

Background: An interaction between androgen and PI3K/AKT pathways has been implicated in prostate cancer cells.
Results: Conditional expression of AR transgene represses PI3K/Akt activation, and Pten loss results in reduced AR expression and transcriptional activity.
Conclusion: Both androgen and PI3K/AKT pathways inversely regulate each other in prostate cancer cells.
Significance: Interplay between androgen and PI3K/AKT pathways may directly contribute to prostate tumorigenesis.
Keywords: Akt PKB, Androgen, Androgen Receptor, Mouse, Phosphatidylinositide 3-Kinase (PI 3-kinase), Prostate Cancer
Abstract
Both androgen action and PI3K medicated signaling pathways have been implicated in prostate tumorigenesis. Our androgen receptor (AR) conditional transgenic mice developed murine prostatic intraepithelial neoplasia (mPIN) and prostatic adenocarcinoma lesions recapitulating human prostate cancer development and progression. Role of transgenic AR contributing to malignancy was demonstrated by high degree of transgenic AR expression in atypical and tumor cells in mPIN as well as prostatic adenocarcinoma lesions of the transgenic mice, but not in adjacent normal tissue. Interestingly, reduced PI3K/Akt activation also appeared in these mouse atypical and tumor cells, suggesting an interaction between androgen and PI3K/AKT pathways. In this study, we further investigated this interaction. We showed that the androgen depletion or knockdown of AR expression results in elevated levels of active phosphorylated AKT in prostate cancer cells. Castration of conditional Pten knock-out mice showed increased Akt, phosphorylated Akt, and pS6 expression in the mouse prostate. Using a series of newly generated Ar reporter and Pten knock-out compound mice, we showed that Pten loss directly represses endogenous Ar expression in prostatic epithelial cells. Moreover, Pten loss and PI3K/Akt activation reduced Ar-mediated transcription in purified Pten-null cells. This study provides novel evidence demonstrating interplay between androgen and PI3K pathways, as well as introduces unique and relevant mouse models for further studies of PI3K and AR pathways in the context of prostate tumorigenesis.
Introduction
The androgen signaling pathway is essential for normal prostate growth and differentiation and for prostate cancer initiation and progression (1, 2). It is mediated through the androgen receptor (AR)2 and its ligands, testosterone, and 5α-dihydrotestosterone (DHT) (3, 4). It is believed that AR is critical for prostate cancer cell proliferation as most prostate cancers are androgen-dependent and androgen ablation therapy (ADT) can result in prostate cancer cell apoptosis (1, 2). Androgen receptor is expressed in most prostate cancer samples before and after androgen ablation therapy (5). AR gene amplification appears mainly in post ADT prostate cancer samples (6). Global gene expression profiling further shows that the AR is one of several genes to be consistently up-regulated in castration-resistant prostate cancer (CRPC) (7, 8), which directly underscores the significance of androgen signaling in disease progression. Additionally, recent integrative genomic analysis revealed a significant enrichment of androgen signaling in early-onset prostate cancers, but not in elderly onset prostate cancers (9), further highlighting the significance of AR across the prostate tumorigenesis spectrum.
The phosphatidylinositol (3,4,5)-phosphate kinase (PI3K) signaling pathway plays a critical role in human tumorigenesis, including prostate cancer (10). The phosphatase and tensin homolog chromosome 10 (PTEN) is one of the most commonly mutated and/or deleted tumor suppressors (11, 12) and its somatic mutations were frequently detected in many sporadic human tumors, including glioblastoma, endometrial cancer, and prostate cancer (13). PTEN functions as a negative regulator of the PI3K pathway by blocking the activation of the kinase AKT/PKB (14, 15). Complete PTEN inactivation has been found in 15% of primary prostate tumors, and in up to 60% of prostate cancer metastases (8, 16–18). The biological significance of Pten in prostate tumorigenesis has been further confirmed in mouse models (19–22). In particular, conditional deletion of Pten in mouse prostate tissues results in invasive prostate cancers (20, 23).
We have previously shown that conditional expression of human AR transgene in the mouse prostate induces the development of prostatic intraepithelial neoplasia (PIN) and prostatic adenocarcinoma lesions in R26hARL/wt:Osr1-Cre mice (24). In this model, transgenic AR expression was detected in atypical and tumor cells in both murine PIN (mPIN) and murine prostatic adenocarcinomas. Interestingly, we also observed decreased Akt activation in atypical and tumor cells of these transgenic mice, suggesting an interaction between the PI3K/Akt and androgen/AR signaling pathways in murine prostate tumorigenesis. In this study, we used a series of in vitro and in vivo experimental approaches to scrutinize this interaction. We showed that the depletion of androgens, presence of antiandrogen, or knockdown of endogenous AR expression results in elevated phosphorylated AKT (pAKT) expression. Castration of conditional Pten knock-out mice elevated total Akt levels, as well as levels of phosphorylated Akt (pAkt), and phosphorylated S6 (pS6) expression in prostate cancer cells. Using the newly developed Ar-IRES-PLAP-IRES-nLacZ/PtenLoxP/LoxP:PB-Cre4 (Ar-IPInL/PtenL/L:PB-Cre) and Ar-IRES-PLAP-IRES-nLacZ/PtenLoxP/LoxP:Osr1-Cre (Ar-IPInL/PtenL/L:Osr1-Cre) mice, we further demonstrated the effect of decreased endogenous Ar expression in Pten-null prostatic cells, suggesting a repressive role for AR-overactivated PI3K signaling under endogenous AR expression conditions. Using another new mouse line, mT/mGloxP/+: PtenloxP/loxP:PB-Cre4, in which Pten deletion and membrane-targeted green fluorescent protein (mG) expression are controlled simultaneously through Cre-mediated recombination (25), we observed increased Akt activation and down-regulation of AR's downstream target gene expression in prostatic Pten-null cells from these mice. Taken together, these data demonstrate a functional interaction between PI3K/AKT and androgen/AR signaling pathways in prostate cancer cells.
EXPERIMENTAL PRODECURES
Mouse Breeding and Castration
Mice homozygous for floxed Pten exon 5, PtenloxP/loxP, on a 129/Balb/c background, were obtained from the Jackson Laboratory (Strain: 004597, Bar Harbor, ME). They were crossed with the Osr1-Cre strain, a kind gift of Dr. Gail Martin at the University of California at San Francisco (26), and PB-Cre4 mice, driven by a modified probasin promoter (ARR2PB) on a C57BL/6xDBA2 background (27). PtenloxP/+:Osr1-Cre mice were backcrossed more than five times with C57BL/6J mice and then used for mating to produce homozygous (PtenloxP/loxP:Osr1-Cre) mice. Using similar mating strategies, we also generated Pten conditional knock-out mice with PB-Cre4 mice. Female mice homozygous for Ar reporters, Ar-IPInL on a C57BL/6 background, in which both nuclear targeted LacZ (nLacZ) and placental alkaline phosphatase (PLAP) were inserted into the 3′-untranslated region of endogenous Ar gene with the internal ribosome entry sites (IRESs) (28), were obtained from Jackson Laboratory (Strain: 012374, Bar Harbor, ME). Ar-IPInL:PtenloxP/loxP:PBCre4 or Ar-IPInL:PtenloxP/loxP:Osr1-Cre mice were generated by crossing PtenloxP/+:PB-Cre4 or PtenloxP/+:Osr1-Cre males, and Ar-IPInL:PtenloxP/+ females. The double-fluorescent mT/mG reporter strain was kindly provided by Dr. Liqun Luo at Stanford University (25), and used it to generate mT/mGloxP/+: PtenloxP/loxP:PB-Cre4 compound mice. R26hARloxP/wt:Osr1-Cre mice were generated as described in our previous report (24). For castration, the mice were anesthetized by IP injection of Ketamine and Xylazine. Both testicles and epididymis were removed through a scrotal approach. The distal end of the spermatic cord was ligated with silk thread as described previously (29). We then examined castrated and age-matched intact mice after 8 weeks. All animal experiments performed in this study were approved by the ethics committee of the Administrative Panel on Laboratory Animal Care at Stanford University.
