

Abstract
Mutations in genes encoding the ERCC3 (XPB), ERCC2 (XPD), and GTF2H5 (p8 or TTD-A) subunits of the transcription and DNA-repair factor TFIIH lead to three autosomal-recessive disorders: xeroderma pigmentosum (XP), XP associated with Cockayne syndrome (XP/CS), and trichothiodystrophy (TTD). Although these diseases were originally associated with defects in DNA repair, transcription deficiencies might be also implicated. By using retinoic acid receptor beta isoform 2 (RARB2) as a model in several cells bearing mutations in genes encoding TFIIH subunits, we observed that (1) the recruitment of the TFIIH complex was altered at the activated RARB2 promoter, (2) TFIIH participated in the recruitment of nucleotide excision repair (NER) factors during transcription in a manner different from that observed during NER, and (3) the different TFIIH variants disturbed transcription by having distinct consequences on post-translational modifications of histones, DNA-break induction, DNA demethylation, and gene-loop formation. The transition from heterochromatin to euchromatin was disrupted depending on the variant, illustrating the fact that TFIIH, by contributing to NER factor recruitment, orchestrates chromatin remodeling. The subtle transcriptional differences found between various TFIIH variants thus participate in the phenotypic variability observed among XP, XP/CS, and TTD individuals.
Introduction
The evolutionarily conserved general transcription factor IIH (TFIIH) plays a key role in maintaining genome stability.1,2 Mammalian TFIIH comprises a core (containing the six subunits ERCC3 [XPB], GTF2H1 [p62], GTF2H4 [p52], GTF2H2 [p44], GTF2H3 [p34], and GTF2H5 [p8 or TTD-A]) bridged by ERCC2 (XPD) to the CDK-activating kinase (CAK) module (composed of the three subunits CDK7, CCNH [cyclin H], and MNAT1 [MAT1]; Figure 1A). In addition to having a function in transcription, TFIIH is also involved in the nucleotide excision repair (NER) pathway, thus illustrating the important interplay between these distinct processes.3,4 NER is responsible for the removal of a variety of bulky DNA adducts, such as those induced by UV irradiation, and is subdivided into two sub-pathways: global genome repair (GGR) is responsible for the removal of DNA lesions from the whole genome, and transcription-coupled repair (TCR) is responsible for the accelerated removal of lesions arising on the transcribed strand of active genes.5,6 In GGR, the XPC-RAD23B complex recognizes the damage-induced DNA distortion, whereas in TCR, RNA polymerase II (pol II) stalled in front of a lesion promotes the recruitment of the TCR-specific proteins ERCC6 (CSB) and ERCC8 (CSA). Both NER sub-pathways then funnel through the TFIIH action that unwinds the DNA via the ATPase and helicase activities of ERCC3 and ERCC2, which are regulated by the GTF2H4-GTF2H5 and GTF2H2 subunits, respectively. XPA and RPA are then recruited to assist the expansion of the DNA bubble around the damage and the arrival of endonucleases ERCC5 (XPG) and ERCC4 (XPF). ERCC5 and ERCC4 then generate cuts in the 3′ and 5′ sides of the lesion, respectively, thereby removing the damaged oligonucleotide before the re-synthesis machinery fills the DNA gap. During transcription of protein-coding genes, TFIIH is involved via its ERCC3 subunit in the opening of the promoter,7,8 whereas its CDK7 kinase phosphorylates serines 5 and 7 of the C-terminal domain (CTD) of pol II, as well as others activators.9,10 Interestingly, NER factors (XPC, ERCC6, XPA, ERCC5, and ERCC4) have been found to also be involved in the regulation of gene expression.11–13 However, it remains to be established whether TFIIH influences the recruitment and the function of NER factors at the promoter of activated genes for chromatin remodeling and accurate transcription.
Figure 1.
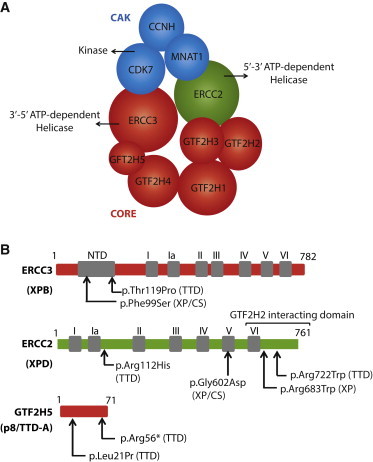
Schematic Representation of TFIIH
(A) The CAK sub-complex (in blue) is bridged to the core TFIIH (in red) by the ERCC2 (XPD) helicase (in green).
(B) Variants of the ERCC3 (XPB), ERCC2 (XPD), and GTF2H5 (TTD-A) subunits are indicated. Black squares indicate the helicase motifs (I–VI). NTD stands for N-terminal domain.
The importance of TFIIH and its relationship with the other NER factors are highlighted by the existence of human genetic disorders with a broad range of clinical features (Table 1).14–20 Indeed, mutations in excision repair complementation group 3 (ERCC3 [MIM 133510]), excision repair complementation group 2 (ERCC2 [MIM 126340]), and general transcription factor IIH polypeptide 5 (GTF2H5 [MIM 608780]) cause three distinct autosomal-recessive disorders: xeroderma pigmentosum (XP [MIM 278730]), XP associated with Cockayne syndrome (XP/CS [MIM 610651]), and trichothiodystrophy (TTD [MIM 610675]), respectively.21,22 XP is characterized by numerous skin abnormalities ranging from excessive freckling and ichthyosis to multiple skin cancers, the frequency of which is about 2,000-fold greater than that in normal individuals. In addition to having hyper-photosensitivity, XP individuals display a progressive neurological degeneration.23 XP/CS individuals display a combination of the cutaneous abnormalities found in XP and the severe neonatal later onset of neurological and developmental anomalies typical of CS. The typical hallmark of TTD is sulfur-deficient brittle hair, caused by a greatly reduced content of cysteine-rich matrix proteins in the hair shafts. Intellectual disability and ichthyosis also characterize TTD individuals,24 and some of them are sensitive to sunlight without any unusual pigmentation changes and severe skin lesions or cancer.
Table 1.
ERCC3, ERCC2, and GTF2H5 Mutations and the Related Clinical XP, XP/CS, and TTD Phenotypes
| Gene Mutation | Protein Variant | Syndrome | Individual | Clinical Features | Reference |
|---|---|---|---|---|---|
|
ERCC3 c.296T>C |
ERCC3 p.Phe99Ser |
XP/CS | male | severe sunburn at 6 weeks of age and later hyper-pigmentation, but no evidence of any malignancy; CS neurological anomalies including cerebellar atrophy, sclerosis of sutures, neuron demyelination, and some hearing difficulties; development of sexual anomalies with age | Scott et al.14 |
|
ERCC3 c.355A>C |
ERCC3 p.Thr119Pro |
TTD | male | congenital ichthyosis (collodion baby); hair with tiger-tail pattern; moderate skin photosensitivity; mild learning disability | Weeda et al.15 |
|
GTF2H5 c.[62T>C];[116C>T] |
GTF2H5 p.[Leu21Pro];[Arg56∗] |
TTD | male | congenital ichthyosis (collodion baby); moderate skin photosensitivity, but no skin cancer; developmental delay, asthmatic attacks, and short stature; severe mental retardation | Stefanini et al.16 |
|
ERCC2 c.335G>A |
ERCC2 p.Arg112His |
TTD | male | moderate skin photosensitivity, but no skin cancer; sulfur-deficient brittle hair and nails; delayed puberty and short stature; neurological anomalies including axial hypotonia and reduced motor coordination | Stefanini et al.17 |
|
ERCC2 c.1805G>A |
ERCC2 p.Gly602Asp |
XP/CS | male | high skin photosensitivity and skin cancer at 2 years of age; CS neurological anomalies; progeroid features | Takayama et al.18 |
|
ERCC2 c.2047C>T |
ERCC2 p.Arg683Trp |
XP | male | high skin photosensitivity and skin cancers; moderate mental retardation | Broughton et al.19 |
|
ERCC2 c.2164C>T |
ERCC2 p.Arg722Trp |
TTD | male | high skin photosensitivity, but no skin cancer; sulfur-deficient brittle hair and nails; severe physical and mental retardation | Taylor et al.20 |
Accumulating evidence suggests that the clinical features of these three disorders cannot be exclusively explained by defects in DNA repair and might also involve transcription deficiencies25 (Table 1).14–20 In this study, we intend to understand how mutations in ERCC3, ERCC2, and GTF2H5 result in impairment of gene expression in order to provide explanations for the large and diverse range of clinical features associated with these mutations. Knowing the close connections between TFIIH and NER factors in the removal of DNA damage,26 we have investigated the consequences of ERCC3, ERCC2, and GTF2H5 variants on the recruitment of the NER factors and their roles in the various steps that lead to accurate RNA synthesis, including histone post-translational modifications (PTMs), DNA breaks, DNA demethylation, and gene-loop formation. In addition to improving our understanding of the cascade of events that drive RNA synthesis, such a systematic approach could help to determine transcriptional default hallmarks that molecularly define different genetic disorders.
