

Abstract
AIM: To observe the effect of vincristine on hepatitis B virus (HBV) replication in vitro and to study its possible mechanisms.
METHODS: Vincristine was added to the cultures of two cell lines stably expressing HBV. Then, the levels of hepatitis B surface antigen (HBsAg), hepatitis B e antigen (HBeAg), and hepatitis B core antigen (HBcAg) in the supernatants or cytoplasm were examined using by enzyme-linked immunosorbent assay and Western blot. The HBV pregenome RNA (pgRNA) was detected using reverse transcription-PCR and real-time fluorescent quantitative PCR (RT-qPCR), and viral DNA was detected using Southern blot and RT-qPCR. Cell proliferation after drug treatment was detected using the BrdU incorporation test and the trypan blue exclusion assay. Cell cycle and cell apoptosis were examined using flow cytometry and Western blot.
RESULTS: Vincristine up-regulated HBV replication directly in vitro in a dose-dependent manner, and 24-h exposure to 0.1 μmol/L vincristine induced more than 4-fold and 3-fold increases in intracellular HBV DNA and the secretion of viral DNA, respectively. The expression of HBV pgRNA, intracellular HBsAg and HBcAg, and the secretion of HBeAg were also increased significantly after drug treatment. Most importantly, vincristine promoted the cell excretion of HBV nucleocapsids instead of HBV Dane particles, and the nucleocapsids are closely related to the HBV pathogenesis. Furthermore, vincristine inhibited the proliferation of cells stably expressing HBV. The higher the concentration of the drug, the more significant the inhibition of the cell proliferation and the stronger the HBV replication ability in cells. Flow cytometry indicated that cell cycle arrest at S-phase was responsible for the cell proliferation inhibition.
CONCLUSION: Vincristine has a strong stimulatory effect on HBV replication and induces cell cycle arrest, and cell proliferation inhibition may be conducive to viral replication.
Keywords: Viral reactivation, Cytotoxic chemotherapy, Cell cycle arrest, Proliferation, Hepatitis B virus
Core tip: Hepatitis B virus (HBV) reactivation is an emerging clinical challenge for HBV carriers receiving anti-cancer chemotherapy. However, the mechanism for the sharp increase in viral replication in the early stages of chemotherapy is still unclear. In this research, we found that vincristine, which is a cytotoxic drug, had a direct stimulatory effect on HBV replication without the involvement of immune factors and that vincristine induced cell cycle arrest; cell proliferation inhibition may be conducive to viral replication, and this may be a novel mechanism of HBV reactivation following cytotoxic chemotherapy.
INTRODUCTION
It has been reported that approximately 2 billion people around the world have been infected with hepatitis B virus (HBV), and approximately 400 million are chronic hepatitis B surface antigen (HBsAg) carriers[1]. In most patients, the virus is quiescent or replicates at a low level. However, chemotherapy may disrupt the balance between the host immune responses and viral replication; of note, enhanced viral replication is a universal phenomenon in clinical cases. Although there are no uniform diagnostic criteria for HBV reactivation, clinical practice suggests that the definition is a 10-fold increase above baseline in serum HBV DNA levels or the reappearance of HBsAg or de novo detection of HBV DNA in HBsAg(-) patients[1,2].
To date, the mechanisms of HBV reactivation have been incompletely understood. Most researchers think that there are at least two underlying mechanisms of HBV reactivation induced by chemotherapy. As the host immune response to the virus plays a pivotal role in controlling HBV infection and replication[3], immune reconstitution following withdrawal of chemotherapy agents should increase viral replication. However, a few anti-cancer agents, such as glucocorticoids, may have a direct stimulatory effect on viral replication. In vitro, corticosteroids increase HBV expression by binding to the glucocorticoid responsive element and augmenting the HBV enhancer I[4,5]. However, steroid-free chemotherapy may also increase HBV replication[6,7]. In some cases, HBV reactivation occurred within the first 2 wk of chemotherapy[6]. It is very likely that HBV reactivation in these cases occurs before immune reconstitution following withdrawal of anti-cancer agents. One possible mechanism for the early reactivation of HBV is that cytotoxic agents may have a direct stimulatory effect on HBV replication.
In this study, we aimed to investigate whether vincristine stimulates HBV replication directly in vitro. If this assumption is valid, it may suggest a novel mechanism of HBV reactivation caused by cytotoxic chemotherapy.
MATERIALS AND METHODS
Cell culture and treatment
HepG2, HepG2-HBV1.1 and HepG2.2.15 cells were cultured in minimum essential medium (MEM, Gibco, Grand Island, CA, United States) with 10% fetal bovine serum (FBS, Hyclone, Shanghai, China) and maintained in a humidified incubator with 5% CO2 at 37 °C. For cytotoxic chemotherapy treatment, cells were treated with vincristine (0-0.5 μmol/L, Pharmachemic Hisun, Zhejiang, China) for 24 h, and the medium was then replaced with fresh serum-free MEM medium for another 48 h of incubation until the cells were harvested.
Production of the stable HBV-expressing cell line HepG2-HBV1.1
HepG2 cells were transfected with pneo-CH9/HBV1.1 vector[8], which contains the cytomegalovirus (CMV) promoter and 1.1 copies of the hepatitis B virus genome, and were then selected with G418 (1200 μg/mL). Isolated cell colonies with the integrated HBV genome were selected after confirmation by Southern blot and Western blot analyses.
Detection of the effect of vincristine on cell viability and proliferation by the trypan blue exclusion assay
HepG2.2.15 (4 × 105 cells/well) and HepG2-HBV1.1 (6 × 105 cells/well) cells were seeded in six-well plates. Following adherence of the cells, vincristine was added to the medium at various concentrations (0-0.5 μmol/L) for 24 h, followed by 48 h of incubation in drug-free culture medium. The cells in each well were washed with phosphate-buffered saline twice following the addition of 200 μL of trypsin and then placed in a 37 °C 5% CO2 incubator for 1-2 min. Subsequently, 800 μL of 10% FBS-containing MEM was added to each well to terminate the reaction, and the cell suspension was pipetted several times to disperse the cells evenly and to count the cells (cell viability = number of live cells/number of total cells). The maximum concentration that did not affect cell viability was chosen as the experimental concentration. For detecting the cell proliferation ability, vincristine at the experimental concentration was added to the six-well plates for 24 h, followed by 0 h, 24 h, 48 h, and 72 h of incubation in drug-free culture medium, and the cells were harvested from each well and counted.
BrdU incorporation test
HepG2.2.15 (4 × 105 cells/well) and HepG2-HBV1.1 (6 × 105 cells/well) cells were seeded in six-well plates. Following adherence of the cells, 0.1 μmol/L vincristine was added to the medium for 24 h, and the cells were then treated with trypsin and seeded in 96-well plates (4 × 103 cells/well) for another 48 h of incubation. The remaining steps were completed based on the manufacturers’ instructions (Cell Proliferation ELISA, BrdU; Roche).