Histological Analyses, Immunohistochemistry, Immunofluorescence, and β-Galactosidase, β-Gal, Staining
Mouse tissues were fixed in 10% neutral-buffered formalin and processed into paraffin, and 5 μm serial sections were cut and processed from xylene to water through a decreasing ethanol gradient followed by 0.1 m PBS for histological, immunohistochemical, and immunofluorescence analyses (23, 24). For immunohistochemistry and immunofluorescence, tissue slides were blocked with 0.3% hydrogen peroxide in methanol for 15 min and 5% goat serum in PBS for 30 min at room temperature. They were then exposed to different first antibody in PBS with 1% goat serum at 4 °C overnight, including anti-human AR (sc-7305, Santa Cruz Biotechnology), anti-mouse/human AR (sc-816, Santa Cruz Biotechnology), anti-pAKT (4060, Cell Signaling), ant-AKT (9272, Cell Signaling), anti-ki67 (NCL-ki67, Novacastra), anti-pS6 (2211, Cell Signaling), anti-FKBP5 (ab46002, Abcam), anti-PHLPP (ab71972, Abcam), and anti-PTEN (9559, Cell Signaling) antibodies. For immunohistochemistry, slides were then incubated with biotinylated anti-rabbit or anti-mouse secondary antibody (BA-1000 or BA-9200, Vector Laboratories) for 1 h, horseradish peroxidase streptavidin (SA-5004, Vector Laboratories) for 30 min, and then visualized by DAB kit (SK-4100, Vector Laboratories) at room temperature. All samples were subsequently counterstained with 5% (w/v) Harris Hematoxylin. For immunofluorescence analyses, slides were incubated with appropriate Alexa Fluor 488- or 594-conjugated secondary antibodies diluted 1:500 in blocking solution for 2 h at room temperature, washed, and mounted on slides with VECTASHIELD Mounting Medium with DAPI (H-1200, Vector Laboratories). Coverslips were mounted using Permount Mounting Medium (Cat SP15-500, Fisher Scientific). All images were obtained using a Nikon Eclipse E800 Epi-fluorescence microscope and Adobe Photoshop CS5.
For β-gal staining, mouse prostate tissues were embedded in OCT compound (Tissue-Tek) for snap freezing. Frozen blocks were sectioned at 7-μm intervals and fixed in 0.2% glutaraldehyde solution at 4 °C for 10 min (30). Slides were washed three times at room temperature for 15 min in buffer (0.1 m phosphate buffer, 2 mm MgCl2, 0.02% Nonidet P-40, 0.01% sodium deoxycholate) and stained with 1 mg/ml X-gal staining solution (washing buffer with 5 mm potassium ferrocyanide and 5 mm potassium ferricyanide) at room temperature overnight. All slides were subsequently counterstained with the Nuclear Fast Red (H-3404, Vector Laboratories).
Prostatic Epithelial Cell Isolation and Purification
Prostate tissues from mT/mGloxP/+:Pten+/+:PB-Cre4, and mT/mGloxP/+:PtenloxP/loxP:PB-Cre4 mice were minced into 1 mm3 pieces, digested in DMEM/Collagenase/FBS for 3 h at 37 °C, and 0.25% trypsin-EDTA (Invitrogen, Carlsbad, CA) on ice for 1 h. Digested cells were dissociated with pipetting and then passed through 70-μm cell strainers (BD Biosciences, San Jose, CA) to make single cell suspensions. GFP-positive cells were sorted through a FACS AriaII cytometer (BD Biosciences).
RNA Isolation and Reverse Transcription (RT)-Quantitative Real-time PCR (qRT-PCR) Assays
Single cell suspensions prepared above were lysed in RNA-Bee (TEL-TEST, Inc., Friendswood, TX) and total RNA was isolated as recommended by the manufactory (31). For reverse transcription, cDNA was synthesized from 10 μg of total RNA with 15 units of avian myeloblastosis virus reverse transcriptase (M5108, Promega) with 0.5 μg of random primer (C1181, Promega) in a total volume of 20 μl. For qRT-PCR, cDNA samples were mixed with SYBR qPCR Super Mix Universal (11762, Invitrogen) with specific primers in the MX 3005P thermocycler (Stratagene). Relative mRNA levels were calculated by ΔΔC(T) method (32). Reactions were done in triplicate, and the values were normalized by the GAPDH expression levels. Primers for FKBP5 (5-TGAGGGCACCAGTAACAATGG-3; 5-CAACATCCCTTTGTAGTGGACAT-3), Probasin (5-ATTGAGAACCTACTTCCGTCACA-3; 5-CAGTTGGCACTTAGTCCCTTTC-3), Nkx3.1 (5-CCGGAGGACCCACCAAGTAT-3; 5-CCTGGATTATGTTCACAGTCCAA-3), Msmb1 (5-TGGCTGGGCAGTCTCTTATTC-3; 5-CAGGGAGTGTTAAGGAAATGCTT-3), Phlpp1 (5-CCCGAAGACCTCGGCTTTAC-3; 5-CTGCCATCGCCTTATCGTCTC-3), Ar (5-TGCCCGAATGCAAAGGTCTT-3; 5-TTGGCGTAACCTCCCTTGAAA-3), and GAPDH (5-AGGTCGGTGTGAACGGATTTG-3; 5-TGTAGACCATGTAGTTGAGGTCA-3) were used in the above qRT-PCR reactions, respectively.
Cell Cultures and Lentivirus Production/Infection
The human embryonic kidney cell line, HEK293, was maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal calf serum (FCS) (HyClone, Denver, CO). LNCaP and LNCaP C4–2 cell lines were maintained as described previously (33, 34). For 5α-dihydrotestosterone (DHT) and Flutamide, an antiandrogen, treatments, the cells were incubated in DMEM supplemented with 5% charcoal-stripped fetal bovine serum (CS-FBS) for 40 h, then further grown in the presence of 10 nm DHT with or without 1 μm Flutamide for 8 h. The AR shRNA constructs were created by inserting double-stranded oligonucleotides corresponding to the human AR cDNA sequences 5′-GGACACTTGAACTGCCGTCT-3′ [amino acids (aa) 335–342, AR shRNA1], and 5′-GGTGTCACTATGGAGCTCTC-3′ (aa 568–575, AR shRNA2) downstream of U6 promoter in the pBS/U6 vector (35). To generate shRNA lentiviruses, pLenti-shRNA vectors, pCMV-dR8.91, and pMD2.G-VSVG plasmids were co-transfected into HEK293T cells at a ratio of 3:2:1 using a Lipofectamine kit (Invitrogen, Carlsbad, CA). The media were replaced at 6 h post-transfection and then collected after 36 to 40 h. The viral supernatant was centrifuged briefly to remove cellular debris and stored at −80 °C. Lentivirus infection was carried out in the presence of 6 mg/ml Polybrene for 8 h.
