

Abstract
Over 20 unstable microsatellite repeats have been identified as the cause of neurological disease in humans. The repeat nucleotide sequences, their location within the genes, the ranges of normal and disease-causing repeat length and the clinical outcomes differ. Unstable repeats can be located in the coding or the non-coding region of a gene. Different pathogenic mechanisms that are hypothesised to underlie the diseases are discussed. Evidence is given both from studies in simple model systems and from studies on human material and in animal models. Since somatic instability might affect the clinical outcome, this is briefly touched on. Available data and theories on the timing and mechanisms of the repeat instability itself are discussed, along with factors that have been observed to affect instability. Finally, the question of why the often harmful unstable repeats have been maintained throughout evolution is addressed.
Keywords: microsatellite instability, neurological disease, repeat instability, trinucleotide repeat
Introduction
In 1991, a (CGG)n in the fragile X mental retardation 1 (FMR1) gene and a (CAG)n in the coding region of the androgen receptor were the first expanded trinucleotide repeats identified as the genetic cause of diseases, namely fragile X syndrome (FRAXA) and X-linked spinal and bulbar muscle atrophy (SBMA: Kennedy’s disease), respectively.(1,2) These repeat expansions form a distinct class of mutations, based on their unusual properties. Since the repeat length is variable and the mutation rate depends on the repeat length, the risk of mutation of an allele is different in each subsequent generation. Therefore, this mechanism was termed ‘dynamic mutation’. Since instability increases with expanding repeat length, the risk of getting the disorder is greater in successive generations. In addition, age of onset is likely to be lower and severity of the disorder may be worse with increasing repeat length. This is known as ‘anticipation’. These trinucleotides repeats are polymorphic and show a normal range, seen in healthy individuals, and a pathological range, above a threshold length, associated with clinical manifestations.(3)
Up to now almost 20 other unstable repeats have been described to be associated with neurological disorders. Clinically these conditions range from congenital syndromes to late-onset neurodegenerative disorders. For a description of the clinical outcome of the diseases described here, we refer to a recent review by Orr and Zoghbi.(4) Although the majority of repeats consist of trinucleotides, tetra- and pentanucleotide repeats can also expand. Although this review mostly focuses on intergenerational repeat instability, somatic instability is touched on briefly, as it might have clinical consequences. The pathogenic mechanisms through which different repeat tracts may cause disease are discussed, as well as the timing of the events and the factors that are thought to play a role in the instability of the repeat tracts.
Pathogenic mechanisms underlying disease
Various pathogenic pathways can underlie trinucleotide repeat-induced disorders. Both loss-of-function and gain-of-function mechanisms are recognised to result from expanded trinucleotide repeats. In loss-of-function mechanisms, the gene product is either not produced, or produced at lower levels. When a disease is associated with an mRNA or protein that has attained a new cellular function, it is considered a gain-of-function mechanism. A gain-of-function can occur at the RNA as well as at the protein level, depending on the location of the trinucleotide repeat within the gene. If an expanded repeat is translated, because it is within the coding sequence, it is likely to affect protein structure and thereby function (reviewed in Ref.(4,5)). When a repeat is located in a non-coding region, it will not change protein structure or function directly. However, as the repeat is transcribed, mRNA function might be altered. All diseases and their pathogenic pathways discussed in this review are summarised in Table 1 and the locations of the repeats within their genes are schematically summarised in Fig. 1.
Table 1.
Characteristics of all microsatellite repeats described in this review and the diseases that they cause, categorised by the pathogenic mechanism thought to underlie these neurological trinucleotide repeat disorders.
| Mechanism | Repeat sequence |
Gene | Chromosomal localisation |
Position within gene |
Normal range |
Disease range |
Disease | Reviewed in Ref. |
|
|---|---|---|---|---|---|---|---|---|---|
| Protein loss-of-function | (CGG)n | FMR1 | Xq27.3 | 5′UTR | 5–55 | >200 | FXS | Fragile X syndrome | (9) |
| (GGC)n | FMR2 | Xq28 | 5′UTR | 6–25 | >200 | FRAXE | FRAXE associated mental retardation | (12) | |
| (GAA)n | FDRA | 9q13-21.1 | Intron 1 | 34–100 | 200–1700 | FRDA | Friedreich ataxia | (14) | |
| RNA gain-of- function | (CTG)n | DMPK | 19q13 | 3′UTR | 5–37 | 50–3000 | DM1 | Myotonic dystrophy type 1 | (16, 17) |
| (CCTG)n | ZNF9 | 3q21 | Intron 1 | <27 | 75–11000 | DM2 | Myotonic dystrophy type 2 | (16, 17) | |
| (CGG)n | FMR1 | Xq27.3 | 5′UTR | 5–55 | 55–200 | FXTAS | Fragile X-associated tremor ataxia syndrome | (39) | |
| Uncertain | (CTG)n | SCA8 | 19q13 | 3′UTR | 16–34 | >100 | SCA8 | Spinocerebellar ataxia type 8 | (42) |
| (ATTCT)n | ATXN10 | 22q13 | Intron 1 | <29 | >800 | SCA10 | Spinocerebellar ataxia type 10 | (17) | |
| (CAG)n | PPP2R2B | 5q32 | 5′UTR | <32 | 51–78 | SCA12 | Spinocerebellar ataxia type 12 | (43, 44) | |
| Protein gain-of-function | (CAG)n | AR | Xq13-21 | Coding region | 9–36 | 38–62 | SBMA | Kennedy’s disease/Spinal and bulbar muscular atrophy | (3, 50) |
| (CAG)n | Huntingtin | 4p16.3 | Coding region | 6–35 | 40–121 | HD | Huntington’s disease | (54) | |
| (CAG)n | SCA1 | 6p23 | Coding region | 6–44 | 39–82 | SCA1 | Spinocerebellar ataxia type 1 | (5) | |
| (CAG)n | SCA2 | 12q24.1 | Coding region | 14–31 | 34–62 | SCA2 | Spinocerebellar ataxia type 2 | (5) | |
| (CAG)n | SCA3 | 14q32.1 | Coding region | 12–~43 | >60 | SCA3 | Spinocerebellar ataxia type 3/Machado-Joseph disease | (5) | |
| (CAG)n | CACNA1A | 19p13 | Coding region | 5–18 | 20–33 | SCA6 | Spinocerebellar ataxia type 6 | (5) | |
| (CAG)n | SCA7 | 3p12-21.2 | Coding region | 7–34 | >37 | SCA7 | Spinocerebellar ataxia type 7 | (5) | |
| (CAG)n | TBP | 6q27 | Coding region | 25–44 | 45–66 | SCA17 | Spinocerebellar ataxia type 17 | (5) | |
| (GCG)n | PABP2 | 5q32 | Coding region | 6 | 8–13 | OPMD | Oculopharyngeal myotonic dystrophy | (59) | |
| (CTG)n | JPH3 | 16q24.3 | Coding region | 6–28 | 40–58 | HDL2 | Huntington’s disease-like 2 | (62) |
Figure 1.
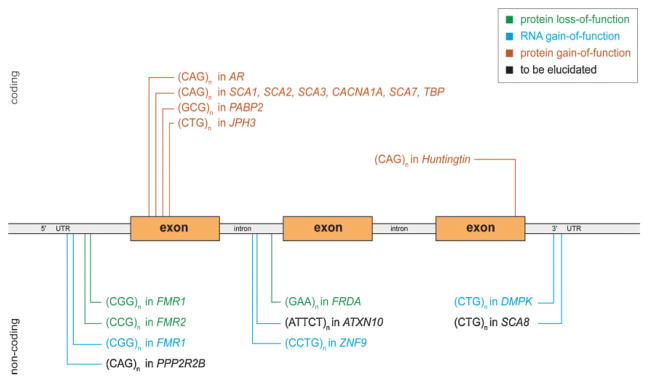
Location of repeats within genes in relation to pathogenic mechanism. This schematic gene shows all microsatellite repeats described in this review, with their location within the gene. The colours indicate which pathogenic mechanism is thought to underlie the associated diseases. All repeats depicted above the gene are located in a coding sequence of the gene, while all repeats below are in non-coding regions. Please refer to the text and Table 1 for further information regarding these repeats, genes and associated disorders.
