

Abstract
Respiratory complex I inhibition by drugs and other chemicals has been implicated as a frequent mode of mitochondria-mediated cell injury. However, the exact mechanisms leading to the activation of cell death pathways are incompletely understood. This study was designed to explore the relative contributions to cell injury of three distinct consequences of complex I inhibition, i.e., impairment of ATP biosynthesis, increased formation of superoxide and, hence, peroxynitrite, and inhibition of the mitochondrial protein deacetylase, Sirt3, due to imbalance of the NADH/NAD+ ratio. We used the antiviral drug efavirenz (EFV) to model drug-induced complex I inhibition. Exposure of cultured mouse hepatocytes to EFV resulted in a rapid onset of cell injury, featuring a no-effect level at 30 µM EFV and submaximal effects at 50 µM EFV. EFV caused a concentration-dependent decrease in cellular ATP levels. Furthermore, EFV resulted in increased formation of peroxynitrite and oxidation of mitochondrial protein thiols, including cyclophilin D (CypD). This was prevented by the superoxide scavenger, Fe-TCP, or the peroxynitrite decomposition catalyst, Fe-TMPyP. Both ferroporphyrins completely protected from EFV-induced cell injury, suggesting that peroxynitrite contributed to the cell injury. Finally, EFV increased the NADH/NAD+ ratio, inhibited Sirt3 activity, and led to hyperacetylated lysine residues, including those in CypD. However, hepatocytes isolated from Sirt3-null mice were protected against 40 µM EFV as compared to their wild-type controls. In conclusion, these data are compatible with the concept that chemical inhibition of complex I activates multiple pathways leading to cell injury; among these, peroxynitrite formation may be the most critical.
Keywords: Sirt3, Efavirenz, Complex I inhibition, Mitochondria, Peroxynitrite, Drug-induced liver injury (DILI)
Abbreviations: CBA, coumarin-7-boronic acid; CYP, cytochrome P450; CypD, cyclophilin D; ETC, electron transport chain; Fe-TCP, Fe(III) tetrakis(4-carboxyphenyl) porphyrin; Fe-TMPyP, Fe(III) tetrakis(1-methyl-4-pyridyl) porphyrin; MitoSOX Red, mitochondria-targeted dihydroethidine; mPT, mitochondrial permeability transition; NQR, NADH:ubiquinone oxidoreductase; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species
Highlights
-
•
The antiviral drug efavirenz (EFV) induces complex I inhibition.
-
•
Complex I inhibition causes ATP depletion, ROS/RNS formation, and Sirt3 inhibition.
-
•
Iron porphyrins protect hepatocytes against EFV-induced lethal injury.
-
•
Genetic ablation of Sirt3 (Sirt3−/− mice) protects against EFV toxicity.
-
•
Peroxynitrite formation is a major mechanism of mitochondria-mediated cell injury.
Introduction
The mitochondrion is a frequent subcellular target that is involved in the toxicity of drugs [1–4]. Multiple initiating mechanisms can contribute to the pathogenesis of cell injury, including drug-mediated inhibition of one or several of the respiratory chain complexes [5]. In particular, complex I is a relatively frequent target, and complex I inhibition induced by drugs has been implicated in causing cell death [6–9].
Complex I (NADH:ubiquinone oxidoreductase, NQR) is the largest among the electron transport chain (ETC) complexes; it is composed of at least 45 subunits encoded by both mitochondrial and nuclear genes [10]. Its major function is the oxidation of NADH, resulting in the transfer of electrons to ubiquinone, while the released energy is utilized to pump protons from the matrix into the intermembrane space, helping maintain the inside negative inner transmembrane gradient. The exact mechanisms of how complex I inhibitors may induce cell injury are still incompletely understood, but three major mechanistic pathways have been identified. First, complex I inhibition may lead to defects in mitochondrial bioenergetics, as a result of reduced electron flow and impairment of oxidative phosphorylation (OXPHOS), especially in tissues with high energy demand [11]. However, complex I has a large reserve capacity, and minor inhibition of complex I may not result in significant deficiencies in ATP biosynthesis [12]. Second, inhibition of NQR activity may result in a backup of electrons and increased reductive stress, causing facilitated reduction of molecular oxygen to form superoxide anion [8]. This can lead to ultrarapid formation of peroxynitrite, which even outcompetes the SOD-catalyzed formation of hydrogen peroxide. In addition, redox-sensitive signaling pathways can be activated, one of them resulting in an increase in the intermembrane pool of cytochrome c, leading to apoptosis [7]. Third, imbalances in the NAD+/NADH ratio, due to impaired substrate oxidation at complex I, could result in inhibition of NAD+-dependent enzymes including the mitochondrial sirtuins (Sirt). Among these, Sirt3 is a soluble mitochondrial protein that is involved in the posttranslational regulation of a number of mitochondrial targets by deacetylating lysine residues [13]. Inhibition of Sirt3 activity will invariably lead to hyperacetylation of these proteins and dysregulation of mitochondrial homeostasis. However, the relative contribution of these three mechanisms to the initiation and progression of cell death is not known.
In this study, we used efavirenz (EFV) to model drug-induced inhibition of complex I. Efavirenz is a non-nucleoside reverse transcriptase inhibitor that is widely used as part of the highly active antiretroviral therapy (HAART) to treat HIV infections. Although considered generally safe, there is evidence that EFV can cause hepatic toxicity; in fact, up to 10% of patients treated with EFV may develop increased plasma ALT activity (a marker for hepatocellular injury), which limits treatment [14,15]. Although the underlying molecular mechanisms have largely remained unclear, it has been reported that EFV inhibits cellular oxygen consumption in isolated rat liver mitochondria energized through complex I (but not through complex II) [16], suggesting that EFV might inhibit complex I activity. More recently, evidence has revealed that EFV indeed inhibits NQR activity in mouse hepatic submitochondrial particles in a concentration-dependent manner, indicating direct inhibition of complex I activity caused by EFV.
The aim of this study was to explore the causal role of both increased reactive oxygen species/reactive nitrogen species (ROS/RNS) and potential inhibition of Sirt3 activity and protein hyperacetylation, as a result of EFV-induced complex I inhibition, in contributing to the pathogenesis of EFV-induced cell death in mouse hepatocytes. We found that peroxynitrite formation plays a mechanistic role in triggering cell death, but that, surprisingly, Sirt3 inhibition seems to protect, rather than promote, hepatocellular injury induced by the complex I inhibitor.
