

Abstract
Fifteen cinnoline analogues and six benzimidazole phosphodiesterase 10A (PDE10A) inhibitors were synthesized as potential PET radiopharmaceuticals and their in vitro activity as PDE10A inhibitors was determined. Nine out of twenty-one compounds were potent inhibitors of PDE10A with IC50 values ranging from 1.5 to 18.6 nM. Notably, the IC50 values of compounds 26a, 26b, and 33c were 1.52 ± 0.18, 2.86 ± 0.10, and 3.73 ± 0.60 nM, respectively; these three compounds also showed high in vitro selectivity (> 1,000-fold) for PDE10A over PDE 3A/3B, PDE4A/4B. The high potency and selectivity of these three compounds suggests that they could be radiolabeled with PET radionuclides for further evaluation of their in vivo pharmacological behavior and ability to quantify PDE10A in the brain.
Keywords: Benzimidazole analogues, Cinnoline analogues, Phosphodiesterase 10A, PET
Phosphodiesterase 10A (PDE10A) is a dual specificity enzyme that hydrolyzes both cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) and is an important modulator of intracellular levels of these cyclic nucleotides. PDE10A has been cloned and characterized from mouse and human tissue; it is highly expressed in striatum with modest expression in other brain regions and low expression outside the central nervous system (CNS).1–4 PDE 10A inhibitors have been investigated as anti-psychotics in schizophrenia and therapeutic agents for treating movement disorders associated with Huntington’s disease and Parkinson disease and other neurological disorders characterized by the decrease in the activity of the neurons in the basal ganglia.5–7 Because positron emission tomography (PET) imaging is a powerful non-invasive tool for quantitatively measuring changes in CNS biomarkers under baseline conditions and after treatment, a potent and selective PDE10A tracer with suitable pharmacokinetic behavior would be valuable in evaluating these therapeutic interventions. 11C-papaverine (IC50 = 36 nM)8, [11C]MP-10 (IC50 = 0.18 nM) and structurally similar analogues have been investigated as PET radiopharmaceuticals in rodents and nonhuman primates, though pharmacokinetic properties and unfavorable metabolism limited their utility in preclinical models of human disease.8–11 The MP-10 analogue 18F-JNJ42259152 was approved for clinical investigation in human subjects. Although preliminary data showed promising kinetics in normal volunteers, the development of kinetic models was challenging due to the presence of brain-penetrating metabolites.12, 13 Despite the lack of correlation between PDE10A expression and clinical severity in a recent exploratory study of 18F-JNJ42259152 in subjects with HD and healthy controls,14 continued efforts to develop structurally diverse PDE10A inhibitors may identify PET ligands with improved in vivo pharmacological behavior. In this manuscript, we report the synthesis and in vitro characterization of analogues having either cinnoline (1) or benzimidazole (2) pharmacophores. (Figure 1) Several new lead compounds reported here and a previously reported cinnoline analogue have IC50 values < 20 nM. The diverse structures represented in these compounds may result in the identification of tracers with kinetic and metabolic profiles suitable for measuring PDE10A in the brain with PET.
Figure 1.
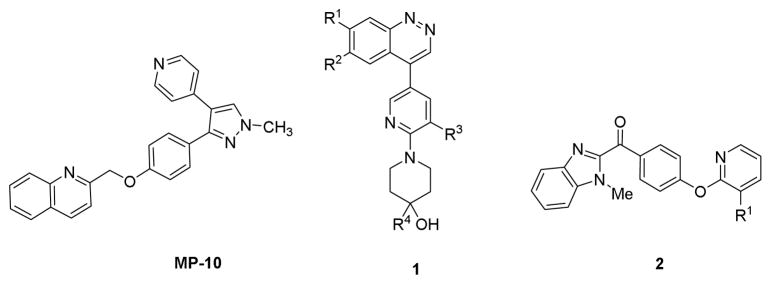
Structures of lead PDE10A inhibitors
The syntheses of cinnoline PDE10A analogues 13a–d and 14a–b were accomplished according to Scheme 1. Commercially available 1-(4-(benzyloxy)-3-methoxyphenyl)-ethanone (3) was nitrated to afford compound 4, then reduced to give amine 6. Either 5, the commercially available 1-(2-amino-4, 5-dimethoxyphenyl)ethanone or the amine 6 underwent diazotization/cyclization, followed by treatment with phosphorus oxychloride to afforded intermediates 9 or 10, respectively. The chloro intermediates were treated with 2-fluoro-3-substituted boronic acid, followed by 4-substituted-4-piperidinol to give the desired cinnoline PDE10A analogues 13a–d. The benzyl group in compounds 13b or 13d was removed by using hydrogen under 10% Pd/C to afford hydroxyl containing compounds 14a or 14b.
