

Abstract
Electrostatic interactions often play key roles in the recognition of small molecules by nucleic acids. An example is aminoglycoside antibiotics, which by binding to ribosomal RNA (rRNA) affect bacterial protein synthesis. These antibiotics remain one of the few valid treatments against hospital-acquired infections by Gram-negative bacteria. It is necessary to understand the amplitude of electrostatic interactions between aminoglycosides and their rRNA targets to introduce aminoglycoside modifications that would enhance their binding or to design new scaffolds. Here, we calculated the electrostatic energy of interactions and its per-ring contributions between aminoglycosides and their primary rRNA binding site. We applied either the methodology based on the exact potential multipole moment (EPMM) or classical molecular mechanics force field single-point partial charges with Coulomb formula. For EPMM, we first reconstructed the aspherical electron density of 12 aminoglycoside-RNA complexes from the atomic parameters deposited in the University at Buffalo Databank. The University at Buffalo Databank concept assumes transferability of electron density between atoms in chemically equivalent vicinities and allows reconstruction of the electron densities from experimental structural data. From the electron density, we then calculated the electrostatic energy of interaction using EPMM. Finally, we compared the two approaches. The calculated electrostatic interaction energies between various aminoglycosides and their binding sites correlate with experimentally obtained binding free energies. Based on the calculated energetic contributions of water molecules mediating the interactions between the antibiotic and rRNA, we suggest possible modifications that could enhance aminoglycoside binding affinity.
Introduction
Since the 1960s, aminoglycoside antibiotics continue to be one of the most valid treatments for hospital-acquired serious bacterial infections (1). From the chemical standpoint, aminoglycosides are often referred to as pseudooligosaccharides. Aminoglycosides are divided into three structural families according to the position of the glycosidic linkages. The two families, 4,5- and 4,6-disubstituted 2-deoxystreptamines (2-DOS) (Fig. 1), have high affinity to the aminoacylated-tRNA binding site in bacterial ribosomal RNA (rRNA).
Figure 1.
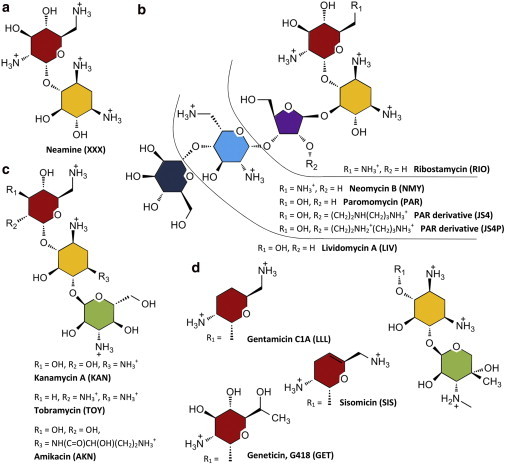
Chemical structures of aminoglycosides considered in this study: neamine (a), 4,5-2-DOS (b), 4,6-2-DOS kanamycin aminoglycosides (c), and 4,6-2-DOS gentamicin aminoglycosides (d). Ring I is red, ring II (2-DOS) yellow, furanose ring III violet, pyranose ring III green, ring IV blue, and ring V dark blue. The numbering style of carbon atoms is shown for 4,5-2-DOS aminoglycosides. To see this figure in color, go online.
The primary mode of action of 2-DOS aminoglycosides against prokaryotic ribosomes arises from their binding to the 30S ribosomal subunit and results in their interference with protein synthesis (2). Aminoglycosides interact with helix 44 of 16S rRNA in the decoding region (A-site) responsible for recognizing cognate aminoacylated-tRNA. The binding of 2-DOS aminoglycosides restricts the conformational freedom of two A-site adenines (A1492 and A1493 using standard Escherichia coli numbering) by locking them in an extrahelical conformation. These adenines assure the fidelity of decoding by acquiring an extrahelical state only if a cognate tRNA binds to the A-site. Since aminoglycosides artificially enforce such a flipped-out adenine conformation, the incorporation of near-cognate tRNAs becomes possible and the accuracy of decoding decreases (3). 2-DOS aminoglycosides have a secondary binding site in the ribosome—helix 69 of the 50S subunit (4).
Due to their polycationic nature, the binding of 2-DOS aminoglycosides to RNA is believed to be driven electrostatically (5–7). The pKas of their amino groups are >7.0 except for the 3-amino group in the 2-DOS ring, whose pKas in neomycin-class aminoglycosides and in amikacin are 5.7 (8) and 6.7 (9), respectively. The amide group in amikacin remains un-ionized at pH >5.4 (9) (Fig. 1). Therefore, aminoglycosides are positively charged in neutral pH and at 25°C (10). However, ionic strength, temperature, and pH of the buffer are important for protonation of their amino groups and energetics of binding (8,11,12). In addition, the binding process at pH >5.5 induces a proton uptake (11), meaning that the RNA-aminoglycoside complexes favor fully protonated antibiotics. Thus, there is interplay between the charge distribution in aminoglycosides and their binding strength. Assuming that the 3-amino group is charged in bound aminoglycosides at neutral pH, their net charge is between +4e and +7e.
Crystallographic techniques confirmed the binding mode of aminoglycosides in bacterial ribosomes and in model RNA oligonucleotides mimicking the A-site (13). It was proven that an isolated RNA oligonucleotide composed of ∼45 nucleotides with two symmetrically located binding sites can be used as an A-site mimic (13) (see Fig. 2 a). Various 2-DOS aminoglycosides bound to such RNA constructs were crystallized (14,15) and their structure was found to be similar to the corresponding site in the ribosome. In addition, A-site-mimicking crystals revealed many water-mediated hydrogen bonds formed between aminoglycosides and RNA (16).
Figure 2.

Secondary structure of 2-DOS aminoglycoside binding sites mimicking the ribosomal A-site. Nucleotides forming non-Watson-Crick basepairs are numbered according to the E. coli 16S rRNA sequence. (a) Aminoglycoside binding site defined in François et al. (16) (blue frame) and the extended binding site used herein, based on distance criteria (red frame). (b) Minimal A-site model of the paromomycin binding site from footprinting studies (2) used in solution studies. To see this figure in color, go online.
A single-stranded model of the A-site shown in Fig. 2 b was exploited in solution studies. The dissociation constants and free enthalpy of binding were determined in thermal denaturation and fluorescence studies (17), UV spectrometry (8,11), isothermal titration calorimetry (8,11,12), differential scanning calorimetry (8), osmotic stress experiments (12), NMR studies (2,11), and surface plasmon resonance experiments (18,19). These experimental studies were complemented by many computational studies. All-atom molecular dynamics (MD) simulations of the A-site concentrated on the functional flexibility of A1492 and A1493 (20,21). MD studies of the A-site with paromomycin (20) and amikacin (22) confirmed that the neamine core (Fig. 1, rings I and II) is an anchor in the binding cleft. The comparison of MD simulations of several 4,5- and 4,6-DOS aminoglycosides (22–24) revealed that the residence of water molecules in certain hydration sites correlates with aminoglycoside binding affinity.