Quantification of HBV DNA copies by real-time fluorescent quantitative PCR
HBV replicative intermediates in the cells were obtained as follows. Cells from one 35-mm diameter dish were lysed with 0.5 mL of lysis buffer containing 10 mmol/L Tris-HCl (pH 8.0), 1 mmol/L EDTA, 1% NP-40, and 2 % sucrose at 37 °C for 10 min. Cell debris and nuclei were removed by centrifugation, and the supernatant was mixed with 200 μL of 35% polyethylene glycol (PEG) 8000 containing 1.5 mol/L NaCl. After incubation on ice for 2 h, viral nucleocapsids were pelleted by centrifugation at 12000 g for 10 min at 4 °C, followed by 24-h digestion at 37 °C in 400 μL of digestion buffer containing 0.5 mg/mL pronase (Takara, Japan), 0.5% sodium dodecyl sulfate (SDS), 10 mmol/L Tris-HCl (pH 8.0), and 10 mmol/L EDTA. The digestion mixture was extracted twice with phenol, and DNA was precipitated with ethanol and dissolved in TE (10 mmol/L Tris-HCl, pH 8.0, 1 mmol/L EDTA) buffer. To collect viral particles instead of free viral DNA in the culture medium, the supernatant was subjected to 35% PEG8000 precipitation overnight (Chi et al, 1998), and the precipitates were then digested according to previously described methods.
The quantification of HBV copies was performed using SYBER-Green assays (Roche, Germany). The primers were designed specifically to amplify the conserved region of the HBV gene: forward primer (F2150), 5’-CCTAGTAGTCAGTTATGTCAAC-3’; reverse primer (R2300), 5’-TCTATAAGCTGGAGGAGTGCGA-3’. The pneo-CH9/HBV1.1 plasmid at different concentrations (5 × 102, 5 × 103, 5 × 104, 5 × 105, 5 × 106, 5 × 107 copies/μL) was used as a template to create the standard curve. The cycling parameters were as follows: initial denaturation at 95 °C for 3 min; 10 cycles of denaturation at 94 °C for 15 s, annealing at 65 °C for 30 s, and extension at 72 °C for 20 s; and 30 cycles of denaturation at 94 °C for 15 s, annealing at 65-55 °C (starting from 65 °C, 1 °C lower after each cycle) for 30 s, and extension at 72 °C for 20 s, with simultaneous fluorescence detection.
Quantification of HBV pregenome RNA (pgRNA) by real-time fluorescent quantitative PCR
Total RNA was extracted using a DNA-free RNA mini extraction kit (Watson, Shanghai, China). One microgram of total RNA was used for cDNA synthesis, which was performed using reverse transcription with the PrimeScript RT reagent kit (Perfect Real Time; Takara, Japan). Relative quantification was performed using SYBER-Green assays (Roche, Germany) for the target genes (HBV 3.5 kb mRNA), with β-actin mRNA as the endogenous control. The expression values of target genes were calculated using the 2-ΔΔCt method.
Southern blot analysis
HBV replicative intermediates were extracted from the cells or the supernatant of the culture medium according to previous methods and then separated on 0.8% agarose gels. DNA samples were transferred onto nylon membranes (Roche, Germany). After ultraviolet crosslinking and prehybridization, the membranes were hybridized with a DIG-labeled HBV-specific probe from a random-primed labeling kit (Roche, Germany). The signal was detected by exposure to an X-ray film and was scanned using the Versa Doc Imaging system (Bio-Rad).
Enzyme-linked immunosorbent assay (ELISA)
The levels of HBsAg and hepatitis B e antigen (HBeAg) in the culture medium and cell extracts were assessed using ELISA following the manufacturer’s protocol (Kehua Biotec Inc., Shanghai, China). The levels of HBV core antigen (HBcAg) in the culture medium were assessed using an ELISA kit (Disease Diagnosis Reagent and Vaccine Engineering Technology Research Center of China Infectious State, Xiamen University, China). The kit contains two types of 96-well plates; one type is coated with HBsAb so that it can capture the Dane particle, and the other is coated with HBcAb and can therefore detect the total HBcAg from the Dane particle and the HBV nucleocapsid. Each experiment was performed in triplicate and independently repeated three times.
Flow cytometry
HepG2.2.15 (4 × 105 cells/well) and HepG2-HBV1.1 (6 × 105 cells/well) cells were seeded in six-well plates. After treatment with 0.1 μmol/L vincristine, the cells were washed twice with PBS. They were then fixed in cold 70% ethanol and stored at 4 °C for 30 min. Ethanol was then removed, and the cells were resuspended in PBS. The fixed cells were then washed with PBS, treated with RNase (100 mg/mL), and stained with propidium iodide (PI, 50 mg/mL; Merck, Germany) in the dark for 30 min at 37 °C. Subsequently, the DNA content and the cell cycle were analyzed by flow cytometry. For the apoptosis assay, treated and untreated cells were resuspended in 100 μL of binding buffer followed by staining with 1.25 μL of annexin V reagent and 10 μL of PI in the dark. The apoptotic cell fraction was analyzed using a FACScan cytometer, and the flow cytometry data were analyzed using FlowJo Software.
Western blot analysis
Cellular proteins were extracted using RIPA buffer supplemented with phenylmethanesulfonyl fluoride. Protein concentrations were determined using a BCA protein concentration determination kit (Beyotime Institute of Biotechnology, Shanghai, China). Equal amounts of samples were separated using 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes, followed by incubation with a primary monoclonal mouse anti-HBcAg antibody (antibody code 6D1D10E4, dilution 1: 150; Huazhong University of Science and Technology, Wuhan, China) or monoclonal rabbit anti-β-actin (dilution 1:2500; Santa Cruz, CA, United States). After incubation, the membranes were washed three times, and a goat anti-rabbit or goat anti-mouse secondary antibody (dilution 1:2500; Santa Cruz) was added for 1 h. After being washed three times with TBST buffer, signals were detected using the chemiluminescence ECL™ detection system (Pierce). For detecting the expression of the apoptotic signaling molecule poly(ADP-ribose) polymerase-1 (PARP-1), the primary antibody was a polyclonal rabbit anti-PARP-1-Ab (dilution 1:1000; Santa Cruz), and the secondary antibody was a monoclonal goat anti-rabbit (dilution 1:2500; Santa Cruz).
Statistical analysis
Data are expressed as the mean and standard deviation of at least three independent experiments. Statistical significance was determined by the independent-samples t-test. Differences were deemed statistically significant at P ≤ 0.05. All analyses were performed using the SPSS statistical software for Windows, version 10.1.4 (SPSS Inc., IL, United States).