Material and Methods
Cell Lines, Culture Conditions, and Transfection
Wild-type (WT)-ERCC3 (XPB) cells, ERCC3-p.Phe99Ser cells, and ERCC3-p.Thr119Pro cells are SV40-transformed human fibroblasts (XPCS2BASV) expressing WT His-ERCC3-HA,27 ERCC3 c.296T>C (RefSeq accession number NM_000122.1), and ERCC3 c.355A>C, respectively.28 GTF2H5 (p8 or TTD-A) cells are SV40-transformed human fibroblasts (TTD1Br-SV) expressing GTF2H5 c.[62T>C];[116C>T], p.[Leu21Pro];[Arg56∗] (RefSeq NM_207118.2).16 WT-GTF2H5 cells (TTD1Br-SV + TTDA-GFP and TTD1Br-SV + TTDA-HA) are rescued TTDBr1-SV cells with pEGFP-N1-TTDA and pCDNA3-HA-TFB5 vectors that stably express GTF2H5-GFP and GTF2H5-HA, respectively.29,30 WT-ERCC2 (XPD) cells (GM637) are SV40-transformed human fibroblasts from a normal 18-year-old female. ERCC2-p.Gly602Asp cells (XPCS2),18 ERCC2-p.Arg112His cells (TTD8PV),16 ERCC2-p.Arg683Trp cells (XP135LO),18 and ERCC2-p.Arg722Trp cells (TTD1BEL)19 are human primary fibroblasts expressing ERCC2 c.1805G>A (RefSeq NM_000400.3), ERCC2 c.335G>A, ERCC2 c.2047C>T, and ERCC2 c.2164C>T, respectively. Cells were incubated with phenol-red-free medium containing charcoal-treated fetal calf serum and 40 μg/ml gentamicin. Cells were treated with 10 μM of all-trans retinoic acid (t-RA, MP Biomedicals). ERCC2-p.Arg112His, ERCC2-p.Gly602Asp, ERCC2-p.Arg683Trp, and ERCC2-p.Arg722Trp cells were transiently transfected with the X-tremeGENE HP DNA transfection reagent (Roche) 48 hr before the t-RA treatment with WT pEGFP-ERCC2.
Antibodies
Mouse monoclonal antibodies toward ERCC3 (1B3), ERCC2 (2F6), GTF2H2 (p44, 1H5), retinoic acid receptor alpha (RARα, 9A6), XPA (1E11), ERCC5 (XPG, 1B5), and pol II (7C2) were produced by the Institut de Génétique et de Biologie Moléculaire et Cellulaire. CDK7 (C-19), general transcription factor IIB (GTF2B [TFIIB], C-18), ERCC4 (XPF, H-300), and BIOTIN (33) antibodies were purchased from Santa-Cruz Biotechnology. CTCF (ab70303) and RNA pol II ser5P (61085) antibodies were purchased from Abcam and Active Motif, respectively. H3K4me2 (9726), H3K9me2 (9753), and H3K9Ac (9671) antibodies were purchased from Cell Signaling Technology.
Reverse Transcriptase and qPCR
Total RNAs were isolated with the GenElute Mammalian Total RNA Miniprep Kit (Sigma) and reverse transcribed with SuperScript II Reverse Transcriptase (Invitrogen). qPCRs were performed with the QuantiTect SYBR Green PCR Kit (QIAGEN) and the LightCycler 480 apparatus (Roche). The primer sequences for retinoic acid receptor beta isoform 2 (RARB2 [MIM 180220]) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH [MIM 138400]) are listed in Table S1. RARB2 mRNA levels were normalized to GAPDH.
Chromatin Immunoprecipitation and BIOTIN-ChIP Assays
Cells were cross-linked at room temperature (RT) for 10 min with 1% formaldehyde. Chromatin was prepared and sonicated on ice for 30 min with a Bioruptor (Diagenode) as previously described.12 Samples were immunoprecipitated with antibodies at 4°C overnight, and protein G Sepharose beads (Upstate) were added, incubated for 4 hr at 4°C, and sequentially washed. Protein-DNA complexes were eluted and de-cross-linked. DNA fragments were purified with the QIAquick PCR Purification Kit (QIAGEN) and analyzed by qPCR with a set of primers targeting the promoter and terminator regions of RARB2 (Table S1).
In parallel, cross-linked cells were permeabilized with cytonin (Trevigen) for 30 min at RT. After extensive washes with PBS, a terminal deoxynucleotidyl transferase (TdT) reaction was performed with Biotin-16-dUTP (Roche) and 60 units of recombinant enzyme rTdT (Promega). The resulting samples were next sonicated and immunoprecipitated with anti-Biotin antibodies and treated as described in the chromatin immunoprecipitation (ChIP) assay. The purified DNA fragments were analyzed by qPCR with the previously described primers (Table S1).
Unmethylated DNA Immunoprecipitation Assays
Genomic DNA was extracted with the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma). Loss of 5-methylcytosine (5mC) at the promoter and terminator regions of RARB2 was measured by digestion of genomic DNA (2 μg) with 10 units of MseI (Fermentas) and with the UnMethylcollector Kit (Active Motif). The unmethylated DNA immunoprecipitation (unMeDIP) kit is based on the affinity of the three zinc-coordinating CXXC domains that specifically bind nonmethylated CpG sites. The resulting samples were selected with magnetic beads conjugated with CXXC domains, extensively washed, and analyzed by qPCR.
Quantitative Chromosome Conformation Capture
The quantitative chromosome conformation capture (q3C) assay was performed as previously described.31 Cells were cross-linked at RT for 10 min with 2% formaldehyde. Cross-linked chromatin was digested in the appropriate restriction buffer with 400 units of HindIII. The digestion was stopped after overnight incubation at 37°C, diluted in ligation buffer, and incubated with the highly concentrated T4 DNA ligase (Roche) for 4 hr at 16°C. The cross-linking was heat reversed, and DNA fragments were purified. Undigested DNA and digested DNA without the ligation step were used as negative controls. Moreover, as an internal positive control, we used the endogenous ERCC3, which has been reported to adopt the same spatial conformation in different tissues.31 All q3C results were normalized by data from ERCC3 analysis, which controlled for changes in nuclear size, chromatin density, and cross-linking efficiency. Primers and probes were designed as follows: a universal sequence-specific Taqman probe and corresponding reverse primer on a fixed restriction fragment (Ter or M1) were used in combination with different forward primers specific to the other restriction fragments (see Figure 4, upper panel). q3C templates (200 ng) were used for the Taqman PCR reaction with the universal PCR Master Mix and the Lightcycler 480 apparatus from Roche.
Figure 4.
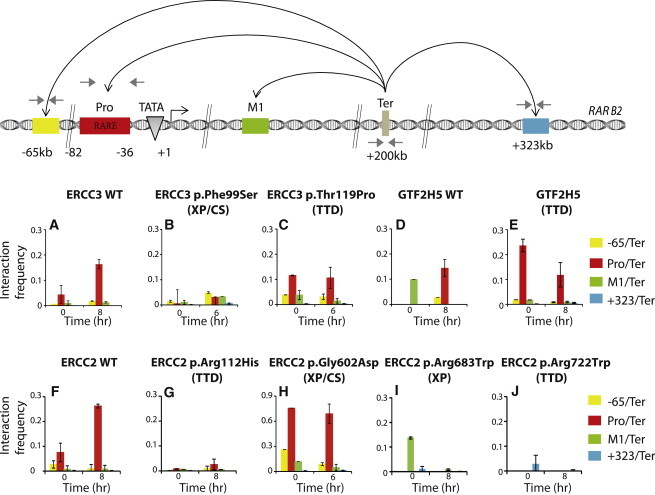
TFIIH Is Involved in RARB2-Loop Formation
(Upper panel) Schematic representation of the primers used in q3C. One primer was designed at the RARB2 termination region (Ter) for investigating the associations between the different elements, including upstream (−65 kb), promoter (Pro), intronic (M1), and downstream (+323 kb) regions as indicated by the black arrows.
(A–J) q3C assays were performed with cross-linked and HindIII-digested chromatin from all cells as indicated at 0 hr and at either 6 or 8 hr after t-RA treatment depending on the timing of the RNA expression peak (see shadowed areas in Figure 2). The bar chart (y axis) shows the PCR product enrichment (%) normalized to the enrichment within human ERCC3. Each PCR was performed at least three times. Signals were normalized to the total amount of DNA used and were estimated with an amplicon located within a HindIII fragment in RARB2. Error bars represent the SD.
