

Abstract
Background
The nucleosome positioning regulates the gene expression and many other DNA-related processes in eukaryotes. Genome-wide mapping of nucleosome positions and correlation of genome-wide nucleosomal remodeling with the changes in the gene expression can help us understanding gene regulation on genome level.
Results
In the present study, we correlate the gene expression and the genomic nucleosomal remodeling in response to salicylic acid (SA) treatment in A. thaliana. We have mapped genome-wide nucleosomes by performing tiling microarray using 146 bp mononucleosomal template DNA. The average nucleosomal coverage is approximately 346 bp per nucleosome both under the control and the SA-treated conditions. The nucleosomal coverage is more in the coding region than in the 5′ regulatory regions. We observe approximately 50% nucleosomal remodeling on SA treatment where significant nucleosomal depletion and nucleosomal enrichment around the transcription start site (TSS) occur in SA induced genes and SA repressed genes respectively in response to SA treatment. Especially in the case of the SA-induced group, the nucleosomal remodeling over the minimal promoter in response to SA is especially significant in the Non-expresser of PR1 (NPR1)-dependent genes. A detailed investigation of npr1-1 mutant confirms a distinct role of NPR1 in the nucleosome remodeling over the core promoter. We have also identified several motifs for various hormonal responses; including ABRE elements in the remodeled nucleosomal regions around the promoter region in the SA regulated genes. We have further identified that the W-box and TGACG/C motif, reported to play an important role in SA-mediated induction, are enriched in nucleosome free regions (NFRs) of the promoter region of the SA induced genes.
Conclusions
This is the first study reporting genome-wide effects of SA treatment on the chromatin architecture of A. thaliana. It also reports significant role of NPR1 in genome-wide nucleosomal remodeling in response to SA.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-014-0404-2) contains supplementary material, which is available to authorized users.
Keywords: Nucleosome, Plants, Chromatin, Salicylic Acid, Arabidopsis thaliana
Background
It is now well known that the position of the nucleosome along a particular sequence of DNA has profound effects on its accessibility for all DNA processes such as transcription regulation, recombination, and repair [1-3]. For example, the nucleosome over the core promoter region of the pathogenesis-related gene (PR-1a) of tobacco is remodeled on induction with salicylic acid (SA) [4]. This suggests that the underlying DNA sequence and histone modifications play their role in gene activation, leading to the sliding of nucleosomes from the PR-1a core promoter. Several reports suggest that the position of the nucleosome is critical in globally regulating the in vivo binding of transcription factors by either allowing or blocking their binding to the nucleosome [5-9]. A nucleosome-depleted region (NDR) close to the transcription start site (TSS) is usually flanked by upstream and downstream positioned nucleosomes (denoted the −1 and +1 nucleosomes, respectively) that are often the starting points for the regular nucleosomal arrays. Protein factors along with the DNA and histone have a necessary role in nucleosome positioning [10-16]. In a previous study, five in vivo packing mechanisms of nucleosome organization have also been proposed for the nucleosomes in yeast [17].
Genome-wide mapping of nucleosome positions of eukaryotic genomes is a prerequisite to understand the basic mechanism of chromosomal organization. Previously genome-wide nucleosome positioning has been mapped and analyzed in Saccharomyces cerevisiae, Drosophila melanogaster, Homo sapiens, Caenorhabditis elegans and A. thaliana [15,18-27]. Different techniques have been used to assay nucleosome positioning in chromatin; for example, whole-genome tiling-microarray using mononucleosomal DNA [28], Micrococcal Nuclease (MNase1)-sequencing [22-25,27], which provides in-depth data on genome-wide nucleosomal positioning, etc.
Previously a study of global nucleosome positioning in A. thaliana has concluded for the first time that the nucleosomes are more densely packed in pericentromeric regions containing heavily methylated DNA than in the euchromatin region [27]. Taking into account the role of nucleosome in regulating eukaryotic transcription, it would be tempting to explore genome-wide nucleosomal dynamics in the context of transcriptional perturbation. Since plants are sessile organisms, they have developed intricate self defense mechanisms that enable them to cope with both biotic and abiotic stresses. The mechanism generally operates by generating the systemic acquired resistance (SAR) response mainly through SA-regulated and Jasmonic acid (JA)-regulated pathways with many other hormones [29,30]. This leads to massive changes in gene expression [31]. NPR1 has emerged as a key component which plays a significant role in regulation of these pathways and their cross-talk [32-34]. The molecule exists in the cytoplasm in multimeric form and is reduced to monomeric form to enter nucleus on being induced with SA. Overall, massive transcriptional reprogramming may be regulated at the chromatin level by nucleosomal remodeling. Thus, the present study aims at understanding the possible relationship between the dynamics of global nucleosomal positioning and SA-mediated transcriptional regulation in A. thaliana.
Briefly, we have mapped global nucleosome positioning in A. thaliana chromosomes under both control and SA-treated conditions. We have also correlated the changes in nucleosome positioning to the global transcriptional response to SA in A. thaliana. Our results report distinct chromatin remodeling in core promoter and 5′ upstream promoter regulatory regions of SA-induced genes. We also find a distinct role of non-expresser of PR1 (NPR1) in the nucleosomal remodeling in SA-regulated genes. For this, we identify SA- regulated NPR1-dependent (ND) and NPR1-independent (NI) genes by performing the whole genome expression analysis under control and SA-treated conditions both in the wild-type and the npr1 mutant plants. The nucleosomal occupancy study of these groups reveals a correlation between SA-regulated ND genes and the dependence of NPR1 for nucleosomal remodeling at their core promoter region. We also find many defense related motifs both in the core promoter region and in the upstream regulatory region of SA regulated genes. While some of these are present in the nucleosome occupied regions (NORs), rest others are present in the nucleosome free region (NFRs).
Methods
Plant growth conditions and treatment with SA
Seed sterilization and plant growth conditions were followed as reported earlier [35] for both A. thaliana Col-0 (used as the wild-type) and npr1-1 mutant. All seeds were grown in vitro in MS (Murashige and Skoog) medium by keeping them on a mesh for stratification at 4°C for 2 days before placing them in the growth chamber. A. thaliana plants were grown under controlled environmental conditions (22 ± 1°C, 120 μmol m−2 sec−1, and 16 h light/8 h dark cycle). After two weeks of growth, the medium was exchanged either with water or with 2 mM SA, followed by 24 h incubation in light at 22 ± 1°C.
Nuclei isolation and digestion with MNase1
The nuclei were isolated as described [36], with slight modifications. Briefly, 10 g of seedlings (Col-0 or npr1-1) were treated either with water or SA, rinsed with water, blotted dry, and ground to powder in liquid nitrogen. Nuclei isolation buffer NIB1 (0.5 M hexylene glycol, 20 mM KCl, 20 mM PIPES (pH 6.5), 0.5 mM EDTA, 0.1% Triton X-100, and 7 mM 2-mercaptoethanol) was added to it. The extract was first filtered through a 4-layered muslin cloth and then sequentially through 80, 60, 40, and 20 μm mesh sieves. The filtrate was centrifuged at 3000 g at 4°C for 10 min. The pellet was suspended in NIB2 (NIB1 without Triton X-100) and centrifuged again. The pellet was suspended in 5% percoll (U.S. Biologicals), loaded on a 20–80% percoll step gradient, and centrifuged. The nuclei were removed from the 20–80% percoll interface, washed in NIB2, and resuspended in NIB1 buffer. The nuclear preparation was incubated with micrococcal nuclease (300 units/μl, Fermentas# EN0181) in a buffer containing 25 mM KCl, 4 mM MgCl2, 1 mM CaCl2, 50 mM Tris-Cl (pH 7.4), and 12.5% glycerol at 37°C for 10 min at the concentration of 1unit/μg of genomic DNA from untreated sample and 0.6unit/μg of genomic DNA from treated sample (see Additional file 1). The reaction was stopped by adding an equal volume of 2% SDS, 0.2 M NaCl, 10 mM EDTA, 10 mM EGTA, and 50 mM Tris-Cl (pH 8); treated with proteinase K (100 μg/ml, Ambion#2546) for 1 h at 55°C, then with RNase A (100 μg/ml, Qiagen#19101) at 37°C for 30 min, extracted by phenol: chloroform, and precipitated in ethanol. The DNA was separated on 1.5% agarose gel, and fragments of an average size of 150 bp were purified (Sigma#NA1020). The experiment was repeated three times each for control and SA-treated seedlings.
The control genomic DNA for the hybridization experiments was prepared from nuclear genomic DNA extraction and sonicated to obtain fragments of an approximate size of 200–500 bp. The experiment was repeated two times.
Tiling array experiment
Isolated mononucleosomal DNA fragments (SA-treated and untreated) and genomic control DNA were amplified with random primers (N6), Klenow fragment (3′ → 5′ exo−; NEB#M0210L) and 25 mM dNTP (with dTTP and dUTP in 4:1 ratio) in 20 μl aliquots for a total volume of 100 μl that was sufficient to obtain approximately 10 μg of purified PCR products for each sample. The PCR products were purified with the PCR cleanup kit (Sigma, #NA1020). Fragmentation and labeling was done as per Affymetrix ChIP Assay Protocol (P/N 702238) using GeneChip® WT Double-Stranded DNA Terminal Labeling Kit (P/N 900812). Hybridization, washing, and staining were done according to the Affymetrix Chromatin Immunoprecipitation Assay Protocol onto GeneChip® A. thaliana Tiling 1.0R array (Reverse) using GeneChip® Hybridization, Wash, and Stain Kit (P/N 900720) on Affymetrix Fluidics station 450 according to the manufacturer’s protocol. Arrays were scanned on an Affymetrix GeneChip® 3000 7G scanner, and the signals were quantified with the Affymetrix GeneChip® Operating Software (GCOS) to generate cel files. The data can be found in NCBI public repository as GSE25553 [37]. The experiments were performed in similar way for all the eight templates (three each for control and SA-treated and two for the genomic DNA).
Processing of tiling array data
In the subsequent analysis, raw data from all probes were mapped onto TAIR10. Tiling Analysis software (TAS) from Affymetrix technology was used to analyze tiling array data (cel files), which included the use of quantile normalization, probe analysis, and two-sample analysis to generate normalized log2 transformed values. The two-sample comparison analysis included datasets, either control versus genomic or SA-treated versus genomic, that could help in identifying consistently differentially enriched regions along the genome in control and SA-treated conditions respectively as compared to background region. Since genomic DNA was randomly sheared DNA, the comparison also yielded negative log2 transformed signal intensity values for certain probe sets in both cases. Throughout this article, this log2 transformed signal intensity value has been directly referred to as enrichment value. These enrichment values were then used for further analysis in a comparative fashion in order to determine the nucleosomal remodeling across the genome.
Definition of stringency level
Seven different enrichment values (≥0.0, ≥−0.01, ≥−0.1, ≥−0.5, ≥−1.0, ≥−1.5 and ≥−2.0) have been used as seven stringency levels. For example, stringency level ≥0.0 means that if a probe has enrichment value ≥0.0, then only it may serve as a nucleosomal signal, not otherwise.
Development of custom scripts
Four different custom scripts were developed to predict nucleosomal occupied regions from the signal intensity data of TAS (Tilling Array analysis) in different steps –
C script to develop the dataset of distinct and fuzzy nucleosomes: Tab separated file containing the genomic positions and the log2 transformed signal intensity values from all the three replicates was used as input for both control and SA treated datasets to get the nucleosomal regions (distinct and fuzzy) at different stringency levels. For a given stringency level, the presence of a nucleosome is inferred, if the enrichment value is ≥ the value of the stringency level continuously at 4 probes (for distinct nucleosomes) or at >4 probes (for fuzzy nucleosomes) in all the three replicates in the dataset under study. For example, for inferring the presence of a distinct nucleosome at stringency level ≥0.0, four continuous probes should have enrichment value ≥0.0.
Perl script to develop a database of global positions of TSS, start codon, stop codon, and mRNA end for all the genes along all the five chromosomes of A. thaliana from TAIR10 tabular files.
C script to find out the remodeled nucleosomal regions and common nucleosomal regions: The nucleosomal regions present either only in control conditions or only in SA treated conditions were considered as remodeled nucleosomal regions and the nucleosomal regions present in both conditions were considered as the common nucleosomal regions for the purpose of input data in this script.
- C script to search every 50 bp window size for the nucleosomal coverage of 1000 bp upstream and downstream from TSS site of the genes in different groups: Two different input files were used –
- The strand position (either forward or reverse) and the TSS (Transcription start site) of gene.
- The nucleosomal dataset both for distinct and fuzzy nucleosomes (coming from first script).