Materials and methods
Chemicals
Efavirenz (EFV) was purchased from Sigma (St. Louis, MO). Fe-TCP (Fe(III) meso-tetra(4-carboxyphenyl)porphyrine chloride) was purchased from Frontier Scientific (Logan, UT). Fe-TMPyP (Fe(III)tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride) was purchased from Cayman (Ann Arbor, MI). All chemicals were obtained at the highest grade available.
Animals and genotyping
The study design and all protocols for animal care and handling were approved by the Institutional Animal Care and Use Committee at the University of Connecticut. Young adult C57BL/6 mice, as well as 129-Sirt3tm1.1Fwa/J (homozygous Sirt3 knockout) mice and their 129S1/SvImJ wild-type controls, were obtained from the Jackson Laboratory (Bar Harbor, ME). Male mice (8–10 weeks of age) were used for all studies. Prior to use, the mice were acclimatized for ≥1 week and kept on a 14/10-h light/dark cycle under controlled environmental conditions. They had free access to mouse chow (Teklad Global Rodent Diet; Harlan Laboratories, Boston, MA) and water. Genotyping was performed by tail biopsy and polymerase chain reaction (PCR) analysis. Briefly, the tissue was lysed in 50 mM Tris–HCl, pH 8.8, containing 1 mM EDTA, 0.5% Tween 20, and 0.6 mg/ml proteinase K, and incubated at 56 °C overnight. Proteinase K was inactivated at 95 °C for 10 min, and the lysates centrifuged at 13,000g for 10 min. Supernatants were used for PCR (Supplementary Table 1). 5 ml PCR reaction mix was loaded onto a 2% agarose gel and run with 1× TBE buffer at 20 mA constant for 2 h.
Primary mouse hepatocyte culture and exposure to drugs
Hepatocytes were isolated by in situ retrograde collagenase perfusion, and subsequently cultured in supplemented Williams' Medium E as described [17]. Briefly, the cells were plated in 48-well plates (8.0×104 cells per well) coated with 50 mg/ml rat tail collagen. The hepatocytes were allowed to attach for 3 h in a humidified atmosphere of 5% CO2/95% air at 37 °C. Subsequently, the cells were washed and incubated in the same medium. After overnight pre-culture, the medium was replaced by fresh serum- and antibiotic-free medium to which the test compounds were added from stock solutions. Efavirenz and other lipophilic compounds were dissolved in DMSO (final DMSO concentrations ≤0.25%). Culture medium was used to dissolve hydrophilic compounds.
Determination of cell injury
Release of cytosolic lactate dehydrogenase (LDH) into the extracellular medium (CytoTox-One Homogeneous Membrane Integrity Assay, Promega, Madison, WI) was used as an indicator of cytotoxicity. The data were expressed as percentage of activity present in the medium as compared to the total intra- and extracellular LDH activity. Total cellular ATP content was measured by chemiluminescence in black 96-well plates (Cell Titer-Glo Luminescent Cell Viability Assay, Promega) and calculated from a standard curve. EFV did not interfere with the luciferin/luciferase reaction.
Measurement of mitochondrial superoxide and peroxynitrite generation in hepatocytes
Mitochondrial generation of superoxide was estimated with the cell-permeable and mitochondria-targeted fluorogenic probe, hydroethidine (HE) linked to triphenylphosphonium (MitoSOX Red, Life Technologies, Carlsbad, CA). The drug-pretreated cells were loaded with MitoSOX Red (25 nM) for 10 min at 37 °C, washed with fresh culture medium, and the mitochondrial 2-hydroxy ethidium-derived fluorescence was determined at 396/580 nm (excitation/emission, respectively) as described [17], in a Safire2 microplate reader (Tecan, Maennedorf, Switzerland). Hepatocellular formation of peroxynitrite was determined with the highly selective fluorogenic probe, coumarin-7-boronic acid (CBA, Cayman, Ann Arbor, MI), which reacts stochiometrically and rapidly with ONOO− several orders of magnitude faster than with H2O2 [18]. Hepatocytes were preloaded with 100 µM CBA for 20 min at 37 °C, and the generation of hydroxycoumarin was determined at 332/450 nm (excitation/emission) in a Safire2 microplate reader.
Determination of CYP3A4 and CYP2B6 activity in baculosomes expressing the human isoforms
Microsomes derived from baculovirus-transfected insect cells expressing recombinant human CYP3A4 or CYP2B6 were incubated with the fluorogenic probe 7-benzyloxy-methyloxy-3-cyanocoumarin (BOMCC, 10 µM) (Vivid® CYP3A4 or CYP2B6 Blue Substrate, Invitrogen), which is a substrate for both cytochrome P4503A4 and 2B6 and which is metabolized to the fluorescent 3-cyano-7-hydroxycoumarin [19]. After preincubation with the metalloporphyrins for 5 min, fluorescence (415/460 nm, excitation/emission) was monitored every 5 min for 20 min. Miconazole (30 µM), a high-affinity inhibitor of CYP2B6 [20] or 1-ABT (100 µM), a CYP inhibitor with a broader isoform selectivity, were used as mechanism-based CYP inhibitor controls.
Immunoblotting
Equal amounts of denatured mitochondrial protein were loaded on each lane, and the proteins were resolved on precast polyacrylamide gels (BioRad, Hercules, CA), under reducing or non-reducing conditions, and subsequently transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA). The membranes were blocked in 5% milk prepared with 0.1% Tween 20 in TBS at room temperature for 1 h. Anti-3-nitrotyrosine (1:3000) (Cell Signaling, Danvers, MA), anti-VDAC (1:3000) (Cell Signaling), anti-acetyl-lysine (1:3000) (Cell Signaling), and CypD (1:5000) (Cell Signaling) were used as primary antibody. The protein bands were visualized by enhanced chemiluminescence (Millipore) after incubation with HRP-conjugated secondary antibody (1:10,000) (BioRad). Anti-VDAC antibody was used as loading control.
Determination of cellular NADH and NAD+ concentrations
NADH and NAD+ concentrations were measured in lysates of 3.2×105 cells, using the NAD/NADH ratio assay kit (e-Enzyme, Gaithersburg, MD) according to the manufacturer’s instructions.
Isolation of hepatic mitochondria and assessment of complex I activity
Hepatic mitochondria were isolated from untreated mice according to standard procedures as previously described [17]. Protein content was determined with the BCR protein assay using albumin as the reference protein. The mitochondria-enriched fractions were kept at −80 °C until analysis. Complex I activity was determined in freeze-thawed (2×) mitochondria according to standard methods [21]. Briefly, complex I was measured as NQR activity in 25 mM potassium phosphate buffer containing 5 mM MgCl2, pH 7.2, and 2.5 mg/ml BSA, 0.13 mM NADH, 2 mg/ml antimycin A, and 65 µM ubiquinone (Q1). NADH oxidation was monitored as decrease in absorbance at 340 nm.
