

Abstract
Long inverted repeats (LIRs) have been shown to induce genomic deletions in yeast. In this study, LIRs were investigated within ±10 kb spanning each breakpoint from 109 human gross deletions, using Inverted Repeat Finder (IRF) software. LIR number was significantly higher at the breakpoint regions, than in control segments (P < 0.001). In addition, it was found that strong correlation between 5′ and 3′ LIR numbers, suggesting contribution to DNA sequence evolution (r = 0.85, P < 0.001). 138 LIR features at ±3 kb breakpoints in 89 (81%) of 109 gross deletions were evaluated. Significant correlations were found between distance from breakpoint and loop length (r = −0.18, P < 0.05) and stem length (r = −0.18, P < 0.05), suggesting DNA strands are potentially broken in locations closer to bigger LIRs. In addition, bigger loops cause larger deletions (r = 0.19, P < 0.05). Moreover, loop length (r = 0.29, P < 0.02) and identity between stem copies (r = 0.30, P < 0.05) of 3′ LIRs were more important in larger deletions. Consequently, DNA breaks may form via LIR-induced cruciform structure during replication. DNA ends may be later repaired by non-homologous end-joining (NHEJ), with following deletion.
Long inverted repeats (LIRs) are imperfect or near to perfect repetitive DNA sequence elements that can form secondary stem-loop structures in prokaryotic and eukaryotic genomes1,2,3. LIRs may induce stem loops through matching complementary repeats placed in inverted orientation, convertible to the hairpins in single stranded DNA or cruciforms in double stranded DNA4,5. It was found that LIRs involved deletion and recombination events in yeast Saccharomyces cerevisiae6,7.
Gross gene deletions are genomic rearrangements that can be observed in many types of human cancers and inherited diseases8,9,10,11,12,13. Deletion and duplication mutations can vary in size from thousands to hundreds of thousands of base pairs in length in the human genome14. It has been proposed that three major mechanisms are responsible for genomic rearrangements, including human genome deletions15. They are non-allelic homologous recombination (NAHR), non-homologous end-joining (NHEJ), and fork stalling and template switching (FoSTeS) models. Some genomic rearrangements are recurrent, with a common size and fixed breakpoints between low copy repeats (LCRs). Recurrent rearrangements are mostly mediated by NAHR between two LCRs16. Conversely, non-recurrent rearrangements have different sizes and distinct breakpoints in each event, and are performed by NHEJ and FoSTeS models15. Gu et al. suggests that the FoSTeS model is a replication-based rearrangement pathway that may operate over long distances (from 120 to 550 kb) through template switching15. Alternatively, it has been proposed that palindrome or cruciform structures may stimulate the FoSTeS model15.
Breakpoints of gross gene deletions coincide with non-B DNA conformations, including hairpin/cruciform structures17. Hairpins are reported to form by direct repeats18. Direct repeats have ranges from 2 to 8 bp, and are associated with small deletion breakpoints in human genetic diseases19. Moreover, retinoblastoma gene deletion involves direct repeats within the deletion breakpoints20.
Short direct repeats were also detected in 15 proximal breakpoints of the dystrophine gene, which has large deletions21. Short IRs and IR inversions were found in 83% of deletions + small insertions, while short direct repeats were detected only in simple deletion breakpoints22.
Two highly homologous Alu repeats in inverted orientations were found in the vicinity of gross deletion breakpoints in the von Willebrand factor (VWF) gene23. Furthermore, LINEs, LTR repetitive elements, and SINEs (including Alus), were enriched at breakpoints of rare pathogenic microdeletions24. Vissers et al. also suggests that microhomology levels of breakpoint junctions play an important role in replication-based mechanisms, such as FoSTeS and microhomology-mediated break-induced replication (MMBIR)24. Zhang et al. also suggested replication fork stalling to initiate FoSTeS25.
Gordenin et al. showed that LIRs cause deletion in Saccharomyces cerevisiae6. Lobachev et al. suggested they form stem-like secondary structures on single stranded DNA during replication, thereby causing deletions26. Warburton et al. found that some IRs are capable of transforming into cruciform structures, with intrastrand double helices, termed stems and unpaired loops forming internal spacers1. The four-way junction of this suggested IR pattern is similar to the Holliday structure. Eichman et al. showed formation of the Holliday junction in synthetic IR DNA using X-ray crystallography27. From this work, it was proposed that IRs may be involved in homologous recombination. Bacolla and Wells indicated that IRs may form cruciform structures, and are often found at genomic rearrangement breakpoints18.
Genomes of many complex organisms have been investigated for larger IRs. It was determined that higher eukaryotic genomes include many imperfect and near-to-perfect LIRs28,29,30,31. In mice, a perfect LIR was shown to create a large deletion32. Subsequently, it was decided that criteria for LIRs in genomic rearrangements involved recombination. In this regards, Wang and Leung reported that LIRs with stem length >30 bp, identity between stem copies (hereafter stem identity) >85% and internal spacer of <2 kb, are recombinogenic in genomes of humans and some other organisms2. Voineagu et al. demonstrated that Alu IRs with 100% sequence homology of stem copies, triggers strong replication blockage3. However, Alu IRs with 75% stem identity between repetitive halves caused mild replication blockage in E. coli cells3.
Potential models referred to as replication slippage and hairpin nicking were proposed by Akgün et al. to explain the mechanism underlying LIR induced deletions4. With these models, many deletions formed inside palindrome stems or loops are explained. However, alternative models are required for clarifying the mechanisms of larger deletions formed in close proximity to palindromes. To understand how gross gene deletions occur in human cancers and inherited diseases, this present study investigated the significance of LIRs on breakpoint regions of human gross gene deletions.
Results
Identification of long inverted repeats in breakpoint regions of gross gene deletions
Sequences from 218 breakpoint regions of 63 gross gene deletions were taken from references 33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89 (see Supplementary Table S1 online) listed in the HGMD90,91 and GRaBD92,93 (Figure 1a). LIRs with stem length >20 bp on surrounding (±10 kb) each breakpoint were investigated using IRF94,95 (Figure 1b). In total, 218 genomic regions, including 5′ and 3′ breakpoints from 109 gross deletions involving 63 different genes (Table 1), were analysed. Total number of LIRs was determined within ±10 kb regions flanking each breakpoint. In the deletion group, a total of 2723 LIRs were detected (see Supplementary Table S2 online). A total of 1345 LIRs were also identified in 20 kb segments from 220 control sequences (see Supplementary Table S2 online).
Figure 1. LIR identification and selection were performed in 218 genomic regions including 5′ and 3′ breakpoints from 109 gross deletions involving 63 human genes using IRF software.

(a) DNA sequences from 5′ and 3′ deletion BPs were obtained from HGMD and GRaBD. Each BP-DNA sequence and corresponding gene was compaired using NCBI BLAST. Deletion BP locations were determined in related genes. 3′ BP sequence from BRCA1 gene was presented for describing BLAST comparing process. (b) LIR identification was done within ±10 kb flanking sequences each of 5′ and 3′ deletion BPs and 20 kb DNA fragments from control groups using IRF. LIRs with SL > 20 bp, IS of 0–10 kb, SID ≥ 70% were included for comparing total LIR number between deletion and control groups using Mann Whitney U test. (c) LIR selection was made within ±3 kb flanking sequences each of 5′ and 3′ BPs in deletion group. LIRs with SL > 20 bp, IS of 0–2.5 kb, SID ≥ 70% were selected for analysing of correlations between LIR features, distance from breakpoint and deletion size using Pearson's coefficient. Abbreviations: Bp, base pair; BP, breakpoint; GRaBD, gross rearrangement breakpoint database; HGMD, human gene mutation database; IRF, inverted repeat finder; IS, internal spacer; kb, kilobase; LIR, long inverted repeat; SID, stem identity; SL, stem length.
Table 1. 5′ and 3′ breakpoint locations from gross gene deletions and detection of long inverted repeats*.
