

Abstract:
Although convention dictates that G protein-coupled receptors localize to and signal at the plasma membrane, accumulating evidence suggests that G protein-coupled receptors localize to and signal at intracellular membranes, most notably the nucleus. In fact, there is now significant evidence indicating that endogenous alpha-1 adrenergic receptors (α1-ARs) localize to and signal at the nuclei in adult cardiac myocytes. Cumulatively, the data suggest that α1-ARs localize to the inner nuclear membrane, activate intranuclear signaling, and regulate physiologic function in adult cardiac myocytes. Although α1-ARs signal through Gαq, unlike other Gq-coupled receptors, α1-ARs mediate important cardioprotective functions including adaptive/physiologic hypertrophy, protection from cell death (survival signaling), positive inotropy, and preconditioning. Also unlike other Gq-coupled receptors, most, if not all, functional α1-ARs localize to the nuclei in adult cardiac myocytes, as opposed to the sarcolemma. Together, α1-AR nuclear localization and cardioprotection might suggest a novel model for compartmentalization of Gq-coupled receptor signaling in which nuclear Gq-coupled receptor signaling is cardioprotective.
Key Words: alpha-1-adrenergic receptor, cardiac myocyte
INTRODUCTION
Alpha-1 adrenergic receptors (α1-ARs) are G protein-coupled receptors (GPCRs) that bind to and are activated by the endogenous catecholamines norepinephrine, and epinephrine. Classically, α1-ARs signal through Gαq/11 leading to the activation of phospholipase Cβ (PLCβ) with the subsequent production of inositol phosphates (IP) and diacylglycerol (DAG) leading to the release of calcium from IP-sensitive stores and the activation of protein kinase C (PKC), respectively.1 The heart expresses all 3 α1-AR subtype mRNAs, α1A, α1B, and α1D,2–5 but at the protein level, cardiac myocytes express only α1A- and α1B-subtype,3,6 fibroblasts do not express α1-ARs,5,7 and coronary vasculature express only the α1D-subtype.6,8 Although α1-ARs account for only 10% of the total adrenergic receptors (ARs) in cardiac myocytes (β-ARs account for 90%),1 studies in cell and animal models indicate that α1-ARs mediate important cardioprotective functions, including adaptive/physiologic hypertrophy, protection from cell death (survival signaling), positive inotropy, and preconditioning (Review: Ref. 1). Combined with the failure of α1-AR antagonists in clinical trials (Review: Ref. 1), these recently identified cardioprotective functions of α1-ARs suggest that α1-AR agonist therapy could represent a novel therapeutic strategy for heart failure (HF).1,9 Although α1-AR cardiac physiology is well studied, the identification of α1-AR signaling mechanisms in cardiac myocytes continues to expand, and more than 70 molecules have been identified as targets of α1-AR signaling thus far.9
Currently, the prevailing dogma is that GPCRs localize to and signal at the plasma membrane in cardiac myocytes (or any cell) to regulate many important processes in health and disease, including hypertrophy, contraction, and cell death. Compartmentalization of GPCR signaling is also important, and examples in cardiac myocytes include the regulation of 3′-5′-cyclic adenosine monophosphate signaling by β-ARs,10 or the influence of neuronal activity on the localization of β-ARs.11 Interestingly, recent studies indicate that several GPCRs localize to intracellular membranes, including the nuclear membrane.12–14 Examples of nuclear GPCRs in adult cardiac myocytes include α1-ARs (reviewed here), as well as β-ARs, endothelin receptors (ET-Rs), and angiotensin receptors (AT-Rs).14a These findings suggest that the nuclei in adult cardiac myocytes represent a novel site for compartmentalized GPCR signaling.
In this review, we delineate recent evidence primarily derived from cultured adult mouse cardiac myocytes indicating that α1-AR signaling is compartmentalized to the nuclei and describe how nuclear α1-AR signaling regulates physiologic function. We propose 3 important criteria that must be met to establish nuclear α1-AR signaling in adult cardiac myocytes: (1) Localization of endogenous α1-ARs to the nuclei, (2) Activation of α1-AR signaling at the nuclei, and (3) Physiologic significance of nuclear α1-AR signaling. Finally, based on our findings that α1-ARs are cardioprotective,1 we propose a novel model for nuclear Gq-coupled GPCR (Gq-receptor) cardioprotective signaling in adult cardiac myocytes.
LOCALIZATION OF ENDOGENOUS Α1-ARS TO THE NUCLEI IN ADULT CARDIAC MYOCYTES
Four lines of evidence suggest that endogenous α1-ARs localize to the nuclei in adult cardiac myocytes. First, radioligand-binding assays on isolated and fractionated adult cardiac myocytes detect 80% of endogenous α1-ARs in the nuclear fraction defined by the nuclear membrane protein lamina-associated polypeptide 2.15 Second, a fluorescent α1-AR ligand, boron-dipyrromethene (BODIPY)-prazosin, labels endogenous α1-ARs at the nuclei, but not the sarcolemma, in cultured adult cardiac myocytes.15,16 Third, in nuclei isolated from adult cardiac myocytes, BODIPY-prazosin labels the nuclear membrane.17 Fourth, reconstitution of α1-ARs in cardiac myocytes lacking endogenous α1A- and α1B-subtypes (α1ABKO) using α1-AR–green fluorescent protein fusion constructs (α1-GFP) recapitulates the nuclear localization of the endogenous α1-ARs.15,17
The finding that α1-ARs localize to the nuclei in adult cardiac myocytes might seem to conflict with previous studies that examined α1-AR localization. For example, α1-AR antibodies (Santa Cruz Biotechnology) detect receptors on the T-tubules and sarcolemma in cultured adult rat cardiac myocytes by immunocytochemistry.17a In contrast, a study performed in 2009 indicates that among 10 commercially available antibodies, none detects a specific signal in tissues of wild-type mice that was absent in α1-AR triple knockout mice.18 A more recent study reports the same result for 3 additional commercially available α1-AR antibodies.19 The lack of specificity of α1-AR antibodies is not unique, and a recent survey shows that among 49 commercially available antibodies against 19 different GPCRs, none are specific.20 In short, these findings raise concern about the use of antibodies to detect GPCR expression and localization.
Avoiding antibodies, other studies find that in cultured neonatal rat ventricular myocytes (NRVM), α1-ARs localize to caveolae.21–23 However, radioligand binding on caveolar fractions isolated from NRVM detect only 27% of total α1-ARs in the caveolar fraction,22 suggesting the majority of α1-ARs in NRVM are in other fractions. This result seems consistent with the finding that 80% of endogenous α1-ARs are at the nuclei in adult cardiac myocytes.15 It is worth noting that NRVM are not phenotypically mature adult cardiac myocytes. Moreover, rat cardiac myocytes express 10-fold more α1-ARs than other species,24 (rat ∼114 fmole/mg heart protein5,24–26; mouse: ∼12 fmole/mg heart protein2,3,27–29). Furthermore, in mice with 170-fold overexpression of an epitope tagged-α1A-subtype in the heart, the α1A-subtype localizes to the sarcolemma in ventricular tissue sections based on immunocytochemistry with an antibody to the epitope tag.27 However, in mice with 5-fold overexpression of an α1A-GFP fusion protein, the α1A-subtype localizes to cardiac myocyte nuclei based on immunocytochemistry with a GFP antibody,15 perhaps suggesting that expression level affects localization.
