

Abstract
L-Threonine aldolases (TAs), a family of enzymes belonging to the fold-type I pyridoxal 5′-phosphate (PLP) dependent enzymes, play a role in catalyzing the reversible cleavage of L-3-hydroxy-α-amino acids to glycine and the corresponding aldehydes. Threonine aldolases have great biotechnological potential for the syntheses of pharmaceutically relevant drug molecules because of their stereospecificity. The pH-dependency of their catalytic activity, affecting reaction intermediates, led us to study the effect of low-pH on E. coli TA (eTA) structure. We report here a low-pH crystal structure of eTA at 2.1 Å resolution, with a noncovalently bound uncleaved L-serine substrate, and a PLP cofactor bound as an internal aldimine. This structure contrasts with other eTA structures obtained at physiological pH that show products or substrates bound as PLP-external aldimines. The non-productive binding at low-pH is due to an unusual substrate serine binding orientation in which the α-amino group and carboxylate group are in the wrong positions (relative to the active site residues) as a result of protonation of the α-amino group of the serine, as well as the active site histidines, His83 and His126. Protonation of these residues prevent the characteristic nucleophilic attack of the α-amino group of substrate serine on C4′ of PLP to form the external aldimine. Our study shows that at low pH the change in charge distribution at the active site can result in substrates binding in a non-productive orientation.
Keywords: pyridoxal 5′-phosphate, threonine aldolase, internal aldimine, transaldimination, PLP-dependent enzymes, protein crystallography
INTRODUCTION
L-Threonine aldolases (TAs) are a group of fold-type I pyridoxal 5′-phosphate (PLP)-dependent enzymes that catalyze the reversible cleavage of L-3-hydroxy-α-amino acids, e.g., L-serine, L-threonine, L-allo-threonine, erythro- and threo-L-3-phenylserine, to glycine and the corresponding aldehydes [1-3]. These enzymes are found in several different species of bacteria and fungi, but also include the family of serine hydroxymethyltransferases from both prokaryotic and eukaryotic sources [4-6]. The addition of an alkyl group to C-3 of the substrate amino acid generates a second asymmetric site resulting in threo and erythro isomers. The lack of strict stereospecificity and the possibility of employing different aldehlydes as substrates suggest the potential of these enzymes catalyzing diastereoselective syntheses of 3-hydroxy amino acids for pharmaceutically relevant drug molecules by the reverse reaction of glycine and the appropriate aldehyde. By understanding the mechanism and active site structure of eTA and using site-directed mutagenesis, it is possible to alter the stereospecificity to favor the synthesis of either the threo- or erythro- forms [4-8].
Threonine aldolase catalysis, as in all other PLP-dependent enzymes, proceeds by a well-studied mechanism where the PLP cofactor is first transferred from an internal aldimine linkage to the ε-amino group of an active site Lys to an external aldimine with the α-amino group of the substrate in a transaldimination reaction (Scheme I). Obtaining structures with reaction intermediates has proved difficult because adding the 3-hydroxy substrate (serine, threonine, etc.) results in rapid conversion in the crystal to an equilibrium mixture of the PLP external aldimine with both the substrate and the glycine product. In this work we describe a very unusual structure of an uncleaved substrate non-covalently bound at the active site of E. coli threonine aldolase (eTA). This structure was obtained as a result of an unproductive binding of the substrate, due to an unusual charge distribution in the active site at pH 5.6 at which eTA is essentially inactive [4].
Scheme 1.

Reaction scheme for external aldimine and quinonoid formation in threonine aldolase
MATERIALS AND METHODS
Crystallization, Data Collection and Structure Determination
E. coli TA (EC 4.1.2.48) was expressed and purified following a previously described procedure [4]. Freshly dialyzed eTA (22 mg/mL in 20 mM potassium phosphate, pH 7.0) was incubated with L-serine (6.25 mM) for five minutes at a final ratio of 1:1 of tetramer eTA to L-serine. The complex was then crystallized by hanging drop vapor diffusion method with precipitant solution containing 0.1 M sodium citrate tribasic dihydrate pH 5.6, 20% v/v 2-propoanol and 20% v/v PEG 4000.
