

Abstract
Background
The regenerative potential of cardiosphere‐derived cells (CDCs) for ischemic heart disease has been demonstrated in mice, rats, pigs, and a recently completed clinical trial (CADUCEUS). CDCs are CD105+ stromal cells of intrinsic cardiac origin, but the antigenic characteristics of the active fraction remain to be defined. CDCs contain a small minority of c‐kit+ cells, which have been argued to be cardiac progenitors, and a variable fraction of CD90+ cells whose bioactivity is unclear.
Methods
We performed a retrospective analysis of data from the CADUCEUS trial and a prospective mouse study to elucidate the roles of c‐kit+ and CD90+ cells in human CDCs. Here, we show, surprisingly, that c‐kit expression has no relationship to CDCs' therapeutic efficacy in humans, and depletion of c‐kit+ cells does not undermine the structural and functional benefits of CDCs in a mouse model of myocardial infarction (MI). In contrast, CD90 expression negatively correlates with the therapeutic benefit of CDCs in humans (ie, higher CD90 expression associated with lower efficacy). Depletion of CD90+ cells augments the functional potency of CDCs in murine MI. CD90− CDCs secrete lower levels of inflammatory cytokines and can differentiate into cardiomyocytes in vitro and in vivo.
Conclusion
The majority population of CDCs (CD105+/CD90−/c‐kit−) constitutes the active fraction, both in terms of therapeutic efficacy and in the ability to undergo cardiomyogenic differentiation. The c‐kit+ fraction is neither necessary for, nor contributory to, the regenerative efficacy of CDCs.
Keywords: cardiosphere‐derived cells, CD90, ckit, myocardial infarction
Introduction
Numerous animal studies1–10 and the first‐in‐human CADUCEUS trial11–12 have demonstrated the regenerative potential of cardiosphere‐derived cells (CDCs) in ischemic cardiomyopathy. CDCs are intrinsic to the heart13 and uniformly express the transforming growth factor beta receptor accessory subunit, CD105 (endoglin), but are negative for the pan‐hematopoietic marker, CD45.7,10 CDCs contain a small fraction of CD117 (c‐kit)‐positive cells, argued to be cardiac stem cells,14–15 and a variable fraction of CD90 (Thy‐1)‐positive cells, which have not been characterized, but may represent a mesenchymal subfraction.16 Thus, the majority population is negative for both CD90 and c‐kit, but the active principle is, as yet, undefined. Purified c‐kit+ cells from CDCs are not as potent as the original CDC mixture in cardiac regeneration in a mouse model of myocardial infarction (MI),7 bringing into question the role of this fraction.
Here, we have investigated the roles of the c‐kit+ and CD90+ fractions in 2 ways: First, we performed a retrospective analysis of multiple patient CDC lines used in the CADUCEUS trial, correlating c‐kit and CD90 expression with the major efficacy endpoint (scar size reduction) in the study. Second, we depleted CD90+ and/or c‐kit+ cells from CDCs and examined whether the absence of 1 or both of these subpopulations affects the functional benefit of CDCs in a mouse model of MI.
Methods
Derivation and Characterization of Human CDCs
Institutional review board (IRB) approval was obtained for all procedures. Human CDCs were generated and expanded, as previously described, from myocardial specimens.10–11 Unless otherwise noted, IMDM basic medium (Gibco, Grand Island, NY) supplemented with 10% FBS (Hyclone, Logan, UT) and 20 mg/mL of gentamycin were used to culture all CDCs. Passage 2 CDCs were used for all in vitro and mouse experiments. CDCs were characterized by flow cytometry (FCM), as previously described.7,10 Briefly, cells were incubated with FITC‐, PE‐, or APC‐conjugated antibodies (Abs; from R&D Systems [Minneapolis, MN] or BD Biosciences [San Jose, CA]) against CD105, CD45, CD90, and CD117 (c‐kit) for 60 minutes. Isotype‐identical Abs served as negative controls. Quantitative analysis was performed using a CyAN flow cytometer with FlowJo software (TreeStar, Inc., Ashland, OR). In addition to FCM analysis, CDCs were seeded onto fibronectin‐coated chamber slides, after which cells were fixed with 4% paraformaldehyde (PFA), blocked/permeabilized with protein block solution (Dako, Carpinteria, CA) containing 1% saponin (Sigma‐Aldrich, St. Louis, MO), and then stained with chicken anti‐CD105 (Sigma‐Aldrich) and rabbit anti‐c‐kit (Sigma‐Aldrich) or rabbit anti‐CD90 (Abcam, Cambridge, MA) Abs. Magnetically activated cell sorting (MACS) was performed using anti‐CD90 and anti‐CD117 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Cell viability was assessed by trypan blue exclusion.