Statistical analysis
All data were expressed as mean±SD. If there was normal distribution, a standard analysis of variance (ANOVA) was used, followed by Dunnett’s test for multiple comparisons versus the control group. When normality failed, a Kruskal–Wallis one-way analysis of variance on ranks was used followed by Dunn's test for multiple comparison versus the control group. A P value of ≤0.05 was considered significant.
Results
EFV causes lethal cell injury in mouse hepatocytes that is insensitive to CYP inhibitors
To determine the concentration-dependent toxic response to efavirenz, we first exposed hepatocytes isolated from C57BL6/J mice to EFV for various periods of time. We found that there was a rapid onset of toxicity (3 h) with a characteristic threshold, as evidenced by increases in LDH leakage and decreases in intracellular ATP concentrations (Fig. 1). Specifically, EFV concentrations ≤30 µM did not induce significant changes from solvent controls, whereas 50 µM EFV caused ~50% LDH leakage and >90% loss of intracellular ATP after 24 h. These data indicate that EFV induces hepatocellular injury in mouse hepatocytes, confirming previous studies in human Hep3B cells [16,22]. The data also suggest that mouse hepatocytes are a good model of EFV-induced liver cell injury.
Fig. 1.
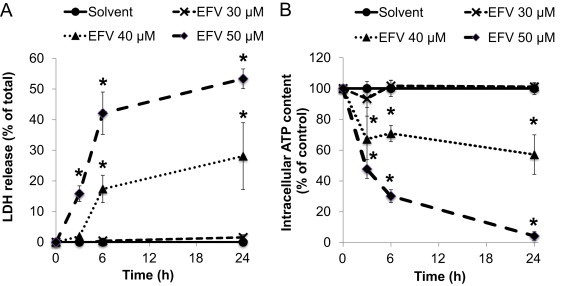
Effects of efavirenz (EFV) on cell injury and intracellular ATP concentrations in cultured mouse hepatocytes. Time course and concentration response of (A) LDH release and (B) intracellular ATP content after treatment with solvent (DMSO, 0.25%) or EFV (30, 40, or 50 µM). Data are mean±SD of three independent hepatocyte preparations using quadruplicate wells. *P<0.05 versus solvent control.
In humans, EFV is metabolized by CYP2B6, resulting in the formation of 8-hydroxy-EFV as the major metabolite (which is subsequently glucuronidated) [23]. In rodents (but not humans) glutathione adducts were detected, originating from the formation of cyclopropane ring hydroxylation [23]. However, the mechanistic role of CYP-mediated bioactivation of EFV in mouse liver is not known. Therefore, we next determined the extent of hepatocellular injury induced by EFV (50 µM) in mouse hepatocytes in the presence or absence of the pan-CYP or CYP2B inhibitor, 1-aminobenzotriazole (ABT) or miconazole, respectively. We found that both ABT and miconazole did not attenuate EFV-induced LDH leakage (Fig. 2), suggesting that the parent EFV, rather than a CYP-mediated reactive metabolite, may directly cause cell injury.
Fig. 2.
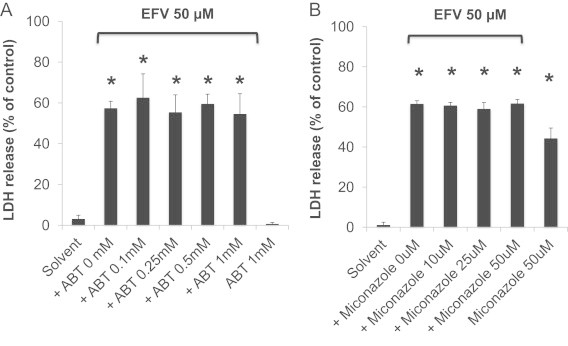
Effect of the pan-cytochrome P450 (CYP) inhibitor or CYP2B inhibitor, 1-aminobenzotriazole (ABT) or miconazole, on EFV-induced cell injury. Hepatocytes were exposed to EFV (50 µM) in the presence or absence of ABT or miconazole, and LDH release was determined after 24 h. Data are mean±SD of three independent hepatocyte preparations using quadruplicate wells. *P<0.05 versus solvent control.
EFV inhibits complex I activity and induces ROS signaling pathways
To confirm and extend previous data demonstrating that EFV inhibits complex I-energized respiration [16] and NQR activity [24], we measured complex I activity in submitochondrial particles isolated from 129S1/SvImJ mouse liver. We found that EFV inhibited NQR activity in a concentration-dependent manner, featuring an IC50 of <10 µM (Supplementary Fig. 1). At ≥30 µM, complex I activity was completely inhibited by EFV. Because of these potent effects on mitochondrial electron transport function, we hypothesized that complex I inhibition might be one of the mechanisms leading to EFV-induced hepatocellular injury. Massive complex I inhibition can have at least three major consequences: first, decreases in ATP biosynthesis rates, resulting in a potential bioenergy crisis; second, increased formation of ROS/RNS due to a blockage of electron transfer at the IQ (ubiquinone-binding) site and facilitated formation of superoxide; and third, inhibition of the catalytic activity of Sirt3, which is a NAD+-dependent deacetylase, as a consequence of a decrease in NADH oxidation resulting in an imbalance of the NADH/NAD+ ratio.
We first evaluated whether EFV caused increases in mitochondrial ROS levels by using a fluorogenic probe that reacts with superoxide to form hydroxyethidine. We found that EFV (50 µM) caused a rapid increase in MitoSOX Red-derived fluorescence (Fig. 3A). Furthermore, because superoxide anion reacts extremely fast with nitric oxide to form the highly toxic species, peroxynitrite (ONOO−), we assessed the formation of peroxynitrite using coumarin-7-boronic acid (CBA), a boronate-based fluorogenic probe that selectively reacts with peroxynitrite [18]. We found that cells preloaded with CBA followed by exposure to EFV (50 µM) developed increases in CBA-derived fluorescence (Fig. 3B). These data suggest that EFV generates increased levels of ROS and RNS through mitochondria, at concentrations that induce massive cell death, but not at concentrations that only marginally induce cell injury (≤30 µM).
Fig. 3.