The X-ray data set was obtained at 100° K on an R-axis IV++ image plate detector using CuKα radiation (λ=1.5417 Å) from a Rigaku Micro-MaxTM -007 X-ray source equipped with Varimax confocal optics operating at 40 kV and 20 mA (Rigaku, The Woodlands, TX). Crystals were cryoprotected in their mother liquor supplemented with 11% glycerol, and diffracted to 2.1 Å resolution. The data set was processed and scaled with Rigaku D*TREK software. Diffraction data statistics are shown in Table 1.
Table 1.
Crystallographic data and refinement statistics for liganded eTA. Values in parentheses refer to the outermost resolution bin.
| Data collection statistics | |
|---|---|
| Space group | P21 |
| Cell dimensions (Å) | a=77.16, b=104.88, c=84.91; β=92.49° |
| Resolution (Å) | 32.80–2.10 (2.18–2.10) |
| No. of measured reflections | 224481 |
| Unique reflections | 77165 (7458) |
| Redundancy | 2.91 (2.77) |
| I/σI | 8.4 (3.8) |
| Completeness (%) | 98.0 (95.4) |
| Rmerge (%)a | 9.3 (26.8) |
|
| |
|
Structure refinement
| |
| Resolution limit (Å) | 29.99–2.10 (2.20–2.10) |
| No. of reflections | 76865 (8917 ) |
| Rwork (%) | 21.3 (32.5) |
| Rfree (%)b | 27.9 (38.0) |
| R.m.s.d. standard geometry | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.5 |
| Dihedral angles (%) | |
| Most favored regions | 91.2 |
| Allowed regions | 9.2 |
| Average B-factors (Å2) | |
| All atoms | 27 |
| Protein alone | 27 |
| Water | 30 |
| Na ions | 23 |
| L-serine substrate | 29 |
| PLP | 27 |
Rmege = ΣhklΣi/Ihkli – <Ihkli>/ ΣhklΣi< Ihkli>.
Rfree was calculated with 5% excluded reflection from the refinement.
The structure was solved by the molecular replacement method with the program AMoRe [9] using the published native eTA homodimer structure (PDB code 4LNJ) that was crystallized at pH 7.5 as a search model [10]. Two dimers forming a homotetramer in the asymmetric unit were obtained with a correlation coefficient of 26% and an R-factor of 53.4%. The catalytic sites showed non-covalently bound L-serine. Several cycles of refinement with CNS and model corrections with coot [11;12], and addition of L-serine molecules (in two possible orientations, each refined at 50% occupancy) at all four catalytic sites, six Na+ atoms and several water molecules resulted in a final Rwork/Rfree of 21.3/27.9 at 2.1 Å.
HINT analysis of interaction between the substrate L-serine and protein
The HINT forcefield [13;14] was used to quantify the interactions between the non-covalently bound L-serine and the active site residues of the eTA structure. The foundation of HINT has been extensively reviewed [13-15], but briefly, it is a scoring tool that exploits the free energy information from partition coefficients of solute transfer between water and 1-octanol (log Po/w) as a forcefield that recognizes hydropathic interactions as above, while inherently encoding entropy and solvation/desolvation.
The HINT calculations were performed both at simulated acidic pH 5.6 (where the active site residues His83, His126, Arg308 and Arg169 should exist predominantly in their protonated forms), and at simulated neutral pH 7.5 (where Arg308 and Arg169 should be positively charged, while His83 and His126 become increasingly unprotonated). The calculation also assumed the substrate’s L-serine carboxylate to be predominantly unprotonated at all pH values, while the amino moiety is predominantly protonated at pH of 5.6 but becomes increasingly unprotonated as the pH is increased to 7.5.
Using the Sybyl 8.1 molecular modelling suite (Tripos LP. www.tripos.com, St. Louis, MO, USA), hydrogen atoms were added to the pH 5.6 eTA-Ser complex structure coordinate containing the two different poses of the substrate, and minimized (Tripos forcefield, with Gasteiger-Hückel charges and distance-dependent dielectric) to a gradient of 0.01 kcal mol−1 Å−1 while the non-hydrogen atoms were treated as an aggregate. Prepared structures were then used for HINT score calculations.