Human Scar Size Data
IRB approval was obtained for all procedures, and all study subjects gave informed consent. Scar size, defined as scar mass divided by total left ventricular (LV) mass, was quantified by contrast‐enhanced cardiac magnetic resonance imaging (MRI), as previously described.11–12 Changes in scar size from baseline study to the 12‐month follow‐up study were quantified in each patient and correlated with the c‐kit+ and CD90+ percentages in each corresponding autologous CDC preparation.
Mouse MI Model and Cell Injection
All animal studies were performed in an American Association for Accreditation of Laboratory Animal Care–accredited facility with approval from the institutional animal care and use committee of the Cedars‐Sinai Health System. Acute MI was created in male severe combined immunodeficiency (SCID)‐beige mice (10 to 12 weeks old), as previously described.7,10 Briefly, immediately after ligation of the left anterior descending artery with 9‐0 prolene, hearts were injected at 4 points in the infarct border zone with a total of 40 μL of one of the following interventions: PBS (control, n=6 mice); 1×105 unsorted CDCs (n=14 mice); 1×105 ckitDEP CDCs (n=14 mice); 1×105 CD90DEP CDCs (n=15 mice); or 1×105 DoubleDEP CDCs (n=12 mice). All animals were analyzed for cardiac function (by echocardiography), but only 5 from each group were randomly selected for histological analysis.
Heart Morphometry
For morphometric analysis, animals in each group were euthanized 3 weeks post‐MI (after cardiac function assessment) and hearts were harvested and frozen in OCT compound. Cryosections every 100 μm (5 μm thick) were prepared. Masson's trichrome staining was performed on 3 heart sections (≈600 μm between 2 sections) from the same heart, as per the manufacturer's instructions (HT15 Trichrome Staining [Masson] Kit; Sigma‐Aldrich). Images were acquired with a PathScan Enabler IV slide scanner (Advanced Imaging Concepts, Princeton, NJ). From the Masson's trichrome‐stained images, infarct wall thickness was measured with ImageJ software (National Institutes of Health [NIH], Bethesda, MD),7,10 and analyses of viable tissue size were performed. Data from the 3 sections were averaged for each individual animal before statistical analysis.
Cardiac Function Assessment
Mice underwent echocardiography 4 hours (baseline) and 3 weeks post‐MI using a Vevo 770™ Imaging System (VisualSonics, Toronto, Ontario, Canada).7,10 Hearts were imaged 2D in long‐axis views at the level of the greatest LV diameter. LV ejection fraction (LVEF) was measured with VisualSonics V1.3.8 software from views taken through the infarcted area. Studies were read and analyzed by an experienced echocardiographer who was blinded as to study‐group identity.
Cardiomyocyte Cycling Assay
Neonatal rat cardiomyocytes (NRCMs) were derived as previously described17–18 and plated onto fibronectin‐coated chamber slides, with or without CDCs at a 1:1 coculture ratio. After 3 days, NRCMs were fixed and stained for cardiac marker (α‐sarcomeric actin [αSA]; Sigma‐Aldrich), proliferation marker (Ki67; Abcam), and 4′,6‐diamidino‐2‐phenylindole (DAPI) for nuclei. Images were taken with a confocal microscope. Ki67+/αSA+ cells per field were counted from the images and quantified as the percentage of total myocytes (αSA+ cells).
Cardiomyocyte Apoptosis Assay
NRCMs were incubated with 50 μmol/L of H2O2 in the medium for 24 hours, with or without CDCs at a 1:1 coculture ratio. Afterward, cells were fixed and apoptotic cells were detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay using the In Situ Cell Death Detection Kit (Roche Diagnostics, Mannheim, Germany), according to the manufacturer's instructions. TUNEL+/αSA+ cells per field were counted from the images and quantified as the percentage of total myocytes (αSA+ cells).