RESULTS
Effect of vincristine on the viability of stable HBV-expressing cell lines
Vincristine as a cytotoxic chemotherapy agent can induce cells apoptosis or necrosis when administered at high concentrations[9,10] through a mechanism that may involve HBV replication. We sought to determine whether vincristine promotes HBV replication at lower concentrations. First, we examined the cytotoxic effect of vincristine on the viability of HepG2.2.15 and HepG2-HBV1.1 cells using the trypan blue exclusive assay and the MTT assay. The results of the two assays (Figure 1) indicated that 0.1 μmol/L vincristine had no significant cytotoxicity to both HepG2.2.15 and HepG2-HBV1.1 cells. In subsequent experiments, 0.1 μmol/L was used as the working concentration of vincristine.
Figure 1.
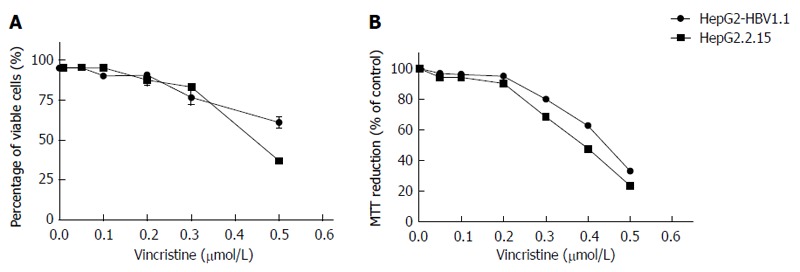
Effect of vincristine on the viability of stable hepatitis B virus-expressing cell lines. A: The effect of vincristine on the viability of HepG2.2.15 and HepG2-HBV1.1 cells based on the trypan blue exclusive assay; B: The effect of vincristine on the viability of HepG2.2.15 and HepG2-HBV1.1 cells based on the MTT assay. HepG2.2.15 (4 × 105/well) and HepG2-HBV1.1 (6 × 105/well) cells were treated with vincristine at various concentrations (0-0.5 μmol/L) for 24 h, followed by 48 h of incubation in drug-free culture medium. The cells were collected 72 h after drug treatment. The results are expressed as the arithmetic mean ± SD from 3 independent experiments.
Dose-dependent increases in HBV DNA and pgRNA in stable HBV-expressing cell lines after treatment with vincristine
HepG2.2.15[11] and HepG2-HBV1.1[12] cells were employed as stable HBV-expressing cell lines. HepG2.2.15 (4 × 105/well) and HepG2-HBV1.1 (6 × 105/well) cells were treated with vincristine at various concentrations (0-0.1 μmol/L) for 24 h, followed by 48 h of incubation in drug-free culture medium. Intracellular HBV nucleocapsids were extracted 72 h after drug treatment, and viral replication intermediates were then measured by real-time fluorescent quantitative PCR (RT-qPCR) and Southern blot analyses. As revealed by RT-qPCR, the HBV DNA copies were up-regulated dose-dependently after vincristine stimulation (Figure 2A). The intracellular HBV DNA copies increased (4.17 ± 0.43)-fold (P = 0.000) and (5.62 ± 0.93)-fold (P = 0.001) in HepG2.2.15 and HepG2-HBV1.1 cells, respectively, after treatment with 0.1 μmol/L vincristine. Similarly, the dose effects of HBV replication in cells activated by vincristine were confirmed by Southern blot analysis (Figure 2B). In addition, we also extracted the HBV DNA from the supernatant of the culture medium after precipitation with PEG8000 and performed RT-qPCR (Figure 2C) and Southern blot (Figure 2D) analyses. As expected, vincristine up-regulated HBV DNA levels in the supernatant in a dose-dependent manner; 0.1 μmol/L vincristine increased the secretion of HBV DNA copies by 4.14 ± 0.60 (P = 0.000) and 3.71 ± 0.28 (P = 0.000) times in HepG2-HBV1.1 cells and HepG2.2.15 cells, respectively. We further determined the effect of vincristine on the levels of HBV RNA. HBV pregenome RNA (pgRNA) was measured by RT-qPCR using primers targeting the 3.5 kb mRNA of the viral genome. As shown in Figure 2E, the levels of HBV pgRNA increased sharply with vincristine treatment (HepG2.2.15 cells: P = 0.010; HepG2-HBV1.1 cells: P = 0.046).
Figure 2.
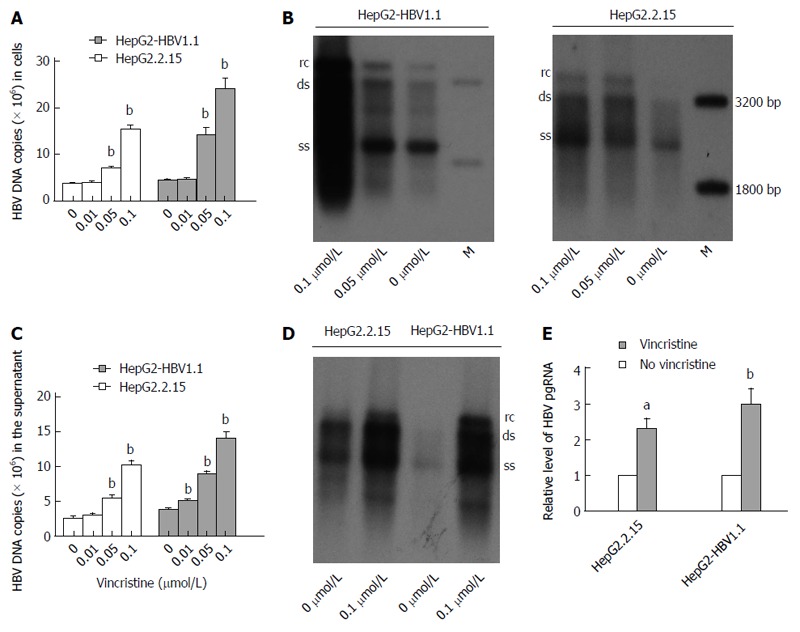
Dose-dependent increases in hepatitis B virus DNA and RNA in stable hepatitis B virus-expressing cell lines after treatment with vincristine. A: Dose-dependent increase in HBV DNA in HepG2-HBV1.1 and HepG2.2.15 cells treated with vincristine based on real-time fluorescent quantitative PCR analysis; B: Dose-dependent increase in HBV DNA in HepG2-HBV1.1 and HepG2.2.15 cells treated with vincristine based on Southern blot analysis; C: Vincristine increases HBV DNA in the supernatants of HepG2-HBV1.1 cells and HepG2.2.15 cells based on real-time fluorescent quantitative PCR analysis; D: Vincristine increases HBV DNA in the supernatants of HepG2-HBV1.1 cells and HepG2.2.15 cells based on Southern blot analysis; E: Vincristine up-regulates HBV pgRNA based on real-time fluorescent quantitative PCR analysis. HepG2-HBV1.1 (6 × 105/well) and HepG2.2.15 (4 × 105/well) cells were treated with 0.1 μmol/L vincristine for 24 h followed by 48 h of incubation in drug-free culture medium. HBV intermediates or total RNA were extracted 72 h after drug treatment. Values represent the mean ± SD of 3 independent experiments, and a representative experiment is shown. aP < 0.05, bP < 0.01 vs control group (no vincristine). rc: Relaxed circular DNA; ds: Double-stranded DNA; ss: Single-stranded DNA. M: DNA ladder; pgRNA: Pregenome RNA.