The electrostatic contributions to aminoglycoside binding in the static A-site (17) and 30S subunit (17,25) were studied with the implicit-solvent Poisson-Boltzmann model of electrostatics (26). The process of association of aminoglycosides with rRNA was studied using Brownian dynamics combined with the Poisson-Boltzmann model (27,28). These studies confirmed the importance of electrostatics to association and formation of aminoglycoside-A-site complexes. The partial charges of aminoglycoside atoms in these computations were fixed single-point charges from classical molecular-mechanics force fields.
Among methods used to approximate electrostatics that go beyond the point-charge description, the approach based on the aspherical atom database (29–31) takes into account the asphericity of atomic electron density, contrary to point charges encountered in classical force fields. It assumes transferability of electron density, meaning that atoms in chemically equivalent vicinities have similarly deformed electron densities. The aspherical electron densities for various atom types are gathered in the University at Buffalo Databank (UBDB) (31) and can be applied to reconstruct the electron density of biomolecules. UBDB contains >200 atom types to cover atoms present in proteins, nucleic acids, and many organic molecules (29). Other similar data banks (32,33) have been developed and are compared in Bąk et al. (34). The latter study showed that the Coulombic intermolecular interaction energies determined by ab initio methods are best reproduced by UBDB combined with the exact potential multipole moment (EPMM) method (35). A few protein complexes have been successfully studied with the UBDB approach, e.g., the interactions between small peptide ligands and glycopeptide antibiotics (36), the PDZ domain from the scaffolding protein syntenin (30), neuraminidase and its inhibitors (37), and protein kinases interacting with sunitinib (38). It was also shown that UBDB effectively reproduces the electrostatic interactions in molecular dimers (39), Watson-Crick basepairs (40), and nucleic acid fragments (41).
Here, we have applied the aspherical electron density approach and UBDB to assess the importance of electrostatics in the binding of aminoglycosides and their rings to rRNA. We used 12 aminoglycosides with different numbers of rings and different net charges. To our knowledge, this study is the first attempt to compare the structures and electrostatic properties of such a large number of different aminoglycoside-A-site complexes. In addition, we have quantified the contributions of the water molecules located in the vicinity of the antibiotics, which mediate the hydrogen bonds with RNA. Based on the water patterns and their electrostatic interactions with the A-site and aminoglycosides, we suggested positions in aminoglycosides that are relevant for modification and might increase their affinity toward RNA.
Materials and Methods
Calculations of electrostatic interaction energy based on electron density
To describe the electron density, we applied Hansen-Coppens formalism (42), in which the atomic electron density for each atom k, , is described by the equation
| (1) |
where , , and are the populations of core, valence electrons, and multipoles, respectively. and denote, respectively, the spherical one-electron normalized core and valence electron densities, and κ and are the contraction-expansion coefficients. The radial functions, , and angular functions, , model the aspherical deformations and are represented by real spherical harmonic functions normalized to the electron density.
The energy of electrostatic interactions, Eel, was calculated from the reconstructed electron density via the EPMM method (35) as a sum of two terms, = + . The energy of short-range interactions, , for the overlapping charge densities, i.e., located at a distance of <4.5 Å, was calculated with the exact potential (EP) using Eq. 2:
| (2) |
where and are the atomic charges, and are the atomic electron densities, and and are the nuclear potentials of molecules A and B, respectively. For long-range interactions, i.e., with nonoverlapping charge densities, the Buckingham-type (multipole moment (MM)) approximation described by Eq. 3 was applied to calculate :
| (3) |
where , etc. are the interaction tensors, and , etc. are the atomic multipole moments as described in Volkov et al. (35).
Preparation of structures
Twelve crystal structures of aminoglycosides complexed with identical rRNA constructs (Fig. 2 a) were taken from the Research Collaboratory for Structural Bioinformatics database: 2ET8 (16) with neamine (XXX), 1MWL (43) with geneticin (GET), 2G5Q (44) with amikacin (AKN), 2ESI (16) with kanamycin (KAN), 4F8V (14) with sisomicin (SIS), 1LC4 (45) with tobramycin (TOY), 2ET3 (16) with gentamicin (LLL), 2ET5 (16) with ribostamycin (RIO), 1J7T (46) with paromomycin (PAR), 2BEE (47) with modified paromomycin (JS4), 2ET4 (16) with neomycin (NMY), and 2ESJ (16) with lividomycin (LIV). The structures contained two binding sites, identical in sequence but slightly different in conformation and composition, e.g., with differently placed water molecules. In all structures, the phosphates on the 5′-termini were removed. Terminal uridines from the 2ESI complex were also removed. Hydrogen atoms were added to aminoglycosides using PyMOL (48) and their partial charges were assigned with antechamber (49) at neutral pH. Aminoglycoside amino groups were protonated according to their pKas (8) and proton uptake was linked with the process of binding (11). The group left neutral was the amide group in the l-HABA (L-(−)-γ-amino-α-hydroxybutyric acid) tail of AKN (9), as shown in Fig. 1. For RNA, the ff99 Amber force field (50) parameters were assigned with Amber11 tools (51). For aminoglycosides, the AM1-BCC method (52) in antechamber was used to reproduce the HF/6-31G∗ RESP partial charges. The hydrogens missing in RNA and oxygens of water molecules were built in LEaP to perform energy minimization with explicit water in Sander. The space around the antibiotic was filled with TIP3P (53) water molecules, but the crystal waters located within 3.5 Å of the antibiotic were kept. For analyses, all water molecules (crystal and TIP3P) within 3.5 Å of both the aminoglycoside and RNA were used, since they were expected to mediate the hydrogen bonds with the antibiotic. The steepest-descent method (8000 cycles) followed by conjugate gradients (2000 cycles) with Cartesian restraints weighting 50 kcal/mol on nonhydrogen atoms excluding water were applied. No counterions were added, since no ions were reported in the crystal structures except for one ion in RIO, which was removed.
Calculations of electrostatic and van der Waals energies
An in-house bash script for calculating electrostatic energy was prepared. Computations included transforming the above prepared PDB files to shelx files and assigning UBDB atom types implemented in the LSDB program (29–31,54). Every nucleotide and aminoglycoside was scaled individually to its formal charge in LSDB. Next, the energies of electrostatic interactions between aminoglycosides and RNA were computed with the XDPROP module in the XD2006 program (55).