RESULTS
Overall Structure
We have co-crystallized eTA with L-serine at pH 5.6 (subsequently referred to as eTA-Ser-pH5.6). Diffraction data, refinement and structural statistics of eTASer-pH5.6 are summarized in Table 1. The structure was determined using the molecular replacement method with the unliganded eTA structure that was crystallized at pH 7.5 (PDB code 4LNJ) as the search model [10]. The structure has been refined to a Rwork/Rfree of 21.3/27.9, and deposited in the PDB with ID code of 4RJY. The asymmetric unit contains two homodimers (A/B and D/C), with each dimer burying a total surface area of ~3600 Å2, forming a stable homotetramer composed of monomers A, B, C and D (Fig. 1A). The previously published eTA structures (all obtained at pH 7.5), i.e. the unliganded structure, the complex with glycine (obtained by co-crystallization of eTA with L-serine), and the complex with L-threonine/L-allo-threonine/glycine (obtained by co-crystallization of eTA with L-threonine) [10], will be used for comparative purposes. These structures will be referred to as eTA-pH7.5 (PDB code 4LNJ), eTA-Ser-pH7.5 (4LNL), and eTA-Thr-pH7.5 (4LNM), respectively. The overall fold of eTA-Ser-pH5.6 is indistinguishable from all three pH 7.5 structures, with root mean square deviation of less than 0.4 Å. Each of the four active sites of eTA-Ser-pH5.6 are contributed to by three subunits, differently from other fold-type I PLP-dependent enzymes that normally utilize 2 subunits to form the active site.
Figure 1.
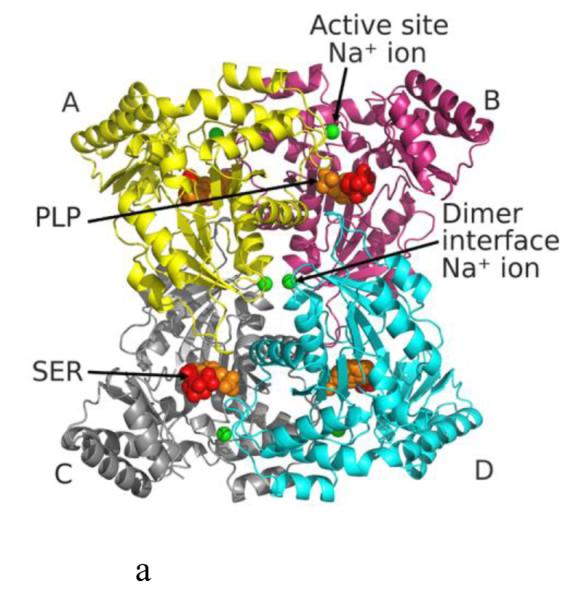
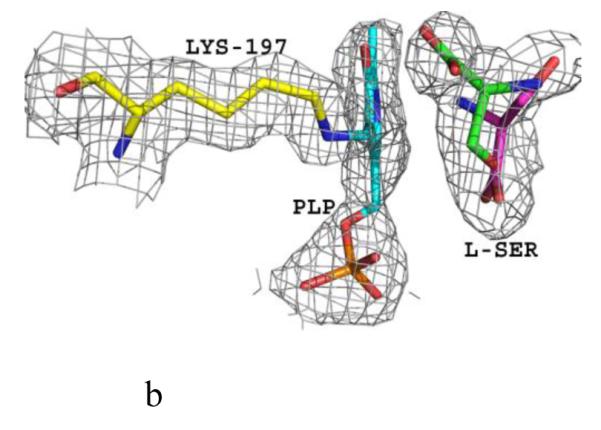
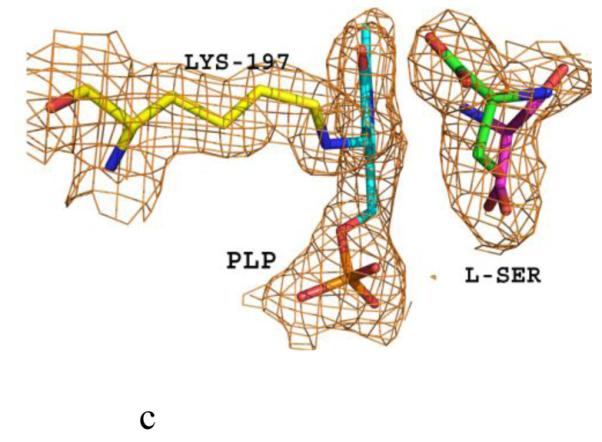
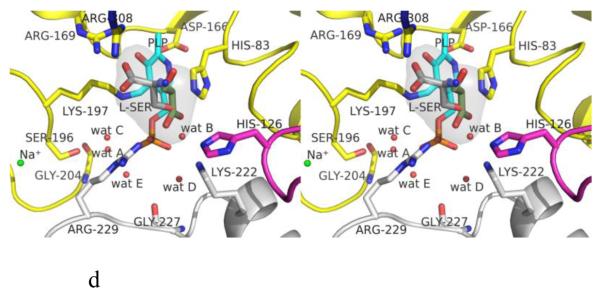
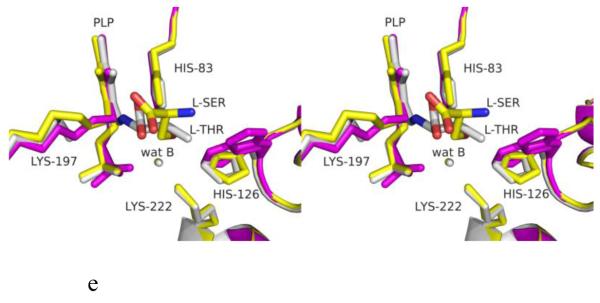
Structure of eTA. (A) Tetrameric structure of eTA-Ser-pH5.6 (in ribbons) in complex with L-serine (red spheres), PLP (orange spheres) and six Na ions (green spheres). Monomers A, B, C and D are in yellow, magenta, grey and cyan, respectively. (B) A 2Fo-Fc omit map (omitting Lys197, PLP and L-serine) shown at 0.9σ level for eTA-Ser-pH5.6. PLP is in the internal aldimine form, as shown by the continuous electron density between Lys197 and the cofactor. The map is superimposed with the final refined model. (C) A final refined 2Fo-Fc map shown at 0.9σ level for eTA-Ser-pH5.6. (D) Stereo-view of the monomer A active site of eTA-Ser-pH5.6 with the internal-aldimine bound PLP (cyan) and noncovalently bound L-serine (grey) in two alternate conformations. Na ion and water molecules are green and red spheres, respectively. (E) Stereo-view comparison of active sites of eTA-ser-pH5.6 (yellow), unliganded eTA-pH7.5 (magenta) and eTA-Thr-pH7.5 (white).