α1-AR Nuclear Localization Is Dictated by Nuclear Localization Signals in the C-terminus of Each Receptor Subtype
Targeting proteins to the nucleus requires a nuclear localization signal (NLS) encoded within the protein. These NLS are short single stretches (monopartite) of basic amino acids, usually lysines and arginines or glycine–arginine repeats or 2 short stretches of basic amino acids separated by a short random stretch of amino acids (bipartite).30–32 Importins, a family of proteins that recognize and bind NLS, facilitate transport of proteins through the nuclear pore complex, and proteins targeting the nucleoplasm and the inner nuclear membrane (INM) use this mechanism.33–35 Examples of GPCRs that use an NLS/importin-based mechanism to localize receptors to the nucleus include the parathyroid hormone receptor and the gonadotropin-releasing hormone receptor.36–38
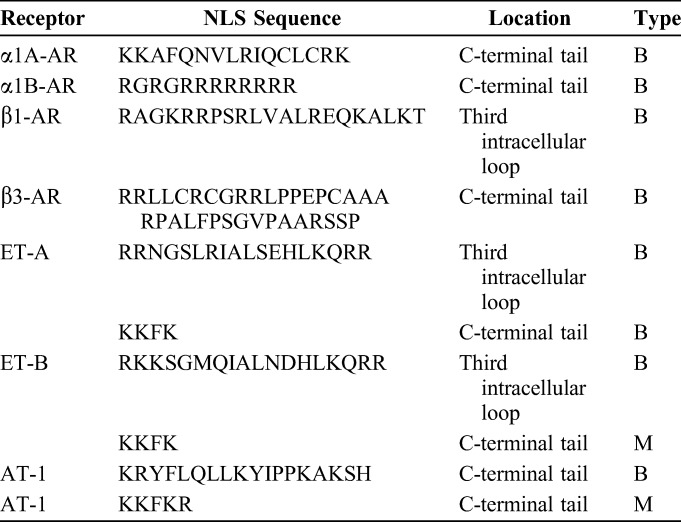
Analysis of the sequence of GPCRs previously localized to the nucleus in cardiac myocytes, including α1-ARs, ET-Rs, AT-Rs, and β-ARs, identifies at least 1 NLS in each receptor (Table 1). Importantly, mutation of NLS in the α1A- or α1B-subtypes (Table 1) results in loss of nuclear localization, although neither receptor redistributes to the sarcolemma.17 Functional NLS in α1-ARs suggest a mechanism for nuclear localization in adult cardiac myocytes, but α1-ARs do not localize exclusively to the nucleus in NRVM21–23 or in other cell types including smooth muscle cells.39 This indicates that an NLS is required but not sufficient for nuclear localization of α1-ARs. However, additional mechanisms required for nuclear localization remain to be determined. The difference in cell-specific localization of α1-ARs could have implications for pharmacologic targeting of receptors on the plasma membrane in smooth muscle cells or at the nuclei in adult cardiac myocytes.
TABLE 1.
Nuclear Localization Sequences of GPCRs
α1-ARs Localize to the Inner Nuclear Membrane Oriented With the C-terminus Facing Inside the Nucleus
The nuclear envelope consists of INM and outer nuclear membranes (ONM) separated by the perinuclear space. The nuclear pore complex forms a junction between the INM and ONM and regulates transport between the nucleus and cytosol. The INM contains functional integral membrane proteins, whereas the ONM is contiguous with the endoplasmic/sarcoplasmic reticulum (Review: Ref. 40). Conceivably, α1-ARs could reside in the INM or ONM, oriented in either direction, which would have a significant impact on α1-AR signaling.
Indirect evidence indicates that some GPCRs likely localize to the INM oriented with the C-terminus facing the nucleoplasm, thus inducing intranuclear signaling. In nuclei isolated from adult cardiac myocytes, α1-ARs, β-ARs, ET-Rs, and AT-Rs activate intranuclear signaling.41–46 Furthermore, α1-ARs and ET-Rs activate nuclear calcium transients, independent of changes in cytosolic calcium in cultured cardiac myocytes.46,47 Yet, without a suitable antibody, direct biochemical evidence for receptor localization and orientation is difficult to obtain. However, using a reconstitution system in α1ABKO myocytes to express the α1A-GFP, with the GFP on the C-terminus, fluorescent confocal microscopy and immunocytochemistry with a GFP antibody in differentially permeabilized myocytes detects α1-ARs in the INM oriented with the C-terminus facing the nucleoplasm.45 This is the first direct biochemical evidence for GPCR localization to the INM, oriented to induce intranuclear signaling in cardiac myocytes.
To our knowledge, biochemical evidence for GPCRs localization to the INM or ONM, aside from α1-ARs, is lacking in adult cardiac myocytes. However, human gonadotropin-releasing hormone receptor type I (GnRH-RI) colocalizes with the inner nuclear membrane protein lamin A/C in HEK293 cells.38 Receptor localization to the ONM or near the nucleus, as shown for the insulin growth factor receptor,48 is entirely possible and cannot be excluded. In adult cardiac myocytes, the insulin growth factor receptor localizes to perinuclear t-tubule invaginations of the plasma membrane and induces nuclear calcium transients.48 Furthermore, ET-Rs activate PLCε-mediated signaling at the ONM in adult cardiac myocytes.49 Therefore, indirect evidence also indicates that some GPCRs could localize to the ONM, and GPCR localization and orientation in the nuclear membrane remains a largely unanswered question.
Localization of α1-AR Signaling Partners at the Nucleus in Adult Cardiac Myocytes
In cardiac myocytes, α1-ARs signal primarily through Gαq and activate PLC as described above. For α1-ARs to signal at the nuclei, the receptors must colocalize with their downstream signaling partners. Nuclei isolated from adult cardiac myocytes express Gαq and PLCβ1,15 and Gαq and PLCβ1 colocalize with α1-ARs only at the nuclei in adult cardiac myocytes based on immunochemical detection of Gαq and PLCβ1 in α1ABKO cardiac myocytes expressing α1-GFP fusion proteins.15 Functionally, α1-ARs induce nuclear calcium transients47 and activation of PKC45 providing indirect evidence for PLC activity at the nuclei (described in detail below). However, direct biochemical evidence for PLCβ activity in the nuclei in adult cardiac myocytes is lacking, although PLCβ activity is found in the nucleus in other cell types (Review: Ref. 50). However, the nuclear membrane lacks phosphatidylinositol 4,5-bisphosphate, the substrate for PLCβ.49 Interestingly, PLCε is bound to muscle-specific A-kinase anchoring protein at the ONM and phosphatidylinositol 4-phosphate, the substrate for PLCε, are both found in the Golgi membrane, which surrounds the nuclei in adult cardiac myocytes, and activation of PLCε generates DAG and activates protein kinase D (PKD).49,51 Furthermore, PLCε knockdown blocks α1-induced hypertrophy, without altering inositol phosphate signaling, and cardiac myocyte-specific knockout of PLCε prevents pressure overload-induced hypertrophy.49 However, PLCε is activated by G12/13, small GTPases, and Gβγ, but is not activated directly by Gαq,52 so if and how α1-ARs activate PLCε is unclear at this time.
In addition to Gq and PLC isoforms, nuclei in adult cardiac myocytes express PKCα, δ, and ε, with PKCδ expression proportionally the highest and only trace amounts of PKCε.45 G protein-coupled receptor kinases (GRK) 2, 3, and 5 are expressed in the heart, but only GRK3 phosphorylates α1-ARs.53 Furthermore, HF does not induce GRK3 expression,54,55 and transgenic overexpression of GRK3ct, an inhibitor of GRK3, improves remodeling after transverse aortic constriction56 consistent with a protective function of α1-ARs. Whether cardiac myocyte nuclei express GRK3 is unknown, although prolonged incubation with an α1-AR ligand induces receptor movement of the α1A-subtype off the nuclei in adult cardiac myocytes, providing circumstantial evidence for receptor phosphorylation and desensitization.15 Interestingly, cardiac myocyte nuclei express GRK5, which surprisingly functions as a histone deacetylase (HDAC) kinase,56a but whether it phosphorylates GPCRs at the INM is unclear.