| GENE/DELETION SIZE | BREAKPOINT LOCATION/NCBI ACCESSION NUMBER | 5′LIR/3′LIR | GENE/DELETION SIZE | BREAKPOINT LOCATION/NCBI ACCESSION NUMBER | 5′LIR/3′LIR |
|---|---|---|---|---|---|
| 1-BRCA1/14 kb | (3′) 16223-30238 (5′)/NT_010755.15 | +/+ | 32-PTEN/662 kb | (5′) 475951-1138023 (3′)/NT_030059.12 | +/− |
| 2-BRCA1/3.8 kb | (3′) 59697-63533 (5′)/NT_010755.15 | +/+ | 33-PTEN/13.6 kb | (5′) 607242-620845 (3′)/NT_030059.12 | +/− |
| 3-BRCA1/36.3 kb | (5′) 29197-65577 (3′)/L78833.1 | +/+ | 34-PTEN/300 kb | (5′) 612328-912640 (3′)/NT_030059.12 | −/− |
| 4-BRCA1/19.8 kb | (5′) 51483-71369 (3′)/L78833.1 | +/+ | 35-MEN1/67.7 kb | (3′) 131486-199212 (5′)/AC000134.14 | +/+ |
| 5-BRCA1/18.3 kb | (5′) 56990-75331 (3′)/L78833.1 | +/+ | 36-BTK/5.9 kb | (5′) 62499-68391 (3′)/U78027.1 | +/+ |
| 6-BRCA1/4.8 kb | (5′) 62115-66940 (3′)/L78833.1 | +/+ | 37-HBB/9.1 kb | (5′) 53257-62324 (3′)/U01317.1 | +/− |
| 7-BRCA2/5.1 kb | (5′) 3806-8872 (3′)/NC_000013.9 | +/+ | 38-ATM/3.4 kb | (5′) 73863-77312 (3′)/U82828.1 | +/+ |
| 8-BRCA2/14.3 kb | (5′) 12323-26644 (3′)/AY436640.1 | +/− | 39-MSH2/36.6 kb | (5′) 99957-136583 (3′)/AC079775.6 | +/+ |
| 9-BRCA2/7.9 kb | (5′) 27566-35513 (3′)/AY436640.1 | −/+ | 40-MSH2/2.2 kb | (5′) 104712-106890 (3′)/AC079775.6 | +/+ |
| 10-BRCA2/10.8 kb | (5′) 45138-55975 (3′)/AY436640.1 | +/+ | 41-MSH2/15.4 kb | (5′) 105900-121280 (3′)/AC079775.6 | +/+ |
| 11-BRCA2/4.9 kb | (5′) 56447-61399 (3′)/AY436640.1 | +/+ | 42-MSH2/6.7 kb | (5′) 124511-131175 (3′)/AC079775.6 | +/+ |
| 12-CDKN/33.8 kb 2A/2B | (3′) 176468-210312 (5′)/NT_008413.17 | −/+ | 43-MLH1/6.1 kb | (3′) 86839-92892 (5′)/AC006583.31 | +/+ |
| 13-NF1/99.6 kb | (5′) 238391-338000 (3′)/NT_010799.14 | −/+ | 44-MLH1/3.5 kb | (3′) 93192-96718 (5′)/AC006583.31 | −/+ |
| 14-NF1/12 kb | (5′) 347607-359613 (3′)/NT_010799.14 | +/− | 45-MLH1/14.3 kb | (5′) 145570-159901 (3′)/AC011816.17 | +/+ |
| 15-RB1/52.3 kb | (5′) 305422-357770 (3′)/NT_024524.13 | −/− | 46-MLH1/12.7 kb | (5′) 147927-160607 (3′)/AC011816.17 | +/+ |
| 16-RB1/178 kb | (5′) 312336-490300 (3′)/NT_024524.13 | +/− | 47-MSH6/11.3 kb | (5′) 5698337-5709648 (3′)/NT_034483.3 | +/+ |
| 17-RB1/201.9 kb | (5′) 356866-558723 (3′)/NT_024524.13 | +/− | 48-MSH6/21.7 kb | (5′) 5699487-5721160 (3′)/NT_034483.3 | +/+ |
| 18-RB1/40 kb | (5′) 365612-405655 (3′)/NT_024524.13 | −/+ | 49-PMS2/1.1 kb | (5′) 53291-54449 (3′)/AC005995.3 | +/+ |
| 19-RB1/3.9 kb | (5′) 378370-382275 (3′)/NT_024524.13 | +/− | 50-HEXA/7.9 kb | (3′) 449987-457933 (5′)/NT_010194.16 | −/+ |
| 20-RB1/2.4 kb | (5′) 462638-465032 (3′)/NT_024524.13 | +/− | 51-HPRT1/13.3 kb | (5′) 297453-310733 (3′)/NT_011786.15 | +/+ |
| 21-APC/435 kb | (5′) 384224-819689 (3′)/NT_034772.5 | −/+ | 52-HPRT1/159 bp | (5′) 314410-314569 (3′)/NT_011786.15 | −/− |
| 22-APC/737 kb | (5′) 529476-1266975 (3′)/NT_034772.5 | −/− | 53-LDLR/7.1 kb | (5′) 191462-198534 (3′)/NT_011295.10 | +/+ |
| 23-TSC2/8.4 kb | (5′) 417616-426044 (3′)/NT_037887.4 | +/− | 54-PAX6/1002 kb | (3′) 750100-1752166 (5′)/NT_009237.17 | −/+ |
| 24-TSC2/10.1 kb | (5′) 432071-442186 (3′)/NT_037887.4 | +/− | 55-PAX6/836 kb | (3′) 905528-1741689 (5′)/NT_009237.17 | −/− |
| 25-TSC2/916 bp | (5′) 437300-438216 (3′)/NT_037887.4 | +/+ | 56-IGHM/732 kb | (3′) 204471-936631 (5′)/NT_026437.11 | −/− |
| 26-TSC1/7.6 kb | (3′) 177985-185621 (5′)/NT_035014.4 | −/+ | 57-PKLR/1.1 kb | (5′) 9489-10630 (3′)/AY316591.1 | +/− |
| 27-ADA/3.2 kb | (3′) 500551-503808 (5′)/NT_011362.9 | +/+ | 58-LHCGR/6.1 kb | (5′) 63641-69726 (3′)/NG_008193.1 | −/+ |
| 28-WT1/7.3 kb | (3′) 32504-39823 (5′)/NC_000011.8 | −/+ | 59-F 8/19.9 kb | (5′) 105125-125067 (3′)/AY769950.1 | +/− |
| 29-DMD/7.8 kb | (3′) 152815-160602 (5′)/NT_011757.15 | +/− | 60-F 8/29.2 kb | (5′) 29349-58524 (3′)/AY769950.1 | −/− |
| 30-DMD/3.8 kb | (3′) 155384-159231 (5′)/NT_011757.15 | −/+ | 61-STS/40.1 kb | (5′) 32594-72652 (3′)/NT_011757.15 | −/− |
| 31-PTEN/453 kb | (5′) 452645-905607 (3′)/NT_030059.12 | +/+ | 62-WAS/1.9 kb | (5′) 2932-4803 (3′)/AF115549.2 | +/+ |
| 63-VWF/61 kb | (5′) 20561-81606 (3′)/NG_009072.1 | +/+ | 87-CFTR/1.5 kb | (5′) 184647-186177 (3′)/NC_000007.12 | −/− |
| 64-VWF/2.3 kb | (5′) 146608-148929 (3′)/NG_009072.1 | −/+ | 88-CFTR/21.1 kb | (5′) 18350-39431 (3′)/NC_000007.12 | +/+ |
| 65-VPS13B/1.8 kb | (5′) 8039-9822 (3′)/NG_007098.2 | +/+ | 89-CHRNE/1.3 kb | (3′) 1335845-1337134(5′)/NT_010823.11 | +/− |
| 66-SMN1/6.7 kb | (5′) 23935-30628 (3′)/NG_008691.1 | +/+ | 90-COL17A1/21 kb | (5′) 11200-32195 (3′)/NG_007069.1 | −/− |
| 67-SLC3A1/30.3 kb | (5′) 101289-131606 (3′)/AC013717.8 | +/+ | 91-CYBB/25.3 kb | (5′) 37250-62577 (3′)/NT_079573.3 | −/− |
| 68-SERPING/2.9 kb | (5′) 4337-7226 (3′)/X54486.1 | +/+ | 92-FANCA/2.1 kb | (3′) 20800-22859 (5′)/NT_010542.15 | +/+ |
| 69-SDHC/8.4 kb | (5′) 85491-93861 (3′)/AL592295.25 | +/+ | 93-FANCA/44.1 kb | (3′) 32415-76544 (5′)/NT_010542.15 | +/+ |
| 70-SALL4/8.9 kb | (3′) 23922-32811 (5′)/AL034420.16 | +/− | 94-FBN1/6.4 kb | (5′) 187965-194400 (3′)/NG_008805.1 | −/− |
| 71-PROS1/4.4 kb | (3′) 89888-94322 (5′)/NC_000003.10 | +/+ | 95-FBN1/7.1 kb | (5′) 197759-204893 (3′)/NG_008805.1 | −/+ |
| 72-PREPL/23.9 kb | (3′) 120275-144148 (5′)/AC013717.8 | −/+ | 96-FERMT1/3.9 kb | (5′) 70250-74168 (3′)/AL118505.17 | +/+ |
| 73-PREPL/38.1 kb | (3′) 123903-162024 (5′)/AC013717.8 | +/+ | 97-FGA/15.2 kb | (3′) 47644-62873 (5′)/AC107385.4 | −/− |
| 74-PKHD1/7.3 kb | (5′) 332887-340206 (3′)/AY129465.1 | −/− | 98-FOXL2/8.2 kb | (3′) 60868-69094 (5′)/AC092947.12 | −/− |
| 75-PKHD1/13.2 kb | (5′) 337078-350281 (3′)/AY129465.1 | −/+ | 99-GAA/8.3 kb | (5′) 97036-105323 (3′)/NT_024871.11 | −/− |
| 76-PKD1/2.9 kb | (5′) 18177-21076 (3′)/L39891.1 | −/− | 100-GBE1/105.7 kb | (3′) 119019-224705 (5′)/NC_000003.10 | −/+ |
| 77-PINK1/4.6 kb | (5′) 18515-23118 (3′)/NG_008164.1 | +/+ | 101-GHR/4.1 kb | (5′) 437912-442010 (3′)/NT_006576.15 | −/− |
| 78-PARK2/156 kb | (5′) 484956-641159 (3′)/NG_008289.1 | +/− | 102-GLA/4.5 kb | (3′) 30733-35251 (5′)/NT_011651.16 | +/+ |
| 79-OCA2/122.6 kb | (3′) 131887-254458 (5′)/NW_925783.1 | +/− | 103-GLA/4.6 kb | (3′) 36143-40797 (5′)/NT_011651.16 | +/+ |
| 80-NSD1/23.9 kb | (5′) 15361-39242 (3′)/NT_023133.12 | +/+ | 104-GLI3/176 kb | (3′)15719-53929(5′)/AC005026.2-AC005158.3 | +/− |
| 81-NSD1/8 kb | (5′) 170994-178991 (3′)/NT_023133.12 | +/+ | 105-GLI3/151 kb | (3′) 64730-77461 (5′)/AC005026.2-AC005158.3 | −/− |
| 82-MLC1/2.1 kb | (5′) 9586-11738 (3′)/NG_009162.1 | +/+ | 106-GLI3/1.01 Mb | (3′)154476-127754(5′)/AC012596.4- AC099798.4 | −/− |
| 83-ATP7A/15.2 kb | (3′) 12883-28093 (5′)/Z94801.1 | −/+ | 107-GLI3/728 kb | (3′) 149442-238453 (5′)/AC012596.4-AC005158.3 | +/− |
| 84-ATP7A/13.7 kb | (3′) 19564-33298 (5′)/Z94801.1 | +/+ | 108-GLI3/6.0 Mb | (3′) 7206-147487 (5′)/AC004844.1-AC005483.1 | −/+ |
| 85-ATP7A/13.7 kb | (3′) 31126-44864 (5′)/Z94801.1 | +/+ | 109-HBA1/11.2 kb | (5′) (31695-31724)-(42846-42867) (3′)/NG_000006.1 | +/+ |
| 86-AVPR2/21.5 kb | (5′) 54052-75566 (3′)/U52112.2 | +/− |
*5′ and 3′ deletion breakpoint sequences were obtained from HGMD and GRaBD. DNA sequences of gene contigs were downloaded from NCBI. DNA sequences of 5′ and 3′ breakpoints and related gene contigs were compared using NCBI-Blast, and breakpoint locations of gross gene deletions determined. LIRs within genomic regions that included gene breakpoint sequences were investigated. Abbreviations: Bp, base pair; GRaBD, gross rearrangement breakpoint database; HGMD, human gene mutation database; kb, kilobase; LIR, long inverted repeat; Mb, megabase.