Paracrine Assay
To compare the production of various growth factors and cytokines, CDCs were seeded in 6‐well culture plates at densities of 1×106 cells/mL in FBS‐free IMDM media for 3 days. The supernatants (conditioned media) were collected and the concentrations of vascular endothelial growth factor, basic fibroblast growth factor, hepatocyte growth factor, insulin‐like growth factor 1, and stromal‐derived factor 1 were measured with human‐specific ELISA kits (R&D Systems), according to the manufacturer's instructions. Matrix metalloproteinase (MMP) activities of CDC‐conditioned media were measured by the InnoZyme™ MMP‐2/MMP‐9 Activity Fluorogenic Assay kit (EMD Chemical Group, Temecula, CA). Secretion of various inflammatory cytokines was visualized by a semiquantitative Ab array (RayBiotech, Norcross, GA), and intensity was determined using ImageJ software (NIH).
Cardiac Differentiation Assay and Heart Histology
CDCs were cultured in cardiomyocyte differentiation media (Millipore, Billerica, MA), after which the cells were fixed with 4% PFA, blocked/permeabilized with protein block solution (Dako) containing 1% saponin (Sigma‐Aldrich), and then stained with chicken anti‐CD105 (Sigma‐Aldrich) and mouse anti‐αSA (Sigma‐Aldrich). FITC or Texas Red secondary Abs were obtained from Abcam. For heart histology, all animals were sacrificed at 3 weeks (after echocardiography study) and excised hearts were cryosectioned (5‐μm thickness). Heart cryosections were then fixed with 4% PFA, blocked/permeabilized, and stained with mouse anti‐human nuclear antigen (HNA; Millipore) and rabbit anti‐αSA (Abcam). Images were taken with a confocal microscope. Detailed Ab information is included in Table S1.
Statistical Analysis
Results are presented as mean±SD, unless specified otherwise. Comparisons between any 2 groups were performed using the 2‐tailed unpaired Student t test. Comparisons among more than 2 groups were performed using 1‐way ANOVA with post‐hoc Bonferroni's correction. Differences were considered statistically significant when P<0.05. Correlation analysis was performed by linear regression using the GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA).
Results
Therapeutic Efficacy of Human CDCs Is Not Affected by c‐Kit+ Percentage but Undermined by Abundant CD90+ Cells in the CADUCEUS Trial
Seventeen patients received autologous CDCs in the CADUCEUS trial (ClinicalTrials.gov: NCT0089336011). As criteria for identity, all patient CDC lines were examined by FCM for CD105 and CD45 expression before final release. CDCs uniformly express CD105 (Figure 1A; range, 93.0% to 99.8%; mean±SD, 97.8±1.7%) and are negative for CD45 (Figure 1B; range, 0.0% to 1.0%; mean±SD, 0.2±0.4%). After the clinical trial was completed, we were able to examine 13 of the 17 patient‐derived CDCs for c‐kit and/or CD90 expression. (The other cell products had been completely infused, leaving no residual for banking.) Only a small fraction of the autologously infused human CDCs express c‐kit, although the relative percentage varies greatly (>20‐fold) from patient to patient (Figure 1C; range, 0.3% to 7.2%; mean±SD, 2.9±2.0%). In contrast, CD90 expression ranges broadly (0.2% to 94.6%; Figure 1D; mean±SD, 25.1±26.9%). Scar size reduction, assessed by contrast cardiac MRI, was the major efficacy endpoint in the CADUCEUS trial. Linear regression analysis, performed in the 10 (of 17) patients' CDC lines for which all 3 relevant variables (c‐kit expression, CD90 expression, and 12‐month scar size data) were available, revealed that c‐kit expression does not correlate with the scar‐reducing ability of CDCs (Figure 1E; R2=0.006, P=0.83). However, CD90 expression negatively correlated with scar reduction (Figure 1F; R2=0.7863, P=0.0006; Table S2), that is, higher CD90 expression levels were associated with lower efficacy (less scar reduction). These results hint that c‐kit+ cells are not an important determinant of the therapeutic efficacy of CDCs, whereas CD90+ cells may undermine the overall benefit of CDC therapy.
Figure 1.