If a continuous stretch of ≥25 bp was predicted to be having nucleosome occupancy in 50 bp window, then it was marked as count 1 (considered as having a nucleosome). On the other hand, if the continuous nucleosome coverage was <25 bp in 50 bp window, then it was marked as count 0 (considered as having no nucleosome).
All of these scripts are available at http://ncgd.nbri.res.in/nucleosome_prediction.aspx.
Identification of genome-wide nucleosome regions in control and SA-treated conditions
A total of 140 datasets were developed over seven stringency levels, two conditions and two types (distinct and fuzzy) of nucleosomes for the five chromosomes using first custom script. Nucleosome regions covering an area of 131–161 bp represented distinct nucleosomes and the nucleosome regions covering an area ≥161 bp represented fuzzy nucleosomes.
Calculation of nucleosomal coverage over different parts of gene
The tiling array data were mapped for all the five chromosomes, and the nucleosome occupancy was calculated in different parts of the genes identified using second custom script. The nucleosomal coverage was calculated at stringency ≥−1.0 as size of the chromosome (in bp) divided by the total region covered by nucleosomes (distinct + fuzzy) for all the five chromosomes of A. thaliana (see Additional file 2).
Identification of remodeled nucleosomes on SA treatment
Nucleosomal regions were identified for the following conditions using third custom script
Regions having nucleosomes shifted/removed under SA-treated condition as compared to control condition.
Regions having nucleosomes added under SA-treated condition as compared to control condition.
Regions retaining nucleosomes under both conditions.
The first two regions taken together (i.e. a + b) formed a remodeled nucleosomal region, whereas all regions taken together formed total nucleosomal coverage (i.e. a + b + c).
RNA extraction
Total RNA was isolated from two week old seedlings (both Col-0 and npr1-1) in two independent biological replicates using Spectrum plant total RNA Kit (Sigma-Aldrich) according to the manufacturer’s instructions and eluted with nuclease-free water. After DNaseI (Ambion) treatment, the RNA was quantified and checked for integrity using a Bioanalyzer (Agilent, Inc., Palo Alto, CA, USA).
RNA amplification and labeling
Direct labeling procedure was used i.e., double-stranded cDNA was synthesized using 250 ng total RNA, either with an oligo-dT primer (for QPCR purpose) or a T7 promoter-containing oligo (dT) primer, followed by in vitro transcription and labeling using a GeneChip® 3′ IVT Express kit (Affymetrix) to generate cRNA as per manufacturer’s protocol.
Microarray hybridization
The cRNA was fragmented to approx. <100 bases, hybridized to ATH1 Genome chip and were incubated at 45°C temperature for 16 hrs at 60 rpm in hybridization chamber. Washing and staining was performed by using an Affymetrix Fluidics Station 450 according to the manufacturer’s instructions (GeneChip expression analysis technical manual). The arrays were scanned using Gene Chip Scanner 3000 7G (Affymetrix). Scanned images were processed and analyzed using GCOS software (Affymetrix). The data has been deposited in NCBI (GSE 51626).
Image acquisition and statistical analysis of salicylic acid treated oligonucleotide microarray data
Arrays Images were captured and then raw cel files were analyzed using affylmGUI biocondcutor R package for Robust Multi-array Average (RMA) algorithm [38] by performing model-based background correction, normalization and summarization across the probe level. Identification of statistically significant (P-values ≤0.05) differentially expressed genes as determined by affylmGUI biocondcutor R package was done using the fold change criteria (see Additional file 1).
Identification of SA induced (SI), SA repressed (SR) and uninduced genes
The fold change criteria along with P-values ≤0.05 has been used to classify the genes in different groups.
Genes with log2 fold change ≥2 were considered as up-regulated, hence were called as SI genes,
Genes with fold change ≤−2 were considered as down-regulated, hence were called as SR group,
Uninduced genes with an equal and consistent expression profile in all the replicates of SA and control array files were grouped into silent (SIL) genes (expression values ≤0) and constitutive (CON) genes (expression values ≥3).
Identification of ND and NI genes
Expression profiling experiments were performed for both wild-type and npr1-1 mutants under untreated and treated conditions (GSE51626) for the identification of ND and NI genes. Thereafter, two-way ANOVA was used under R script developed by Pavlidis et al. [39] for selecting statistically significant SA-regulated ND and SA-regulated NI genes. Normalized log2 transformed values of all replicates of untreated and treated Col-0 and npr1-1 mutant array files were processed for ANOVA analysis. Genes with adjusted P-value ≤0.05 were considered as SA regulated genes (as performed by Q-value [40] package implemented in R). For SA-regulated ND genes, the genes with P-value ≤0.0001 (for NPR1-dependence) were selected while for SA-regulated NI genes P-value ≥0.1 were selected.
Further, SA-regulated ND genes were classified into two groups i.e. SI-ND and SR-ND genes based on the fold change value of the genes upon SA treatment in wild-type plants. Similarly, SA-regulated NI genes were classified into two groups i.e. SI-NI and SR-NI genes.
Quantitative PCR for the analysis of nucleosomal occupancy and transcript abundance
For validating the ≥−1.0 stringency level, primers were designed both from the regions showing the presence of the nucleosome in both control and SA-treated conditions (exonic regions) and from the regions showing the absence of the nucleosome in both control and SA-treated conditions (intergenic-UTR region for At1g53790, exon-UTR region for At2g28650, intergenic region for At3g55430 and the exonic region for At4g07408), as depicted in our tiling array analysis (see Additional files 1 and 3) using Primer Express software (Applied Biosystems). The PCR efficiency of all the primers was checked by using the A. thaliana genomic DNA as a template. QPCR was carried out using ABI Taqman chemistry with untreated mononucleosomal DNA template on ABI Prism 7000 sequence detection system (Applied Biosystems). The Ct (threshold cycle) value for each gene was determined and QPCR data analysis was done by comparing Ct values [41,42]. The experiments were repeated twice with at least 3 biological replicates.
To assess the effect of NPR1 on nucleosomal occupancy on the promoter region in all the six groups, mononucleosomes were prepared as described earlier from both treated and untreated wild-type and npr1-1 mutant seedlings. Next, QPCR was performed for the assessment of nucleosomal occupancy at the promoter loci of three representative genes for each class (total six classes; i.e. SI-ND genes– AT1G28480, AT1G80590, AT3G56400; SR-ND genes– AT5G38700, AT4G37800, AT5G13220; SI-NI genes– AT1G05700, AT1G09380, AT1G68450; SR-NI genes– AT4G21850, AT1G66100, AT4G34410; CON genes – AT5G02380, AT5G02500, AT5G60390 and SIL genes – AT1G27380, AT4G23780, AT3G26130). For the internal control set, five exonic regions (AT4G04570, AT5G64905, AT1G24020, AT4G29030, and AT1G14150) were selected; the selected exons did not show nucleosomal remodeling on SA treatment. Moreover, they did not belong to any groups such as SI, SR, ND, or NI. The average Ct values of each of the three genes was normalized to the average Ct values of a complete set of internal control genes to obtain ΔCt values. Similar treatment was followed for all the sets; that is, wild-type Control (Wt-Ct), wild-type SA treated (Wt-SA), npr1-1 Control (NPR1-Ct), and npr1-1 SA treated (NPR1-SA). Thereafter, the average ΔCt value for the set of three genes was determined and average fold change was calculated and plotted along with standard error (SE). Student’s t-test was applied to test the significance of the changes under both conditions in wild-type versus mutant plants in a pair-wise manner.
Analysis of the relative transcript abundance of each group (for the same genes used for the nucleosomal occupancy analysis in different groups) was done using total RNA for each group prepared under similar conditions and using the same analyses strategy as done for studying nucleosomal occupancy described earlier.
Mapping of the nucleosomal coverage
Only the regions consisting of −1000 bp to +1000 bp with respect to TSS of a particular gene were considered for this purpose. Overlapping coordinates or genomic regions were filtered from −1000 to +1000 bp region with respect to TSS in all genes in the constitutive (CON), silent (SIL), SA induced (SI), SA repressed (SR), SA induced-ND (SI-ND), SA repressed-ND (SR-ND), SA induced-NI (SI-NI), and SA repressed-NI (SR-NI) groups. These regions were then removed from each group. The regions were mapped for the nucleosomal coverage (at stringency ≥−1.0 as earlier calculated using custom script 1) using fourth custom script. Statistical significance analysis at a P-value ≤0.05 was performed using Student’s t-test (N = 3, null hypothesis of no significance difference) for each window in all the groups. Only the adjusted P-values ≤0.05 (calculated using Q-value package) were considered as significant.
Calculation of nucleosomal remodeling
The presence or absence of a nucleosome in −1000 to +1000 bp-long region was calculated on per gene basis for all genes for both control and SA-treated conditions in different groups (CON, SIL, SI and SR, SI-ND, SR-ND, SI-NI and SR-NI). Based on the remodeling at each position, percentage of the genes showing remodeling was calculated for each position for all groups. A graph representing the percentage of genes having nucleosome at each position was drawn along with the statistical significance data for comparison between different groups. Statistical analysis was done for −1000 to +1000 bp region with respect to TSS over a window of 150 bp (so as to cover one nucleosome long region) to find differences in the form of nucleosomal enrichment and nucleosomal depletion at the proximal promoter region among SI versus unaffected groups (i.e. CON and SIL groups together) and SR versus unaffected groups, SI-ND versus SI-NI and SR-ND versus SR-NI. For this, Student’s t-test (N = 3, null hypothesis of no significance difference) was applied along with the Q-value package for multiple hypothesis testing correction. Only the adjusted P-values ≤0.05 were considered as significant.
Gene Ontology analysis
All the groups were analyzed for their enriched gene ontologies using agriGO software [43] under SEA (singular enrichment analysis) using Fischer statistical test under adjusted P-values ≤0.05 using Yekutieli method.
Prediction of nucleosomal occupied, nucleosomal free and remodeled nucleosomal regions
Third custom script was used to count the number of nucleosomal occupied, nucleosomal free and remodeled nucleosomal regions in the dataset of different groups. Nucleosomal bases were calculated in control as well as SA treated condition. Common nucleosomal bases present in the both the conditions were also calculated. Remodeled nucleosomal bases were calculated by subtracting the common nucleosomal bases from total nucleosomal bases (Control and SA treated).
Motif finding methods
a) For SI genes: Remodeled nucleosomal regions (199) (Total bp = 28360) of 1000 bp upstream regions from SI genes (111) were extracted from TAIR10 Database. Motif analysis was done for these regions. MEME (ver. 4.2) [44] was deployed using ZOOPS model (with minimum motif width = 6 bases and maximum motif width = 10 bases and maximum number of motifs to return = 10) to these regions. It identified several interesting motifs which were annotated with PLACE [45] database.
B) For SR genes: The promoter regions −600 to −800 upstream TSS (200 bp) of SI and SR genes were extracted from TAIR10 database. Additionally, as control dataset, the remodeled nucleosomal regions (191695 regions; Total bp = 32052083) from whole genome of A. Thaliana (W) were used to identify novel cis-regulatory elements specifically enriched in SR genes (with respect to SI genes) present in the remodeled regions. A database was constructed consisting of all possible combinations of 6 bases length. Thereafter, the motif analysis was done using following two methods:
-
i)
Lift method
Lift [46] was calculated for the database containing all the possible combinations in SI versus W and SR versus W. Finally, the ratio of both was calculated to find out the enriched motif of SR with respect to SI.
-
ii)
Motif enrichment (OE) method
The expected occurrence of a particular hexameric motif was counted by the method used for counting W-Box and TGA motifs. Thereafter, the ratios between observed and expected occurrence for each motif found in the SI and W remodeled regions were calculated. Similarly, the ratios between observed and expected occurrence for each motif found in the SR and W remodeled regions was also calculated. Finally, ratio of motifs enriched in SR as compared to W and the motif enriched in SI as compared to W was calculated to obtain enriched motif in SR genes with respect to SI genes.
Nucleosome coverage on W box and TGA motifs
Nucleosomal regions from the 1000 bp upstream TSS (promoter regions) were extracted for all the groups. In these regions, the enrichment of cis-regulatory W-box motif (TTGAC) and TGA motif (TGACG/C) was independently calculated for the following four regions:
total promoter region,
region having the nucleosome occupied under both conditions (NOR),
region lacking the nucleosome under any of the two conditions ( NFR), and
regions with remodeled nucleosomes.
First of all, the local nucleotide frequency (F) of a base was calculated in the above mentioned regions as per the formula –
Say, T = total no of all bases in a particular region
Then F = (count of the a particular base in a region)/T
Thereafter, the expected frequency of occurrence of a particular cis-regulatory motif was calculated based on the local nucleotide frequency of each constituent base of the motif under study using the following formula:
E = Expected occurrence of the motif, N = Number of the constituent bases in the motif.