Mean ranks of LIR numbers were compared between gross deletion breakpoints and control sequences using the Mann Whitney U test.The mean LIR number was significantly higher at the breakpoint regions from gross gene deletions, than in control group (P < 0.001).
In addition, associations between 5′ and 3′ LIR numbers within ±10 kb regions flanking each breakpoint were determined using Pearson's correlation coefficients. Positive, strongly significant association was found between LIR numbers from 5′ and 3′ breakpoints in 109 gross deletions (r = 0.85, P < 0.001).
Additionally, Spearman's correlation showed that a negative moderately significant associations were found between deletion size and 5′ LIR number (rs = −0.30, P < 0.003), and 3′ LIR number (rs = −0.30, P < 0.002) in 109 gross deletions respectively.
Features of LIRs selected within ±3 kb genomic regions flanking 5′ and 3′ deletion breakpoints
Next, LIRs were selected using appropriate criteria (outlined in Materials and Methods) (Figure 1c). Properties of these selected LIRs from 5′ and 3′ breakpoints were analysed. In total, 138 LIRs at distance of 0–3 kb from breakpoints, with stem length >20 bp, internal spacer of 0–2.5 kb, and stem identity >70% were detected (see Supplementary Table S3 online). The stem lengths and identities, internal spacer (loop) lengths and distances from breakpoints of LIRs were determined at the breakpoint locations of genes involved in gross deletions, including PINK1 (NG_008164.1; 5001-23057), ATM (U82828.1; 10722-156953), PTEN (NT_030059.12; 613176-718513) and BRCA1 (L78833.1; 3344-84436) (Figure 2).
Figure 2. Breakpoint regions of PINK1, ATM, PTEN and BRCA1 genes.

Sizes of LIR features, e.g. stem length, stem identity and internal spacer (loop length) are shown. NCBI accession numbers of each gene are provided. Coordinates correspond to GenBank sequences. (a) 3′ breakpoint sequence of the PINK1 deletion is in the downstream of gene. The LIR of PINK1 is located at the upstream of 1785 bp from 3′ breakpoint, has a stem length of 292 bp, internal spacer of 1736 bp, and stem identity of 82.65%. (b) 3′ breakpoint sequence of the ATM deletion is within the gene. The LIR of ATM is located at the 222 bp downstream of 3′ breakpoint, and has a stem length of 291 bp, internal spacer of 207 bp, and stem identity of 75.07%. (c) 5′ breakpoint sequence of the PTEN deletion is in the upstream of gene. The LIR of PTEN includes 5′ breakpoint, and has a stem length of 220 bp, internal spacer of 83 bp, and stem identity of 87.94%. (d) 5′ breakpoint sequence of the BRCA1 deletion is within the gene. The LIR of BRCA1 is located at the upstream of 632 bp from 5′ breakpoint, and has a stem length of 299 bp, internal spacer of 454 bp, and stem identity of 84.38%. Abbreviation: Bp, base pair; BP, breakpoint; LIR, long inverted repeat.
Distribution of LIRs at the 5′ and 3′ breakpoint regions from gross gene deletions
LIR distribution was also examined at the 218 genomic locations, including 5′ and 3′ breakpoint regions from 109 gross gene deletions (see Supplementary Figure S1 online). From these 218 locations, 138 LIRs were detected at the 5′ and/or 3′ breakpoints from 89 gross gene deletions (Figure 3a). Moreover, 49 of 89 gross deletions had 98 LIRs in both 5′ and 3′ deletion breakpoints (Fig. 3b), and 40 of 89 gross deletion had LIR at the 5′ or 3′ breakpoint sites (Figure 3c).
Figure 3. Distribution of 138 LIRs at the 5′ and 3′ BP regions of 109 gross gene deletions.

(a) LIRs were detected in 89 (81%) gene deletions. (b) In 49 of these deletions, LIRs were located at both 5′ and 3′ BP regions. (c) Among the 40 deletions with LIRs at one of the breakpoint regions, in 21, the LIRs were at the 5′ BP region, and in 19 deletions, at the 3′ BP region. Abbreviations: BP, breakpoint; LIR, long inverted repeat.
Correlations between LIR features such as length and identity of stem, internal spacer length and distance from breakpoint, and also deletion size in 89 gross gene deletions
In all identified 138 LIRs, at the 5′ and/or 3′ breakpoints in 89 (81%) of 109 gross deletions had stem lengths between 24 and 973 bp (see Supplementary Fig. S2 online), with stem identities between 70.54 and 100% (see Supplementary Figure S3 online). These LIRs were located at the distance of ±0–2,539 bp from 5′ and 3′ breakpoints (see Supplementary Fig. S4 online), with internal spacer lengths between 0 and 2,435 bp (see Supplementary Figure S5 online).
Associations between features of these LIRs were examined using Pearson's correlation coefficient. Low to moderately significant correlations were found between certain LIR features (e.g. stem length and identity, internal spacer length and distance from breakpoint). In all 138 LIRs located at the regions including 5′ and 3′ breakpoints, negative correlations were found between stem length and stem identity (r = −0.49, P < 0.001), internal spacer length and distance from breakpoint (r = −0.18, P < 0.05), stem length and distance from breakpoint (r = −0.18, P < 0.05), and internal spacer length and stem identity (r = −0.17, P < 0.05). Conversely, a moderately positive correlation was found between internal spacer length and stem length (r = 0.27, P < 0.002). No correlation was found between stem identity and distance from breakpoint (r = −0.008, P > 0.1).
Moreover, associations between gross gene deletion size and features of the 138 LIRs were analysed by Pearson's correlation coefficient. It was found that positive significant correlation between internal spacer length and deletion size (r = 0.19, P < 0.05). However, no correlations were found between deletion size and three other LIR features, specifically, stem length (r = 0.01, P > 0.1), stem identity (r = −0.06, P > 0.1), and distance from breakpoint (r = 0.08, P > 0.1).
In addition, 5′ and 3′ LIR features from 89 gross deletions were re-examined individually. Thus, associations between properties of 70 and 68 LIRs located on 5′ and 3′ breakpoints, respectively, and deletion size, were analysed by Pearson's coefficient. Negative moderate to strong correlations were found between internal spacer length and stem identity (r = −0.28, P < 0.02), and stem length and stem identity (r = −0.57, P < 0.001) for LIRs within 5′ breakpoints.
A positive moderate correlation was found between internal spacer length and stem length (r = 0.35, P < 0.004) for LIRs within 3′ breakpoints. In addition, negative moderate correlations were found between stem identity and stem length (r = −0.40, P < 0.002), and distance from breakpoint and stem length (r = −0.31, P < 0.02).
Furthermore, positive moderately significant correlation was found between internal spacer length of 3′ LIRs and deletion size (r = 0.29, P < 0.02). However, no correlation was found between internal spacer length of 5′ LIRs and deletion size (r = −0.16, P > 0.1). No correlations were found between deletion size and three other LIR features, specifically, stem length (5′: r = −0.06, P > 0.1; 3′: r = 0.04, P > 0.1), stem identity (5′: r = 0.12, P > 0.1; 3′: r = −0.10, P > 0.1), and distance from breakpoint (5′: r = 0.22, P > 0.05; 3′: r = 0.08, P > 0.1).
