF-kappaB is important for TNF-alpha-induced lipolysis in human adipocytes. Journal of Lipid Research. 2007;48:1069–1077. doi: 10.1194/jlr.M600471-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Hotamisligil G. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 9.Weisberg S.P., McCann D., Desai M., Rosenbaum M., Leibel R.L., Ferrante A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. Journal of Clinical Investigation. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feuerer M., Herrero L., Cipolletta D., Naaz A., Wong J., Nayer A. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nature Medicine. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishimura S., Manabe I., Nagasaki M., Eto K., Yamashita H., Ohsugi M. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nature Medicine. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 12.Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 13.Shapiro H., Lutaty A., Ariel A. Macrophages, meta-inflammation, and immuno-metabolism. ScientificWorldJournal. 2011;11:2509–2529. doi: 10.1100/2011/397971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sica A., Mantovani A. Macrophage plasticity and polarization: in vivo veritas. Journal of Clinical Investigation. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lumeng C.N., Bodzin J.L., Saltiel A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. Journal of Clinical Investigation. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolattukudy P.E., Niu J. Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circulation Research. 2012;110:174–189. doi: 10.1161/CIRCRESAHA.111.243212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicholls H.T., Kowalski G., Kennedy D.J., Risis S., Zaffino L.A., Watson N. Hematopoietic cell-restricted deletion of CD36 reduces high-fat diet-induced macrophage infiltration and improves insulin signaling in adipose tissue. Diabetes. 2011;60:1100–1110. doi: 10.2337/db10-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez-Ambrosi J., Catalan V., Rodriguez A., Ramirez B., Silva C., Gil M.J. Involvement of serum vascular endothelial growth factor family members in the development of obesity in mice and humans. Journal of Nutritional Biochemistry. 2010;21:774–780. doi: 10.1016/j.jnutbio.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Silha J., Krsek M., Sucharda P., Murphy L. Angiogenic factors are elevated in overweight and obese individuals. International Journal of Obesity. 2005;29:1308–1314. doi: 10.1038/sj.ijo.0802987. [DOI] [PubMed] [Google Scholar]
- 20.Wada H., Ura S., Kitaoka S., Satoh-Asahara N., Horie T., Ono K. Distinct characteristics of circulating vascular endothelial growth factor-A and C levels in human subjects. PLoS One. 2011;6:e29351. doi: 10.1371/journal.pone.0029351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alitalo K. The lymphatic vasculature in disease. Nature Medicine. 2011;17:1371–1380. doi: 10.1038/nm.2545. [DOI] [PubMed] [Google Scholar]
- 22.Cueni L., Detmar M. The lymphatic system in health and disease. Lymphatic Research and Biology. 2008;6:109–122. doi: 10.1089/lrb.2008.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rockson S.G. Update on the biology and treatment of lymphedema. Current Treatment Options in Cardiovascular Medicine. 2012;14:184–192. doi: 10.1007/s11936-012-0170-0. [DOI] [PubMed] [Google Scholar]
- 24.Greene A.K., Grant F.D., Slavin S.A. Lower-extremity lymphedema and elevated body-mass index. New England Journal of Medicine. 2012;366:2136–2137. doi: 10.1056/NEJMc1201684. [DOI] [PubMed] [Google Scholar]
- 25.Blum K.S., Karaman S., Proulx S.T., Ochsenbein A.M., Luciani P., Leroux J.C. Chronic high-fat diet impairs collecting lymphatic vessel function in mice. PLoS One. 2014;9:e94713. doi: 10.1371/journal.pone.0094713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elias I., Franckhauser S., Ferre T., Vila L., Tafuro S., Munoz S. Adipose tissue overexpression of vascular endothelial growth factor protects against diet-induced obesity and insulin resistance. Diabetes. 2012;61:1801–1813. doi: 10.2337/db11-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun K., Wernstedt Asterholm I., Kusminski C.M., Bueno A.C., Wang Z.V., Pollard J.W. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5874–5879. doi: 10.1073/pnas.1200447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lijnen H.R., Frederix L., Van Hoef B., Dewerchin M. Deficiency of vascular endothelial growth factor-D does not affect murine adipose tissue development. Biochemical and Biophysical Research Communications. 2009;378:255–258. doi: 10.1016/j.bbrc.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 29.Mäkinen T., Jussila L., Veikkola T., Karpanen T., Kettunen M., Pulkkanen K. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nature Medicine. 2001;7:199–205. doi: 10.1038/84651. [DOI] [PubMed] [Google Scholar]
- 30.Aschen S.Z., Farias-Eisner G., Cuzzone D.A., Albano N.J., Ghanta S., Weitman E.S. Lymph node transplantation results in spontaneous lymphatic reconnection and restoration of lymphatic flow. Plastic and Reconstructive Surgery. 2014;133:301–310. doi: 10.1097/01.prs.0000436840.69752.7e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alitalo A.K., Proulx S.T., Karaman S., Aebischer D., Martino S., Jost M. VEGF-C and VEGF-D blockade inhibits inflammatory skin carcinogenesis. Cancer Research. 2013;73:4212–4221. doi: 10.1158/0008-5472.CAN-12-4539. [DOI] [PubMed] [Google Scholar]