Western Blotting
Cells were harvested and then lysed in a buffer containing 0.5% Nonidet P-40, 150 mm NaCl, 2 mm MgCl2, 50 mm HEPES-KOH (pH 7.4), 1 mm EDTA, 5% glycerol, 1 mm dithiothreitol, 0.5 mm phenylmethylsulfonyl fluoride, 25 mm NaF, and 1 mm sodium orthovanadate. After SDS-PAGE, proteins were transferred to nitrocellulose (Schleicher and Schuell) and blocked in TBS-T (50 mm Tris-HCl, 150 mm NaCl, 0.08% Tween 20) with 5% dry nonfat milk. Membranes were probed with AR (sc-816, Santa Cruz Biotechnology), pAKT (4060, Cell Signaling) or AKT (9272, Cell Signaling) antibody. Anti-rabbit IgG conjugated to horseradish peroxidase were used as secondary antibodies (Promega). Detection was performed with ECL reagents according to the manufacturer's protocol using ECL Hyperfilm (Amersham Biosciences). Densitometry of the protein bands was performed using ImageJ software, and the relative numbers were reported as OD units of pAKT/OD units of AKT.
Statistical Analyses
Data are shown as the mean ± S.D. Differences between groups were examined by 2-tailed Student's t test or 2-way ANOVA for comparisons among multiple groups. For all analyses, p < 0.05 was considered statistically significant.
RESULTS
Conditional Expression of AR Transgene Results in Decreased Akt Activation in Mouse Prostatic Tumor Cells
The Osr1 (odd-skipped-related 1) promoter activates at E11.5 in urogenital sinus epithelium and retains its activity in prostatic epithelium throughout development (26). Conditional expression of human AR transgene in the prostates of R26hARL/wt:Osr1-Cre mice displayed PIN and prostatic adenocarcinoma lesions (24). To fully understand the role of transgenic AR in prostate oncogenesis, we performed a series of experiments to establish additional cell signaling pathways that promote oncogenic transformation in the prostate of R26hARL/wt:Osr1-Cre mice. We observed that transgenic AR expression in prostatic adenocarcinoma cells in R26hARL/wt:Osr1-Cre mice (Fig. 1, A to B′) have robust Ki67 expression (Fig. 1, C and C′) and expression of Akt also appears in these cells (Fig. 1, D and D′). However, these tumor cells show very weak or no immunoreactivity with antibodies against either pAkt or pS6, a downstream target of the Akt pathway (Fig. 1, E to F′), suggesting inactivated Akt signaling in prostatic adenocarcinoma cells of R26hARL/wt:Osr1-Cre mice. A reciprocal regulation between PI3K and androgen signaling pathways has been demonstrated in prostate cancer cells and in the prostates of conditional Pten knock-out mice (36, 37). Recent studies demonstrated that an androgen-regulated gene FKBP5 promotes PHLPP dephosphorylation of AKT and as a result suppresses AKT activity (37, 38). To establish whether transgenic AR expression also increases the expressions of FKBP5 and PHLPP proteins, we examined both protein expressions in prostate cancer cells of R26hARL/wt:Osr1-Cre mice. We observed high levels of the Fkbp5 and Phlpp proteins in prostate cancer cells of R26hARL/wt:Osr1-Cre mice (Fig. 1, G1 to H2′). Our data are consistent with the previous observation and suggest that AR-induced Fkbp5 and Phlpp expression is involved in repressing pAkt expression.
FIGURE 1.

Reduced Akt phosphorylation in prostatic epithelial cells with conditionally expressed trangenic AR. H&E staining (A) and immunohistochemistry (B–H) were performed on prostate tissues from prostatic adenocarcinoma regions of R26hARL/+:Osr1-Cre mice. Adjacent prostate tissue sections were analyzed with different antibodies as marked, including the human AR (B), Ki67 (C), Akt (D), pAkt (E), pS6 (F), FKBP5 (G1 and H1), and PHLPP (G2 and H2). Corresponding high power images (400×) are shown in A′–H′.
Depletion of Androgens Elevates AKT Activity in Prostate Cancer Cells
We next evaluated the role of androgen in the regulation of AKT signaling activation in prostate cancer cells. We first analyzed the effects of androgens on AKT expression and activation in LNCaP cells, an AR-positive and Pten-null prostate cancer cell line (11, 12). LNCaP cells were cultured in the presence or absence of DHT, or in the presence of DHT and Flutamide, an antiandrogen. Western blotting analyses showed no significant change in both endogenous AR and AKT expression in the whole cell lysates isolated from LNCaP cells among different treatments (Fig. 2, A and E1). However, decreased expression of pAKT was detected in cells cultured in the presence of DHT, suggesting a repressive role of androgens in AKT activation. We then analyzed the effect of androgens on AKT expression and activation in LNCaP C4–2 cells, which are derived from LNCaP cells passaged in a castrated host (39). Consistent with previous reports we found a slightly higher AR expression in samples in the presences of DHT (Fig. 2B) (39). However, there is no significant change in either AKT or pAKT expression between samples with different treatment (Fig. 2, B and E2). To further explore the effects of AR on AKT expression and activation, we performed knockdown experiments using two short hairpin RNAs against AR. Both LNCaP and LNCaP C4–2 cells were transduced with either AR shRNA or Swabled shRNA lentiviruses as a control. A significant reduction of endogenous AR expression was shown in cells transduced with AR shRNA lentiviruses (Fig. 2, C and D). While there is no notable change in AKT expression levels between samples transduced with either AR shRNA or controls in both LNCaP and C4–2 cells, we observed an increase of pAKT in LNCaP cells transduced with AR shRNA viruses (bottom panel, Fig. 2C). Densitometry of both AKT and pAKT protein bands in each sample was performed, and the relative numbers were reported as OD units of pAKT/OD units of AKT (Fig. 2, E1–4). Interestingly, addition of antiandrogen, and knockdown of AR expression increase AKT activation in LNCaP but not C4–2 cells, suggesting an alternative role for AR in these prostate cancer cells.
FIGURE 2.

Androgen signaling negatively regulates AKT phosphorylation in prostate cancer cells. A and B, LNCaP (A) and LNCaP C4–2 (B) cells were grown in hormone-deprived medium supplemented with 5% charcoal-stripped fetal bovine serum for 40 h, then incubated for further 8 h in the presence of 10 nm DHT with or without 1 μm Flutamide, then harvested for Western blotting assays with anti-AR, anti-AKT, and anti-pAKT antibodies. C and D, LNCaP (C) and LNCaP C4–2 (D) cells were infected with lentivirus enconding shRNA against AR (AR shRNA1 or shRNA2). The infected cells were cultured for 48 h, then harvested for Western blotting assays with AR, AKT, and pAKT antibodies. E, densitometry of AKT and pAKT protein bands was performed using ImageJ software, and the relative numbers were reported as OD units of pAKT/OD units of AKT.
As a tumor suppressor, PTEN can counteract PI3 kinase in repressing AKT signaling in cell growth and tumor formation (14, 15). Conditional deletion of Pten in the prostate of PtenL/L:Osr1-Cre mice resulted in high-grade PIN and invasive prostatic tumor development (23). We further evaluated the effect of androgen signaling on Akt expression and activation in prostate tissues isolated from castrated mice for 8 weeks and age-matched intact controls. As shown in Fig. 3, A and A′, deletion of Pten was confirmed in both intact and castrated prostate tissues isolated from PtenL/L:Osr1-Cre mice. Using specific antibodies against Akt and pAkt, we assessed expression and activity of Akt in the above prostate tissues. While both Akt and pAkt staining appears in the lesions of both intact and castrated mice, more intense immunoreactivity to Akt and pAkt antibodies is observed in castrated mice (Fig. 3, B′ and C′ versus 3, B and C). Staining of the above samples with an antibody against pS6, also showed strong immunoreactivity in the lesions of castrated mice (Fig. 3, D′ versus D). We also observed a typical nuclear staining pattern of AR in tumor cells of intact mice (Fig. 3E), and in contrast a diffuse and cytoplasmic staining pattern of AR in castrated mice (Fig. 3E′). There is no significant change in the staining of Ki67 between castrated and intact mice (Fig. 3, F and F′). These data demonstrate that castration elevates Akt and pAkt expression in prostate cancer cells of PtenL/L:Osr1-Cre mice.