Correlations between stem length and identity, internal spacer length and distance from breakpoint, and also deletion size in 49 gross gene deletions including LIRs at both of 5′ and 3′ breakpoints
In addition, 98 LIRs were identified in both 5′ and 3′ breakpoints from 49 of 89 gross gene deletions (Figure 3b). These LIRs had stem identities of 71.65–100%, stem lengths of 27–603 bp, and internal spacer lengths of 0–2,435 bp (see Supplementary Table S3 and Figure S6 online). Features of these 98 LIRs were analysed using Pearson's correlation coefficient. Low to moderately significant correlations were found between certain LIR features, including stem length, stem identity, loop length and distance from breakpoint. Positive correlation was found between internal spacer length and stem length (r = 0.23, P < 0.05). Negative moderate correlations were found between stem identity and stem length (r = −0.39, P < 0.001), and distance from breakpoint and stem length (r = −0.31, P < 0.003). However, no correlations were found between internal spacer length and stem identity (r = −0.08, P > 0.1), internal spacer length and distance from breakpoint (r = −0.13, P > 0.1), and stem identity and distance from breakpoint (r = 0.06, P > 0.1).
Furthermore, 5′ and 3′ breakpoint regions of these 98 LIRs were examined individually. Associations between LIR features from 5′ and 3′ breakpoint locations and gross gene deletion size in 49 gross deletions were analysed by Pearson's correlation method. A negative moderate correlation was found between stem length and distance from breakpoint for LIRs in 5′ breakpoint regions (r = −0.30, P < 0.05). Negative moderate correlation was also found between stem length and distance from breakpoint for LIRs in 3′ breakpoint regions (r = −0.33, P < 0.05). Strong negative correlation was found between stem length and stem identity from 3′ LIRs (r = −0.51, P < 0.001). Positive moderate correlation was found between stem length and internal spacer length from 3′ LIRs (r = 0.36, P < 0.02).
In addition, the relationship between 5′ and 3′ LIRs were analysed. Positive moderate correlation was found between distance from breakpoint for 5′ LIRs and stem identity of 3′ LIRs, involving 49 gross gene deletion regions (r = 0.28, P < 0.05).
Associations between deletion size and 5′ and 3′ LIR features from these 49 gross deletions were also analysed by Pearson's correlation method. Negative moderate correlation was found between stem identity of 5′ LIRs and deletion size (r = −0.40, P < 0.005). Positive moderate correlation was found between stem identity of 3′ LIRs and deletion size (r = 0.30, P < 0.05). However, no correlations were found between deletion size and loop length (5′: r = −0.04, P > 0.1; 3′: r = 0.04, P > 0.1), stem length (5′: r = 0.17, P > 0.1; 3′: r = −0.18, P > 0.1), and distance from breakpoint (5′: r = 0.11, P > 0.1; 3′: r = −0.10, P > 0.1).
Correlations between length and identity of stem, internal spacer length and deletion size in 40 gross gene deletions including LIRs spanning 5′ or 3′ breakpoints
LIRs were also identified at the other 40 gross gene deletions containing LIRs in 5′ or 3′ breakpoints. In total, 21 and 19 of the 40 LIRs from 5′ and 3′ breakpoint sites, respectively, were analysed (Figure 3c). LIRs had stem identities of 70.54–100%, stem lengths of 24–973 bp, and internal spacer lengths of 0–2,422 bp, and were located at the distance of 0–2,311 bp from breakpoints (see Supplementary Table S3 online).
Associations between LIR features were analysed by Pearson's correlation coefficient. Moderate to strong significant correlations were found between LIR features, including stem length and stem identity, and internal spacer length and distance from breakpoint. In 40 LIRs, a positive moderate correlation was found between internal spacer length and stem length (r = 0.34, P < 0.05). In addition, negative correlations were found between internal spacer length and stem identity (r = −0.33, P < 0.05), and stem length and stem identity (r = −0.61, P < 0.001). However, no correlations were found between distance from breakpoint and internal spacer length (r = −0.25, P > 0.1), stem length (r = −0.02, P > 0.1), or stem identity (r = −0.12, P > 0.1).
Deletion size and LIR features were also analysed by Pearson's correlation method. A positive moderate correlation was found between internal spacer length of LIRs and deletion size (r = 0.35, P < 0.05). However, no correlations were found between deletion size and three other LIR features, specifically, stem length (r = 0.00, P > 0.1), stem identity (r = −0.08, P > 0.1), and distance from breakpoint (r = 0.09, P > 0.1).
Re-examination of LIRs detected in only one of regions spanning 5′ and 3′ breakpoints of gross gene deletions
The 40 gross gene deletions containing LIRs in only one of genomic regions spanning 5′ and 3′ breakpoints were re-examined in terms of their ability to form new LIRs between breakpoints with LIRs and non LIRs in related deletion regions. LIRs with distance of 0–10 kb from breakpoints, stem identity >70% and stem length >150 bp were analysed.
In 24 of the 40 gross deletions, new LIRs between 5- and 10-kb genomic segments from 5′ and 3′ breakpoints containing LIR or no LIR, respectively, were found (see Supplementary Table S4 online; Figure 4). From these 24 gross deletions, LIR stem identities and lengths were determined to be 70.19–86.66% and 173–1789 bp, respectively (Figure 5). In addition, these LIRs were located at distance of ±42–9,330 bp from breakpoints (Figure 5).
Figure 4. Identification of new LIRs between breakpoint regions of gross gene deletions including LIR at only one of the 5′ and 3′ BPs.
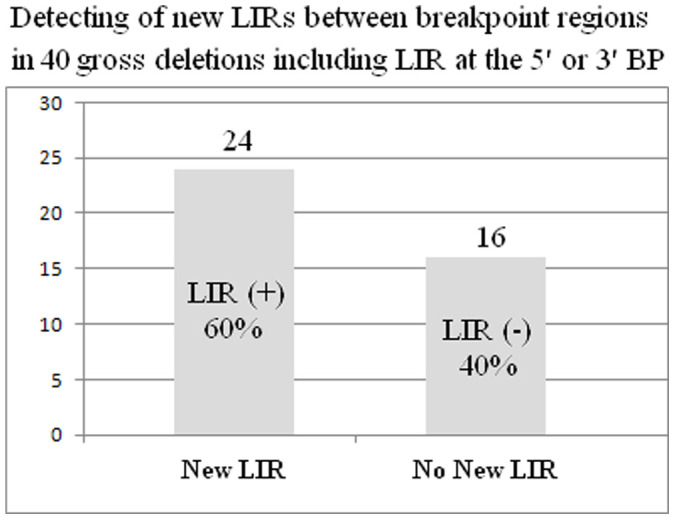
New LIRs were detected between genomic sequences flanking breakpoints in 24 of the 40 gross deletions including LIR at the 5′ or 3′ BP. Abbreviations: BP, breakpoint; LIR, long inverted repeat.
Figure 5. In 24 gross deletion, new identified LIRs with stem identity, stem length and distance from breakpoint were shown.
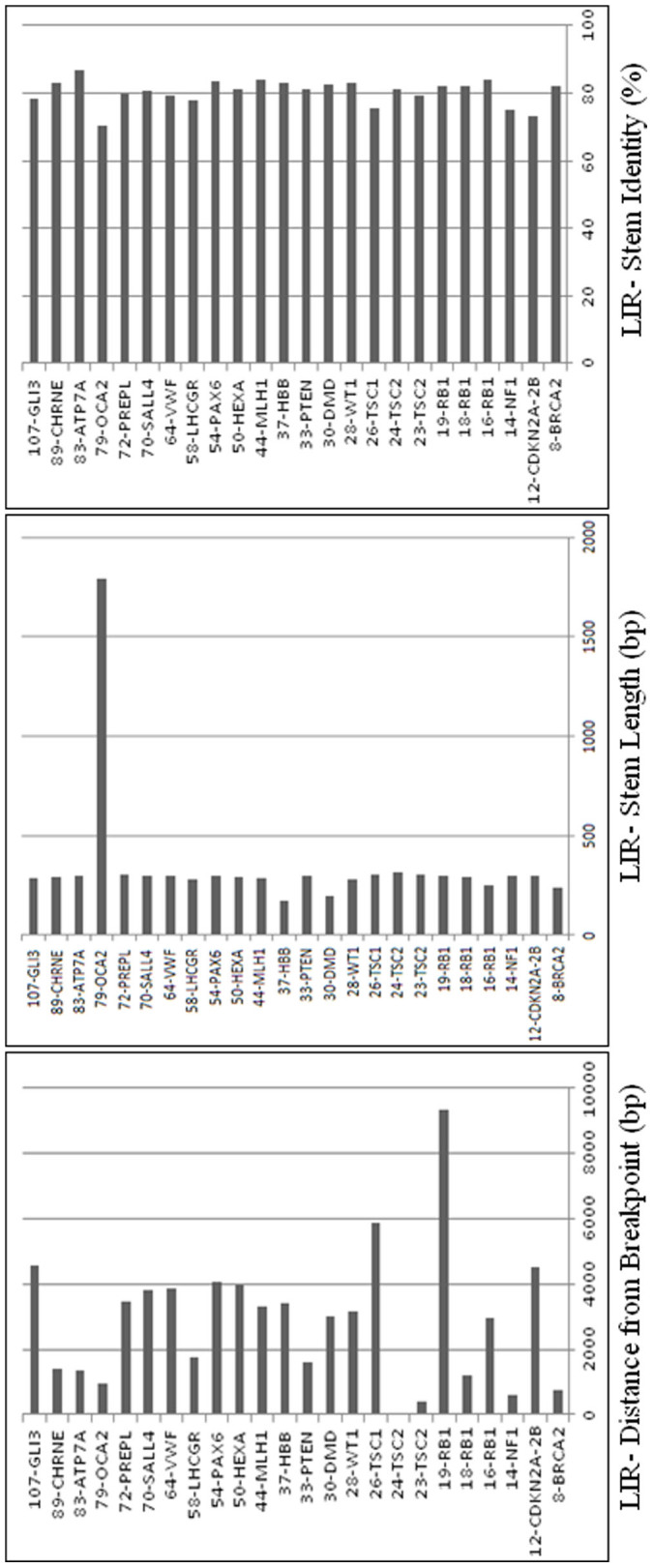
Black bars indicate stem length, stem identity and distance from breakpoint of LIRs found between distant sites. Abbreviations: Bp, base pair; LIR, long inverted repeat.