Summary
In adult cardiac myocytes, α1-ARs localize to the INM oriented with C-terminus facing the nucleoplasm such that activation of α1-ARs would induce intranuclear signaling. Although binding assays on fractionated myocytes detect a small fraction of receptors in a nominal plasma membrane fraction, BODIPY-prazosin staining fails to detect receptors at the sarcolemma in cultured adult cardiac myocytes indicating the majority if not all α1-ARs localize to the nuclei in adult cardiac myocytes. Finally, α1-AR signaling partners including Gαq, an yet to be defined PLC isozyme, PKCα, δ, and ε, and possibly GRK3 localize to the nuclei in adult cardiac myocytes, providing the necessary components for nuclear α1-AR signaling.
ACTIVATION OF Α1-AR SIGNALING AT THE NUCLEI IN ADULT CARDIAC MYOCYTES
Although conventional models of GPCR signaling suggest activation of signaling at the plasma membrane and transduction of signaling inside the cell, a so-called “outside-in” model, significant evidence indicates that α1-ARs induce intranuclear signaling that exits the nucleus to activate cytosolic targets or an “inside-out” model. Four lines of evidence support α1-AR signaling at the nuclei in adult cardiac myocytes including: (1) activation of α1-AR signaling at the nucleus in adult cardiac myocytes, (2) requirement for α1-AR nuclear localization to induce signaling, (3) failure to detect α1-AR signaling at the sarcolemma, and (4) rapid catecholamine uptake in a time frame consistent with signaling.
Activation of α1-AR and Ca2+ Signaling at the Nucleus in Adult Cardiac Myocytes
As mentioned above, nuclei from adult cardiac myocytes express PKCα, δ, and ε, and PKCδ is the most abundant isoform (relatively).45 Interestingly, in nuclei isolated from adult cardiac myocytes, the α1-AR agonist phenylephrine activates PKCδ, based on phosphorylation at the activation site Thr505 and increases total PKC activity, based on increased phosphorylation of the PKC target myristoylated alanine-rich PKC substrate.45 This would also support the idea that PLC signaling is activated at the nucleus, which is required to produce DAG needed to activate PKC. Furthermore, in NRVM, phenylephrine activates nuclear calcium transients that have the ability to trigger cytosolic calcium transients,47 consistent with the idea that α1-ARs signal at the nucleus in cardiac myocytes. Other Gq-coupled receptors signal at the nucleus in adult cardiac myocytes, including ET-Rs and AT-Rs. For example, endothelin induces calcium transients through activation of the ETB-R subtype in nuclei isolated from adult cardiac myocytes41 and intact cultured adult cardiac myocytes.46 Interestingly, endothelin-induced nuclear calcium transients arise from the nuclear–perinuclear space, which is contiguous with the sarcoplasmic reticulum. Moreover, these transients arise in the presence of ryanodine, suggesting either the presence of nuclear ryanodine receptors46 or nuclear IP3 receptors.46,57 Similarly, angiotensin induces calcium transients through activation of AT1-R subtype in nuclei isolated from adult cardiac myocytes.43 Collectively, these finding indicate that nuclei from adult cardiac myocytes contain machinery sufficient to induce α1-AR signaling specifically and Gq-coupled GPCR signaling in general. Furthermore, activation of intranuclear GPCR signaling supports the localization of α1-ARs to the INM with the C-terminus facing the nucleoplasm.45
Requirement for Nuclear Localization to Induce α1-AR Signalingin Adult Cardiac Myocytes
Mutation of putative NLS in the C-terminus of both the α1A- and α1B-subtypes leads to a loss of nuclear localization in adult cardiac myocytes.17 Importantly, neither NLS overlaps with sequences required for ligand binding in the third and fifth transmembrane domains or G-protein activation in the third intracellular loop.58–60 Furthermore, in HeLa cells, where α1-ARs do not localize to the nucleus (despite the NLS), activation of α1A- or α1B-NLS mutant receptors induces phosphorylation of extracellular signal-regulated kinase (ERK), indicating that the α1-NLS mutant receptors are functional.45 However, neither α1-NLS mutant receptor (α1A or α1B) can reconstitute α1-AR–induced phosphorylation of ERK17 or phosphorylation of cardiac troponin I (cTnI)45 in α1ABKO cardiac myocytes. This demonstrates a requirement for nuclear localization to induce α1-AR signaling and suggests that the α1-NLS mutant receptors function as localization knockouts in adult cardiac myocytes.17,45
Surprisingly, nuclear α1-ARs induce phosphorylation of ERK in caveolae at the sarcolemma15 and cTnI at the sarcomere.45 However, pretreatment of adult cardiac myocytes with the nuclear export inhibitor leptomycin B inhibits phenylephrine-mediated phosphorylation of ERK and cTnI.17,45 This indicates that α1-AR signaling arises in the nucleus and exits to activate cytosolic targets, supporting localization of α1-ARs in the INM, activation of intranuclear signaling, and providing evidence for an “inside-out” signaling mechanism.
Failure to Detect α1-AR Signaling at the Sarcolemma in Adult Cardiac Myocytes
Although BODIPY-prazosin failed to detect α1-ARs at the sarcolemma, binding assays of fractionated adult cardiac myocytes have detected a small portion of receptor (<20%) in the nominal plasma membrane fraction.15 Regardless, the implication is that some α1-AR signaling could occur at the sarcolemma in adult cardiac myocytes. However, a membrane impermeant α1-AR antagonist fails to block α1-AR signaling in adult cardiac myocytes. Specifically, CGP-12177A, a membrane impermeant β-AR antagonist that can block α1-ARs at higher concentrations, fails to block phenylephrine-induced phosphorylation of ERK at concentrations as high as 200 μM. In contrast, prazosin, a membrane permeable α1-AR antagonist, does block phenylephrine-induced phosphorylation of ERK.15 This suggests that there might not be functional α1-ARs at the sarcolemma in adult cardiac myocytes.
Rapid Catecholamine Uptake Mediated by Organic Cation Transporters 3 in Adult Cardiac Myocytes
To activate α1-ARs at the nuclear membrane, endogenous catecholamines, norepinephrine, and epinephrine, must cross the sarcolemma and ONM to the reach the perinuclear space to bind to and activate α1-ARs in the INM in a time frame (seconds) consistent with signaling. The slc22A gene family of organic cation transporters (OCT1-3) mediates catecholamine uptake in cardiac myocytes and smooth muscle cells,61,62 a process termed norepinephrine uptake-2. Importantly, the heart expresses high levels of OCT3, with lower levels of OCT1,63 and based on immunocytochemistry, adult cardiac myocytes express OCT3 on the sarcolemma and nuclear membrane.15
In uptake assays with a fluorescent catecholamine analog, OCT3-mediated catecholamine uptake occurs rapidly in adult cardiac myocytes, on the order of seconds, steadily increasing for several minutes, peaking by 30 minutes, and is inhibited by preincubation with unlabeled norepinephrine.15 The biophysical properties of OCT3 also support rapid catecholamine uptake. The uptake kinetics of recombinant OCT3 expressed in HEK293 cells indicate a Vmax of ∼30,000 pmole·mg−1 protein per minute and kilometer of ∼900 μM for norepinephrine, and ∼13,000 and ∼500 for epinephrine respectively.64 In this system, OCT3 catecholamine uptake occurs in seconds, with a half maximal response of 2 minutes.64 These biophysical properties are likely consistent between cells with expression level dictating the amount of uptake, and the heart expresses high levels of OCT3.63
In mice, cardiac uptake of the cation methyl-4-phenylpyridinium acetate happens quickly (∼4000 ng/g heart tissue in 5 minutes) but is reduced 75% in OCT3 knockout mice.63 In addition, OCT3 knockout mice mimic α1ABKO mice with a trend towards a small heart (OCT3 KO, 10% smaller heart, n = 7 NS, α1ABKO, 17% smaller heart, P < 0.05, n = 27–33).3,63 In cultured adult cardiac myocytes, blockade of OCT3 uptake with corticosterone inhibits α1-AR–mediated phosphorylation (activation) of ERK.15 Furthermore, unlike β-AR inotropic responses, which occur rapidly, α1-AR inotropy and calcium transients are delayed, with a latency of 2–5 minutes in isolated myocytes,17a,65–70 consistent with catecholamines having to cross the membrane and reach the nucleus to activate signaling.
