

Abstract
We present primary results from the Sequencing Quality Control (SEQC) project, coordinated by the United States Food and Drug Administration. Examining Illumina HiSeq, Life Technologies SOLiD and Roche 454 platforms at multiple laboratory sites using reference RNA samples with built-in controls, we assess RNA sequencing (RNA-seq) performance for junction discovery and differential expression profiling and compare it to microarray and quantitative PCR (qPCR) data using complementary metrics. At all sequencing depths, we discover unannotated exon-exon junctions, with >80% validated by qPCR. We find that measurements of relative expression are accurate and reproducible across sites and platforms if specific filters are used. In contrast, RNA-seq and microarrays do not provide accurate absolute measurements, and gene-specific biases are observed, for these and qPCR. Measurement performance depends on the platform and data analysis pipeline, and variation is large for transcript-level profiling. The complete SEQC data sets, comprising >100 billion reads (10Tb), provide unique resources for evaluating RNA-seq analyses for clinical and regulatory settings.
Technological advances have made deep RNA sequencing feasible, expanding our view of the transcriptome1 and promising to permit quantitative profiling with large dynamic range.2 Recent comparisons of RNA-seq with established technologies for differential expression analysis have found good overall agreement between RNA-seq, qPCR and microarrays. In general, RNA-seq has provided increased detection sensitivity and opened new avenues of research in transcriptome analyses, such as the study of gene fusions, allele-specific expression and novel alternative transcripts. However, it has been shown that RNA-seq data exhibit measurement noise, which is a direct consequence of the random sampling process inherent to the assay. Assessments of RNA-seq have been limited to individual sequencing platforms and experiments, which may explain the variation of conclusions by study (see Supplementary Table 1 and Supplementary Notes section 2.4 for discussion)3–6 Moreover, new platforms and protocols for RNA-seq have emerged in recent years. With the widespread adoption of RNA-seq, including the completion of large projects, such as ENCODE,7 TCGA8 and ICGC,9 a comprehensive multi-site cross-platform analysis of RNA-seq performance is timely. Reproducibility across laboratories, in particular, is a crucial requirement for any new experimental method in research and clinical applications, and can only be tested in extensive comparisons of different sites and platforms. As in phase-I of the MicroArray Quality Control project (MAQC-I),10 which tested agreement across sites and platforms for gene-expression microarrays, the FDA has coordinated the Sequencing Quality Control project (SEQC/MAQC-III), a large-scale community effort to assess the performance of RNA-seq across laboratories and to test different sequencing platforms and data analysis pipelines.
Here we report a multi-site cross-platform analysis of RNA-seq measurement performance in a controlled setting. We sequenced commercially available reference RNA samples spiked with synthetic RNA from the External RNA Control Consortium. Two distinct samples were assessed individually and also combined in known ratios. This allowed us to examine how well truths built into the study design, such as known relationships between samples within and across sites, could be recovered from measurements. With no independent ‘gold standard’ feasible, these ‘known truths’ support an objective assessment of performance. To this end, we examined a multitude of properties, including complementary metrics of reproducibility, accuracy and information content. Such a multi-dimensional characterization is critical for the development of more powerful analyses of the underlying biological mechanisms in complex data sets because often there is a trade-off between one desirable property and another, such as accuracy versus precision. Analyses focusing on measurement quality metrics51, spike-in controls and limits of detection52 and the effects of analytic pipeline choice53 are presented in separate studies.
The SEQC project also involved studies assessing RNA-seq in several research applications (Fig. 1a), including a performance analysis of neuroblastoma outcome prediction54, a comparative investigation of toxicogenomic samples testing chemicals with different modes of action,55 and a comprehensive survey of tissue-specific gene expression in rat.47 In total, >100 billion reads (10 terabases) of RNA-seq data were produced and studied, which to our knowledge represents the largest effort to date to generate and analyze comprehensive reference data sets. A rigorous dissection of sources of noise and signal indicates that, given appropriate data treatment and analysis, RNA-seq can be highly reproducible, particularly in differential gene-expression analysis.
Figure 1.

The SEQC (MAQC-III) project and experimental design. (a) Overview of projects. We report on a group of studies assessing different sequencing platforms in real-world use cases, including transcriptome annotation and other research applications, as well as clinical settings. This paper focuses on the results of a multi-center experiment with built-in ground truths. (b) Main study design. Similar to the MAQC-I benchmarks, we analysed RNA samples A to D: Samples C and D were created by mixing the well-characterized samples A and B in 3:1 and 1:3 ratios, respectively. This allows tests for titration consistency (c) and the correct recovery of the known mixing ratios (d). Synthetic RNAs from the External RNA Control Consortium (ERCC) were both pre-added to samples A and B before mixing and also sequenced separately to assess dynamic range (samples E and F). Samples were distributed to independent sites for RNA-seq library construction and profiling by Illumina’s HiSeq 2000 (3 official + 3 inofficial sites) and Life Technologies’ SOLiD 5500 (3 official sites + 1 inofficial site). Unless mentioned otherwise, data presented shows results from the three official sites (italics). In addition to the four replicate libraries each for samples A to D per site, for each platform, one vendor-prepared library A5…D5 was being sequenced at the official sites, giving a total of 120 libraries. At each site, every library has a unique bar-code sequence and all libraries were pooled before sequencing, so each lane was sequencing the same material, allowing a study of lane specific effects. To support a later assessment of gene models, we sequenced samples A and B by Roche 454 (3x, no replicates, see Supplementary Notes Section 2.5). (c) Schema illustrating tests for titration order consistency. Four examples are shown. The dashed lines represent the ideal mixture of samples A and B (blue and red) expected for samples D and C (magenta and dark purple). (d) Schema illustrating a consistency test for recovering the expected sample mixing ratio. The yellow lines mark a 10% deviation from the expected response (black) for a perfect mixing ratio. Both tests (c) and (d) will reflect both systemic distortions (bias) and random variation (noise).
RESULTS
Study design
We used the well-characterized reference RNA samples A (Universal Human Reference RNA) and B (Human Brain Reference RNA) from the MAQC consortium,10 adding spike-ins of synthetic RNA from the External RNA Control Consortium (ERCC).11 We then mixed A and B in known ratios, 3:1 and 1:3, to construct samples C and D, respectively (Supplementary Fig. 1). All samples were distributed to several independent sites for RNA-seq library construction and profiling by Illumina’s HiSeq 2000 and Life Technologies’ SOLiD 5500 instruments. In addition, vendors created their own cDNA libraries, which were distributed to each test site to examine ‘site effects’ independent of the library preparation process (Fig. 1b). To support an assessment of gene models, three sites also independently sequenced samples A and B using the Roche 454 GS FLX platform, providing longer reads. In total, for samples A to D, 108 libraries were sequenced on a HiSeq 2000, another 68 libraries on SOLiD, and 6 libraries on a Roche 454.
To compare technologies, we also examined expression profiles of the same reference samples generated from Affymetrix HGU133Plus2.0 microarrays in the MAQC-I study, and profiled and analysed these samples on several current microarray platforms (Methods). Besides RNA-seq and microarrays, we also considered qPCR-based protocols: we examined 843 TaqMan assays from the MAQC-I study, and in addition performed 20,801 PrimePCR reactions.
Read depth dependency of gene detection and junction discovery
Because efficient quantitative expression profiling takes advantage of known gene models3 the choice of a reference annotation can considerably affect results, including performance assessments. Our data showed that the number of reads mapped to known genes depends on the accuracy and completeness of the gene models. Among all 23.2 billion reads that could be mapped to genes other than those encoding mitochondrial or ribosomal RNAs, 85.9% were mapped to RefSeq,12 whereas 92.9% mapped to GENCODE,13 and 97.1% to NCBI AceView14 (Fig. 2a). This is not a property of the samples examined (A, B, C and D), as a similar trend is seen when adding all reads from the SEQC neuroblastoma project (Supplementary Notes Section 1.2). The higher read fraction unique to AceView is genuinely due to the higher accuracy of its gene models (Fig. 2a), as AceView annotated exons cover fewer bases than does GENCODE (191 Mb versus 203 Mb; Supplementary Notes Section 1.2).
Figure 2.
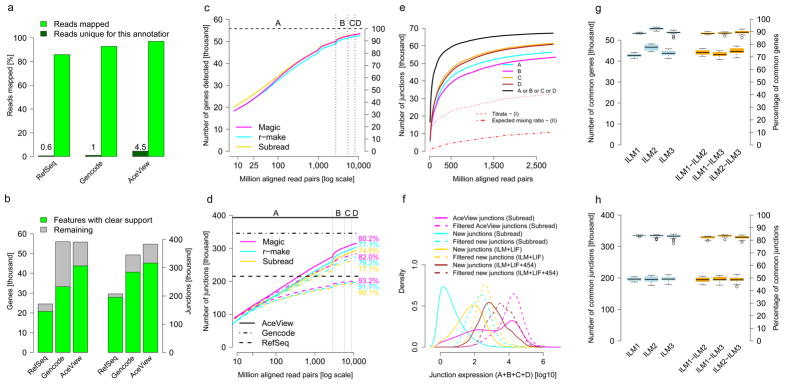
Gene detection and junction discovery. (a) The fraction of all reads aligned to gene models from different annotations, RefSeq, Encode and NCBI AceView (green). Reads aligning only to specific annotations are shown in dark green. (b) Known genes (left) and exon junctions (right) supported by at least 16 HiSeq 2000 or SOLiD reads are in green; genes or junctions annotated but not observed at this threshold are shown in grey. (c–e) show sensitivity as a function of read depth. (c) Known genes detected. We show the number and percentage of all AceView annotated genes detected for three RNA-seq analysis pipelines, Subread (yellow), r-make (cyan) and Magic (magenta). The x-axis marks cumulative aligned fragments from all replicates and sites. Vertical lines indicate boundaries between samples A through D. (d) Known junctions detected. The numbers and percentages of all exon-exon junctions (supported by 8 or more reads) are shown for different gene model databases (line style). Horizontal lines show the respective total numbers of annotated junctions. (e) Unannotated junctions supported by multiple platforms and pipelines. Subsets of unannotated junctions have expression levels with correct titration orders and mixing ratios (cf. Figs 1b–d and 4a,b). (f) Distribution of junction expression levels. Unannotated junctions, then unannotated junctions supported by multiple platforms and pipelines, and known junctions show increasing expression levels (colors). Subsets expressed with mutual information about the samples and correct titration order and mixing ratio display a further shift towards higher expression levels (dashed lines). (g, h) Intra- (blue) and inter-site reproducibility (orange) of detected known genes (g) and junctions (h). Pairwise agreement is shown by boxplots, where the second set of boxplots (upper group) indicates percentages.