Loss-of-function at the protein level
The most common syndrome in which protein function is lost is FRAXA which occurs when the (CGG)n in the 5′UTR of the FMR1 gene exceeds 200 CGGs (full mutation (FM)).(1) Beyond this length, the CpG island within the promoter region as well as the repeat become methylated and histone acetylation decreases. This leads to transcriptional silencing. The gene product FMRP is normally highly expressed in brain neurons and regulates mRNA translation in dendrites, thereby modulating synaptic function. Its absence leads to the mental retardation seen in FRAXA.(6)
A closely related disease is due to a (CCG)n at the FRAXE locus, which lies just downstream of the FRAXA locus. The expanded (CCG)n lies in the 5′-untranslated region (5′UTR) of the FMR2 gene. Similar to FRAXA, when the repeat exceeds 200 triplets, FMR2 is silenced due to methylation and protein function is lost. The FMR2 protein is a nuclear protein thought to act as a transcription factor,(7) but its cellular function and the role in the disease have not yet been elucidated.
Friedreich ataxia (FRDA) is caused by expansion in the range of 66–1,700 of a (GAA)n in the first intron of the FDRA gene. FRDA is an autosomal recessive disorder. The so-called premutation (PM; 34–100 GAA) alleles can expand to up to ten-fold in length to give the disease-causing alleles.(8) In FRDA, the (GAA)n expansion results in decreased mRNA levels so that the level of the gene product, Frataxin, is greatly decreased. Frataxin appears to control iron availability in mitochondria and has a role in biogenesis of iron–sulphur clusters and haem. Low levels of frataxin decrease energy production and increase the release of free radicals.(9,10)
RNA gain-of-function
An expanded repeat located in a non-coding region causes cellular toxicity in myotonic dystrophy (DM), fragile X-associated tremor/ataxia syndrome (FXTAS) and spinocerebellar ataxias type 8, 10 and 12 (SCA8, SCA10 and SCA12). The repeat is transcribed, but not translated, therefore the proteins do not play a central role in the development of the disease.
Myotonic dystrophy
Two forms of DM exist: DM1 is caused by a (CTG)n in the 3′UTR of the DMPK gene encoding DM protein kinase (DMPK), while DM2 results from expanded tetranucleotide repeat [(CCTG)n] in intron 1 of the zinc finger protein 9 gene (ZNF9). Both DM1 and DM2 show childhood and adult onset, but congenital forms only occur for DM1.(11,12)
Although the two genes involved in DM1 and DM2 are functionally different, the syndromes share a toxic RNA gain-of-function mechanism. The first evidence pointing in this direction was that almost all of the expanded repeat-containing mRNAs were retained in nuclear foci.(13) This is thought to occur because expanded RNA (CUG)n molecules form secondary structures such as hairpins.(14) In the foci, RNA molecules sequester RNA-binding proteins (RNA-BPs), which bind specifically to (CUG)n (CUG-binding proteins (CUG-BPs)).(15,16) Since mRNA processing, including splicing, is normally regulated by a dynamic complex of RNA-BPs, the function of these proteins in the presence of expanded (CUG)n was investigated. (4)
CUG-BP1 and muscleblind-like protein 1 (MBNL1) bind (CUG)n. CUG-BP1 is a heterogeneous nuclear ribonucleoprotein (hnRNP) and a member of the CUG-BP/ETR-3-like family (CELF) of proteins. These regulate alternative splicing, editing and translation. Splicing of the repeat-containing DMPK and ZNF9 mRNAs occurs normally. It is proposed that CUG-BPs are sequestered to expanded DMPK-(CUG)n, which causes aberrant expression of other transcripts that are normally regulated by these proteins.(17) MBNL1 is a specific CUG-BP, homologous to the Drosophila muscleblind proteins that are essential in terminal differentiation of muscle and photoreceptor cells. It accumulates in the nuclear foci in DM1 cells, so that MBNL1 cannot exert its normal function during a critical period of cell differentiation.(16)
MBNL1 and CUG-BP1 are antagonistic splicing regulators of specific transcripts affected in DM.(18) Relative activities of CELF proteins and human muscleblind control an important developmental switch in splice pattern. CELF proteins promote an embryonic splice pattern for certain transcripts, while an adult pattern is seen when MBNL1 activity predominates. In DM, the switch to an adult splice pattern does not occur (summarised in Ref.(19)).
Evidence supporting this hypothesis is now accumulating from murine models.(18) In addition, Drosophila expressing a non-coding expanded (CUG)n show several DM features, including altered CUG-bp1 and mbnl1 levels and distribution. Overexpression of human MBNL1 suppressed the (CUG)n-induced toxicity, whereas higher levels of human CUG-BP1 worsen the phenotype.(20) However, how expansion of the C(C)UG-repeats alters the activities of CUG-BP1 and MBNL is still not known.
Fragile X-associated tremor/ataxia syndrome
Elderly carriers of (CGG)55–200 (PM) in the 5′UTR of the FMR1 gene can develop FXTAS.(21,22) In PM carriers, FMR1 mRNA levels are elevated, while the gene product FMRP is present at levels that are close to normal, suggesting that toxicity originates at the RNA rather than the protein level. Postmortem immunohistochemical studies on FXTAS brains revealed the presence of ubiquitin-positive intranuclear inclusions in neurons and astrocytes in many brain regions. The presence of inclusions is related to FXTAS.(23,24) Also FMR1 mRNA has been detected within the intranuclear inclusions, further supporting the hypothesis that FMR1 mRNA plays a central role in the pathogenesis of FXTAS.(24)
FXTAS has been proposed to result from a pathogenic mechanism similar to that for DM.(25) MBNL1, among many other proteins, has been identified in the intranuclear inclusions in human FXTAS brain.(26) No downstream effects on splicing patterns as in DM have been described to occur in FXTAS. Some signs of cellular toxicity are observed in cellular models.(27,28) Studies are currently focussing on finding possible CGG-BPs that cause cellular dysfunction. Indeed, studies in Drosophila have identified hnRNP A2/B1 and pur alpha as CGG-BP. Overexpressing these proteins suppresses the expanded (CGG)n-induced neurodegenerative eye phenotype.(29,30) Although these findings support a pathogenesis model that predicts that PM-sized (CGG)n disturb cellular function by sequestering RNA-BPs, a role for these proteins in humans and the precise mechanism through which they cause clinical symptoms remains to be confirmed.
About 20% of the female PM carriers develop premature ovarian failure (POF: cessation of menstruation at or before 40 years of age).(31) POF has also been postulated to result from a similar toxic RNA gain-of-function mechanism.(31,32)
A clear difference between FXTAS and DM is that in FXTAS FMR1 transcript levels are increased, while DMPK and ZNF9 are normally expressed. The explanation for this discrepancy might be that disease-associated C(C)UG-repeats are much longer (several thousands), as opposed to 55–200 CGGs in FMR1. Shorter, but more abundant repeat tracts in FXTAS might produce the same effect. Penetrance of FXTAS appears dependent on (CGG)n length,(33) and severity of the (CGG)n-induced eye neurodegeneration in Drosophila depends both on expression level and (CGG)n length.(34) Thus, a threshold might exist for the number of RNA-BPs bound to the expanded repeat at which cellular toxicity to become apparent.
Uncertainty of pathogenic mechanisms in the spinocerebellar ataxias
Expanding repeats also cause a group of SCA disorders. Nine SCAs have been described to date, of which three (SCA8, -10 and -12) are caused by repeats in non-coding regions of different genes. SCA8 is the result of a (CTG)n in the 3′UTR of the gene coding for Ataxin-8, SCA10 involves an (ATTCT)n in intron 1 of the ATXN10 (ataxin-10) gene, and SCA12 is caused by a (CAG)n in the 5′UTR of the PPP2R2B gene (protein phosphatase 2 (formerly 2A), regulatory subunit B (PR 52), β-isoform).