Construction of Baculoviruses and Purification of Complexes
Baculoviruses overexpressing the FLAG-ERCC3, FLAG-ERCC2, GTF2H1 (p62), GTF2H4 (p52), GTF2H2, GTF2H3 (p34), FLAG-CDK7, CCNH (cyclin H), MNAT1 (MAT1), and GTF2H5 subunits of TFIIH were produced as previously described.32 The cDNAs encoding altered FLAG-ERCC3 and FLAG-ERCC2 were obtained by PCR-site-directed mutagenesis. The resulting vectors were recombined with baculovirus DNA (BD BaculoGold, Pharmingen). The recombinant viruses were purified from isolated plaques, and viral stocks were prepared by three-step growth amplification.
We infected Sf21 insect cells with the different baculoviruses in order to separately obtain core-IIH (with or without an ERCC3 variant), CAK, and ERCC2 (with or without a variant). The different whole-cell extracts were incubated for 4 hr at 4°C with anti-M2-FLAG antibody bound to agarose beads. After extensive washings, the immunoprecipitated fractions were eluted. The recombinant TFIIH was made by a mixture of purified core-IIH, CAK, and ERCC2, allowing the preparation of the different TFIIH complexes containing either ERCC2 or ERCC3 variants.
In Vitro Transcription Assays
Run-off transcription assays were performed with recombinant GTF2B, TFIIE, TFIIF, TBP, endogenous pol II, and the different TFIIH complexes (recombinant IIHs) as previously described.32 Pol II phosphorylation was carried out as a classical run-off transcription assay as previously described.33 Hypo-phosphorylated (IIA) and hyper-phosphorylated (IIO) forms of pol II were resolved by SDS-PAGE and detected with a monoclonal antibody (7C2).
Results
Mutations in ERCC2 and ERCC3 Compromise the Formation of the Transactivation Complex
To determine the transcriptional defects due to the mutations in ERCC3, ERCC2, and GTF2H5, we analyzed cells derived from XP, XP/CS, and TTD individuals bearing different mutations as indicated (Table 1; Figure 1B). We used RARB2 as a model to investigate the transcription process. A few hours after t-RA treatment of these mutated cells, we observed that their patterns of RARB2 mRNA synthesis were different from those of the respective WT-ERCC3 (XPB), WT-ERCC2 (XPD), and WT-GTF2H5 (p8 or TTD-A) cells (Figures 2A1–2J1). The amount of RARB2 mRNA was significantly lower for ERCC3-p.Phe99Ser and ERCC3-p.Thr119Pro cells than for WT-ERCC3 cells, which peaked at 8 hr post-treatment (Figures 2A1–2C1). Compared to the cells rescued with an overexpressing WT ERCC2, the four cell lines bearing mutations in ERCC2 showed similarly reduced RARB2 induction (Figures 2F1–2J1). Compared to rescued cells, GTF2H5 cells did not show a reduction of the RARB2 mRNA level (Figures 2D1 and 2E1).
Figure 2.
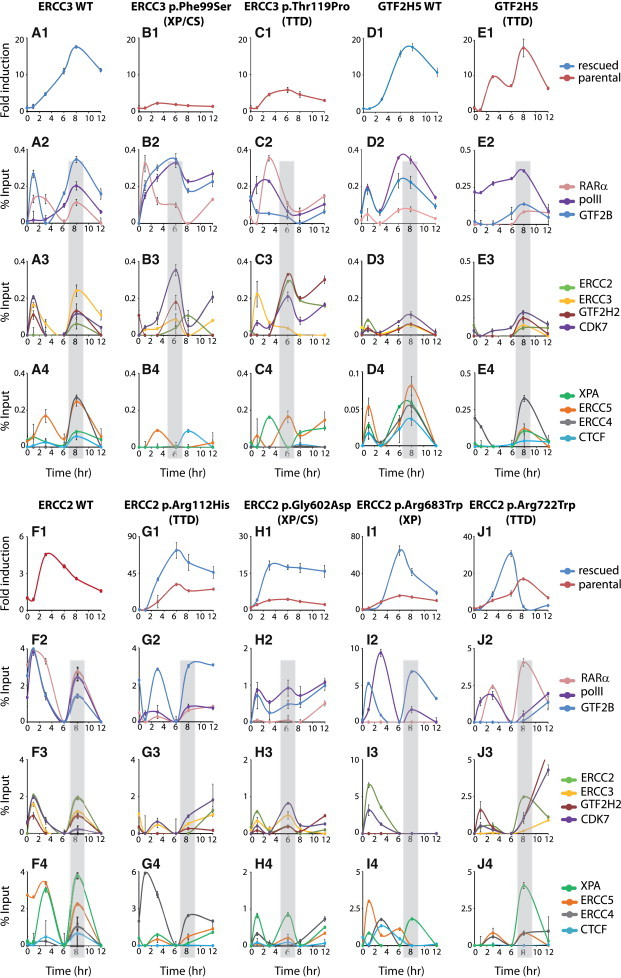
RARB2 mRNA Expression and Transcriptional-Machinery Recruitment on the RARB2 Promoter Are Disturbed in Cells Bearing Mutations in Genes Encoding TFIIH Subunits
(A1–J1) Relative RARB2 mRNA expression monitored by qPCR over time from t-RA-treated (1) WT-ERCC3, ERCC3-p.Phe99Ser, and ERCC3-p.Thr119Pro cells, which are SV40-transformed human fibroblasts (XPCS2BASV) expressing WT His-ERCC3-HA,27ERCC3 c.296T>C, and ERCC3 c.355A>C, respectively; (2) GTF2H5 (p8 or TTD-A) cells, which are SV40-transformed human fibroblasts (TTD1Br-SV) expressing GTF2H5 c.[62T>C];[116C>T], p.[Leu21Pro];[Arg56∗]; (3) WT-GTF2H5 cells (TTD1Br-SV + TTDA-HA), which are rescued TTD1Br-SV cells stably expressing GTF2H5-HA; and (4) WT-ERCC2 (XPD) cells (GM637), ERCC2-p.Gly602Asp cells (XPCS2), ERCC2-p.Arg112His cells (TTD8PV), ERCC2-p.Arg683Trp cells (XP135LO),18 and ERCC2-p.Arg722Trp cells (TTD1BEL), which are human primary fibroblasts expressing WT ERCC2, ERCC2 c.1805G>A, ERCC2 c.335G>A, ERCC2 c.2047C>T, and ERCC2 c.2164C>T, respectively. Red curves show the mRNA expression of RARB2 in the parental cells, including cells bearing mutations in ERCC2 and ERCC3 and the WT cell line (GM637). Blue curves show mRNA expression of RARB2 in the rescued cells (expressing WT ERCC3 and WT GTF2H5) and cells overexpressing WT GFP-ERCC2 (ERCC2 p.Arg112His, ERCC2 p.Gly602Asp, ERCC2 p.Arg683Trp, and ERCC2 p.Arg722Trp). Error bars represent the SD of three independent experiments.
(A2–J4) ChIP monitoring of the t-RA-dependent recruitment of RARα, pol II, GTF2B (A2–J2), ERCC3, ERCC2, GTF2H2, and CDK7 subunits of TFIIH (A3–J3) and XPA, ERCC5, ERCC4, and CTCF (A4–J4) on the RARB2 promoter. Each series of ChIP is representative of at least two independent experiments. Values are expressed as the percentage of the input. Error bars represent the SD.
We next performed ChIP assays to evaluate the dynamic recruitment of pol II partners at the RARB2 promoter over time. We observed a concomitant recruitment of RARα, pol II, and GTF2B at 8 hr after t-RA treatment in WT-ERCC3, WT-ERCC2, and WT-GTF2H5 cells (Figures 2A2, D2, and F2, respectively). At this time, TFIIH was also recruited, as seen by the presence of its ERCC3, ERCC2, GTF2H2 (p44), and CDK7 subunits (Figures 2A3, 2D3, and 2F3). We also detected the simultaneous presence of XPA, ERCC5 (XPG), and ERCC4 (XPF) (Figures 2A4, 2D4, and 2F4).
Each mutation in ERCC3, ERCC2, and GTF2H5 led to different and specific deregulation of recruitment of the components of the transactivation complex at both the promoter and the terminator (Figure 2; Figure S1). Whereas RARα was detected at early time points in ERCC3-p.Phe99Ser cells, pol II and GTF2B only accumulated at the promoter after 1 hr of t-RA induction (Figure 2B2). We also noticed a non-concomitant recruitment of the TFIIH subunits and NER factors. Whereas ERCC5 was recruited at 3 hr in ERCC3-p.Phe99Ser cells, XPA and ERCC4 were not detected over a 12-hr period (Figure 2B4). In ERCC3-p.Thr119Pro cells, RARα, pol II, and GTF2B were detected at the promoter around the 3-hr mark (Figure 2C2). The ERCC3 subunit was found at the promoter at 1 hr, whereas the ERCC2, GTF2H2, and CDK7 subunits were detected later (Figure 2C3). Similarly, XPA arrival (1 hr) preceded ERCC5 (6 hr), whereas ERCC4 was not detected (Figure 2C4). In cells bearing mutations in GTF2H5, we observed a concomitant recruitment of TFIIH subunits and NER factors with the transcriptional machinery at 8 hr after t-RA treatment; a similar result was observed with the corresponding rescued cell types (Figures 2D2–2D4 and 2E2–2E4).