Scheme 1. Synthesis of cinnoline analogues.

Reagents and conditions:
(a) HNO3, SnCl4, CH2Cl2, −70°C; (b) Fe, NH4HCO3, toluene, water, reflux; (c) NaNO2, acetic acid, 70%H2SO4, Et3N; (d) POCl3, PCl5; (e) 2-fluoropyridine-5-boronic acid, Pd(PPh3)4, Cs2CO3, diglyme; (f) i. 4-(pyridin-3-yl)piperidin-4-ol or 4-methylpiperidin-4-ol, K2CO3, DMSO, reflux; or ii. K2CO3, DMSO, reflux, then H2, 10% Pd/C, methanol, 75 psi.
Compounds 26a–i, which possess substitution groups in the pyridine ring of cinnoline, were synthesized as shown in Scheme 2. Compound 26a was previously reported by Hu et al.15 The synthesis of 26a–f and 26h–i used procedures similar to those described above for 13a–b and 14a–b. The synthesis of compound 26g was accomplished by the conversion of 4-(pyridin-3-yl)piperidin-4-ol hydrochloride to compound 25 by alkylation and Miyaura borylation, followed by Suzuki reaction to afford the final product, 26g.
Scheme 2. Synthesis of cinnoline derivatives with substitution in the pyridine ring.

Reagents and conditions:
(a) H2 (75 psi), 10% Pd/C, MeOH; (b) 2-(bromomethyl)quinoline or 1-bromo-3-fluoropropane, NaH, THF.(c) 2-fluoro-3-methylpyridine-5-boronic acid, Pd(PPh3)4, Cs2CO3, 1,2-dimethoxyethane; (d) 5-bromo-2,3-dichloro-pyridin, K2CO3, DMSO, 110 °C; (e) bis(pinacolato)diboron, KOAc, (1,1′-bis(diphenylphosphino)ferrocene)Pd(II) dichloride, dioxane, 120 °C; (f) piperidinol, K2CO3, DMSO, 100 °C; (g) 9, Pd(PPh3)4, Cs2CO3, 1,2-dimethoxyethane.
The syntheses of benzimidazole PDE10A analogues 33a–f were accomplished according to Scheme 3. Treatment of benzimidazole 27 with 4-methoxy benzoyl chloride under basic conditions to generate compound 28, followed by N-methylation of benzimidazole afforded compound 29. Deprotection of the anisole with boron tribromide afforded phenol intermediate 30, which was treated with 3-bromo-2-chloropyridine to give key intermediate 31. Compounds 33a–c were obtained by Suzuki coupling of intermediate 31 with substituted boronic acids. Benzimidazole derivatives 33d–e were synthesized by two steps: 31 was first converted into the stannyl containing compound 32; this was followed by Stille coupling of compound 32 with substituted acyl chlorides to give compounds 33d–e.
Scheme 3. Syntheses of new benzimidazole analogues.

Reagents and conditions:
(a) 4-Methoxybenzoyl chloride, pyridine, Et3N, NaOH, reflux; (b) CH3I, NaH, DMF; (c) BBr3, DCM; (d) 3-bromo-2-chloropyridine, K2CO3, Cs2CO3, DMSO; (e) boronic acid ester, K2CO3, Pd(PPh3)4; (f) hexamethyltin, K2CO3, Pd(PPh3)4, toluene; (g) substituted acyl chloride or aromatic or heteroaromatic boronic acid, palladium.