Coulomb interaction energies in vacuum between all atom pairs, , were calculated using the single-point fixed partial charges described earlier (AM1-BCC for aminoglycosides and ff99 fixed Amber charges for RNA) and a dielectric constant of 1. Similar computations were performed with the electrostatic potential (ESP) charges for aminoglycosides, , obtained with Gaussian 09 (56) using HF/6-31G∗. The van der Waals energy between aminoglycosides and their binding sites was approximated by the Lennard-Jones 6-12 potential, truncated at the 20 Å cutoff distance, with the same parameters as in the Amber ff99 force field used in the minimization procedure.
Superposition of aminoglycoside-RNA complexes
Different aminoglycosides bind the A-site while maintaining the neamine (XXX) core in the same position. Fig. 3 shows the superposition of the complexes using least-squares fitting. The average total root mean-square deviation (RMSD), measured between the phosphorus atoms of the aligned complexes and 1J7T was 0.60 ± 0,05 Å for binding site 1 and 0,62 ± 0,06 Å for site 2. The RNA binding site for the electrostatic analyses (Fig. 2 a, red outline) was chosen according to the distance criteria. Nucleotides contacting any aminoglycoside at a distance of ≤4 Å were included. A discussion on the binding-site choice is presented in Section S1 and Figs. S1 and S2 in the Supporting Material. Each aminoglycoside located in site 1 or 2 was considered separately (Fig. 2 a).
Figure 3.
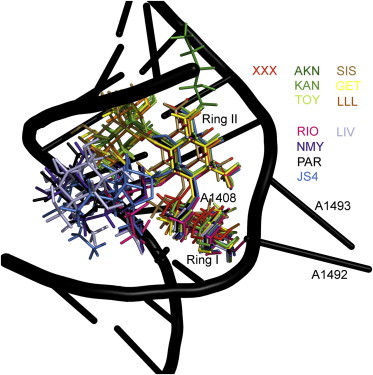
Positions of aminoglycosides bound specifically in the first binding site after superposing the RNA complexes onto the phosphorus atoms of the 1J7T reference structure. The aminoglycosides used were neamine (XXX); the 4,5-DOS aminoglycosides ribostamycin (RIO), neomycin (NMY), paromomycin (PAR), lividomycin (LIV), and modified paromomycin (JS4); and the 4,6-DOS aminoglycosides sisomicin (SIS), geneticin (GET), gentamicin (LLL), amikacin (AKN), kanamycin (KAN), and tobramycin (TOY). Antibiotic abbreviations are as in the crystal structures. For clarity, only the RNA backbone of the 1J7T complex with PAR is shown. To see this figure in color, go online.
Results and Discussion
Correlations of electrostatic interaction energies in aminoglycoside-RNA complexes with experimental binding free energies
Experiments do not provide the electrostatic contribution to binding, since it cannot be decoupled from other contributions. Typically, the equilibrium association binding constants, Ka, and Gibbs energies are determined. Therefore, we checked whether the calculated electrostatic interaction energies (gathered in Table S1) reproduce the experimentally determined order of binding of different aminoglycosides and correlated with four sets of experimental binding free energies (12,17–19). The correlation plots for the UBDB-based interaction energies, Eel, are presented in Fig. 4. Note that each experiment was performed using a different technique and buffer, so we compared them separately. Also, because binding of aminoglycosides is salt- and pH-dependent, this comparison needs to be treated with caution. The R2 values are >0.8, which is a surprisingly good result, bearing in mind the limitations of the method (discussed in the Conclusions) and small number of experimental data. The R2 values between the experimental s and the Coulomb interaction energies (, calculated with partial charges) were 0.803, 0.890, and 0.898 for the three assays, respectively.
Figure 4.
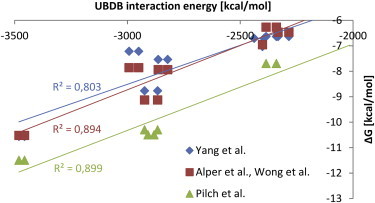
Correlation between the UBDB-based electrostatic energies of interaction (Eel) of various aminoglycosides with RNA and the corresponding Gibbs energies () obtained from fluorescence experiments (Yang et al. (17), pH 7.5, 150 mM Na+, 0.5 mM EDTA, and 20 mM HEPES buffer), surface plasmon resonance (Alper et al. (19), Wong et al. (18), pH 7.4, 150 mM NaCl, 3.4 mM EDTA, and 10 mM HEPES), and isothermal titration calorimetry and melting temperature assays (Pilch et al. (12), pH 5.5, 150 mM Na+, 0.1 mM EDTA, and 10 mM sodium cacodylate). To see this figure in color, go online.
The comparison between the UBDB-computed Eel and Coulomb-computed is shown in Fig. S3 and discussed in Section S2 in the Supporting Material. The differences between the electrostatic interaction energies of the more sophisticated method and simple Coulomb approach were surprisingly small, suggesting that the monopole term dominates electrostatics in these highly charged complexes. Note that for the aminoglycosides bound specifically to the energies were less negative than , but, for the nonspecific antibiotics, this trend was reversed. Also, the interaction energies (see Table S1), computed using AM1BCC charges () were very similar to the ones calculated with the presumably more exact ESP charges (). Further, we present only the results of the UBDB approach.
We also compared the van der Waals interaction energies between aminoglycosides and their binding sites (see Table S1 and Fig. S4). These energies do not correlate with experimental , which further confirms that the electrostatic contribution is an important factor in aminoglycoside binding.
Electrostatic energy contributions to binding from individual aminoglycoside rings
The electrostatic energy contributions to binding partitioned into individual aminoglycoside rings presented in Fig. 5 show that total Eel values are similar for two mirrored binding sites. Antibiotics bound nonspecifically (RIO∗ and KAN∗) can be recognized because their Eels are less favorable. Overall, Eels are dominated by the contributions from the positively charged amino groups. For example, in NMY and PAR complexes, in which aminoglycosides differ by one amino group substituted by one hydroxyl group, the electrostatic energy term varies by ∼500 kcal/mol. A similar change in Eel is observed in the complexes with TOY and KAN, in which, apart from one amino group replaced with a hydroxyl group, there is an additional hydroxyl group in KAN. Eel also depends on the protonation state of an amino group, as shown for paromomycin derivatives JS4 and JS4P. Their secondary amino group in the side chain of the furanose ring was either neutral (JS4) or protonated (JS4P) (Fig. 1 b), which resulted in the change in Eel for ring III.
Figure 5.
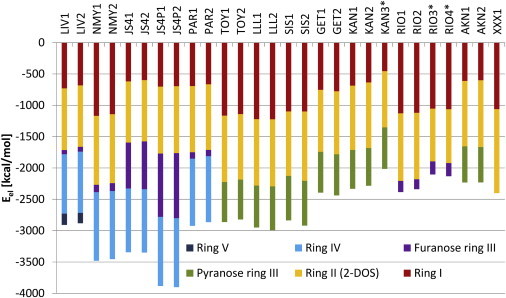
The electrostatic interaction energy between an aminoglycoside and A-site partitioned into aminoglycoside rings obtained from the aspherical charge approach (Eel). Numbers 1 and 2 after the PDB ID denote the order in which the antibiotic appears in the PDB file and correspond to binding sites 1 and 2. The asterisk indicates antibiotics bound nonspecifically. Coloring of rings is the same as in Fig. 1. To see this figure in color, go online.