In summary, the high level of OCT3 expression in the heart, the kinetics of catecholamine uptake in adult cardiac myocyte, the reduced cation uptake with the trend toward a small heart in OCT3 KO mice, the ability to block α1-AR signaling by inhibiting OCT3 in adult cardiac myocytes, and the latency of α1-AR contractile function all suggest that catecholamine uptake is required for α1-AR signaling.
Summary
Evidence from studies with isolated nuclei and whole cells indicates that α1-ARs induce intranuclear signaling in adult cardiac myocytes. In isolated nuclei, α1-ARs activate PKC indicating that nuclei contain machinery sufficient to induce α1-AR (GPCR) signaling and supporting the observation that α1-ARs localize to the INM oriented to induce intranuclear signaling. Furthermore, the failure of α1-AR localization mutants (α1-NLS mutants) to reconstitute signaling in α1ABKO cardiac myocytes demonstrates a requirement for nuclear localization. The inability of the membrane impermeant α1-AR antagonist CGP-12177A to block α1-AR signaling suggests little or no functional α1-AR signaling at the sarcolemma in adult cardiac myocytes. Finally, consistent with nuclear α1-AR signaling, OCT3 mediates rapid uptake of catecholamines in cardiac myocytes, which is required for signaling.
PHYSIOLOGIC SIGNIFICANCE OF NUCLEAR Α1-AR SIGNALING IN ADULT CARDIAC MYOCYTES
Establishing the physiologic significance of nuclear GPCR signaling is paramount to validating a model of nuclear α1-AR signaling. In the heart, α1-ARs mediate adaptive/physiologic hypertrophy, positive inotropic responses, prevent cell death, and induce preconditioning.1,9 A confounding factor for many nuclear GPCRs, for example ET-Rs, is that only a fraction of the total receptor population localizes to the nucleus (5% for ET-Rs).41 However, α1-ARs localize primarily to the nuclei in adult cardiac myocytes, and the lack of functional α1-ARs at the sarcolemma15 simplifies ascribing physiologic function of nuclear α1-ARs.
Requirement for Nuclear Localization of α1-ARs to Induce Inotropic Responses in Adult Cardiac Myocytes
In cardiac myocytes, α1-ARs induce positive inotropic responses, but in the basal state, α1-AR–mediated inotropic responses are relatively minor. In HF, where β-ARs are downregulated, α1-AR inotropy becomes more significant (Review: Ref. 1). In fact, α1-AR–mediated inotropy can equal β-AR–mediated inotropy in muscle strips isolated from failing human hearts.71 In mice, transgenic overexpression of the α1A-subtype induces a basal hypercontractile phenotype that is protective against pathologic stress induced by ischemic injury or pressure overload.72,73
α1-ARs induce inotropic response through a variety of mechanisms including altering K+ and Ca2+ currents, intracellular pH, and myofilament Ca2+ sensitivity (Review: Ref. 1). Interestingly, in adult cardiac myocytes, phenylephrine induces an inotropic response correlated with phosphorylation of cTnI at a putative PKCδ site, threonine 144 (T144). However, in α1ABKO cardiac myocytes, both responses are lost. Reconstitution of the α1A-sbutype, but not the α1B-subtype, restores phenylephrine-induced inotropy and phosphorylation of cTnI at T144, whereas the α1A-NLS “localization mutant” fails to restore function.45 This demonstrates that nuclear localization is required for α1-AR–mediated inotropy in adult cardiac myocytes.45
Requirement for Nuclear Localization of α1-AR to Induce Survival Signaling in Adult Cardiac Myocytes
In addition to regulating inotropic signaling, the α1A-subtype prevents cardiac myocyte death in response to pathologic stress. In adult cardiac myocytes, an α1A-subtype ERK signaling pathway prevents cell death (survival signaling),16 and the absence of this survival signaling pathway might help explain the pathologic response to pressure overload in α1ABKO mice.74 In adult cardiac myocytes, phenylephrine induces phosphorylation of ERK, but not in α1ABKO cardiac myocytes. However, reconstitution of the α1A- or α1B-subtypes restores phenylephrine-induced phosphorylation of ERK in α1ABKO cardiac myocytes, whereas both α1A-NLS “localization mutants” fail to restore signaling.17 This implies that nuclear localization is also required for α1-AR–mediated survival signaling.
α1-AR Signal Transduction Out of the Nucleus
The most significant remaining question is how α1-AR–mediated intranuclear signals exit the nuclei to reach cytosolic targets. Phenylephrine activates PKCδ in cardiac myocyte nuclei45 and induces phosphorylation of cTnI at T144,45 a putative PKCδ site,75 together suggesting that PKCδ is exported to the sarcomere upon activation by α1-ARs. Translocation of PKC from the cytoplasm to the plasma membrane is a hallmark of its activation in many cell types, but PKC translocation is more complex in cardiac myocytes.76 In NRVM, PKCδ and PKCε localize to the nucleus in the basal state, and α1-AR stimulation induces PKCδ translocation to the perinuclear space and PKCε translocation to the sarcomere,77 consistent with α1-AR–mediated activation and export of intranuclear PKCδ in adult cardiac myocytes.45 Mechanistically, PKC export could involve binding to a chaperone protein, such as 14-3-3. For example, α1-AR hypertrophic signaling involves PKD-mediated phosphorylation of HDAC5, binding of HDAC5 to 14-3-3 and nuclear export of the HDAC5: 14-3-3 complex, thereby inducing hypertrophic gene transcription.78,79 Whether 14-3-3 mediated export of signaling proteins represents a general mechanism whereby α1-ARs export signals from the nucleus remains to be determined.
Activation of Hypertrophic Genes by α1-ARs and “Inside-in” Signaling
In cardiac myocytes, α1-ARs induce the expression of hypertrophic genes, such as skeletal actin and α-myosin heavy chain.1 Convention dictates that GPCR signaling initiates at the sarcolemma with second messengers and signaling molecules diffusing to the nucleus. However, the idea of “excitation–transcription coupling” could challenge this convention. Here, localized Ca2+ release from nuclear IP3 receptors within the perinuclear space activates transcriptional regulators, such as HDAC5.57 Potentially, this nuclear localized release of Ca2+ is mediated, at least in some cases, by nuclear GPCRs leading to transcriptional activation. In fact, in nuclei isolated from adult cardiac myocytes, β-ARs and ET-Rs induce gene transcription,44,57,80 suggesting that the nuclei might contain the machinery sufficient to induce a hypertrophic response. By extension, the localization of functional α1-ARs to the nucleus in adult cardiac myocytes could suggest an α1-AR “inside-in” hypertrophic signaling mechanism, whereby α1-AR hypertrophic signaling is regulated predominantly in the nucleus, an interesting, but as of yet unproven hypothesis.
Summary
The failure of α1-NLS “localization” mutants to reconstitute α1-AR inotropic function and survival signaling in adult cardiac myocytes demonstrates a requirement for nuclear localization to regulate physiologic function in adult cardiac myocytes. Although the requirement for nuclear localization appears clear, the exact mechanisms whereby α1-ARs regulate extranuclear signaling remain unclear, and how signals are exported from the nucleus remain to be determined.