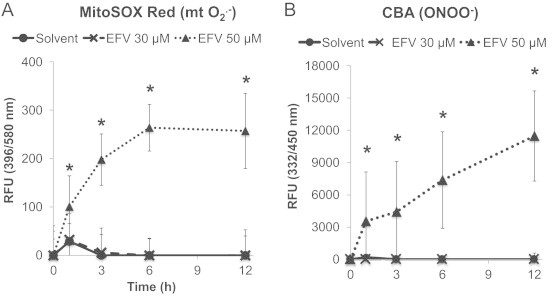
Time course of EFV-induced superoxide anion and peroxynitrite generation in hepatocytes. Hepatocytes were preloaded with MitoSOX red or CBA and exposed to EFV (30 or 50 µM). (A) MitoSOX Red-derived fluorescence, indicative of increased superoxide generation. (B) Hydroxycoumarin fluorescence generated from peroxynitrite-mediated CBA oxidation was serially recorded with a plate reader. Data are mean±SD of three independent hepatocyte preparations using quadruplicate wells. *P<0.05 versus solvent control.
Iron porphyrins protect hepatocytes against EFV-induced lethal injury
To determine whether the increase in superoxide anion and peroxynitrite are causally involved in cell injury, rather than simply being a consequence of cell death, we explored the effects of two distinct ferroporphyrins on the extent of toxicity. Specifically, we used the cell-permeable SOD mimetic, Fe-TCP, and the peroxynitrite decomposition catalyst, Fe-TMPyP. Both compounds are cationic metalloporphyrins that are taken up into the mitochondrial matrix [25]. Other inhibitors, such as superoxide dismutase or catalase, are difficult to be introduced into cells and, specifically, into mitochondria in cell cultures. We found that both Fe-TCP and Fe-TMPyP attenuated the EFV-induced fluorescence of MitoSOX Red and CBA (Fig. 4). Importantly, both Fe-TCP and Fe-TMPyP prevented the increases in LDH leakage caused by EFV alone after 24 h (Fig. 5). These data suggest that the mitochondrial generation of ROS/RNS may be causally involved in the pathogenesis of EFV-induced cell injury.
Fig. 4.
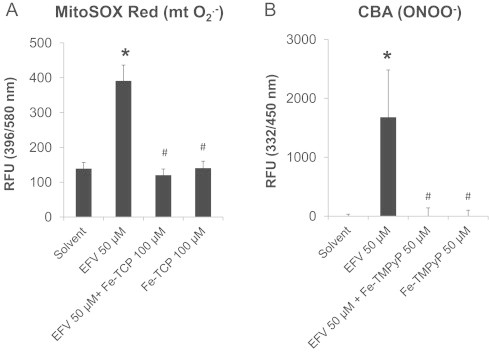
Effects of Fe-TCP or Fe-TMPyP on EFV-induced superoxide anion and peroxynitrite generation in hepatocytes. Fe-TCP, a SOD mimetic, or Fe-TMPyP, a peroxynitrite decomposition catalyst, were used to demonstrate the selectivity of the assay for mitochondrial superoxide anion (mtO2−) and peroxynitrite (ONOO−) in hepatocytes exposed to EFV (50 µM) for 6 h. Data are mean±SD of three independent hepatocyte preparations using quadruplicate wells. *P<0.05 versus solvent control; #P<0.05 versus EFV alone.
Fig. 5.
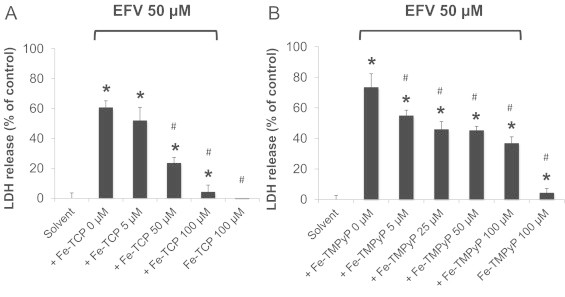
Effect of Fe-TCP or Fe-TMPyP on EFV-induced cell injury. Hepatocytes were exposed to EFV (50 µM) in the presence or absence of (A) Fe-TCP or (B) Fe-TMPyP for 24 h. Data are mean±SD of three independent hepatocyte preparations using quadruplicate wells. *P<0.05 versus solvent control; #P<0.05 versus EFV alone.
An unexpected finding was that both ferroporphyrins were potent inhibitors of CYP2B6 and CYP3A4 activity in baculosomes expressing the human isoforms (Fig. 6). Because other CYP inhibitors did not attenuate the toxicity of EFV in mouse hepatocytes, it is unlikely that the protective effect provided by the ferroporphyrins was due to an inhibition of CYP-mediated bioactivation. However, because iron porphyrins have been used in various other experimental models, the interpretation of results needs increased attention to a potential inhibition of drug bioactivation.
Fig. 6.
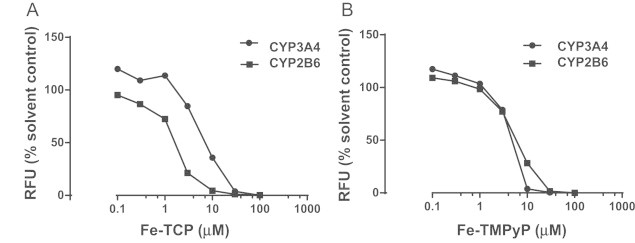
Inhibitory effects of ferroporphyrins on CYP3A4 and CYP2B6 activity. Baculosomes expressing the recombinant human CYP isoforms were incubated with Fe-TCP (A) or Fe-TMPyP (B) and the fluorogenic substrate, BOMCC (10 µM). The rates of formation of the fluorescent reaction product, 3-cyano-7-hydroxycoumarin, was determined (λex 415/λem 460 nm). Data points are mean±SD of triplicate determinations (SDs are smaller than the symbols). Fe-TCP or Fe-TMPyP did not significantly interfere with the assay readouts.
Peroxynitrite causes oxidative modification of mitochondrial proteins
Peroxynitrite can modify tyrosine residues in proteins by nitration, and/or it can directly oxidize sulfhydryl groups. To first analyze whether EFV-induced oxidant/nitrative stress was associated with nitration, we explored changes in global mitochondrial protein nitration in an immunoblotting approach, using a nitrotyrosine antibody, as a biomarker of nitration. We found that the abundance of major immunoreactive mitochondrial protein bands did not significantly differ between EFV-exposed cells and solvent controls (not shown). In contrast, we found that EFV caused oxidative modification of target proteins involved in mitochondria-mediated cell death pathways. Specifically, we explored the role of oxidative modification of cyclophilin D (CypD), a modulator of the mitochondrial permeability transition (mPT) [26]. We found by Western immunoblotting that in solvent controls there was a distinct band of 18 kDa representing the CypD monomer (Fig. 7). In contrast, EFV treatment of hepatocytes resulted in loss of the 18 kDa band and in the formation of CypD protein aggregation, which was reversed to the monomers under reducing conditions (reduction of disulfide bonds by β-mercaptoethanol). These data suggest that EFV causes thiol oxidation in CypD and probably other proteins targeted by peroxynitrite.