FIGURE 3.
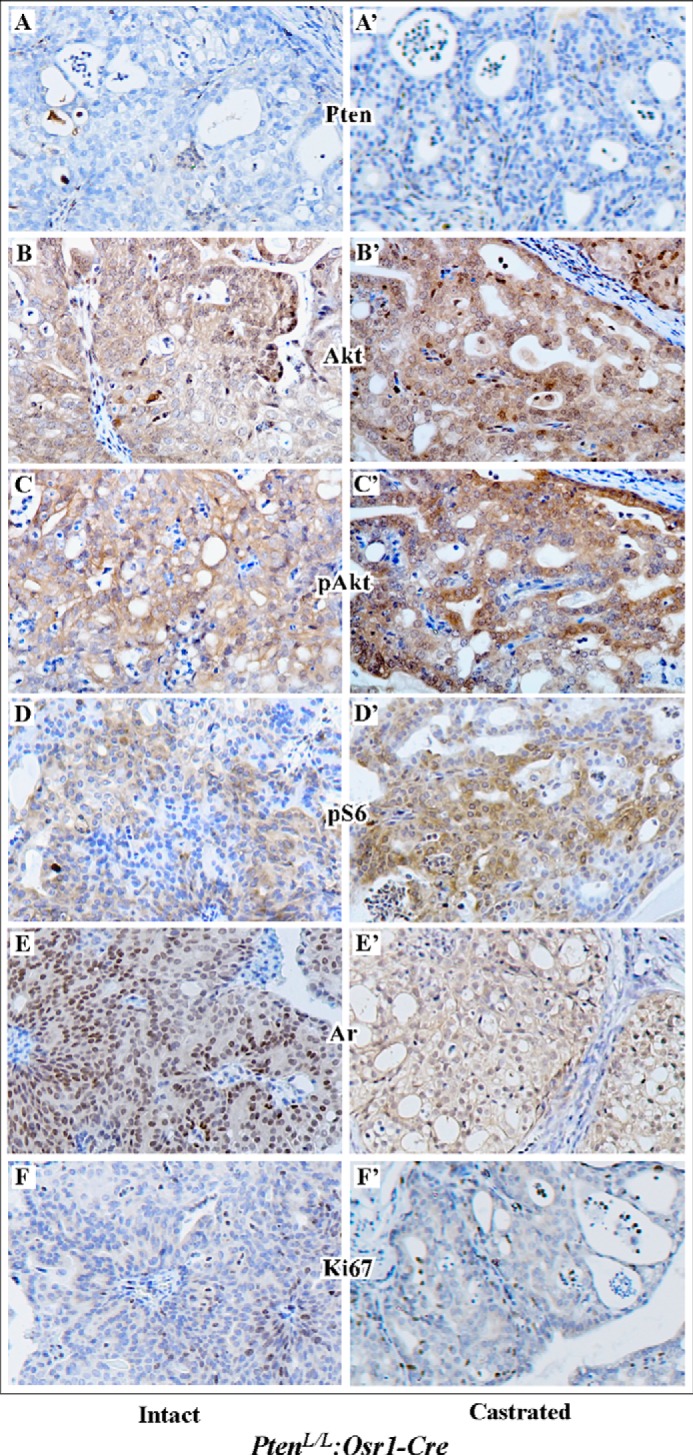
Androgens affect Akt expression and activation in Pten null prostatic cancer cells. PtenloxP/loxP:Osr1-Cre mice were castrated at 12 month for 8 weeks, and then analyzed with age-matched intact controls. Immunohistochemistry was carried out to address PI3K signaling in Pten-null prostate tumors isolated from intact (A–F) and castrated (A′–F′) mice. Adjacent prostate tissue slides were analyzed with the antibodies as labeled in the figure.
Reduced Ar Expression in Pten-null Prostate Tumor Cells
Previous studies have shown that Pten loss attenuates androgen signaling (36, 37). However, the effect of Pten loss on endogenous AR expression has not been fully investigated in a biologically relevant system. For this reason, we intercrossed PtenL/L:Osr1-Cre or PtenL/L:PB-Cre mice to Ar-IRES-PLAP-IRES-nlacZ (Ar-IPIL) mice (Fig. 4A), in which both nuclear-targeted LacZ (nLacZ) and placental alkaline phosphatase (PLAP) were inserted into the 3′-untranslated region of endogenous Ar gene with the internal ribosome entry sites (IRESs) (28). These new mouse models enabled us to directly assess the effect of Pten loss on Ar expression. We first analyzed 6-week-old Ar-IPIL/PtenL/L:PB-Cre4 mice. As shown in Fig. 4, B and C, strong LacZ staining was observed in normal prostate glands. In contrast, adjacent atypical prostate glands showed much weaker staining (Fig. 4, B′ and C′). At high magnification, robust nuclear and cytoplasmic LacZ staining was observed in normal prostatic luminal cells (blue arrows, Fig. 4, B and C) in comparison to few scattered blue atypical cells in mPIN areas (pink arrows, Fig. 4, B′ and C′). With immunohistochemistry, we analyzed endogenous Ar and Pten expression using adjacent sections of the above tissues. We observed stronger nuclear expression of Ar protein in normal luminal cells than atypical cells (Fig. 4, D and D′). In contrast, Pten expression appears much weaker in atypical cells than normal luminal epithelial cells (Fig. 4, E and E′). Consistently, we detected much stronger pAkt staining in atypical cells of PIN lesions than in prostatic luminal cells of adjacent normal glands (Fig. 4, F and G′). Taken together, these data demonstrate that Pten loss significantly reduces endogenous AR expression in prostatic atypical cells.
FIGURE 4.