Vincristine promotes HBV protein expression
To elucidate the effect of vincristine on the expression of HBV proteins, we detected the levels of HBeAg, HBcAg and HBsAg using an ELISA assay or Western blot analysis. After treatment with 0.1 μmol/L vincristine, HBeAg in the supernatant (Figure 3A) was up-regulated based on the ELISA assay (HepG2.2.15 cells: P = 0.002; HepG2-HBV1.1 cells: P = 0.021), and HBcAg expression in the cytoplasm increased greatly based on the Western blot analysis (Figure 3B). Interestingly, vincristine increased the HBsAg expression in the cytoplasm, as shown in Figure 3C (HepG2.2.15 cells: P = 0.024; HepG2-HBV1.1 cells: P = 0.000), while the secretion of HBsAg was not affected (Figure 3D).
Figure 3.
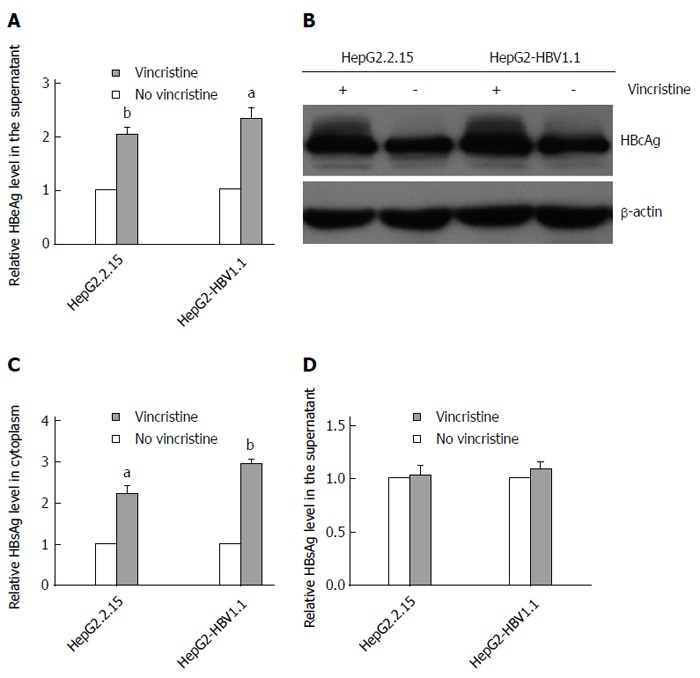
Vincristine promotes hepatitis B virus protein expression in HepG2-HBV1.1 and HepG2.2.15 cells. A: Vincristine up-regulates HBeAg expression. HBeAg in the supernatant was analyzed using an ELISA assay; B: Vincristine increases cellular HBcAg expression based on Western blot analysis; C: The effect of vincristine on intracellular HBsAg expression was analyzed using an ELISA assay; D: The effect of vincristine on HBsAg expression in the supernatant was analyzed using an ELISA assay. HepG2-HBV1.1 (6 × 105/well) and HepG2.2.15 (4 × 105/well) cells were treated with vincristine at a concentration of 0.1 μmol/L for 24 h followed by 48 h of incubation in drug-free culture medium. Values represent the mean ± SD of 3 independent experiments. aP < 0.05, bP < 0.01 vs control group (no vincristine). HBeAg: Hepatitis B e Antigen; HBcAg: Hepatitis B c Antigen; HBsAg: Hepatitis B surface antigen.
Vincristine promotes stable HBV-expressing cell excretion of HBV nucleocapsids instead of HBV Dane particles
To elucidate why the secretion of HBsAg did not increase after treatment with vincristine, we detected the HBcAg levels in the supernatant using an ELISA assay. Because the HBV virus cannot secrete free HBcAg into the culture medium, the HBcAg in the supernatant can only originate from HBV Dane particles or HBV nucleocapsids. Therefore, the HBcAg in the supernatant represents the level of HBV Dane particles or HBV nucleocapsids. Unexpectedly, the level of Dane particles in the supernatant did not change significantly after vincristine treatment; however, the overall levels of HBcAg from Dane particles and nucleocapsids in the supernatant increased sharply after treatment with 0.1 μmol/L vincristine, as shown in Figure 4 (P = 0.000). This result suggested that vincristine promotes HepG2-HBV1.1 cell excretion of HBV nucleocapsids instead of HBV Dane particles.
Figure 4.
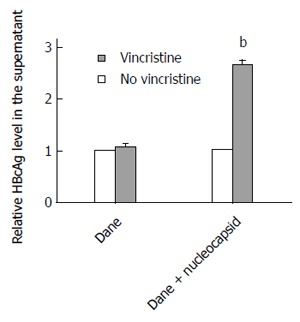
Vincristine promotes HepG2-HBV1.1 cell excretion of hepatitis B virus nucleocapsids instead of hepatitis B virus Dane particles. HepG2-HBV1.1 cells (6 × 105/well) were treated with vincristine at a concentration of 0.1 μmol/L for 24 h followed by 48 h of incubation in drug-free culture medium. The HBcAg from HBV Dane particles or nucleocapsids in the supernatant was analyzed using an ELISA assay. Dane: HBcAg from HBV Dane particles; Dane + nucleocapsid: HBcAg from HBV Dane particles and nucleocapsids. Values represent the mean ± SD of 3 independent experiments. aP < 0.05, bP < 0.01 vs control group (no vincristine). HBeAg: Hepatitis B e Antigen.
Vincristine dose-dependently inhibits the proliferation of stable HBV-expressing cells
HepG2.2.15 (4 × 105/well) cells and HepG2-HBV1.1 (6 × 105/well) cells were treated with vincristine at a concentration of 0.1 μmol/L for 24 h, and the medium was then changed to a drug-free culture medium. The cells were harvested 1, 2, 3, 4, and 5 d after vincristine treatment, and the cell counts were detected using the trypan blue exclusion assay (Figure 5A and B). On the first day after drug treatment, there was almost no difference in the cell counts between the control group and the drug group. However, there was a notable difference from the second day after drug treatment (P = 0.000). The proliferation rate declined greatly in the drug group, and the ratio of the cell counts in the vincristine group to the control group was approximately 60% in both stable HBV-expressing cell lines. The BrdU incorporation test also confirmed that the ratio of the proliferation ability in the vincristine group to the control group was approximately 55% at 72 h after drug treatment in the HepG2.2.15 cells and HepG2-HBV1.1 cells (Figure 5C). As shown in Figure 5D, the cells were treated with vincristine at a concentration of 0, 0.01, 0.05, and 0.1 μmol/L for 24 h, followed by 48 h of incubation in drug-free culture medium. The cell counts were down-regulated in a dose-dependent manner after drug stimulation based on the trypan blue exclusion assay.