Fig. 7.
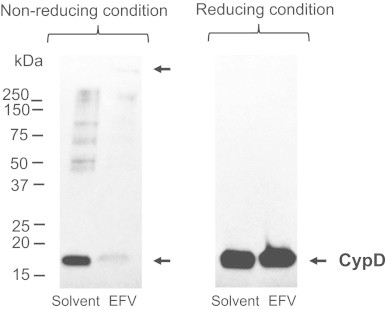
Effect of EFV exposure on mitochondrial protein thiol oxidation. Hepatocytes were treated with EFV (50 µM) or solvent for 4.5 h, and mitochondrial proteins were resolved and probed with anti-CypD antibody by western immunoblotting techniques in the presence (reducing conditions) or absence (non-reducing conditions) of β-mercaptoethanol.
EFV causes shifts in the NADH/NAD+ ratio and induces mitochondrial protein hyperacetylation via inhibition of Sirt3
To explore whether EFV altered the ratio of NADH/NAD+ in favor of NADH, due to the decreased NQR activity at complex I, we determined NADH and NAD+ concentrations in hepatocytes exposed to EFV. We found that EFV indeed increased the NADH/NAD+ ratio by ~50% (Fig. 8A). Based on these data, we hypothesized that the reduced capacity of mitochondria to provide NAD+ to Sirt3, which utilizes NAD+ as a required cofactor, might inhibit Sirt3 activity. Sirt3 activity cannot be measured with a Sirt3 assay because NAD+ added in excess is part of the assay; however, we measured Sirt3 activity indirectly by analyzing the degree of protein hyperacetylation with immunochemical techniques. Again, we chose CypD as one target protein that is regulated by Sirt3 via deacetylation. CypD was isolated by immunoprecipitation with an anti-acetyl-lysine antibody, followed by immunoblotting and probing with a CypD antibody. We found that EFV treatment resulted in increased levels of acetylated CypD, as compared to solvent controls (Fig. 8B), suggesting that Sirt3 activity (protein lysine deacetylation) was partially inhibited.
Fig. 8.
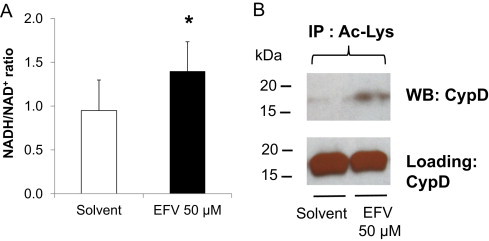
Effects of EFV on the hepatocellular NADH/NAD+ ratio and on mitochondrial protein acetylation. (A) Hepatocytes were exposed to EFV (50 µM) for 3 h, and NADH/NAD+ levels were determined. *P<0.05 versus solvent control. (B) Abundance of acetylated lysine (Ac-Lys) of CypD in mitochondria isolated from solvent controls or EFV (50 µM)-treated hepatocytes (4.5 h exposure). The Ac-Lys antibody was used to immunoprecipitate mitochondrial proteins, and the immunoblot analysis was performed with anti-CypD antibody. The CypD immunoblot analysis shows equal loading of samples.
Genetic ablation of Sirt3 (Sirt3−/− mice) protects against EFV toxicity
Evidence suggests that mitochondrial protein hyperacetylation, due to inhibition of Sirt3 activity, may sensitize cells to the induction of mPT and aggravation of cell death [27]. To determine whether inhibition of Sirt3 activity and the resulting protein hyperacetylation might contribute to the modulation of the degree of toxicity, we chose a genetic approach to eliminate Sirt3. Mice with a homozygous deletion of Sirt3 (Sirt3−/−) on a 129S1/SvImJ background were genotyped. Hepatocytes were isolated from both Sirt3−/− mice and their wild-type controls (Sirt3+/+), and overnight cultures were exposed to EFV. Both genotypes exhibited the same steep onset of toxicity between 30 and 50 µM as observed in the C57BL/6J mice (Fig. 9). However, the Sirt3-null mice were clearly protected against 40 µM EFV. At higher concentrations (50 µM), this protection was lost. These data suggest that genetic ablation of Sirt3 provides partial protection against the mitochondrial toxicity of EFV (but not necessarily against other, nonmitochondrial pathways of toxicity). However, the findings were unexpected as we had initially speculated that inhibition of Sirt3 activity or genetic ablation would promote, rather than protect against, EFV toxicity.
Fig. 9.
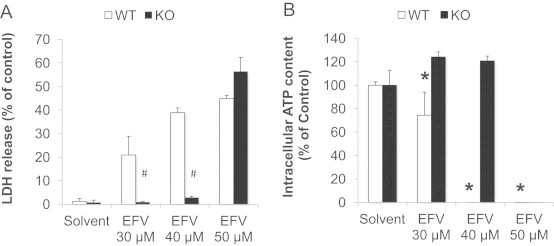
EFV-induced cell injury in hepatocytes isolated from Sirt3+/+ (WT) or Sirt3−/− (KO) mice. LDH release (A) and intracellular ATP content (B) were measured after treatment with solvent or EFV for 24 h. Data are mean±SD of three independent hepatocyte preparations using quadruplicate wells. *P<0.05 versus solvent control; #P<0.05 KO versus WT.
To ascertain whether the protective effects of Sirt3 deletion may be caused by different levels of complex I expression and activity in the Sirt3−/− mice, we next measured complex I activity in mitochondria isolated from both genotypes. We found that complex I activity in the Sirt3−/− mice did not exhibit any significant differences as compared to wild-type controls (Supplementary Fig. 1). The activity data were normalized to VDAC protein levels to correct for potential changes in mitochondrial mass. Furthermore, to assess whether the protection against EFV toxicity in Sirt3−/− mice might be caused by differential response to the inhibitory action of EFV on complex I, as a possible result of differential degrees of acetylation of complex I, another target for Sirt3-mediated deacetylation [9,28] we exposed hepatocytes from both genotypes to various concentrations of EFV. We found that EFV added to submitochondrial particles from Sirt3−/− mice caused a similar concentration-dependent inhibition of complex I activity as that in wild-type controls. These data indicate that the protective effect of Sirt3 depletion on EFV-induced toxicity is unlikely a consequence of smaller protein expression levels of complex I or decreased sensitivity to EFV on complex I activity.