(CTG)>100 in the SCA8 locus is usually associated with the disease. The function of the SCA8 gene is unknown. However, its overlap with the 5′ end of a gene (Kelch-like 1, encoding an actin-binding protein) that is transcribed in the opposite direction suggests transcriptional regulation of this gene. Since transgenic mice and flies show parallels to some aspects of DM and FXTAS, an RNA gain-of-function mechanism has been proposed to underlie SCA8, although further studies will have to confirm this.(35)
SCA10 is the only human disease known to be caused by an expansion of a pentanucleotide repeat. Repeats shorter than 29 units are present in normal alleles of the ATXN10 gene. (ATTCT)>800 are considered SCA10 mutations. The function of the ATAXN10 protein product and the pathogenic mechanism that causes SCA10 are at present unknown. Since transcript levels are not reduced in patient cell lines, an RNA gain-of-function mechanism has been proposed.(12)
SCA12 is a very rare disease. Normal alleles have up to 32 trinucleotides in the gene coding for phosphatase PP2ABb. The lower border of disease-causing repeat length is unclear, but affected patients have alleles in the range of 51–78 CAGs. The gene PPP2R2B encodes a brain-specific regulatory subunit of the trimeric phosphatase PP2A. The repeat seems to affect transcriptional efficiency, as transcript levels are increased. Protein levels are also elevated. Many splice isoforms exist and it is conceivable that the SCA12 repeat expansion disrupts splice regulation, thereby altering the function of PP2A. PP2A is a ubiquitously expressed enzyme and is involved in many cellular processes, including apoptosis. A toxic role for (CAG)n-containing transcripts cannot be excluded in the development of SCA12.(36) It is also possible that a toxic polyglutamine (polyQ)-containing protein exists.
Gain-of-function at the protein level
The most common trinucleotide repeat causing disease by altering protein physiology is the (CAG)n. When translated, this results in a polyQ tract. Many different disorders share a (CAG)n in the coding region of a gene. Although expansion sizes, structures, cellular localisation and functions of the resulting proteins differ, all (CAG)n-induced diseases are neurodegenerative disorders.
All disorders are associated with neuronal aggregates that contain the disease-causing gene product. PolyQ stretches have an inherent ability to aggregate. Apart from binding to many other proteins, glutamine also shows self-interaction.(37) Once polyQ stretches exceed a certain length, they are no longer soluble and form aggregates. The threshold length, above which in vitro aggregation takes place, is strikingly similar to the threshold that causes disease (~35–40 CAGs).(38) Although it is tempting to hold the aggregates responsible for the development of disease, some evidence exists that the large aggregates are not the primary cause of cell toxicity. Some disease models have indeed shown cell toxicity in the absence of aggregates, or that toxicity decreased as aggregate density increased.(39)
Nine expanded (CAG)n disease-associated genes have been identified. However, these are not the only polyQ-encoding sequences in the genome. Getting to know the normal function of polyQ tracts might facilitate understanding how expansion of the (CAG)n tracts and loss of the normal protein function causes disease.(37)
Kennedy’s disease
Kennedy’s disease was the first disorder known to result from expansion of a polymorphic (CAG)n in the coding region of the androgen receptor gene located on the X-chromosome. Patients have 38–62 CAGs as opposed to 9–36 in normal controls.(2) The androgen receptor is a nuclear receptor that regulates transcription of hormone-responsive genes upon androgen binding. The polyQ tract is located in the trans-activation domain. Presence of an expanded (CAG)n decreases receptor expression and alters transcriptional activation. This may account for the androgen insensitivity seen in SBMA patients, but it does explain motor neuron toxicity. Therefore, it has been proposed that the mutant androgen receptor causes motor neuron dysfunction by a toxic gain of function.(40)
Huntington’s disease
In Huntington’s disease (HD), the expanded (CAG)n is located at the N terminus of the gene that codes for a protein named Huntingtin. Although the precise function of normal Huntingtin has not yet been established, this protein is known to be involved in several cellular processes such as vesicular transport, cytoskeletal anchoring and clathrin-mediated endocytosis.(41) Although loss-of-function of Huntingtin may account for some of the pathogenesis seen in HD, evidence points to a more prominent role for a toxic gain-of-function of mutant Huntingtin. Inclusion bodies containing N-terminal Huntingtin and ubiquitin have been found in postmortem brain material of HD patients. Abnormal protein interactions with mutant Huntingtin could take place, possibly leading to coaggregation of other proteins. However, the precise mechanism by which inclusions lead to pathology, and whether they are a prerequisite for pathology remains to be elucidated. Evidence also exists that inclusions might exert a neuroprotective effect by sequestering the more toxic forms of the aggregated protein.(42) However, in transgenic mouse models the presence of inclusions was clearly correlated to disease pathology.(43) A conditional mouse model showed reversal of neuropathology and disappearance of inclusion bodies, when mutant Huntingtin expression was turned off.(44)
Spinocerebellar ataxias
Expanded (CAG)n in coding regions are the cause of SCA1, 2, 3, 6, 7 and 17 (summarised in Ref.(4)). Other than sharing a polyQ tract, the gene products of the different SCA genes bear no homology. SCA1–3 and 7 are caused by Ataxin-1–3 and −7, respectively. SCA6 involves the α1A-subunit of the voltage-dependent calcium channel (CACNA1A), and SCA17 is the result of an expanded polyQ tract in the gene coding for TATA-binding protein (TBP). Much remains to be elucidated in these disease pathways.(4)
Formation of inclusions suggests that the cellular quality control mechanisms fail to deal appropriately with mutant polyQ-containing proteins. Research focussing on the precise role for chaperones and the ubiquitin proteasome system in these diseases could lead to promising therapeutical strategies.
It cannot be ruled out that in some polyQ diseases loss-of-function of the polyQ-containing protein contributes to the pathogenesis.(4) Alternatively, it may be that the repeat-containing RNA also contributes to pathogenesis in polyQ-mediated disease.(45)
Other trinucleotide repeats in coding regions
Another coding repeat that causes neurological disease is a (GCG)n, coding for a polyalanine (poly(A)) tract, in the poly(A)-binding protein 2 (PABP2). This leads to oculopharyngeal DM (OPMD). The (GCG)n represents the first short disease-causing trinucleotide repeat discovered, i.e. 8–13 repeat units in the OPMD families, as opposed to the normal allele of 6 GCGs. Furthermore, the (GCG)n does not show the features of a dynamic mutation.(46) The poly(A) tract was postulated to cause pathologically expanded PABP2 to accumulate as filament inclusions in nuclei, which are seen in OPMD patients.(47)
An expanded (CTG)n in an alternatively spliced exon of the junctophilin-3 (JPH3) gene leads to HD-like 2 (HDL2). Two out of the three splice isoforms result in the presence of the repeat in the coding region, leading to a poly(A) or a polyleucine tract. Intranuclear protein aggregates are seen in brain, but the exact effect of the poly(A) and polyleucine tracts in the pathogenesis of HDL2 is unclear. Formation of RNA inclusions has been postulated, which might suggest a role played by toxic transcripts.(48)
Timing of repeat instability
Timing of repeat instability remains surrounded by question marks. Attempts have been made to gain insight into what happens around fertilisation, although the availability of human material is limited.
Timing of expansion in the fragile X mutation
Expansion to a fragile X FM occurs only upon maternal transmission of a PM allele, while daughters of FM males are clinically and cytogenetically normal. This is due to the sole presence of PM alleles in sperm of FM carriers.(49) Two models that consider the moment of instability have been proposed to explain this. First, the prezygotic model (Fig. 2) predicts that an expansion of PM to FM could occur during maternal meiosis, with the FM contracting to a PM in the gametes of male offspring. PM gametes might have some selection advantage either due to the presence of FMRP, or against the presence of an expanded CGG repeat.(50) Furthermore, FMs are responsible for a delay in replication of the FMR1 gene during the cell cycle.(51) Thus, primordial germ cells (PGCs) with a PM might have a proliferative advantage, thereby outgrowing FM cells. However, if multiple contraction events can take place, it is puzzling why only one distinct PM band is mostly seen in sperm of FM men.(49)
Figure 2.

The prezygotic model of expansion of the fragile X mutation. This model assumes that an expansion of a maternal PM to an FM takes place during meiosis. The fertilised oocyte carries an FM allele. After separation from the embryo proper, the PGCs have an FM. Some alleles will contract to PMs. To explain why FMs are only transmitted through females, some selection must exist against FMs in the male germ line during spermatogenesis. In the mature testes, PM alleles predominate. In somatic cells and the female germ line, this selection does not take place. Cells with a PM are shown in green and cells with an FM are depicted in orange.