In vitro assays were performed following standard literature techniques developed by other investigators; the procedure used here and in our prior studies is detailed in the supplementary methods.7, 16 Briefly, a scintillation proximity assay (SPA) to was used to measure the IC50 values of all inhibitors. The tritiated cyclic nucleotide substrate concentration used in the enzyme assay was 1/3 of the Km. The potency of each analogue as a PDE inhibitor was determined by measuring the activity of a fixed amount of recombinant human PDE enzyme in the presence of varying inhibitor concentrations. After background subtraction, counts for samples containing the inhibitors were plotted against inhibitor concentration to calculate the IC50 values using non-linear regression analysis. All compounds were independently assayed at least three times.
The IC50 values of the fifteen cinnoline analogues (13a–d, 14a–b, and 26a–i) and six benzimidazole analogues (33a–f) for PDE10A are shown in Table 1 and Table 2. Among the cinnoline analogues, for compounds 13a, 13b, and 14a, the potency of substituted inhibitors for PDE10A showed in a decreasing order OCH3> OBn > OH; the IC50 values of 13a, 13b, and 14a were 18.4, 4,250, and 10,800 nM, respectively. Although compounds 13c, 13d, and 14b have a pyridine-3-yl group instead of a methyl group at the R3 position, a similarly decreasing trend for PDE10A inhibition was observed for compounds 13c, 13d, and 14b with IC50 values of 14, 1,550, and 6,620 nM, respectively. The known compound 13c17 was also synthesized and its IC50 value was determined to ensure the tests results are comparable with published values. Compound 26a has methyl group instead of the –H group at the R3 position compound 13c; this led to the 9.2-fold increased potency of the IC50 value from 14.0 nM for 13c to 1.52 nM for 26a. The pyridine-3-yl group at R4 position of 26a was replaced by a 2-fluoro-pyrindin-5-yl group to give 26b; the chlorine-substituted analogue 26e was also synthesized. The IC50 values of 26b and 26e for PDE10A were 2.86 and 9.80 nM respectively, which are similar to the IC50 value we determined for 26a (1.52 nM). When the methoxyl group at R1 in 26b was replaced by a benzyloxyl group in 26f, the potency decreased with IC50 values increasing from 2.86 nM for 26b to 350 nM for 26f. This result was consistent with the results observed for 13a and 13b, 13c and 13d. When methoxyl groups at R1 and R2 were either replaced by a cyclized group (-OCH2O-) in 26c or removed in 26d, compounds 26c and 26d lost inhibition activity for PDE10A completely. In comparing 26g and 13c, when the –H group in R3 was substituted with a –Cl group the potency of the inhibitor improved slightly from 14.0 nM for 13c to 5.52 nM for 26g. Compounds 26h and 26i were obtained by replacing the methoxyl group of R1 in 26b with a fluoroproxyl and 2-(oxymethyl)-quinloinyl group, respectively. The IC50 values increased from 2.86 nM for 12b to 518 and 79.3 nM for 26h and 26i, respectively. This result is consistent with the results observed for compounds 13a, 13b, 13c and 13d. Throughout these measurements, the methoxyl group at R1 position of cinnoline structural analogues was critical in retaining high potency for inhibiting PDE10A. The replacement of a methoxyl group at R1 with other groups such as –H, -OCH2O-, -OBn, fluoropropoxy, 2-(oxymethyl)-quinolinyl led to a significant reduction or a total loss in PDE10A inhibition activity.
Table 1.