POTENTIAL IMPLICATIONS OF NUCLEAR Α1-AR SIGNALING: COMPARTMENTALIZATION OF GQ-COUPLED GPCR SIGNALING IN ADULT CARDIAC MYOCYTES
Convention indicates that GPCRs that signal through Gαq (Gq-receptors) worsen HF. Yet, accumulating evidence now indicates that not all Gq-receptors induce pathology, and in fact, α1-ARs are cardioprotective.1,9 Additionally, other studies now show that Gq-receptors localize to different subcellular compartments in cardiac myocytes. This could suggest a new model for compartmentalization of Gq-receptor signaling in adult cardiac myocytes (Fig. 1). In this model, Gq-receptor signaling at the nucleus is cardioprotective in adult cardiac myocytes, with α1-ARs as the prototypical cardioprotective nuclear Gq-receptors, as alluded to in previous reports.17,50,81
FIGURE 1.
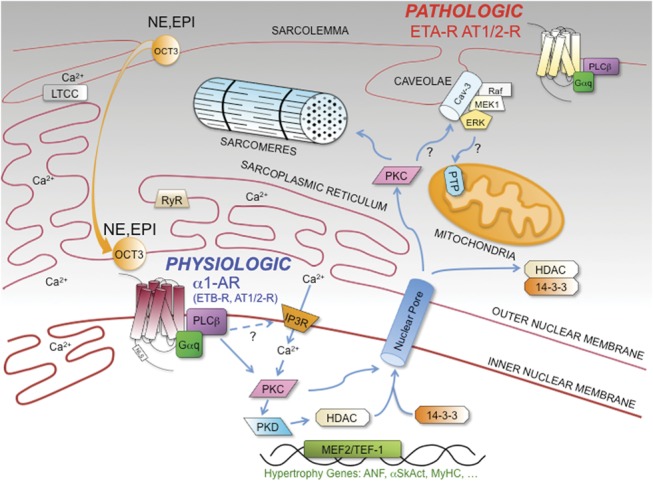
Compartmentalization of Gq-receptor signaling in adult cardiac myocytes. In adult cardiac myocytes, Gq-receptors are compartmentalized. In this model, α1-ARs are the prototypical cardioprotective nuclear Gq-receptors, although the ETB-R and AT1/2-Rs are also at the nucleus. ETA-R and AT1/2-R are primarily at the sarcolemma and mediate pathologic remodeling in HF. α1-AR, α1-adrenergic receptor; ET-R, endothelin receptor; AT-R, angiotensin receptor; NE, norepinephrine; EPI, epinephrine; OCT3, organic cation transporter 3; PLCb, phospholipase Cb; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3-receptor; PKC, protein kinase C; PKD, protein kinase D; HDAC, histone deacetylase; LTCC, L-type calcium channel; RYR, ryanodine receptor; PTP, mitochondrial permeability transition pore; Cav-3, caveolin-3; ERK, extracellular regulated kinase; MEK1, mitogen-activated protein kinase; raf1, MAP kinase; MEF2, myocyte enhancer factor-2; TEF-1; transcriptional enhancer factor-1.
Gq-receptor Pathophysiology in HF
HF is a clinical syndrome and the common end stage of many cardiovascular diseases, including ischemic heart disease and hypertension. The structural and functional changes in the heart during HF, a process termed remodeling, include hypertrophy, contractile dysfunction (systolic and/or diastolic dysfunction), cell death, and fibrosis (interstitial and/or replacement fibrosis). Previous models suggest that Gq-receptors, including α1-ARs, ET-R, and AT-R, activate important physiologic responses that worsen remodeling. Several lines of evidence support this idea including (1) Gq-receptors in NRVM induce hypertrophy with re-expression of the fetal gene program, a marker of pathologic remodeling,82 (2) overexpression of Gαq in NRVM induces hypertrophy and a constitutively active mutant of Gαq induces apoptosis,83 (3) increasing levels of cardiac-specific overexpression of Gαq produces progressively more pathologic phenotypes in mice,83,84 (4) cardiac-specific and conditional knockout of Gαq and Gα11 prevents pathologic remodeling in mice,85 and (5) Gαq-inhibitor peptides block pathologic remodeling.86 Therapeutically, this suggests that inhibiting Gq-receptor signaling in HF would improve outcomes. Although AT-R antagonists are now used in human HF (Review: Ref. 87), ET-R antagonists show no benefit (Review: Ref. 88) and α1-AR antagonists in hypertension double the risk of HF and increase mortality (Review: Ref. 1), suggesting that cardiac Gq-receptors are functionally unique.
Gq-receptor Function: α1-ARs Are Cardioprotective, Whereas ET-Rs and AT-Rs Worsen Pathologic Remodeling in HF
As mentioned above, studies in cell and animal models indicate that α1-ARs mediate cardioprotective functions including an adaptive/physiologic hypertrophy, protection from cell death, positive inotropy, and preconditioning (Review: Refs. 1 and 9). Furthermore, α1-ARs levels are increased approximately 2-fold in HF, suggesting a compensatory role.1 Clinically, α1-AR antagonists for HF (prazosin, V-HeFT) or hypertension (doxazosin, ALLHAT trial) fail to show efficacy (Review: Ref. 1), leading to termination of the ALLHAT trial due to increased mortality and a doubling in the risk of HF.89,90
Both ET-R subtypes, ETA and ETB, are expressed in cardiac myocytes and fibroblasts (Review: Ref. 91). Mouse models indicate that ET overexpression induces HF,92 but gene-deletion models show less consistent results with global knockouts inducing embryonic lethality,93–95 and cardiac-specific knockouts suggesting direct and indirect effects to induce HF.96 Clinically, ET antagonists for both acute and chronic HF show no benefit.88
Both AT-R subtypes, AT1 and AT2, are expressed in the cardiac myocytes and fibroblasts. In mouse models, overexpression of AT1-R induces hypertrophy and HF,97,98 whereas knockout of the AT2-R, but not the AT1-R, attenuates the pathologic remodeling.99,100 Clinically, AT-R antagonists (ARBs) are used in the treatment of hypertension, and ARBs have consistently proven efficacious in treatment of HF (Review: Ref. 87).
Compartmentalized Gq-receptor Signaling
Several lines of evidence support the hypothesis for compartmentalization of Gq-receptors. Quantitatively, binding assays on fractionated adult cardiac myocytes detect 80% of endogenous α1-ARs in the nuclear fraction, whereas 95% of endogenous ET-Rs localize to the sarcolemma,41 (AT-Rs below the level of detection, unpublished result Wright CD and O'Connell TD, November 2009). However, AT-Rs localize to both the sarcolemma and the nuclei in adult cardiac myocytes based on immunostaining, but quantitative data on their distribution is lacking. Interestingly, ARBs do not cross the sarcolemma101 or block intracrine angiotensin signaling43 suggesting that AT-R pathologic signaling probably initiates at the sarcolemma. Functionally, α1-ARs and ET-Rs differentially activate PKD and ET-Rs activate PKD at the sarcolemma, whereas α1-ARs activate PKD at the nuclei in adult cardiac myocytes.102 Furthermore, AT-Rs and ET-Rs activate calmodulin kinase II through different phosphorylation sites than α1-ARs.103
Alternatively, it is entirely possible that functional differences between Gq-receptors in cardiac myocytes exist due to intrinsic differences in how receptors activate subsequent downstream signaling pathways and interact with Gαq. In this case, differential activation of signaling pathways or biased signaling could play a significant role in the physiologic outcome of Gq-receptor signaling. Therefore, future studies will need to define the functional significance of nuclear GPCR signaling in adult cardiac myocytes.
FINAL SUMMARY
There is now significant evidence demonstrating that α1-ARs localize to and signal at the nuclei in adult cardiac myocytes as follows: (1) identification of endogenous α1-ARs at the nuclei in adult cardiac myocytes based on radio-ligand binding assays on fractionated myocytes or fluorescent ligands in cultured myocytes provide the basis for this novel model, (2) demonstration of signaling in isolated nuclei, failure of α1-AR “localization” mutants to signal, failure to detect α1-AR signaling at the plasma membrane, and rapid and specific catecholamine uptake support the nuclear α1-AR signaling model, and (3) evidence that nuclear localization is required for α1-AR–mediated inotropy and survival signaling identifies a physiologic significance to the localization of α1-ARs at the nuclei in adult cardiac myocytes. Finally, unlike other Gq-receptors, α1-ARs are cardioprotective and localize primarily to the nuclear membrane, suggesting that compartmentalized signaling at the nuclei in adult cardiac myocytes could be cardioprotective.