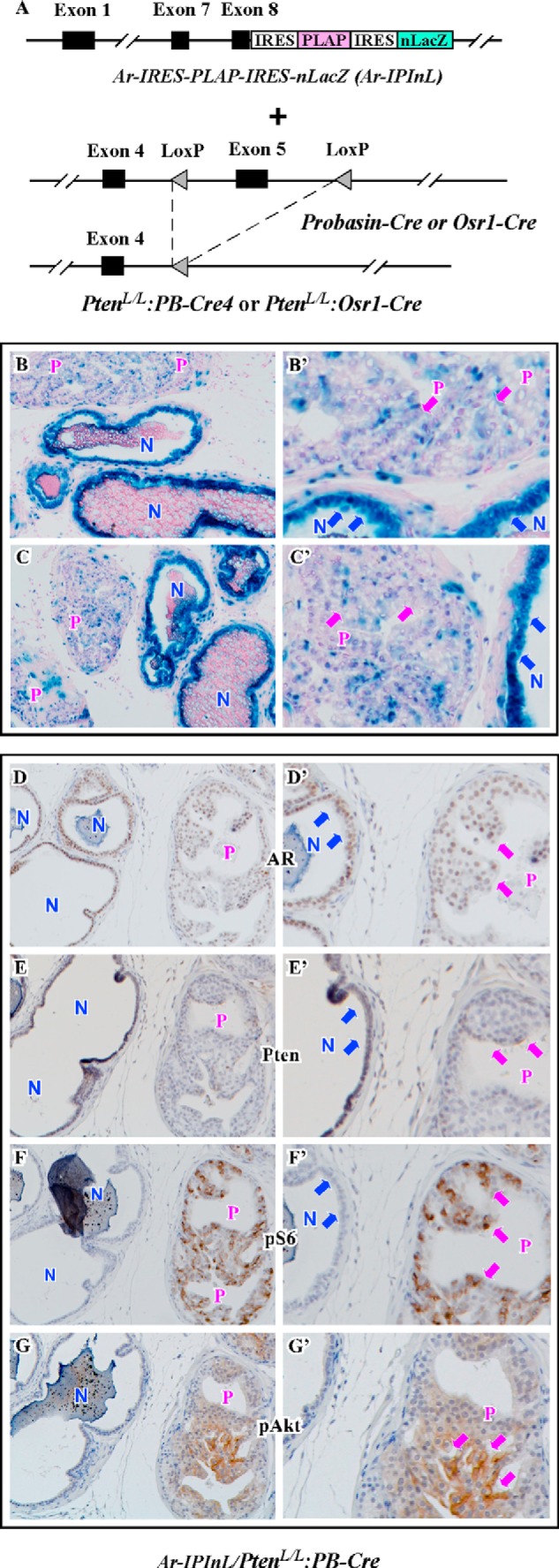
Pten loss represses endogenous Ar expression in the mouse prostate. A, scheme of the PLAP and nLacZ expression at the AR locus in Ar-IPInL mice is shown on an upper figure. A first IRES was inserted in the 3′-UTR of the AR gene, addressing expression of PLAP. A second IRES was placed 3′ of the PLAP gene to activate expression of nLacZ. The diagram for conditional Pten knock-out strategy is shown on a bottom figure. In PtenloxP/loxP mice, LoxP sites were placed into the endogenous Pten locus flanking exon 5. Two different Cre transgenic lines carrying Osr1-Cre or PB-Cre4 transgene were crossed to PtenloxP/loxP mice for the generation of PtenloxP/loxP:Osr1-Cre and PtenloxP/loxP:PB-Cre4 mice. B and C, frozen sections of prostate tissues isolated from 6-week-old Ar-IPInL:PtenloxP/loxP:PB-Cre4 mice were stained for β-gal activity. Corresponding high power images (400×) are shown in B′–C′. D–G, serially adjacent sections from each mouse were stained with the antibodies against AR, Pten, pS6, and pAkt. Corresponding high power images (400×) are shown in D′–G′. N indicates normal glands. P indicates PIN lesions.
The PB-Cre4 mice have been frequently used in the field of prostate cancer research, in which a composite promoter, ARR2PB, drives Cre transgene expression, which is androgen dependent and derived from the rat prostate-specific probasin gene (27). This introduces a possibility that the PB-Cre4 induced Pten loss may be influenced diversely by autonomous Ar expression. For this reason, we further assessed the effect of Pten loss in endogenous Ar expression using another prostatic conditional Pten deletion mouse model, Ar-IPIL/PtenL/L:Osr1-Cre. The Osr1 promoter is not androgen dependent and is activated at E11.5 in the urogenital sinus epithelium (26). PtenLoxP/LoxP:Osr1-Cre mice developed high-grade PINs with high penetrance as early as one-month of age, and locally invasive prostatic tumors after 12 months of age (23). Histological analyses showed intracystic carcinoma lesions in the prostates of Ar-IPIL/PtenLoxP/LoxP:Osr1-Cre mice (Fig. 5, A and A′), which were very similar to the changes that we previously observed in PtenLoxP/LoxP:Osr1-Cre mice (23). Consistently, Pten staining is much weaker in tumor areas than in normal prostatic glands (Fig. 5, F and F′). To evaluate endogenous Ar expression in the mice, we first examined LacZ expression and observed a robust staining in normal prostatic cells but no or very weak staining in tumor cells (Fig. 5, B and B′). Immunohistochemical analyses also showed a strong Ar signal in normal prostatic cells in comparison to tumor cells (Fig. 5, C and C′). Again, in contrast, strong expression of pAkt and pS6 appears in prostatic intracystic tumor cells but weak or no expression is observed in normal prostatic epithelial cells (Fig. 5, D to E′). Taken together, our observation of decreased expression of endogenous Ar in Pten-null prostatic tumor cells in Ar-IPIL/PtenLoxP/LoxP:Osr1-Cre mice further demonstrates a role of Pten loss in endogenous Ar expression.
FIGURE 5.
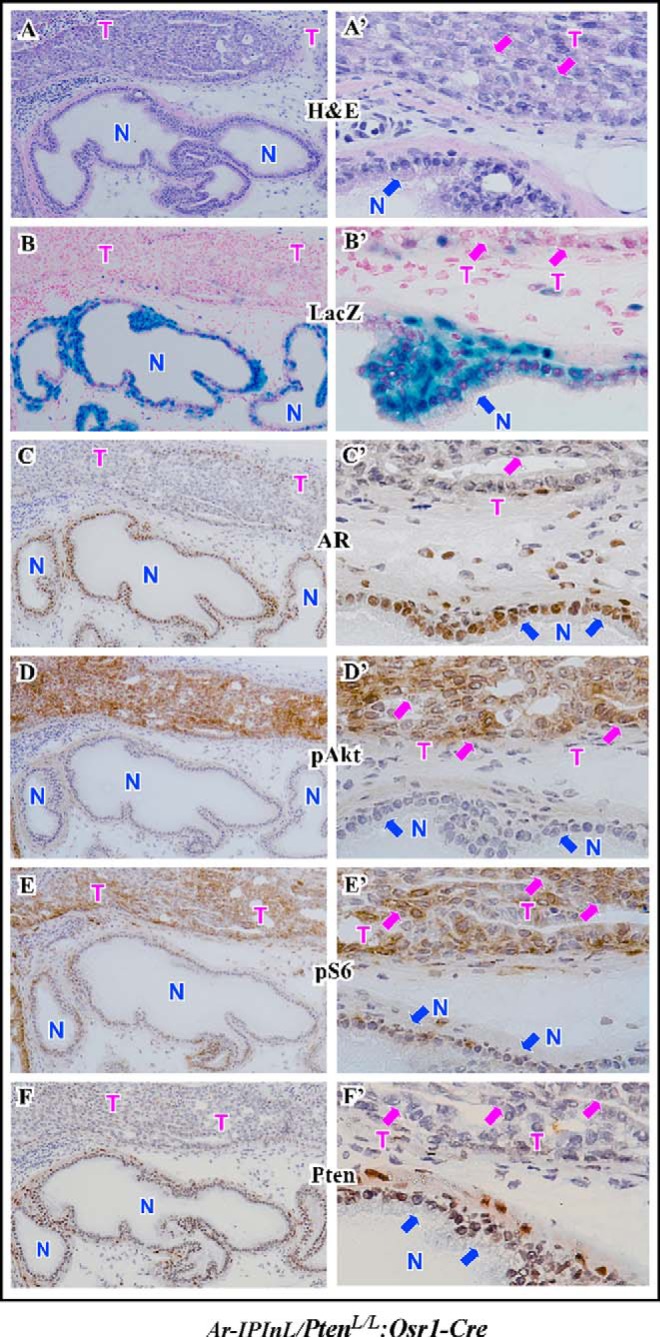
Analysis of endogenous Ar expression and Akt expression and activation in the mouse prostate. Serially adjacent frozen sections of prostate tissues isolated from 5-week-old Ar-IPInL:PtenloxP/loxP:Osr1-Cre mice were stained with hematoxylin and eosin (A), stained for β-gal activity (B), and stained with the antibodies against AR, pAkt, pS6, and Pten (C–F). Corresponding high power images (400×) are shown in A′–F′. N indicates normal glands. T indicates tumor areas.