The PDE10A inhibition activity (IC50 nM) of cinnoline analogues
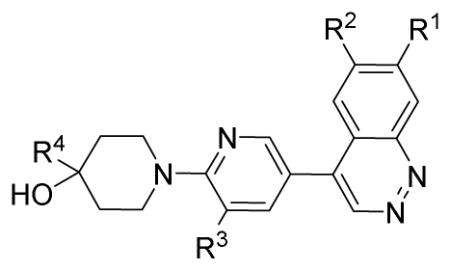
| |||||
|---|---|---|---|---|---|
| Compound# | R3 | R2 | R3 | R4 | IC50 (nM)a |
| 13a | -OCH3 | -OCH3 | -H | CH3 | 18.4 ± 6.0 |
| 13b | -OBn | -OCH3 | -H | CH3 | 4,250 ± 520 |
| 13c | -OCH3 | -OCH3 | -H | pyridin-3-yl | 14.0 ± 2.0 |
| 13d | -OBn | -OCH3 | -H | pyridin-3-yl | 1,550 ± 260 |
| 14a | -OH | -OCH3 | -H | CH3 | 10,800 ± 1,500 |
| 14b | -OH | -OCH3 | -H | pyridine-3-yl | 6,620 ± 600 |
| 26a | -OCH3 | -OCH3 | -CH3 | pyridine-3-yl | 1.52 ± 0.18 |
| 26b | -OCH3 | -OCH3 | -CH3 | 2-fluoro-pyridin-5-yl | 2.86 ± 0.10 |
| 26c | -O-CH2-O- | -CH3 | 2-fluoro-pyridin-5-yl | NAb | |
| 26d | -H | -H | -CH3 | 2-fluoro-pyridin-5-yl | NAb |
| 26e | -OCH3 | -OCH3 | -CH3 | 2-chloro-pyridin-5-yl | 9.8 ± 1.3 |
| 26f | -OBn | -OCH3 | -CH3 | 2-fluoro-pyridin-5-yl | 350 ± 23 |
| 26g | -OCH3 | -OCH3 | -Cl | pyridin-3-yl | 5.52 ± 0.33 |
| 26h | 2-(oxymethyl)-quinolinyl | -OCH3 | -CH3 | 2-fluoro-pyridin-5-yl | 518 ± 86 |
| 26i | fluoropropoxy | -OCH3 | -CH3 | 2-fluoro-pyridin-5-yl | 79.3 ± 3.6 |
IC50 value (mean ± SD nM) were determined in at least three experiments;
NA = no inhibition activity
Table 2.
The PDE10A inhibition activity (IC50 nM) of benzimidazole analogues
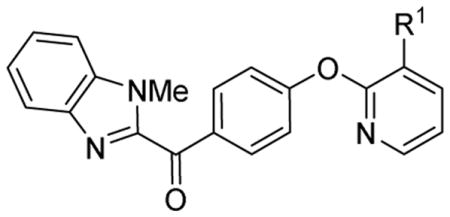
| ||
|---|---|---|
| Compound # | R1 | IC50 (nM)a |
| 33a | phenyl | 18.6 ± 5.3 |
| 33b | 4-methoxyphenyl | 8.00 ± 1.38 |
| 33c | pyridine-3-yl | 3.73 ± 0.60 |
| 33d | benzoyl | NAa |
| 33e | 4-methoxybenzoyl | NAa |
| 33f | N-acetylpiperidin-4-ethanoyl | >1.9×103 |
IC50 value (mean ± SD nM) were determined in at least three experiments;
NA = no inhibition activity.
The benzimidazole analogues are structurally distinct from cinnoline analogues; other benzimidazole inhibitors with high potency toward PDE10A have been previously reported.18 Six benzimidazole analogues (33a–f) were synthesized and their IC50 values for PDE10A were determined. In vitro data indicated that compounds 33a, 33b, and 33c possessing a phenyl, 4-methoxyphenyl or heteroaryl substitution group for R1 had higher inhibition potency for PDE10A; the IC50 values were 18.6 ± 5.3, 8.0 ± 1.38, and 3.73 ± 0.60 nM for compounds 33a, 33b, and 33c, respectively. Optimization of the substituted group in R1 may improve potency for PDE10A. However, imposing a carbonyl group between the aryl or heteroaryl group and the skeleton of the benzimidazole resulted in significantly decreased activity toward PDE10A. Both 33d and 33e lost inhibition activity for PDE10A; the IC50 value of 33f increased to 1900 nM as shown in Table 2.