ACKNOWLEDGMENTS
The authors thank Brian C. Jensen, MD, Departments of Medicine and Pharmacology and The McAllister Heart Institute, The University of North Carolina for insightful editorial comments during the writing of this manuscript.
Footnotes
Supported by the National Institute of Health (grant P20 RR-017662), and The American Heart Association, Greater Midwest Affiliate.
The authors reports no conflicts of interest.
REFERENCES
- 1.O'Connell TD, Jensen BC, Baker AJ, et al. Cardiac alpha1-adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol Rev. 2014;66:308–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cavalli A, Lattion AL, Hummler E, et al. Decreased blood pressure response in mice deficient of the alpha1b-adrenergic receptor. Proc Natl Acad Sci U S A. 1997;94:11589–11594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Connell TD, Ishizaka S, Nakamura A, et al. The alpha (1A/C)- and alpha (1B)-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J Clin Invest. 2003;111:1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rokosh DG, Bailey BA, Stewart AF, et al. Distribution of alpha 1C-adrenergic receptor mRNA in adult rat tissues by RNase protection assay and comparison with alpha 1B and alpha 1D. Biochem Biophys Res Commun. 1994;200:1177–1184. [DOI] [PubMed] [Google Scholar]
- 5.Stewart AF, Rokosh DG, Bailey BA, et al. Cloning of the rat alpha 1C-adrenergic receptor from cardiac myocytes. alpha 1C, alpha 1B, and alpha 1D mRNAs are present in cardiac myocytes but not in cardiac fibroblasts. Circ Res. 1994;75:796–802. [DOI] [PubMed] [Google Scholar]
- 6.Turnbull L, McCloskey DT, O'Connell TD, et al. Alpha 1-adrenergic receptor responses in alpha 1AB-AR knockout mouse hearts suggest the presence of alpha 1D-AR. Am J Physiol Heart Circ Physiol. 2003;284:H1104–H1109. [DOI] [PubMed] [Google Scholar]
- 7.O'Connell TD, Rokosh DG, Simpson PC. Cloning and characterization of the mouse alpha1C/A-adrenergic receptor gene and analysis of an alpha1C promoter in cardiac myocytes: role of an MCAT element that binds transcriptional enhancer factor-1 (TEF-1). Mol Pharmacol. 2001;59:1225–1234. [DOI] [PubMed] [Google Scholar]
- 8.Jensen BC, Swigart PM, Laden ME, et al. The alpha-1D Is the predominant alpha-1-adrenergic receptor subtype in human epicardial coronary arteries. J Am Coll Cardiol. 2009;54:1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen BC, O'Connell TD, Simpson PC. Alpha-1-adrenergic receptors: targets for agonist drugs to treat heart failure. J Mol Cell Cardiol. 2011;51:518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikolaev VO, Bunemann M, Schmitteckert E, et al. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ Res. 2006;99:1084–1091. [DOI] [PubMed] [Google Scholar]
- 11.Shcherbakova OG, Hurt CM, Xiang Y, et al. Organization of beta-adrenoceptor signaling compartments by sympathetic innervation of cardiac myocytes. J Cell Biol. 2007;176:521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bkaily G, Avedanian L, Jacques D. Nuclear membrane receptors and channels as targets for drug development in cardiovascular diseases. Can J Physiol Pharmacol. 2009;87:108–119. [DOI] [PubMed] [Google Scholar]
- 13.Boivin B, Vaniotis G, Allen BG, et al. G protein-coupled receptors in and on the cell nucleus: a new signaling paradigm? J Recept Signal Transduct Res. 2008;28:15–28. [DOI] [PubMed] [Google Scholar]
- 14.Gobeil F, Fortier A, Zhu T, et al. G-protein-coupled receptors signalling at the cell nucleus: an emerging paradigm. Can J Physiol Pharmacol. 2006;84:287–297. [DOI] [PubMed] [Google Scholar]
- 14a.Tadevosyan A, Vaniotis G, Allen BG, et al. G protein-coupled receptor signalling in the cardiac nuclear membrane: evidence and possible roles in physiological and pathophysiological function. J Physiol. 2012;590:1313–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wright CD, Chen Q, Baye NL, et al. Nuclear alpha1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ Res. 2008;103:992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang Y, Wright CD, Merkwan CL, et al. An alpha1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation. 2007;115:763–772. [DOI] [PubMed] [Google Scholar]
- 17.Wright CD, Wu SC, Dahl EF, et al. Nuclear localization drives alpha1-adrenergic receptor oligomerization and signaling in cardiac myocytes. Cell Signal. 2012;24:794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17a.O-Uchi J, Sasaki H, Morimoto S, et al. Interaction of α1-adrenoceptor subtypes with different G proteins induces opposite effects on cardiac L-type Ca2+ channel. Circ Res. 2008;102:1378–1388. [DOI] [PubMed] [Google Scholar]
- 18.Jensen BC, Swigart PM, Simpson PC. Ten commercial antibodies for alpha-1-adrenergic receptor subtypes are nonspecific. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohmer T, Pfeiffer N, Gericke A. Three commercial antibodies against alpha1-adrenergic receptor subtypes lack specificity in paraffin-embedded sections of murine tissues. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:703–706. [DOI] [PubMed] [Google Scholar]
- 20.Michel MC, Wieland T, Tsujimoto G. How reliable are G-protein-coupled receptor antibodies? Naunyn Schmiedebergs Arch Pharmacol. 2009;379:385–388. [DOI] [PubMed] [Google Scholar]
- 21.Fujita T, Toya Y, Iwatsubo K, et al. Accumulation of molecules involved in alpha1-adrenergic signal within caveolae: caveolin expression and the development of cardiac hypertrophy. Cardiovasc Res. 2001;51:709–716. [DOI] [PubMed] [Google Scholar]
- 22.Lanzafame AA, Turnbull L, Amiramahdi F, et al. Inositol phospholipids localized to caveolae in rat heart are regulated by alpha1-adrenergic receptors and by ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2059–H2065. [DOI] [PubMed] [Google Scholar]
- 23.Ogata T, Naito D, Nakanishi N, et al. MURC/Cavin-4 facilitates recruitment of ERK to caveolae and concentric cardiac hypertrophy induced by alpha1-adrenergic receptors. Proc Natl Acad Sci U S A. 2014;111:3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinfath M, Chen YY, Lavicky J, et al. Cardiac alpha 1-adrenoceptor densities in different mammalian species. Br J Pharmacol. 1992;107:185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michel MC, Hanft G, Groß G. Radioligand binding studies of α1-adrenoceptor subtypes in rat heart. Br J Pharmacol. 1994;111:533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noguchi H, Muraoka R, Kigoshi S, et al. Pharmacological characterization of alpha 1-adrenoceptor subtypes in rat heart: a binding study. Br J Pharmacol. 1995;114:1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin F, Owens WA, Chen S, et al. Targeted alpha (1A)-adrenergic receptor overexpression induces enhanced cardiac contractility but not hypertrophy. Circ Res. 2001;89:343–350. [DOI] [PubMed] [Google Scholar]
- 28.Rokosh DG, Simpson PC. Knockout of the alpha 1A/C-adrenergic receptor subtype: the alpha 1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc Natl Acad Sci U S A. 2002;99:9474–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang M, Reese J, Cotecchia S, et al. Murine alpha1-adrenoceptor subtypes. I. Radioligand binding studies. J Pharmacol Exp Ther. 1998;286:841–847. [PubMed] [Google Scholar]
- 30.Hock R, Scheer U, Bustin M. Chromosomal proteins HMG-14 and HMG-17 are released from mitotic chromosomes and imported into the nucleus by active transport. J Cell Biol. 1998;143:1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dono R, James D, Zeller R. A GR-motif functions in nuclear accumulation of the large FGF-2 isoforms and interferes with mitogenic signalling. Oncogene. 1998;16:2151–2158. [DOI] [PubMed] [Google Scholar]
- 32.Ishidate T, Yoshihara S, Kawasaki Y, et al. Identification of a novel nuclear localization signal in Sam68. FEBS Lett. 1997;409:237–241. [DOI] [PubMed] [Google Scholar]
- 33.Cook A, Bono F, Jinek M, et al. Structural biology of nucleocytoplasmic transport. Annu Rev Biochem. 2007;76:647–671. [DOI] [PubMed] [Google Scholar]