The substrate L-serine in eTA-Ser-pH5.6 binds in a non-covalent fashion at all four active sites (in two alternate conformations; each refined at 50% occupancy), while the PLP cofactor binds as an internal aldimine with the fold-type 1 PLP-dependent enzyme conserved Lys197 (Fig. 1B-D). This is in contrast to eTA-Ser-pH7.5 and eTA-Thr-pH7.5 structures that show either the substrate glycine or a mixture of glycine/threonine/allo-threonine, respectively, covalently attached to PLP (Fig. 1E) as a result of the enzyme-catalyzed reaction [10;16]. Thus, despite the presence of bound ligand at the active site of eTA-SerpH5.6, there appears to be no enzymatic reaction that could lead to external aldimine complex formation and retro-aldol cleavage.
The positions of the amino acid residues at the active sites of the four comparing structures do not change much with the exception of the side-chains of Lys197 and His126. The former residue makes a covalent interaction (internal aldimine) with the C4’ of PLP in both eTA-pH7.5 and eTA-Ser-pH5.6, but has moved significantly with elimination of this interaction in eTA-Ser-pH7.5 and eTA-Thr-pH7.5. His126 is located on an active site loop contributed by a subunit that is part of the other dimer and recently had been proposed to play an important role in substrate recognition [10]. His126 was observed to be highly disordered in eTA at pH 7.5 when ligands are absent (eTA-pH7.5) or when glycine is bound as an external aldimine (eTA-Ser-pH7.5); where in both structures, there is no associated interaction between His126 and a substrate. In contrast, in eTA-Ser-pH5.6 or eTA-Thr-pH7.5 the bound amino acid ligand (attached either covalently or non-covalently) hydroxyl moiety makes hydrogen-bond interactions with His126, stabilizing its position (Fig. 1E).
Six Na+ ions from the crystallization buffer are present in the eTA-Ser-pH5.6 structure. Four of the metal ions are located adjacent to the active site, while the other two are found at the interface of the two dimers. The metal ions have previously been observed in the other eTA structures with conserved protein interactions that are proposed to stabilize the active site and tetrameric structure, respectively [10].