As the data constitute the deepest sequencing of any set of samples yet reported and include a total of 12 billion mapped HiSeq 2000 RNA-seq fragments, we examined how well the known genes could be detected as a function of aggregate read depth, taking all replicate libraries, sites and samples together. We report read depth as the number of sequenced fragments because the mapping and counting of paired ends are highly correlated; single-ended reads can thus be used when the additional long-range information from read pairs is not required. At a sequencing depth of 10 million aligned fragments, about 35,000 of the 55,674 genes annotated in AceView14 were found by at least one read. Some of these are due to background noise, for instance, from genomic DNA contamination (Supplementary Notes). For a comparison of alternative pipelines, annotations and the effect of read depth, we next focused on genes with strong support (16 or more reads). At this stringency, we found that about 20,000 genes were detected at a sequencing depth of 10 million aligned fragments, which covered the majority of strongly expressed genes. Detection increased to >30,000 genes at 100 million fragments, and finally to >45,000 at about one billion fragments. Although the number of additional known genes detected successively decreased for each doubling of read depth, additional genes were still being detected even at high read depths of >1 billion fragments, indicative of low expression levels per cell or small numbers of cells expressing these genes (Fig. 2c for HiSeq 2000 and Supplementary Figure 2 for SOLiD).
We examined the detection of exon-exon junctions as a function of read depth for RefSeq, GENCODE and AceView annotation (Fig. 2d). In general, with each doubling of the read depth, many additional known junctions were detected for the more comprehensive annotations, even at high read depths exceeding one billion reads. As samples A and B in the study are very different, we expected to see more transcriptional complexity when combining samples. Indeed, combining biologically distinct samples contributes more to exploring the complex transcriptome space than merely increasing read depth (Fig. 2d)47,54. The number of additional known junctions decreased fastest for RefSeq, which provides the least complex annotation (Supplementary Figure 3), with practically all annotated junctions observed at the highest read depth. In contrast, the AceView database is the most comprehensive and has the highest number of junctions supported by reads from this study, reaching over 300,000 junctions at the maximum read depth, more than three times the number detected at 10 million reads. Although GENCODE and AceView have similar total numbers of genes and similar footprints on the genome, considerably fewer annotated genes and junctions in GENCODE were supported by the observed reads (Fig. 2b and Supplementary Figure 4). Therefore, all subsequent analyses presented in this manuscript are based on AceView, unless stated otherwise.
We next analyzed the reproducibility of detecting genes and junctions across measurement sites, platforms and analysis pipelines, as a key strength of RNA-seq is its inherent ability to identify splice sites de novo. To test this ability of RNA-seq to discover junctions, we first examined the HiSeq 2000 data because of the greater read length and depth. We considered three independent pipelines for de novo discovery of junctions independent of existing gene models: NCBI Magic,14 r-make (which uses STAR15) and Subread.16 All pipelines reported millions of junctions, with r-make predicting about 50% more than Subread and Magic, although almost all junctions found by Subread or Magic were also found by r-make. We also observed substantial but 12% lower agreement with TopHat2, regardless of whether it was run with or without gene model guided alignment17 (Supplementary Figure 5a), giving a total of 1,110,550 junctions consistently found by all five analysis variants. In total, 2.6 million previously unannotated splice junctions were called by at least one of the five analysis pipelines, yet only 820,727 (32%) were consistently predicted by all the methods (Supplementary Figure 5b), illustrating the considerable difficulty of reliably detecting splice junctions de novo with current analysis tools.
We then examined whether unannotated junctions were independently discovered in both HiSeq 2000 and SOLiD data, as junctions only found by a single platform or library preparation protocol could be technical artifacts (Supplementary Figure 6). Junction discoveries from the SOLiD data reflected the lower read length and lower read depth as expected (simulation results in Supplementary Table 2). In particular we discovered 87,117 unannotated potential junctions in the SOLiD data, of which 74,561 (86%) were also independently discovered from HiSeq 2000 reads using the Subread aligner. The number of these new junctions found in each of the four samples (Fig. 2e) followed the order expected corresponding to sample complexity: B<A<D<C.
We then used the built-in truths of the benchmark measurements to examine the accuracy of the sample-specific levels of support of the unannotated junctions in terms of their ability to capture the expected A/B sample mixing ratio and yield titration order consistency. For example (Fig. 1c), if a gene is more strongly expressed in sample A than in sample B (‘A > B’) then we expect A > C > D > B because C = ¾ A + ¼ B and D = ¼ A + ¾ B, and we expect the inverse order if B > A. This consistency test is affected both by systemic distortions reducing accuracy and random variations reducing reproducibility. Another complementary test assesses the A/B mixing ratio recovery in the samples C (3:1) and D (1:3), which can be examined in a plot of log2(C/D) vs log2(A/B). Deviations from the ideal line (Fig. 1d) are also affected by systemic distortions, which reduce accuracy, and by random variations, which reduce reproducibility. Because both tests only reflect reproducibility for genes with similar expression levels in samples A and B, we also require a clear differential signal as assessed by the mutual information, a measure of information content (Online Methods).
We observed that requiring a consistent titration order and the correct A/B mixing ratio clearly enriched for junctions with higher expression levels (Fig. 2f), which were the easiest to measure and quantify. A comparison with junctions detected in Roche 454 data confirmed that the more abundant junctions could reliably be detected across different sequencing platforms (Supplementary Figure 7).
For an examination of how well junctions discovered by RNA-seq could be independently confirmed by a different technology, we performed qPCR with primer pairs designed to specifically validate 173 detected junctions. We randomly selected 136 well-supported junctions that had been discovered de novo by all three RNA-seq analysis pipelines in both HiSeq 2000 and SOLiD data. These junctions were chosen so that they log-uniformly covered a range of ~10–3,000 supporting reads, and so that half of them met all consistency tests. In addition, we also tested 13 known AceView junctions as positive controls and 24 unannotated junctions that had only been discovered by a single analysis pipeline in the HiSeq 2000 or SOLiD data, despite having support by many reads (~300–3,000). Only five assays were non-informative (Online Methods). Notably, in the remaining assays, all of the 13 positive controls and all of the 133 well-supported junctions were reliably identified by qPCR, with the numbers of supporting RNA-seq reads largely reflecting estimates of expression levels by qPCR (slope 0.95, Pearson (Spearman) correlation 0.74 (0.77), N = 146), even for junctions with low read numbers or not meeting all consistency tests (Supplementary Figure 8). Moreover, 18 of the 22 pipeline specific junctions (>80%) could be confirmed at least qualitatively. For the most comprehensive surveys of potential new junctions, one may therefore want to consider all discovered junctions, although it makes sense to prioritize well-supported junctions found consistently (Fisher p < 4×10−4).
In the complete data set comprising SEQC samples A, B, C and D, we consistently detected ~44,000 known genes (Fig. 2g) and ~310,000 known exons across pairs of replicate sites (Supplementary Figure 9), constituting about 79% and 47% of all known genes and exons, respectively. Nearly 200,000 splice junctions were seen consistently (Fig. 2h), making up about 50% of all known junctions. This corresponds to about 90%of all detected known genes, 87% of detected known exons and 83% of detected known junctions, consistent with the explanation that larger features aggregating more reads are easier to measure reproducibly. The fluctuations in the detection of sequence features stemmed largely from sequencing depth–dependent sampling noise, which was reflected in the very similar intra- and inter-site agreements in the detection of known genes (Fig. 2g), exons (Supplementary Figure 9) and junctions (Fig. 2h). Considering the low technical variance in addition to the unavoidable sampling noise, these results emphasize the value of biological replicates.
Improving differential expression analysis reliability
Studies on microarrays have shown that results of typical statistical differential expression tests thresholded by p-value need to be filtered and sorted by effect strength (fold-change) in order to attain robust comparisons across platforms and sites.10 We thus sought to identify corresponding requirements for RNA-seq, examining the reproducibility of rank ordered lists of differentially expressed genes (DEGs), as well as the False Discovery Rate reflecting the information content of the measurements. Because the same samples were profiled at all sites, the number of true DEGs is zero when comparing the same sample between any two sites. Any DEGs found for self–self comparisons thus represent technical differences and can be considered ‘false positives’ (shown as dots in Fig. 3a). We examined the number of inter-site A versus A ‘false positives’ relative to the number of DEGs in A versus B comparisons, giving an empirical estimate of the False Discovery Rate (eFDR). We tested several RNA-seq data analysis pipelines for the six HiSeq 2000 sites, focusing on the set of 23,437 genes present on the Affymetrix HGU133Plus2.0 microarrays for comparison.
Figure 3.
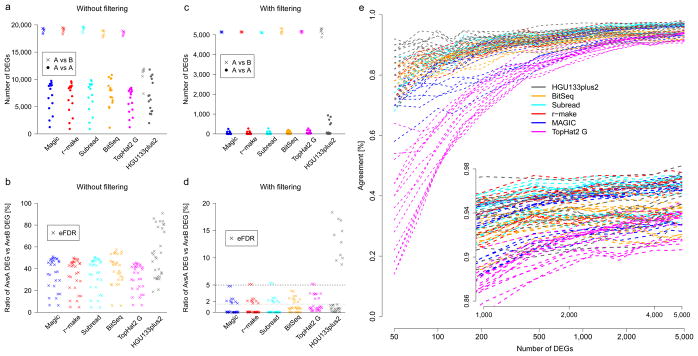
Sensitivity, specificity and reproducibility of differential expression calls. Robust cross-site analyses depend on pipeline choice and appropriate filter rules. Results are shown for five MAQC-III RNA-seq pipelines and the MAQC-I Affymetrix microarray platform (color). Panels (a and b) show results without filtering, panels (c to e) show results using appropriate filters. (a and c) Number of standardized differential expression calls. All possible pairwise inter-site A versus A comparisons (●) are shown next to all intra-site A versus B comparisons (x) as indicators for specificity and sensitivity. (b and d) Ratios of A versus A calls and A versus B calls give an estimate of the false discovery rate (eFDR). For all platforms and pipelines, differential expression calls identify thousands of differences in inter-site comparisons of identical samples. These can be controlled for microarray by additional filters for effect size. In addition, RNA-seq also requires filters for expression strength due to the high sampling fluctuations at lower read counts. These were set to give similar numbers of A versus B expression calls (b), improving the eFDR to <1.5% except for several outliers. (e) Inter-site reproducibility of differential expression calls. Comparing the identities and the directions of change for differentially expressed genes (DEGs) across sites, agreement is plotted for lists including the top-ranked genes as sorted by effect size (x-axis). The observed response curves depend on pipeline and filter choice, showing more variation for shorter lists. The performance of several RNA-seq pipelines was comparable to that of differential expression profiling using the microarray measurements from the MAQC-I study, with the microarrays showing higher inter-site reproducibility when considering only the differentially expressed genes with the strongest fold change (left side of e).