Repressing Ar Transcriptional Activity in Pten-null Tumor Cells
In the double-fluorescent mT/mG Cre reporter mouse strain, expression of membrane targeted tandem dimer Tomato (mT), or membrane-targeted green fluorescent protein (mG) is controlled by Cre-mediated recombination (25). Therefore, with an appropriate Cre expression, mT/mG expression can be employed to trace and isolate specific cell populations that have undergone Cre-driven recombination (25). In this study, we developed mT/mGLoxP/+/PtenLoxP/LoxP:PB-Cre4 mice that enable us to label and isolate mG positive and Pten null prostatic tumor cells (Fig. 6A). We first analyzed mT/mGLoxP/+/PtenLoxP/LoxP:PB-Cre4 and mT/mGLoxP/+/Pten+/+:PB-Cre4 mice as controls using co-immunofluorescence microscopy. We observed specific mG protein expression in normal prostatic epithelial cells and atypical cells in PIN lesions in mT/mGLoxP/+/Pten+/+:PB-Cre4 or mT/mGLoxP/+/PtenLoxP/LoxP:PB-Cre4 mice, respectively (Fig. 6, B1 and C1). However, robust expression of pAkt was only observed in atypical cells in PIN lesions of mT/mGLoxP/+/PtenLoxP/LoxP:PB-Cre4 mice (Fig. 6C2), which fully overlapped with mG expression (Fig. 6C3). However, we did not detect pAkt staining in the prostate of mT/mGLoxP/+/Pten+/+:PB-Cre4 mice (Fig. 6, B2 and B3). We proceeded to isolate these mG positive epithelial cells from both mT/mGLoxP/+/PtenLoxP/LoxP:PB-Cre4 and mT/mGLoxP/+/Pten+/+:PB-Cre4 mice and collected total RNA to determine the effect of Pten loss on androgen signaling. Approximately 180,000 mG-positive cells were obtained through cell sorting (Fig. 6, D and E). Subsequent qRT-PCR analysis revealed that endogenous Ar expression was reduced in samples isolated from mG-positive and Pten-null cells in comparison to ones from control mG positive cells (Fig. 6F). Expression of Fkbp5, Nkx3.1, Probasin, Msmb, and Phlpp1, all established Ar downstream targets, was significantly less in mG-positive and Pten-null cells than in control mG positive cells (Fig. 6F). These data further reveal that Pten loss results in PI3K/Akt activation and subsequently represses endogenous Ar expression and its transcriptional activity in prostatic cells.
FIGURE 6.

Decreased AR transcriptional activity in Pten-null prostatic cells. A, schematic illustration of labeling PtenloxP/loxP:PB-Cre4-positive cells with the mT/mG reporter. B and C, immunofluorescence analysis was performed on prostate tissues from 6-weeks-old mT/mGL/+:Pten+/+:PB-Cre4 (B1–B3) or mT/mGL/+:PtenloxP/loxP:PB-Cre4 (C1–C3) mice with the GFP and pAkt antibodies. D and E, dissociated prostatic cells from 6-week-old mT/mGL/+:Pten+/+:PB-Cre4 or mT/mGL/+:PtenloxP/loxP:PB-Cre4 mice were analyzed for FACS sorting of GFP-expressing cells. The dot plots show the prostatic cell population before (D) and after FACS sorting (E). F, sorted GFP-positive cells from mT/mGL/+:Pten+/+:PB-Cre4 or mT/mGL/+:PtenloxP/loxP:PB-Cre4 prostates were harvested for real-time qRT-PCR. The levels of Ar, Fkbp5, Nkx3.1, Probasin, Msmb, and Phlpp1 were normalized to that of GAPDH mRNA. The relative mRNA levels from each sample are presented as the mean ± S.D. of the triplicate reactions.
DISCUSSION
The androgen signaling pathway is essential in the pathogenesis of prostate cancer (40, 41). AR is a member of the nuclear hormone receptor superfamily (3, 4) and regulates its downstream target expression in a ligand-dependent manner (42, 43). This in turn controls prostate development and tumorigenesis. Thus, androgen deprivation therapy has been developed and routinely used to treat prostate cancer (44). Despite substantial effort that has been devoted in past decades, the precise molecular mechanisms by which androgen signaling promotes prostate cancer initiation and progression still remain largely unknown. As reported previously, we developed a conditional AR transgenic mouse line, R26hARL/wt:Osr1-Cre, in which the human AR transgene with a LoxP-stop-loxP (LSL) cassette was activated through Osr-1 promoter mediated Cre activity (24). These transgenic mice developed both PIN and prostatic adenocarcinoma lesions in a manner akin to human prostate cancer. In the process of searching for additional cell signaling players in the context of AR mediated oncogenic transformation in this mouse model, we observed that transgenic AR expression inactivated Akt signaling in both prostatic atypical and tumor cells. Our observation demonstrated a possible role of androgen signaling in repressing PI3K/AKT activation.
Within the in vitro scope of this work, we examined the effect of AR/androgen signaling in phosphorylated AKT in two PTEN-null prostate cell lines, LNCaP and LNCaP C4–2. Interestingly, we observed that either depleting androgens or adding Flutamide, an antiandrogen, induces phosphorylation of AKT in LNCaP, but not in LNCaP C4–2 cells, which are derived from LNCaP cells passaged in a castrated host (39). Furthermore knockdown of endogenous AR expression with two different AR shRNAs also increased phosphorylated AKT in LNCaP cells, but not in LNCaP C4–2 cells. Our findings suggest a novel mechanism for prostate cancer progression and CRPC development in which AR signaling is also a mechanism of PI3K/AKT pathway repression.
We further supported our in vitro findings and explored the effect of androgen signaling in Akt activation in vivo. We analyzed Akt expression and activation in the prostates of 10 month old castrated PtenL/L:Osr1-Cre mice that contain invasive prostatic tumors (23). Interestingly, although there was no significant regression of tumor lesions in castrated mice, an increase in Akt, pAkt, and pS6 expression was observed in tumor cells of castrated mice. Although the precise mechanisms of how AR/androgens regulate Akt expression and activation in prostate cancer cells are currently unclear, decreased Ar in the nucleus in tumor cells of castrated mice suggests the action mediated through nuclear AR protein may contribute to this cellular event. Because AR expression has been observed in most androgen-insensitive prostate cancer cells (7), it would be of great value to establish AR's role in androgen insensitive tumor cells. Data generated from these lines of research should further clarify an important but unclear question regarding the interaction between androgen and PI3K/AKT pathways during prostate cancer initiation and progression, leading toward identifying new therapeutic targets for future prostate cancer treatments.
A reciprocal regulation between PI3K and androgen signaling pathways has been implicated in prostate tumorigenesis (36, 37). However, the molecular mechanism underlying this regulation remains unclear. In this study, we used a series of technically advanced and biologically relevant mouse models to further determine whether and how Pten loss regulates AR/androgen action in the mouse prostate. We first generated Ar-IPIL/PtenLoxP/LoxP:PB-Cre and Ar-IPIL/PtenLoxP/LoxP:Osr1-Cre mice (28). Because both nuclear targeted LacZ (nLacZ) and placental alkaline phosphatase (PLAP) reporter genes were inserted into the 3′-untranslated region of the endogenous Ar gene with internal ribosome entry sites (IRESs) (28), we could evaluate endogenous Ar expression by examining LacZ reporter expression in either Pten containing or Pten null prostatic cells in a direct and sensitive way. We observed robust LacZ staining in normal prostatic epithelial cells in both of the transgenic mice (Figs. 4 and 5). In contrast, only a few scattered LacZ positive atypical or tumor cells appeared in PIN or prostatic adenocarcinoma lesions of PtenloxP/loxP:PB-Cre4 or PtenloxP/loxP:Osr1-Cre mice, respectively indicative of reciprocal Ar suppression accompanying Pten loss. Using immunohistochemical approaches, we further confirmed reduced endogenous Ar staining in atypical and tumor cells in those mice. As expected, Akt, pAkt, and pS6 staining appeared much stronger in Pten null atypical and tumor cells than in adjacent normal prostatic epithelial cells. Given that nuclear targeted LacZ was inserted into the 3′-untranslated region of the endogenous Ar gene in the above mouse models (28), our observation that both endogenous Ar and LacZ reporter expression was reduced in Pten-null prostatic atypical and tumor cells suggests that Pten loss/Akt activation likely suppress endogenous Ar expression at transcriptional level. These newly generated mouse models present a unique opportunity to further interrogate the mechanisms and pathways involved in Pten/Akt-mediated endogenous Ar expression and vice versa.