The other model considers a postzygotic expansion, after separation of the germ line. It assumes that the FM allele has never been present in male or female gametes.(52) However, oocytes of a foetal female FM carrier had only FM alleles.(50) A postzygotic expansion mechanism occurring only for a PM allele on the maternal X-chromosome would require an imprinting mechanism to distinguish it from a paternally derived PM, which would not expand.(49)
Based on a simulation study and analysis of tissues of affected foetuses, the prezygotic model is favoured.(53) Naturally, a final conclusion can only be drawn after analysis of oocytes from a PM carrier. This material is not available for obvious reasons. Expanded (CGG)n knock-in mouse models have been developed,(54,55) and these show intergenerational repeat instability.(55,56) These models will be valuable in answering the remaining questions.
Timing of expansion in other trinucleotide repeat disorders
The exact timing of expansion is also still under debate for other trinucleotide repeat disorders. Intergenerational expansion in DM1 was suggested to result from initial expansion, followed by somatic instability.(57) Studies in tissues of monozygotic twin pairs suggest that this first instability event happens at very early embryonic stages, as the twins showed identical expansion patterns. Monozygotic twinning happens at the latest on day 10.5 of embryonic development.(57) Expanded DMPK alleles are already detectable in oocytes (including the germinal vesicle stage)(58) and embryos of mothers with expanded (CTG)n, who underwent in vitro fertilisation (IVF) with pre-implantation diagnostics for DM1.(58) (CAG)n expansions have also been detected in oocytes in a transgenic mouse model for SCA1, i.e. after meiotic DNA replication and prior to fertilisation.(59) Thus, oogenesis seems the most likely moment of (CTG)n expansion.(58) A study in a DM mouse model did not show significant differences between spermatozoa and spermatogonia, implying that germinal expansions are already present very early in spermatogenesis.(60)
Instability of normal DMPK alleles can occur during male gametogenesis or early embryogenesis. It not yet known whether expansions in the normal range are the result of the same mechanism as that causing larger expansions. Further studies will also need to clarify whether (CTG)n expansion continues after fertilisation, during embryonic cell division.(58)
Instability of repeats in coding regions has been less well characterised. Expansions of the (CAG)n have been observed in sperm of HD patients,(61) as well as transgenic mouse models for HD.(62) Testicular germ cells of two men who died from HD showed that repeat expansions were already present before the end of the first meiotic division, and in some cells even before the start of meiosis. However, the majority of the larger expansions was found in the postmeiotic cell population, suggesting that instability processes may continue after meiosis.(63)
Somatic repeat instability
Despite the intergenerational repeat instability that links all diseases described in this review, the extent of somatic instability varies. Very little, if any, somatic (CGG)n instability occurs over the course of life in FRAXA. It has been proposed that CpG methylation stabilises the repeat.(64)
Other trinucleotide repeat disorders show substantial somatic instability. In particular, the (CTG)n in DM1 is highly unstable and displays tissue-specific rates of instability.(65) Muscle cells consistently show longer repeats than peripheral blood leukocytes, suggesting somatic mosaicism.(66,67) To date no studies have been published that correlate tissue repeat length to the severity of the phenotype. Ongoing expansions in somatic cells may contribute to the progressive and tissue-specific nature of DM.(68)
Somatic (CTG)n instability is dependent on repeat size and age. Thus, older patients show a longer average repeat size.(68) In a knock-in mouse model for DM1, which reproduces somatic instability of the (CTG)n, it was observed that once mutant alleles had expanded, they continued to increase in length, while the stable alleles remained of a constant length.(69)
Somatic (CAG)n length heterogeneity within and between tissues has been observed in HD patients. Interestingly, region-specific repeat length analysis revealed dramatic increases in repeat length in the striatum, which is the brain region that is primarily affected in HD both in a mouse model for HD(70) and in early stage HD patients.(71) Somatic (CAG)n instability has also been seen in SCA1, SCA3, SCA7, SBMA and FRDA.(72)
Molecular mechanisms of repeat instability
One of the first proposed mechanisms involved in repeat instability at the molecular level was slippage of the replication fork during replication. Unpaired bases form loops, which result in expansions or contractions in a next round of replication, depending on whether the looped repeats are in the newly synthesised or template strand (Fig. 3).(73) However, slippage alone cannot explain all aspects of repeat expansions.
Figure 3.

Replication fork progression and repeat instability. During normal replication, helicases break the hydrogen bonds that keep the two DNA strands together, which yield the replication fork. DNA polymerase can only synthesise a new strand in a 5′ to 3′ direction. Hence, on one strand (leading strand) DNA polymerase reads the DNA and adds nucleotides to the nascent strand in a continuous manner. On the other strand (lagging strand) the complementary strand is synthesised in short segments (Okazaki fragments) at a time, which are later joined together by DNA ligase. Formation of secondary structures, such as hairpins can form in one of the strands. This can impair normal replication fork processing. The sequence of the strands, together with the position of the origin of replication with respect to the repeat sequence, determine which strand is more prone to form hairpins. Whether a hairpin is present in the template or nascent strand, in turn determines whether contraction or expansion results in the next round of replication.
Recognition of the unusual structural properties of trinucleotide repeats yielded new insights. Disease-causing repeats are almost exclusively formed of (CNG)n triplets. Single-stranded (CNG)n can form hairpin-like structures that can have both Watson–Crick and mismatched base pairs. Due to their different sequences, the leading and lagging strand have different tendencies to form hairpins. The secondary structures are likely to affect recognition and subsequent repair or recombination of this structure.(5,74)
Unusual DNA structures may stall DNA polymerases. Studies in yeast replication mutants showed a marked increase in frequency of repeat instability. Proteins involved in lagging strand synthesis, coordination between synthesis of the leading and the lagging strand and restarting of the replication fork play a role in the stability of microsatellite repeats (reviewed in Ref.(5)). A complex model based on replication fork stalling and restarting is described in detail by Mirkin.(75) This model can explain all observations on trinucleotide instability on a molecular level. For instance, anticipation could be the result of progressively increased instability due to consecutive replication stalls and restarts within longer repeat tracts. Also, the threshold length of the repeat tract for the formation of secondary structures coincides with the length of the average eukaryotic Okazaki initiation zone (~200 bases), which might explain why various repeats show similar thresholds for expansion.(5)
Modifiers of repeat instability
Initial studies in simple model systems as Escherichia coli and Saccharomyces cerevisiae have provided important insight into the dynamics of triplet repeat instability. They are, however, of limited value since repeats have a tendency to contract in bacteria and yeast, as opposed to human expansions. In addition, germ line and somatic instability cannot be studied. Transgenic and knock-in mouse models have been shown to mimic human repeat dynamics more closely.(72)
The repetitive nature of the sequence of a trinucleotide repeat is a primary determinant of its unstable nature, but many other factors also play a role. Results obtained in the different model systems are discussed below, divided in cis-and trans-acting factors. Cis-acting refers to factors that are directly associated with the repeat, whereas elements that interact with the repeat can be considered trans-acting elements.
Cis-acting factors
The most obvious factor involved is the length of the repeat tract. Most trinucleotide repeat disorders show a stable length range, as well as a threshold above which they become unstable.(72)
Repeat sequence
The precise sequence of a repeat affects instability. When a different trinucleotide interrupts a repeat tract, it is more stable than a pure repeat. Interruptions determine the chances of folding into hairpins and their stability. For example, AGG interruptions greatly stabilise expanded (CGG)n tracts.(76) Also the position of the AGG interruption influences stability as most (CGG)n-length variations are seen at the 3′ end of the repeat in the FMR1 gene.(77) A similar stabilising effect of interruptions on (CAG)n has been described in humans(78) and mice.(59)
A sequence-specific effect can also be concluded from observations that (CTA)n and (TAG)n did not show detectable expansions in a yeast model that did show (CAG)n and (CTG)n instability, possibly related to the impossibility of forming secondary structures for the former repeat sequences.(79)
Flanking sequences
The first attempts to mimic intergenerational repeat instability in mouse models expressing expanded trinucleotide repeats failed. Transgenic models expressing (CGG)n of lengths that cause instability in humans showed stable transmission to next generations.(80)
The observation that almost all C/G-rich trinucleotide repeat-associated genes have a CpG island in close proximity was seen as a clue that flanking regions and chromatin structure near the triplets might play a role.(81) The relative expandability of a repeat is associated with the GC content of the flanking region. GC-rich chromatin might affect the flexibility of the trinucleotide repeat and the stability of secondary structures formed by the repeat. This could affect DNA replication and transcription processes.(82) The observation that a fragile site can be induced at the FRAXA locus might also hint at an abnormal chromatin structure,(83) although (CAG)n and (CTG)n expansions have not been associated with fragile sites.