Compounds 26a, 26b, and 33c are very potent inhibitors of PDE10A with IC50 values < 5 nM, so further in vitro assays were conducted to determine their selectivity. It has been reported that inhibition of PDE3A/B may cause arrhythmia and increased mortality,19, 20 inhibition of PDE4A may increase heart and respiratory rates.21 Although such undesirable side-effects pose fewer safety concerns for clinical radiotracers in comparison with therapeutic agents, specificity is important. Therefore, the IC50 values of 26a, 26b and 33c for PDE3A, PDE3B, PDE4A, and PDE4B were measured to determine their selectivity as PDE10A inhibitors. The enzyme assays revealed that all three lead compounds had very weak inhibition activity toward PDE3A/3B, PDE4A/B with > 1000-fold selectivity for PDE10A vs. PDE3A/B and PDE4A/B, as shown in Table 3.
Table 3.
The inhibition activity (IC50 nM) of compounds 26a, 26b, and 33c towards PDE 3A&B, PDE4A & B*a
| Compound # | PDE10A (nM) | PDE3A (nM) | PDE3B (nM) | PDE4A (nM) | PDE4B (nM) |
|---|---|---|---|---|---|
| 26a | 1.52 ± 0.18 | 3,070 ± 180 | 4,110 ± 360 | 7,830 ± 800 | 8,730 ± 730 |
| 26b | 2.86 ± 0.10 | 3,700 ± 250 | 6,540 ± 520 | 25,400 ± 3,600 | 30,600 ± 400 |
| 33c | 3.73 ± 0.60 | 167,000 ± 52,000 | 234,000 ± 66,000 | 403,00 ± 6,700 | 25,900 ± 2,900 |
IC50 value (mean ± SD nM) were determined in at least three experiments
Overall, fifteen cinnoline and six benzimidazole analogues were synthesized and screened for their in vitro activity as PDE10A inhibitors. Nine of the twenty-one compounds showed high in vitro potency for PDE10A with IC50 values ranging from 1.52 to 18.6 nM. The three most potent compounds 26a, 26b, and 33c have IC50 values of 1.52 ± 0.18, 2.86 ± 0.10, and 3.73 ± 0.60 nM, respectively; in addition, these three compounds have > 1000-fold selectivity for PDE10A over PDE3A/B and PDE4A/B and are structurally suitable compounds for labeling with PET radionuclides. Further studies will be needed to characterize the pharmacological behavior of the PET ligands in vivo and determine their potential as PET tracers for assessing PDE10A in living subjects.
Supplementary Material
Acknowledgments
This study was supported by the National Institute of Mental Health (NIMH) and the National Institute of Neurological Disorders (NINDS) under MH092797, NS061025 and NS075527, and through funding from the Mallinckrodt Institute of Radiology (MIR) at the Washington University School of Medicine. The authors would also like to thank Lynne Jones for assistance in preparing the manuscript.
Footnotes
Supplementary material associated with this article including the detailed chemical synthesis, yields, NMR data and the in vitro assay methods are provided in the supporting online information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Fujishige K, Kotera J, Michibata H, Yuasa K, Takebayashi S, Okumura K, Omori K. J Biol Chem. 1999;274:18438. doi: 10.1074/jbc.274.26.18438. [DOI] [PubMed] [Google Scholar]
- 2.Loughney K, Snyder PB, Uher L, Rosman GJ, Ferguson K, Florio VA. Gene. 1999;234:109. doi: 10.1016/s0378-1119(99)00171-7. [DOI] [PubMed] [Google Scholar]