The occurrence of these motifs has been further corrected for their expected frequency of occurrences and for the differences due to database sizes for total number of promoters, NORs, remodeled nucleosomes, and NFRs. The corrected frequency of occurrence of a particular motif was calculated by normalizing the observed frequency with the expected frequency of their occurrence. Next, graphs showing the significance of each motif in all groups were plotted at 95% binomial confidence interval with respect to the total promoter region.
Results and discussion
Global mapping of nucleosome positioning on the A. thaliana genome
The MNase1 digestion strategy is widely used to map nucleosome position and occupancy, based on its ability to produce sequence-dependent cleavages in DNA preferentially within linker regions. An earlier publication [47] claims that the mapping of nucleosome positioning using MNase1 generates a sequence-dependent bias in the resulting fragments, which may affect sequence-dependent models for nucleosome positioning. Still, it is unlikely that such biasness is differentially influenced by SA treatment. Hence, it can be safely used to understand differential nucleosome positioning in A. thaliana on SA treatment. To determine the changes in the nucleosome positions that eventually lead to changes in the gene expression, 2-week old control or SA-treated A. thaliana seedlings were used for mononucleosomal DNA preparation (see Additional file 1). The mononucleosomal DNA was then hybridized to Affymetrix GeneChip® Arabidopsis Tiling 1.0R Array. Custom scripts were developed and used for assigning nucleosomes where nucleosomes were identified at different stringency levels (i.e. log2 transformed signal intensity levels termed as enrichment values, described in Methods). PR1 (pathogenesis-related gene1) is a marker gene of the SA pathway. Earlier, it was demonstrated by us in case of Nicotiana tabacum that a distinct nucleosome spans the TATA-box and TSS of PR-1a minimal promoter at the resting state, which gets remodeled and removed on activation with SA [4]. PR1 in A. thaliana, similar to tobacco PR-1a, is also an SA-induced NPR1-dependent gene; thus, PR1 was selected to validate the results obtained by tiling microarray on SA-induced NPR1-dependent genes. The A. thaliana PR1 gene (At2g14610) is located on the reverse strand of chromosome 2; its genic region spans from position 6,241,704 to 6,243,463 (1760 bp) encompassing the promoter region which spans from 6,242,463 to 6,243,463 with distinct TATA-box at position 6,242,491 (−28 bp with respect to TSS; Figure 1A). The nucleosome positioning around PR1 gene in the tiling microarray data was examined. At stringency level ≥ −1.0 under control condition, nucleosome signals were observed spanning from 6,242,452 to 6,242,879; covering AUG, TSS, and TATA-box (−416 to +11 bp with respect to TSS); and extending further into both the promoter region and the region downstream TSS, indicating the presence of many nucleosomes in that region. After SA induction, distinct MNase1 sensitivity was observed in this region and, thus, lack of hybridization signals around the region −221 to +78 bp, indicating the nucleosomal remodeling and removal of the nucleosome from the region (Figure 1A). The nucleosomal coverage on PR1 at −24 position showed a conspicuous decline from −0.04 (control) to −2.67 (SA; Figure 1A), reconfirming depletion of the nucleosome at the core promoter in SA-induced genes. Since the results were validated at the stringency level of ≥ −1.0 enrichment value by finding a nucleosome at the core promoter of the PR1 gene, it was subjectively decided to use the stringency of ≥ −1.0 enrichment value for subsequent analysis.
Figure 1.
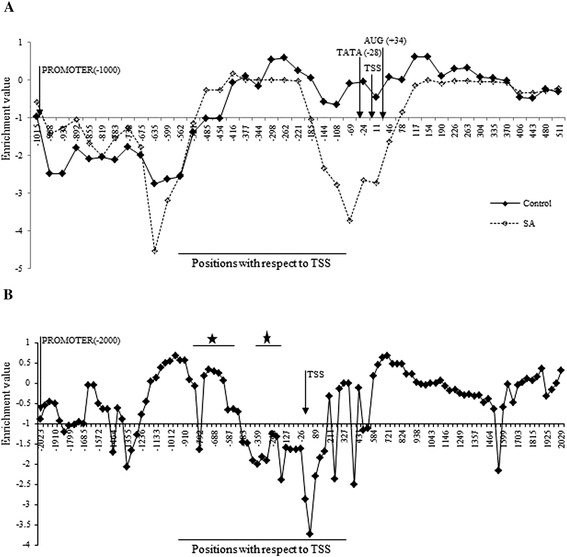
Graph showing nucleosome positioning at At2g14610 and At1g77120 loci to determine stringency criteria. A– depicts the nucleosomal remodeling around At2g14610 promoter regulatory region. The X-axis denotes the position with respect to Transcription start site (TSS) on chromosome 2 and Y-axis denotes the enrichment values. B– Validation of nucleosomal stringency level by mapping on Adh gene (AT1G77120, previously mapped). The X-axis denotes the position with respect to TSS on chromosome 1 and Y-axis denotes the enrichment values. The asterisk shows the regions having nucleosome according to the previous mapping done by DNase assay (Vega-Palas and Ferl, 1995).
This stringency level was cross-validated by mapping the nucleosome occupancy over −2000 to 2000 bp region with respect to TSS of Alcohol dehydrogenase1 gene (Adh; AT1G77120; Figure 1B). This region of the gene has earlier been mapped [48] and is shown to contain three nucleosomes in the promoter region (0 to −1000 bp with respect to TSS), where one nucleosome is loosely positioned at −130 to −350 bp with a best fit at −230 bp and other two nucleosomes span −500 to −800 bp. The downstream region is also occupied by clear nucleosome ladders. A clear nucleosome ladder was found both downstream and upstream TSS in the map generated by us, where the nucleosome occupied −800 to −495 bp region with respect to TSS. The only minor observable difference was that a loosely positioned nucleosome reported at −130 to −350 bp position could not be interpreted clearly at the stringency level of ≥−1.0 enrichment values in our data. However, if the stringency level is reduced from ≥−1.0 to ≥−2.0 in our data, even this nucleosome can also be identified. Thus, our results are in good agreement with an already published nucleosome map of Adh gene [48], thereby justifying the ≥−1.0 stringency criteria experimentally, to assign nucleosome positioning genome wide (Figure 1B).
The chosen stringency level was subjected to further verification using QPCR analysis over four loci having a nucleosome under control conditions and four other loci lacking any nucleosome under control conditions. The results are concurrent with the interpretation made for each gene locus based on the tilling microarray experiments (see Additional files 1, 3 and 4). Thus, in our analysis, the presence of four continuous probes, consistently in three independent experiments, with enrichment value ≥−1.0 marks a distinct nucleosome and likewise, the presence of more than four continuous probes marks a fuzzy nucleosome (See Methods). Our results show that fuzzy nucleosome signals are several folds higher than those of the distinct nucleosomes at the chosen stringency level. Additional file 5 shows the number of bases that covered as distinct nucleosomes and the number of bases covered as fuzzy nucleosomal regions identified at the different stringency levels (See Methods). The number of distinct and fuzzy nucleosomal regions obtained is almost consistent in all the chromosomes under both control and SA-treated conditions. The fuzzy nucleosomal regions were approximately four times higher than the distinct nucleosomes at the most stringent level (≥0) in terms of the number of nucleosomes (and 8-10 times higher in terms of number of bases covered) and more than ten-time higher at the least stringent level (≥−2.0) in terms of the number of nucleosomes (and approximately 60-110 time higher in terms of number of bases covered). At ≥−1.0 stringency level, the fuzzy nucleosomal regions are 8-10 folds higher than the distinct nucleosomes in terms of the number of nucleosomes (and 36-39 times higher in terms of number of bases covered). Below −1.0, the fuzzy nucleosomal regions extend so much that the coverage by distinct nucleosome (both in term of bases and in term of the number of nucleosomes) decreases. This extension also affects the number of fuzzy nucleosomes negatively. While the overall coverage as fuzzy region increases, the number of fuzzy nucleosome decreases, indicating that the fuzzy regions eventually merge to increase their span. In conclusion, the table shows the gregariousness of data expected if stringency level below ≥−1.0 were to be used for this study. The higher abundance of the fuzzy nucleosomal region can be partially attributed to the presence of more than one nucleosome that cannot be resolved by tiling microarray due to its limited resolution of 35 bp [49] and partially, to the use of complete A. thaliana seedlings (a composite tissue) for extracting mononucleosomes, thus interpreting only an average position of a nucleosome rather than the distinct nucleosome present in a specific cell lineage, as also observed in the case of Caenorhabditis elegans [25].
Global nucleosomal coverage follows similar pattern across the genomes
The overall nucleosomal coverage obtained in our study is approximately 346 bp per nucleosome on all the chromosomes of A. thaliana (see Additional file 2). Thus the number of nucleosomes per 1000 bp in our study is much less than the number of nucleosomes per 1000 bp with the nucleosomal coverage of 211 bp per nucleosome obtained by Yuan et al. in yeast [15] and 200 bp per nucleosome obtained by Schones et al. in the human T4 cell line [23]. In addition, our study shows approximately 50% nucleosomal remodeling on SA treatment (see Additional file 6). This means that although the nucleosome coverage is approximately 42-44% under either of the two conditions and that approximately 56-58% region of the genome remains free under either condition, 56.34% of the genome is the NOR when both conditions are taken together. Approximately 50% of this NOR (26.9% of the total genome) either gains or loses the nucleosomes on SA treatment and hence is unique to each subset. Remaining approximately 50% NOR (29.44% out of the total genome) is common to both conditions. Further, the estimate of approximately 56-58% NFR may be an over estimation, considering the limitation of using GeneChip® Arabidopsis Tiling 1.0R Array, which covers not more than 88% of A. thaliana genome. Further, the nucleosomal remodeling post SA treatment does not show any bias for a specific chromosome (see Additional file 6).
Next, the nucleosomal coverage in non-coding regulatory regions, namely the promoter region, 5′UTR, and 3′UTR and the coding region was examined on each chromosome. The coding regions show a significantly higher nucleosomal percentage (more than 60%) than the regulatory region (around 40%, see Additional file 7). These observations are consistent with earlier studies in the case of Saccharomyces cerevisae, Caenorhabditis elegans, and Homo sapiens [15,23,25,50], where lower nucleosomal occupancy in the regulatory region has been reported.
Nucleosomal coverage was also compared in pericentromeric and centromeric regions along the five chromosomes in A. thaliana by plotting the nucleosomal coverage across the 10000 bp window (Figure 2). It was found that the nucleosomal coverage is consistently higher over entire chromosome (marginally peaking at the pericentromeric regions of all chromosomes, being 4000-5000 bp per 10000 bp) as compared with the centromere with <1000 bp coverage (see Additional file 8). The depleted nucleosomal coverage over the centromeric region is solely because of poor probe density over the region. There is approximately equal distribution of probes throughout the genome except over the centromeric and pericentromeric regions which have rather thin density of probes. Additionally, the nucleosomal coverage is directly correlated to the probe density over both the centromeric and pericentromeric regions, except for chromosome 2 (see Additional file 8, correlation data). These results agree with earlier reports in which the authors have found high content of nucleosome in pericentromeric heterochromatin regions arranged as a regularly spaced array [27].
Figure 2.