A transgenic mouse model expressing a 45-kb genomic segment with a (CTG)55 showed moderate intergenerational and somatic repeat instability. It was suggested that the threshold for large increases may be higher in transgenic mice.(84) Indeed, mice expressing (CTG)>300 exhibited dramatically increased instability, as compared to the (CTG)55 line, again confirming that instability strongly depends on repeat length.(85)
A knock-in mouse model expressing a human (CGG)PM tract in the Fmr1 gene, thus preserving the genomic context, showed substantial repeat instability,(55,56) while mice transgenically expressing an expanded (CGG)n with some flanking sequences only showed moderate instability.(86) Thus, the insertion site also determines repeat instability. This was confirmed by different mouse lines expressing the same transgene, albeit differently integrated, that showed different repeat instability.(84,87)
Orientation of the repeat with respect to the replication fork
The orientation of the repeat relative to an origin of replication largely determines the stability of the repeat,(88) possibly as a result of the temporary existence of single-stranded DNA (ssDNA) during lagging strand synthesis. This could allow hairpin formation. An origin of replication has recently been identified adjacent to the FMR1 promoter. The position of this origin of replication with respect to the (CGG)n is such that it favours contraction, since the CGG sequence, which is more prone to form stable hairpins than is (CCG)n, is in the lagging strand template.(89) Use of an alternative origin of replication downstream of the repeat would cause the (CGG)n to be in the nascent Okazaki fragment, which could lead to expansions.(45)
Trans-acting factors
Not only characteristics of the repeat itself or the direct vicinity thereof play a role, but also other factors, such as DNA metabolism or the function of other gene, have been described to affect trinucleotide repeat stability.
Parental effects
Different trinucleotide repeat disorders have different patterns of transmission. For instance, a paternal bias for transmission of expansions is seen in HD, while expansions into the fragile X-range occur solely with alleles of maternal origin.(90) Observations that the intergenerational mutability of some repeats depends on the sex of the parent suggests that sex-specific factors might induce mosaicism in the male and female germ line, such as differences in DNA metabolism or the number of rounds of replication during gametogenesis (summarised in Ref.(72)). In females, instability might occur when oocytes are arrested in meiosis I, after meiotic DNA replication, which gives a large time window of opportunity for DNA damage to occur. Cells at this stage have only undergone a limited number of cell divisions, which would point to a role for DNA repair in quiescent cells.(91) When considering the number of cell cycles, it is not surprising that age of the transmitting parent also appears to have an effect on instability. In fact, expansions can accumulate in sperm over time(60) and the magnitude of (CAG)n expansions correlates with the age of the transmitting father.(92)
Tissue-specific factors
The differences in repeat length found between tissues point towards tissue-specific factors influencing repeat stability. Mosaicism has been seen in both proliferating and quiescent tissues, suggesting a replication-independent mechanism.(93) Mouse models for DM also show somatic instability throughout life. It is interesting to note that different individual mice of the same mouse line presented the same pattern of mosaicism. This means that somatic instability is not a random process, but that some, as yet unknown, deterministic processes underlie these tissue differences (reviewed in Ref.(72)).
DNA replication and repair
Despite the abundant presence of repetitive DNA in the genome, no general microsatellite instability occurs in the diseases described. Thus, repeat instability is limited to the disease locus. It was therefore concluded that expansions do not result primarily from aberrant trans-acting factors involved in DNA replication, repair and recombination. However, evidence is currently accumulating that factors involved in DNA repair and replication do in fact influence trinucleotide instability.
Disturbed replication fork dynamics have been shown to play a role in (CTG)n instability.(94) Trinucleotide repeats with a high G–C content impair replication. Flap endonuclease 1 (FEN1) has endonuclease activities acting specifically on 5′ flaps that are created by strand displacement during Okazaki fragment maturation. FEN1 deficiency led to increased (intergenerational) instability of (CAG)n/(CTG)n.(95)
However, somatic instability has also been seen in non-dividing cells.(57,96) Thus, if intergenerational instability is the result of the same mechanism as somatic instability, it cannot solely depend on replication-dependent mechanisms.
DNA repair pathways ought to maintain genome integrity and stability, thus they are likely candidates for modifying trinucleotide repeat instability. Aberrant mismatch repair (MMR) systems may cause repeats to become unstable. In MMR, heterodimers are formed of MutS homologue 2 (MSH2) with either MSH3 or MSH6, which can then recognise single-base mismatches or short unpaired regions. Transgenic mice expressing (CTG)>300 were crossed with mice knockout for Msh2. Lack of Msh2 changed the direction of both inter-generational and somatic instability, with only contractions taking place, as opposed to mostly expansions in a wild-type Msh2 background.(97) Mouse models for HD have also shown increased contraction frequencies in an Msh2-deficient background.(98) Msh3 is also necessary for (CTG)n expansion, while Msh6 does not play a key role.(99)
The influence of genes involved in replication and repair on instability of the (CGG)n repeat in Fmr1 gene has also been investigated in a mouse model for FXTAS. Only ataxia-telangiectasia and rad3-related kinase (ATR) deficiency was found to influence (CGG)n instability. ATR responds to stalled replication forks and bulky DNA adducts. Maternal ATR heterozygosity led to more expansions, and an additional effect was seen for ATR heterozygosity in the offspring. This suggests that expansion can occur prior to fertilisation of the oocyte, and subsequently in the haploid oocyte.(100)
Mutant screens have been conducted in yeast to search for genes that have a modulating effect on (CAG)n instability. Mediator of the replication checkpoint protein 1 (Mrc1) was revealed as a mediator of instability. Mrc1 and associated proteins have a role in preventing development of too-long stretches of ssDNA that are likely to form aberrant secondary structures. Mrc1 also has a function as a checkpoint protein after secondary structure formation, aiming at prevention of establishing the mutagenic event.(101)
Taken together, it is clear that disturbed replication and repair processes increase the frequency of repeat instability events. These processes can be difficult to distinguish, as replication can be part of normal mitosis, or repair dependent. Whether expansion in human disease is the result of normal or repair-induced replication(102) remains to be elucidated.
Epigenetic factors
Transcription of repeats might promote formation of secondary structures, which then attract various chromatin-modifying complexes.(103,104) This is in line with the stabilising effect of CpG methylation that has been reported for the (CGG)n in the FMR1 gene, since DNA methylation causes transcriptional silencing.(64) Other epigenetic changes may also occur in the vicinity of long (CTG)n that may trigger formation and spreading of heterochromatin, affecting transcription. In a mouse model for SCA1, heterozygosity for the maintenance DNA (cytosine-5-)methyltransferase 1 (Dnmt1), indeed promoted intergenerational (CAG)n expansion. (105)
A transgenic mouse model expressing a construct containing an origin of replication of SV40, followed by a (CGG)26, showed large intergenerational expansions, into the FRAXA FM range.(106) SV40 origin of replication is known to exclude nucleosomes, which is likely the cause of the large instability seen in these mice, since expanded (CGG)n have also been shown to exclude nucleosomes in experiments in vitro. This also hints at chromatin characteristics having an influence on repeat instability.(107)
Concluding remarks
This review has dealt solely with the negative consequences of the presence of expanded trinucleotide repeats, so the question remains ‘why have these repetitive elements evolved, and even more striking, been maintained’? Transcriptome-wide database analysis has identified several hundreds of transcripts containing triplet repeat tracts (>6 repeat units). Repeats are overrepresented in 5′UTRs. 5′UTRs often play a role in regulation of translation. The preferential localisation of the repeats within the mRNA might hint at their biological role. Many repeat-containing transcripts are indeed involved in cell signalling or transcription and translation. Expansion of the repeat may impair these functions.(108)
Concerning polyQ tracts, it is noteworthy that glutamine is encoded for by both CAG and CAA. Therefore, if there were some selectional advantage for a polyQ tract, it would not need a pure CAG tract, as is generally seen. Evidence exists that both (CTG)n- and (CAG)n-repeat, but not non-repeat, sequences form a functional component of an insulator element, thereby influencing regulation of gene expression. (CTG)n and (CAG)n repeats have been found to act as strong nucleosome positioning elements in vitro(109,110) and also (CGG)n has been shown to exclude nucleosomes in in vitro studies.(107)
Thus, trinucleotide repeats do not only generate negative effects. However, negative effects might prevail once they expand beyond a certain threshold. Future studies might shed light on beneficial characteristics, which will explain why they have been maintained over the course of evolution. Naturally, future studies should also focus on how instability, as well as the negative effects of expansion, can be prevented.