To support our assertions on transcriptional level we developed another new mouse model, mT/mGLoxP/+/PtenLoxP/LoxP:PB-Cre4 mice, to directly and precisely determine the effect of Pten loss on endogenous Ar-mediated transcription. In this elegant double fluorescent mT/mG Cre reporter mouse strain, expression of membrane targeted tandem dimer Tomato (mT) or membrane-targeted green fluorescent protein (mG) is controlled by Cre-mediated recombination (25). Therefore, with the probasin promoter driven Cre expression, mT/mG expression and Pten deletion can occur simultaneously in prostatic epithelial cells, which enables us to trace and isolate Pten-null cells. In these mice we observed a significant decrease in the expression of a series of AR downstream target genes in purified mG-positive and Pten-null prostatic epithelial cells in comparison to mG positive only prostatic epithelial cells (Fig. 6). These data clearly demonstrated that Pten loss represses AR-mediated transcription. Our findings complement published observations in prostate cancer cell lines and human prostate tissues showing that Pten loss and PI3K/AKT activation can repress AR transcriptional activity through either AR co-regulators, EGR1, c-Jun, and EZH2, or HER3-mediated pathways (36, 37). However, our data suggest an additional mechanism by which Pten loss may directly reduce AR expression, directly resulting in decreased AR transcriptional activity. More investigation using the above newly developed mouse models should be pursued to define the mechanisms underlying Pten loss and PI3K/AKT activation in AR expression and transcriptional activation as well as other potential synergistic mechanisms that contribute to prostate tumorigenesis.
In this study, we used a series of relevant and advanced mouse models to investigate the interaction between androgen and PI3K/Akt signaling pathways in Pten null prostatic epithelial cells and animal models. Our data provide several lines of novel evidence demonstrating a reciprocal regulation between PI3K and androgen signaling pathways during prostate cancer initiation and progression. We also provided more comprehensive evidence pointing to a direct role of Pten loss and PI3K/AKT activation in repressing AR expression and activation using biologically relevant mouse models. These mouse models will be further utilized to investigate cellular and molecular events by which the loss of Pten contributed to prostate tumorigenesis. Indubitably, further study of the regulation of the interaction between PI3K and androgen signaling pathways in prostate cancer cells is necessary and should provide fresh insight into the pathogenesis of prostate cancer. This and subsequent work in these presented animal models may enable the identification of novel pathways that can be targeted for prostate cancer prevention and treatment.
Acknowledgments
We thank the Sun laboratory members for technical assistance and scientific inputs.
This work was supported by Public Health Service Grants R01CA070297, R01CA151623, U01CA166894, and R21CA190021 from the National Cancer Institute.
- AR
- androgen receptor
- mPIN
- murine prostatic intraepithelial neoplasia
- DHT
- 5α-dihydrotestosterone
- ADT
- androgen ablation therapy
- CRPC
- castration-resistant prostate cancer
- PTEN
- phosphatase and tensin homolog chromosome 10.
REFERENCES
- 1. Balk S. P. (2002) Androgen receptor as a target in androgen-independent prostate cancer. Urology 60, 132–138; discussion 138–139 [DOI] [PubMed] [Google Scholar]
- 2. Gelmann E. P. (2002) Molecular biology of the androgen receptor. J. Clin. Oncol. 20, 3001–3015 [DOI] [PubMed] [Google Scholar]
- 3. Chang C. S., Kokontis J., Liao S. T. (1988) Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science 240, 324–326 [DOI] [PubMed] [Google Scholar]
- 4. Lubahn D. B., Joseph D. R., Sar M., Tan J., Higgs H. N., Larson R. E., French F. S., Wilson E. M. (1988) The human androgen receptor: complementary deoxyribonucleic acid cloning, sequence analysis and gene expression in prostate. Mol. Endocrinol. 2, 1265–1275 [DOI] [PubMed] [Google Scholar]
- 5. Heinlein C. A., Chang C. (2004) Androgen receptor in prostate cancer. Endocr. Rev. 25, 276–308 [DOI] [PubMed] [Google Scholar]
- 6. Koivisto P., Kononen J., Palmberg C., Tammela T., Hyytinen E., Isola J., Trapman J., Cleutjens K., Noordzij A., Visakorpi T., Kallioniemi O. P. (1997) Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 57, 314–319 [PubMed] [Google Scholar]
- 7. Chen C. D., Welsbie D. S., Tran C., Baek S. H., Chen R., Vessella R., Rosenfeld M. G., Sawyers C. L. (2004) Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 10, 33–39 [DOI] [PubMed] [Google Scholar]
- 8. Baca S. C., Prandi D., Lawrence M. S., Mosquera J. M., Romanel A., Drier Y., Park K., Kitabayashi N., MacDonald T. Y., Ghandi M., Van Allen E., Kryukov G. V., Sboner A., Theurillat J. P., Soong T. D., Nickerson E., Auclair D., Tewari A., Beltran H., Onofrio R. C., Boysen G., Guiducci C., Barbieri C. E., Cibulskis K., Sivachenko A., Carter S. L., Saksena G., Voet D., Ramos A. H., Winckler W., Cipicchio M., Ardlie K., Kantoff P. W., Berger M. F., Gabriel S. B., Golub T. R., Meyerson M., Lander E. S., Elemento O., Getz G., Demichelis F., Rubin M. A., Garraway L. A. (2013) Punctuated evolution of prostate cancer genomes. Cell 153, 666–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weischenfeldt J., Simon R., Feuerbach L., Schlangen K., Weichenhan D., Minner S., Wuttig D., Warnatz H. J., Stehr H., Rausch T., Jäger N., Gu L., Bogatyrova O., Stütz A. M., Claus R., Eils J., Eils R., Gerhäuser C., Huang P. H., Hutter B., Kabbe R., Lawerenz C., Radomski S., Bartholomae C. C., Fälth M., Gade S., Schmidt M., Amschler N., Hass T., Galal R., Gjoni J., Kuner R., Baer C., Masser S., von Kalle C., Zichner T., Benes V., Raeder B., Mader M., Amstislavskiy V., Avci M., Lehrach H., Parkhomchuk D., Sultan M., Burkhardt L., Graefen M., Huland H., Kluth M., Krohn A., Sirma H., Stumm L., Steurer S., Grupp K., Sultmann H., Sauter G., Plass C., Brors B., Yaspo M. L., Korbel J. O., Schlomm T. (2013) Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 23, 159–170 [DOI] [PubMed] [Google Scholar]
- 10. Cantley L. C., Neel B. G. (1999) New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 4240–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li J., Yen C., Liaw D., Podsypanina K., Bose S., Wang S. I., Puc J., Miliaresis C., Rodgers L., McCombie R., Bigner S. H., Giovanella B. C., Ittmann M., Tycko B., Hibshoosh H., Wigler M. H., Parsons R. (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 [DOI] [PubMed] [Google Scholar]
- 12. Steck P. A., Pershouse M. A., Jasser S. A., Yung W. K., Lin H., Ligon A. H., Langford L. A., Baumgard M. L., Hattier T., Davis T., Frye C., Hu R., Swedlund B., Teng D. H., Tavtigian S. V. (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362 [DOI] [PubMed] [Google Scholar]