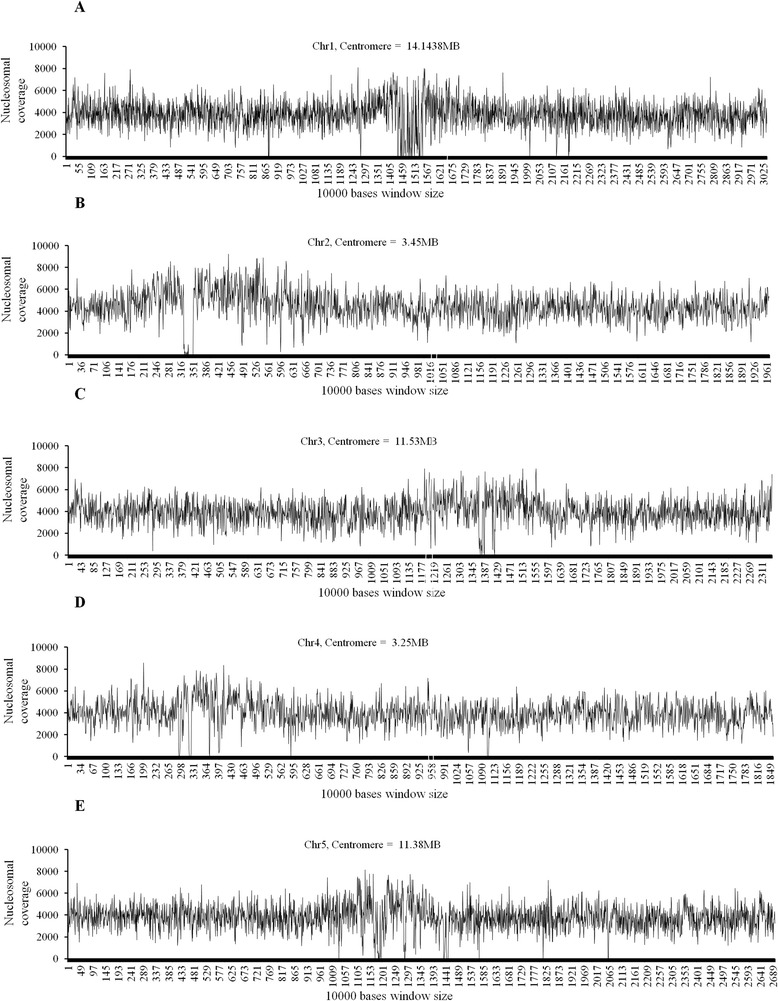
Distribution of Nucleosome Coverage in the chromosomes. X axis (A-E) represents the 10000 bases window size in chromosomes 1–5 respectively and Y axis (A-E) represents the nucleosomal coverage in the window size of 10000 bases.
Genome-wide changes occur in nucleosome positioning in response to SA
To understand the genome-wide nucleosomal remodeling in response to SA in A. thaliana, global gene expression profiling was examined using A. thaliana ATH1 chip (GSE51626) in conjunction to our nucleosomal mapping experiment. SI, SR, CON and SIL groups were identified (see Additional file 9). Average nucleosome coverage around the −1000 to +1000 bp regions with respect to TSS was examined in the genes of the groups specified earlier in a window of 50 bp. All the groups show considerably higher nucleosomal coverage in the coding region ranging from 0.6 to 0.7 as compared to the promoter region, where it ranges from 0.1 to 0.4 (Figure 3A-D).
Figure 3.
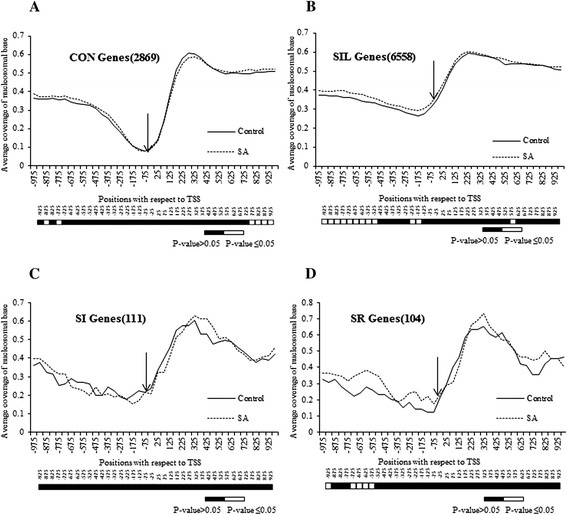
Nucleosomal occupancy is always depleted at Transcription start site (TSS). Coverage of -1000 bp to +1000 bp long region by nucleosome was done for different groups of genes (A – constitutive-CON; B – silent-SIL; C – SA induced-SI and D – SA repressed-SR). X-axis represents the positions with respect to TSS (where −75 means −51 to −100) and Y-axis represents average coverage of nucleosomal base in each figure. The numbers of genes taken in each group is given in parentheses. The −25 position is shown with arrow. Bar below each figure shows the comparative statistical significance difference for each window.
Another distinct feature commonly observed in all classes is the presence of a nucleosomal valley around the minimal promoter region within -200 bp upstream of TSS (Figure 3). This is consistent with the previous findings in other studies [15,17,51], where also the TSS has been shown to be free of the nucleosome. In Saccharomyces cerevisae, the authors have consistently found NFRs in the promoter region [15,17]. Likewise, the promoters of actively expressing genes in Homo sapiens also possess nucleosome-free TSS flanked by positioned nucleosomes [51]. Thus, the average low nucleosomal coverage at TSS seems to be a ubiquitous property of all the genes across the genomes. In our study, the average coverage at this valley always remains below 0.23 (Figure 3A-D) in all groups (reaching as low as 0.09 in the CON group), except the SIL group where it is above 0.35 (Figure 3B). For this group, the valley is considerably shallower than others, indicating a dense chromatin structure around the minimal promoter region. A possible reason accounting for this shallowness may be that since these genes are not expressed, they maintain the presence of nucleosomes at their minimal promoter region. It should be noted that the average nucleosomal occupancy at the −25 window also remains nearly insensitive to SA treatment in all groups.
To gain better insights about the correlation of nucleosomal remodeling in promoters with the SA-mediated response, specific change in nucleosomal remodeling (i.e. either depletion or enrichment) at the TSS was ascertained in all these groups on SA treatment. For this, the total numbers of genes, showing nucleosomal occupancy under any condition, were retrieved and the percentage of the genes showing either nucleosomal depletion or enrichment on SA treatment around −1000 to +1000 bp region with respect to TSS was calculated. Mapping of the cumulative data showed interesting patterns. It was found that the region downstream of TSS show comparatively less remodeling in all the four groups (Figure 4). The CON and SIL groups show less nucleosomal remodeling even at the upstream region (−1000 bp with respect to TSS), where the percentage of genes undergoing nucleosomal depletion or enrichment increases only marginally near TSS (Figure 4). The SI and SR groups show a significant and mutually opposite pattern. The SI and SR groups show nucleosomal depletion (Figure 4A) and nucleosomal enrichment (Figure 4B), respectively, at the complete promoter region as compared to CON and SIL groups, which are statistically significant at P-value ≤0.05. Interestingly, the SI group also shows minimum nucleosomal enrichment at TSS (Figure 4B), and the SR group shows minimum nucleosomal depletion at TSS (Figure 4A) on SA treatment. Thus, the analysis establishes a correlation between the nucleosomal remodeling at proximal promoters and their expression on SA treatment.
Figure 4.
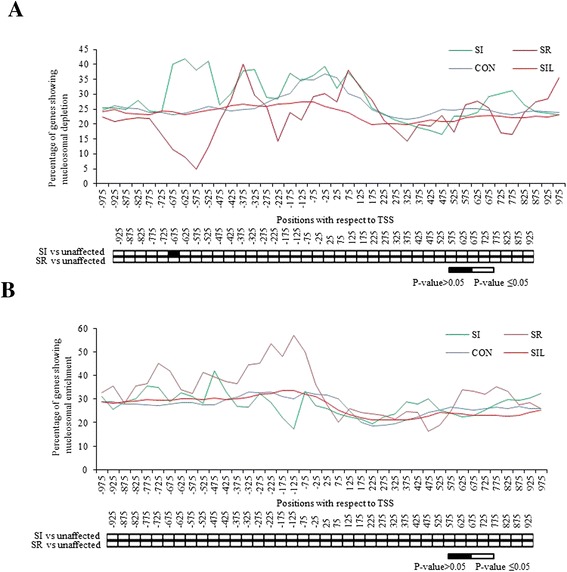
Salicylic acid (SA) induced nucleosomal remodeling is more frequent in the SA responsive genes. The figure represents percentage of genes showing remodeled nucleosome out of total nucleosomes under any of the two conditions (A shows the nucleosomal depletion and B shows the nucleosomal enrichment respectively upon SA treatment) in −1000 bp to +1000 bp long regions in all the four groups. X-axis represents the positions with respect to Transcription start site (TSS) where −75 means −51 to −100. Y-axis represents percentage of genes having remodeled nucleosome at the particular position. Below the graph is the statistical significance data for each position when compared for different groups as mentioned beside the map (Unaffected group constitutes ‘CON’ and ‘SIL’ groups together) at P-value ≤0.05.
To understand the reason behind distinct nucleosomal remodeling profiles of different groups, collective expression profile of these groups was obtained by annotating these groups on the basis of their functions. It is found that most of the CON genes were significantly expressed in different metabolic pathways, while most of the SIL genes were significantly expressed in reproductive and floral development pathways (see Additional file 10). This indicates that the SIL genes, in general, may be involved in some specific pathways, thus allowing minimum disturbance at their promoter by this treatment. Similarly, the SI genes show significant involvement in SAR pathway, SA mediated signaling pathways, ROS pathway, response to hypoxia and other relevant pathways while SR genes showed involvement in amino acid biosynthesis and metabolic processes, photosynthesis etc.
Nucleosome remodels at the promoter locus of PR1
The data analysis at the minimal promoter region of the PR1 gene shows the presence of a nucleosome upstream TSS, on TATA-box which is removed under SA-treated conditions (Figure 1A). The absence of the PR1 core promoter nucleosome is depicted due to the MNase1 digestion strategy which preferentially digests the open chromatin in regions such as promoters of active genes [15,50,52]. Transcriptional activation requires the assembly of pre-initiation complex (PIC) upstream TSS, and the nucleosomal removal is sometimes required for active transcription [53]. In the past, studies have shown that the nucleosome is lost in the promoter region of Pho5 (encoding a repressible acid phosphatase in yeast) gene during transcription activation in yeast [15,52,54,55] and in humans [50,51] where it is hyperacetylated before being lost. Similarly, Interferon β-promoter is activated on nucleosome sliding by viral infection [56]. Li et al. have also shown the same for the beta-phaseolin gene [36]. Till date, the nucleosomal loss has been shown to accompany gene activation by either sliding of nucleosomes or eviction of nucleosomes.
This data analysis was validated by QPCR analysis using primers designed around the core promoter region at the PR1 locus using a mononucleosomal template (see Additional file 3, primer sequences cover a −22 to −147 bp region with respect to TSS). The analysis shows a 5- to 6-fold reduction in amplification in case of the SA-treated conditions as compared to the control sample. This indicates a loss of nucleosomal signal from the PR1 promoter locus (Figure 5A). Thus indeed, the QPCR results agree with the model deduced using tiling microarray at the stringency level of ≥−1.0 (Figure 5B).
Figure 5.
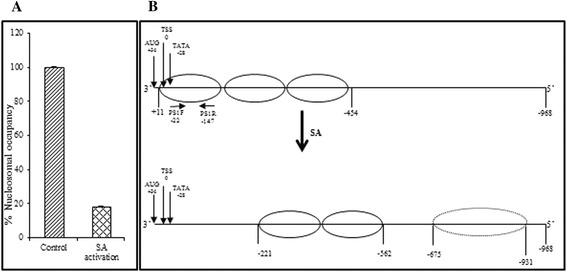
Nucleosome remodeling at the At2g14610 locus prior and post Salicylic acid induction. A– Validation of the PR1 promoter nucleosome remodeling around At2g14610 promoter by QPCR. The bar with diagonal stripes represents percentage of nucleosomal occupancy around TATA-box assigned as 100% and the checkered bar represents the percentage nucleosomal occupancy around TATA-box upon SA activation. B– Schematic representation of nucleosomal remodeling around At2g14610 promoter regulatory region as per Figure 1A and 5A. PS1F (−22) and PS1R (−147) are the primer pairs used for the QPCR validation.
The nucleosomal repositioning in the 5′ regulatory region is found not only in PR1; rather, it is reproducible in other genes of the SI group. The -1000 bp to 1000 bp region of all the SI genes was mapped for the remodeling of nucleosomes (both depleted and enriched) over the entire region on SA treatment. The data (see Additional file 1) show that while the TSS remains largely depleted of nucleosomes even under uninduced conditions (represented as black colour), about two-third of the remaining nucleosomes undergo remodeling on SA treatment (represented as green colour for depletion and red colour for enrichment), with only a few genes showing no change in the presence of the nucleosome around TSS (represented as cyan colour). Thus, the results indicate that the nucleosome remodeling does not limit only to the PR1 gene but is applicable to the complete SI group in A. thaliana.