Discussion
This study was designed to explore the relative contribution of distinct molecular pathways in the induction of cell injury following inhibition of mitochondrial complex I by drugs. To model pharmacologic inhibition of complex I, we used EFV, which has been reported earlier to be a potent inhibitor of complex I [16,24], which was confirmed and extended in this study. Specifically, we analyzed three major consequences of complex I inhibition; ATP depletion, ROS/RNS formation, and Sirt3 inhibition (Fig. 10). We found that ROS/RNS formation had the greatest impact as a causal factor in the pathogenesis of cell injury. This conclusion was supported by a number of findings.
Fig. 10.
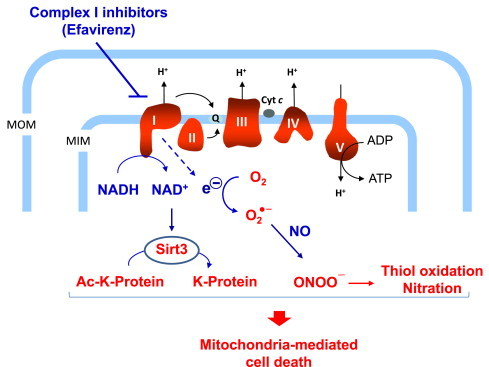
Schematic representation of the putative mechanisms involved in EFV-mediated inhibition of mitochondrial complex I. The major consequences of complex I inhibition are alterations in the NADH/NAD+ balance and the generation of peroxynitrite stress, resulting in mitochondrial protein hyperacetylation and thiol oxidation, respectively. MOM, mitochondrial outer membrane; MIM, mitochondrial inner membrane; Q, ubiquinone; Ac−K, acetylated-lysine; ONOO−, peroxynitrite.
First, one of the major direct consequences of EFV-induced complex I inhibition is a disruption of cellular bioenergetics, due to severely impaired ETC function. However, it is unlikely that this is the major mode of toxicity, at least at lower EFV concentrations. One of the reasons is that, in organs with a high energy demand, ATP is present at millimolar levels; therefore, even if there was a 50% reduction in ATP concentration, ATP levels would still be way above the km of almost all intracellular enzymes [29]. However, it is possible that, at higher EFV concentrations (≥50 µM), ATP would be depleted to dangerously low levels.
Second, it has been shown previously that exposure of hepatocytes to EFV causes an increase in mitochondrial superoxide formation and decreases in cellular GSH levels [22]. In addition, it has been recently reported that EFV increases mitochondrial nitric oxide production [30]. Here, we provide evidence that EFV rapidly induced the formation of peroxynitrite (demonstrated with the highly selective boronate probe, CBA). Although the subcellular source of peroxynitrite has not been determined, it likely has mitochondrial origin because superoxide generated in mitochondria cannot easily cross biomembranes. Importantly, the SOD mimetic Fe-TCP, or the peroxynitrite decomposition catalyst Fe-TMPyP, not only reduced the net formation of superoxide/peroxynitrite, respectively, but they also attenuated the cytotoxic response in hepatocytes in a concentration-dependent manner. Because both of the Fe-porphyrins may also affect other cellular processes, apart from decreasing superoxide/peroxynitrite, we explored their potential role in inhibiting CYP activity. However, as reported in this study, the protective effects of Fe-TCP or Fe-TMPyP are unlikely related to CYP-mediated EFV bioactivation in the mouse model. Taken together, the data suggest that peroxynitrite might play a major role in the pathways leading to cell death. However, it cannot be excluded that peroxynitrite-independent pathways may also be involved. The exact mode of cell death is not clear; however, peroxynitrite can cause oxidative modification of proteins and the formation of aggregates, which can lead to mPT pore formation. This type of pore is insensitive to CsA (“unregulated mPT”). Indeed, in our mouse hepatocyte model, CsA (1 µM) did not protect cells against EFV-induced cell death (data not shown). It is possible that, especially at high concentrations of EFV (50 µM and greater), other, non-mitochondrial effects may be involved, as demonstrated with neuronal cells [31].
Third, we hypothesized that if complex I-targeting drugs (including EFV) increased the NADH/NAD+ ratio (as a consequence of inhibition of electron flow from NADH to ubiquinone), this would inhibit the NAD+-dependent Sirt3 activity. Sirt3 is a NAD+-dependent deacetylase localized to the mitochondrial matrix [32,33] and expressed at high levels in a number of tissues including liver [13]. Like other sirtuins, Sirt3 removes the acetyl group from N-ε-lysine residues. Because decreased Sirt3 activity leads to hyperacetylation of a number of mitochondrial proteins, we sought to determine whether EFV-induced inhibition of Sirt3 would contribute to the precipitation of cell injury. However, surprisingly, Sirt3-null mice were not more sensitive to EFV toxicity; rather, they exhibited partial protection against EFV in a narrow concentration range, although overall protein acetylation was increased.
The molecular targets of Sirt3-mediated deacetylation of mitochondrial lysine residues include several proteins; in fact, it has been demonstrated that ~35% of all mitochondrial proteins have at least one acetylation site [34]. One important candidate protein is CypD [27,35]. CypD is acetylated at K166; this highly conserved site is adjacent to the CsA-binding pocket of CypD, suggesting that acetylation/deacetylation might regulate the mPT [35]. Other Sirt3 targets include the SdhA subunit of complex II [36] and the Ndufa9 subunit of complex I [29]. However, the mechanistic role of these altered respiratory complexes in drug-induced Sirt3 inhibition is not known. While we did not find any differences in complex I activity between Sirt3−/− mice and their wild-type controls, another report found that Sirt3 knockout mice exhibited slightly decreased respiration rates in mitochondria energized with glutamate/malate as compared to wild-type controls [29].
The mechanisms underlying the protection against EFV toxicity in Sirt3-null mice are not known. Because Sirt3 is a lysine deacetylase, it is likely that the differential lysine acetylation status of certain critical proteins is involved. In line with this is a recent report demonstrating that Sirt3 KO mice were protected from acetaminophen hepatotoxicity because of a specific role of mitochondrial aldehyde dehydrogenase, which is a target of Sirt3 [37]. Because Sirt3 localizes to the mitochondrial matrix, the mode of protection afforded by the loss of Sirt3 may be operating at the mitochondrial level. According to this concept, it is possible that 30 and 40 µM EFV-treated cells from Sirt3 KO mice are protected via a mitochondrial mechanism, whereas cells exposed to 50 µM EFV are no longer protected because of a more generalized extramitochondrial stress.