We observed a relatively high Eel between the smallest aminoglycoside (neamine (XXX)) and its binding site. Further, the electrostatic contribution from neamine, but as a part of larger aminoglycosides (RIO and NMY, which have the same functional groups as XXX in rings I and II), becomes less favorable in the presence of additional rings III and IV (see Figs. 5 and S5). This may be a result of attenuating the of neamine’s amino groups. Overall, for all aminoglycosides, the Eel for the neamine core (rings I and II) is less favorable than for neamine alone. This happens even though the neamine core is stably bound, in contrast to rings III, IV, and V, as shown in MD studies (20,23); the contacts formed by the other rings with the A-site were more transient than the contacts formed by neamine.
The differences in functional groups between aminoglycoside rings and the corresponding Eel are shown in Table S2. Ring I has the largest variety of substituents that give up to twofold differences in the electrostatic interaction energies depending on the aminoglycoside. However, it is not only the number of charged amino groups, but also the presence of uncharged moieties, that matters. Placing the hydroxyl groups instead of hydrogen atoms in the 3′ and 4′ positions decreases the strength of the electrostatic interaction between a ring and its binding site (e.g., LLL and TOY in Table S2). This effect corresponds with the experimental observation that the removal of hydroxyl groups lowers the basicity of neighboring amines (5). Adding a 4′-5′ double bond in ring I as in SIS gives a similar effect. The electrostatic importance of the 2′ amino group is visible by comparing the Eel of ring I for NMY, RIO, and XXX with that for KAN and AKN, which are deprived of this group. In a similar way, the importance of the 5′ amino group is emphasized by comparing the Eel of ring I for NMY with that for PAR.
Among the 2-DOS rings (ring II), the highest contribution to Eel comes from the XXX ring type (Table S2). The differences in the interaction energies between 4,5- and 4,6-2-DOS substituted rings II are not significant. Adding the l-HABA group to ring II in AKN does not change the electrostatic energy of interaction, which is in accord with our findings that the binding of AKN (specifically the l-HABA group) is also entropically driven (22). The six-membered pyranose ring III in LLL, SIS, and GET complexes has a higher Eel than does the analogous ring III in TOY, KAN, or AKN, which differs only by one hydroxymethyl and two methyl groups. Significantly lower Eel values are observed for the five-membered uncharged furanose ring III. JS4(P) is an exception, but it is charged, and the 10-fold difference in Eel is caused by two amino groups in the 2″ substituent. Rings IV are identical in all aminoglycosides equipped with this ring. On average, ring IV gives high electrostatic contribution, similar to rings I and II. MD simulations of the complex of the A-site with PAR (20) and the superposition shown in Fig. 3 suggest that ring IV is flexible, so its high Eel is somewhat surprising. The contribution of ring V is significantly lower and comparable to the contribution of the unmodified furanose ring.
From the electrostatic interaction energy standpoint (Table S2), one could indicate a beneficial combination of functional groups that would contribute to high electrostatic interaction energy. A good example would be JS4P with modifications introduced in ring I—a change of the 3′ group to H, 4′ to H, and 5′ to (Fig. 1). However, the proposed modifications follow only from the observed correlations between the computed Eel and experimental and do not take into account the influence of neigboring rings and entropy effects.
Water-mediated interactions in the binding site
The locations of water molecules with high attractive electrostatic interaction energies are interesting because they could be replaced with functional groups attached to an aminoglycoside to improve its binding affinity. We analyzed the electrostatic interactions of the water molecules located within 3.5 Å of both the aminoglycoside and the RNA. Next, we selected only those water molecules whose Eels with the closest RNA nucleotide or aminoglycoside ring were <−15 kcal/mol. Their calculated electrostatic interaction energies, distances between heavy atoms, and angles of hydrogen bonds are shown in Section S3 in the Supporting Material and Tables S3–S6. We found a few repetitive hydrogen-bond patterns in which the interactions between the antibiotic and RNA are through a water molecule. The highest number of mediating waters appeared around JS4P, LIV, RIO, and XXX, whereas water mediation around SIS, GET, and KAN was almost negligible. Among the water molecules that interact most strongly with RNA or aminoglycoside, we describe the ones that overlap in different complexes.
The most common hydration pattern emerges in six complexes (RIO, JS4P, LIV, NMY, PAR, and TOY) with a hydrogen-bond network termed Pattern 1 (Fig. 6, left; also, see Section S3 in the Supporting Material and Tables S3–S6). All these complexes, except TOY, contain 4,5-DOS aminoglycosides. A water molecule is trapped between ring I and the RNA with two hydrogen bonds assuring its stability. The length of one hydrogen bond formed between the water oxygen and A1493:OP1 oxygen of the phosphate group varies from 2.7 to 3.1 Å, depending on the complex (the respective angle range is 170–179°). The second common hydrogen bond, between the water oxygen and N2′ of the amino group, is 2.8–2.9 Å long and the angle range is 154–168°. The Eels between these waters and either aminoglycoside or RNA oscillate between −60 and −85 kcal/mol. In two complexes (PAR and JSP4), an additional, third interaction is present between the water oxygen and the O3′ hydroxyl group of PAR and JS4P. Although the bonding distance for this third hydrogen bond is longer (∼3.6 Å), this water molecule was previously shown to be stable in MD simulations of a PAR-A-site complex (20,23,46).
Figure 6.
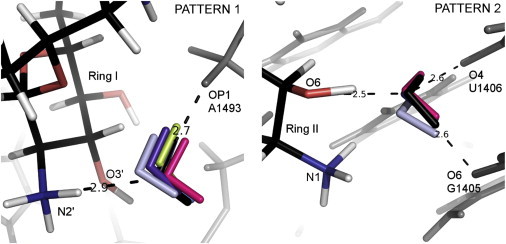
Patterns 1 (left) and 2 (right). Superposition of the A-site complexes showing water molecules mediating the interactions of RNA (gray) with ribostamycin (RIO (pink)), neomycin (NMY (violet)), paromomycin (PAR (black)), lividomycin (LIV (light gray)), modified paromomycin (JS4 (blue)), and tobramycin (TOY (light green)). PAR atoms are shown in black (carbon atoms), red (oxygen), blue (nitrogen), and white (hydrogen). The distances indicated by the dashed lines correspond to the PAR complex. To see this figure in color, go online.