NPR1 plays distinct role in the nucleosomal remodeling around the core promoter region of the SA-regulated genes
The SAR pathway is regulated by chromatin remodeling [57] and works in both an NPR1-dependent (ND) and NPR1-independent (NI) manner downstream to SA induction. Since PR1 is an ND gene, it was interesting to see whether the nucleosome depletion at the −25 window (i.e. 0 to -50 bp with respect to TSS) in the SI genes is affected by the presence of NPR1. Thus, SA-regulated -ND and -NI genes were identified (GSE 51626, see Additional file 9) [39]. Further, it was observed that ~80% of the SA-regulated genes used to study nucleosomal remodeling patterns (Figure 3C and D) are also present in the list of SA-regulated genes selected by two-way ANOVA (at P-value ≤0.05). A list showing the functional GO groups to which each gene significantly contribute was generated, when these genes were subjected to annotation followed by singular enrichment analysis under agriGO software (see Additional file 11). The analysis confirms the expression profile of each subset of the genes to their respective group; that is, while both groups are involved in the defense pathways; ND genes show significantly more involvement in defense pathways than the NI genes (see Additional file 11). Each group is further divided into two: SI and SR. Interestingly, the examination of nucleosomal coverage in these subgroups in the −1000 to +1000 bp regions with respect to TSS in a window of 50 bp shows that the average nucleosomal coverage at −25 window does not change significantly under SA-treated condition as compared with the control condition in all four groups (i.e. SI-ND, SR-ND, SI-NI, and SR-NI) (Figure 6A-D). So, the graphs showing percentage of genes undergoing either nucleosomal depletion (Figure 6E) or nucleosomal enrichment (Figure 6F) on SA treatment were further plotted. Statistically significant nucleosomal depletion just above the TSS (−25 window) in SI-ND genes as compared with the SI-NI genes was observed, which indeed, extends from 75 to −175 windows at P-value ≤0.05 (Figure 6E). Interestingly, significant nucleosome depletion in SR-ND genes around TSS as compared to SI-NI genes (−75 to 25 windows) was also observed at P-value ≤0.05; however, not in comparison to SR-NI genes. Similarly, statistically significant nucleosomal enrichment above the TSS (−75 to −175 windows) in SR-ND genes as compared to the SR-NI genes was observed at P-value ≤0.05. No nucleosomal enrichment was observed in SI-ND genes; rather, a significant decline in nucleosome enrichment in SI-ND genes (−125 to −225 windows) was observed as compared to SI-NI genes at P-value ≤0.05. Thus, the results show that both the nucleosomal depletion in SI-ND genes and the nucleosomal enrichment in SR-ND genes in response to SA induction correlate to the presence of NPR1. Overall, it suggests a distinct role of NPR1 in chromatin remodeling in SA-responsive genes in A. thaliana. However, the nucleosome depletion observed in SR-ND genes over SI-NI genes does not seem to correlate to the presence of NPR1.
Figure 6.
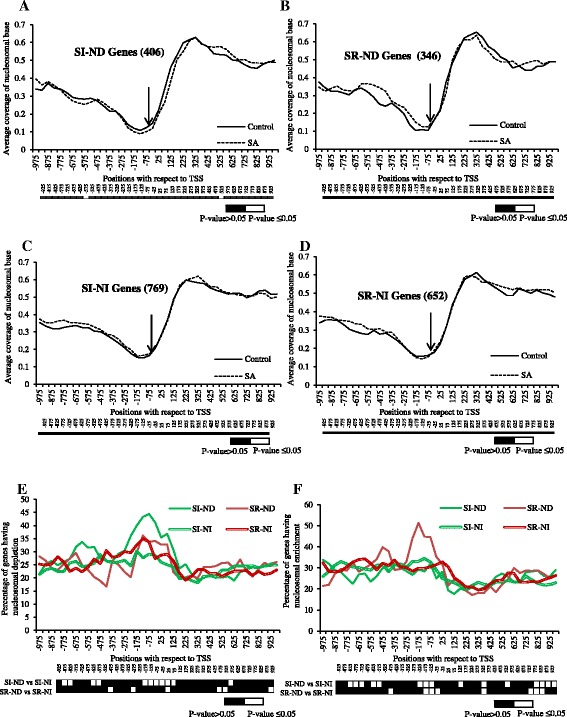
NPR1 dependent genes function by nucleosome remodeling. Coverage of −1000 bp to +1000 bp long region by nucleosome was done for SA induced NPR1 dependent – SI-ND (A), SA repressed NPR1 dependent – SR-ND (B), SA induced NPR1 independent – SI-NI (C) and SA repressed NPR1 independent – SR-ND genes (D). The numbers of genes taken in each group is given in parentheses. The −25 position is shown with arrow. Below each figure is shown the comparative statistical significance difference for each window. E) and F) represent percentage of genes showing nucleosomal depletion and enrichment, respectively, out of total nucleosomes upon SA treatment in −1000 bp to +1000 bp long regions in all the four groups (A-D). X-axis represents the positions with respect to TSS where −75 means −51 to −100 and Y-axis represents either average coverage of nucleosomal base (A-D) or percentage of genes getting remodeled at each position (E-F). Below the graph is the statistical significance data for each position when compared for different groups as mentioned beside the map (Unaffected group constitutes ‘CON’ and ‘SIL’ groups together) at P-value <0.05.
The result based on the role of NPR1 in nucleosomal remodeling around the core promoter region has been further cross-validated. The nucleosomal occupancy at the core promoter and corresponding relative transcript abundance of three representative genes each from all the six categories under control and SA-treated conditions in two-week old seedlings of both wild-type (Col-0) and npr1-1 mutant (Figure 7 and Additional file 1) has been checked. Based on the normalization with the internal control genes set, each group shows average expression profile according to its class. Our results show that the remodeling of the nucleosome at the promoter region in these different groups has characteristic differences (Figure 7 and Additional file 12). First, the CON, SIL, and SR-NI groups do not show any statistically significant change in nucleosomal occupancy at the minimal promoter in both wild-type and npr1-1 mutant under either control or SA-treated condition at P-value ≤ 0.05 (Figure 7). SI-ND and SI-NI genes show significant remodeling of the nucleosome at minimal promoter post SA treatment either in the presence or in the absence of NPR1 at P-value ≤ 0.05; however, SR-ND genes show significant remodeling of the nucleosome at minimal promoter post SA treatment only in the absence of NPR1. Both SR-ND and SR-NI classes do not show any differences in the nucleosome occupancy at minimal promoter in Col-0 post SA treatment. Interestingly, in all the four SA-regulated classes (SI-ND, SR-ND, SI-NI, and SR-NI), treatment of SA causes a statistically significant increase in nucleosomal occupancy over the minimal promoter loci in the absence of NPR1 at P-value ≤0.05. NPR1 has long been known to mediate the plant defense pathways through SA and JA cross-talk [58]. Further, it is well understood that NPR1 accumulates in the nucleus post SA treatment [59] and associates with TGA transcription factors to regulate SA-mediated transcription [60,61]. An increase in the nucleosome occupancy in SA-regulated genes in the absence of NPR1 (Figure 7) indicates that NPR1 has a general role in chromatin remodeling in all SA-regulated genes post SA treatment. However, exact role of NPR1 in SA-mediated response is not clear from the present study and needs further investigation to augment our current understanding about the role of NPR1 in the global nucleosomal remodeling in response to SA.
Figure 7.
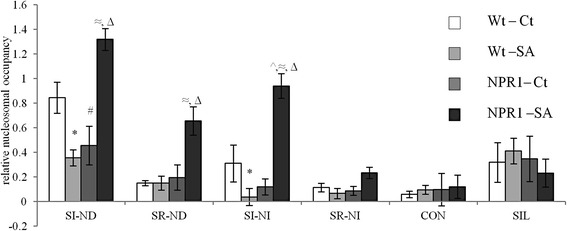
NPR1 plays distinct role in the nucleosomal remodeling around the core promoter region of the SA regulated genes. Figure shows average relative nucleosomal occupancy in Col-0 (c) and npr1-1 mutants (n) under untreated (w) and treated (s) conditions of three genes from each group after normalization with a set of five internal control genes and with SE. ‘*, #, ^, ≈ and Δ’ denote the significance of the change in nucleosomal occupancy values among Wt-Ct–Wt-SA, Wt-Ct–NPR1-Ct, Wt-Ct–NPR1-SA, Wt-SA–NPR1-SA and NPR1-Ct–NPR1-SA pairs respectively, as per Student’s t-test at p ≤0.05. No significant changes were found in the values among Wt-SA–NPR1-Ct pair in all groups. Complete Student’s t-test table has been given for all pair-wise comparisons among Wt-Ct, Wt-SA, NPR1-Ct and NPR1-SA for each gene class as Additional file 12.
ABRE elements and MYB elements are enriched in NORs of SI genes, while W-box and TGACG/C motif are enriched in NFRs
Next, some cis-regulatory motifs were identified in the remodeled nucleosomal region in the promoters (1 kb upstream of TSS) of 111 SI genes (See Methods) and annotated as per PLACE database. These motifs include, at the top, motif 4, similar to GT1GMSCAM4, which interacts with GT-1 transcription factor to activate the pathogen- and salt-inducible gene SCaM-4 in Glycine max [62]. Motifs 1, 3, 5, 8 and 9 are similar to different ABRE motifs (Figure 8A) like ABRECE1HVA22, ABAREG2 element, ABREOSRAB21, PROXBBNNAPA and ABRELATERD1 (involved in ABA specific gene expression in barley [63] during plant embryogenesis and seed germination [64,65], as consensus sequence in wheat, rice and Brassica napa genes [66-68], and in regulating etiolation-induced activity in A. thaliana [69]) respectively. Motif 2, 6 and 7 are similar to MYB family motifs like MYBCOREATCYCB1, AGMOTIFNTMYB2 and MYBPLANT (involved in cell-cycle phase independent cyclin B1 gene activation [70], in the activation of a wound responsive gene [71], as a consensus sequence in phenylpropanoid biosynthesis pathway gene promoters [72]) respectively (Figure 8A). Motif 10 is similar to LTREHVBLT, a low temperature response element in the barley blt4.9 promoter [73]. The preferred presence of these motifs cannot be attributed to the biasness caused by the use of MNase1, as then it would not be found specifically in the SI genes.
Figure 8.
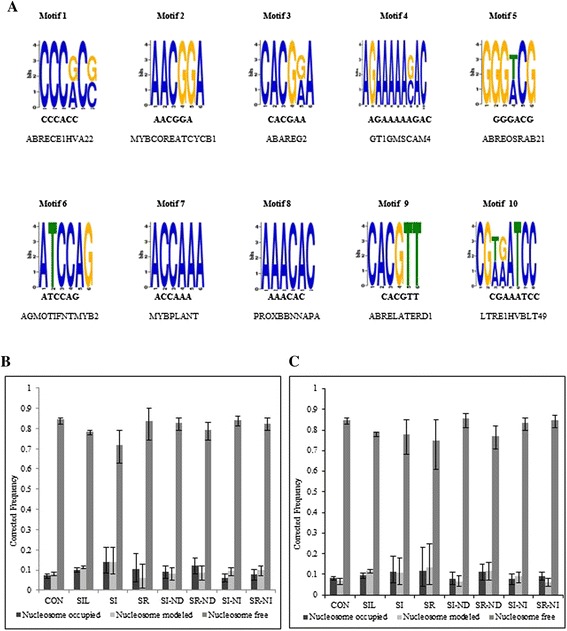
Motif analysis in the promoter region of the Salicylic Acid Induced group. A – The top 10 motifs generated using MEME software. B and C– The W Box and TGA box occupancies in different groups. X-axis shows different groups of genes analyzed. The error bar represents 95% Binomial Confidence Interval and Y-axis shows the relative corrected frequency of respective motifs in each group with respect to the corrected frequency for complete promoter regions.
Promoters of the genes co-regulated with PR1 in the SAR pathway have been reported to be highly enriched in W-box motif TTGAC [31], providing binding sites to WRKY transcription factors. Another pentameric motif TGACG/C, overlapping core W motif TGAC, is not very common in PR1 regulon genes. However, both TGA factors and WRKY transcription factors are reported to regulate SA response by binding TGACG motif and W-box, respectively, in the PR1 promoter [61,74] and globally [75,76]. Therefore, it was interesting to identify the actual presence of both these motifs in context of nucleosomes. Surprisingly, none of these motifs was observed as enriched in the remodeled region by MEME (Figure 8A). So, the presence of the W-box motif and TGAGC/C motif was examined in the promoter region of all groups, namely, CON, SIL, SI, SR, SI-ND, SR-ND, SI-NI, and SR-NI. Both of the motifs were identified as enriched in the NFRs in the promoters of all the groups (Figure 8B-C and Additional file 13) invariably. Only a very small fraction of W-box or TGA motifs was present in either the nucleosomal or remodeled region. Interestingly, enrichment of the TGA motif was considerably higher in both ND and NI groups as compared to other groups (Figure 8C). Both motifs were significantly depleted in the SIL group as compared with other groups (Figure 8B-C), reflecting the functional significance of these motifs that are enriched in the active group. Thus, it is concluded that both the W-box and TGA motifs are significantly enriched in NFR. TGA factors are involved in regulating basal expression of SAR genes by either repressing the locus or promoting its basal expression [77,78], and they physically interact with as 1-like elements for PR-1a gene regulation [79]. Hence, the presence of W-box and TGACG motifs in NFR is interesting and needs to be validated regarding the affinity of WRKY transcription factors and TGA factors to bind their cognate sequence in nucleosomal and non-nucleosomal templates, respectively.