Active Site Structure
The active site of monomer A, which is formed by subunits A, B and D, will be used for detailed discussion of active site interactions. The internal aldimine-bound PLP in eTA-Ser-pH5.6 makes similar protein interactions (with protein subunits A and B) as the previously reported eTA structures (Fig. 2A) [10]. The side-chains of His83 and Ala168 from subunit A make hydrophobic interactions at the re-face and si-face of the PLP ring, respectively. The phenolic oxygen and the pyridine nitrogen of PLP make hydrogen-bond interactions with the guanidinium group of Arg169 and the carboxylate group of Asp166 from subunit A, respectively. The PLP phosphate group makes several hydrogen-bond interactions with the peptide or side-chain atoms of Gly58 and Thr59 from subunit A, as well as with Arg229 from subunit B. There are also several water-mediated interactions (water molecules A-E; Figs. 1D, 2A) between the PLP phosphate and the protein. Most of these water molecules are conserved in the eTA structures obtained at pH 7.5 [10]. In eTA-SerpH7.5 and eTA-Thr-pH7.5, the external aldimine-bound ligands have displaced the amino group of Lys197 from PLP to form a Schiff-base interaction with the cofactor [10].
Figure 2.



Scheme of two-dimensional contacts between ligands, protein residues and the structural water molecules discussed in the text. Dotted lines indicate hydrogen-bond interactions and their length (in Å), and broad dashed lines represent hydrophobic interactions. (A) PLP contacts. (B) L-serine (with the carboxylate facing the arginines) contacts. (C) L-serine (with the carboxylate facing the histidines) contacts.
As noted above, although in eTA-Ser-pH5.6 the enzyme was co-crystallized with L-serine, it is evident that the amino acid did not undergo enzyme-catalysed cleavage, as the serine is observed to bind non-covalently (Fig. 1B-D). It is also notable that, despite the presence of ligand in the active site of eTA-Ser-pH5.6, the PLP stays bound as an internal aldimine with Lys197 as observed in the unliganded eTA-pH7.5 structure (Fig. 1B-D and 2A). These findings make eTA-Ser-pH5.6 the first reported PLP-dependent enzyme structure that has its substrate trapped in the active site in an unreactive abortive manner when the enzyme is co-crystallized with the substrate. We note that another PLP-dependent enzyme, Citrobacter freundii methionine γ-lyase was also recently shown to non-covalently bind the amino acids L-1-amino-3-methylthiopropylphosphinic acid or S-ethyl-L-cysteine. However, unlike the eTA study in this report, the methionine γ-lyase complex structure was obtained by soaking the native crystal with a large excess of the amino acid substrates [17].
In fold-type I enzymes, the formation of an external aldimine between PLP and the amino acid ligand is accompanied by a 10-30° rotation of the PLP ring from its internal aldimine position in the unliganded native structure [10;16;18]. As expected from its internal aldimine position, the PLP ring in eTA-Ser-pH5.6 has also rotated by about 25° from the external aldimine position, close to the unliganded position (Fig. 1E).
L-serine binding at pH 5.6
The electron density map in Figure 1B and C clearly shows that at pH 5.6 the substrate L-serine is bound at the active site, however it does not form an external aldimine with PLP, which remains bound to Lys197 as an internal aldimine. Electron density of serine may be fitted in two possible conformations, each refined at 50% occupancy (Figs. 1B-D; 2B and 2C). One conformation has the α-carboxylate group pointing toward and making hydrogen-bond/salt-bridge interactions with the positively charged Arg169 and Arg308, while the β-hydroxyl forms a hydrogen-bond interaction with His126 (Figs. 1D and 2B). We note that external aldimine bound amino acids in fold-type I PLP-dependent enzymes also show the α-carboxylate making interaction with Arg169 and/or Arg308. In the alternate conformer, serine is flipped about 180° from the first conformer so that the serine α-carboxylate group makes hydrogen-bond/salt-bridge interactions with the positively charged His83 and His126 (Figs. 1D and 2C). In this orientation, the serine hydroxyl group makes a hydrogen-bond interaction with Arg308. The two arginines and the histidines are conserved in threonine aldolases, while only Arg308 is conserved across all PLP-dependent enzymes. Interestingly, even though all four active sites show two alternate bound serine conformations as described above, the binding modes are slightly different, especially when it comes to the location of the serine α-amino group of one of the alternate conformers. Specifically, in all the active sites, the conformer with the α-carboxylate facing Arg169 and Arg308 shows the α-amino group pointing away from the C4’ of PLP, where it would normally make a nucleophilic attack to form a geminal diamine; the first intermediate in forming the external aldimine (Scheme I). On the other hand, the conformer with the α-carboxylate facing His83 and His126, shows differing α-amino group positions, especially between the monomer A bound serine and the other three active site bound ligands. In monomer A, the serine α-amino group is about 3.4 Å from the C4’ of PLP (as shown in Fig. 1B-D), and it is apparent from the electron density map that the two do not make any observable covalent interaction. For the other three active sites, the serine α-amino groups is about 4-6Å from the C4’ of PLP, pointing in a ~90° direction. In any position in the four active sites, the serine α-amino group makes hydrogen-bond interactions with water molecules; some of these water molecules mediate interactions with the protein (Fig. 2B and C). Such asymmetric binding of ligands is a characteristic feature of many PLP-dependent enzymes [10;19].