The next common water mediation of ring II is conserved in four analyzed structures, PAR, NMY, RIO, and LIV (Pattern 2; Fig. 6, right, and see Section S3 in the Supporting Material and Tables S3, S5, and S6). The electrostatic interaction energy of this water with RNA varies from −32 kcal/mol to −83 kcal/mol, whereas its electrostatic interaction energy with aminoglycosides is about −130 kcal/mol. In this pattern, three hydrogen bonds are formed with the water molecule, and it also weakly interacts with the N1 amino group of ring II (except in NMY).
In the second binding site in the crystal complex with PAR (1J7T), the above interactions, between the O6 hydroxyl group of the antibiotic and RNA oxygen atoms O4 (U1406) and O6 (G1405), are held by not one but two different crystal water molecules. However, MD simulations either reveal only one water molecule contacting the U1406:O4 atom (23) or do not detect any higher water density in this site (20). In the NMY and LIV crystal structures, the water contacting U1406:O4 is present, but there is no direct water contact with G1405:O6. Another MD study that focused on NMY, PAR, and RIO again showed long-lasting water mediating contact with the U1406:O4 atom (24).
The pattern repeated in the PAR, LIV, and JS4P complexes, termed Pattern 3, is shown in Fig. S6 (for a description, see Section S3 in the Supporting Material and Tables S4–S6). The water-mediated interaction is with two phosphates (A1492 and G1491) and an amino group N2′ of ring I. High water content was observed in this site in MD simulations with PAR (20,23). The interaction of this water molecule with G1491 has a favorable Eel of between −49 and −87 kcal/mol; its Eel with A1492 is between −23 and −78 kcal/mol, and that with the amino group is between −99 and −112 kcal/mol.
Three water molecules preserving similar contacts with RNA were detected in the complexes with NMY, RIO, and XXX (Pattern 4 in Fig. 7, left, and Tables S3, S5, S6). The water molecules are close to the N3 and N6′ aminoglycoside groups and the A1493 phosphate. This water-mediated interaction with the phosphate group and two amino groups was also observed in MD simulations (23). A similar scheme is present in the PAR complex, but the water molecule mediates the interactions between the phosphate of A1493 and the hydroxyl group O6′ of PAR (which is in a location similar to that of the amino group N6′ in this example).
Figure 7.
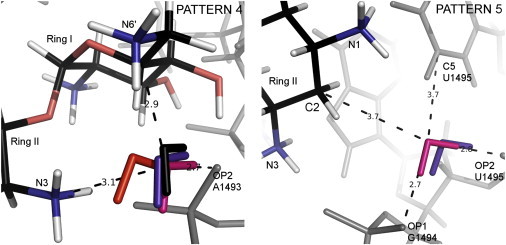
Patterns 4 (left) and 5 (right). Water molecules mediating hydrogen bonds in the complexes of ribostamycin (RIO (pink)), neomycin (NMY (violet)), paromomycin (PAR (black)), and neamine (XXX (red)) with A-site RNA (gray). RIO atoms are colored black, red, blue, and white according to atom type, as in Fig. 6. The distances (dashed lines) correspond to the RIO complex. To see this figure in color, go online.
In Fig. 7, right, we show a water molecule located close to the C2 atom of ring II in the NMY and RIO complexes (Pattern 5 in Tables S3 and S5) that interacts with G1494:OP1 and U1495:OP2 and with the U1495:C5 atom of the base. The Eel between ring II and this water molecule is >−50 kcal/mol, which means that this water is not crucial for aminoglycoside binding.
In the NMY complex, a water molecule located in the same area as the hydroxyl group of ring III in KAN, TOY, SIS, and LLL was detected (see Fig. S7 and Pattern 6 in Table S5). The hydrogen bonding between this water molecule and the N1 amino nitrogen reflects the intramolecular interaction formed by the O2″ and the N1 group of KAN. This pattern is a nice example of how some water molecules mediating hydrogen bonds between aminoglycosides and RNA could be successfully substituted with a functional group, mimicking these interactions.
Modifications proposed based on water-mediated interactions
From the analysis of water-mediated interaction patterns (distances between hydrogen-bond donors and acceptors and the directionality and spatial localization of these bonds) and the electrostatic interactions of waters with RNA and aminoglycoside in the studied complexes, we propose a few modifications that could enhance aminoglycoside binding affinity. Note that our analysis does not take into account such effects as impact on solvation, influence of the modifications on pKas of other groups, and entropic contribution. The easiest modifications would simply substitute the water molecules detected in different complexes, which take part in similar mediating hydrogen-bond patterns, for amino or hydroxyl groups attached via a flexible linker to the aminoglycoside. However, water molecules that interact strongly and repetitively with RNA are not necessarily good candidates for replacement with an aminoglycoside extension, because their release from RNA (even though entropically favorable for binding) probably would not compensate the enthalpic cost. Pattern 1 is an example of how attempts to replace the water molecule with any functional group attached to aminoglycoside would probably be inefficient due to the presence of this water in many complexes and its strong electrostatic interaction with RNA. The interactions from a modified aminoglycoside would need to be enthalpically much stronger than those via water, which would be difficult to achieve. Therefore, the best candidates for substitution are those water molecules that interact strongly with aminoglycoside and weakly with RNA.
In 4,5-DOS aminoglycosides, the water molecule in Pattern 2 in Fig. 6 might be a good candidate for replacement. The O6 hydroxyl group of ring II could be substituted with a functional group with a more basic character, for example, , which would keep the existing connection with G1405:O6 and U1406:O4 and create an overall stronger hydrogen-bond network. This modification should be feasible, because this water molecule interacts strongly with an aminoglycoside and weakly with RNA (Section S3 in the Supporting Material and Tables S3, S5, and S6). Moreover, the 4,6-DOS aminoglycosides contain a pyranose ring III at this site and they do maintain the antibacterial potency.
A promising site for a modification could be based on the repeated water arrangement in NMY and RIO complexes (Pattern 5 in Fig. 7). The attachment of the group to the C2 carbon could be beneficial. Based on the structures of the complexes, this additional group should introduce new contacts between an aminoglycoside and G1494 and U1495 nucleotides.
The hydrogen-bond pattern shown in Fig. 7 (Pattern 4 in Tables S3, S5, S6) also suggests a modification. Based on this interaction pattern, we propose a locked aminoglycoside, which would join rings I and II through the amino groups N3 and N6′. One could connect these rings using the path appointed by the mediating water molecule to obtain a conformationally restricted aminoglycoside. This modification should stiffen the aminoglycoside and lower the necessary conformational adaptation of rings I and II upon binding. However, other conformationally restricted derivatives of PAR and NMY appeared to have affinities similar to those of the unmodified aminoglycosides (57), so the conformational restriction does not necessarily mean overall affinity improvement.