Upstream activator region in the SR genes harbours many potential motifs involved in the plant defense pathway
The SR group shows characteristic nucleosomal remodeling by enrichment on SA treatment at an upstream region, that is, −600 to –800 bp upstream of TSS (Figure 3D). Using Lift and Motif enrichment methods, 40 common motifs were found to be enriched in SR over the SI group in the −600 to –800 bp promoter region, with enrichment fold change ≥2 (Table 1). On annotation of these motifs using PLACE database, it was found that many of these are involved in the plant defense pathway. Among such important motifs include three motifs similar to LS5ATPR1, one motif each similar to LS7ATPR1, AS1LIKECSHPRA, TGA1NTPR1A and PR2GCNT and two motifs similar to WBOXNTCHN48 (Table 1). LS5 and LS7 have been reported to provide binding sites to TGA2 and TGA1 respectively in A. thaliana [61,80]. TGA1NTPR1A and AS1LIKECSHPRA are reported as as1-like elements in tobacco and cucumber [81,82] respectively. Similarly, PR2GCNT and WBOXNTCHN48 are reported to be present in stress- and developmentally-regulated pathogenesis-related acidic tobacco β-1,3-glucanase gene [83] and an ethylene inducible tobacco class1 chitinase48 gene [84] respectively. Three motifs each similar to AGCBOXNPGLB and QARBNEXTA were also found, which were earlier reported in tobacco β-1,3-glucanase gene enhancer element similar to GCC element required for ethylene regulated transcription of extA gene in B. napa in response to wounding and tensile stress [85,86]. Many motifs similar to ABRE elements were found like ABREDISTBBNNAPA, ABREMOTIFIIIOSRAB16B, ABREMOTIFIOSRAB16B, ABRECE1HVA22, ABRE3HVA22 and ABRETAEM earlier identified in B. napa, rice, barley and wheat in relation to abscissic acid regulated transcription [64,65,68,87,88]. Likewise, several motifs similar to MYB2 binding elements were found — AGMOTIFNTMYB2, MYB26PS and MYBPZM (Table 1) which were linked to many defense associated pathways like phenylpropanoid pathway, flavonoid biosynthetic pathway, wound inducible pathway etc. [71,89,90]. Two motifs similar to the SORLIP2AT element were also found in our analysis [91]. Motifs similar to GARE2OSREP1, GAREHVAMY1, NDEGMSAUR, AUXREPSIAA4, D4GMAUX28, DRECRTCOREAT and DRE1COREZMRAB1 involved in other stress response pathways like auxin response pathway, gibberellic acid pathway and dehydration response pathway [92-101] were also identified. The cross-talk between SA, JA, auxin, gibberellin, and ABA has also been reported earlier to fine tune the plant stress responses [29,102-106]. Our study also indicates that SA induction is responsible for chromatin remodeling over the cis-regulatory elements that are involved in both the SA-mediated response of SA-induced genes and the JA- and ABA-mediated response of SA-repressed genes.
Table 1.
Overrepresented motifs in Salicylic acid repressed (SR) group in the -600 bp to -800 bp region upstream TSS
| Consensus motif | OE | LIFT | PLACE ID |
|---|---|---|---|
| GCCATG | 33 | 6.404850381 | NDEGMSAUR |
| CGATCC | 27.5 | 5.337375318 | AGMOTIFNTMYB2 |
| CACTCG | 27.5 | 5.337375318 | ABREDISTBBNNAPA |
| ACCCGT | 22 | 4.269900254 | QARBNEXTA |
| CACGCT | 22 | 4.269900254 | ABREMOTIFIIIOSRAB16B |
| GTCGAC | 18 | 6.404850381 | DRECRTCOREAT |
| CGATGG | 15 | 5.337375318 | ABRECE1HVA22 |
| TGTACG | 13.2 | 6.404850381 | ABRE3HVA22 |
| AAGGTA | 12.1 | 11.7422257 | GARE2OSREP1 |
| ACGGGT | 12 | 4.269900254 | QARBNEXTA |
| CGTACC | 12 | 4.269900254 | ABRE3HVA22 |
| CGTGAC | 12 | 4.269900254 | LS5ATPR1 |
| GCACTG | 12 | 4.269900254 | D4GMAUX28 |
| ACTGAC | 11 | 5.337375318 | WBOXNTCHN48 |
| CGTAGC | 10.5 | 7.472325445 | LS5ATPR1 |
| GTCACG | 10.5 | 7.472325445 | LS5ATPR1 |
| TAGCCG | 10.5 | 3.736162722 | AGCBOXNPGLB |
| TATACG | 9.9 | 9.607275572 | RSRBNEXTA |
| TGGACC | 9 | 6.404850381 | UPRE1AT |
| TAGGTC | 8.4 | 7.472325445 | MYB26PS |
| AGGCCC | 8 | 8.539800509 | SORLIP2AT |
| CTAGAC | 7.7 | 3.736162722 | SURECOREATSULTR11 |
| CACACG | 7.5 | 5.337375318 | QARBNEXTA |
| CGGCTT | 7.5 | 5.337375318 | PR2GCNT |
| GCAGCA | 7.5 | 5.337375318 | D4GMAUX28 |
| TGTGCC | 7.5 | 5.337375318 | RNFG2OS |
| ATGGCC | 7.5 | 5.337375318 | CAATBOX2 |
| GTTCAG | 7.2 | 6.404850381 | WBOXNTCHN48 |
| GCATAG | 7.2 | 6.404850381 | LS7ATPR1 |
| TATCGG | 7.2 | 6.404850381 | GAREHVAMY1 |
| AATGCG | 6.6 | 3.202425191 | AS1LIKECSHPRA |
| GTATGC | 6.6 | 3.202425191 | SORLIP4AT |
| CCCATC | 6 | 4.269900254 | AUXREPSIAA4 |
| CTCTCG | 6 | 4.269900254 | DRE1COREZMRAB17 |
| AACCGC | 6 | 4.269900254 | AGCBOXNPGLB |
| ACAGGC | 6 | 4.269900254 | ANAERO5CONSENSUS |
| AGACGC | 6 | 4.269900254 | AGCBOXNPGLB |
| AGGGCA | 6 | 4.269900254 | ANAERO5CONSENSUS |
| CGTTGG | 6 | 4.269900254 | NDEGMSAUR |
| GGACCA | 6 | 4.269900254 | ABREMOTIFIOSRAB16B |
| GGTACT | 6 | 5.337375318 | ABREMOTIFIOSRAB16B |
| GTGTCC | 6 | 4.269900254 | ABRETAEM |
| GTTAGG | 6 | 4.269900254 | MYB26PS |
| TCTCGA | 6 | 5.337375318 | TGA1ANTPR1A |
Conclusions
Perturbations change the transcriptional profile of an organism. These changes require both silencing and activation of certain genes and, thus, demand chromatin remodeling of such gene loci. In our study, conspicuous nucleosomal remodeling is found in the promoter region of salicylic acid regulated genes, where, under any of the two conditions, 40% of the chromatin is occupied by nucleosomes. The nucleosomal coverage is higher in 3′ coding regions as compared to the non-coding regions. Likewise, the nucleosome coverage is higher over the entire chromosome except the centromeric region with marginally higher coverage in the pericentromeric regions. The nucleosomal remodeling at the promoter region in constitutive and silent groups remains insensitive to the salicylic acid treatment, whereas both salicylic acid induced and salicylic acid repressed groups show nucleosomal depletion and enrichment, respectively, on salicylic acid treatment. The nucleosomal depletion in the promoter region of the salicylic acid induced genes is a general feature of the group. A distinct role of Non-expresser of PR1 (NPR1) in salicylic acid regulated remodeling is observed. The salicylic acid induced-NPR1dependent genes operate through nucleosomal depletion, and salicylic acid repressed-NPR1 dependent genes operate through nucleosomal enrichment in response to salicylic acid induction in an NPR1-dependent manner. All the four groups, that is, salicylic acid induced-NPR1 dependent, salicylic acid repressed-NPR1 dependent, salicylic acid induced-NPR1 independent, and salicylic acid repressed-NPR1 independent, show significant nucleosomal enrichment in the absence of NPR1 on salicylic acid treatment.
The remodeled nucleosomal regions in salicylic acid induced groups harbour many important motifs that are known to be enriched in the genes involved in defense mechanisms such as ABRE elements, MYB box etc. Nevertheless, prominent motifs in the salicylic acid pathway, that is, W-box and TGACG/C, are enriched in nucleosome free regions of the promoter region. Even the upstream region of salicylic acid repressed groups harbours many motifs that are involved in defense pathways.
Overall, this study reports, for the first time, the genome-wide effects of salicylic acid treatment on the chromatin architecture of A. thaliana. It also interprets the enrichment of relevant motifs with respect to overall nucleosomal positioning in the promoter regions of salicylic acid regulated genes.
Availability of supporting data
The datasets supporting the results of this article are available in the NCBI GEO repository as, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE25553 and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE51626.
Acknowledgement
We acknowledge Prof. Randall Morse, Wadsworth Centre, USA for critical reading and suggestions on our Manuscript. We are also thankful to Dr. Ashis Nandi, JNU, India for providing us npr1-1 mutant. We would also like to thank the anonymous reviewers for their valuable comments and suggestions to improve the quality of the paper.
Funding
This work was supported by Council of Scientific and Institutional Research under CSIR EMPOWER (OLP79) programme.
Abbreviations
- SA
Salicylic acid
- PR-1a
Pathogenesis related gene-1a (in tobacco)
- PR1
Pathogenesis related gene 1 (in A. thaliana)
- MNase1
Micrococcal nuclease1
- TSS
Transcription start site
- SAR
Systemic acquired resistance
- NPR1
Non- expresser of PR1
- CON
Constitutive
- SIL
Silent
- SI
SA induced
- SR
SA repressed
- ND
NPR1-dependent
- NI
NPR1- independent
- SI-ND
SA induced-NPR1 dependent
- SR-ND
SA repressed-NPR1 dependent
- SI-NI
SA induced-NPR1 independent
- SR-NI
SA repressed-NPR1 independent
- NOR
Nucleosome occupied region
- NFR
Nucleosome free region
Additional files
Additional Information.pdf containing description of additional figures as follows: Figure S1. Mononucleosomes preparation from 14 days old untreated or Salicylic acid treated A. thaliana seedlings. Figure S2. Box plots of log2 transformed values for control and Salicylic acid treated replicate arrays used for the classification of four different groups. Figure S3. Schematic representation of nucleosomes either present or absent in both the cases and their validation of nucleosome occupancy in A. thaliana genome using QPCR analysis. Figure S4. Validation of nucleosomal remodeling by mapping of Salicylic acid induced genes. Figure S5. NPR1 plays a distinct role in the nucleosomal remodeling around the core promoter region of the Salicylic acid regulated genes.
Nucleosomal Coverage for all the five chromosomes in A. thaliana.
Sequences of the probes and primers used in this study.
Regions showing presence and absence of nucleosomes used for QPCR verification of nucleosomal occupancy over four loci having a nucleosome under control conditions and four other loci lacking any nucleosome under control conditions as per Additional file 1 : Figure S3.
Number of distinct and fuzzy nucleosome at different stringent levels.
Nucleosome remodeling post Salicylic acid (SA) induction.
Percentage of Nucleosomal occupied Regions (NORs) in different region of total genes of A. thaliana Genomes.
Nucleosome coverage at the centromeric and pericentromeric region on all the chromosomes of A. thaliana .
List of Accession number of the genes under each category as per Figures 3 and 6 .
GO analysis of the four groups of genes as per Figure 3 .
GO analysis of NPR1-dependent and NPR1-independent group of genes as per Figure 6 .
Student t -test values for all the pairs for Figure 7 .
Calculation of the TTGAC and TGACG/C motif enrichment in promoters of different group of genes.
Footnotes
Mala Singh and Sumit Kumar Bag contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MS and AR conducted the tiling microarray experiments. MS conducted the expression microarray experiments, mononucleosomes-QPCR and RT-QPCR analysis and contributed in manuscript preparation. AR performed mononucleosomes-QPCR analysis and contributed in manuscript preparation. SB, AB and SM conducted all the bioinformatics analysis and SB contributed in manuscript preparation. AB also conducted both tiling array and expression array data analyses. DN conducted initial tiling microarray data analysis. SVS and YKS designed the research, analyzed the data and contributed in manuscript preparation. All authors read and approved the final manuscript.