We found that unfiltered data for both RNA-seq and microarrays show many DEGs, both for false positives (A versus A) and the likely ‘real’ DEGs in the A versus B comparison (Fig. 3a), with the ratio of false positives versus true positives unacceptably high for both platforms (Fig. 3b). Also, we observed that different analysis pipelines vary in these measures of performance (Figs 3a–e). Next, we applied the |log2 fold-change|>1 filter advocated in the MAQC study on microarrays,10 and for microarrays observed a reduction in the eFDR to below 1.5%, save for one outlier site (Figs 3c,d). Notably, we found that applying pipeline-dependent filters for p-value, fold-change and expression-level (lowest third of all examined AceView genes, Supplementary Tables 3 and 4) successfully reduced the RNA-seq eFDR across sites without sacrificing sensitivity compared to the arrays (Fig. 3d). We note that results for SOLiD were very similar (Supplementary Figures 10 and 11), with expression level thresholds reflecting the lower read depth for that platform.
After applying these filters, we found that most (but not all) RNA-seq pipelines achieved high inter-site reproducibility of differential expression calls with up to 95% concordance in DEGs (Fig. 3e and Supplementary Figure 12). This concordance between sites was highest for the most strongly expressed genes. Moreover, the filters resulted in a good agreement of differential expression calls across platforms (for example, A versus B on HiSeq 2000 compared with A versus B on SOLiD, Supplementary Figures 13 and 14), suggesting that differential expression analyses from different platforms can be combined—for example, to extend existing studies with additional samples.
Relative but not absolute expression measures satisfy tests
After an examination of the reliability of differential expression analysis of genes, we next examined the quantification of RNA using four consistency tests exploiting ground truths built into the study design (Fig. 4). First, we considered titration-order consistency as introduced in Figure 1c. This metric is affected both by systemic distortions reducing accuracy and random variations reducing reproducibility. The majority of genes (59%) titrated correctly (Fig. 4a), with little disagreement between platforms (Supplementary Table 5). Genes with large differential expression performed best, with all genes showing consistent titration in several HiSeq 2000 and SOLiD sites, and no contradiction regarding the direction of change (blue curve). For the second built-in truth, we examined the A/B mixing ratio recovery (Fig. 1d) as another test reflecting accuracy and reproducibility. We observed the correct ratio for the majority of genes (Fig. 4b), with better agreement at higher expression levels (top 25%). Notably, the scatter of genes marked as titrating in this plot indicates that consistent titration does not guarantee a reliable recovery of the mixing ratio (and vice versa).
Figure 4.

Built-in truths for assessing RNA-seq. (a) Titration order A, C, D, B. Log2 fold-change is related to cross-platform titration consistency. At sufficiently strong log2 fold-change, reliable titration is also found across platforms: The dark blue line represents the 22,074 ‘unmissable’ genes showing the correct titration order with no contradiction in at least 14 HiSeq 2000 and 6 SOLiD samples. Most genes with high differential expression are in this class. (b) Known A/B mixing ratios in samples C and D. The yellow solid line traces the expected values after mRNA/total-RNA shift correction. The 1%, 10% and 25% most highly expressed genes are shown in red, cyan and magenta, respectively. On average, the most strongly expressed genes recover the expected mixing ratio best. Genes with inconsistent titration (cf. a) are colored grey. Black and grey symbols intermixing indicates that consistent titration (black) does not guarantee reliable recovery of the mixing ratio (and vice versa). (c) ERCC spike-in ratios can be recovered increasingly well at higher expression levels. From the response curves, one can calculate signal thresholds for the detection of a change.50 (d) Variation of the total amounts of detected ERCC spikes. The lack of reliable titration indicates that the considerable differences between libraries of a given site and protocol are random, implying limits for absolute expression level estimates, in general, and using spike-ins for the calibration of absolute quantification, in particular. The observed variations likely arise in library construction, as the vendor-prepared libraries (colored cyan or grey) gave constant results across different sites. For (a) and (b), all 55,674 AceView genes tested.
The third and fourth built-in truths leveraged the ERCC spike-ins.11 These analyses complement work examining fold-change recovery for these synthetic RNAs52. Across platforms, we observed that with sufficiently high expression levels (log2[conc]>3), the expected ratios of ½, ⅔, 1 and 4 were accurately recovered using about 90 million mapped fragments (Fig. 4c), with high precision indicating good reproducibility. Finally, we examined the ERCC absolute titration levels, as the ERCC RNAs were spiked into the samples A and B before the C and D samples were created (Supplementary Figure 1). We observed, however, that the fraction of reads aligning to ERCC spike-ins for a given sample varied widely between libraries and platforms, with ranges of measured ERCCs between 1–2.5% for HiSeq 2000 and 2.5–4.7% for SOLiD, with a clear ‘library effect’ observed for all sites and platforms, affecting reproducibility. (Fig. 4d and Supplementary Figure 15). Indeed, when using the vendor-prepared library as the cross-site control, we observed very consistent measurements of the percentages of reads mapping to ERCCs, which indicates a large degree of variation from the preparation of libraries even at the same site. The resulting lack of meaningful absolute expression level measurements is moreover not specific to the ERCC spike-ins, as similar variation and substantial platform specific differences could also be observed for human genes (Supplementary Figure 16).
In addition, when the 92 ERCC spike-in RNAs are compared to their nominal concentrations, some of them (e.g., ERCC-116) are systematically measured up to 10 times below or above their expected concentrations, perturbing even the order of the ERCC scale. These discrepancies are highly reproducible, suggesting that the bias is sequence dependent. Although marginal trends could be observed as functions of GC content and average sequence region unfolding probabilities (Supplementary Figures 17 and 18), these did not pass tests for statistical significance. No consistent trend could be observed as function of mRNA length (Supplementary Figure 19), and the majority of the deviations is not explained by any of these co-variates, indicating a need for further investigations of such distortions and their possible sources. We observed, however, that the effect is also protocol dependent and is reduced in the absence of poly-A selection (Supplementary Figure 20). This is in line with results in other studies, further underscoring the impact of protocol choice on quantification51,52, where fragmentation time, poly-A enrichment by columns, beads, or ribo-depletion, hexamer or oligo-dT priming, library isolation by gel or beads, different ligation efficiency and RNA quality at the start of library preparation have all been shown to have an effect. Consequently, just as for microarrays, absolute measurements by RNA-seq using a particular protocol are well reproducible but not very accurate. This observation implies that the use of external spike-in controls to accurately infer absolute expression levels of a gene of biological interest will remain challenging as long as these deviations are not better understood and can thus be avoided or quantitatively modeled.
Relative gene expression measurements agree across platforms
Because we observed good performance for RNA-seq in consistency tests of relative expression levels, we then sought to compare alternative measurement platforms. We first examined the differential expression of 843 genes measured by TaqMan for samples A and B in the MAQC-I study. Although more strongly expressed than typical AceView genes, these genes nevertheless span a wide range of expression levels (Supplementary Figure 21). We found good and comparable agreement among different platforms (Fig. 5a, where Pearson and Spearman correlation coefficients are given; Supplementary Figure 22). The HiSeq 2000 and SOLiD sequencing platforms showed the highest correlation to one another. This is consistent with other comparisons of relative expression measures.4,6,18,19
Figure 5.

Cross-platform agreement of expression levels. (a) Comparison of log2 fold-change estimates for 843 selected genes. Good and similar concordances were observed between relative expression measures from the MAQC-III HiSeq 2000 and SOLiD sequencing platforms, MAQC-I TaqMan and the MAQC-III Affymetrix HuGene2 arrays (Pearson and Spearman correlation coefficients are shown; cf. Supplementary Figure 22). (b) Comparison of absolute expression levels from HiSeq 2000 and SOLiD in a rank scatter density plot. Expression level ranks for sample A are shown on the x-axis for HiSeq 2000, and on the y-axis for SOLiD. Genes are represented by dots, and areas with several genes are shown in blue, with darker blue corresponding to a higher gene density in the area. Large cross-platform deviations are seen even for highly expressed genes and these variations are systematic. The genes in the vertical ‘spur’, for instance, are not detected by SOLiD RNA-seq but show strong expression levels in HiSeq 2000 RNA-seq, with an analog comparison to 20,801 qPCR measurements giving a similar picture (Supplementary Figure 25). The ERCC spike-ins are shown as red symbols (+). ERCC spike-in signals are systematically lower in the HiSeq 2000 data, which may be explained by their shorter poly-A tails and differences in the library construction protocols. (c) The same plot as (b) but removing the 11,066 genes that can be affected by the non-stranded nature of the applied Illumina protocol. Although the actual number of genes in the vertical spur that are not detected by SOLiD but show strong expression levels in the HiSeq 2000 is now smaller, it is still substantial. (d) Comparison of TaqMan and PrimePCR for 843 selected genes. Expression estimates vary considerably for individual genes, with some genes showing high expression in one platform but are not detected at all by the other.
For absolute expression levels, correlations to TaqMan were slightly better for RNA-seq than for microarrays (Spearman correlation 0.83 versus 0.79, p = 0.02), and the average trend follows a more linear shape (Supplementary Figure 23b). Although we observed that, on average, absolute expression levels agreed between different platforms, there were substantial deviations across the entire dynamic range for large numbers of individual genes. These deviations are systematic—that is, they are not a question of reproducibility, but rather reflect the accuracy of absolute expression measures. In particular, by comparing expression level estimates from HiSeq 2000 and SOLiD RNA-seq (Fig. 5b), we observed 5,056 genes that were expressed according to one platform but not the other (9%). This effect is only partly due to the non-stranded nature of the Illumina protocol used here, and the presence of 11,066 genes antisense to genes annotated on the opposite strand (Fig. 5c).
As an independent additional test, we generated 20,801 PrimePCR measurements of the SEQC samples A, B, C and D. We again observed that more than a thousand genes (5%) were not considered expressed by one platform but were clearly expressed according to the other (Supplementary Figures 24 and 25). Although qPCR based methods have traditionally been used as a reference ‘gold’ standard owing to their high sensitivity and dynamic range, it is noteworthy in this context that specific primer selection and protocol calibration are challenging in their own right.20 PCR is affected by GC bias21, and considerable differences in expression level measurements from different PCR based assays can be observed (Fig. 5d).
Performance assessment is metric dependent
A major promise of RNA-seq is the extension of expression profiling to the discovery and quantification of alternative transcripts. For transcript-specific profiling, however, no large-scale expression data from other technologies are available as an external reference point. The SEQC data represent an opportunity for the multi-platform comparison of transcript-specific measurements.