- 32.Livingston J.N., Cuatrecasa P., Lockwood D.H. Insulin insensitivity of large fat cells. Science. 1972;177:626–628. doi: 10.1126/science.177.4049.626. [DOI] [PubMed] [Google Scholar]
- 33.Wallace T.M., Levy J.C., Matthews D.R. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487–1495. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 34.Lumeng C.N., DelProposto J.B., Westcott D.J., Saltiel A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008;57:3239–3246. doi: 10.2337/db08-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang K., Reilly S.M., Karabacak V., Gangl M.R., Fitzgerald K., Hatano B. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metabolism. 2008;7:485–495. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Constant V.A., Gagnon A., Landry A., Sorisky A. Macrophage-conditioned medium inhibits the differentiation of 3T3-L1 and human abdominal preadipocytes. Diabetologia. 2006;49:1402–1411. doi: 10.1007/s00125-006-0253-0. [DOI] [PubMed] [Google Scholar]
- 37.Lacasa D., Taleb S., Keophiphath M., Miranville A., Clement K. Macrophage-secreted factors impair human adipogenesis: involvement of proinflammatory state in preadipocytes. Endocrinology. 2007;148:868–877. doi: 10.1210/en.2006-0687. [DOI] [PubMed] [Google Scholar]
- 38.Permana P.A., Menge C., Reaven P.D. Macrophage-secreted factors induce adipocyte inflammation and insulin resistance. Biochemical and Biophysical Research Communications. 2006;341:507–514. doi: 10.1016/j.bbrc.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 39.Poulos S.P., Dodson M.V., Hausman G.J. Cell line models for differentiation: preadipocytes and adipocytes. Experimental Biology and Medicine. 2010;235:1185–1193. doi: 10.1258/ebm.2010.010063. [DOI] [PubMed] [Google Scholar]
- 40.Linde N., Lederle W., Depner S., van Rooijen N., Gutschalk C.M., Mueller M.M. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. Journal of Pathology. 2012;227:17–28. doi: 10.1002/path.3989. [DOI] [PubMed] [Google Scholar]
- 41.Skobe M., Hawighorst T., Jackson D.G., Prevo R., Janes L., Velasco P. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nature Medicine. 2001;7:192–198. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- 42.Schoppmann S., Birner P., Stockl J., Kalt R., Ullrich R., Caucig C. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. American Journal of Pathology. 2002;161:947–956. doi: 10.1016/S0002-9440(10)64255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stepanova O.I., Krylov A.V., Lioudyno V.I., Kisseleva E.P. Gene expression for VEGF-A, VEGF-C, and their receptors in murine lymphocytes and macrophages. Biochemistry (Moscow) 2007;72:1194–1198. doi: 10.1134/s0006297907110041. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y., Lu Y., Ma L., Cao X., Xiao J., Chen J. Activation of vascular endothelial growth factor receptor-3 in macrophages restrains TLR4-NF-kappaB signaling and protects against endotoxin shock. Immunity. 2014;40:501–514. doi: 10.1016/j.immuni.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 45.Skobe M., Hamberg L.M., Hawighorst T., Schirner M., Wolf G.L., Alitalo K. Concurrent induction of lymphangiogenesis, angiogenesis, and macrophage recruitment by vascular endothelial growth factor-C in melanoma. American Journal of Pathology. 2001;159:893–903. doi: 10.1016/S0002-9440(10)61765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuniga L.A., Shen W.J., Joyce-Shaikh B., Pyatnova E.A., Richards A.G., Thom C. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. Journal of Immunology. 2010;185:6947–6959. doi: 10.4049/jimmunol.1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim D.H., Sandoval D., Reed J.A., Matter E.K., Tolod E.G., Woods S.C. The role of GM-CSF in adipose tissue inflammation. American Journal of Physiology. Endocrinology and Metabolism. 2008;295:E1038–E1046. doi: 10.1152/ajpendo.00061.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Persaud K., Tille J.C., Liu M., Zhu Z., Jimenez X., Pereira D.S. Involvement of the VEGF receptor 3 in tubular morphogenesis demonstrated with a human anti-human VEGFR-3 monoclonal antibody that antagonizes receptor activation by VEGF-C. Journal of Cell Science. 2004;117:2745–2756. doi: 10.1242/jcs.01138. [DOI] [PubMed] [Google Scholar]
- 49.Meissburger B., Ukropec J., Roeder E., Beaton N., Geiger M., Teupser D. Adipogenesis and insulin sensitivity in obesity are regulated by retinoid-related orphan receptor gamma. EMBO Molecular Medicine. 2011;3:637–651. doi: 10.1002/emmm.201100172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hara A., Radin N.S. Lipid extraction of tissues with a low-toxicity solvent. Analytical Biochemistry. 1978;90:420–426. doi: 10.1016/0003-2697(78)90046-5. [DOI] [PubMed] [Google Scholar]
- 51.Karpanen T., Heckman C.A., Keskitalo S., Jeltsch M., Ollila H., Neufeld G. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. FASEB Journal. 2006;20:1462–1472. doi: 10.1096/fj.05-5646com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.