One of the requirements for successful improvement of aminoglycosides is to design a modification that would make the compound resistant to enzymatic modifications by aminoglycoside-modifying enzymes. Enzymatic modification of aminoglycoside functional groups is the major way by which bacteria fight these antibiotics (58). Thus, avoiding the functional groups of aminoglycosides, which are prone to enzymatic modification by bacterial enzymes, appears to be a testable solution. Our results show that aminoglycosides with hydrogen atoms instead of hydroxyl groups in the 3′ and 4′ positions of ring I interact more strongly with their binding sites (e.g., compare LLL with RIO in Table S2). At the same time, these two hydroxyl groups are frequently modified by aminoglycoside phosphotransferase APH(3′) and nucleotidyltransferase ANT(4′) (59).
On the other hand, it has been shown that to preserve the efficacy of aminoglycosides, certain functional groups are necessary, even though they are susceptible to aminoglycoside-modifying enzymes. For example, in 4,5-DOS aminoglycosides, the N2′ amino group in ring I (Fig. 1, red rings) determines the mobility of A1492 and A1493 bases and therefore is essential for antibacterial activity (60). Our computational result corroborates this finding, because the contribution of this amino group to the interaction energy between aminoglycosides and their binding site is high (e.g., compare rings I of KAN and NMY in Table S2). However, a modification of this site is possible, according to the location of a water molecule in Pattern 3 (Fig. S6). If we replace this 2′-amino group with, e.g., , then contacts between the new amino group and G1491 and A1492 phosphates could be created. This modification should prevent the acetylation of this group by aminoglycoside acetyltransferase AAC(2′)-I, present in Gram-negative bacteria and Mycobacterium (61).
Conclusions
We performed a structural and electrostatic comparison of 12 4,5- and 4,6-aminoglycoside crystal complexes with the RNA model of the ribosomal A-site. We used two approaches to calculate the electrostatic energy of interactions between aminoglycosides and RNA: single-point partial atomic charges from the classical all-atom force fields and charge densities reconstructed with the databank of aspherical pseudoatoms (UBDB). Both approaches gave a similarly high correlation of electrostatic interaction energies between aminoglycosides and A-site, with experimental Gibbs energies corroborating previous data showing that electrostatics is a crucial factor in aminoglycoside binding. The corresponding van der Waals energies did not show any correlations with .
The reasons for the roughly similar electrostatic interaction energies obtained from the multipole-based approach and single-point charges that assume spherical electron densities may be that for the charged antibiotics and RNA, the net monopole term dominates. From the calculations, we have reproduced and indicated the rings and functional groups in aminoglycosides that are crucial for their binding to the A-site. The electrostatic contributions of water molecules mediating the hydrogen bonds between aminoglycosides and RNA pointed to a few fragments of aminoglycosides that are potential candidates for modification.
Overall, the electrostatic interaction energy between 2-DOS aminoglycosides and RNA increases with the number of protonated amino groups. This is in accord with findings from a study showing that modification of the amino groups to hydroxyl groups decreases the binding affinities of aminoglycosides for hammerhead ribozyme (6). Introducing even more amino groups should strengthen the electrostatics but could decrease the ability to access the bacterial cell, so it would not be a therapeutically effective modification.
The electrostatic contribution depends also on the number of rings. Rings I and II are sufficient for specific binding to the A-site (as shown for XXX) and rings III and IV were proven to contribute weakly to the specificity of aminoglycoside binding (62). However, if rings III and IV are removed, the affinity decreases (12,17–19). The contributions from individual rings (Fig. 5) suggest that the highest electrostatic contribution is for aminoglycosides containing four rings. The exceptions are the three-ring antibiotics AKN and KAN, whose Eel is less favorable than that for the two-ring neamine XXX, even though these aminoglycosides have the same net charge of +4e. Our recent studies on amikacin-A-site complexes suggest that the binding of AKN (due to the l-HABA extension) is mainly entropically driven, so this less favorable Eel seems reasonable (22). Also, adding the fifth ring in LIV decreases the overall in comparison with the four-ring antibiotics.
The methodology applied here is not without limitations. The crystal structures that were used represent a solid packed state, whereas aminoglycoside-RNA interactions should be analyzed in solvent. We have partially solved this problem through energy minimization of these structures in a box of water molecules. However, desolvation and entropy effects were omitted. An analysis of electrostatics in the course of MD simulations in explicit solvent would probably better represent the aminoglycoside environment. The calculated Eels have to be treated as an electrostatic contribution to the total free energy of binding, and only their relative values are meaningful. Also, comparing them with experimental must be treated with caution, because contain multiple contributions beyond the electrostatic one. Notwithstanding the limitations, both single-point and multipole approaches work well for this charged system.
Acknowledgments
We acknowledge support from the University of Warsaw (CeNT/BST), the National Science Centre (DEC-2012/05/B/NZ1/00035), the Foundation for Polish Science (TEAM/2009-3/8 project cofinanced by the European Regional Development Fund operated within the Innovative Economy Operational Programme). M.J. and A.M.G. were supported in part by the European Union through the European Social Fund, contract number UDA-POKL.04.01.01-00-072/09-00. A.M.G. also has been cofinanced with European Union funds by the European Social Fund 08.02.02_133/ES/ZS-III/W-POKL/14. Calculations were performed using resources at the ICM, University of Warsaw (ICM/KDM/G31-4).