Figure 5.
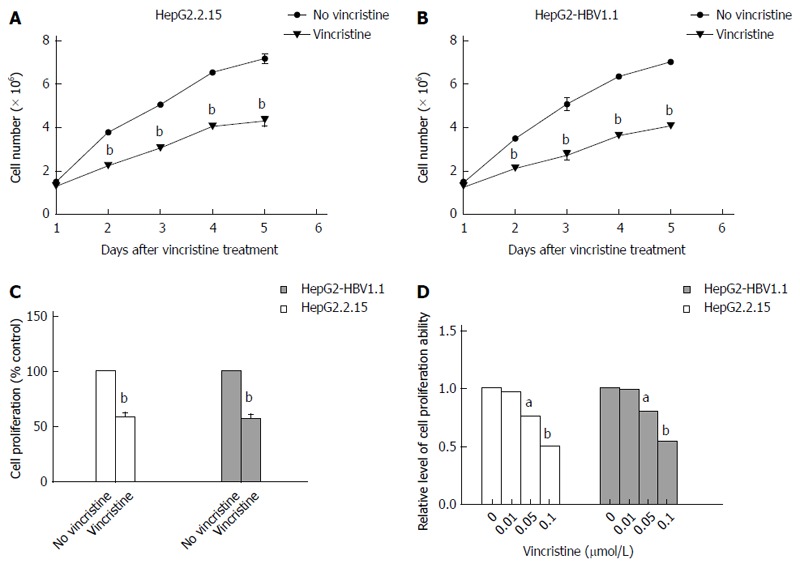
Vincristine inhibits the proliferation of HepG2.2.15 cells and HepG2-HBV1.1 cells. HepG2.2.15 (4 × 105/well) and HepG2-HBV1.1 (6 × 105/well) cells were treated with vincristine at a concentration of 0.1 μmol/L for 24 h, and the medium was then changed to a drug-free culture medium. The cells were harvested 1, 2, 3, 4, and 5 d after vincristine treatment, respectively. A: The proliferation ability of HepG2.2.15 cells was examined using the trypan blue exclusion assay; B: The proliferation ability of the HepG2-HBV1.1 cells was examined using the trypan blue exclusion assay; C: Vincristine inhibits the proliferation of HepG2.2.15 cells and HepG2-HBV1.1 cells based on the BrdU incorporation test. Cells were treated with 0.1 μmol/L vincristine for 24 h and were transferred to 96-well plates (4 × 103/well) with drug-free culture medium for another 48 h incubation; D: Vincristine dose-dependently inhibited the proliferation of cells based on the trypan blue exclusion assay. HepG2.2.15 (4 × 105/well) and HepG2-HBV1.1 (6 × 105/well) cells were treated with vincristine at concentrations of 0, 0.01, 0.05, and 0.1 μmol/L for 24 h, respectively, followed by 48 h of incubation in drug-free culture medium. Values represent the mean ± SD of 3 independent experiments. aP < 0.05, bP < 0.01 vs control group (no vincristine).
S-phase cell cycle arrest induced by vincristine is responsible for the cell proliferation inhibition
To elucidate why the cell proliferation was inhibited by vincristine, the cell cycle was examined using flow cytometry. HepG2.2.15 (4 × 105/well) and HepG2-HBV1.1 (6 × 105/well) cells were treated with vincristine at a concentration of 0.1 μmol/L for 24 h, and the medium was then changed to a drug-free culture medium. The cells were harvested 12, 24, 48, and 72 h after vincristine treatment; the ratios of cells in S-phase to G2-phase after vincristine treatment were 1.0, 2.2, 4.2, and 3.5, respectively, in the HepG2.2.15 cells and 1.0, 2.2, 4.0, and 2.6, respectively, in the HepG2-HBV1.1 cells (Figure 6A). This result suggested that vincristine induced cell cycle arrest at S-phase, and this phenomenon was most apparent at 48 h after drug treatment. In Figure 6B, the cells in each phase of the cell cycle at 48 h after drug treatment are displayed. Both the numbers of cells at G1/G0 and G2/M phases decreased, while the number of cells at S-phase increased significantly (P < 0.01). A diagram of the cell cycle for each phase based on flow cytometry (Figure 7) also shows that the number of cells at S-phase increased greatly 48 h after drug treatment in both stable HBV-expressing cell lines.
Figure 6.
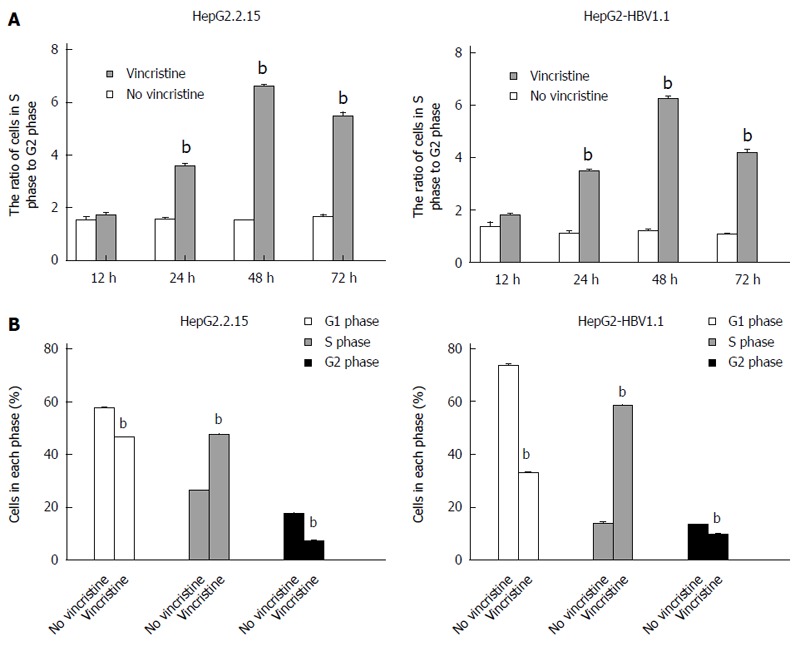
Vincristine induces the S-phase cell cycle arrest by flow cytometry. HepG2.2.15 (4 × 105/well) and HepG2-HBV1.1 (6 × 105/well) cells were treated with vincristine at a concentration of 0.1 μmol/L for 24 h, and the medium was then changed to drug-free culture medium. A: The ratio of the cells in S-phase to G2-phase after vincristine treatment was calculated. The cells were harvested 12, 24, 48, and 72 h after vincristine treatment, respectively; B: Vincristine significantly increased the number of S-phase cells. The cells were harvested 48 h after vincristine treatment. Values represent the mean ± SD of 3 independent experiments. bP < 0.01 vs control group (no vincristine).
Figure 7.