We next focused on the cells bearing mutations in ERCC2 and resulting in three different phenotypes (Figure 1B; Table 1). In ERCC2-p.Arg112His cells, the recruitment of transcription and NER factors was temporarily correlated. All these factors were found in a second recruitment cycle that peaked at 8 hr, remained present until 12 hr after t-RA treatment, and paralleled RARB2 mRNA induction (Figures 2G1 and 2G2–2G4). In ERCC2-p.Gly602Asp cells, the recruitment pattern at 1 hr and 6 hr after t-RA treatment of pol II, GTF2B, TFIIH, and NER factors at the promoter was comparable to that in WT-ERCC2 cells (Figures 2H2–2H4). The ERCC2 p.Gly602Asp variant, which affects the helicase motif V, does not seem to disturb TFIIH architecture.32 In ERCC2-p.Arg683Trp cells, the recruitment of TFIIH and NER factors was highly deregulated at early time points (Figures 2I2–2I4). At 8 hr, only pol II, GTF2B, and XPA were detected. In ERCC2-p.Arg722Trp cells, in which the aberration is located in the C-terminal unfolded domain of ERCC2, we observed the recruitment of both transcription and NER factors at 8 hr (Figures 2J2–2J4); a much higher and continuous accumulation of pol II, GTF2B, and some of the TFIIH subunits also occurred at 12 hr. In these cells, the ERCC2 p.Arg683Trp and p.Arg722Trp variants weaken the interaction with the GTF2H2 subunit and consequently destabilize the architecture of TFIIH.32
All together, our data suggest that mutations in ERCC3 and ERCC2 disturb RARB2 activation by impeding the formation of the pre-initiation complex at the promoter. Although the dysregulation was different depending on the mutation, we noticed a compromised integrity of the TFIIH complex and, in some cases, the absence of NER factors at the activated promoter.
Mutations in Genes Encoding TFIIH Subunits Affect Chromatin Remodeling
Previous works have underlined the sequential recruitment of the transcription and NER components at the promoter of activated genes and their role in bringing about chromatin modifications, e.g., histone PTMs, DNA breaks, and DNA demethylation.11,13 Among the histone hallmarks characterizing the chromatin status, we have focused and compared euchromatin histone PTMs (acetylated lysine 9 histone H3 [H3K9Ac] and dimethylated lysine 4 histone H3 [H3K4me2]) to the heterochromatin mark (dimethylated lysine 9 histone H3 [H3K9me2]) previously observed at the RARB2 promoter.12,34
In WT-ERCC3 and WT-ERCC2 cells treated with t-RA, ChIP showed the characteristic histone PTM signature of gene activation (increased H3K9ac and H3K4me2 and decreased H3K9me2; Figures 3A1 and 3F1), correlating with the RARB2 mRNA induction peak at 8 hr. In all mutated cells tested so far (Figures 3B1, 3C1, and 3G1–3J1), we observed a persistence of active histone PTMs around the RARB2 promoter. As an example, in ERCC2-p.Arg112His cells, in which the transcription and NER factors were shown to be recruited at the promoter by 8 hr post-treatment, H3K9me3 remained low; however, we noticed high acetylation and methylation of H3K9 and H3K4, respectively (Figure 2G1). In both GTF2H5 and rescued cells, H3K9me2 was hardly detectable, whereas H3K9ac and H3K4me2 were visible (Figure 3D1 and E1).
Figure 3.
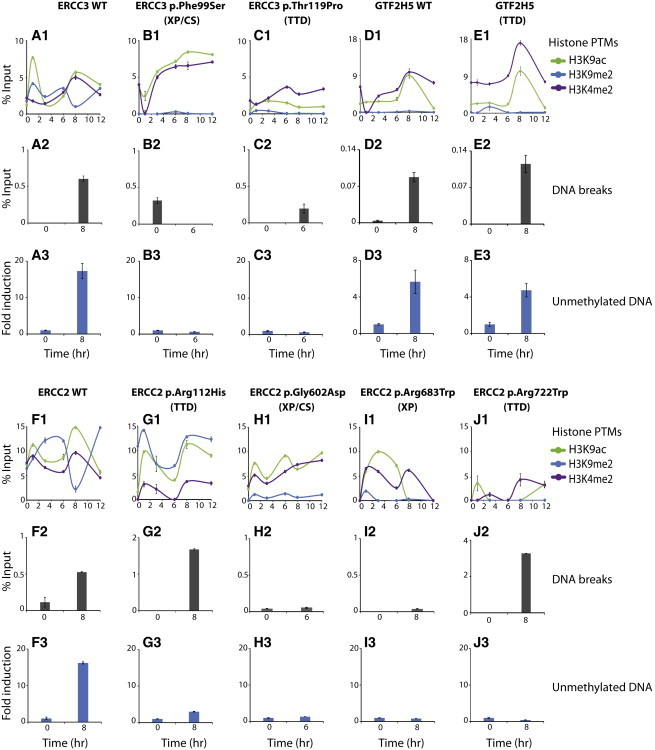
Mutations in Genes Encoding TFIIH Subunits Disrupt Histone PTMs, DNA Breaks, and Active DNA Demethylation on the RARB2 Promoter
(A1–J1) ChIP monitoring of the t-RA-dependent presence of H3K4me2, H3K9me2, and H3K9ac on the RARB2 promoter over time in all the mentioned cell lines. Each series of ChIP is representative of at least two independent experiments.
(A2–J2) Detection of DNA breaks at the RARB2 promoter at 0 hr and at either 6 or 8 hr after t-RA treatment (depending on the timing of the RNA expression peak; see shadowed areas in Figure 2). Each series of BioChIP is representative of three independent experiments, and values are expressed as the percentage of the input. Error bars represent the SD.
(A3–J3) UnMeDIP experiments. Samples containing unmethylated DNA on the RARB2 promoter were analyzed by qPCR. Each series of unMeDIP is representative of two independent experiments, and values are expressed as the percentage of the input. Error bars represent the SD.
The formation of transient DNA breaks upon gene activation is linked to endonucleases ERCC5 and ERCC4 and has previously been reported at the RARB2 promoter.35,36 We thus evaluated the formation of such DNA breaks in the cells bearing mutations in ERCC3, ERCC2, and GTF2H5 by performing a BioChIP assay that measured the incorporation of biotinylated dUTP within broken DNA. We observed a concomitant increase in DNA cleavage at both the promoter and the terminator of all WT cells upon t-RA activation (Figures 3A2, 3D2, and 3F2; Figure S2). In these cells, we noticed a parallel among the presence of ERCC5, ERCC4, and DNA breaks at both promoters and terminators. Except for in GTF2H5, ERCC2-p.Arg112His, ERCC2-p.Arg722Trp, and ERCC3-p.Thr119Pro cells (although to a much lower extent) in which ERCC5 and ERCC4 were still detected (Figures 3C2, 3E2, 3G2, and 3J2), a significant induction of DNA breaks near the RARB2 promoter was not detected in mutated cell lines (Figures 3B2, 3H2, and 3I2). It should be noted that the DNA breaks were observed around the RARB2 terminator in WT and GTF2H5 cells, whereas in all the other mutated cell lines, no DNA breaks were detected (Figure S2).
Studies have documented a relationship between ERCC5 and DNA demethylation upon transcription.11 Using an unMeDIP approach, we measured the removal of 5mC at the RARB2 promoter. We found that DNA demethylation occurred at the promoter by 8 hr after t-RA treatment and perfectly paralleled the recruitment of the entire transcription machinery in the three WT cell lines (Figures 3A3, 3D3, and 3F3). On the contrary, there was a complete lack of DNA demethylation in all cells bearing mutations in ERCC2 and ERCC3 (Figures 3B3, 3C3, and 3G3–3J3), but not in GTF2H5 cells (Figure 3E3).
Taken together, the above data strongly support an involvement of TFIIH in chromatin remodeling, including the histone PTMs, the formation of DNA breaks, and active DNA demethylation.