- 34.King MC, Lusk CP, Blobel G. Karyopherin-mediated import of integral inner nuclear membrane proteins. Nature. 2006;442:1003–1007. [DOI] [PubMed] [Google Scholar]
- 35.Lusk CP, Blobel G, King MC. Highway to the inner nuclear membrane: rules for the road. Nat Rev Mol Cell Biol. 2007;8:414–420. [DOI] [PubMed] [Google Scholar]
- 36.Pickard BW, Hodsman AB, Fraher LJ, et al. Type 1 parathyroid hormone receptor (PTH1R) nuclear trafficking: association of PTH1R with importin alpha1 and beta. Endocrinology. 2006;147:3326–3332. [DOI] [PubMed] [Google Scholar]
- 37.Pickard BW, Hodsman AB, Fraher LJ, et al. Type 1 parathyroid hormone receptor (PTH1R) nuclear trafficking: regulation of PTH1R nuclear-cytoplasmic shuttling by importin-alpha/beta and chromosomal region maintenance 1/exportin 1. Endocrinology. 2007;148:2282–2289. [DOI] [PubMed] [Google Scholar]
- 38.Re M, Pampillo M, Savard M, et al. The human gonadotropin releasing hormone type I receptor is a functional intracellular GPCR expressed on the nuclear membrane. PLoS one. 2010;5:e11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackenzie JF, Daly CJ, Pediani JD, et al. Quantitative imaging in live human cells reveals intracellular alpha (1)-adrenoceptor ligand-binding sites. J Pharmacol Exp Ther. 2000;294:434–443. [PubMed] [Google Scholar]
- 40.Stewart CL, Roux KJ, Burke B. Blurring the boundary: the nuclear envelope extends its reach. Science. 2007;318:1408–1412. [DOI] [PubMed] [Google Scholar]
- 41.Boivin B, Chevalier D, Villeneuve LR, et al. Functional endothelin receptors are present on nuclei in cardiac ventricular myocytes. J Biol Chem. 2003;278:29153–29163. [DOI] [PubMed] [Google Scholar]
- 42.Boivin B, Lavoie C, Vaniotis G, et al. Functional beta-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc Res. 2006;71:69–78. [DOI] [PubMed] [Google Scholar]
- 43.Tadevosyan A, Maguy A, Villeneuve LR, et al. Nuclear-delimited angiotensin receptor-mediated signaling regulates cardiomyocyte gene expression. J Biol Chem. 2010;285:22338–22349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaniotis G, Glazkova I, Merlen C, et al. Regulation of cardiac nitric oxide signaling by nuclear beta-adrenergic and endothelin receptors. J Mol Cell Cardiol. 2013;62:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu SC, Dahl EF, Wright CD, et al. Nuclear localization of a1A-adrenergic receptors is required for signaling in cardiac myocytes: an “inside-out” a1-AR signaling pathway. J Am Heart Assoc. 2014;3:e000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merlen C, Farhat N, Luo X, et al. Intracrine endothelin signaling evokes IP3-dependent increases in nucleoplasmic Ca in adult cardiac myocytes. J Mol Cell Cardiol. 2013;62:189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo D, Yang D, Lan X, et al. Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5-trisphosphate receptors in neonatal rat cardiomyocytes. Cell Calcium. 2008;43:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ibarra C, Vicencio JM, Estrada M, et al. Local control of nuclear calcium signaling in cardiac myocytes by perinuclear microdomains of sarcolemmal insulin-like growth factor 1 receptors. Circ Res. 2013;112:236–245. [DOI] [PubMed] [Google Scholar]
- 49.Zhang L, Malik S, Pang J, et al. Phospholipase cepsilon hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell. 2013;153:216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ibarra C, Vicencio JM, Varas-Godoy M, et al. An integrated mechanism of cardiomyocyte nuclear Ca signaling. J Mol Cell Cardiol. 2014;75:40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L, Malik S, Kelley GG, et al. Phospholipase C epsilon scaffolds to muscle-specific A kinase anchoring protein (mAKAPbeta) and integrates multiple hypertrophic stimuli in cardiac myocytes. J Biol Chem. 2011;286:23012–23021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lopez I, Mak EC, Ding J, et al. A novel bifunctional phospholipase c that is regulated by Galpha 12 and stimulates the Ras/mitogen-activated protein kinase pathway. J Biol Chem. 2001;276:2758–2765. [DOI] [PubMed] [Google Scholar]
- 53.Vinge LE, Andressen KW, Attramadal T, et al. Substrate specificities of g protein-coupled receptor kinase-2 and -3 at cardiac myocyte receptors provide basis for distinct roles in regulation of myocardial function. Mol Pharmacol. 2007;72:582–591. [DOI] [PubMed] [Google Scholar]
- 54.Monto F, Oliver E, Vicente D, et al. Different expression of adrenoceptors and GRKs in the human myocardium depends on heart failure etiology and correlates to clinical variables. Am J Physiol Heart Circ Physiol. 2012;303:H368–H376. [DOI] [PubMed] [Google Scholar]
- 55.Vinge LE, Oie E, Andersson Y, et al. Myocardial distribution and regulation of GRK and beta-arrestin isoforms in congestive heart failure in rats. Am J Physiol Heart Circ Physiol. 2001;281:H2490–H2499. [DOI] [PubMed] [Google Scholar]
- 56.von Lueder TG, Gravning J, How OJ, et al. Cardiomyocyte-restricted inhibition of G protein-coupled receptor kinase-3 attenuates cardiac dysfunction after chronic pressure overload. Am J Physiol Heart Circ Physiol. 2012;303:H66–H74. [DOI] [PubMed] [Google Scholar]
- 56a.Martini JS, Raake P, Vinge LE, et al. Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc Natl Acad Sci U S A. 2008;105:12457–12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu X, Zhang T, Bossuyt J, et al. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cotecchia S, Stanasila L, Diviani D, et al. Structural determinants involved in the activation and regulation of G protein-coupled receptors: lessons from the alpha1-adrenegic receptor subtypes. Biol Cell. 2004;96:327–333. [DOI] [PubMed] [Google Scholar]
- 59.Hwa J, Graham RM, Perez DM. Identification of critical determinants of a1-adrenergic receptor subtype selective agonist binding. J Biol Chem. 1995;270:23189–23195. [DOI] [PubMed] [Google Scholar]
- 60.Wu D, Jiang H, Simon MI. Different a1-adrenergic receptor sequences required for activating different Ga subunits of Gq class of G proteins. J Biol Chem. 1995;270:9828–9832. [DOI] [PubMed] [Google Scholar]
- 61.Jonker JW, Schinkel AH. Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3). J Pharmacol Exp Ther. 2004;308:2–9. [DOI] [PubMed] [Google Scholar]
- 62.Nies AT, Koepsell H, Damme K, et al. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011;201:105–167. [DOI] [PubMed] [Google Scholar]
- 63.Zwart R, Verhaagh S, Buitelaar M, et al. Impaired activity of the extraneuronal monoamine transporter system known as uptake-2 in Orct3/Slc22a3-deficient mice. Mol Cell Biol. 2001;21:4188–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duan H, Wang J. Selective transport of monoamine neurotransmitters by human plasma membrane monoamine transporter and organic cation transporter 3. J Pharmacol Exp Ther. 2010;335:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gambassi G, Spurgeon HA, Ziman BD, et al. Opposing effects of alpha 1-adrenergic receptor subtypes on Ca2+ and pH homeostasis in rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 1998;274:H1152–H1162. [DOI] [PubMed] [Google Scholar]
- 66.Tohse N, Nakaya H, Hattori Y, et al. Inhibitory effect mediated by alpha 1-adrenoceptors on transient outward current in isolated rat ventricular cells. Pflugers Arch. 1990;415:575–581. [DOI] [PubMed] [Google Scholar]
- 67.Terzic A, Puceat M, Clement O, et al. Alpha 1-adrenergic effects on intracellular pH and calcium and on myofilaments in single rat cardiac cells. J Physiol. 1992;447:275–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O-Uchi J, Komukai K, Kusakari Y, et al. Alpha1-adrenoceptor stimulation potentiates L-type Ca2+ current through Ca2+/calmodulin-dependent PK II (CaMKII) activation in rat ventricular myocytes. Proc Natl Acad Sci U S A. 2005;102:9400–9405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deleted in proof.