Supporting Material
References
- 1.Avent M.L., Rogers B.A., Paterson D.L. Current use of aminoglycosides: indications, pharmacokinetics and monitoring for toxicity. Intern. Med. J. 2011;41:441–449. doi: 10.1111/j.1445-5994.2011.02452.x. [DOI] [PubMed] [Google Scholar]
- 2.Recht M.I., Fourmy D., Puglisi J.D. RNA sequence determinants for aminoglycoside binding to an A-site rRNA model oligonucleotide. J. Mol. Biol. 1996;262:421–436. doi: 10.1006/jmbi.1996.0526. [DOI] [PubMed] [Google Scholar]
- 3.Houghton J.L., Green K.D., Garneau-Tsodikova S. The future of aminoglycosides: the end or renaissance? ChemBioChem. 2010;11:880–902. doi: 10.1002/cbic.200900779. [DOI] [PubMed] [Google Scholar]
- 4.Borovinskaya M.A., Pai R.D., Cate J.H. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat. Struct. Mol. Biol. 2007;14:727–732. doi: 10.1038/nsmb1271. [DOI] [PubMed] [Google Scholar]
- 5.Wang H., Tor Y. Electrostatic interactions in RNA aminoglycosides binding. J. Am. Chem. Soc. 1997;119:8734–8735. [Google Scholar]
- 6.Tor Y. Targeting RNA with small molecules. ChemBioChem. 2003;4:998–1007. doi: 10.1002/cbic.200300680. [DOI] [PubMed] [Google Scholar]
- 7.Blount K.F., Tor Y. Using pyrene-labeled HIV-1 TAR to measure RNA-small molecule binding. Nucleic Acids Res. 2003;31:5490–5500. doi: 10.1093/nar/gkg755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaul M., Pilch D.S. Thermodynamics of aminoglycoside-rRNA recognition: the binding of neomycin-class aminoglycosides to the A site of 16S rRNA. Biochemistry. 2002;41:7695–7706. doi: 10.1021/bi020130f. [DOI] [PubMed] [Google Scholar]
- 9.Kane R.S., Glink P.T., Whitesides G.M. Basicity of the amino groups of the aminoglycoside amikacin using capillary electrophoresis and coupled CE-MS-MS techniques. Anal. Chem. 2001;73:4028–4036. doi: 10.1021/ac010173m. [DOI] [PubMed] [Google Scholar]
- 10.Walter F., Vicens Q., Westhof E. Aminoglycoside-RNA interactions. Curr. Opin. Chem. Biol. 1999;3:694–704. doi: 10.1016/s1367-5931(99)00028-9. [DOI] [PubMed] [Google Scholar]
- 11.Kaul M., Barbieri C.M., Pilch D.S. Coupling of drug protonation to the specific binding of aminoglycosides to the A site of 16 S rRNA: elucidation of the number of drug amino groups involved and their identities. J. Mol. Biol. 2003;326:1373–1387. doi: 10.1016/s0022-2836(02)01452-3. [DOI] [PubMed] [Google Scholar]
- 12.Pilch D.S., Kaul M., Kerrigan J.E. Thermodynamics of aminoglycoside-rRNA recognition. Biopolymers. 2003;70:58–79. doi: 10.1002/bip.10411. [DOI] [PubMed] [Google Scholar]
- 13.Vicens Q., Westhof E. Molecular recognition of aminoglycoside antibiotics by ribosomal RNA and resistance enzymes: an analysis of x-ray crystal structures. Biopolymers. 2003;70:42–57. doi: 10.1002/bip.10414. [DOI] [PubMed] [Google Scholar]
- 14.Kondo J., Koganei M., Kasahara T. Crystal structure and specific binding mode of sisomicin to the bacterial ribosomal decoding site. ACS Med Chem Lett. 2012;3:741–744. doi: 10.1021/ml300145y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kondo J., Koganei M., Hanessian S. Crystal structures of a bioactive 6′-hydroxy variant of sisomicin bound to the bacterial and protozoal ribosomal decoding sites. ChemMedChem. 2013;8:733–739. doi: 10.1002/cmdc.201200579. [DOI] [PubMed] [Google Scholar]
- 16.François B., Russell R.J., Westhof E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005;33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang G., Trylska J., McCammon J.A. Binding of aminoglycosidic antibiotics to the oligonucleotide A-site model and 30S ribosomal subunit: Poisson-Boltzmann model, thermal denaturation, and fluorescence studies. J. Med. Chem. 2006;49:5478–5490. doi: 10.1021/jm060288o. [DOI] [PubMed] [Google Scholar]
- 18.Wong C.-H., Hendrix M., Greenberg W.A. Specificity of aminoglycoside antibiotics for the A-site of the decoding region of ribosomal RNA. Chem. Biol. 1998;5:397–406. doi: 10.1016/s1074-5521(98)90073-4. [DOI] [PubMed] [Google Scholar]
- 19.Alper P.B., Hendrix M., Wong C.-H. Probing the specificity of aminoglycoside-ribosomal RNA interactions with designed synthetic analogs. J. Am. Chem. Soc. 1998;120:1965–1978. [Google Scholar]
- 20.Romanowska J., Setny P., Trylska J. Molecular dynamics study of the ribosomal A-site. J. Phys. Chem. B. 2008;112:15227–15243. doi: 10.1021/jp806814s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaiana A.C., Sanbonmatsu K.Y. Stochastic gating and drug-ribosome interactions. J. Mol. Biol. 2009;386:648–661. doi: 10.1016/j.jmb.2008.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dudek M., Romanowska J., Trylska J. Interactions of amikacin with the RNA model of the ribosomal A-site: computational, spectroscopic and calorimetric studies. Biochimie. 2014;102:188–202. doi: 10.1016/j.biochi.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Vaiana A.C., Westhof E., Auffinger P. A molecular dynamics simulation study of an aminoglycoside/A-site RNA complex: conformational and hydration patterns. Biochimie. 2006;88:1061–1073. doi: 10.1016/j.biochi.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Chen S.-Y., Lin T.-H. A molecular dynamics study on binding recognition between several 4,5 and 4,6-linked aminoglycosides with A-site RNA. J. Mol. Recognit. 2010;23:423–434. doi: 10.1002/jmr.1008. [DOI] [PubMed] [Google Scholar]
- 25.Ma C., Baker N.A., McCammon J.A. Binding of aminoglycoside antibiotics to the small ribosomal subunit: a continuum electrostatics investigation. J. Am. Chem. Soc. 2002;124:1438–1442. doi: 10.1021/ja016830+. [DOI] [PubMed] [Google Scholar]
- 26.Honig B., Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 27.Długosz M., Antosiewicz J.M., Trylska J. Association of aminoglycosidic antibiotics with the ribosomal A-site studied with Brownian dynamics. J. Chem. Theory Comput. 2008;4:549–559. doi: 10.1021/ct700210n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Długosz M., Trylska J. Aminoglycoside association pathways with the 30S ribosomal subunit. J. Phys. Chem. B. 2009;113:7322–7330. doi: 10.1021/jp8112914. [DOI] [PubMed] [Google Scholar]
- 29.Jarzembska K.N., Dominiak P.M. New version of the theoretical databank of transferable aspherical pseudoatoms, UBDB2011—towards nucleic acid modelling. Acta Crystallogr. A. 2012;68:139–147. doi: 10.1107/S0108767311042176. [DOI] [PubMed] [Google Scholar]
- 30.Dominiak P.M., Volkov A., Coppens P. A theoretical databank of transferable aspherical atoms and its application to electrostatic interaction energy calculations of macromolecules. J. Chem. Theory Comput. 2007;3:232–247. doi: 10.1021/ct6001994. [DOI] [PubMed] [Google Scholar]
- 31.Volkov A., Li X., Coppens P. Ab initio quality electrostatic atomic and molecular properties including intermolecular energies from a transferable theoretical pseudoatom databank. J. Phys. Chem. A. 2004;108:4283–4300. [Google Scholar]