Contributor Information
Mala Singh, Email: singhmala21@gmail.com.
Sumit Kumar Bag, Email: sumit.bag@nbri.res.in.
Archana Bhardwaj, Email: archana2287@gmail.com.
Amol Ranjan, Email: amolranjan@yahoo.co.in.
Shrikant Mantri, Email: shrikant@nabi.res.in.
Deepti Nigam, Email: deept_mbi@yahoo.co.in.
Yogesh Kumar Sharma, Email: yogesh_s26@yahoo.com.
Samir Vishwanath Sawant, Email: samirsawant@nbri.res.in.
References
- 1.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10(3):161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Radman-Livaja M, Rando OJ. Nucleosome positioning: how is it established, and why does it matter? Dev Biol. 2010;339(2):258–266. doi: 10.1016/j.ydbio.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Segal E, Widom J. What controls nucleosome positions? Trends Genet. 2009;25(8):335–343. doi: 10.1016/j.tig.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lodhi N, Ranjan A, Singh M, Srivastava R, Singh SP, Chaturvedi CP, et al. Interactions between upstream and core promoter sequences determine gene expression and nucleosome positioning in tobacco PR-1a promoter. Biochim Biophys Acta. 2008;1779(10):634–644. doi: 10.1016/j.bbagrm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 5.Vinayachandran V, Pusarla RH, Bhargava P. Multiple sequence-directed possibilities provide a pool of nucleosome position choices in different states of activity of a gene. Epigenetics Chromatin. 2009;2(1):4. doi: 10.1186/1756-8935-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Z, Zheng C, Hayes JJ. The core histone tail domains contribute to sequence-dependent nucleosome positioning. J Biol Chem. 2007;282(11):7930–7938. doi: 10.1074/jbc.M610584200. [DOI] [PubMed] [Google Scholar]
- 7.Hansen L, Marino-Ramirez L, Landsman D. Many sequence-specific chromatin modifying protein-binding motifs show strong positional preferences for potential regulatory regions in the Saccharomyces cerevisiae genome. Nucleic Acids Res. 2009;38(6):1772–1779. doi: 10.1093/nar/gkp1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X, Lee CK, Granek JA, Clarke ND, Lieb JD. Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection. Genome Res. 2006;16(12):1517–1528. doi: 10.1101/gr.5655606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Truss M, Bartsch J, Schelbert A, Hache RJ, Beato M. Hormone induces binding of receptors and transcription factors to a rearranged nucleosome on the MMTV promoter in vivo. EMBO J. 1995;14(8):1737–1751. doi: 10.1002/j.1460-2075.1995.tb07163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartley PD, Madhani HD. Mechanisms that specify promoter nucleosome location and identity. Cell. 2009;137(3):445–458. doi: 10.1016/j.cell.2009.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ioshikhes IP, Albert I, Zanton SJ, Pugh BF. Nucleosome positions predicted through comparative genomics. Nat Genet. 2006;38(10):1210–1215. doi: 10.1038/ng1878. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458(7236):362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peckham HE, Thurman RE, Fu Y, Stamatoyannopoulos JA, Noble WS, Struhl K, et al. Nucleosome positioning signals in genomic DNA. Genome Res. 2007;17(8):1170–1177. doi: 10.1101/gr.6101007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, et al. A genomic code for nucleosome positioning. Nature. 2006;442(7104):772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, et al. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309(5734):626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, Kadonaga JT, et al. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol. 2009;16(8):847–852. doi: 10.1038/nsmb.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Z, Wippo CJ, Wal M, Ward E, Korber P, Pugh BF. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science. 2011;332(6032):977–980. doi: 10.1126/science.1200508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Field Y, Fondufe-Mittendorf Y, Moore IK, Mieczkowski P, Kaplan N, Lubling Y, et al. Gene expression divergence in yeast is coupled to evolution of DNA-encoded nucleosome organization. Nat Genet. 2009;41(4):438–445. doi: 10.1038/ng.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Field Y, Kaplan N, Fondufe-Mittendorf Y, Moore IK, Sharon E, Lubling Y, et al. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comp Biol. 2008;4(11):e1000216. doi: 10.1371/journal.pcbi.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, et al. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet. 2007;39(10):1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- 21.Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, et al. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008;18(7):1073–1083. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453(7193):358–362. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132(5):887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shivaswamy S, Bhinge A, Zhao Y, Jones S, Hirst M, Iyer VR. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008;6(3):e65. doi: 10.1371/journal.pbio.0060065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18(7):1051–1063. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. Chromatin remodelling at promoters suppresses antisense transcription. Nature. 2007;450(7172):1031–1035. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- 27.Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466(7304):388–392. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gregory BD, Yazaki J, Ecker JR. Utilizing tiling microarrays for whole-genome analysis in plants. Plant J. 2008;53(4):636–644. doi: 10.1111/j.1365-313X.2007.03320.x. [DOI] [PubMed] [Google Scholar]
- 29.Robert-Seilaniantz A, Grant M, Jones JDG. Hormone crosstalk in plant disease and defense: more than just jasmonate-salicylate antagonism. Annu Rev Phytopathol. 2011;49:317–343. doi: 10.1146/annurev-phyto-073009-114447. [DOI] [PubMed] [Google Scholar]
- 30.Thomma BPHJ, Penninckx IAMA, Cammue B, Broekaert WF. The complexity of disease signaling in Arabidopsis. Curr Opin Immunol. 2001;13(1):63–68. doi: 10.1016/S0952-7915(00)00183-7. [DOI] [PubMed] [Google Scholar]
- 31.Maleck K, Levine A, Eulgem T, Morgan A, Schmid J, Lawton KA, et al. The transcriptome of Arabidopsis thaliana during systemic acquired resistance. Nat Genet. 2000;26(4):403–410. doi: 10.1038/82521. [DOI] [PubMed] [Google Scholar]
- 32.Van Verk MC, Gatz C, Linthorst HJM. Transcriptional regulation of plant defense responses. Adv Bot Res. 2009;51:397–438. doi: 10.1016/S0065-2296(09)51010-5. [DOI] [Google Scholar]
- 33.Cao H, Bowling SA, Gordon AS, Dong X. Characterization of an Arabidopsis mutant that is nonresponsive to inducers of systemic acquired resistance. Plant Cell. 1994;6(11):1583–1592. doi: 10.1105/tpc.6.11.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao H, Glazebrook J, Clarke JD, Volko S, Dong X. The Arabidopsis NPR1 Gene That Controls Systemic Acquired Resistance Encodes a Novel Protein Containing Ankyrin Repeats. Cell. 1997;88(1):57–63. doi: 10.1016/S0092-8674(00)81858-9. [DOI] [PubMed] [Google Scholar]
- 35.Hardtke CS, Deng XW. The cell biology of the COP/DET/FUS proteins. Regulating proteolysis in photomorphogenesis and beyond? Plant Physiol. 2000;124(4):1548–1557. doi: 10.1104/pp.124.4.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li G, Chandler SP, Wolffe AP, Hall TC. Architectural specificity in chromatin structure at the TATA box in vivo: nucleosome displacement upon beta-phaseolin gene activation. Proc Natl Acad Sci USA. 1998;95(8):4772–4777. doi: 10.1073/pnas.95.8.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mishra H, Singh N, Misra K, Lahiri T. An ANN-GA model based promoter prediction in Arabidopsis thaliana using tilling microarray data. Bioinformation. 2011;6(6):240. doi: 10.6026/97320630006240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 39.Pavlidis P. Using ANOVA for gene selection from microarray studies of the nervous system. Methods. 2003;31(4):282–289. doi: 10.1016/S1046-2023(03)00157-9. [DOI] [PubMed] [Google Scholar]
- 40.Dabney A, Storey JD, Warnes GR. qvalue: Q-value estimation for false discovery rate control. R package 2010; version1.38.0.
- 41.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 42.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 43.Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 2010; gkq310. [DOI] [PMC free article] [PubMed]
- 44.Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34(Web Server issue):W369–373. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Higo K, Ugawa Y, Iwamoto M, Higo H. PLACE: a database of plant cis-acting regulatory DNA elements. Nucleic Acids Res. 1998;26(1):358–359. doi: 10.1093/nar/26.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doi K, Hosaka A, Nagata T, Satoh K, Suzuki K, Mauleon R, et al. Development of a novel data mining tool to find cis-elements in rice gene promoter regions. BMC Plant Biol. 2008;8:20. doi: 10.1186/1471-2229-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung HR, Dunkel I, Heise F, Linke C, Krobitsch S, Ehrenhofer-Murray AE, et al. The effect of micrococcal nuclease digestion on nucleosome positioning data. PLoS One. 2010;5(12):e15754. doi: 10.1371/journal.pone.0015754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vega-Palas MA, Ferl RJ. The Arabidopsis Adh gene exhibits diverse nucleosome arrangements within a small DNase I-sensitive domain. Plant Cell. 1995;7(11):1923–1932. doi: 10.1105/tpc.7.11.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell. 2006;126(6):1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 50.Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet. 2004;36(8):900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- 51.Ozsolak F, Song JS, Liu XS, Fisher DE. High-throughput mapping of the chromatin structure of human promoters. Nat Biotechnol. 2007;25(2):244–248. doi: 10.1038/nbt1279. [DOI] [PubMed] [Google Scholar]
- 52.Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL. Global nucleosome occupancy in yeast. Genome Biol. 2004;5(9):R62. doi: 10.1186/gb-2004-5-9-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lorch Y, LaPointe JW, Kornberg RD. Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones. Cell. 1987;49(2):203–210. doi: 10.1016/0092-8674(87)90561-7. [DOI] [PubMed] [Google Scholar]
- 54.Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Nucleosomes unfold completely at a transcriptionally active promoter. Mol Cell. 2003;11(6):1587–1598. doi: 10.1016/S1097-2765(03)00231-4. [DOI] [PubMed] [Google Scholar]
- 55.Reinke H, Horz W. Histones are first hyperacetylated and then lose contact with the activated PHO5 promoter. Mol Cell. 2003;11(6):1599–1607. doi: 10.1016/S1097-2765(03)00186-2. [DOI] [PubMed] [Google Scholar]
- 56.Lomvardas S, Thanos D. Modifying gene expression programs by altering core promoter chromatin architecture. Cell. 2002;110(2):261–271. doi: 10.1016/S0092-8674(02)00822-X. [DOI] [PubMed] [Google Scholar]
- 57.van den Burg HA, Takken FL. Does chromatin remodeling mark systemic acquired resistance? Trends Plant Sci. 2009;14(5):286–294. doi: 10.1016/j.tplants.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 58.Rayapuram C, Baldwin IT. Increased SA in NPR1-silenced plants antagonizes JA and JA-dependent direct and indirect defenses in herbivore-attacked Nicotiana attenuata in nature. Plant J. 2007;52(4):700–715. doi: 10.1111/j.1365-313X.2007.03267.x. [DOI] [PubMed] [Google Scholar]