Features of these 24 LIRs were analysed by Spearman's correlation method. A strong significantly negative correlation was found between stem length and stem identity (rs = −0.51, P < 0.02). No correlations were found between distance from breakpoint and stem length (rs = −0.08, P > 0.1) or stem identity (rs = −0.08, P > 0.1).
Discussion
Deletion breakpoints are often associated with Alu and non-B DNA-forming elements such as short direct and inverted repeats, and inversions of inverted repeats in human genomic rearrangements17,19,20,21,22,23. In this study, LIRs within ±10 kb regions flanking 218 breakpoint sequences from gross gene deletions in human cancers and inherited diseases, were investigated by using IRF94,95 software. As a program that uses an algorithm presented by Benson95, IRF software can efficiently detect two or more contiguous approximate inverted repeats in sizes up to 700 kb at the same location on DNA sequences without the need to specify either the pattern or pattern size. In this way, IRF software served that present study accurately analyzes significance of relationship between LIR numbers and breakpoint regions in human gross gene deletions.
This work showed that the mean LIR number was significantly higher at the breakpoint regions of gross gene deletions, than in control group (P < 0.001). In addition, strongly significant positive correlation was found between 5′ and 3′ LIR numbers from breakpoint regions (r = 0.85, P < 0.001). In this regards, increasing LIR numbers can cause or induce chromosomal rearrangements (including duplication, recombination and/or deletion) in human genome during evolutionary process.
Furthermore, negative moderately significant associations were found between deletion size and 5′ and 3′ LIR numbers (rs = −0.30, P < 0.003; rs = −0.30, P < 0.002) in 109 gross deletions, respectively. This result indicates that increasing 5′ or 3′ LIR numbers at the breakpoints cause smaller deletion sizes. Over-LIR intensity may impede efficiency, strengthens and further kinetic properties of inverted repeats because of competing LIRs with each other. Consequently, these findings suggest that DNA sequence evolution may also be prosecuted by LIRs in human genome.
In Saccharomyces cerevisiae, Saini et al. reported that IRs induce mutagenesis by break formation at distant sites (up to 8 kb)96. Similarly, Lobachev et al. suggested that LIRs may stimulate recombination and deletion by forming secondary structures on the single strand DNA during replication26. In addition, Bacolla and Wells indicated that repetitive DNA motifs may fold into non-B DNA structures including cruciforms/hairpins, leading to genomic rearrangements associated with neurodegenerative and genomic disorders18.
In 138 LIRs identified in 89 gross deletion, significant associations were found between internal spacer length and distance from breakpoint (r = −0.18, P < 0.05), stem length and distance from breakpoint (r = −0.18, P < 0.05). These associations suggest DNA strand breaks potentially in locations close to larger LIRs. Similarly, Lobachev et al. reported that stimulation of deletions was positively correlated with IR size26. In addition, Lim et al. reported that IRs ≥ 800 bp are required for gene deletion effectiveness in Saccharomyces cerevisiae, showing IRs improve gene deletion efficiency up to 1.2 kb97.
In addition, a positive significant correlation between internal spacer length and deletion size in 138 LIRs was found (r = 0.19, P < 0.05), suggesting LIRs with bigger loops cause larger deletions at fragile DNA sites. Weiss and Wilson reported that loops with 25–247 nucleotides (nt) were efficiently and accurately repaired during homologous recombination98. It was suggested that bigger loops (>247 nt) cannot repair and excise in homologous recombination accurately, therefore cells with these loops may be subject to either apoptosis or NHEJ. If cells cannot induce apoptosis, it was suggested that LIRs > 247 nt may break DNA, and be repaired by NHEJ.
In conclusion, larger deletions may more efficiently form by LIRs with larger loops at 5′ or 3′ breakpoints in human cancers and inherited diseases. DNA end may gain further kinetic properties, and match with distant brekpoint site (Figure 6).
Figure 6. A model mechanism for single LIR-mediated gene deletion.

LIR forming cruciform structure in the single strand DNA nearing 3′ breakpoint of the gross gene deletion during replication is shown. After the first break is occurred in the vicinity of 3′ LIR, second break is induced by back-folded stem-loop structures forming with homolog sequences between distant 5′ and 3′ breakpoint sites. Free DNA ends may combine via 53BP1-mediated NHEJ. Abbreviations: LIR, long inverted repeat; NHEJ, non-homologous end-joining.
Moreover, correlation between distance from breakpoint and stem length (r = −0.31, P < 0.02) was observed in 3′ LIRs from 89 gross deletions. These data suggest that DNA strand is potentially broken in locations closer to 3′ LIRs with larger stem lengths. In addition, a positive moderately significant correlation was found between deletion size and internal spacer length of 3′ LIRs (r = 0.29, P < 0.02), with no correlation between internal spacer length of 5′ LIRs (r = −0.16, P > 0.1). These results show that 3′ LIRs with bigger loops are more important than 5′ LIRs, for larger gross deletions in human genome.
Similarly, associations between deletion size and stem identities of 5′ (r = −0.40, P < 0.005) and 3′ (r = 0.30, P < 0.05) LIRs were found in 49 gross deletions including LIR on the both of 5′ and 3′ breakpoints. These data suggest that 3′ LIRs with greater stem identities cause larger deletion sizes, while similar 5′ LIRs cause smaller deletion sizes. Furthermore, a association between distance from breakpoint of 5′ LIRs and stem identity of 3′ LIRs (r = 0.28, P < 0.05) was also found, suggesting 3′ LIRs with greater stem identities are more likely to induce DNA breakage than 5′ LIRs.
Consequently, LIRs may induce DNA breakages at the nearby locations through forming cruciform structures. Free DNA ends between distant sites may come together by NHEJ, with following gene deletion (Figure 7). Similarly, Varga and Aplan reported that DNA breaks produced various deletions exhibiting NHEJ features in the human monocytic cell line, U93799. They showed that aberrant double-strand break repair by NHEJ may lead to gross chromosomal rearrangements including interstitial deletion and large insertions.
Figure 7. A model mechanism for 5′ and 3′ LIRs-mediated gene deletion.

Cruciform structures of LIRs are formed on DNA strands during replication, with breaks potentially occurring inside LIR or near locations. LIR-induced breakages at the 5′ and 3′ breakpoint sequences may cause gene deletion by enabling free DNA ends to recombine via 53BP1-mediated NHEJ. Abbreviations: LIR, long inverted repeat; NHEJ, non-homologous end-joining.
In 40 gross deletions containing 5′ or 3′ LIR, positive moderate correlation between internal spacer length and deletion size (r = 0.35, P < 0.05) was found, similar to the group that included 138 LIRs. In addition, in 24 of 40 gross deletions, new LIRs between distant free ends containing LIR and no LIR were detected (Figures 4 and 5). These results show that LIRs with bigger loops cause larger deletions in human genome, suggesting that larger loops may give rise to greater stress and transition activity on the DNA strand during replication. Moreover, it was reported that bigger inverted repeats can dominate strand separation and B-Z transition, with Zhabinskaya and Benham, showing that long IRs occupy clinically important chromosomal breakpoints corresponded closely with translocation frequencies through probably cruciform extrusion100.
In conclusion, these results suggest that a LIR found in 5′ or 3′ breakpoints, may break DNA strand via cruciform structure and match with homolog sequences in other breakpoint site, resulting in a back-folded stem-loop structure during replication (Figure 6). In this way, DNA breakage may also occur in other breakpoint location containing no LIR. After double-strand breakages are formed at 5′ and 3′ breakpoints, DNA ends between distant sites may combine by NHEJ, with following gene deletion.
As presented in Fig. 6, this model is supported with a study carried out in Saccharomyces cerevisiae101. In this study, IRs with internal spacer of 21 kb were placed into Saccharomyces cerevisiae chromosome. After double-strand break was induced, large dicentric inverted dimers were observed, leading to gross chromosomal rearrangements during anaphase stage. In addition, it has been suggested that p53-binding protein 1 (53BP1) combines free DNA ends between distant sites for NHEJ102.