To support a balanced performance study of gene-level and transcript-specific expression profiling, we combined multiple metrics for a robust characterization of platforms, sites and data processing options: (i) average measurement precision,3 which directly assesses reproducibility, (ii) titration order consistency22 and (iii) recovery of the expected A/B mixing ratio, providing two complementary assessments reflecting both measurement accuracy and reproducibility, as well as (iv) differential expression and (v) the mutual information of sample titration, which capture different aspects of information content (Online Methods). For a summary view, we first focused on genes with a clear directional signal—that is, those that allowed an ordered discrimination of the samples A to D, as indicated by the mutual information metric (v). We then counted how many of these genes also satisfied a second requirement, for each of the metrics (i)–(iv). Such an integration of tests through counting genes that fulfill multiple assay criteria allows a comprehensive consideration of all the genes instead of restricting comparisons to a common subset of genes always identified as expressed. This is necessary for a meaningful comparison of pipelines and platforms with varying degrees of sensitivity (Supplementary Fig. 26a,b). The resulting four combined assays for the respective metrics (i)–(iv) are complementary—that is, a gene satisfying one does not generally satisfy the others (Supplementary Figure 27). The average of the four assays then provides a consistency score for robust characterization of measurement performance (Supplementary Figure 28).
For gene-level profiling, pipelines showed similar performances on average (Fig. 6a). Providing known gene models always considerably improved results (cf. our results for standard and gene model guided TopHat2). Relatively lower scores for transcript-level profiling indicate that the discrimination of alternative transcripts is more difficult, which is also reflected in stronger effects of pipeline choice (Fig. 6b).
Figure 6.

Multiple performance metrics for the quantification of genes and alternative transcripts. The y-axes show a Consistency Score. Secondary y-axes mark the percentage of the maximal possible score. Panels show the three official HiSeq 2000 and SOLiD sites and compare a few analysis variants: Green, TopHat2; magenta, TopHat2 guided by known gene models; cyan, Subread; yellow, BitSeq; blue, Magic. Panels a and b consider all AceView annotated genes. Panels c and d focus on a subset of expressed complex genes with multiple alternative transcripts where comparison to a high-resolution test microarray (rightmost bar) can be conducted. (e) Comparison of RNA-seq to four different microarrays and data-processing methods (red bars) by plotting the mutual information (y-axes) at different read depths (x-axes). For the microarrays, the number of probes used is shown. The numbers given for RNA-seq state the number of fragments mapped to genes as well as the [total fragments]. SOLiD and HiSeq 2000 performed similarly well for comparable effective read depths (Supplementary Figure 33a). HiSeq 2000 data is plotted here. Each bar shows the minima and maxima across the three official sites. The read depth for which average RNA-seq performance met or exceeded that of the array is marked by a cyan bar. The corresponding read depths varied widely from 5 M (HGU133plus2 with MAS5) to about 50 M fragments (PrimeView with gcRMA/affyPLM), showing the strong effect of the reference gene set implied by the probes on the respective arrays and the employed microarray data-processing methods. Results are shown for the Subread pipeline. Alternative RNA-seq data analysis pipelines, however, can require up to double the number of fragments (TopHat2+Cufflinks, Supplementary Figure 35). See Supplementary Figures 33 and 34 for comparisons of other platforms and read depths.
RNA-seq has sparked an interest in transcript-specific profiling and the development of advanced algorithms for estimating alternative transcript abundances. With known gene models, similar approaches can now also be applied for microarrays. We thus next focused on a test set of 782 genes with multiple alternative transcripts of varying complexity and specifically selected to represent the full subset of spliced genes in AceView (Online Methods). Covering 5,691 alternative transcripts, this test set allows a first comparison of transcript-specific expression level estimates from RNA-seq and high-resolution transcript-level microarray data. We found that efficient transcript-specific measurements with good precision on microarrays for quantitative expression profiling (Fig. 6d, Supplementary Figures 28 and 29) could complement the power of RNA-seq in the discovery and identification of new alternative transcripts (Fig. 2). In other words, the novel transcripts found by RNA-seq can lead to efficient measurements with good precision on microarrays, which can in turn aid in the confirmation and functional study of new transcript variants.
Finally, each metric showed a different and platform-specific response to signal strength (Supplementary Figures 30 and 31), which for RNA-seq increases with transcript expression level and read depth. The read depth at which average RNA-seq performance meets or exceeds that of another platform thus directly depends on the chosen metric and the distribution of expression strength and differential signal in the samples measured. As a result, it also depends on the set of tested genes, over which the average performance is being computed. We show results here for the mutual information metric (Supplementary Figure 32), which is of direct relevance for classifier performance. As expected, RNA-seq performance improved with increasing numbers of mapped fragments (Fig. 6e). In particular, Life Technologies’ SOLiD and Illumina’s HiSeq 2000 performed similarly well for comparable effective read depths (Supplementary Figure 33a). The choice of reference platform considerably affects the number of RNA-seq reads required for obtaining comparable mutual information per gene (Supplementary Figure 34). For some of the microarrays and data-processing methods tested, as few as 5 million mapped RNA-seq fragments were more than sufficient (HGU133plus2 with MAS5), whereas ~50 million mapped fragments were required for others (PrimeView with gcRMA/affyPLM). The choice of RNA-seq pipeline also had an effect, with some tools requiring up to twice as many aligned fragments (cf. TopHat2+Cufflinks,23 Supplementary Figure 35).
DISCUSSION
In a multi-site cross-platform study led by the US FDA, four well-characterized reference RNA sample mixtures with built-in truths were profiled to test RNA-seq reproducibility, accuracy and information content in a detailed analysis of >30 billion reads on the reference samples alone. To our knowledge, the data presented here provide the deepest molecular characterization of any RNA samples to date.
We leveraged this deep data set to test the reliability and power of RNA-seq in exploring the complexity of the transcriptome. We studied the detection of known splice junctions and the discovery of unannotated junctions. De novo junction discovery was robust across sites both at low and at high sequencing depths, even beyond 10 billion aligned fragments. Many unannotated splice junctions were detected by multiple platforms and pipelines, with concordance directly reflecting the junction expression level. Similar to observations by ENCODE at the gene level,7 we observed three distinct classes of expression levels for splice junctions: highly expressed known junctions, known and unannotated junctions at medium levels, and many unannotated junctions found only at low expression levels. Although it has been proposed that an abundance of weakly expressed transcripts may reflect biological noise,25 the lower expression of these junctions may alternatively be the reason that they have so far received less attention in traditional experiments. With alternative transcript expression being dependent on cell type and experimental conditions, deep RNA-seq will continue to play a key role in fully exploring the transcriptomic repertoire, including the construction of extensive maps of alternative transcripts3 highlighting splicing variants as well as alternative start and polyadenylation sites.7 De novo discovery constitutes a key strength of RNA-seq, and is reflected in the expansive transcriptional landscapes observed from different cells and tissues in the transcriptome re-annotation projects for human and rat47,56 and the rich profiles collected in clinical and toxicogenomic applications in which many terabases of additional RNA-seq data were collected and analyzed54,55. Future studies can be conducted to identify novel alternative transcripts through full gene models, which allow the filtering of spurious junctions that cannot be explained by expression levels of alternative transcripts consistent with the distribution of reads that map to exons.
Our read-mapping results underscore how crucial comprehensive gene model annotations are to accurate expression profiling.3 The human genome now has >55,000 well-validated genes, and the majority of them are not protein coding.7 Almost all human multi-exon genes exhibit alternative splicing, and spliced human genes have on average over nine alternative transcribed forms.7,14 Additional genes and transcripts are still being discovered, even beyond the already expansive gene annotation from ENCODE.13 Notably, the NCBI AceView14 database, which has >50,000 genes annotated from cDNA evidence, holds by far the largest and most-extensively validated set of splice junctions, with >300,000 well supported by the RNA-seq data reported in this study alone. Transcripts that may explain a particular phenotype may be missed by less-extensive annotations, stressing that the most comprehensive annotation for expression profiling is vital to accurate clinical research54,57. The characterization and, particularly, the quantification of alternative transcripts, however, still require further research. Although expression profiling of alternative transcripts is feasible, reattributing measurements to a set of alternative transcripts requires knowledge of all the alternative transcript forms of a gene, and involves combining information across the transcripts. For genome-scale RNA-seq, this is particularly difficult because of the sampling noise from low read counts for many transcripts, with recent work observing 300 million sequenced fragments to be required for the detection of a specific human alternative splicing event with 80% power.28
This also highlights the value of targeted RNA-seq.29 Although simpler organisms such as C. elegans may be less affected by the difficulties of reliably attributing reads to alternative transcripts,24 analogous considerations will apply to research on mouse, rat and other complex transcriptomes. The need for longer-range information is a consequence of the fact that certain complex gene models cannot be resolved by the local information provided by either microarray probes or individual short RNA-seq reads alone. Although read pairs of size-controlled fragments can improve on this, they also limit the recovery of shorter transcripts. Full alternative transcript profiling will thus greatly benefit from longer RNA-seq reads, which may eventually approach the full length of complex cDNAs. Combining deep RNA-seq for alternative transcript discovery with modern high-resolution microarrays for genome-scale quantification may provide an efficient approach for systematic transcript-level expression profiling.18
Although none of the technologies we tested could provide reliable absolute quantification, relative expression measures agreed well across platforms, including RNA-seq, qPCR and microarrays. The majority of genes satisfied constraints based on the truths built into the study design. Going beyond earlier platform comparisons that considered individual performance metrics,3–6,18,19,27 we combined complementary metrics for a robust characterization of measurement performance that can be combined with further assays such as tests for strandedness, considerations based on the ERCC spike-in response, and tests for the exclusion of non-specific background. Notably, the important but difficult task of estimating and removing background noise (Supplementary Figure 36b), typically improves accuracy at the expense of precision (Supplementary Figure 29).
Considering the substantial disagreements even between different types of qPCR-based assays, we conclude that there is no single ‘gold standard.’ Although our cross-platform comparisons reveal common trends, drastic systemic differences remain. Reference data sets such as the compendium presented here are invaluable for a systematic characterization of measurements, which is critical for reliable conclusions from large-scale experiments. Specifically, a closer examination of the varying amount of detected ERCCs per sample indicated substantial differences and inconsistencies even across libraries prepared from the same sample at the same site and sequenced by the same machine. This implies inherent limitations for the read out of absolute expression level estimates and absolute quantification.26 As the vendor-prepared libraries gave very uniform results across sites (Fig. 4d), the observed variations likely arose in library construction, and may partially be explained by platform-specific differences in kit chemistry and varying degrees of sample poly-adenylation26. Therefore, in RNA-seq experiments, multiple libraries per examined condition or sample should be profiled.