- 32.Dittrich B., Koritsánszky T., Luger P. A simple approach to nonspherical electron densities by using invarioms. Angew. Chem. Int. Ed. Engl. 2004;43:2718–2721. doi: 10.1002/anie.200353596. [DOI] [PubMed] [Google Scholar]
- 33.Domagała S., Jelsch C. Optimal local axes and symmetry assignment for charge-density refinement. J. Appl. Crystallogr. 2008;41:1140–1149. [Google Scholar]
- 34.Bąk J.M., Domagała S., Dominiak P.M. Verification of structural and electrostatic properties obtained by the use of different pseudoatom databases. Acta Crystallogr. A. 2011;67:141–153. doi: 10.1107/S0108767310049731. [DOI] [PubMed] [Google Scholar]
- 35.Volkov A., Koritsanszky T., Coppens P. Combination of the exact potential and multipole methods (EP/MM) for evaluation of intermolecular electrostatic interaction energies with pseudoatom representation of molecular electron densities. Chem. Phys. Lett. 2004;391:170–175. [Google Scholar]
- 36.Li X., Volkov A.V., Coppens P. Interaction energies between glycopeptide antibiotics and substrates in complexes determined by x-ray crystallography: application of a theoretical databank of aspherical atoms and a symmetry-adapted perturbation theory-based set of interatomic potentials. Acta Crystallogr. D Biol. Crystallogr. 2006;62:639–647. doi: 10.1107/S0907444906013072. [DOI] [PubMed] [Google Scholar]
- 37.Dominiak P.M., Volkov A., Coppens P. Combining crystallographic information and an aspherical-atom data bank in the evaluation of the electrostatic interaction energy in an enzyme-substrate complex: influenza neuraminidase inhibition. Acta Crystallogr. D Biol. Crystallogr. 2009;65:485–499. doi: 10.1107/S0907444909009433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malińska M., Jarzembska K.N., Dominiak P.M. Sunitinib: from charge-density studies to interaction with proteins. Acta Crystallogr. D Biol. Crystallogr. 2014;70:1257–1270. doi: 10.1107/S1399004714002351. [DOI] [PubMed] [Google Scholar]
- 39.Kumar P., Bojarowski S.A., Dominiak P.M. A comparative study of transferable aspherical pseudoatom databank and classical force fields for predicting electrostatic interactions in molecular dimers. J. Chem. Theory Comput. 2014;10:1652–1664. doi: 10.1021/ct4011129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Czyżnikowska Z., Góra R.W., Leszczynski J. Structural variability and the nature of intermolecular interactions in Watson-Crick B-DNA base pairs. J. Phys. Chem. B. 2010;114:9629–9644. doi: 10.1021/jp101258q. [DOI] [PubMed] [Google Scholar]
- 41.Jarzembska K.N., Goral A.M., Dominiak P.M. Hoogsteen-Watson-Crick 9-methyladenine:1-methylthymine complex: charge density study in the context of crystal engineering and nucleic acid base pairing. Cryst. Growth Des. 2013;13:239–254. [Google Scholar]
- 42.Hansen N.K., Coppens P. Electron population analysis of accurate diffraction data. VI. Testing aspherical atom refinements on small-molecule data sets. Acta Crystallogr. A. 1978;34:909–921. [Google Scholar]
- 43.Vicens Q., Westhof E. Crystal structure of geneticin bound to a bacterial 16S ribosomal RNA A site oligonucleotide. J. Mol. Biol. 2003;326:1175–1188. doi: 10.1016/s0022-2836(02)01435-3. [DOI] [PubMed] [Google Scholar]
- 44.Kondo J., François B., Westhof E. Crystal structure of the bacterial ribosomal decoding site complexed with amikacin containing the γ-amino-α-hydroxybutyryl (haba) group. Biochimie. 2006;88:1027–1031. doi: 10.1016/j.biochi.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 45.Vicens Q., Westhof E. Crystal structure of a complex between the aminoglycoside tobramycin and an oligonucleotide containing the ribosomal decoding a site. Chem. Biol. 2002;9:747–755. doi: 10.1016/s1074-5521(02)00153-9. [DOI] [PubMed] [Google Scholar]
- 46.Vicens Q., Westhof E. Crystal structure of paromomycin docked into the eubacterial ribosomal decoding A site. Structure. 2001;9:647–658. doi: 10.1016/s0969-2126(01)00629-3. [DOI] [PubMed] [Google Scholar]
- 47.François B., Szychowski J., Hanessian S. Antibacterial aminoglycosides with a modified mode of binding to the ribosomal-RNA decoding site. Angew. Chem. Int. Ed. Engl. 2004;43:6735–6738. doi: 10.1002/anie.200462092. [DOI] [PubMed] [Google Scholar]
- 48.Schrödinger, LLC, 2010. The PyMOL Molecular Graphics System, Version 1.3r1.
- 49.Wang J., Wang W., Case D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 50.Wang J., Cieplak P., Kollman P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 51.Case, D. A., T. Darden, …, P. A. Kollman. Amber 11. Users Manual. University of California, San Francisco.
- 52.Jakalian A., Bush B.L., Bayly C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 53.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 54.Volkov A., Messerschmidt M., Coppens P. Improving the scattering-factor formalism in protein refinement: application of the University at Buffalo Aspherical-Atom Databank to polypeptide structures. Acta Crystallogr. D Biol. Crystallogr. 2007;63:160–170. doi: 10.1107/S0907444906044453. [DOI] [PubMed] [Google Scholar]
- 55.Koritsanszky T., Macchi P., Richter T. Department of Chemistry, SUNY at Buffalo; Buffalo, NY: 2006. XD2006: A Computer Program for Multipole Refinement, Topological Analysis of Charge Densities and Evaluation of Intermolecular Energies from Experimental or Theoretical Structure Factors. [Google Scholar]
- 56.Gaussian 09, Revision D.01. Gaussian, Wallingford CT 2009.
- 57.Blount K.F., Zhao F., Tor Y. Conformational constraint as a means for understanding RNA-aminoglycoside specificity. J. Am. Chem. Soc. 2005;127:9818–9829. doi: 10.1021/ja050918w. [DOI] [PubMed] [Google Scholar]
- 58.Jana S., Deb J.K. Molecular understanding of aminoglycoside action and resistance. Appl. Microbiol. Biotechnol. 2006;70:140–150. doi: 10.1007/s00253-005-0279-0. [DOI] [PubMed] [Google Scholar]
- 59.Ramirez M.S., Tolmasky M.E. Aminoglycoside modifying enzymes. Drug Resist. Updat. 2010;13:151–171. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barbieri C.M., Kaul M., Pilch D.S. Defining the molecular forces that determine the impact of neomycin on bacterial protein synthesis: importance of the 2′-amino functionality. Antimicrob. Agents Chemother. 2007;51:1760–1769. doi: 10.1128/AAC.01267-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vetting M.W., Hegde S.S., Roderick S.L. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat. Struct. Biol. 2002;9:653–658. doi: 10.1038/nsb830. [DOI] [PubMed] [Google Scholar]
- 62.Fourmy D., Recht M.I., Puglisi J.D. Binding of neomycin-class aminoglycoside antibiotics to the A-site of 16 S rRNA. J. Mol. Biol. 1998;277:347–362. doi: 10.1006/jmbi.1997.1552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.