Mutations in Genes Encoding TFIIH Subunits Affect Gene Loop Formation
The detection of the basal transcription machinery together with the NER factors at both the promoter and the terminator of RARB2 has previously been correlated with a chromatin-loop formation mediated by the CCCTC-binding factor (CTCF) chromatin organizer.35 Such loop formation was shown to parallel DNA demethylation and DNA breaks at both regions. We performed q3C assays to analyze the interactions between the promoter and the terminator, as well as intronic (M1), upstream (−65 kb), and downstream (+323 kb) regions of RARB2 (Figure 4, upper scheme). Using the terminator and M1 as bait, we observed that the promoter specifically and significantly interacted with the terminator by 8 hr in t-RA-treated WT cells (Figures 4A, 4D, and 4F), paralleling the recruitment of the entire transcriptional apparatus to both regions (Figure 2; Figure S1). By contrast, in all cells bearing mutations in genes encoding TFIIH subunits, including GTF2H5 cells, no significant increase in the frequency of terminator-promoter interactions occurred upon RARB2 transactivation (Figures 4B–4J). In addition, we also observed that the absence of loop formation correlated with the absence or delayed recruitment of CTCF at both the promoter and the terminator of RARB2 (Figures 2B4, 2C4, 2G4, 2H4, and 2J4; Figure S1). In GTF2H5 cells, we did not observe an increase in the interaction frequency between the terminator and the promoter upon transcription, even though there were no perturbations in recruitment of transcriptional or NER machinery, nor of other tested chromatin-remodeling events (Figure 4E). No specific interactions were observed between the intronic M1 bait and the promoter or between all the other analyzed fragments upon t-RA treatment (Figure S3).
By impeding the recruitment of NER factors at the activated RARB2 promoter, TFIIH variants further disturbed the chromatin-loop formation required for optimal gene expression.
Mutations in Genes Encoding TFIIH Subunits Impair Some of Its Enzymatic Activities
We next addressed the contribution of ERCC3 and ERCC2 activities to the formation of an accurate transcription-initiation complex, a prerequisite for optimal RNA synthesis. We first generated recombinant IIH6 (rIIH6, the core TFIIH containing GTF2H1, GTF2H4, GTF2H2, GTF2H3, GTF2H5, and either WT or altered ERCC3), WT or altered ERCC2, and CAK. These rIIH6 sub-complexes were added to an in vitro transcription assay containing the adenoviral major late promoter (run-off of 309 nt), all the basal transcription factors, and pol II,32 along with CAK and ERCC2, either alone or in combination. When added to the transcription assay containing all the factors (including the ERCC2-CAK sub-complex), ERCC3-p.Phe99Ser rIIH6 exhibited a much weaker basal transcription activity than did WT-ERCC3 rIIH6 and ERCC3-p.Thr119Pro rIIH6 (Figure 5A). The addition of CAK together with WT ERCC2 and ERCC3-p.Phe99Ser rIIH6 did not improve RNA synthesis (lanes 5–7), contrary to what occurred with WT-ERCC3 rIIH6 and ERCC3-p.Thr119Pro rIIH6, which absolutely required the CAK sub-complex for optimal RNA synthesis (lanes 1–3 and 8–10). Because the variant weakens the contact with the GTF2H4 regulatory subunit within TFIIH,37 it results in a defect in the unwinding of the RARB2 promoter by ERCC3 and a defect in RNA synthesis. Interestingly, we also noticed that the absence of WT ERCC2 resulted in very weak RNA synthesis (lane 4 in Figure 5A and lanes 1 and 2 in Figure 5B). Because the ERCC2-GTF2H2 interaction allows the anchoring of CAK to the core TFIIH,38 we next investigated the transcription activity of TFIIHs containing ERCC2 variants. The addition of CAK and increasing amounts of WT ERCC2 to our transcription assay, which already contained WT rIIH6, stimulated RNA synthesis (lanes 3 and 4 in Figure 5B); this was also observed in the presence of ERCC2 p.Arg112His or ERCC2 p.Gly602Asp (lanes 5–8). On the contrary, when either ERCC2 p.Arg683Trp or ERCC2 p.Arg722Trp was added, no significant increase in RNA synthesis was observed (lanes 9–12).
Figure 5.
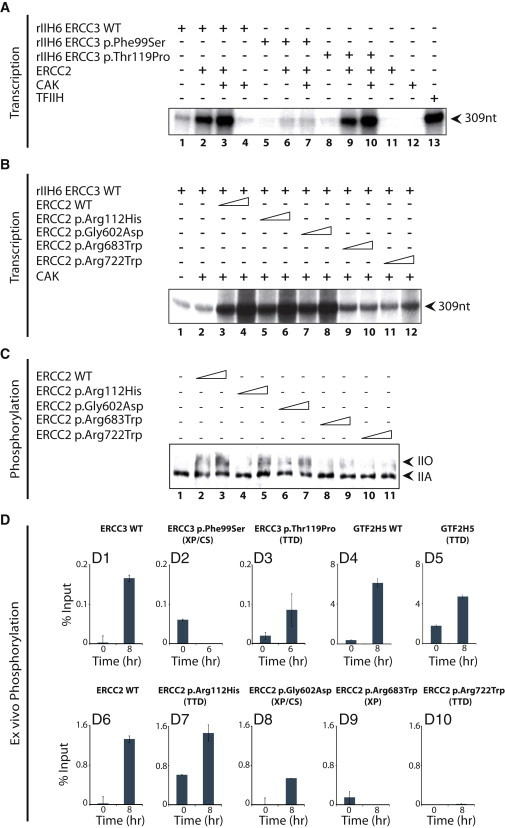
Effect of the TFIIH Alterations during In Vitro RNA Synthesis and Pol II Phosphorylation
(A) In vitro transcription assays with rIIH6 including WT ERCC3, ERCC3 p.Phe99Ser, or ERCC3 p.Thr119Pro. When indicated (+), these rIIH6 were incubated either alone or in combination with ERCC2 and/or CAK.
(B) WT rIIH6 was incubated either alone or in association with CAK and WT ERCC2, ERCC2 p.Arg112His, ERCC2 p.Gly602Asp, ERCC2 p.Arg683Trp, or ERCC2 p.Arg722Trp as indicated. The size (309 nt) of the transcript is indicated on the right side of each panel.
(C) Phosphorylation of pol II during in vitro transcription assays in the presence of WT rIIH6, CAK, and increasing amounts of ERCC2 p.Arg112His, ERCC2 p.Gly602Asp, ERCC2 p.Arg683Trp, and ERCC2 p.Arg722Trp as indicated. Arrows indicate hypo-phosphorylated (IIA) and hyper-phosphorylated (IIO) forms of pol II.
(D) ChIP monitoring the t-RA-dependent occupancy of the serine 5 phosphorylated pol II on the RARB2 promoter from the different indicated cells. Each series of ChIP is representative of at least two independent experiments. Values are expressed as the percentage of the input. Error bars represent the SD.
We then investigated the impact of CAK on the phosphorylation status of pol II during a classical run-off transcription experiment (see Material and Methods; Figure 5C). We observed that the hyper-phosphorylated form of pol II (IIO) was prevalent in the presence of WT ERCC2, ERCC2 p.Arg112His, or ERCC2 p.Gly602Asp (lanes 2–7) and paralleled the increase in RNA synthesis (Figure 5B, lanes 3–8). On the contrary, in the presence of ERCC2 p.Arg683Trp or ERCC2 p.Arg722Trp, which is deficient in stimulating RNA synthesis (Figure 5B, lanes 9–12), pol II was not hyper-phosphorylated (Figure 5C, lanes 8–11). Moreover, ChIP experiments demonstrated that in WT cells—as well as in GTF2H5, ERCC3-p.Thr119Pro, ERCC2-p.Arg112His, and ERCC2-p.Gly602Asp cells—phosphorylated pol II was detected at the RARB2 promoter (Figures 5D1 and 5D3–5D8). On the contrary, in ERCC3-p.Phe99Ser, ERCC2-p.Arg683Trp, and ERCC2-p.Arg722Trp cells, phosphorylated pol II was not detected (Figure 5D2, 5D9, and 5D10). The effects of the ERCC2 variants on pol II phosphorylation were thus similar in both in vitro and ex vivo contexts.
All together, the above data suggest that the pol II phosphorylation defects might contribute to the gene-expression deregulation observed in some cells bearing mutations in ERCC3 and ERCC2.
Discussion
After the assembly of the pre-initiation machinery (including TFIIA, GTF2B, TFIID, TFIIE, TFIIF, and RNA pol II), TFIIH unwinds the DNA around the proximal promoter through its ERCC3 (XPB) helicase subunit7 and phosphorylates the CTD of the largest subunit of pol II via its CDK7 kinase,9,39 allowing promoter escape and RNA elongation.40 In addition to regulating pol II, TFIIH regulates other components of the transcription machinery (such as nuclear receptors).41–43 Conversely, some of them—including TFIIE, ERCC5 (XPG), and the Mediator complex—regulate the activity of TFIIH, highlighting the pivotal role played by this complex in transcription.44–47
By investigating the mechanistic defects resulting from mutations in genes encoding TFIIH subunits, the present study intends to go deeper in the understanding of the transcriptional role played by TFIIH at the cellular level. Here, we document the involvement of TFIIH in pol II phosphorylation, as well as in promoter unwinding, by showing that TFIIH influences (1) the concomitant recruitment of NER factors, (2) histone PTMs, (3) DNA breaks, (4) DNA demethylation, and (5) gene-loop formation, all of which are essential for accurate RNA synthesis (Figure 6).