We show that filters can improve robustness of differential expression calls and consistency across sites and platforms. For RNA-seq, removing small fold-changes as well as excluding low-expression measurements reduced the false discovery rate considerably and, in general, gave an improvement over microarrays10 at similar sensitivity. These filters also achieved good inter-site agreement of lists of differentially expressed genes, with the performance of several (but not all) RNA-seq pipelines becoming comparable to that of microarrays (Fig. 3e). Even though a direct comparison of absolute expression levels across platforms was not possible, the filters yielded good agreement of differential expression calls between platforms (for example, A versus B on HiSeq 2000 compared to A versus B on SOLiD, Supplementary Figures 13 and 14), suggesting that differential expression analyses from different platforms could be combined.
Importantly, the observed sensitivity of results to pipeline choice suggests that substantial improvements in short-read RNA-seq analysis are still required, particularly for transcript identification and quantification. The data we collected in this multi-center study can serve as a benchmark set for further advances. Some recent progress can be directly attributable to the impact of more-successful read mappers.16,54 In addition, although systematic and sample-specific variations in GC bias,30 sequence bias and non-specific signal (Supplementary Figure 36) can contribute to unwanted or missed differential expression calls, continued study of the confounding factors in RNA-seq can be expected to improve signal quality,3,19,30,31 just as methodological developments have improved microarray signal read out.32–38 Conversely, with several microarray designs tested here probing less than half of all known AceView genes, new microarray designs can take advantage of updated gene annotations and models refined by RNA-seq,18 as we have shown here with pilot microarrays.
Already today, RNA-seq can be used as a versatile tool for relative expression profiling, with comparable or superior performance to microarrays in many applications given sufficient read depth and appropriate choice of analysis pipeline. An effective sequencing depth is clearly contingent on the experimental goals, with simple gene-level expression profiling only requiring 5–50 M single-ended reads for an appropriate analysis pipeline (cf. Supplementary Figures 11 and 34, Fig. 6e). A comprehensive characterization of alternative transcript expression benefits from the longer-range information of read pairs and requires considerably deeper sequencing. In our data set, at five million mapped fragments, >15,000 AceView genes could already be detected with strong support (16 reads), including ~10,000 RefSeq genes (Fig. 2c). Moreover, 10 million mapped fragments sufficed for differential expression analysis of the most strongly expressed genes in our study, reliably across sites (see Supplementary Figure 11 for adapted filter parameters). Other applications may require deeper sequencing, as is reflected by different metrics responding differently to an increase in reads for the samples and genes studied. Classifier performance, for instance, is directly related to the mutual information metric. Although 5 million mapped fragments easily gave a mutual information per gene comparable to that of HGU133Plus2.0 microarrays with MAS5, performance comparable to newer arrays and processing methods required about 50 million mapped fragments in this study (Fig. 6e and Supplementary Figure 34), or even required considerably more, depending on the RNA-Seq analysis pipeline (cf. TopHat2+Cufflinks, Supplementary Figure 35). In addition, the required read depth is also dependent on the genome size, transcriptional complexity,24 cellular distribution of stored versus active RNAs, biological noise48 and the panoply of all other factors of cell biology and RNA dynamics. Our comprehensive multi-site cross-platform benchmark measurements under controlled settings thus complement and extend comparisons for individual biological experiments (Supplementary Table 1, Supplementary Notes Section 2.4 for further discussion).
In summary, the study and data collection presented here form an important milestone in the development and dissection of RNA-seq as a method for transcriptome profiling. The results based on data sets of this size and complexity and an array of independent measures as introduced by this study will contribute to a better understanding of the power and limitations of RNA-seq. This work is complemented by SEQC companion studies analyzing the application of RNA-seq to specific biological research and clinical questions47,51–57. The cumulative SEQC data sets with >100 billion reads (10 Tb) provide a key resource for testing future developments of RNA-seq, as required in clinical and regulatory settings.
METHODS
Study design and data
The SEQC (MAQC-III) main study design is based on the well-characterized MAQC-I RNA samples: the Universal Human Reference RNA (UHRR, from 10 pooled cancer cell lines, Agilent Technologies, Inc.) and the Human Brain Reference RNA (HBRR, from multiple brain regions of 23 donors, Life Technologies, Inc.).10 To these, two different ERCC spike-in mixes were added11 (50 μl of ERCC mix was spiked into 2500 μl of total RNA) to give: Sample A – UHRR with ERCC spike-in mix E, and Sample B – HBRR with ERCC spike-in mix F. These were then combined in ratios of 3:1 and 1:3, respectively, to generate samples C and D (Fig. 1b and Supplementary Figure 1).
Each platform vendor designated three ‘official sites’ before samples were distributed, and these are marked with a star (*) below. Data produced by the official sites were used in all of the performed analyses. In addition, data produced by the non-official sites were incorporated in some analyses, e.g., the analysis of gene detection and junction discovery as a function of read depth (Figure 2, Supplementary Figures 2–4, 6, 7 and 37) and the study of sensitivity, specificity, and reproducibility of differential expression calls (Figure 3, Supplementary Figures 10–14 and 38–40).
Illumina HiSeq 2000 data were provided by 6 sites (Supplementary Tables 6 and 7): 1) Australian Genome Research Facility; 2) Beijing Genomics Institute*; 3) City of Hope; 4) Weill Cornell Medical College*; 5) Mayo Clinic*; and 6) Novartis, generating 100+100 nt read-pairs.
Life Technologies SOLiD 5500 data were provided by 4 sites (Supplementary Tables 8 and 9): 1) Liverpool; 2) Northwestern University*; 3) Penn State University*; and 4) SeqWright Inc.*, generating 51+36 nt read-pairs, except for Liverpool which applied a protocol variant giving single 76 nt reads.
All official sites created four replicate measurements of each sample A to D, and also sequenced a vendor-prepared fifth replicate (Figure 1b). The other HiSeq 2000 sites sequenced four replicate libraries of each sample A to D. In Liverpool, one site-prepared library and one vendor-provided library of each of the samples A to D were sequenced.
For comparisons of gene-level expression profiling, samples A to D were also hybridized to a variety of commercial microarray platforms: 1) Affymetrix HuGene2.0 (one site: Stanford); 2) Affymetrix PrimeView (one site: Stanford); 3) Agilent 60k (one site: Boku University Vienna); and 4) Illumina Bead arrays (two sites: City of Hope, and University of Texas Southwestern Medical Center). In addition, MAQC-I Affymetrix HGU133Plus2.0 data from six sites were reanalyzed. Providing another independent platform, 20,801 PrimePCR measurements were also performed, with at least 10 qPCR reactions per assay to assure good specificity, efficiency, linear dynamic range, and background from negative controls (see Supplementary Notes Section 3.5 for more detail).
For comparisons of transcript-level profiling, exploring the potential of high-density microarrays for alternative transcript specific quantification, an Agilent 1M feature microarray was tested at Boku University Vienna. The microarray contained 1 million probes of length 60 nt, covering 782 AceView genes with 5,691 alternative transcripts, and including the ERCC spike-ins, averaging: 33 probes per exon (7x coverage, 9 nt spacing) and 55 probes per junction (about 1 nt spacing). The set of genes was selected to: 1) show expression in one of the samples in an SEQC RNA-seq pilot study; 2) have a similar average expression distribution as the full set of AceView genes in the pilot study; 3) have a similar differential expression distribution as the full set of AceView genes in the pilot study; and 4) have a similar distribution of the number of transcripts per gene as the full set of genes annotated in AceView. These and similar microarrays can be ordered from Agilent, and the design of the test microarray is published together with this paper. Affymetrix also manufactures high-density transcriptome microarrays, which were released early 2013, not in time to be included in the SEQC study.
Roche 454 GS FLX data were provided by: 1) the Medical Genomes Project; 2) the New York University Medical Center; and 3) SeqWright Inc.. At each site, one replicate of samples A and B was sequenced (two runs).
Reads were mapped to a human reference and the ERCC spike-in sequences. Depending on the pipeline, genomic DNA (hg19) or transcript sequences were used as human reference. Unless otherwise stated, results for the gene model annotation of AceView 2010 are shown. Other annotations considered included RefSeq v104 and GENCODE v15.
The HiSeq 2000 sites produced on average 110 million read-pairs per replicate, for a total of 2,200 million per site (Supplementary Tables 6 and 7). The official SOLiD sites produced on average 50 million read-pairs per replicate, for a total of 980 million per site (Supplementary Table 8). Liverpool generated 545 million single reads (Supplementary Table 9). The Roche 454 sites produced on average 1 million reads per replicate, for a total of about 2.1 million reads per site (Supplementary Table 10).
For the validation of junctions discovered by RNA-seq, for a random selection of 173 junctions to test, qPCR measurements were performed with primers designed to specifically validate the particular junction, running 2 qPCR reactions per assay for all samples A…D (see Supplementary Notes Section 3.6 for more detail). Specificity was confirmed by analyses of PCR product lengths. This allowed the identification of non-specific assays, identifying the target but also picking up additional unintended targets due to unexpected or unavoidable cross-reactivity (giving a qualitative validation but no meaningful quantitative readout) and of non-informative assays, failing to pick up the target but picking up unintended targets. We provide information on RNA-seq read coverage flanking all 250 candidate junctions considered for validation in file Supplementary Data 1. Supplementary Data 2 file provides the employed qPCR primer sequences, qPCR results and expression level estimates, as well as the corresponding RNA-seq expression level estimates for the 173 performed assays.
Data processing – assessing expression estimates
A variety of tools/pipelines to process RNA-seq data were compared (see file Supplementary Protocol for used pipeline parameters):
TopHat2 G: TopHat v2.0.0 with-G parameter (providing the reference GTF file) + CuffDiff v2.0.0.
Magic: NCBI AceView MAGIC14.
Subread: Subread 1.3.016 The Subread pipeline uses the subjunc function to identify exon-exon junctions and the featureCounts41 function to obtain count summaries for each gene and spike-in transcript (see Supplementary Notes Section 3.1 for more detail)
r-make: Cornell’s r-make pipeline incorporating STAR15 (http://physiology.med.cornell.edu/faculty/mason/lab/r-make/). For LifeTech reads, all alignments were processed in color space.
Applying consistency tests based on truths built into the study design to expression levels of individual junctions, we consider the number of reads hitting a specific exon-exon junction as indicator of expression level.
Except for r-make, which provides raw read counts, each pipeline already has a built-in approach to normalization. In order to analyze Agilent 1M microarray data, a variance stabilizing normalization (vsn)33 was used. Probe sequences-specific signals have been modeled using established methods, saturation effects detrended, and outlier probes downweighted.35–37 Transcript variant expression levels have been estimated using a hierarchical Bayesian approach similar to modern methods applied for RNA-seq data analysis (Stegle et al. in preparation) (see ‘Transcript quantification for Agilent high-density microarrays’ section).