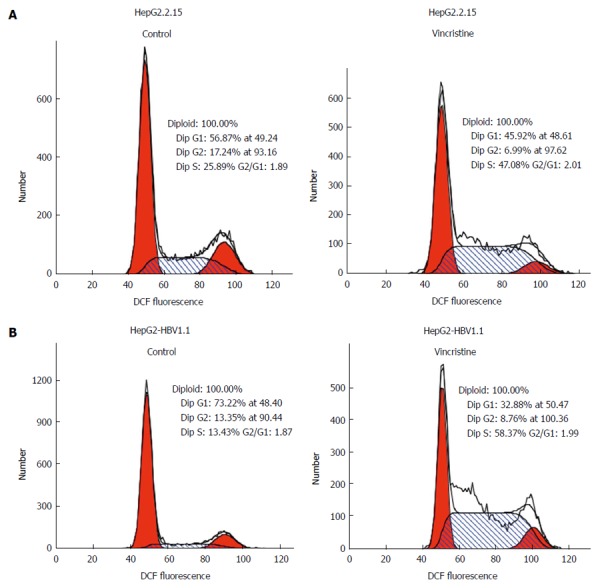
Diagram of the cell cycle for each phase based on flow cytometry. HepG2.2.15 (4 × 105/well) and HepG2-HBV1.1 (6 × 105/well) cells were treated with vincristine at a concentration of 0.1 μmol/L for 24 h, followed by 48 h of incubation in drug-free culture medium. A: HepG2.2.15 cells; B: HepG2-HBV1.1 cells.
Vincristine at the working concentration does not induce cell apoptosis
Another factor related to the proliferation inhibition was cell apoptosis. In the flow cytometry apoptosis diagram, the upper-right (UR) quadrant is representative of early apoptotic cells and the lower-right (LR) quadrant is representative of late apoptotic cells. Therefore, the total number of apoptotic cells was equal to the sum of the fluorescence intensities in the UR quadrant and LR quadrant. As shown in Figure 8A and B, there was no significant difference between the control group and the drug group, and the proportion of apoptotic cells in both groups was less than 5%. In the apoptotic state, the 113-kDa protein, PARP, is cut into an 89-kDa fragment and a 24-kDa fragment. Western blot analysis did not detect these two bands (Figure 8C).
Figure 8.
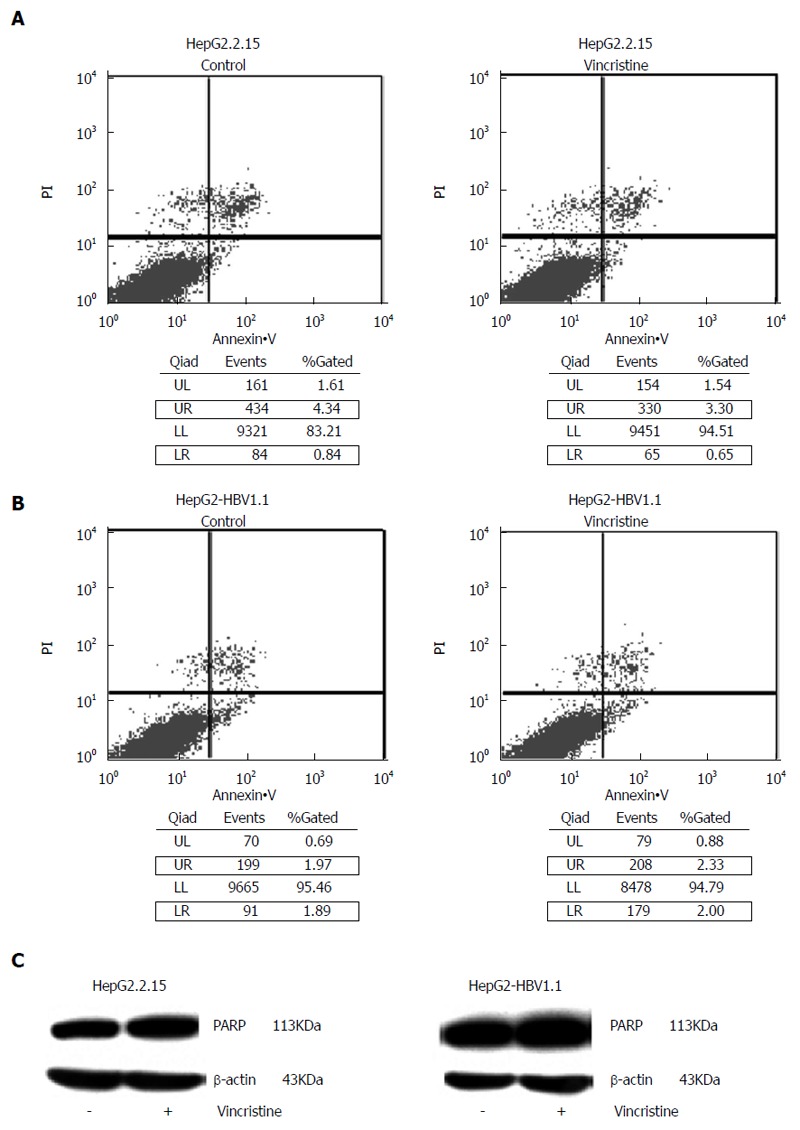
Vincristine at 0.1 μmol/L does not cause significant cell apoptosis. HepG2.2.15 (4 × 105/well) and HepG2-HBV1.1 (6 × 105/well) cells were treated with vincristine at a concentration of 0.1 μmol/L for 24 h, followed by 48 h of incubation in drug-free culture medium. A: The level of apoptosis in the HepG2.2.15 cells was detected based on flow cytometry; B: The level of apoptosis in the HepG2-HBV1.1 cells was detected based on flow cytometry; C: Western blot analysis of the PARP protein. PARP: Poly(ADP-ribose) polymerase; LR: Lower-right; UR: Upper-right.
DISCUSSION
Chronic HBV infection is a worldwide problem, and HBV reactivation after anti-cancer chemotherapy has become an emerging clinical challenge. All types of anti-cancer drugs used in chemotherapy have been related to HBV reactivation, including the classic cytostatics, monoclonal antibodies and steroids[13]. Patients with lymphoma appear to be particularly at risk, as the incidence of HBV reactivation following chemotherapy for lymphoma is between 24% and 67%[14,15] with a mortality of 4%-41%[16,17]. In part, this high incidence may be explained by the intensive chemotherapy necessary for lymphoma. CHOP (cytophosphamide, doxorubicin, vincristine, and prednisone) is the standard chemotherapy regimen for lymphoma. As is widely known, steroids such as prednisone can increase HBV DNA and RNA production directly by augmenting the HBV enhancer I[4]. In addition, cyclophosphamide, doxorubicin, and vincristine as cytotoxic drugs have immunosuppressive activity that is thought to be the main mechanism of HBV reactivation. In addition to the above, it is not known whether there are any other mechanisms that are responsible for this phenomenon. In vitro, HBV DNA secretion in culture medium was dose-dependently increased by doxorubicin[18]. One-hour exposure of cells to 1 μmol/L doxorubicin induced more than a 10-fold and 3-fold increase in HBV DNA and HBsAg, respectively. Therefore, we hypothesized that vincristine, another commonly used anti-cancer drug for lymphoma, plays a similar role to doxorubicin in directly promoting HBV replication.