Figure 6.
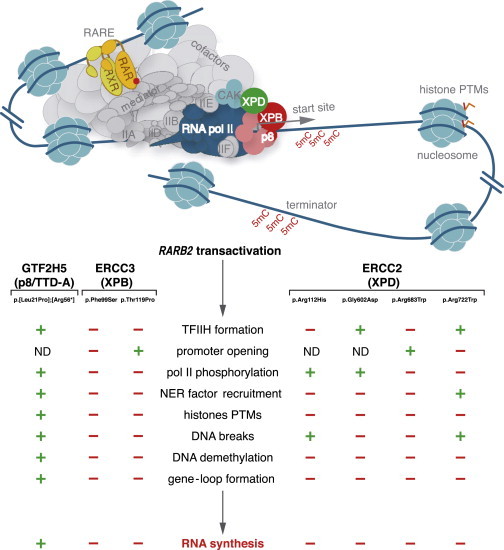
Mutations in Genes Encoding TFIIH Subunits Specifically Disrupt Transcription
(Upper panel) Upon t-RA ligand induction, the transactivation complex is formed once RARα and RXR targets its responsive element; co-activators and the Mediator complex are assembled at the RARB2 promoter together with the pre-initiation complex. NER factors (XPC, ERCC6 [CSB], XPA, ERCC5 [XPG], and ERCC4 [XPF]) are then sequentially recruited and regulate RNA synthesis by participating in chromatin remodeling. These include histone PTMs, induction of DNA breaks, and active demethylation of 5mC, all together necessary for the gene-loop formation between the promoter and the terminator of RARB2.
(Lower panel) Cells bearing mutations in genes encoding GTF2H5 p.[Leu21Pro];[Arg56∗], ERCC3 p.Phe99Ser, ERCC3 p.Thr119Pro, ERCC2 p.Arg112His, ERCC2 p.Gly602Asp, ERCC2 p.Arg683Trp, and ERCC2.Arg722Trp differently disrupt the RARB2 mRNA synthesis by deregulating several transcriptional steps. When indicated (−), each variant specifically affects (1) the formation of TFIIH, (2) its enzymatic activities (such as promoter opening and pol II phosphorylation), (3) the recruitment of NER factors, and (4) chromatin remodeling. ND stands for not determined.
Plasticity of TFIIH Recruitment at Activated Promoter
In both WT and GTF2H5 (p8 or TTD-A) cells, we observed a coordinated recruitment of all TFIIH subunits at the RARB2 promoter upon t-RA induction (Figure 2). Conversely, the TFIIH subunits were no longer recruited together in cells bearing mutations in ERCC3 and ERCC2. In particular, the recruitment of the TFIIH subunits appeared disorganized and took place over a longer period of time in ERCC2 (XPD)-p.Arg683Trp and ERCC2-p.Arg722Trp cells (Figures 2I2–2I4 and 2J2–2J4). These observations suggest that TFIIH is not recruited as a whole in the presence of subunit variants that affect its integrity. Interestingly, this observation illustrates the plasticity existing within TFIIH. Immunofluorescence experiments have previously shown that GTF2H5 is present in two distinct kinetic pools: one bound to TFIIH and a free fraction that shuttles between the cytoplasm and nucleus.29 Distinct movements of TFIIH subunits were also observed in Rift-Valley-fever-virus-infected cells, in which the GTF2H2 (p44) and ERCC3 subunits were sequestered in nuclear filaments formed by the viral protein NSs, whereas ERCC2 and GTF2H1 (p62) were maintained in the cytoplasm and proteolyzed in the nucleus, respectively.48,49 Moreover, it has been shown that a dynamic composition of TFIIH occurs during NER, in which the arrival of XPA at damaged DNA is subsequent to the removal of the CAK sub-complex.50 This might explain at least partially why CAK is also found free in cellular extracts. The ERCC2 subunit that can be found associated with CAK51,52 might also be implicated in a complex other than TFIIH, such as MMXD, a TFIIH-independent ERCC2-MMS19 protein complex involved in chromosome segregation.53 Although each subunit and/or sub-complex can act independently, our data suggest that the activity of TFIIH in pol-II-dependent transcription results from the coordinated assembly of all its subunits.
TFIIH: A Platform to Recruit NER Factors during Transcription
In some cases, failure in the integrity and/or formation of TFIIH affects the enzymatic activities of this complex. In particular, the ERCC3 p.Phe99Ser variant weakens the interaction between ERCC3 and its regulatory GTF2HH4 (p52) subunit,37 disrupting the unwinding of activated promoters, which is a key point during transcription initiation. Similarly, the ERCC2 p.Arg683Trp and p.Arg722Trp variants are known to weaken the binding between ERCC2 and GTF2H2 (p44)32 and thus affect the ability of CAK to phosphorylate pol II (Figure 5), a key step for promoter escape. Accordingly, the transcriptional defects associated with these ERCC2 mutations might be more related to disruption of pol II phosphorylation than to ERCC2 helicase deficiencies, reinforcing the idea that ERCC2 helicase activity is not crucial for transcription.
The gene-expression defects observed in cells bearing mutations in ERCC2 and ERCC3 could not, however, be solely explained by deficiencies in the enzymatic activities of TFIIH. This is well illustrated by the fact that ERCC3 p.Thr119Pro, ERCC2 p.Arg112His, and ERCC2 p.Gly602Asp did not affect basal transcription in an in vitro context, whereas the expression of RARB2 was defective in cells expressing the same variants (Figures 5A and 5B and Figures 2C1, 2G1, and 2H1). It is likely that TFIIH variants might disturb the accurate recruitment and/or positioning of components required for RNA synthesis. Our results suggest that such recruitment of NER factors is either incomplete or unsynchronized with the general transcription machinery (Figure 2). In particular, although ERCC5 stabilizes TFIIH and contributes to its transactivation function, no simultaneous recruitment of ERCC5, TFIIH, or the transcription machinery was observed in ERCC2-p.Arg683Trp cells (Figures 2I2–2I4); this might be related to the inability of ERCC5 to interact with this aberrant form of ERCC2.46 Furthermore, our results showed that the recruitment of NER factors during transcription was differently affected depending on the nature of the TFIIH variants. For instance, in ERCC3-p.Thr119Pro cells, ERCC4 (XPF) was not detected at the RARB2 promoter, on which the recruitment of all basal transcription machinery, including TFIIH, was deregulated (Figures 2C2–2C4). However, in ERCC2-p.Gly602Asp cells, TFIIH subunits and NER factors XPA and ERCC5, but not ERCC4, were correctly recruited (Figures 2H2–2H4).
All together, our results show that TFIIH promotes the recruitment of NER factors during transcription in a manner different from that observed during NER. This was well observed in GTF2H5 cells, in which the recruitment of NER factors was normal during transcription (Figures 2E2–2E4), whereas the GTF2H5 variant prevented the recruitment of these factors during the NER pathway.54 In parallel, the recruitment of the NER factors during transcription occurred normally in cells expressing ERCC2 p.Gly602Asp (Figures 2H2–2H4), which is known to prevent damaged DNA from opening and the recruitment of XPA in NER.32 The molecular aspects that differently influence the NER factors during transcription and DNA repair are currently unclear.