CustomCDFs (v16, re-mapped to the latest AceView) were used for an analysis of the Affymetrix data (HGU133Plus2.0, PrimeView, and HuGene2.0),42 respectively covering 24,623, 17,984, and 29,879 genes. PrimeView and HuGene2.0 data were analyzed using established methods (correction for probe sequence specific effects by gcRMA,34 conservative normalization across arrays by vsn,33 and robust probe set summarization by affyPLM), whereas for the HGU133Plus2.0 microarrays, a combination of more recent tools that appeared to be more efficient were used (correction for probe specific saturation effects by Hook,36 conservative normalization across arrays by vsn,33 and factor based probe set summarization by FARMS35).
For Illumina Bead microarrays and Agilent 60K microarrays variance stabilization normalization (vsn33) was applied.
Discovery of transcriptome complexity at high read depth
The detection and discovery of junctions was performed by Subread using data from all 6 HiSeq 2000 sites as well as all 4 SOLiD sites, and compared to results by r-make using data from all 6 HiSeq 2000 sites.
We applied consistency tests for the truths built into the study design to junction expression levels. The sample expression levels of almost ⅓ of all known AceView junctions (and 38% of all detected) follow the expected titration order while also correctly yielding the expected A/B mixing ratio and show a clear differential signal as assessed by the mutual information, a measure of information content (Supplementary Figure 37 and Supplementary Table 11). Of the well-supported new junctions, 5,189 passed these rigorous filters. Conservatively assuming a 1:3 ratio as in the AceView junctions, there may easily be three times as many new junctions, thereby adding over 15,000 likely new junctions to the already extensive AceView annotation. Furthermore, considering that we essentially required junctions to be independently detected in by SOLiD, where 98% of junctions were detected by a single site (LIV) and thus corresponding to just about 1/65 of the total HiSeq 2000 sequencing volume, and detection power being about 2x lower (Supplementary Table 2), there may well be up to 5,000×3×65×2 = 2 million new junctions to be discovered in samples A and B alone. Even more junctions than in samples A and B were discovered in the SEQC neuroblastoma study.15
Transcript quantification for Agilent high-density microarrays
Quantification of transcript expression from the probe level information was carried out using a linear mixed model independently for each gene and sample. Denoting the expression level of probe p as yp, we model the probe expression as the sum of effects from transcripts with a probe-match:
The Kronecker delta δt,p is one exactly if the probe p is matching the transcript t, and zero otherwise, whereas xt denotes the unknown abundance for transcript t. Further, we assume Gaussian additive and multiplicative error variances. Probe-level noise tends to exhibit a strong spatial correlation structure, which we account for by using a latent Gaussian process function.43 We employ a squared exponential covariance function where the probe distance in transcript space is used to parameterize the covariance.
Inference is performed by maximizing the joint marginal likelihood of all considered probes given with respect to the hidden transcript abundances (xt) and the noise covariance parameters. To mitigate the computational complexity of Gaussian process models (cubical scaling in the number of probes), we randomly choose probes for each gene selecting a subset of at most 700 probes, including probes falling onto junctions.
Discrete nature of RNA-seq data
With the discrete nature of RNA-seq data and considering that most analysis tools work on a log2 scale, consistent ways need to be found for dealing with not expressed features, which are supported by zero reads. As the lowest positive expression is just a single read, a common approach is the addition of a pseudo-count (e.g., 0.5 in voom46). An alternative well established for microarray data analysis is the application of asinh as a variance stabilizing transform assuming an additive-multiplicative error model. The transform is approximately linear for small values; for larger values it is well approximated by a logarithm. Another approach (natively applied e.g. in Magic14) is to use a threshold below which measurements are considered below detection sensitivity. Here that threshold has been set to the highest minimum read count of all measurement samples adjusted for library size (the total number of reads), and this threshold is then applied consistently as floor to all expression levels. We have applied this approach to improving consistency in our studies to the data from each pipeline and platform (and thus we were not adding the pseudo-count of 0.5 reads when using voom).
In order to identify genes, transcripts, and junctions with clear support of sequence reads, thresholds were applied: Support was considered sufficient when at least 16, 16, or 8 at reads were observed, respectively. For Figure 2b, expression above background level as determined by the Magic pipeline was additionally required for genes.
Sensitivity, specificity, and reproducibility of differential expression calls
In this part of the study the subset of 23,420 AceView genes (of the version frozen for the SEQC study) present on the MAQC-I Affymetrix HGU133Plus2.0 microarray was used.
As the array data were already processed and normalized with state-of-the-art methods (see ‘Data processing’ sections) no further processing was required. For RNA-seq data, weighted trimmed mean of log fold-change normalization31 improved results (data not shown), in agreement with a recent performance comparison of normalization methods.44 For this normalization step, the TMM implementation provided in the Bioconductor R package edgeR45 was employed.
Several of the examined RNA-seq pipelines exploit multi-mapping reads. This increases power51 and, more generally, also allows the analysis of alternative transcripts. Those pipelines report expression-level estimates rather than read counts. For a uniform approach to differential expression analysis, RNA-seq data were therefore analyzed using an established approach supporting such pipelines. Precision-based weights were attached to normalized expression estimates on the log-scale to account for higher variability at low expression levels using voom46 of the limma package.32 The voom function has been developed to account for different variances as a function of signal intensity. For count data, such a variation is expected by theory, whereas for expression level estimates it is empirically justified. So we in general apply the voom model for expression level dependent variance to account for the different platform specific noise characteristics as a function of the expression level (Supplementary Figure 38).
Differential expression was then assessed for both microarray and sequencing platforms using the empirical Bayes moderated t-statistic of the limma package.32 A p-value threshold of 0.01 unadjusted for multiple testing was used, as suggested in the MAQC-I study.10 As the number of differentially expressed genes (DEGs) was similar for p<0.01 and the qBY<5%, where qBY is the Benjamini-Yekutieli adjusted False Discovery Rate, downstream analysis is not qualitatively affected by this choice (Supplementary Figure 39 vs Figure 40).
An estimate of the empirical False Discovery Rate (eFDR) was computed by comparing the number of DEGs for intra-site A vs B and inter-site A vs A comparisons. For each A vs A analysis two eFDRs were calculated (using the A vs B comparison of the two sites considered in the matching A vs A comparison).
Further filters were applied in order to control the eFDR, with parameters chosen to give similar numbers of A vs B differential expression calls:
AFX: |log2(fold-change)| > 1
MAGIC: |log2(fold-change)|> 1.7 and AveExp>32%
r-make: |log2(fold-change)|>1.7 and AveExp>33%
Subread: |log2(fold-change) |>1.7 and AveExp>32%
BitSeq: |log2(fold-change)|>2 and AveExp>19%
TopHat2-G |log2(fold-change)|>2 and AveExp>23%
When filtering for average log2 expression level (AveExp), the stated fraction of weakly expressed genes to remove also included in that percentage the genes that were not observed at all. This is to allow comparisons across different pipelines observing varying numbers of genes.
Metrics for a robust characterization of platforms, sites, and data processing options
The complementary metrics examined react differently to rescaling, shifts, and other consequences of data-processing. As a result, individual pipelines can show varying performance in specific assays. For a robust performance characterization we combine the complementary metrics.
Different analysis pipelines and platforms, however, identify varying numbers of targets (Supplement). With this number not constant, performance needs to be assessed in terms of actual counts or fractions of all genes, rather than fractions of observed genes. An increased sensitivity of some pipelines and platforms can be demonstrated by not limiting analysis only to genes observed by all pipelines and platforms.
We count a gene as preserving titration monotonicity, when A > B and, as expected A ≤ C ≤ D ≤ B or, conversely, for A < B.
A gene is considered precise for a sample, if the standard error across technical replicates is below 10%.
-
A deviation less than 10% from the expected behavior of
where the correction z of the known mixing coefficients k1 = 3z/{3z + 1} and k2 = z/{z + 3} arising out of different ratios of mRNA versus total RNA in the samples A and B has been determined by a non-linear robust fit (nlrob) from an independent RNA-seq library (library #5). The obtained value, 1.45±0.01 is very much in line with the experimental estimate22 of 1.43±0.10. The plots and statistics shown in the paper give the same picture with either value. To ensure pipeline independence, the experimental value 1.43 is being used.
For the purpose of this metric, we call a gene differentially expressed if it is significant at a Benjamini-Yekutieli corrected FDR of 5% in an empirical Bayes moderated t-test across the expression level estimates of samples A and B (limma).
Finally, we calculate the mutual information of sample titration by extending the approach introduced for two state measurements.38 The mutual information between gene or transcript expression and titration requires modeling the probability of a measurement being from sample A, C, D, or B, under the constraint that these labels are ordered. To avoid a dependency of our assessment on choosing mutual information of sample titration as evaluation measure, we complemented this assay with three other alternative measures. For that purpose, we evaluated the mutual information for discriminating A vs B, and the mutual information for discriminating C vs D using an established approach.38 In order to add a further measure which does not depend on modeling assumptions, for all genes and transcripts, we also calculated a non-parametric estimate of the probability that the respective measurement fulfils the order constraint which is implied by the titration experiment. All four measures are illustrated in Supplementary Figure 41 for the official HiSeq 2000 and SOLiD sites and a number of different quantification pipelines. Although the complementary measures suggest different numbers of ‘good’ transcripts and genes, they qualitatively agree with no exception on how they rank the different platforms and pipelines. This confirms that we can select the mutual information of sample titration to represent this class of information preserving measures to add an independent robust viewpoint for characterizing quantification performance in Figure 6.
Supplementary Material
Acknowledgments
All SEQC (MAQC-III) participants freely donated their time and reagents for the completion and analyses of the project. Many participants contributed to the sometimes-heated discussions on the topic of this paper during numerous e-mail exchanges, teleconferences, and face-to-face project meetings. The common conclusions and recommendations reported in this paper evolved from this extended discourse. The authors gratefully acknowledge support by the NCBI’s Supercomputing Center, the FDA’s Supercomputing Center, China’s National Supercomputing Center of Tianjin, the Vienna Scientific Cluster HPCF (VSC), the Vienna Science and Technology Fund (WWTF), Baxter AG, the Austrian Institute of Technology, and the Austrian Centre of Biopharmaceutical Technology. This work was supported in part by China’s Program of Global Experts. This work was supported in part by the National Institutes of Health (NIH) grants R01CA163256, R01HG006798, R01NS076465, R44HG005297, U54CA119338, PO1HG00205, R24GM102656 and the Intramural Research Program of the NIH, National Library of Medicine, National Institute of Environmental Health Sciences (NIEHS) Z01 ES102345–04, Shriners Research Grant 85500, an Australia National Health and Medical Research Council (NH&MRC) Project Grant (1023454) and Victorian State Government Operational Infrastructure Support (Australia), the National 973 Key Basic Research Program of China (2010CB945401), the National Natural Science Foundation of China (31240038 and 31071162), and the Science and Technology Commission of Shanghai Municipality (11DZ2260300). We greatly appreciate SAS Institute, Inc. for kindly hosting several face-to-face meetings of the SEQC (MAQC-III) project.