- 59.Kinkema M, Fan W, Dong X. Nuclear localization of NPR1 is required for activation of PR gene expression. Plant Cell. 2000;12(12):2339–2350. doi: 10.1105/tpc.12.12.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson C, Mhatre A, Arias J. NPR1 preferentially binds to the DNA-inactive form of Arabidopsis TGA2. Biochim Biophys Acta. 2008;1779(10):583–589. doi: 10.1016/j.bbagrm.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 61.Despres C, DeLong C, Glaze S, Liu E, Fobert PR. The Arabidopsis NPR1/NIM1 protein enhances the DNA binding activity of a subgroup of the TGA family of bZIP transcription factors. Plant Cell. 2000;12(2):279–290. doi: 10.1105/tpc.12.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park HC, Kim ML, Kang YH, Jeon JM, Yoo JH, Kim MC, et al. Pathogen- and NaCl-induced expression of the SCaM-4 promoter is mediated in part by a GT-1 box that interacts with a GT-1-like transcription factor. Plant Physiol. 2004;135(4):2150–2161. doi: 10.1104/pp.104.041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen Q, Ho TH. Functional dissection of an abscisic acid (ABA)-inducible gene reveals two independent ABA-responsive complexes each containing a G-box and a novel cis-acting element. Plant Cell. 1995;7(3):295–307. doi: 10.1105/tpc.7.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thomas TL. Gene expression during plant embryogenesis and germination: an overview. Plant Cell. 1993;5(10):1401–1410. doi: 10.1105/tpc.5.10.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Busk PK, Pages M. Regulation of abscisic acid-induced transcription. Plant Mol Biol. 1998;37(3):425–435. doi: 10.1023/A:1006058700720. [DOI] [PubMed] [Google Scholar]
- 66.Marcotte WR, Jr, Russell SH, Quatrano RS. Abscisic acid-responsive sequences from the em gene of wheat. Plant Cell. 1989;1(10):969–976. doi: 10.1105/tpc.1.10.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mundy J, Chua NH. Abscisic acid and water-stress induce the expression of a novel rice gene. EMBO J. 1988;7(8):2279–2286. doi: 10.1002/j.1460-2075.1988.tb03070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ezcurra I, Ellerstrom M, Wycliffe P, Stalberg K, Rask L. Interaction between composite elements in the napA promoter: both the B-box ABA-responsive complex and the RY/G complex are necessary for seed-specific expression. Plant Mol Biol. 1999;40(4):699–709. doi: 10.1023/A:1006206124512. [DOI] [PubMed] [Google Scholar]
- 69.Simpson SD, Nakashima K, Narusaka Y, Seki M, Shinozaki K, Yamaguchi-Shinozaki K. Two different novel cis-acting elements of erd1, a clpA homologous Arabidopsis gene function in induction by dehydration stress and dark-induced senescence. Plant J. 2003;33(2):259–270. doi: 10.1046/j.1365-313X.2003.01624.x. [DOI] [PubMed] [Google Scholar]
- 70.Planchais S, Perennes C, Glab N, Mironov V, Inze D, Bergounioux C. Characterization of cis-acting element involved in cell cycle phase-independent activation of Arath;CycB1;1 transcription and identification of putative regulatory proteins. Plant Mol Biol. 2002;50(1):111–127. doi: 10.1023/A:1016018711532. [DOI] [PubMed] [Google Scholar]
- 71.Sugimoto K, Takeda S, Hirochika H. Transcriptional activation mediated by binding of a plant GATA-type zinc finger protein AGP1 to the AG-motif (AGATCCAA) of the wound-inducible Myb gene NtMyb2. Plant J. 2003;36(4):550–564. doi: 10.1046/j.1365-313X.2003.01899.x. [DOI] [PubMed] [Google Scholar]
- 72.Sablowski RW, Moyano E, Culianez-Macia FA, Schuch W, Martin C, Bevan M. A flower-specific Myb protein activates transcription of phenylpropanoid biosynthetic genes. EMBO J. 1994;13(1):128–137. doi: 10.1002/j.1460-2075.1994.tb06242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dunn MA, White AJ, Vural S, Hughes MA. Identification of promoter elements in a low-temperature-responsive gene (blt4.9) from barley (Hordeum vulgare L.) Plant Mol Biol. 1998;38(4):551–564. doi: 10.1023/A:1006098132352. [DOI] [PubMed] [Google Scholar]
- 74.Lebel E, Heifetz P, Thorne L, Uknes S, Ryals J, Ward E. Functional analysis of regulatory sequences controlling PR-1 gene expression in Arabidopsis. Plant J. 1998;16(2):223–233. doi: 10.1046/j.1365-313x.1998.00288.x. [DOI] [PubMed] [Google Scholar]
- 75.Lam E, Benfey PN, Gilmartin PM, Fang RX, Chua NH. Site-specific mutations alter in vitro factor binding and change promoter expression pattern in transgenic plants. Proc Natl Acad Sci USA. 1989;86(20):7890–7894. doi: 10.1073/pnas.86.20.7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thibaud-Nissen F, Wu H, Richmond T, Redman JC, Johnson C, Green R, et al. Development of Arabidopsis whole-genome microarrays and their application to the discovery of binding sites for the TGA2 transcription factor in salicylic acid-treated plants. Plant J. 2006;47(1):152–162. doi: 10.1111/j.1365-313X.2006.02770.x. [DOI] [PubMed] [Google Scholar]
- 77.Kesarwani M, Yoo J, Dong X. Genetic interactions of TGA transcription factors in the regulation of pathogenesis-related genes and disease resistance in Arabidopsis. Plant Physiol. 2007;144(1):336–346. doi: 10.1104/pp.106.095299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Y, Tessaro MJ, Lassner M, Li X. Knockout analysis of Arabidopsis transcription factors TGA2, TGA5, and TGA6 reveals their redundant and essential roles in systemic acquired resistance. Plant Cell. 2003;15(11):2647–2653. doi: 10.1105/tpc.014894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Butterbrodt T, Thurow C, Gatz C. Chromatin immunoprecipitation analysis of the tobacco PR-1a- and the truncated CaMV 35S promoter reveals differences in salicylic acid-dependent TGA factor binding and histone acetylation. Plant Mol Biol. 2006;61(4–5):665–674. doi: 10.1007/s11103-006-0039-2. [DOI] [PubMed] [Google Scholar]
- 80.Xiang C, Miao Z, Lam E. DNA-binding properties, genomic organization and expression pattern of TGA6, a new member of the TGA family of bZIP transcription factors in Arabidopsis thaliana. Plant Mol Biol. 1997;34(3):403–415. doi: 10.1023/A:1005873500238. [DOI] [PubMed] [Google Scholar]
- 81.Strompen G, Gruner R, Pfitzner UM. An as-1-like motif controls the level of expression of the gene for the pathogenesis-related protein 1a from tobacco. Plant Mol Biol. 1998;37(5):871–883. doi: 10.1023/A:1006003916284. [DOI] [PubMed] [Google Scholar]
- 82.Jin G, Davey MC, Ertl JR, Chen R, Yu ZT, Daniel SG, et al. Interaction of DNA-binding proteins with the 5′-flanking region of a cytokinin-responsive cucumber hydroxypyruvate reductase gene. Plant Mol Biol. 1998;38(5):713–723. doi: 10.1023/A:1006034932322. [DOI] [PubMed] [Google Scholar]
- 83.Ohme-Takagi M, Shinshi H. Structure and expression of a tobacco beta-1,3-glucanase gene. Plant Mol Biol. 1990;15(6):941–946. doi: 10.1007/BF00039434. [DOI] [PubMed] [Google Scholar]
- 84.Yamamoto S, Nakano T, Suzuki K, Shinshi H. Elicitor-induced activation of transcription via W box-related cis-acting elements from a basic chitinase gene by WRKY transcription factors in tobacco. Biochim Biophys Acta. 2004;1679(3):279–287. doi: 10.1016/j.bbaexp.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 85.Hart CM, Nagy F, Meins F., Jr A 61 bp enhancer element of the tobacco beta-1,3-glucanase B gene interacts with one or more regulated nuclear proteins. Plant Mol Biol. 1993;21(1):121–131. doi: 10.1007/BF00039623. [DOI] [PubMed] [Google Scholar]
- 86.Elliott KA, Shirsat AH. Promoter regions of the extA extensin gene from Brassica napus control activation in response to wounding and tensile stress. Plant Mol Biol. 1998;37(4):675–687. doi: 10.1023/A:1005918816630. [DOI] [PubMed] [Google Scholar]
- 87.Ono A, Izawa T, Chua NH, Shimamoto K. The rab16B promoter of rice contains two distinct abscisic acid-responsive elements. Plant Physiol. 1996;112(2):483–491. doi: 10.1104/pp.112.2.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guiltinan MJ, Marcotte WR, Jr, Quatrano RS. A plant leucine zipper protein that recognizes an abscisic acid response element. Science. 1990;250(4978):267–271. doi: 10.1126/science.2145628. [DOI] [PubMed] [Google Scholar]
- 89.Uimari A, Strommer J. Myb26: a MYB-like protein of pea flowers with affinity for promoters of phenylpropanoid genes. Plant J. 1997;12(6):1273–1284. doi: 10.1046/j.1365-313x.1997.12061273.x. [DOI] [PubMed] [Google Scholar]
- 90.Grotewold E, Drummond BJ, Bowen B, Peterson T. The myb-homologous P gene controls phlobaphene pigmentation in maize floral organs by directly activating a flavonoid biosynthetic gene subset. Cell. 1994;76(3):543–553. doi: 10.1016/0092-8674(94)90117-1. [DOI] [PubMed] [Google Scholar]
- 91.Hudson ME, Quail PH. Identification of promoter motifs involved in the network of phytochrome A-regulated gene expression by combined analysis of genomic sequence and microarray data. Plant Physiol. 2003;133(4):1605–1616. doi: 10.1104/pp.103.030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li Y, Liu ZB, Shi X, Hagen G, Guilfoyle TJ. An auxin-inducible element in soybean SAUR promoters. Plant Physiol. 1994;106(1):37–43. doi: 10.1104/pp.106.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sutoh K, Yamauchi D. Two cis-acting elements necessary and sufficient for gibberellin-upregulated proteinase expression in rice seeds. Plant J. 2003;34(5):635–645. doi: 10.1046/j.1365-313X.2003.01753.x. [DOI] [PubMed] [Google Scholar]
- 94.Skriver K, Olsen FL, Rogers JC, Mundy J. cis-acting DNA elements responsive to gibberellin and its antagonist abscisic acid. Proc Natl Acad Sci U S A. 1991;88(16):7266–7270. doi: 10.1073/pnas.88.16.7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ballas N, Wong LM, Theologis A. Identification of the auxin-responsive element, AuxRE, in the primary indoleacetic acid-inducible gene, PS-IAA4/5, of pea (Pisum sativum) J Mol Biol. 1993;233(4):580–596. doi: 10.1006/jmbi.1993.1537. [DOI] [PubMed] [Google Scholar]
- 96.Klinedinst S, Pascuzzi P, Redman J, Desai M, Arias J. A xenobiotic-stress-activated transcription factor and its cognate target genes are preferentially expressed in root tip meristems. Plant Mol Biol. 2000;42(5):679–688. doi: 10.1023/A:1006332708388. [DOI] [PubMed] [Google Scholar]
- 97.Nagao RT, Goekjian VH, Hong JC, Key JL. Identification of protein-binding DNA sequences in an auxin-regulated gene of soybean. Plant Mol Biol. 1993;21(6):1147–1162. doi: 10.1007/BF00023610. [DOI] [PubMed] [Google Scholar]
- 98.Suzuki M, Ketterling MG, McCarty DR. Quantitative statistical analysis of cis-regulatory sequences in ABA/VP1- and CBF/DREB1-regulated genes of Arabidopsis. Plant Physiol. 2005;139(1):437–447. doi: 10.1104/pp.104.058412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dubouzet JG, Sakuma Y, Ito Y, Kasuga M, Dubouzet EG, Miura S, et al. OsDREB genes in rice, Oryza sativa L., encode transcription activators that function in drought-, high-salt- and cold-responsive gene expression. Plant J. 2003;33(4):751–763. doi: 10.1046/j.1365-313X.2003.01661.x. [DOI] [PubMed] [Google Scholar]
- 100.Diaz-Martin J, Almoguera C, Prieto-Dapena P, Espinosa JM, Jordano J. Functional interaction between two transcription factors involved in the developmental regulation of a small heat stress protein gene promoter. Plant Physiol. 2005;139(3):1483–1494. doi: 10.1104/pp.105.069963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Busk PK, Jensen AB, Pages M. Regulatory elements in vivo in the promoter of the abscisic acid responsive gene rab17 from maize. Plant J. 1997;11(6):1285–1295. doi: 10.1046/j.1365-313X.1997.11061285.x. [DOI] [PubMed] [Google Scholar]
- 102.Spoel SH, Dong X. Making sense of hormone crosstalk during plant immune responses. Cell Host Microb. 2008;3(6):348–351. doi: 10.1016/j.chom.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 103.Bari R, Jones JDG. Role of plant hormones in plant defence responses. Plant Mol Biol. 2009;69(4):473–488. doi: 10.1007/s11103-008-9435-0. [DOI] [PubMed] [Google Scholar]
- 104.Pieterse CMJ, Leon-Reyes A, Van der Ent S, Van Wees SCM. Networking by small-molecule hormones in plant immunity. Nat Chem Biol. 2009;5(5):308–316. doi: 10.1038/nchembio.164. [DOI] [PubMed] [Google Scholar]
- 105.Wang D, Pajerowska-Mukhtar K, Culler AH, Dong X. Salicylic acid inhibits pathogen growth in plants through repression of the auxin signaling pathway. Curr Biol. 2007;17(20):1784–1790. doi: 10.1016/j.cub.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 106.Navarro L, Bari R, Achard P, Lisón P, Nemri A, Harberd NP, et al. DELLAs control plant immune responses by modulating the balance of jasmonic acid and salicylic acid signaling. Curr Biol. 2008;18(9):650–655. doi: 10.1016/j.cub.2008.03.060. [DOI] [PubMed] [Google Scholar]