An algorithm such as internal spacer <2 kb, stem copy identity >85% and stem length >30 bp for recombinogenic LIRs in human and other organism genomes was suggested2. In the present study, only 35 (25.36%) of 138 LIRs located close to the 5′ and 3′ breakpoints from 89 gross deletions, correspond to this criteria (see Supplementary Table S3 online). However, the present findings indicate that significant relationship between LIR numbers and breakpoint regions of gross gene deletions. There is also a strongly positive correlation between 5′ and 3′ LIR numbers on breakpoint regions. On the other hand, 5′ and 3′ LIRs may have converse effects on deletion size. However, over-LIR intensity on 5′ or 3′ breakpoint locations cause smaller deletion sizes. In addition, this study showed that 3′ LIRs may be more active than 5′ LIRs in deletional and recombinational events. Moreover, internal spacer length affects breakage site and deletion size in the gross deletions. Therefore, the present study suggests necessity of a new algorithm for LIRs in breakpoint regions of gross gene deletions associated with human cancers and inherited genetic diseases.
Consequently, LIRs detected in genomic regions including breakpoint sequences of many gross gene deletions, may lead to cruciform structure formation during DNA replication and break DNA strand. After double-strand breaks occur in 5′ and 3′ breakpoints, gene deletions may be formed by combining free DNA ends with 53BP1 for NHEJ.
Methods
Gross gene deletions and breakpoint regions
In total, 109 gross gene deletions involving 63 genes, were obtained from the Human Gene Mutation Database (HGMD)90,91 (see Supplementary Table S1 online). Base sequences of 5′ and 3′ deletion breakpoints were taken from references 33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89 listed in the HGMD90, or obtained from the Gross Rearrangement Breakpoint Database (GRaBD)92,93 (see Supplementary Table S1 online). Sequences of genes associated with deletions were downloaded from NCBI103. Gene accession numbers are provided (Table 1). Each deletion breakpoint sequence and corresponding genes were compared using NCBI BLAST104, and breakpoint locations matched with related genes (Figure 1a). For each gene deletion, nucleotide positions of 5′ and 3′ breakpoints are shown (Table 1). Sequences (±10 kb) spanning 5′ and 3′ breakpoints of gross gene deletions were included in the deletion group (see Supplementary Table S2 online). In total, 218 breakpoint sequences from 109 gross gene deletions were examined for LIR identification (Figure 1b).
For the control group, the DNA sequences of 68 different genes were downloaded from NCBI103 to be selected randomly (see Supplementary Table S2 online). Searching the HGMD90 site confirmed that selected control genes were not associated with deletions. Subsequently, 20 kb segments of DNA sequence from each control gene were included in the control group. In total, 220 control sequences were examined for LIR identification.
LIR identification
Identification of LIRs was performed within genomic regions (including the 218 breakpoint sequences from 109 gross gene deletions of 63 genes, and 220 control sequences from 68 genes) using IRF94,95 software (Figure 1b). The 2, 3, 5 and 40 (match, mismatch, indel and minimum score) parameters of IRF94 were selected for identification.
LIRs with stem length >20 bp, internal spacer of 0–10 kb, stem identity ≥70%, and within ±10 kb fragments flanking each of the 5′ and 3′ breakpoint sequences of human gross gene deletions, or 20 kb segments of control genes, were investigated (Figure 1b). Total LIR numbers were determined (see Supplementary Table S2 online) and statistically compared between control and deletion groups. In addition, associations between LIR numbers on 5′ and 3′ breakpoints and also deletion size were statistically investigated.
Recently, Wang and Leung reported that LIRs with stem length >30 bp, stem identity >85% and internal spacer <2 kb were highly recombinogenic in humans and other organisms2. It was also shown that long Alu IRs with 75% stem identity caused mild replication blockage in E. coli3. Thus, LIRs with distance of 0–3 kb from breakpoints, stem length >20 bp, internal spacer of 0–2.5 kb, and stem identity ≥70%, were selected for determining associations between LIR features, distances from breakpoint and deletion size (see Supplementary Table S3 online; Figure 1c). At this stage, if many LIRs were observed in the same breakpoint region, the one which best fits the above criteria was chosen.
In addition, 40 of 109 gross gene deletions containing LIRs in only one of regions flanking 5′ and 3′ breakpoints, were further examined. The capacity to form new LIRs between breakpoints with LIRs and other breakpoint sites (including non LIRs of related deletion regions) was researched using IRF94.
For this, 5 kb of DNA sequence from breakpoints containing LIRs, and 10 kb of DNA sequence including other breakpoints but containing no LIRs, were combined before scanning for LIRs using IRF94. During this process, deleted gross genes were excluded and combined DNA sequences used. LIRs with stem length >150 bp and >70% stem identity were selected for determining associations between LIR features and distance from breakpoints (see Supplementary Table S4 online).
Statistical analysis
Mann-Whitney U test was used for statistical comparison of mean ranks of LIR numbers between gross gene deletion and control groups. Pearson's (r) and Spearman's (rs) correlation coefficients were used to examine associations between LIR features (stem length and identity, and loop length), and distance from breakpoint and gene deletion size. In addition, Pearson's and Spearman's correlation coefficients were also used for determining associations between deletion size and 5′ and 3′ LIR numbers within ±10 kb sequence spanning each breakpoint in 109 gross deletions. Correlation coefficients (r, rs) were classified according to criteria as low (0.00–0.24), moderate (0.25–0.49), strong (0.50–0.74) and strongly (0.75–1.00)105. Two-sided P values < 0.05 were considered statistically significant. All analyses were performed using SPSS 11.0 software (Chicago, USA).
Author Contributions
N.A. conceived the study and performed the bioinformatics methods and analysed the statistical data using SPSS 11.0 software, wrote the manuscript and revised it, and prepared all figures and tables.
Supplementary Material
Supplementary information
Acknowledgments
I thank Ozden Ulker for his critical review. In addition, I thank to EDANZGROUPG (www.edanzediting.com) for copy editing of the manuscript.
References
- Warburton P. E., Giordano J., Cheung F., Gelfand Y. & Benson G. Inverted repeat structure of the human genome: The X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 14, 1861–1869 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. & Leung F. C. C. Long inverted repeats in eukaryotic genomes: Recombinogenic motifs determine genomic plasticity. FEBS Lett. 580, 1277–1284 (2006). [DOI] [PubMed] [Google Scholar]
- Voineagu I., Narayanan V., Lobachev K. S. & Mirkin S. M. Replication stalling at unstable inverted repeats: Interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. USA 105, 9936–9941 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akgün E. et al. Palindrome resolution and recombination in the mammalian germ line. Mol. Cell Biol. 17, 5559–5570 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham C. J., Savitt A. G. & Bauer W. R. Extrusion of an imperfect palindrome to a cruciform in superhelical DNA: Complete determination of energetics using a statistical mechanical model. J. Mol. Biol. 316, 563–581 (2002). [DOI] [PubMed] [Google Scholar]
- Gordenin D. A. et al. Inverted DNA repeats: a source of eukaryotic genomic instability. Mol. Cell Biol. 13, 5315–5322 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag D. K. & Kurst A. A 140-bp-long palindromic sequence induces double-strand breaks during meiosis in the yeast Saccharomyces cerevisiae. Genetics 146, 835–847 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya M. et al. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum. Genet. 102, 591–597 (1998). [DOI] [PubMed] [Google Scholar]
- Albrecht P. et al. Spectrum of gross deletions and insertions in the RB1 gene in patients with retinoblastoma and association with phenotypic expression. Hum. Mutat. 26, 437–445 (2005). [DOI] [PubMed] [Google Scholar]
- Férec C. et al. Gross genomic rearrangements involving deletions in the CFTR gene: characterization of six new events from a large cohort of hitherto unidentified cystic fibrosis chromosomes and meta-analysis of the underlying mechanisms. Eur. J. Hum. Genet. 14, 567–576 (2006). [DOI] [PubMed] [Google Scholar]
- Preisler-Adams S. et al. Gross rearrangements in BRCA1 but not BRCA2 play a notable role in predisposition to breast and ovarian cancer in high-risk families of German origin. Cancer Genet. Cytogenet. 168, 44–49 (2006). [DOI] [PubMed] [Google Scholar]
- Ashton E. J., Yau S. C., Deans Z. C. & Abbs S. J. Simultaneous mutation scanning for gross deletions, duplications and point mutations in the DMD gene. Eur. J. Hum. Genet. 16, 53–61 (2008). [DOI] [PubMed] [Google Scholar]
- Owens M., Ellard S. & Vaidya B. Analysis of gross deletions in the MEN1 gene in patients with multiple endocrine neoplasia type 1. Clin. Endocrinol. (Oxf) 68, 350–354 (2008). [DOI] [PubMed] [Google Scholar]