HINT analysis predicts substrate binding orientation at different pH values
HINT (Hydropathic INTeractions) is an empirical force field that exploits partition coefficient properties; which encodes all types of interactions expected to be found in bimolecular systems, giving qualitative and quantitative analysis of interactions. Higher HINT scores indicate more favourable interactions. We thus used this tool to quantitatively evaluate the binding energetics of L-serine at pH 5.6 using eTA-Ser-pH5.6; first with the non-covalently bound serine conformer which has its carboxylate facing the two protonated histidines, His83 and His126. We next repeated the analysis with the second conformer with its carboxylate facing the protonated arginines, Arg308 and Arg169. The calculation assumed that the amino group of the serine is essentially in the protonated form. The above calculations were repeated, this time assuming a pH of 7.5, where the putative serine amino group, as well as the histidines become increasingly unprotonated, while the arginines are protonated.
We obtained a favourable HINT score of +1200 with the substrate serine carboxylate facing protonated His83 and His126 at pH 5.6; a favourable HINT score of +1431 with the substrate serine carboxylate facing protonated Arg308 and Arg169 at pH 5.6; a favourable HINT score of +1522 with the substrate serine carboxylate facing protonated Arg308 and Arg169 at pH 7.5 (as observed in neutral-pH structures); and an unfavourable HINT score of −727 with the substrate serine carboxylate facing unprotonated His83 and His126 at pH 7.5 (control experiment). This analysis suggests that at pH 5.6, both orientations are possible consistent with the structural studies that show equal occupancies for the two orientations (Fig. 1B-D); at pH 7.5, however, HINT clearly suggests that the only orientation possible is with the carboxylate interacting with Arg169 and Arg308, consistent with this exclusive orientation observed at external aldimine bound amino acids in PLP-dependent enzymes at neutral pH values.
DISCUSSION
Previous kinetic studies showed eTA to be essentially inactive at pH 5.6 [4]. Enzymes exhibit a pH optimum and become inactive at either high or low pH. The inactivity at low pH is usually suggested to be the result of the conversion of a critical active site base (like a His residue) to its conjugate acid form or to significant changes in protein structure. Neither of these proposals appears to be the reason for the inactivity of eTA at pH 5.6.
In order to elucidate the molecular reason for eTA inactivity at low pH, we determined its crystal structure at pH 5.6 with L-serine substrate. Unlike previous eTA complex structures that were crystallized at neutral pH and showed covalently bound amino acid substrates, the eTA complex structure at pH 5.6 shows an exclusively non-covalently bound substrate in non-productive orientations in which the α-amino group and carboxyl group of the serine are in the wrong orientations. The α-amino group of the serine points away from the C4’ of PLP cofactor, and is not able to form the external aldimine intermediate and therefore carry out catalysis. Interestingly, unlike eTA, the α-amino group of the noncovalently bound amino acid substrates in Citrobacter freundii methionine γ-lyase are directly facing the C4’ of PLP [17].
Previous inhibition, spectral and rapid reaction studies with the PLP-dependent enzyme, serine hydroxymethyltransferase, which catalyzes all of the same reactions as eTA, showed that the substrates serine and glycine bind in the anionic form (neutral amino group) at the active site [19;21]. The substrate α-amino group needs to be in its unprotonated form in order to react with C4’ of PLP to initiate the transaldimination reaction [22;23]. At pH 5.6 the α-amino group of serine is highly protonated and unable to act as a nucleophile in the first step of the transaldimination step (Scheme 1). Thus, the most likely reason for the α-amino group of serine to be pointing away from C4’ of PLP is repulsion by the positive charge of the protonated internal aldimine at the bottom of the enzyme active site.