- 13. Bonneau D., Longy M. (2000) Mutations of the human PTEN gene. Hum Mutat 16, 109–122 [DOI] [PubMed] [Google Scholar]
- 14. Maehama T., Dixon J. E. (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378 [DOI] [PubMed] [Google Scholar]
- 15. Myers M. P., Pass I., Batty I. H., Van der Kaay J., Stolarov J. P., Hemmings B. A., Wigler M. H., Downes C. P., Tonks N. K. (1998) The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc. Natl. Acad. Sci. U.S.A. 95, 13513–13518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cairns P., Okami K., Halachmi S., Halachmi N., Esteller M., Herman J. G., Jen J., Isaacs W. B., Bova G. S., Sidransky D. (1997) Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 57, 4997–5000 [PubMed] [Google Scholar]
- 17. Suzuki H., Freije D., Nusskern D. R., Okami K., Cairns P., Sidransky D., Isaacs W. B., Bova G. S. (1998) Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 58, 204–209 [PubMed] [Google Scholar]
- 18. Wang S. I., Parsons R., Ittmann M. (1998) Homozygous deletion of the PTEN tumor suppressor gene in a subset of prostate adenocarcinomas. Clin. Cancer Res. 4, 811–815 [PubMed] [Google Scholar]
- 19. Di Cristofano A., Pesce B., Cordon-Cardo C., Pandolfi P. P. (1998) Pten is essential for embryonic development and tumour suppression. Nat. Genet. 19, 348–355 [DOI] [PubMed] [Google Scholar]
- 20. Wang S., Gao J., Lei Q., Rozengurt N., Pritchard C., Jiao J., Thomas G. V., Li G., Roy-Burman P., Nelson P. S., Liu X., Wu H. (2003) Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4, 209–221 [DOI] [PubMed] [Google Scholar]
- 21. Ma X., Ziel-van der Made A. C., Autar B., van der Korput H. A., Vermeij M., van Duijn P., Cleutjens K. B., de Krijger R., Krimpenfort P., Berns A., van der Kwast T. H., Trapman J. (2005) Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res. 65, 5730–5739 [DOI] [PubMed] [Google Scholar]
- 22. Backman S. A., Ghazarian D., So K., Sanchez O., Wagner K. U., Hennighausen L., Suzuki A., Tsao M. S., Chapman W. B., Stambolic V., Mak T. W. (2004) Early onset of neoplasia in the prostate and skin of mice with tissue-specific deletion of Pten. Proc. Natl. Acad. Sci. U.S.A. 101, 1725–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwak M. K., Johnson D. T., Zhu C., Lee S. H., Ye D. W., Luong R., Sun Z. (2013) Conditional deletion of the Pten gene in the mouse prostate induces prostatic intraepithelial neoplasms at early ages but a slow progression to prostate tumors. PLoS One 8, e53476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu C., Luong R., Zhuo M., Johnson D. T., McKenney J. K., Cunha G. R., Sun Z. (2011) Conditional expression of the androgen receptor induces oncogenic transformation of the mouse prostate. J. Biol. Chem. 286, 33478–33488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muzumdar M. D., Tasic B., Miyamichi K., Li L., Luo L. (2007) A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 [DOI] [PubMed] [Google Scholar]
- 26. Grieshammer U., Agarwal P., Martin G. R. (2008) A Cre transgene active in developing endodermal organs, heart, limb, and extra-ocular muscle. Genesis 46, 69–73 [DOI] [PubMed] [Google Scholar]
- 27. Wu X., Wu J., Huang J., Powell W. C., Zhang J., Matusik R. J., Sangiorgi F. O., Maxson R. E., Sucov H. M., Roy-Burman P. (2001) Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev. 101, 61–69 [DOI] [PubMed] [Google Scholar]
- 28. Shah N. M., Pisapia D. J., Maniatis S., Mendelsohn M. M., Nemes A., Axel R. (2004) Visualizing sexual dimorphism in the brain. Neuron 43, 313–319 [DOI] [PubMed] [Google Scholar]
- 29. Sugimura Y., Cunha G. R., Donjacour A. A. (1986) Morphological and histological study of castration-induced degeneration and androgen-induced regeneration in the mouse prostate. Biol. Reprod. 34, 973–983 [DOI] [PubMed] [Google Scholar]
- 30. Johnson D. T., Luong R., Lee S. H., Peng Y., Shaltouki A., Lee J. T., Lin D., Wang Y., Sun Z. (2013) Deletion of leucine zipper tumor suppressor 2 (lzts2) increases susceptibility to tumor development. J. Biol. Chem. 288, 3727–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee J., Beliakoff J., Sun Z. (2007) The novel PIAS-like protein hZimp10 is a transcriptional co-activator of the p53 tumor suppressor. Nucleic Acids Res. 35, 4523–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 33. Tu W. H., Zhu C., Clark C., Christensen J. G., Sun Z. (2010) Efficacy of c-Met inhibitor for advanced prostate cancer. BMC Cancer 10, 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Verras M., Lee J., Xue H., Li T. H., Wang Y., Sun Z. (2007) The androgen receptor negatively regulates the expression of c-Met: implications for a novel mechanism of prostate cancer progression. Cancer Res. 67, 967–975 [DOI] [PubMed] [Google Scholar]
- 35. Li T. H., Zhao H., Peng Y., Beliakoff J., Brooks J. D., Sun Z. (2007) A promoting role of androgen receptor in androgen-sensitive and -insensitive prostate cancer cells. Nucleic Acids Res. 35, 2767–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carver B. S., Chapinski C., Wongvipat J., Hieronymus H., Chen Y., Chandarlapaty S., Arora V. K., Le C., Koutcher J., Scher H., Scardino P. T., Rosen N., Sawyers C. L. (2011) Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19, 575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mulholland D. J., Tran L. M., Li Y., Cai H., Morim A., Wang S., Plaisier S., Garraway I. P., Huang J., Graeber T. G., Wu H. (2011) Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 19, 792–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brognard J., Sierecki E., Gao T., Newton A. C. (2007) PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 25, 917–931 [DOI] [PubMed] [Google Scholar]
- 39. Thalmann G. N., Anezinis P. E., Chang S. M., Zhau H. E., Kim E. E., Hopwood V. L., Pathak S., von Eschenbach A. C., Chung L. W. (1994) Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 54, 2577–2581 [PubMed] [Google Scholar]
- 40. Kyprianou N., Isaacs J. T. (1988) Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology 122, 552–562 [DOI] [PubMed] [Google Scholar]
- 41. Abate-Shen C., Shen M. M. (2000) Molecular genetics of prostate cancer. Genes Dev. 14, 2410–2434 [DOI] [PubMed] [Google Scholar]
- 42. Sanchez E. R., Faber L. E., Henzel W. J., Pratt W. B. (1990) The 56–59-kilodalton protein identified in untransformed steroid receptor complexes is a unique protein that exists in cytosol in a complex with both the 70- and 90-kilodalton heat shock proteins. Biochemistry 29, 5145–5152 [DOI] [PubMed] [Google Scholar]
- 43. Sullivan W. P., Vroman B. T., Bauer V. J. (1992) Isolation of steroid receptor binding protein from chicken oviduct and production of monoclonal antibodies. J. Steroid Biochem. Mol. Biol. 43, 37–41 [DOI] [PubMed] [Google Scholar]
- 44. Huggins C., Hodges C. V. (2002) Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J. Urol. 168, 9–12 [DOI] [PubMed] [Google Scholar]