TFIIH Orchestrates Chromatin Remodeling
According to the chromatin signatures observed in WT cells, it seems that TFIIH variants alter chromatin remodeling upon transcription by disturbing histone PTMs, DNA demethylation, and gene-loop formation. These variants prevent the establishment of permissive chromatin by causing an intermediate environment that includes euchromatic histone PTMs (H3K4me and H3K9ac) and heterochromatin hallmarks (methylated DNA and impaired gene-loop formation). In cells bearing mutations in genes encoding TFIIH subunits, we observed that H3K4me and H3K9ac occurred all along the time course and that these modifications did not follow the pattern of RNA synthesis (Figures 2 and 3). In most cases, H3K9me was strongly reduced, suggesting that the deficiency might particularly concern the methylation or demethylation process of the lysine residue. Furthermore, mutations in ERCC3 and ERCC2 seem to profoundly alter the tight connection that exists between histone PTMs and the methylated status of DNA,55 which is well illustrated by the concomitant presence of H3K4me2 and H3K9ac and the methylated DNA at the promoter (Figure 3). Indeed, in addition to the histone PTM disturbance, the active DNA demethylation that occurs upon transcription was abolished for all of cells bearing mutations in ERCC2 and ERCC3. Several recent studies have documented the involvement of DNA-repair factors, including the endonuclease ERCC5, in the regulation of DNA demethylation.11,56 We have notably correlated DNA breaks involving ERCC5 with the 5mC sites that were demethylated.35 Interestingly, ERCC3 p.Phe99Ser, ERCC2 p.Gly602Asp, and ERCC2 p.Arg683Trp, which affect ERCC5 recruitment, might contribute to the absence of DNA breaks surrounding the RARB2 promoter (Figures 2B4, 2H4, and 2I4 and Figures 3B2, 3H2, and 3I2). Conversely, DNA breaks were maintained in GTF2H5, ERCC2-p.Arg112His, and ERCC2-p.Arg722Trp cells, in which ERCC5 was normally recruited but without the expected co-detection of unmethylated DNA (Figures 3D2, 3D3, 3G2, 3G3, 3J2, and 3J3). DNA breaks therefore occur even in the absence of DNA demethylation, indicating that the presence of ERCC5 and the related cuts are necessary but not sufficient to achieve such a process. It should be noted that unmethylated DNA influences long-range chromosomal interactions by being targeted by chromatin organizers such as CTCF, as previously observed for the imprinted Igf2-H19.57,58 Consequently, the defect of DNA demethylation observed in cells bearing mutations in genes encoding TFIIH subunits might contribute in part to the absence of an inducible CTCF-dependent chromatin loop between the promoter and the terminator of RARB2 (Figure 4).
Taken together, our data underline the key role of TFIIH in the transcription process, in which its primary enzymatic activities are combined with the recruitment of NER factors to further orchestrate events such as histone PTMs, DNA breaks, DNA methylation, and loop formation. Although each mutation in genes coding for TFIIH subunits specifically affects transcription, it seems that, in addition to causing DNA-repair deficiencies, subtle differences in the transcription defects might contribute to the phenotypic heterogeneity observed among XP, XP/CS, and TTD individuals.
Acknowledgments
We thank F. Coin for fruitful discussion, T. Sexton and F. Costanzo for critical reading of the manuscript, C. Braun for help in in vitro experiments, and the Institute of Genetics and Molecular and Cellular Biology Cell Culture Facility. This study was supported by grants from the European Research Council Advanced Scientists, l’Association de la Recherche contre le Cancer, l’Agence Nationale de Recherche, la Ligue Nationale contre le Cancer “Equipe Labellisée Ligue,” l’Association Nationale des Membres de l’Ordre National du Mérite ANMONM, and the Korean National Research Foundation for international collaboration (Global Research Laboratory program). A.S. was supported by Advanced European Research Council grant and by la Ligue Nationale contre le Cancer fellowship for young scientists.
Contributor Information
Nicolas Le May, Email: nlemay@igbmc.fr.
Jean-Marc Egly, Email: egly@igbmc.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
OMIM, http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Compe E., Egly J.M. TFIIH: when transcription met DNA repair. Nat. Rev. Mol. Cell Biol. 2012;13:343–354. doi: 10.1038/nrm3350. [DOI] [PubMed] [Google Scholar]
- 2.Zurita M., Merino C. The transcriptional complexity of the TFIIH complex. Trends Genet. 2003;19:578–584. doi: 10.1016/j.tig.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Schaeffer L., Roy R., Humbert S., Moncollin V., Vermeulen W., Hoeijmakers J.H., Chambon P., Egly J.M. DNA repair helicase: a component of BTF2 (TFIIH) basic transcription factor. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- 4.Feaver W.J., Svejstrup J.Q., Bardwell L., Bardwell A.J., Buratowski S., Gulyas K.D., Donahue T.F., Friedberg E.C., Kornberg R.D. Dual roles of a multiprotein complex from S. cerevisiae in transcription and DNA repair. Cell. 1993;75:1379–1387. doi: 10.1016/0092-8674(93)90624-y. [DOI] [PubMed] [Google Scholar]
- 5.Schärer O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013;5:a012609. doi: 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanawalt P.C., Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 7.Holstege F.C., van der Vliet P.C., Timmers H.T. Opening of an RNA polymerase II promoter occurs in two distinct steps and requires the basal transcription factors IIE and IIH. EMBO J. 1996;15:1666–1677. [PMC free article] [PubMed] [Google Scholar]
- 8.Coin F., Bergmann E., Tremeau-Bravard A., Egly J.M. Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J. 1999;18:1357–1366. doi: 10.1093/emboj/18.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu H., Zawel L., Fisher L., Egly J.M., Reinberg D. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature. 1992;358:641–645. doi: 10.1038/358641a0. [DOI] [PubMed] [Google Scholar]
- 10.Rochette-Egly C., Adam S., Rossignol M., Egly J.M., Chambon P. Stimulation of RAR alpha activation function AF-1 through binding to the general transcription factor TFIIH and phosphorylation by CDK7. Cell. 1997;90:97–107. doi: 10.1016/s0092-8674(00)80317-7. [DOI] [PubMed] [Google Scholar]
- 11.Barreto G., Schäfer A., Marhold J., Stach D., Swaminathan S.K., Handa V., Döderlein G., Maltry N., Wu W., Lyko F., Niehrs C. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- 12.Le May N., Mota-Fernandes D., Vélez-Cruz R., Iltis I., Biard D., Egly J.M. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell. 2010;38:54–66. doi: 10.1016/j.molcel.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Schmitz K.M., Schmitt N., Hoffmann-Rohrer U., Schäfer A., Grummt I., Mayer C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol. Cell. 2009;33:344–353. doi: 10.1016/j.molcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 14.Scott R.J., Itin P., Kleijer W.J., Kolb K., Arlett C., Muller H. Xeroderma pigmentosum-Cockayne syndrome complex in two patients: absence of skin tumors despite severe deficiency of DNA excision repair. J. Am. Acad. Dermatol. 1993;29:883–889. doi: 10.1016/0190-9622(93)70263-s. [DOI] [PubMed] [Google Scholar]
- 15.Weeda G., Rossignol M., Fraser R.A., Winkler G.S., Vermeulen W., van ’t Veer L.J., Ma L., Hoeijmakers J.H., Egly J.M. The XPB subunit of repair/transcription factor TFIIH directly interacts with SUG1, a subunit of the 26S proteasome and putative transcription factor. Nucleic Acids Res. 1997;25:2274–2283. doi: 10.1093/nar/25.12.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stefanini M., Vermeulen W., Weeda G., Giliani S., Nardo T., Mezzina M., Sarasin A., Harper J.I., Arlett C.F., Hoeijmakers J.H.J. A new nucleotide-excision-repair gene associated with the disorder trichothiodystrophy. Am. J. Hum. Genet. 1993;53:817–821. [PMC free article] [PubMed] [Google Scholar]
- 17.Stefanini M., Giliani S., Nardo T., Marinoni S., Nazzaro V., Rizzo R., Trevisan G. DNA repair investigations in nine Italian patients affected by trichothiodystrophy. Mutat. Res. 1992;273:119–125. doi: 10.1016/0921-8777(92)90073-c. [DOI] [PubMed] [Google Scholar]
- 18.Takayama K., Salazar E.P., Lehmann A., Stefanini M., Thompson L.H., Weber C.A. Defects in the DNA repair and transcription gene ERCC2 in the cancer-prone disorder xeroderma pigmentosum group D. Cancer Res. 1995;55:5656–5663. [PubMed] [Google Scholar]
- 19.Broughton B.C., Steingrimsdottir H., Weber C.A., Lehmann A.R. Mutations in the xeroderma pigmentosum group D DNA repair/transcription gene in patients with trichothiodystrophy. Nat. Genet. 1994;7:189–194. doi: 10.1038/ng0694-189. [DOI] [PubMed] [Google Scholar]