- Lupski J. R. & Stankiewicz P. Genomic disorders: Molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 1, e49; 10.1371/pgen0010049 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W., Zhang F. & Lupski J. R. Mechanisms for human genomic rearrangements. Pathogenetics 1, 4 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw C. J. & Lupski J. R. Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum. Mol. Genet. 13, R57–R64 (2004). [DOI] [PubMed] [Google Scholar]
- Bacolla A. et al. Breakpoints of gross deletions coincide with non-B DNA conformations. Proc. Natl. Acad. Sci. USA 101, 14162–14167 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A. & Wells R. D. Non-B DNA conformations as determinants of mutagenesis and human disease. Mol. Carcinog. 48, 273–285 (2009). [DOI] [PubMed] [Google Scholar]
- Krawczak M. & Cooper D. N. Gene deletions causing human genetic disease: mechanisms of mutagenesis and the role of the local DNA sequence environment. Hum. Genet. 86, 425–441 (1991). [DOI] [PubMed] [Google Scholar]
- Canning S. & Dryja T. P. Short, direct repeats at the breakpoints of deletions of the retinoblastoma gene. Proc. Natl. Acad. Sci. USA 86, 5044–5048 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton J. C. et al. Is gene deletion in eukaryotes sequence-dependent? A study of nine deletion junctions and nineteen other deletion breakpoints in intron 7 of the human dystrophin gene. Gene 222, 41–51 (1998). [DOI] [PubMed] [Google Scholar]
- Chuzhanova N., Abeysinghe S. S., Krawczak M. & Cooper D. N. Translocation and gross deletion breakpoints in human inherited disease and cancer II: Potential involvement of repetitive sequence elements in secondary structure formation between DNA ends. Hum. Mutat. 22, 245–251 (2003). [DOI] [PubMed] [Google Scholar]
- Xie F. et al. A novel Alu-mediated 61-kb deletion of the von Willebrand factor (VWF) gene whose breakpoints co-locate with putative matrix attachment regions. Blood Cells Mol. Dis. 36, 385–391 (2006). [DOI] [PubMed] [Google Scholar]
- Vissers L. E. L. M. et al. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 18, 3579–3593 (2009). [DOI] [PubMed] [Google Scholar]
- Zhang F., Carvalho C. M. B. & Lupski J. R. Complex human chromosomal and genomic rearrangements. Trends Genet. 25, 298–307 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobachev K. S. et al. Factors affecting inverted repeat stimulation of recombination and deletion in Saccharomyces cerevisiae. Genetics 148, 1507–1524 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichman B. F., Vargason J. M., Mooers B. H. M. & Ho P. S. The Holliday junction in an inverted repeat DNA sequence: Sequence effects on the structure of four-way junctions. Proc. Natl. Acad. Sci. USA 97, 3971–3976 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck C. M., Rinehart F. P. & Schmid C. W. A ubiquitous family of repeated DNA sequences in the human genome. J. Mol. Biol. 132, 289–306 (1979). [DOI] [PubMed] [Google Scholar]
- Biezunski N. Structure and distribution of inverted repeats (palindromes). I. Analysis of DNA of Drosophila melanogaster. Chromosoma 84, 87–109 (1981a). [DOI] [PubMed] [Google Scholar]
- Biezunski N. Structure and distribution of inverted repeats (palindromes). II. Analysis of DNA of the mouse. Chromosoma 84, 111–129 (1981b). [DOI] [PubMed] [Google Scholar]
- Russell G. C. & Mann N. H. Analysis of inverted repeat DNA in the genome of Rhodomicrobium vannielii. J. Gen. Microbiol. 132, 325–330 (1986). [DOI] [PubMed] [Google Scholar]
- Collick A. et al. Instability of long inverted repeats within mouse transgenes. EMBO J. 15, 1163–1171 (1996). [PMC free article] [PubMed] [Google Scholar]
- Montagna M. et al. Genomic rearrangements account for more than one-third of the BRCA1 mutations in northern Italian breast/ovarian cancer families. Hum. Mol. Genet. 12, 1055–1061 (2003). [DOI] [PubMed] [Google Scholar]
- Tancredi M. et al. Haplotype analysis of BRCA1 gene reveals a new gene rearrangement: characterization of a 19.9 kbp deletion. Eur. J. Hum. Genet. 12, 775–777 (2004). [DOI] [PubMed] [Google Scholar]
- Agata S. et al. Prevalence of BRCA1 genomic rearrangements in a large cohort of Italian breast and breast/ovarian cancer families without detectable BRCA1 and BRCA2 point mutations. Genes Chromosomes Cancer 45, 791–797 (2006). [DOI] [PubMed] [Google Scholar]
- Agata S. et al. Large genomic deletions inactivate the BRCA2 gene in breast cancer families. J. Med. Genet. 42, e64; 10.1136/jmg032789 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier I. et al. Significant contribution of germline BRCA2 rearrangements in male breast cancer families. Cancer Res. 64, 8143–8147 (2004). [DOI] [PubMed] [Google Scholar]
- Vandenbroucke I. et al. Genetic and clinical mosaicism in a patient with neurofibromatosis type 1. Hum. Genet. 114, 284–290 (2004). [DOI] [PubMed] [Google Scholar]
- Takahashi M. et al. Detection of APC gene deletion by double competitive polymerase chain reaction in patients with familial adenomatous polyposis. Int. J. Oncol. 29, 413–421 (2006). [PubMed] [Google Scholar]
- Longa L. et al. TSC1 and TSC2 deletions differ in size, preference for recombinatorial sequences, and location within the gene. Hum. Genet. 108, 156–166 (2001). [DOI] [PubMed] [Google Scholar]
- Tadokoro K. et al. Intragenic homozygous deletion of the WT1 gene in Wilms' tumor. Oncogene 7, 1215–1221 (1992). [PubMed] [Google Scholar]
- Gualandi F. et al. Multiple exon skipping and RNA circularisation contribute to the severe phenotypic expression of exon 5 dystrophin deletion. J. Med. Genet. 40, e100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibon F. et al. Contribution of PTEN large rearrangements in Cowden disease: a multiplex amplifiable probe hybridisation (MAPH) screening approach. J. Med. Genet. 45, 657–665 (2008). [DOI] [PubMed] [Google Scholar]
- Kikuchi M., Ohkura N., Yamaguchi K., Obara T. & Tsukada T. Gene dose mapping delineated boundaries of a large germline deletion responsible for multiple endocrine neoplasia type 1. Cancer Lett. 208, 81–88 (2004). [DOI] [PubMed] [Google Scholar]
- Jo E. K. et al. Identification of mutations in the Bruton's tyrosine kinase gene, including a novel genomic rearrangements resulting in large deletion, in Korean X-linked agammaglobulinemia patients. J. Hum. Genet. 48, 322–326 (2003). [DOI] [PubMed] [Google Scholar]
- Palena A., Blau A., Stamatoyannopoulos G. & Anagnou N. P. Eastern European (delta beta) zero-thalassemia: molecular characterization of a novel 9,1-kb deletion resulting in high levels of fetal hemoglobin in the adult. Blood 83, 3738–3745 (1994). [PubMed] [Google Scholar]
- Mitui M. et al. Independent mutational events are rare in the ATM gene: haplotype prescreening enhances mutation detection rate. Hum. Mutat. 22, 43–50 (2003). [DOI] [PubMed] [Google Scholar]
- Li L. et al. Distinct patterns of germ-line deletions in MLH1 and MSH2: the implication of Alu repetitive element in the genetic etiology of Lynch syndrome (HNPCC). Hum. Mutat. 27, 388 (2006). [DOI] [PubMed] [Google Scholar]
- van der Klift H. et al. Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC). Genes Chromosomes Cancer 44, 123–138 (2005). [DOI] [PubMed] [Google Scholar]
- Myerowitz R. & Hogikyan N. D. A deletion involving Alu sequences in the beta-hexosaminidase alpha-chain gene of French Canadians with Tay-Sachs disease. J. Biol. Chem. 262, 15396–15399 (1987). [PubMed] [Google Scholar]
- Lauderdale J. D., Wilensky J. S., Oliver E. R., Walton D. S. & Glaser T. 3′ deletions cause aniridia by preventing PAX6 gene expression. Proc. Natl. Acad. Sci. USA 97, 13755–13759 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zelm M. C. et al. Gross deletions involving IGHM, BTK, or Artemis: a model for genomic lesions mediated by transposable elements. Am. J. Hum. Genet. 82, 320–332 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baronciani L. & Beutler E. Molecular study of pyruvate kinase deficient patients with hereditary nonspherocytic hemolytic anemia. J. Clin. Invest. 95, 1702–1709 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromoll J., Eiholzer U., Nieschlag E. & Simoni M. Male hypogonadism caused by homozygous deletion of exon 10 of the luteinizing hormone (LH) receptor: differential action of human chorionic gonadotropin and LH. J. Clin. Endocrinol. Metab. 85, 2281–2286 (2000). [DOI] [PubMed] [Google Scholar]
- Shibata M. et al. An alloantibody recognizing the FVIII A1 domain in a patient with CRM reduced haemophilia A due to deletion of a large portion of the A1 domain DNA sequence. Thromb. Haemost. 84, 442–448 (2000). [PubMed] [Google Scholar]
- Imai K. et al. Clinical course of patients with WASP gene mutations. Blood 103, 456–464 (2004). [DOI] [PubMed] [Google Scholar]
- Peake I. R. et al. Severe type III von Willebrand's disease caused by deletion of exon 42 of the von Willebrand factor gene: family studies that identify carriers of the condition and a compound heterozygous individual. Blood 75, 654–661 (1990). [PubMed] [Google Scholar]