In conclusion, EFV at lower concentrations (<50 µM) caused toxicity to cultured mouse hepatocytes by inhibiting complex I and causing increased oxidant stress via the formation of peroxynitrite. At higher concentrations (>50 µM), the massive and sustained ATP depletion may directly lead to cell death by disrupting the cellular bioenergetics. However, such high EFV concentrations are less relevant if extrapolated to the clinical situation [38]. Sirt3 depletion did not result in exacerbation of cell injury but, at EFV concentrations <50 µM, afforded protection against the cytotoxic effects of EFV. Taken together, the data are compatible with the concept that peroxynitrite formation, resulting from mitochondrial complex I inhibition, is a major mechanism of mitochondria-mediated cell injury.
Acknowledgments
This work was supported by a grant from Connecticut Innovations (11SCDIS02, to U.A.B.) and by the Boehringer Ingelheim Endowed Chair in Mechanistic Toxicology at UCONN.
Appendix A. Supplementary materials
Effects of EFV on complex I activity in Sirt3+/+ and Sirt3−/− mice. (A) Isolated liver mitochondria from both genotypes were used to determine the effects of EFV on NQR activity. Rotenone (ROT, 20 µM) was used as a positive control. Data are mean±SD of three independent mitochondrial preparations using duplicate determinations. (B) Mitochondrial proteins were resolved and probed with anti-VDAC antibody by western immunoblotting techniques under reducing conditions. GAR; goat anti-rabbit antibody conjugated with HRP.
Supplementary Data
References
- 1.Chan K., Truong D., Shangari N., O’Brien P.J. Drug-induced mitochondrial toxicity. Expert Opinion on Drug Metabolism and Toxicology. 2005;1(4):655–669. doi: 10.1517/17425255.1.4.655. 16863431 [DOI] [PubMed] [Google Scholar]
- 2.Dykens J.A., Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discovery Today. 2007;12(17–18):777–785. doi: 10.1016/j.drudis.2007.07.013. 17826691 [DOI] [PubMed] [Google Scholar]
- 3.Pereira C.V., Moreira A.C., Pereira S.P., Machado N.G., Carvalho F.S., Sardão V.A., Oliveira P.J. Investigating drug-induced mitochondrial toxicity: a biosensor to increase drug safety? Current Drug Safety. 2009;4(1):34–54. doi: 10.2174/157488609787354440. 19149524] [DOI] [PubMed] [Google Scholar]
- 4.Pessayre D., Fromenty B., Berson A., Robin M.A., Lettéron P., Moreau R., Mansouri A. Central role of mitochondria in drug-induced liver injury. Drug Metabolism Reviews. 2012;44(1):34–87. doi: 10.3109/03602532.2011.604086. 21892896] [DOI] [PubMed] [Google Scholar]
- 5.Nadanaciva S., Bernal A., Aggeler R., Capaldi R., Will Y. Target identification of drug induced mitochondrial toxicity using immunocapture based OXPHOS activity assays. Toxicology in Vitro. 2007;21(5):902–911. doi: 10.1016/j.tiv.2007.01.011. 17346924] [DOI] [PubMed] [Google Scholar]
- 6.Degli Esposti M. Inhibitors of NADH-ubiquinone reductase: an overview. Biochimica et Biophysica Acta. 1998;1364(2):222–235. doi: 10.1016/s0005-2728(98)00029-2. 9593904 [DOI] [PubMed] [Google Scholar]
- 7.Perier C., Tieu K., Guégan C., Caspersen C., Jackson-Lewis V., Carelli V., Martinuzzi A., Hirano M., Przedborski S., Vila M. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proceedings of the National Academy Sciences of the United States of America. 2005;102(52):19126–19131. doi: 10.1073/pnas.0508215102. 16365298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fato R., Bergamini C., Bortolus M., Maniero A.L., Leoni S., Ohnishi T., Lenaz G. Differential effects of mitochondrial complex I inhibitors on production of reactive oxygen species. Biochimica et Biophysica Acta. 2009;1787(5):384–392. doi: 10.1016/j.bbabio.2008.11.003. 19059197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karamanlidis G., Lee C.F., Garcia-Menendez L., Kolwicz S.C., Suthammarak W., Gong G., Sedensky M.M., Morgan P.G., Wang W., Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metabolism. 2013;18(2):239–250. doi: 10.1016/j.cmet.2013.07.002. 23931755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirst J. Mitochondrial complex I. Annual Review of Biochemistry. 2013;82:551–575. doi: 10.1146/annurev-biochem-070511-103700. 23527692 [DOI] [PubMed] [Google Scholar]
- 11.Neubauer S. The failing heart − an engine ou of fuel. New England Journal of Medicine. 2007;356(11):1140–1151. doi: 10.1056/NEJMra063052. 17360992 [DOI] [PubMed] [Google Scholar]
- 12.Davey G.P., Peuchen S., Clark J.B. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. Journal of Biological Chemistry. 1998;273(21):12753–12757. doi: 10.1074/jbc.273.21.12753. 9582300 [DOI] [PubMed] [Google Scholar]
- 13.Lombard D.B., Alt F.W., Cheng H.L., Bunkenborg J., Streeper R.S., Mostoslavsky R., Kim J., Yancopoulos G., Valenzuela D., Murphy A., Yang Y., Chen Y., Hirschey M.D., Bronson R.T., Haigis M., Guarente L.P., Farese R.V., Weissman S., Verdin E., Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Molecular and Cellular Biology. 2007;27(24):8807–8814. doi: 10.1128/MCB.01636-07. 17923681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sulkowski M.S., Thomas D.L., Chaisson R.E., Moore R.D. Hepatotoxicity associated with antiretroviral therapy in adults infected with human immunodeficiency virus and the role of hepatitis C or B virus infection. The Journal of the American Medical Association. 2000;283(1):74–80. doi: 10.1001/jama.283.1.74. [DOI] [PubMed] [Google Scholar]
- 15.Jones M., Núñez M. Liver toxicity of antiretroviral drugs. Seminars in Liver Disease. 2012;32(2):167–176. doi: 10.1055/s-0032-1316472. 22760656 [DOI] [PubMed] [Google Scholar]
- 16.Blas-García A., Apostolova N., Ballesteros D., Monleón D., Morales J.M., Rocha M., Victor V.M., Esplugues J.V. Inhibition of mitochondrial function by efavirenz increases lipid content in hepatic cells. Hepatology. 2010;52(1):115–125. doi: 10.1002/hep.23647. 20564379 [DOI] [PubMed] [Google Scholar]
- 17.Lee K.K., Fujimoto K., Zhang C., Schwall C.T., Alder N.N., Pinkert C.A., Krueger W., Rasmussen T., Boelsterli U.A. Isoniazid-induced cell death is precipitated by underlying mitochondrial complex I dysfunction in mouse hepatocytes. Free Radical Biology and Medicine. 2013;65:584–594. doi: 10.1016/j.freeradbiomed.2013.07.038. 23911619 [DOI] [PubMed] [Google Scholar]