- 3.Soderling SH, Bayuga SJ, Beavo JA. Proc Natl Acad Sci USA. 1999;96:7071. doi: 10.1073/pnas.96.12.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hebb AL, Robertson HA, Denovan-Wright EM. Neuroscience. 2004;123:967. doi: 10.1016/j.neuroscience.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Menniti FS, Chappie TA, Humphrey JM, Schmidt CJ. Curr Opin Invest Dr. 2007;8:54. [PubMed] [Google Scholar]
- 6.Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. PlOS ONE. 2010;5:e13417. doi: 10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt CJ, Chapin DS, Cianfrogna J, Corman ML, Hajos M, Harms JF, Hoffman WE, Lebel LA, McCarthy SA, Nelson FR, Proulx-LaFrance C, Majchrzak MJ, Ramirez AD, Schmidt K, Seymour PA, Siuciak JA, Tingley FD, 3rd, Williams RD, Verhoest PR, Menniti FS. J Pharm Exp Ther. 2008;325:681. doi: 10.1124/jpet.107.132910. [DOI] [PubMed] [Google Scholar]
- 8.Tu Z, Xu JB, Jones LA, Li SH, Mach RH. Nucl Med Biol. 2010;37:509. doi: 10.1016/j.nucmedbio.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tu Z, Fan JD, Li SH, Jones LA, Cui JQ, Padakanti PK, Xu JB, Zeng DX, Shoghi KI, Perlmutter JS, Mach RH. Bioorg Med Chem. 2011;19:1666. doi: 10.1016/j.bmc.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plisson C, Salinas C, Weinzimmer D, Labaree D, Lin SF, Ding YS, Jakobsen S, Smith PW, Eiji K, Carson RE, Gunn RN, Rabiner EA. Nucl Med Biol. 2011;38:875. doi: 10.1016/j.nucmedbio.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Celen S, Koole M, De Angelis M, Sannen I, Chitneni SK, Alcazar J, Dedeurwaerdere S, Moechars D, Schmidt M, Verbruggen A, Langlois X, Van Laere K, Andres JI, Bormans G. J Nucl Med. 2010;51:1584. doi: 10.2967/jnumed.110.077040. [DOI] [PubMed] [Google Scholar]
- 12.Van Laere K, Ahmad RU, Hudyana H, Celen S, Dubois K, Schmidt ME, Bormans G, Koole M. Eur J Nucl Med Mol Imaging. 2013;40:254. doi: 10.1007/s00259-012-2270-1. [DOI] [PubMed] [Google Scholar]
- 13.Van Laere K, Ahmad RU, Hudyana H, Dubois K, Schmidt ME, Celen S, Bormans G, Koole M. J Nucl Med. 2013;54:1285. doi: 10.2967/jnumed.112.118679. [DOI] [PubMed] [Google Scholar]
- 14.Ahmad R, Bourgeois S, Postnov A, Schmidt ME, Bormans G, Van Laere K, Vandenberghe W. Neurology. 2014;82:279. doi: 10.1212/WNL.0000000000000037. [DOI] [PubMed] [Google Scholar]
- 15.Hu E, Kunz RK, Rumfelt S, Chen N, Bürli R, Li C, Andrews KL, Zhang J, Chmait S, Kogan J, Lindstrom M, Hitchcock SA, Treanor J. Bioorg Med Chem Lett. 2012;22:2262. doi: 10.1016/j.bmcl.2012.01.086. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Jin H, Zhou H, Rothfuss J, Tu Z. MedChemComm. 2013;4:443.2. doi: 10.1039/C2MD20239E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chappie T, Humphrey J, Menniti F, Schmidt C. Curr Opin Drug Discovery Dev. 2009;12:458. [PubMed] [Google Scholar]
- 18.Asproni B, Murineddu G, Pau A, Pinna GA, Langgard M, Christoffersen CT, Nielsen J, Kehler J. Bioorg Med Chem. 2011;19:642. doi: 10.1016/j.bmc.2010.10.038. [DOI] [PubMed] [Google Scholar]
- 19.Mager G, Klocke RK, Kux A, Hopp HW, Hilger HH. Am Heart J. 1991;121:1974. doi: 10.1016/0002-8703(91)90834-5. [DOI] [PubMed] [Google Scholar]
- 20.Movsesian M, Stehlik J, Vandeput F, Bristow MR. Heart Fail Rev. 2009;14:255. doi: 10.1007/s10741-008-9130-x. [DOI] [PubMed] [Google Scholar]
- 21.Heaslip RJ, Evans DY. Eur J Pharmacol. 1995;286:281. doi: 10.1016/0014-2999(95)00457-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.