Project Coordination
US Food and Drug Administration
Project lead
Manuscript lead
Scientific management
David P Kreil2,85, Christopher E Mason3,4, Weida Tong1, and Leming Shi86,87,88,1
Corresponding authors
David P Kreil2,85, Christopher E Mason3,4, and Leming Shi86,87,88,1
The following authors contributed equally
The following authors contributed to project leadership
Zhenqiang Su1, Paweł P Łabaj2, Sheng Li3,4, Jean Thierry-Mieg5, Danielle Thierry-Mieg5, Wei Shi6, Charles Wang7, Gary P Schroth8, Robert A Setterquist9, John F Thompson10, Wendell D Jones11, Wenzhong Xiao12, Weihong Xu13, Roderick V Jensen14, Reagan Kelly1, Joshua Xu1, Ana Conesa15, Cesare Furlanello16, Hanlin Gao17, Huixiao Hong1, Nadereh Jafari18, Stan Letovsky19, Yang Liao6, Fei Lu20, Edward J Oakeley21, Zhiyu Peng22, Craig A Praul23, Javier Santoyo-Lopez24, Andreas Scherer25,26, Tieliu Shi27, Gordon K Smyth6, Frank Staedtler21, Peter Sykacek2, Xin-Xing Tan20, E Aubrey Thompson28, Jo Vandesompele29, May D Wang30, Jian Wang31, Russell D Wolfinger32, Jiri Zavadil33,34,35, Weida Tong1, David P Kreil2,85, Christopher E Mason3,4, and Leming Shi86,87,88,1
SEQC/MAQC-III Consortium
Zhenqiang Su1, Paweł P Łabaj2, Sheng Li3,4, Jean Thierry-Mieg5, Danielle Thierry-Mieg5, Wei Shi6, Charles Wang7, Gary P Schroth8, Robert A Setterquist9, John F Thompson10, Wendell D Jones11, Wenzhong Xiao12, Weihong Xu13, Roderick V Jensen14, Reagan Kelly1, Joshua Xu1, Ana Conesa15, Cesare Furlanello16, Hanlin Gao17, Huixiao Hong1, Nadereh Jafari18, Stan Letovsky19, Yang Liao6, Fei Lu20, Edward J Oakeley21, Zhiyu Peng22, Craig A Praul23, Javier Santoyo-Lopez24, Andreas Scherer25,26, Tieliu Shi27, Gordon K Smyth6, Frank Staedtler21, Peter Sykacek2, Xin-Xing Tan20, E Aubrey Thompson28, Jo Vandesompele29, May D Wang30, Jian Wang31, Russell D Wolfinger32, Jiri Zavadil33,34,35, Scott S Auerbach36, Wenjun Bao32, Hans Binder37, Thomas Blomquist38, Murray H Brilliant39, Pierre R Bushel36, Weimin Cai40, Jennifer G Catalano41, Ching-Wei Chang1, Tao Chen1, Geng Chen40, Rong Chen42, Marco Chierici16, Tzu-Ming Chu32, Djork-Arné Clevert43, Youping Deng44, Adnan Derti45, Viswanath Devanarayan46, Zirui Dong22, Joaquin Dopazo15,47 Tingting Du40, Hong Fang1, Yongxiang Fang48, Mario Fasold37, Anita Fernandez21, Matthias Fischer49, Pedro Furió-Tari15, James C Fuscoe1, Stan Gaj50, Jorge Gandara3, Huan Gao22, Weigong Ge1, Yoichi Gondo51, Binsheng Gong1, Meihua Gong22, Zhuolin Gong22, Bridgett Green1, Chao Guo52, Lei Guo1, Li-Wu Guo1, James Hadfield53, Jan Hellemans29, Sepp Hochreiter43, Meiwen Jia40, Min Jian22, Charles D Johnson54, Suzanne Kay48, Jos Kleinjans50, Samir Lababidi55, Shawn Levy56, Quan-Zhen Li57, Li Li32, Li Li42, Peng Li27, Yan Li1, Haiqing Li58, Jianying Li36, Shiyong Li22, Simon M Lin59, Francisco J López24, Xin Lu60, Heng Luo61, Xiwen Ma62, Joseph Meehan1, Dalila B Megherbi63, Nan Mei1, Bing Mu58, Baitang Ning1, Akhilesh Pandey64,65, Javier Pérez-Florido24, Roger G Perkins1, Ryan Peters66, John H Phan30, Mehdi Pirooznia67, Feng Qian1, Tao Qing40, Lucille Rainbow48, Philippe Rocca-Serra68, Laure Sambourg69, Susanna-Assunta Sansone68, Scott Schwartz54, Ruchir Shah 70, Jie Shen1, Todd M Smith71, Oliver Stegle72, Nancy Stralis-Pavese2, Elia Stupka73, Yutaka Suzuki74, Lee T Szkotnicki20, Matthew Tinning25, Bimeng Tu22, Joost van Delft50, Alicia Vela24, Elisa Venturini33, Stephen J Walker75, Liqing Wan1, Wei Wang76, Jinhui Wang17, Jun Wang22,77,78,79, Eric D Wieben80, James C Willey37, Po-Yen Wu81, Jiekun Xuan1, Yong Yang31, Zhan Ye58, Ye Yin22, Ying Yu40, Yate-Ching Yuan58, John Zhang82, Ke K Zhang83, Wenqian Zhang22, Wenwei Zhang22, Yanyan Zhang22, Chen Zhao40, Yuanting Zheng40, Yiming Zhou84, Paul Zumbo3,4, Weida Tong1, David P Kreil2,85, Christopher E Mason3,4, and Leming Shi86,87,88,1[L1]
Footnotes
FDA/NCTR, Jefferson, Arkansas, U.S.A.
Chair of Bioinformatics, Boku University Vienna, Austria
Department of Physiology and Biophysics, Weill Cornell Medical College, New York, U.S.A.
The HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute for Computational Biomedicine, Weill Cornell Medical College, New York, U.S.A.
NIH/NCBI, Bethesda, Maryland, U.S.A.
Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, Melbourne, Victoria, Australia
Center for Genomics and Division of Microbiology & Molecular Genetics, School of Medicine, Loma Linda University, Loma Linda, California, U.S.A.
Illumina Inc., Hayward, California, U.S.A.
Life Technologies Corporation, Austin, Texas, U.S.A.
Nabsys Inc., Providence, Rhode Island, U.S.A.
Expression Analysis Inc., Durham, North Carolina, U.S.A.
Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts, U.S.A.
Stanford Genome Technology Center, Palo Alto, California, U.S.A.
Department of Biological Sciences, Virginia Tech, Blacksburg, Virginia, U.S.A.
Centro de Investigación Príncipe Felipe (CIPF), Computational Genomics Program, Valencia, Spain
Fondazione Bruno Kessler (FBK), Trento Povo (TN), Italy
DNA Sequencing/Solexa Core, Beckman Research Institute, City of Hope Comprehensive Cancer Center, City of Hope National Medical Center, Duarte, California, U.S.A.
Center for Genetic Medicine, Feinberg School of Medicine, Northwestern University, Chicago, Illinois, U.S.A.
SynapDx Corporation, Lexington, Massachusetts, U.S.A.
SeqWright Inc., Houston, Texas, U.S.A.
Novartis Institutes for Biomedical Research (NIBR), Basel, Switzerland
BGI-Shenzhen, Bei Shan Industrial Zone, Yantian District, Shenzhen, Guangdong, China
The Pennsylvania State University, University Park, Pennsylvania, U.S.A.
Medical Genome Project (MGP), INSUR Building, Andalusian Human Genome Sequencing Center (CASEGH), Sevilla, Spain
Australian Genome Research Facility Ltd., The Walter and Eliza Hall Institute of Medical Research, Melbourne, Victoria, Australia
Spheromics, Joensuu Science Park, Finland
Center for Bioinformatics and Computational Biology, Shanghai Key Laboratory of Regulatory Biology, the Institute of Biomedical Sciences and School of Life Sciences, East China Normal University, Shanghai, China
Department of Cancer Biology, Mayo Clinic Jacksonville, Jacksonville, Florida, U.S.A.
Biogazelle NV, Zwijnaarde, Belgium
Department of Biomedical Engineering, GeorgiaTech and Emory University, Atlanta, Georgia, U.S.A.
Research Informatics, Lilly Corporate Center, Eli Lilly and Company, Indianapolis, Indiana, U.S.A.
SAS Institute Inc., Cary, North Carolina, U.S.A.
NYU Genome Technology Center, New York University Langone Medical Center, New York, U.S.A.
NYU Center for Health Informatics and Bioinformatics, Department of Pathology, New York University Langone Medical Center, New York, U.S.A.
Molecular Mechanisms and Biomarkers Group, International Agency for Research on Cancer, Lyon, France
NIH/NIEHS, National Institute of Environmental Health Sciences, NIH, Research Triangle Park, North Carolina, U.S.A.
Interdisciplinary Centre for Bioinformatics, University of Leipzig, Leipzig, Germany
University of Toledo Health Sciences Campus, Division of Pulmonary and Critical Care Medicine, Department of Medicine, Medical College of Ohio, Toledo, Ohio, U.S.A.
Center of Human Genetics, Marshfield Clinic Research Foundation, Marshfield, Wisconsin, U.S.A.
School of Pharmacy, Fudan University, Shanghai, China
Office of Cellular, Tissue, and Gene Therapies, FDA/CBER, Bethesda, Maryland, U.S.A.
Department of Genetics and Genomic Sciences, Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, U.S.A.
Institute of Bioinformatics, Johannes Kepler University Linz, Linz, Austria
Department of Internal Medicine, Rush University Cancer Center, Chicago, Illinois, U.S.A.
Novartis Institutes for Biomedical Research, Novartis, Cambridge, Massachusetts, U.S.A.
AbbVie Inc., Global Pharmaceutical R&D, Souderton, Pennsylvania, U.S.A.
CIBER de Enfermedades Raras (CIBERER) and Functional Genomics Node (INB) at CIPF, Valencia, Spain
Centre for Genomic Research, University of Liverpool, Liverpool, England
Department of Pediatric Oncology and Hematology and Center for Molecular Medicine (CMMC), University of Cologne, Cologne, Germany
Department of Toxicogenomics, Maastricht University, Maastricht, The Netherlands
RIKEN BioResource Center, Tsukuba, Ibarak, Japan
Functional Genomics Core, Beckman Research Institute, City of Hope National Medical Center, Duarte, California, U.S.A.
Cancer Research UK Cambridge Institute, University of Cambridge, Li Ka Shing Centre, Cambridge, U.K.