- 18.Zielonka J., Sikora A., Hardy M., Joseph J., Dranka B.P., Kalyanaraman B. Boronate probes as diagnostic tools for real time monitoring of peroxynitrite and hydroperoxides. Chemical Research in Toxicology. 2012;25(9):1793–1799. doi: 10.1021/tx300164j. 22731669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marks B.D., Goossens T.A., Braun H.A., Ozers M.S., Smith R.W., Lebakken C., Trubetskoy O.V. High-throughput screening assays for CYP2B6 metabolism and inhibition using fluorogenic vivid substrates. AAPS PharmSciTech. 2003;5(2):E18. doi: 10.1208/ps050218. 12866948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W., Ramamoorthy Y., Kilicarslan T., Nolte H., Tyndale R.F., Sellers E.M. Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metabolism and Disposition. 2002;30(3):314–318. doi: 10.1124/dmd.30.3.314. 11854151 [DOI] [PubMed] [Google Scholar]
- 21.Birch-Machin M.A., Turnbull D.M. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods in Cell Biology. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. 11381612 [DOI] [PubMed] [Google Scholar]
- 22.Apostolova N., Gomez-Sucerquia L.J., Moran A., Alvarez A., Blas-Garcia A., Esplugues J.V. Enhanced oxidative stress and increased mitochondrial mass during efavirenz-induced apoptosis in human hepatic cells. British Journal of Pharmacology. 2010;160(8):2069–2084. doi: 10.1111/j.1476-5381.2010.00866.x. 20649602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mutlib A.E., Chen H., Nemeth G.A., Markwalder J.A., Seitz S.P., Gan L.S., Christ D.D. Identification and characterization of efavirenz metabolites by liquid chromatography/mass spectrometry and high field NMR: species differences in the metabolism of efavirenz. Drug Metabolism and Disposition. 1999;27:1319–1333. [PubMed] [Google Scholar]
- 24.Lee K.K., Boelsterli U.A. Bypassing the compromised mitochondrial electron transport with methylene blue alleviates efavirenz/isoniazid-induced oxidant stress and mitochondria-mediated cell death in mouse hepatocytes. Redox Biology. 2014;2C:599–609. doi: 10.1016/j.redox.2014.03.003. 25460728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi I.Y., Lee S.J., Nam W., Park J.S., Ko K.H., Kim H.C., Shin C.Y., Chung J.H., Noh S.K., Choi C.R., Shin D.H., Kim W.K. Augmented death in immunostimulated astrocytes deprived of glucose: inhibition by an iron porphyrin FeTMPyP. Journal of Neuroimmunology. 2001;112(1–2):55–62. doi: 10.1016/s0165-5728(00)00382-9. 11108933 [DOI] [PubMed] [Google Scholar]
- 26.Linard D., Kandlbinder A., Degand H., Morsomme P., Dietz K.J., Knoops B. Redox characterization of human cyclophilin D: identification of a new mammalian mitochondrial redox sensor? Archives of Biochemistry and Biophysics. 2009;491(1–2):39–45. doi: 10.1016/j.abb.2009.09.002. 19735641 [DOI] [PubMed] [Google Scholar]
- 27.Shulga N., Wilson-Smith R., Pastorino J.G. Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. Journal of Cell Science. 2010;123(6):894–902. doi: 10.1242/jcs.061846. 20159966 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Shulga N., Pastorino J.G. Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-D mediated by sirtuin-3. Journal of Cell Science. 2010;123(23):4117–4127. doi: 10.1242/jcs.073502. 21062897 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Ahn B.H., Kim H.S., Song S., Lee I.H., Liu J., Vassilopoulos A., Deng C.X., Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(38):14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Apostolova N., Funes H.A., Blas-Garcia A., Alegre F., Polo M., Esplugues J.V. Involvement of nitric oxide in the mitochondrial action of efavirenz: a differential effect on neurons and glial cells. The Journal of Infectious Disease. 2014 doi: 10.1093/infdis/jiu825. [DOI] [PubMed] [Google Scholar]
- 31.Purnell P.R., Fox H.S. Efavirenz induces neuronal autophagy and mitochondrial alterations. Journal of Pharmacology and Experimental Therapeutics. 2014;351(2):250–258. doi: 10.1124/jpet.114.217869. 25161171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Onyango P., Celic I., McCaffery J.M., Boeke J.D., Feinberg A.P. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13653–13658. doi: 10.1073/pnas.222538099. 12374852] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwer B., North B.J., Frye R.A., Ott M., Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. Journal of Cell Science. 2002;158(4):647–657. doi: 10.1083/jcb.200205057. 12186850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson K.A., Hirschey M.D. Mitochondrial protein acetylation regulates metabolism. Essays in Biochemistry. 2012;52:23–35. doi: 10.1042/bse0520023. 22708561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hafner A.V., Dai J., Gomes A.P., Xiao C.Y., Palmeira C.M., Rosenzweig A., Sinclair D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging. 2010;2(12):914–923. doi: 10.18632/aging.100252. 21212461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cimen H., Han M.J., Yang Y., Tong Q., Koc H., Koc E.C. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010;49(2):304–311. doi: 10.1021/bi901627u. 20000467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu Z., Bourdi M., Li J.H., Aponte A.M., Chen Y., Lombard D.B., Gucek M., Pohl L.R., Sack M.N. SIRT3-dependent deacetylation exacerbates acetaminophen hepatotoxicity. EMBO Reports. 2011;12(8):840–846. doi: 10.1038/embor.2011.121. 21720390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burger D., van der Heiden I., La Porte C., van der Ende M., Groeneveld P., Richter C., Koopmans P., Kroon F., Sprenger H., Lindemans J., Schenk P., van Schaik R. Interpatient variability in the pharmacokinetics of the HIV non-nucleoside reverse transcriptase inhibitor efavirenz: the effect of gender, race, and CYP2B6 polymorphism. British Journal of Clinical Pharmacology. 2006;61(2):148–154. doi: 10.1111/j.1365-2125.2005.02536.x. 16433869 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of EFV on complex I activity in Sirt3+/+ and Sirt3−/− mice. (A) Isolated liver mitochondria from both genotypes were used to determine the effects of EFV on NQR activity. Rotenone (ROT, 20 µM) was used as a positive control. Data are mean±SD of three independent mitochondrial preparations using duplicate determinations. (B) Mitochondrial proteins were resolved and probed with anti-VDAC antibody by western immunoblotting techniques under reducing conditions. GAR; goat anti-rabbit antibody conjugated with HRP.
Supplementary Data