Acknowledgments
This study was financially supported by the Prinses Beatrix Fonds (J. R. B.: MAR03-0208), NIH RL1 NS062411-01 (R. W.) and NIH ROI HD38038 (B. A. O).
Abbreviations
- ATR
ataxia-telangiectasia and rad3-related kinase
- ATXN10
ataxin-10
- CACNA1A
α1A-subunit of the voltage-dependent calcium channel
- CELF
CUG-BP/ETR-3-like family
- CUG-BP1
CUG-binding protein
- DM
myotonic dystrophy
- DMPK
DM protein kinase
- Dnmt1
DNA (cytosine-5-)-methyltransferase 1
- FEN1
flap endonuclease 1
- FM
full mutation
- FMR1
fragile X mental retardation 1
- FRAXA
fragile X syndrome
- FRDA
Friedreich ataxia
- FXTAS
fragile X-associated tremor/ataxia syndrome
- HD
Huntington’s disease
- HDL2
Huntington disease-like 2
- hnRNP
heterogeneous nuclear ribonucleoprotein
- IVF
in vitro fertilisation
- JPH3
junctophilin-3
- MBNL1
muscleblind-like protein 1
- MMR
mismatch repair
- Mrc1
mediator of the replication checkpoint protein 1
- MSH
MutS homologue
- OPMD
oculopharyngeal DM
- PABP2
poly(A)-binding protein 2
- PM
premutation
- POF
premature ovarian failure
- PPP2R2B
protein phosphatase 2 (formerly 2A), regulatory subunit B (PR 52), β-isoform
- RNA-BP
RNA-binding protein
- SBMA
spinal and bulbar muscle atrophy
- SCA
spinocerebellar ataxia
- ssDNA
single-stranded DNA
- TBP
TATA-binding protein
- UTR
untranslated region
- ZNF9
zinc finger protein 9
References
- 1.Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 2.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 3.Richards RI, Sutherland GR. Heritable unstable DNA sequences. Nat Genet. 1992;1:7–9. doi: 10.1038/ng0492-7. [DOI] [PubMed] [Google Scholar]
- 4.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 5.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 6.Willemsen R, Oostra BA, Bassell GJ, Dictenberg J. The fragile X syndrome: from molecular genetics to neurobiology. Ment Retard Dev Disabil Res Rev. 2004;10:60–67. doi: 10.1002/mrdd.20010. [DOI] [PubMed] [Google Scholar]
- 7.Gecz J, Bielby S, Sutherland GR, Mulley JC. Gene structure and subcellular localization of FMR2, a member of a new family of putative transcription activators. Genomics. 1997;44:201–213. doi: 10.1006/geno.1997.4867. [DOI] [PubMed] [Google Scholar]
- 8.De Biase I, Rasmussen A, Bidichandani SI. Evolution and instability of the GAA triplet-repeat sequence in Friedreich’s Ataxia. In: Wells RD, Ashizawa T, editors. Genetic instabilities and neurological diseases. Burlington: Academic Press; 2006. pp. 305–319. [Google Scholar]
- 9.Karthikeyan G, Santos JH, Graziewicz MA, Copeland WC, Isaya G, et al. Reduction in frataxin causes progressive accumulation of mitochondrial damage. Hum Mol Genet. 2003;12:3331–3342. doi: 10.1093/hmg/ddg349. [DOI] [PubMed] [Google Scholar]
- 10.Yoon T, Cowan JA. Iron-sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J Am Chem Soc. 2003;125:6078–6084. doi: 10.1021/ja027967i. [DOI] [PubMed] [Google Scholar]
- 11.Cho DH, Tapscott SJ. Myotonic dystrophy: emerging mechanisms for DM1 and DM2. Biochim Biophys Acta. 2007;1772:195–204. doi: 10.1016/j.bbadis.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 12.Ashizawa T, Harper P. Myotonic dystrophies: an overview. In: Wells R, Ashizawa T, editors. Genetic instabilities and neurological diseases. Burlington: Academic Press; 2006. pp. 21–36. [Google Scholar]
- 13.Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci USA. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Napierala M, Krzyzosiak WJ. CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J Biol Chem. 1997;272:31079–31085. doi: 10.1074/jbc.272.49.31079. [DOI] [PubMed] [Google Scholar]
- 15.Fardaei M, Larkin K, Brook JD, Hamshere MG. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res. 2001;29:2766–2771. doi: 10.1093/nar/29.13.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, et al. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 19.Pascual M, Vicente M, Monferrer L, Artero R. The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation. 2006;74:65–80. doi: 10.1111/j.1432-0436.2006.00060.x. [DOI] [PubMed] [Google Scholar]
- 20.de Haro M, Al-Ramahi I, De Gouyon B, Ukani L, Rosa A, et al. MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum Mol Genet. 2006;15:2138–2145. doi: 10.1093/hmg/ddl137. [DOI] [PubMed] [Google Scholar]
- 21.Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 22.Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72:869–878. doi: 10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- 24.Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biol. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- 25.Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective. Am J Hum Genet. 2004;74:805–816. doi: 10.1086/386296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, et al. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129:256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 27.Arocena DG, Iwahashi CK, Won N, Beilina A, Ludwig AL, et al. Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum Mol Genet. 2005;14:3661–3671. doi: 10.1093/hmg/ddi394. [DOI] [PubMed] [Google Scholar]
- 28.Handa V, Goldwater D, Stiles D, Cam M, Poy G, et al. Long CGG-repeat tracts are toxic to human cells: implications for carriers of Fragile X premutation alleles. FEBS Lett. 2005;579:2702–2708. doi: 10.1016/j.febslet.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Jin P, Duan R, Qurashi A, Qin Y, Tian D, et al. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;55:556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sofola OA, Jin P, Qin Y, Duan R, Liu H, et al. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007;55:565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherman SL. Premature ovarian failure among fragile X premutation carriers: parent-of-origin effect? Am J Hum Genet. 2000;67:11–13. doi: 10.1086/302985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oostra BA, Willemsen R. A fragile balance: FMR1 expression levels. Hum Mol Genet. 2003;12:R249–R257. doi: 10.1093/hmg/ddg298. [DOI] [PubMed] [Google Scholar]
- 33.Jacquemont S, Leehey MA, Hagerman RJ, Beckett LA, Hagerman PJ. Size bias of fragile X premutation alleles in late-onset movement disorders. J Med Genet. 2006;43:804–809. doi: 10.1136/jmg.2006.042374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin P, Zarnescu DC, Zhang F, Pearson CE, Lucchesi JC, et al. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in drosophila. Neuron. 2003;39:739–747. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- 35.Ikeda Y, Dick KA, Day JW, Ranum LPW. Molecular genetics of spinocerebellar ataxia type 8. In: Wells RD, Ashizawa T, editors. Genetic instabilities and neurological disease. Burlington: Academic Press; 2006. pp. 417–431. [Google Scholar]
- 36.Holmes SE, O’Hearn E, Cortez-Apreza N, Hwang HS, Ross CA, et al. Spinocerebellar ataxia type 12. In: Wells RD, Ashizawa T, editors. Genetic instabilities and neurological diseases. Burlington: Academic Press; 2006. pp. 461–473. [Google Scholar]
- 37.Wetzel R. Chemical and physical properties of polyglutamine repeat sequences. In: Wells R, Ashizawa T, editors. Genetic instabilities and neurological disease. Burlington: Academic Press; 2006. pp. 517–534. [Google Scholar]
- 38.Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, et al. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci USA. 1999;96:4604–4609. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ross CA, Wood JD, Peters MF, Schilling G, Nucifora FC, et al. Polyglutamine pathogenesis, potential role of protein interactions, proteolytic processing and nuclear localization. In: Harper P, Perutz M, editors. Glutamine repeats and neurodegenerative diseases: molecular aspects. New York: Oxford University Press, Inc; 2001. pp. 93–112. [Google Scholar]