- Seifert W. et al. Mutational spectrum of COH1 and clinical heterogeneity in Cohen syndrome. J. Med. Genet. 43, e22; 10.1136/jmg039867 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B. et al. Quantitative analysis of survival motor neuron copies: Identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am. J. Hum. Genet. 64, 1340–1356 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeken J. et al. Deletion of PREPL, a gene encoding a putative serine oligopeptidase, in patients with hypotonia-cystinuria syndrome. Am. J. Hum. Genet. 78, 38–51 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche O. et al. Hereditary angioedema: the mutation spectrum of SERPING1/C1NH in a large Spanish cohort. Hum. Mutat. 26, 135–144 (2005). [DOI] [PubMed] [Google Scholar]
- Baysal B. E. et al. An Alu-mediated partial SDHC deletion causes familial and sporadic paraganglioma. J. Med. Genet. 41, 703–709 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borozdin W. et al. SALL4 deletions are a common cause of Okihiro and acro-renal-ocular syndromes and confirm haploinsufficiency as the pathogenic mechanism. J. Med. Genet. 41, e113; 10.1136/jmg019901 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidel D. K. et al. A 5,3-kb deletion including exon XIII of the protein S α gene occurs in two protein S-deficient families. Blood 77, 551–559 (1991). [PubMed] [Google Scholar]
- Bergmann C. et al. Multi-exon deletions of the PKHD1 gene cause autosomal recessive polycystic kidney disease (ARPKD). J. Med. Genet. 42, e63; 10.1136/jmg032318 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R. et al. Identification of mutations in the repeated part of the autosomal dominant polycystic kidney disease type 1 gene, PKD1, by long-range PCR. Am. J. Hum. Genet. 65, 39–49 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology 64, 1955–1957 (2005). [DOI] [PubMed] [Google Scholar]
- Clarimon J. et al. Defining the ends of Parkin exon 4 deletions in two different families with Parkinson's disease. Am. J. Med. Genet. B. Neuropsychiatr Genet. 133B, 120–123 (2005). [DOI] [PubMed] [Google Scholar]
- Yi Z. et al. A 122,5-kilobase deletion of the P gene underlies the high prevalence of oculocutaneous albinism type 2 in the Navajo population. Am. J. Hum. Genet. 72, 62–72 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas J. et al. Partial NSD1 deletions cause 5% of Sotos syndrome and are readily identifiable by multiplex ligation dependent probe amplification. J. Med. Genet. 42, e56; 10.1136/jmg031930 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leegwater P. A. et al. Identification of novel mutations in MLC1 responsible for megalencephalic leukoencephalopathy with subcortical cysts. Hum. Genet. 110, 279–283 (2002). [DOI] [PubMed] [Google Scholar]
- Poulsen L. et al. X-linked recessive Menkes disease: identification of partial gene deletions in affected males. Clin. Genet. 62, 449–457 (2002). [DOI] [PubMed] [Google Scholar]
- Schöneberg T. et al. Compound deletion of the rhoGAP C1 and V2 vasopressin receptor genes in a patient with nephrogenic diabetes insipidus. Hum. Mutat. 14, 163–174 (1999). [DOI] [PubMed] [Google Scholar]
- Audrézet M. P. et al. Genomic rearrangements in the CFTR gene: extensive allelic heterogeneity and diverse mutational mechanisms. Hum. Mutat. 23, 343–357 (2004). [DOI] [PubMed] [Google Scholar]
- Dörk T. et al. Characterization of a novel 21-kb deletion, CFTRdele2,3(21 kb), in the CFTR gene: a cystic fibrosis mutation of Slavic origin common in Central and East Europe. Hum. Genet. 106, 259–268 (2000). [DOI] [PubMed] [Google Scholar]
- Abicht A. et al. A newly identified chromosomal microdeletion and an N-box mutation of the AChR epsilon gene cause a congenital myasthenic syndrome. Brain 125, 1005–1013 (2002). [DOI] [PubMed] [Google Scholar]
- Huber M. et al. Deletion of the cytoplasmatic domain of BP180/collagen XVII causes a phenotype with predominant features of epidermolysis bullosa simplex. J. Invest. Dermatol. 118, 185–192 (2002). [DOI] [PubMed] [Google Scholar]
- Morgan N. V., Tipping A. J., Joenje H. & Mathew C. G. High frequency of large intragenic deletions in the Fanconi anemia group A gene. Am. J. Hum. Genet. 65, 1330–1341 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping A. J. et al. Molecular and genealogical evidence for a founder effect in Fanconi anemia families of the Afrikaner population of South Africa. Proc. Natl. Acad. Sci. USA 98, 5734–5739 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Schrijver I., Brenn T., Furthmayr H. & Francke U. Multi-exon deletions of the FBN1 gene in Marfan syndrome. BMC Med. Genet. 2, 11 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Has C. et al. Molecular basis of Kindler syndrome in Italy: novel and recurrent Alu/Alu recombination, splice site, nonsense, and frameshift mutations in the KIND1 gene. J. Invest. Dermatol. 126, 1776–1783 (2006). [DOI] [PubMed] [Google Scholar]
- Spena S. et al. Congenital afibrinogenaemia caused by uniparental isodisomy of chromosome 4 containing a novel 15-kb deletion involving fibrinogen Aα-chain gene. Eur. J. Hum. Genet. 12, 891–898 (2004). [DOI] [PubMed] [Google Scholar]
- Beysen D. et al. Deletions involving long-range conserved nongenic sequences upstream and downstream of FOXL2 as a novel disease-causing mechanism in blepharophimosis syndrome. Am. J. Hum. Genet. 77, 205–218 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huie M. L., Anyane-Yeboa K., Guzman E. & Hirschhorn R. Homozygosity for multiple contiguous single-nucleotide polymorphisms as an indicator of large heterozygous deletions: identification of a novel heterozygous 8-kb intragenic deletion (IVS7-19 to IVS15-17) in a patient with glycogen storage disease type II. Am. J. Hum. Genet. 70, 1054–1057 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay S. K. et al. Fatal infantile neuromuscular presentation of glycogen storage disease type IV. Neuromuscul. Disord. 14, 253–260 (2004). [DOI] [PubMed] [Google Scholar]
- Besson A. et al. Primary GH insensitivity '(Laron syndrome) caused by a novel 4 kb deletion encompassing exon 5 of the GH receptor gene: effect of intermittent long-term treatment with recombinant human IGF-I. Eur. J. Endocrinol. 150, 635–642 (2004). [DOI] [PubMed] [Google Scholar]
- Kornreich R., Bishop D. F. & Desnick R. J. α-galactosidase A gene rearrangements causing Fabry disease. Identification of short direct repeats at breakpoints in an Alu-rich gene. J. Biol. Chem. 265, 9319–9326 (1990). [PubMed] [Google Scholar]
- Johnston J. J. et al. Clinical and molecular delineation of the Greig cephalopolysyndactyly contiguous gene deletion syndrome and its distinction from acrocallosal syndrome. Am. J. Med. Genet. 123A, 236–242 (2003). [DOI] [PubMed] [Google Scholar]
- Jia S. Q. et al. α0 thalassaemia as a result of a novel 11,1 kb deletion eliminating both of the duplicated α globin genes. J. Clin. Pathol. 57, 164–167 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Gene Mutation Database (HGMD) (2007). Available at: http://www.hgmd.cf.ac.uk/ac/index.php (Accessed: 3rd January 2009).
- Stenson P. D. et al. The human gene mutation database: 2008 update. Genome Med. 1, 13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross Rearrangement Breakpoint Database (GRaBD) (2004). Available at: http://www.uwcm.ac.uk/uwcm/mg/grabd/. (Accessed: 25th December 2008). [DOI] [PubMed]
- Abeysinghe S. S., Chuzhanova N., Krawczak M., Ball E. V. & Cooper D. N. Translocation and gross deletion breakpoints in human inherited disease and cancer I. Nucleotide composition and recombination-associated motifs. Hum. Mutat. 21, 229–244 (2003). [DOI] [PubMed] [Google Scholar]
- Inverted Repeat Finder (IRF) (2006). Available at: https://tandem.bu.edu/cgi-bin/irdb/irdb.exe. (Accessed: 9th January 2009).
- Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N. et al. Fragile DNA motifs trigger mutagenesis at distant chromosomal loci in Saccharomyces cerevisiae. PLOS Genet. 9, e1003551; 10.1371/pgen1003551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C. et al. Size of gene specific inverted repeat-dependent gene deletion in Saccharomyces cerevisiae. PLoS One 8, e72137; 10.1371/pone0072137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss U. & Wilson J. H. Repair of single-stranded loops in heteroduplex DNA transfected into mammalian cells. Proc. Natl. Acad. Sci. USA 84, 1619–1623 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga T. & Aplan P. D. Chromosomal aberrations induced by double strand DNA breaks. DNA Repair 4, 1038–1046 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhabinskaya D. & Benham C. J. Competitive superhelical transitions involving cruciform extrusion. Nucleic Acids Res. 41, 9610–9621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanHulle K. et al. Inverted DNA repeats channel repair of distant double-strand breaks into chromatid fusions and chromosomal rearrangements. Mol. Cell Biol. 27, 2601–2614 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gent D. C. Reaching out for the other end with p53-binding protein 1. Trends Biochem. Sci. 34, 226–229 (2009). [DOI] [PubMed] [Google Scholar]
- National Center for Biotechnology Information (NCBI) (1988). Available at: http://www.ncbi.nlm.nih.gov. (Accessed: 20th December 2008).
- NCBI BLAST (1990). Available at: http://www.ncbi.nlm.nih.gov/blast. (Accessed: 4th January 2009).
- Aksakoglu G. [Korelasyon ve Regresyon (Correlation and Regression)] Sağlıkta araştırma teknikleri ve analiz yöntemleri (Research techniques and analysis methods in health) [Aksakoglu G., (1st ed.)] [306–320] (Dokuz Eylul University, Izmir, 2001).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information