In this report, we observed the effect of vincristine on HBV replication in HepG2.2.15 and HepG2-HBV1.1 cells. HBV DNA copies were dose-dependently increased by vincristine not only in the cytoplasm but also in the supernatant of the culture medium. We further found that intracellular HBsAg and HBcAg and the secretion of HBeAg also increased after treatment with vincristine. However, vincristine did not affect the secretion of HBsAg. This result indicated that the abundant secretion of HBV DNA copies stimulated by the drug could not be extracted from HBV Dane particles, as HBsAg was the main component of its outer layer. Based on the detection of the level of HBcAg in the supernatant of the cell culture medium, we showed that vincristine promotes cell excretion of HBV nucleocapsids instead of HBV Dane particles. This result indicated that vincristine increases HBV DNA, RNA production and protein expression but did not affect secreted virus. Similar to this phenomenon, corticosteroids increased the total intracellular levels of HBV DNA, RNA and HBsAg, but secreted HBsAg was not affected[19,20].
HBV nucleocapsids are closely related to HBV pathogenesis. The outer layer of HBV nucleocapsids consists of HBcAg, which is the main factor responsible for HBV-associated acute liver failure (ALF). Studies have suggested that HBcAg directly activates B cells to produce specific antibodies (anti-HBc IgG1 and IgM) without the help of T lymphocytes. This implicates a major role of B cell immunity in the pathogenesis of HBV-associated ALF[21,22]. In the present study, vincristine was found to stimulate stable HBV-expressing cell excretion of HBV nucleocapsids directly, which may be the principle factor in severe liver damage induced by cytotoxic anti-cancer drugs.
In this study, we found that the cell proliferation rate decreased significantly after vincristine treatment. This might be due to the cell cycle arrest or cell apoptosis. However, the results only indicated that the S/G2-phase cell cycle arrest was involved in this phenomenon. On one hand, studies have shown that the HBV or HBx gene inhibits cell proliferation[23,24]. On the other hand, some studies have found that cell cycle arrest was beneficial to viral replication. The CMV replication rate decreased significantly in p53-knockout fibroblast cell lines[25]. Huang et al[26] used four different approaches, including serum-free medium, all-trans retinoic acid, dimethyl sulfoxide, or sodium butyrate, to treat HepG2.2.15 cells. These approaches inhibited cell proliferation and cell cycle arrest at G0/G1 phase, whereas the number of cells expressing HBV DNA, HBsAg, and HbeAg increased. Therefore, cell proliferation inhibition was a manifestation of viral replication. When the virus replication accelerated, intracellular organelles and nutrients preferentially served the virus so that cell proliferation was suspended or greatly reduced.
Furthermore, it is clear that the recovering immune system following withdrawal of cytotoxic chemotherapy drugs plays a major role in liver damage. However, the mechanisms underlying the sharp increase in HBV replication in the early stage of the anti-cancer treatment are still unclear. In this study, we first reported that vincristine increased HBV DNA, RNA and protein expression and promoted HBV nucleocapsid secretion directly under cytotoxic stress in vitro. The possible link between cytotoxic stress and the activation of HBV replication is that the cell cycle arrest was induced by vincristine, and this may be a novel mechanism of HBV reactivation in HBV carriers receiving anti-cancer chemotherapy.
COMMENTS
Background
Hepatitis B virus (HBV) reactivation is an emerging clinical challenge to HBV carriers receiving anti-cancer chemotherapy. However, the mechanisms of HBV reactivation are incompletely understood. The host immune response to the virus is thought to play a pivotal role in HBV reactivation, but it cannot explain why HBV reactivation occurred within the first 2 wk of chemotherapy in some cases.
Research frontiers
All types of anti-cancer drugs used in chemotherapy have been related to HBV reactivation, including the classic cytostatics, monoclonal antibodies and steroids. In vitro, HBV DNA secretion into the culture medium was dose-dependently increased by doxorubicin. Vincristine as a plant alkaloid is a cytotoxic anti-cancer drug. The cancers treated with vincristine include acute leukemia, Hodgkin’s and non- Hodgkin’s lymphoma, neuroblastoma, and rhabdomyosarcoma.
Innovations and breakthroughs
This study is the first to report that vincristine has a direct stimulatory effect on HBV replication and promotes HBV nucleocapsid secretion without the involvement of immunity in vitro. This drug induced cell cycle arrest, and cell proliferation inhibition may be conducive to viral replication; this may be a novel mechanism of HBV reactivation following cytotoxic chemotherapy.
Applications
This study provides a possible novel mechanism of HBV reactivation following cytotoxic chemotherapy, which is beneficial to scientists seeking to avoid the HBV reactivation after cytotoxic drug treatment.
Terminology
HBV reactivation: HBV replication being enhanced after cytotoxic chemotherapy is a universal phenomenon in clinical cases. Although there are no uniform diagnostic criteria for HBV reactivation, clinical practice suggests that the definition is an increase in serum HBV DNA levels 10-fold higher than baseline, the reappearance of HBsAg or de novo detection of HBV DNA in HBsAg(-) patients.
Peer-review
This paper is well written and easy to understand, and the data seem to be convincing. The results suggest that vincristine as a cytotoxic drug has a direct stimulatory effect on HBV replication without the involvement of immunity in vitro, and this may be a novel mechanism of HBV reactivation following cytotoxic chemotherapy.