- 40.Chen CJ, Fischbeck KH. Clinical features and molecular biology of Kennedy’s disease. In: Wells R, Ashizawa T, editors. Genetic instabilities and neurological disease. Burlington: Academic Press; 2006. pp. 211–220. [Google Scholar]
- 41.Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 42.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Li SH, Cheng AL, Mangiarini L, Bates GP, et al. Ultrastructural localization and progressive formation of neuropil aggregates in Huntington’s disease transgenic mice. Hum Mol Genet. 1999;8:1227–1236. doi: 10.1093/hmg/8.7.1227. [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 45.Mirkin SM, Smirnova EV. Positioned to expand. Nat Genet. 2002;31:5–6. doi: 10.1038/ng0502-5. [DOI] [PubMed] [Google Scholar]
- 46.Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- 47.Tome FM, Fardeau M. Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol. 1980;49:85–87. doi: 10.1007/BF00692226. [DOI] [PubMed] [Google Scholar]
- 48.Margolis RL, Holmes SE, Rudnicki D, O’Hearn E, Ross CA, et al. Huntington’s disease-like 2. In: Wells R, Ashizawa T, editors. Genetic instabilities and neurological diseases. Burlington: Academic Press; 2006. pp. 261–273. [Google Scholar]
- 49.Reyniers E, Vits L, De Boulle K, Van Roy B, Van Velzen D, et al. The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nat Genet. 1993;4:143–146. doi: 10.1038/ng0693-143. [DOI] [PubMed] [Google Scholar]
- 50.Malter HE, Iber JC, Willemsen R, De Graaff E, Tarleton JC, et al. Characterization of the full fragile X syndrome mutation in fetal gametes. Nat Genet. 1997;15:165–169. doi: 10.1038/ng0297-165. [DOI] [PubMed] [Google Scholar]
- 51.Hansen RS, Canfield TK, Lamb MM, Gartler SM, Laird CD. Association of fragile X syndrome with delayed replication of the FMR1 gene. Cell. 1993;73:1403–1409. doi: 10.1016/0092-8674(93)90365-w. [DOI] [PubMed] [Google Scholar]
- 52.Wöhrle D, Hennig I, Vogel W, Steinbach P. Mitotic stability of fragile X mutations in differentiated cells indicates early post-conceptional trinucleotide repeat expansion. Nat Genet. 1993;4:140–142. doi: 10.1038/ng0693-140. [DOI] [PubMed] [Google Scholar]
- 53.Moutou C, Vincent MC, Biancalana V, Mandel JL. Transition from premutation to full mutation in fragile X syndrome is likely to be prezygotic. Hum Mol Genet. 1997;6:971–979. doi: 10.1093/hmg/6.7.971. [DOI] [PubMed] [Google Scholar]
- 54.Bontekoe CJ, Bakker CE, Nieuwenhuizen IM, van Der Linde H, Lans H, et al. Instability of a (CGG)(98) repeat in the Fmr1 promoter. Hum Mol Genet. 2001;10:1693–1699. doi: 10.1093/hmg/10.16.1693. [DOI] [PubMed] [Google Scholar]
- 55.Entezam A, Biacsi R, Orrison B, Saha T, Hoffman GE, et al. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;395:125–134. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brouwer JR, Mientjes EJ, Bakker CE, Nieuwenhuizen IM, Severijnen LA, et al. Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated fragile X full mutation. Exp Cell Res. 2007;313:244–253. doi: 10.1016/j.yexcr.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jansen G, Willems P, Coerwinkel M, Nillesen W, Smeets H, et al. Gonosomal mosaicism in myotonic dystrophy patients: involvement of mitotic events in (CTG)n repeat variation and selection against extreme expansion in sperm. Am J Hum Genet. 1993;54:575–585. [PMC free article] [PubMed] [Google Scholar]
- 58.Dean NL, Tan SL, Ao A. Instability in the transmission of the myotonic dystrophy CTG repeat in human oocytes and preimplantation embryos. Fertil Steril. 2006;86:98–105. doi: 10.1016/j.fertnstert.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 59.Kaytor MD, Burright EN, Duvick LA, Zoghbi HY, Orr HT. Increased trinucleotide repeat instability with advanced maternal age. Hum Mol Genet. 1997;6:2135–2139. doi: 10.1093/hmg/6.12.2135. [DOI] [PubMed] [Google Scholar]
- 60.Savouret C, Garcia-Cordier C, Megret J, te Riele H, Junien C, et al. MSH2-dependent germinal CTG repeat expansions are produced continuously in spermatogonia from DM1 transgenic mice. Mol Cell Biol. 2004;24:629–637. doi: 10.1128/MCB.24.2.629-637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leeflang EP, Tavare S, Marjoram P, Neal CO, Srinidhi J, et al. Analysis of germline mutation spectra at the Huntington’s disease locus supports a mitotic mutation mechanism. Hum Mol Genet. 1999;8:173–183. doi: 10.1093/hmg/8.2.173. [DOI] [PubMed] [Google Scholar]
- 62.Kovtun IV, McMurray CT. Trinucleotide expansion in haploid germ cells by gap repair. Nat Genet. 2001;27:407–411. doi: 10.1038/86906. [DOI] [PubMed] [Google Scholar]
- 63.Yoon SR, Dubeau L, de Young M, Wexler NS, Arnheim N. Huntington disease expansion mutations in humans can occur before meiosis is completed. Proc Natl Acad Sci USA. 2003;100:8834–8838. doi: 10.1073/pnas.1331390100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nichol Edamura K, Pearson CE. DNA methylation and replication: implications for the “deletion hotspot” region of FMR1. Hum Genet. 2005;118:301–304. doi: 10.1007/s00439-005-0037-5. [DOI] [PubMed] [Google Scholar]
- 65.Wöhrle D, Kennerknecht I, Wolf M, Enders H, Schwemmle S, et al. Heterogeneity of DM kinase repeat expansion in different fetal tissues and further expansion during cell proliferation in vitro: evidence for a causal involvement of methyl-directed DNA mismatch repair in triplet repeat stability. Hum Mol Genet. 1995;4:1147–1153. doi: 10.1093/hmg/4.7.1147. [DOI] [PubMed] [Google Scholar]
- 66.Ashizawa T, Dubel JR, Harati Y. Somatic instability of CTG repeat in myotonic dystrophy. Neurology. 1993;43:2674–2678. doi: 10.1212/wnl.43.12.2674. [DOI] [PubMed] [Google Scholar]
- 67.Monckton DG, Wong LJ, Ashizawa T, Caskey CT. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum Mol Genet. 1995;4:1–8. doi: 10.1093/hmg/4.1.1. [DOI] [PubMed] [Google Scholar]
- 68.Wong LJC, Ashizawa T, Monckton DG, Caskey CT, Richards CS. Somatic heterogeneity of the CTG repeat in myotonic dystrophy is age and size dependent. Am J Hum Genet. 1995;56:114–122. [PMC free article] [PubMed] [Google Scholar]
- 69.van den Broek WJ, Wansink DG, Wieringa B. Somatic CTG*CAG repeat instability in a mouse model for myotonic dystrophy type 1 is associated with changes in cell nuclearity and DNA ploidy. BMC Mol Biol. 2007;8:61. doi: 10.1186/1471-2199-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kennedy L, Shelbourne PF. Dramatic mutation instability in HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington’s disease? Hum Mol Genet. 2000;9:2539–2544. doi: 10.1093/hmg/9.17.2539. [DOI] [PubMed] [Google Scholar]
- 71.Kennedy L, Evans E, Chen CM, Craven L, Detloff PJ, et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. 2003;12:3359–3367. doi: 10.1093/hmg/ddg352. [DOI] [PubMed] [Google Scholar]
- 72.Gomes-Pereira M, Foiry L, Gourdon G. Transgenic mouse models of unstable trinucleotide repeats: toward an understanding of disease-associated repeat size mutation. In: Wells R, Ashizawa T, editors. Genetic instabilities and neurological diseases. Burlington: Academic Press; 2006. pp. 563–583. [Google Scholar]
- 73.Kunkel TA. Nucleotide repeats. Slippery DNA and diseases. Nature. 1993;365:207–208. doi: 10.1038/365207a0. [DOI] [PubMed] [Google Scholar]
- 74.Pearson CE, Tam M, Wang YH, Montgomery SE, Dar AC, et al. Slipped-strand DNAs formed by long (CAG)*(CTG) repeats: slipped-out repeats and slip-out junctions. Nucleic Acids Res. 2002;30:4534–4547. doi: 10.1093/nar/gkf572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mirkin SM. DNA structures, repeat expansions and human hereditary disorders. Curr Opin Struct Biol. 2006;16:351–358. doi: 10.1016/j.sbi.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 76.Eichler EE, Holden J, Popovich BW, Reiss AL, Snow K, et al. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat Genet. 1994;8:88–94. doi: 10.1038/ng0994-88. [DOI] [PubMed] [Google Scholar]