At low pH, the active site residues His83 and His126 are being converted from largely neutral molecules into their positively charged conjugate acid. The only significant change in position of active site residues, occurring at pH 5.6 in comparison to pH 7.5, is the movement of His126. Previous studies with eTA have proposed His83 and/or His126 as the putative base for the retro-aldol cleavage mechanism [4;16]. However, recent structural and site-directed mutation studies of wild-type and active-site mutant forms of eTA clearly showed that neither residue is the catalytic base, but rather the base is a conserved active site water molecule, its basicity enhanced by several coordinating basic residues, including His126 and His83 [10]. It was also suggested that the two histidine residues were important in substrate binding and specificity [10]. The structure at pH 5.6 shows that His126 is important in binding and positioning of the substrate in a specific orientation. At low pH, the likely protonated His126, and for that matter His83, which is capable of forming strong H-bonds and electrostatic interactions with the carboxylate moiety of the substrate, account for the observed alternate substrate conformation where the carboxylate group is pointing toward the two His residues. As the pH increases to neutral, the histidine residues and the α-amino group of serine become increasingly unprotonated, allowing the substrate to re-orient to its catalytic position that brings the carboxylate facing Arg169 and Arg308 and the neutral α-amino group facing C4’ of PLP. This interpretation is supported by the high HINT scores for the two orientations at pH 5.6. In summary, the reason for the inactivity of the enzyme at pH 5.6 is the change in the charge distribution where two critical His residues and the serine α-amino group become positively charged instead of neutral. Our study explains on a molecular level why the activity of eTA is quite different at different pH values; information important from protein engineering point of view, and more specifically may be useful in developing eTA as a biocatalyst.
Highlights.
Structure of e. coli threonine aldolase with non-covalently bound serine substrate
Non-catalytic orientation of serine due to an unusual charge distribution at low pH
Amine of serine and C4’ of PLP repels due to protonation of both species at low pH
PLP binds as an internal aldimine despite the presence of serine substrate
ACKNOWLEDGMENT
The structural biology resources used in this study were provided in-part by the National Cancer Institute of the National Institutes of Health to the VCU Massey Cancer Center [Grant CA 16059-28].
Footnotes
Conflict of interest We certify that there is no conflict of interest with any financial organization regarding the material discussed in this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duckers N, Baer K, Simon S, Groger H, Hummel W. Threonine aldolases-screening, properties and applications in the synthesis of non-proteinogenic beta-hydroxy-alpha-amino acids. Appl. Microbiol. Biotechnol. 2010;88:409–424. doi: 10.1007/s00253-010-2751-8. [DOI] [PubMed] [Google Scholar]
- 2.Strijbis K, van Roermund CW, Hardy GP, van den Burg J, Bloem K, de Haan J, van Vlies N, Wanders RJ, Vaz FM, Distel B. Identification and characterization of a complete carnitine biosynthesis pathway in Candida albicans. Faseb j. 2009;23:2349–2359. doi: 10.1096/fj.08-127985. [DOI] [PubMed] [Google Scholar]
- 3.Sagui F, Conti P, Roda G, Contestabile R, Riva S. Enzymatic synthesis of ω-carboxy-β-hydroxy-(L)-α-amino acids. Tetrahedron. 2008;64:5079–5084. [Google Scholar]
- 4.Contestabile R, Paiardini A, Pascarella S, di Salvo ML, D’Aguanno S, Bossa F. l-Threonine aldolase, serine hydroxymethyltransferase and fungal alanine racemase. A subgroup of strictly related enzymes specialized for different functions. Eur. J. Biochem. 2001;268:6508–6525. doi: 10.1046/j.0014-2956.2001.02606.x. [DOI] [PubMed] [Google Scholar]