Texas A&M AgriLife Research, College Station, Texas, U.S.A.
FDA/CBER, Rockville, Maryland, U.S.A.
HudsonAlpha Institute for Biotechnology, Huntsville, Alabama, U.S.A.
University of Texas Southwestern Medical Center (UTSW), Dallas, Texas, U.S.A.
Bioinformatics Core, Beckman Research Institute, City of Hope National Medical Center, Duarte, California, U.S.A.
Biomedical Informatics Research Center, Marshfield Clinic Research Foundation, Marshfield, Wisconsin, U.S.A.
AbbVie Inc., Global Pharmaceutical R&D, North Chicago, Illinois, U.S.A.
UALR/UAMS Joint Bioinformatics Graduate Program, University of Arkansas at Little Rock, Little Rock, Arkansas, U.S.A.
Discovery Statistics, Lilly Corporate Center, Eli Lilly and Company, Indianapolis, Indiana, U.S.A.
CMINDS Research Center, Department of Electrical and Computer Engineering, University of Massachusetts at Lowell, Lowell, Massachusetts, U.S.A.
McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland, U.S.A.
Institute of Bioinformatics, Bangalore, India
Partek Inc., Saint Louis, Missouri, U.S.A.
School of Medicine, Department of Psychiatry, Johns Hopkins University, Baltimore, Maryland, U.S.A.
Oxford e-Research Centre, University of Oxford, Oxford, U.K.
UJF-Grenoble 1/CNRS/TIMC-IMAG UMR 5525, Computational and Mathematical Biology (BCM), Grenoble, France
SRA International Inc., Durham, North Carolina, U.S.A.
Geospiza Inc., Seattle, Washington, U.S.A.
EMBL Outstation - Hinxton, European Bioinformatics Institute, Hinxton, Cambridge, U.K.
San Raffaele Scientific Institute, Center for Translational Genomics and Bioinformatics, Milano, Italy
Department of Medical Genome Sciences, The University of Tokyo, 5-1-5 Kashiwa Kashiwanoha Chiba, Japan
Wake Forest Institute for Regenerative Medicine, Wake Forest University, Health Sciences, Winston-Salem, North Carolina, U.S.A.
Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, New Jersey, U.S.A.
Department of Biology, University of Copenhagen, Copenhagen, Denmark
King Abdulaziz University, Jeddah, Saudi Arabia
The Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen
Department of Biochemistry and Molecular Biology, Mayo Clinic Rochester, Rochester, Minnesota, U.S.A.
School of Electrical and Computer Engineering, Georgia Institute of Technology, Atlanta, Georgia, U.S.A.
Center of Big Data Research, Zhijiang College, Zhejiang University of Technology, Hangzhou, Zhejiang, China
School of Medicine, Department of Pathology, University of North Dakota, Grand Forks, North Dakota, U.S.A.
Digomics LLC, Brookline, Massachusetts, U.S.A.
University of Warwick, Coventry, U.K.
State Key Laboratory of Genetic Engineering and MOE Key Laboratory of Contemporary Anthropology, Schools of Life Sciences and Pharmacy, Fudan University, Shanghai, China
Fudan-Zhangjiang Center for Clinical Genomics, Shanghai, China
Zhanjiang Center for Translational Medicine, Shanghai, China
DISCLAIMER
The views presented in this article are those of their authors and do not necessarily reflect current or future opinion or policy of the authors’ institutions or agencies. Any mention of commercial products is for clarification and not intended as endorsement.
COMPETING FINANCIAL INTERESTS
Some of the SEQC (MAQC-III) consortium members are employed by companies that provide services or manufacture products or equipment related to gene expression profiling, as can be seen from the provided affiliations of the manuscript authors.
ACCESSION CODES
All SEQC (MAQC-III) data sets are available through GEO (series accession number: GSE47792).
References
- 1.Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 3.Łabaj PP, et al. Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics. 2011;27:i383–i391. doi: 10.1093/bioinformatics/btr247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu S, Lin L, Jiang P, Wang D, Xing Y. A comparison of RNA-Seq and high-density exon array for detecting differential gene expression between closely related species. Nucleic Acids Res. 2011;39:578–588. doi: 10.1093/nar/gkq817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McIntyre LM, et al. RNA-seq: technical variability and sampling. BMC Genomics. 2011;12:293. doi: 10.1186/1471-2164-12-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toung JM, Morley M, Li M, Cheung VG. RNA-sequence analysis of human B-cells. Genome Res. 2011;21:991–998. doi: 10.1101/gr.116335.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLendon R, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(Chairperson) TJH, et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi L, et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker SC, et al. The External RNA Controls Consortium: a progress report. Nat Methods. 2005;2:731–734. doi: 10.1038/nmeth1005-731. [DOI] [PubMed] [Google Scholar]
- 12.Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2011;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrow J, et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts. Genome Biol. 2006;7:S12. doi: 10.1186/gb-2006-7-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012 doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu W, et al. Human transcriptome array for high-throughput clinical studies. Proc Natl Acad Sci. 2011;108:3707–3712. doi: 10.1073/pnas.1019753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.VanGuilder H, Vrana K, Freeman W. Twenty-five years of quantitative PCR for gene expression analysis. BioTechniques. 2008;44(Supplement):619–626. doi: 10.2144/000112776. [DOI] [PubMed] [Google Scholar]
- 21.Aird D, et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011;12:R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shippy R, et al. Using RNA sample titrations to assess microarray platform performance and normalization techniques. Nat Biotechnol. 2006;24:1123–1131. doi: 10.1038/nbt1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agarwal A, et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 2010;11:383. doi: 10.1186/1471-2164-11-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pickrell JK, Pai AA, Gilad Y, Pritchard JK. Noisy Splicing Drives mRNA Isoform Diversity in Human Cells. Plos Genet. 2010;6:e1001236. doi: 10.1371/journal.pgen.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qing T, Yu Y, Du T, Shi L. mRNA enrichment protocols determine the quantification characteristics of external RNA spike-in controls in RNA-seq studies. Sci China Life Sci. 2013;56:134–142. doi: 10.1007/s11427-013-4437-9. [DOI] [PubMed] [Google Scholar]
- 27.Raghavachari N, et al. A systematic comparison and evaluation of high density exon arrays and RNA-seq technology used to unravel the peripheral blood transcriptome of sickle cell disease. BMC Med Genomics. 2012;5:28. doi: 10.1186/1755-8794-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, et al. Evaluating the Impact of Sequencing Depth on Transcriptome Profiling in Human Adipose. Plos One. 2013;8:e66883. doi: 10.1371/journal.pone.0066883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levin JZ, et al. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol. 2009;10:R115. doi: 10.1186/gb-2009-10-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benjamini Y, Speed TP. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012;40:e72–e72. doi: 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smyth GK. In: Bioinforma Comput Biol Solutions Using R Bioconductor. Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editors. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- 33.Huber W, Heydebreck A, von Sültmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18:S96–S104. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- 34.Wu Z, Irizarry R, Gentleman R, Murillo FM, Spencer F. A Model Based Background Adjustment for Oligonucleotide Expression Arrays. Johns Hopkins Univ Dept Biostat Work Pap. 2004 [Google Scholar]
- 35.Hochreiter S, Clevert DA, Obermayer K. A new summarization method for affymetrix probe level data. Bioinformatics. 2006;22:943–949. doi: 10.1093/bioinformatics/btl033. [DOI] [PubMed] [Google Scholar]
- 36.Fasold M, Stadler PF, Binder H. G-stack modulated probe intensities on expression arrays- sequence corrections and signal calibration. BMC Bioinformatics. 2010;11:207. doi: 10.1186/1471-2105-11-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mueckstein U, Leparc GG, Posekany A, Hofacker I, Kreil DP. Hybridization thermodynamics of NimbleGen Microarrays. BMC Bioinformatics. 2010;11:35. doi: 10.1186/1471-2105-11-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sykacek P, et al. The impact of quantitative optimization of hybridization conditions on gene expression analysis. BMC Bioinformatics. 2011;12:73. doi: 10.1186/1471-2105-12-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.David M, Dzamba M, Lister D, Ilie L, Brudno M. SHRiMP2: Sensitive yet Practical Short Read Mapping. Bioinformatics. 2011;27:1011–1012. doi: 10.1093/bioinformatics/btr046. [DOI] [PubMed] [Google Scholar]
- 40.Glaus P, Honkela A, Rattray M. Identifying differentially expressed transcripts from RNA-seq data with biological variation. Bioinformatics. 2012;28:1721–1728. doi: 10.1093/bioinformatics/bts260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 42.Dai M, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175–e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rasmussen CE. Gaussian processes for machine learning. MIT Press; 2006. [Google Scholar]
- 44.Dillies MA, et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2013;14:671–683. doi: 10.1093/bib/bbs046. [DOI] [PubMed] [Google Scholar]
- 45.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Law CW, et al. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu Y, et al. A rat RNA-seq transcriptomic Bodymap across eleven organs and four developmental stages. Nature Comm. 2013 doi: 10.1038/ncomms4230. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rapaport F, et al. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013;14:R95. doi: 10.1186/gb-2013-14-9-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.‘t Hoen PA, et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat Biotechnol. 2013;31:1015–1022. doi: 10.1038/nbt.2702. [DOI] [PubMed] [Google Scholar]
- 50.Liu S, et al. A comparison of RNA-Seq and high-density exon array for detecting differential gene expression between closely related species. Nucleic Acids Res. 2011;39:578–588. doi: 10.1093/nar/gkq817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li S, et al. Detecting and Ameliorating Systematic Variation from Large-scale RNA Sequencing. 2013 submitted. [Google Scholar]
- 52.Munro SA, et al. Assessing technical performance in RNA-Seq experiments with external spike-in RNA controls. 2013 doi: 10.1038/ncomms6125. Submitted. [DOI] [PubMed] [Google Scholar]
- 53.Phan JH, et al. Impact of RNA-Seq Data Analysis Algorithms on Gene Expression Estimation and Downstream Prediction. 2014 doi: 10.1038/s41598-020-74567-y. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang W, et al. RNA-Seq reveals an unprecedented complexity of the neuroblastoma transcriptome and is suitable for clinical endpoint prediction. 2013 Submitted. [Google Scholar]
- 55.Wang C, et al. A comprehensive study design reveals treatment- and abundance-dependent concordance between RNA-Seq and microarrays. 2013 Submitted. [Google Scholar]
- 56.Li P, et al. Landscape of human proteome revealed by large-scale tandem mass spectrometry and RNA-Seq data. 2013 Submitted. [Google Scholar]
- 57.Zhao C, et al. A comprehensive rat transcriptome built from large scale RNA-Seq based annotation. 2013 doi: 10.1093/nar/gkaa638. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.