- 77.Kunst CB, Warren ST. Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell. 1994;77:853–861. doi: 10.1016/0092-8674(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 78.Chung MY, Ranum LPW, Duvick LA, Servadio A, Zoghbi HY. Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I. Nat Genet. 1993;5:254–258. doi: 10.1038/ng1193-254. [DOI] [PubMed] [Google Scholar]
- 79.Miret JJ, Pessoa-Brandao L, Lahue RS. Orientation-dependent and sequence-specific expansions of CTG/CAG trinucleotide repeats in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1998;95:12438–12443. doi: 10.1073/pnas.95.21.12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bontekoe CJM, de Graaff E, Nieuwenhuizen IM, Willemsen R, Oostra BA. FMR1 premutation allele is stable in mice. Eur J Hum Genet. 1997;5:293–298. [PubMed] [Google Scholar]
- 81.Gourdon G, Dessen P, Lia AS, Junien C, Hofmann-fRadvanyi H. Intriguing association between disease associated unstable trinucleotide repeat and CpG island. Ann Genet. 1997;40:73–77. [PubMed] [Google Scholar]
- 82.Brock GJ, Anderson NH, Monckton DG. Cis-acting modifiers of expanded CAG/CTG triplet repeat expandability: associations with flanking GC content and proximity to CpG islands. Hum Mol Genet. 1999;8:1061–1067. doi: 10.1093/hmg/8.6.1061. [DOI] [PubMed] [Google Scholar]
- 83.Sutherland GR. Fragile sites on human chromosomes: demonstration of their dependence on the type of tissue culture medium. Science. 1977;197:265–266. doi: 10.1126/science.877551. [DOI] [PubMed] [Google Scholar]
- 84.Gourdon G, Radvanyi F, Lia AS, Duros C, Blanche M, et al. Moderate intergenerational and somatic instability of a 55-CTG repeat in transgenic mice. Nat Genet. 1997;15:190–192. doi: 10.1038/ng0297-190. [DOI] [PubMed] [Google Scholar]
- 85.Seznec H, Lia-Baldini AS, Duros C, Fouquet C, Lacroix C, et al. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the CM CTG repeat intergenerational and somatic instability. Hum Mol Genet. 2000;9:1185–1194. doi: 10.1093/hmg/9.8.1185. [DOI] [PubMed] [Google Scholar]
- 86.Peier A, Nelson D. Instability of a premutation-sized CGG repeat in FMR1 YAC transgenic mice. Genomics. 2002;80:423–432. doi: 10.1006/geno.2002.6849. [DOI] [PubMed] [Google Scholar]
- 87.Monckton DG, Coolbaugh MI, Ashizawa KT, Siciliano MJ, Caskey CT. Hypermutable myotonic dystrophy CTG repeats in transgenic mice. Nat Genet. 1997;15:193–196. doi: 10.1038/ng0297-193. [DOI] [PubMed] [Google Scholar]
- 88.Kang S, Jaworski A, Ohshima K, Wells RD. Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat Genet. 1995;10:213–218. doi: 10.1038/ng0695-213. [DOI] [PubMed] [Google Scholar]
- 89.Gray SJ, Gerhardt J, Doerfler W, Small LE, Fanning E. An origin of DNA replication in the promoter region of the human fragile X mental retardation (FMR1) gene. Mol Cell Biol. 2007;27:426437. doi: 10.1128/MCB.01382-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cleary JD, Pearson CE. The contribution of cis-elements to disease-associated repeat instability: clinical and experimental evidence. Cytogenet Genome Res. 2003;100:25–55. doi: 10.1159/000072837. [DOI] [PubMed] [Google Scholar]
- 91.Pearson CE. Slipping while sleeping? Trinucleotide repeat expansions in germ cells. Trends Mol Med. 2003;9:490–495. doi: 10.1016/j.molmed.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y, Monckton DG, Siciliano MJ, Connor TH, Meistrich ML. Age and insertion site dependence of repeat number instability of a human DM1 transgene in individual mouse sperm. Hum Mol Genet. 2002;11:791–798. doi: 10.1093/hmg/11.7.791. [DOI] [PubMed] [Google Scholar]
- 93.Thornton CA, Griggs RC, Moxley R. Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol. 1994;35:269–272. doi: 10.1002/ana.410350305. [DOI] [PubMed] [Google Scholar]
- 94.Yang Z, Lau R, Marcadier JL, Chitayat D, Pearson CE. Replication inhibitors modulate instability of an expanded trinucleotide repeat at the myotonic dystrophy type 1 disease locus in human cells. Am J Hum Genet. 2003;73:1092–1105. doi: 10.1086/379523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Spiro C, McMurray CT. Nuclease-deficient FEN-1 blocks Rad51/ BRCA1-mediated repair and causes trinucleotide repeat instability. Mol Cell Biol. 2003;23:6063–6074. doi: 10.1128/MCB.23.17.6063-6074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gonitel R, Moffitt H, Sathasivam K, Woodman B, Detloff PJ, et al. DNA instability in postmitotic neurons. Proc Natl Acad Sci USA. 2008;105:3467–3472. doi: 10.1073/pnas.0800048105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Savouret C, Brisson E, Essers J, Kanaar R, Pastink A, et al. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J. 2003;22:2264–2273. doi: 10.1093/emboj/cdg202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wheeler VC, Lebel LA, Vrbanac V, Teed A, te Riele H, et al. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet. 2003;12:273–281. doi: 10.1093/hmg/ddg056. [DOI] [PubMed] [Google Scholar]
- 99.Foiry L, Dong L, Savouret C, Hubert L, te Riele H, et al. Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum Genet. 2006;119:520–526. doi: 10.1007/s00439-006-0164-7. [DOI] [PubMed] [Google Scholar]
- 100.Entezam A, Usdin K. ATR protects the genome against CGG*CGG-repeat expansion in Fragile X premutation mice. Nucleic Acids Res. 2007;36:1050–1056. doi: 10.1093/nar/gkm1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Razidlo DF, Lahue RS. Mrc1, Tof1 and Csm3 inhibit CAG. CTG repeat instability by at least two mechanisms. DNA Repair (Amst) 2008;7:633–640. doi: 10.1016/j.dnarep.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kovtun IV, McMurray CT. Features of trinucleotide repeat instability in vivo. Cell Res. 2008;18:198–213. doi: 10.1038/cr.2008.5. [DOI] [PubMed] [Google Scholar]
- 103.Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–1859. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- 104.Lin Y, Wilson JH. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2007;27:6209–6217. doi: 10.1128/MCB.00739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dion V, Lin Y, Hubert L, Jr, Waterland RA, Wilson JH. Dnmt1 deficiency promotes CAG repeat expansion in the mouse germ-line. Hum Mol Genet. 2008;17:1306–1317. doi: 10.1093/hmg/ddn019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baskaran S, Datta S, Mandal A, Gulati N, Totey S, et al. Instability of CGG repeats in transgenic mice. Genomics. 2002;80:151. doi: 10.1006/geno.2002.6813. [DOI] [PubMed] [Google Scholar]
- 107.Wang YH, Griffith J. Methylation of expanded CCG triplet repeat DNA from fragile X syndrome patients enhances nucleosome exclusion. J Biol Chem. 1996;271:22937–22940. [PubMed] [Google Scholar]
- 108.Jasinska A, Michlewski G, de Mezer M, Sobczak K, Kozlowski P, et al. Structures of trinucleotide repeats in human transcripts and their functional implications. Nucleic Acids Res. 2003;31:5463–5468. doi: 10.1093/nar/gkg767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Godde JS, Kass SU, Hirst MC, Wolffe AP. Nucleosome assembly on methylated CGG triplet repeats in the Fragile X Mental Retardation gene 1 promoter. J Biol Chem. 1996;271:24325–24328. doi: 10.1074/jbc.271.40.24325. [DOI] [PubMed] [Google Scholar]
- 110.Wang YH, Amirhaeri S, Kang S, Wells RD, Griffith JD. Preferential nucleosome assembly at DNA triplet repeats from the myotonic dystrophy gene. Science. 1994;265:669–671. doi: 10.1126/science.8036515. [DOI] [PubMed] [Google Scholar]