- 5.Schirch V, Shostak K, Zamora M, Guatam-Basak M. The origin of reaction specificity in serine hydroxymethyltransferase. J. Biol. Chem. 1991;266:759–764. [PubMed] [Google Scholar]
- 6.Shostak K, Schirch V. Serine hydroxymethyltransferase: mechanism of the racemization and transamination of D- and L-alanine. Biochemistry. 1988;27:8007–8014. doi: 10.1021/bi00421a006. [DOI] [PubMed] [Google Scholar]
- 7.Schmid A, Dordick JS, Hauer B, Kiener A, Wubbolts M, Witholt B. Industrial biocatalysis today and tomorrow. Nature. 2001;409:258–268. doi: 10.1038/35051736. [DOI] [PubMed] [Google Scholar]
- 8.Liu JQ, Dairi T, Kataoka M, Shimizu S, Yama H. Diversity of microbial threonine aldolases and their application. J Mol Catalysis B: Enzymatic. 2000;10:107–115. [Google Scholar]
- 9.Trapani S, Navaza J. AMoRe: classical and modern. Acta Crystallogr. 2008;D64:11–16. doi: 10.1107/S0907444907044460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.di Salvo ML, Remesh SG, Vivoli M, Ghatge MS, Paiardini A, D’Aguanno S, Safo MK, Contestabile R. On the catalytic mechanism and stereospecificity of Escherichia coli L-threonine aldolase. Febs j. 2014;281:129–145. doi: 10.1111/febs.12581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 12.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarkar A, Kellogg GE. Hydrophobicity--shake flasks, protein folding and drug discovery. Curr. Top. Med. Chem. 2010;10:67–83. doi: 10.2174/156802610790232233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eugene Kellogg G, Abraham DJ. Hydrophobicity: is LogP(o/w) more than the sum of its parts? Eur. J. Med. Chem. 2000;35:651–661. doi: 10.1016/s0223-5234(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 15.Cozzini P, Fornabaio M, Marabotti A, Abraham DJ, Kellogg GE, Mozzarelli A. Free energy of ligand binding to protein: evaluation of the contribution of water molecules by computational methods. Curr. Med. Chem. 2004;11:3093–3118. doi: 10.2174/0929867043363929. [DOI] [PubMed] [Google Scholar]
- 16.Kielkopf CL, Burley SK. X-ray structures of threonine aldolase complexes: structural basis of substrate recognition. Biochemistry. 2002;41:11711–11720. doi: 10.1021/bi020393+. [DOI] [PubMed] [Google Scholar]
- 17.Revtovich SV, Morozova EA, Khurs EN, Zakomirdina LN, Nikulin AD, Demidkina TV, Khomutov RM. Three-dimensional structures of noncovalent complexes of Citrobacter freundii methionine γ-lyase with substrates. Biochemistry (Mosc) 2011;76:564–570. doi: 10.1134/S0006297911050063. [DOI] [PubMed] [Google Scholar]
- 18.Kirsch JF, Eichele G, Ford GC, Vincent MG, Jansonius JN, Gehring H, Christen P. Mechanism of action of aspartate aminotransferase proposed on the basis of its spatial structure. J. Mol. Biol. 1984;174:497–525. doi: 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- 19.Szebenyi DM, Musayev FN, di Salvo ML, Safo MK, Schirch V. Serine hydroxymethyltransferase: role of glu75 and evidence that serine is cleaved by a retroaldol mechanism. Biochemistry. 2004;43:6865–6876. doi: 10.1021/bi049791y. [DOI] [PubMed] [Google Scholar]
- 20.Schirch LV, Diller A. Serine transhydroxymethylase. Affinity of the active site for substrates, substrate analogues, and anions. J. Biol. Chem. 1971;246:3961–3966. [PubMed] [Google Scholar]
- 21.Schirch L. Serine transhydroxymethylase. Relaxation and transient kinetic study of the formation and interconversion of the enzyme-glycine complexes. J. Biol. Chem. 1975;250:1939–1945. [PubMed] [Google Scholar]
- 22.Di Salvo ML, Scarsdale JN, Kazanina G, Contestabile R, Schirch V, Wright HT. Structure-based mechanism for early PLP-mediated steps of rabbit cytosolic serine hydroxymethyltransferase reaction. Biomed. Res. Int. 2013;2013:458571. doi: 10.1155/2013/458571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vivoli M, Angelucci F, Ilari A, Morea V, Angelaccio S, di Salvo ML, Contestabile R. Role of a conserved active site cation-pi interaction in Escherichia coli serine hydroxymethyltransferase. Biochemistry. 2009;48:12034–12046. doi: 10.1021/bi901568b. [DOI] [PubMed] [Google Scholar]