- 70.Zhang S, Hiraoka M, Hirano Y. Effects of alpha1-adrenergic stimulation on L-type Ca2+ current in rat ventricular myocytes. J Mol Cell Cardiol. 1998;30:1955–1965. [DOI] [PubMed] [Google Scholar]
- 71.Skomedal T, Borthne K, Aass H, et al. Comparison between alpha-1 adrenoceptor-mediated and beta adrenoceptor- mediated inotropic components elicited by norepinephrine in failing human ventricular muscle. J Pharmacol Exp Ther. 1997;280:721–729. [PubMed] [Google Scholar]
- 72.Du XJ, Fang L, Gao XM, et al. Genetic enhancement of ventricular contractility protects against pressure-overload-induced cardiac dysfunction. J Mol Cell Cardiol. 2004;37:979–987. [DOI] [PubMed] [Google Scholar]
- 73.Du XJ, Gao XM, Kiriazis H, et al. Transgenic alpha1A-adrenergic activation limits post-infarct ventricular remodeling and dysfunction and improves survival. Cardiovasc Res. 2006;71:735–743. [DOI] [PubMed] [Google Scholar]
- 74.O'Connell TD, Swigart PM, Rodrigo MC, et al. Alpha1-adrenergic receptors prevent a maladaptive cardiac response to pressure overload. J Clin Invest. 2006;116:1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burkart EM, Sumandea MP, Kobayashi T, et al. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem. 2003;278:11265–11272. [DOI] [PubMed] [Google Scholar]
- 76.Sabri A, Steinberg SF. Protein kinase C isoform-selective signals that lead to cardiac hypertrophy and the progression of heart failure. Mol Cell Biochem. 2003;251:97–101. [PubMed] [Google Scholar]
- 77.Disatnik M-H, Buraggi G, Mochly-Rosen D. Localization of protein kinase C isozymes in cardiac myocytes. Exp Cell Res. 1994;210:287–297. [DOI] [PubMed] [Google Scholar]
- 78.Harrison BC, Kim MS, van Rooij E, et al. Regulation of cardiac stress signaling by protein kinase d1. Mol Cell Biol. 2006;26:3875–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vega RB, Harrison BC, Meadows E, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vaniotis G, Del Duca D, Trieu P, et al. Nuclear beta-adrenergic receptors modulate gene expression in adult rat heart. Cell Signal. 2011;23:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bers DM. Membrane receptor neighborhoods: snuggling up to the nucleus. Circ Res. 2013;112:224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dorn GW, 2nd, Brown JH. Gq signaling in cardiac adaptation and maladaptation. Trends Cardiovasc Med. 1999;9:26–34. [DOI] [PubMed] [Google Scholar]
- 83.Adams JW, Sakata Y, Davis MG, et al. Enhanced Galphaq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci U S A. 1998;95:10140–10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.D'Angelo DD, Sakata Y, Lorenz JN, et al. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A. 1997;94:8121–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wettschureck N, Rutten H, Zywietz A, et al. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat Med. 2001;7:1236–1240. [DOI] [PubMed] [Google Scholar]
- 86.Akhter SA, Luttrell LM, Rockman HA, et al. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998;280:574–577. [DOI] [PubMed] [Google Scholar]
- 87.Shearer F, Lang CC, Struthers AD. Renin-angiotensin-aldosterone system inhibitors in heart failure. Clin Pharmacol Ther. 2013;94:459–467. [DOI] [PubMed] [Google Scholar]
- 88.Kohan DE, Cleland JG, Rubin LJ, et al. Clinical trials with endothelin receptor antagonists: what went wrong and where can we improve? Life Sci. 2012;91:528–539. [DOI] [PubMed] [Google Scholar]
- 89.Cohn JN. The Vasodilator-heart failure trials (V-HeFT). Mechanistic data from the VA Cooperative studies. Introduction. Circulation. 1993;87:VI1–VI4. [PubMed] [Google Scholar]
- 90.ALLHAT, CRG. Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). Jama. 2000;283:1967–1975. [PubMed] [Google Scholar]
- 91.Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol. 2001;41:851–876. [DOI] [PubMed] [Google Scholar]
- 92.Yang LL, Gros R, Kabir MG, et al. Conditional cardiac overexpression of endothelin-1 induces inflammation and dilated cardiomyopathy in mice. Circulation. 2004;109:255–261. [DOI] [PubMed] [Google Scholar]
- 93.Clouthier DE, Hosoda K, Richardson JA, et al. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125:813–824. [DOI] [PubMed] [Google Scholar]
- 94.Kurihara Y, Kurihara H, Suzuki H, et al. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368:703–710. [DOI] [PubMed] [Google Scholar]
- 95.Yanagisawa H, Yanagisawa M, Kapur RP, et al. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–836. [DOI] [PubMed] [Google Scholar]
- 96.Zhao XS, Pan W, Bekeredjian R, et al. Endogenous endothelin-1 is required for cardiomyocyte survival in vivo. Circulation. 2006;114:830–837. [DOI] [PubMed] [Google Scholar]
- 97.Hein L, Stevens ME, Barsh GS, et al. Overexpression of angiotensin AT1 receptor transgene in the mouse myocardium produces a lethal phenotype associated with myocyte hyperplasia and heart block. Proc Natl Acad Sci U S A. 1997;94:6391–6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Paradis P, Dali-Youcef N, Paradis FW, et al. Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc Natl Acad Sci U S A. 2000;97:931–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harada K, Komuro I, Shiojima I, et al. Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation 1998;97:1952–1959. [DOI] [PubMed] [Google Scholar]
- 100.Senbonmatsu T, Ichihara S, Price E, Jr, et al. Evidence for angiotensin II type 2 receptor-mediated cardiac myocyte enlargement during in vivo pressure overload. J Clin Invest. 2000;106:R25–R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Singh VP, Le B, Bhat VB, et al. High-glucose-induced regulation of intracellular ANG II synthesis and nuclear redistribution in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H939–H948. [DOI] [PubMed] [Google Scholar]
- 102.Bossuyt J, Chang CW, Helmstadter K, et al. Spatiotemporally distinct protein kinase D activation in adult cardiomyocytes in response to phenylephrine and endothelin. J Biol Chem. 2011;286:33390–33400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Erickson JR, Patel R, Ferguson A, et al. Fluorescence resonance energy transfer-based sensor Camui provides new insight into mechanisms of calcium/calmodulin-dependent protein kinase II activation in intact cardiomyocytes. Circ Res. 2011;109:729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]