- 20.Taylor E.M., Broughton B.C., Botta E., Stefanini M., Sarasin A., Jaspers N.G., Fawcett H., Harcourt S.A., Arlett C.F., Lehmann A.R. Xeroderma pigmentosum and trichothiodystrophy are associated with different mutations in the XPD (ERCC2) repair/transcription gene. Proc. Natl. Acad. Sci. USA. 1997;94:8658–8663. doi: 10.1073/pnas.94.16.8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cleaver J.E. Splitting hairs—discovery of a new DNA repair and transcription factor for the human disease trichothiodystrophy. DNA Repair (Amst.) 2005;4:285–287. doi: 10.1016/j.dnarep.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 22.Lehmann A.R., Norris P.G. DNA repair and cancer: speculations based on studies with xeroderma pigmentosum, Cockayne’s syndrome and trichothiodystrophy. Carcinogenesis. 1989;10:1353–1356. doi: 10.1093/carcin/10.8.1353. [DOI] [PubMed] [Google Scholar]
- 23.Bradford P.T., Goldstein A.M., Tamura D., Khan S.G., Ueda T., Boyle J., Oh K.S., Imoto K., Inui H., Moriwaki S. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J. Med. Genet. 2011;48:168–176. doi: 10.1136/jmg.2010.083022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itin P.H., Sarasin A., Pittelkow M.R. Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes. J. Am. Acad. Dermatol. 2001;44:891–920. doi: 10.1067/mjd.2001.114294. quiz 921–924. [DOI] [PubMed] [Google Scholar]
- 25.Brooks P.J., Cheng T.F., Cooper L. Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage? DNA Repair (Amst.) 2008;7:834–848. doi: 10.1016/j.dnarep.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yokoi M., Masutani C., Maekawa T., Sugasawa K., Ohkuma Y., Hanaoka F. The xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J. Biol. Chem. 2000;275:9870–9875. doi: 10.1074/jbc.275.13.9870. [DOI] [PubMed] [Google Scholar]
- 27.Winkler G.S., Vermeulen W., Coin F., Egly J.M., Hoeijmakers J.H., Weeda G. Affinity purification of human DNA repair/transcription factor TFIIH using epitope-tagged xeroderma pigmentosum B protein. J. Biol. Chem. 1998;273:1092–1098. doi: 10.1074/jbc.273.2.1092. [DOI] [PubMed] [Google Scholar]
- 28.Riou L., Zeng L., Chevallier-Lagente O., Stary A., Nikaido O., Taïeb A., Weeda G., Mezzina M., Sarasin A. The relative expression of mutated XPB genes results in xeroderma pigmentosum/Cockayne’s syndrome or trichothiodystrophy cellular phenotypes. Hum. Mol. Genet. 1999;8:1125–1133. doi: 10.1093/hmg/8.6.1125. [DOI] [PubMed] [Google Scholar]
- 29.Giglia-Mari G., Miquel C., Theil A.F., Mari P.O., Hoogstraten D., Ng J.M., Dinant C., Hoeijmakers J.H., Vermeulen W. Dynamic interaction of TTDA with TFIIH is stabilized by nucleotide excision repair in living cells. PLoS Biol. 2006;4:e156. doi: 10.1371/journal.pbio.0040156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giglia-Mari G., Coin F., Ranish J.A., Hoogstraten D., Theil A., Wijgers N., Jaspers N.G., Raams A., Argentini M., van der Spek P.J. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat. Genet. 2004;36:714–719. doi: 10.1038/ng1387. [DOI] [PubMed] [Google Scholar]
- 31.Vernimmen D., De Gobbi M., Sloane-Stanley J.A., Wood W.G., Higgs D.R. Long-range chromosomal interactions regulate the timing of the transition between poised and active gene expression. EMBO J. 2007;26:2041–2051. doi: 10.1038/sj.emboj.7601654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dubaele S., Proietti De Santis L., Bienstock R.J., Keriel A., Stefanini M., Van Houten B., Egly J.M. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol. Cell. 2003;11:1635–1646. doi: 10.1016/s1097-2765(03)00182-5. [DOI] [PubMed] [Google Scholar]
- 33.Dubois M.F., Vincent M., Vigneron M., Adamczewski J., Egly J.M., Bensaude O. Heat-shock inactivation of the TFIIH-associated kinase and change in the phosphorylation sites on the C-terminal domain of RNA polymerase II. Nucleic Acids Res. 1997;25:694–700. doi: 10.1093/nar/25.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zentner G.E., Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013;20:259–266. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 35.Le May N., Fradin D., Iltis I., Bougnères P., Egly J.M. XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol. Cell. 2012;47:622–632. doi: 10.1016/j.molcel.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 36.Ju B.G., Lunyak V.V., Perissi V., Garcia-Bassets I., Rose D.W., Glass C.K., Rosenfeld M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 37.Coin F., Oksenych V., Egly J.M. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol. Cell. 2007;26:245–256. doi: 10.1016/j.molcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 38.Coin F., Marinoni J.C., Rodolfo C., Fribourg S., Pedrini A.M., Egly J.M. Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat. Genet. 1998;20:184–188. doi: 10.1038/2491. [DOI] [PubMed] [Google Scholar]
- 39.Feaver W.J., Gileadi O., Li Y., Kornberg R.D. CTD kinase associated with yeast RNA polymerase II initiation factor b. Cell. 1991;67:1223–1230. doi: 10.1016/0092-8674(91)90298-d. [DOI] [PubMed] [Google Scholar]
- 40.Dvir A., Conaway R.C., Conaway J.W. A role for TFIIH in controlling the activity of early RNA polymerase II elongation complexes. Proc. Natl. Acad. Sci. USA. 1997;94:9006–9010. doi: 10.1073/pnas.94.17.9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keriel A., Stary A., Sarasin A., Rochette-Egly C., Egly J.M. XPD mutations prevent TFIIH-dependent transactivation by nuclear receptors and phosphorylation of RARalpha. Cell. 2002;109:125–135. doi: 10.1016/s0092-8674(02)00692-x. [DOI] [PubMed] [Google Scholar]
- 42.Kim M., Suh H., Cho E.J., Buratowski S. Phosphorylation of the yeast Rpb1 C-terminal domain at serines 2, 5, and 7. J. Biol. Chem. 2009;284:26421–26426. doi: 10.1074/jbc.M109.028993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Traboulsi H., Davoli S., Catez P., Egly J.M., Compe E. Dynamic partnership between TFIIH, PGC-1α and SIRT1 is impaired in trichothiodystrophy. PLoS Genet. 2014;10:e1004732. doi: 10.1371/journal.pgen.1004732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohkuma Y., Roeder R.G. Regulation of TFIIH ATPase and kinase activities by TFIIE during active initiation complex formation. Nature. 1994;368:160–163. doi: 10.1038/368160a0. [DOI] [PubMed] [Google Scholar]
- 45.Akoulitchev S., Chuikov S., Reinberg D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature. 2000;407:102–106. doi: 10.1038/35024111. [DOI] [PubMed] [Google Scholar]
- 46.Ito S., Kuraoka I., Chymkowitch P., Compe E., Takedachi A., Ishigami C., Coin F., Egly J.M., Tanaka K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol. Cell. 2007;26:231–243. doi: 10.1016/j.molcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 47.Esnault C., Ghavi-Helm Y., Brun S., Soutourina J., Van Berkum N., Boschiero C., Holstege F., Werner M. Mediator-dependent recruitment of TFIIH modules in preinitiation complex. Mol. Cell. 2008;31:337–346. doi: 10.1016/j.molcel.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 48.Le May N., Dubaele S., Proietti De Santis L., Billecocq A., Bouloy M., Egly J.M. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell. 2004;116:541–550. doi: 10.1016/s0092-8674(04)00132-1. [DOI] [PubMed] [Google Scholar]
- 49.Kalveram B., Lihoradova O., Ikegami T. NSs protein of rift valley fever virus promotes posttranslational downregulation of the TFIIH subunit p62. J. Virol. 2011;85:6234–6243. doi: 10.1128/JVI.02255-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coin F., Oksenych V., Mocquet V., Groh S., Blattner C., Egly J.M. Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol. Cell. 2008;31:9–20. doi: 10.1016/j.molcel.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 51.Drapkin R., Le Roy G., Cho H., Akoulitchev S., Reinberg D. Human cyclin-dependent kinase-activating kinase exists in three distinct complexes. Proc. Natl. Acad. Sci. USA. 1996;93:6488–6493. doi: 10.1073/pnas.93.13.6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reardon J.T., Ge H., Gibbs E., Sancar A., Hurwitz J., Pan Z.Q. Isolation and characterization of two human transcription factor IIH (TFIIH)-related complexes: ERCC2/CAK and TFIIH. Proc. Natl. Acad. Sci. USA. 1996;93:6482–6487. doi: 10.1073/pnas.93.13.6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ito S., Tan L.J., Andoh D., Narita T., Seki M., Hirano Y., Narita K., Kuraoka I., Hiraoka Y., Tanaka K. MMXD, a TFIIH-independent XPD-MMS19 protein complex involved in chromosome segregation. Mol. Cell. 2010;39:632–640. doi: 10.1016/j.molcel.2010.07.029. [DOI] [PubMed] [Google Scholar]
- 54.Coin F., Proietti De Santis L., Nardo T., Zlobinskaya O., Stefanini M., Egly J.M. p8/TTD-A as a repair-specific TFIIH subunit. Mol. Cell. 2006;21:215–226. doi: 10.1016/j.molcel.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 55.Cedar H., Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 56.Williams K., Christensen J., Helin K. DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep. 2012;13:28–35. doi: 10.1038/embor.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murrell A., Heeson S., Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat. Genet. 2004;36:889–893. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- 58.Wang H., Maurano M.T., Qu H., Varley K.E., Gertz J., Pauli F., Lee K., Canfield T., Weaver M., Sandstrom R. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012;22:1680–1688. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.