Footnotes
Supported by National Natural Science Foundation of China, No. 81201282 and No. 30901280; Grants from Chongqing Natural Science Foundation, No. cstc2012jjA10047; and the PhD Programs Foundation of the Ministry of Education of China, No. 20125503120004.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 8, 2014
First decision: June 27, 2014
Article in press: September 19, 2014
P- Reviewer: Chiang TA, Komatsu H, Sugimura H S- Editor: Qi Y L- Editor: Wang TQ E- Editor: Liu XM
References
- 1.Liu CJ, Chen PJ, Chen DS, Kao JH. Hepatitis B virus reactivation in patients receiving cancer chemotherapy: natural history, pathogenesis, and management. Hepatol Int. 2011:Epub ahead of print. doi: 10.1007/s12072-011-9279-6. [DOI] [PubMed] [Google Scholar]
- 2.Lubel JS, Angus PW. Hepatitis B reactivation in patients receiving cytotoxic chemotherapy: diagnosis and management. J Gastroenterol Hepatol. 2010;25:864–871. doi: 10.1111/j.1440-1746.2010.06243.x. [DOI] [PubMed] [Google Scholar]
- 3.Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 4.Tur-Kaspa R, Burk RD, Shaul Y, Shafritz DA. Hepatitis B virus DNA contains a glucocorticoid-responsive element. Proc Natl Acad Sci USA. 1986;83:1627–1631. doi: 10.1073/pnas.83.6.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liaw YF. Hepatitis viruses under immunosuppressive agents. J Gastroenterol Hepatol. 1998;13:14–20. doi: 10.1111/j.1440-1746.1998.tb00539.x. [DOI] [PubMed] [Google Scholar]
- 6.Cheng AL, Hsiung CA, Su IJ, Chen PJ, Chang MC, Tsao CJ, Kao WY, Uen WC, Hsu CH, Tien HF, et al. Steroid-free chemotherapy decreases risk of hepatitis B virus (HBV) reactivation in HBV-carriers with lymphoma. Hepatology. 2003;37:1320–1328. doi: 10.1053/jhep.2003.50220. [DOI] [PubMed] [Google Scholar]
- 7.Galbraith RM, Eddleston AL, Williams R, Zuckerman AJ. Fulminant hepatic failure in leukaemia and choriocarcinoma related to withdrawal of cytotoxic drug therapy. Lancet. 1975;2:528–530. doi: 10.1016/s0140-6736(75)90897-1. [DOI] [PubMed] [Google Scholar]
- 8.Nassal M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J Virol. 1992;66:4107–4116. doi: 10.1128/jvi.66.7.4107-4116.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muscarella DE, Bloom SE. The contribution of c-Jun N-terminal kinase activation and subsequent Bcl-2 phosphorylation to apoptosis induction in human B-cells is dependent on the mode of action of specific stresses. Toxicol Appl Pharmacol. 2008;228:93–104. doi: 10.1016/j.taap.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu X, Fu A, Luo KQ. A high-throughput fluorescence resonance energy transfer (FRET)-based endothelial cell apoptosis assay and its application for screening vascular disrupting agents. Biochem Biophys Res Commun. 2012;418:641–646. doi: 10.1016/j.bbrc.2012.01.066. [DOI] [PubMed] [Google Scholar]
- 11.Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci USA. 1987;84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Liu X, Chen J, Su H, Luo Q, Ye J, Tang N, Zhang W, Chen W, Ko BC, et al. Heparin sulphate D-glucosaminyl 3-O-sulfotransferase 3B1 plays a role in HBV replication. Virology. 2010;406:280–285. doi: 10.1016/j.virol.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 13.Manzano-Alonso ML, Castellano-Tortajada G. Reactivation of hepatitis B virus infection after cytotoxic chemotherapy or immunosuppressive therapy. World J Gastroenterol. 2011;17:1531–1537. doi: 10.3748/wjg.v17.i12.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeo W, Zee B, Zhong S, Chan PK, Wong WL, Ho WM, Lam KC, Johnson PJ. Comprehensive analysis of risk factors associating with Hepatitis B virus (HBV) reactivation in cancer patients undergoing cytotoxic chemotherapy. Br J Cancer. 2004;90:1306–1311. doi: 10.1038/sj.bjc.6601699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeo W, Chan PK, Zhong S, Ho WM, Steinberg JL, Tam JS, Hui P, Leung NW, Zee B, Johnson PJ. Frequency of hepatitis B virus reactivation in cancer patients undergoing cytotoxic chemotherapy: a prospective study of 626 patients with identification of risk factors. J Med Virol. 2000;62:299–307. doi: 10.1002/1096-9071(200011)62:3<299::aid-jmv1>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 16.Lok AS, Liang RH, Chiu EK, Wong KL, Chan TK, Todd D. Reactivation of hepatitis B virus replication in patients receiving cytotoxic therapy. Report of a prospective study. Gastroenterology. 1991;100:182–188. doi: 10.1016/0016-5085(91)90599-g. [DOI] [PubMed] [Google Scholar]
- 17.Hsu C, Hsiung CA, Su IJ, Hwang WS, Wang MC, Lin SF, Lin TH, Hsiao HH, Young JH, Chang MC, et al. A revisit of prophylactic lamivudine for chemotherapy-associated hepatitis B reactivation in non-Hodgkin’s lymphoma: a randomized trial. Hepatology. 2008;47:844–853. doi: 10.1002/hep.22106. [DOI] [PubMed] [Google Scholar]
- 18.Hsu CH, Hsu HC, Chen HL, Gao M, Yeh PY, Chen PJ, Cheng AL. Doxorubicin activates hepatitis B virus (HBV) replication in HBV-harboring hepatoblastoma cells. A possible novel mechanism of HBV reactivation in HBV carriers receiving systemic chemotherapy. Anticancer Res. 2004;24:3035–3040. [PubMed] [Google Scholar]
- 19.Lau JY, Bain VG, Smith HM, Alexander GJ, Williams R. Modulation of hepatitis B viral antigen expression by immunosuppressive drugs in primary hepatocyte culture. Transplantation. 1992;53:894–898. doi: 10.1097/00007890-199204000-00034. [DOI] [PubMed] [Google Scholar]
- 20.McMillan JS, Shaw T, Angus PW, Locarnini SA. Effect of immunosuppressive and antiviral agents on hepatitis B virus replication in vitro. Hepatology. 1995;22:36–43. [PubMed] [Google Scholar]
- 21.Farci P, Diaz G, Chen Z, Govindarajan S, Tice A, Agulto L, Pittaluga S, Boon D, Yu C, Engle RE, et al. B cell gene signature with massive intrahepatic production of antibodies to hepatitis B core antigen in hepatitis B virus-associated acute liver failure. Proc Natl Acad Sci USA. 2010;107:8766–8771. doi: 10.1073/pnas.1003854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu W, Chen Z, Cheng N, Watts NR, Stahl SJ, Farci P, Purcell RH, Wingfield PT, Steven AC. Specificity of an anti-capsid antibody associated with Hepatitis B Virus-related acute liver failure. J Struct Biol. 2013;181:53–60. doi: 10.1016/j.jsb.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang T, Zhao R, Wu Y, Kong D, Zhang L, Wu D, Li C, Zhang C, Yu Z, Jin X. Hepatitis B virus induces G1 phase arrest by regulating cell cycle genes in HepG2.2.15 cells. Virol J. 2011;8:231. doi: 10.1186/1743-422X-8-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gearhart TL, Bouchard MJ. Replication of the hepatitis B virus requires a calcium-dependent HBx-induced G1 phase arrest of hepatocytes. Virology. 2010;407:14–25. doi: 10.1016/j.virol.2010.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J Virol. 2006;80:8390–8401. doi: 10.1128/JVI.00505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang YQ, Wang LW, Yan SN, Gong ZJ. Effects of cell cycle on telomerase activity and on hepatitis B virus replication in HepG2 2.2.15 cells. Hepatobiliary Pancreat Dis Int. 2004;3:543–547. [PubMed] [Google Scholar]