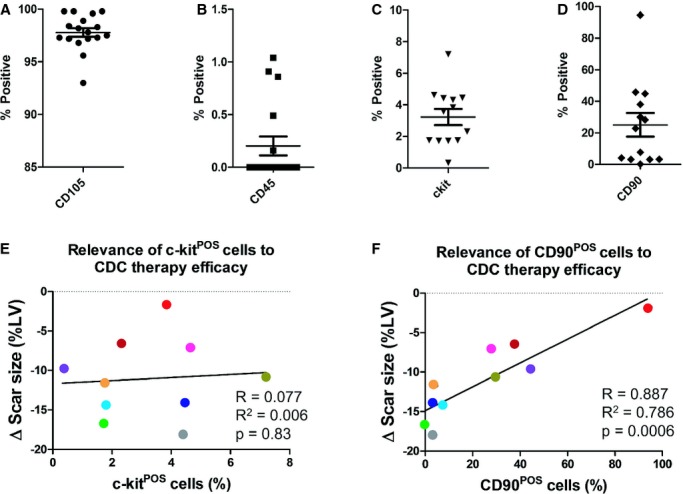
Correlations of c‐kit and CD90 expression with therapeutic efficacy of CDCs in the CADUCEUS trial. A through C, Flow cytometry analysis of CD105, CD45, c‐kit, and CD90 expression in the patient CDCs used in the CADUCEUS trial. Each colored bar represents an individual patient's CDC line. Black bars represent an average from all the patients. E and F, Linear regression analysis is performed to reveal the relationship between c‐kit or CD90 expression and the changes in the patients' cardiac scar size (ie, scar mass divided by total left ventricular mass) by contrast‐enhanced cardiac MRI over the 12 month follow‐up. Each dot represents an individual patient and its color matches that of the bars in C and D. Error bars=SDs. CDCs indicates cardiosphere‐derived cells; LV, left ventricular; MRI, magnetic resonance imaging.
Generating c‐Kit‐ and CD90‐Depleted CDCs by MACS
We further tested the roles of c‐kit+ and CD90+ cells by performing selective depletion experiments using MACS (Figures 2A and S1). The efficiency of cell sorting was confirmed by fluorescent microscopy and FCM (Figure 2B and 2C). The c‐kit‐ and CD90‐depleted CDCs are hereafter referred to as c‐kitDEP and CD90DEP CDCs. We also prepared CDCs depleted for both c‐kit+ and CD90+ fractions; these cells are abbreviated as DoubleDEP CDCs. The MACS process did not affect cell viability, as confirmed by trypan blue assay (Figure 2D), nor did it affect the expression of other cell‐surface markers (eg, CD105 and CD45; Figure 2E).
Figure 2.
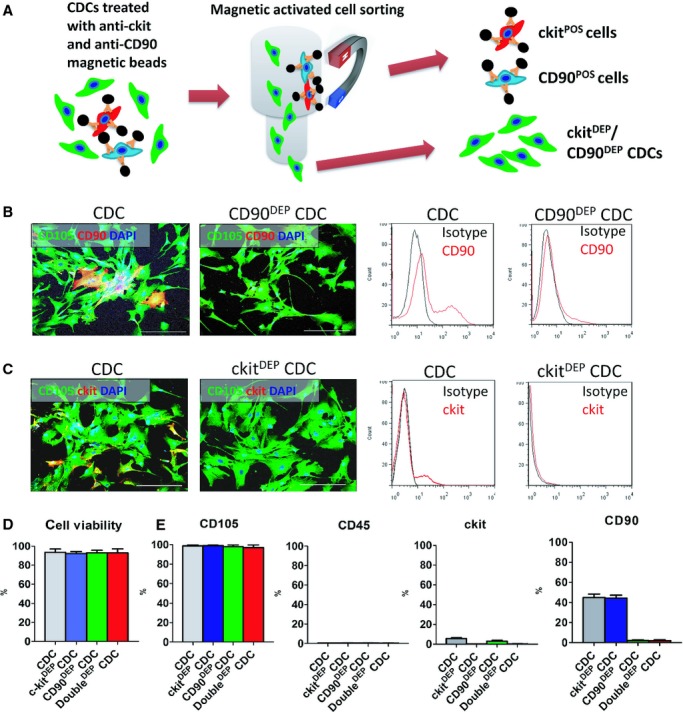
Generation of ckitDEP, CD90DEP, and DoubleDEP cardiosphere‐derived cells (CDCs) by magnetic‐activated cell sorting (MACS). A, Schematic diagram depicting the MACS process. B and C, Representative fluorescent micrographs and flow cytometry plots showing the expression of c‐kit and CD90 before and after MACS. D, CDC viability was determined by trypan blue exclusion assay. E, MACS sorting of c‐kit+ and/or CD90+ cells did not affect other surface markers (eg, CD105, CD45). Data were obtained with 3 MACS sorting experiments from a CDC sample with ≈40% CD90 positivity. Bars=50 μm. Error bars=SDs. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Depleting CD90+ Cells Enhances the Functional Benefit of CDCs
SCID mice underwent coronary ligation and were randomized to receive intramyocardial injection of one of the following: (1) control (PBS vehicle); (2) unsorted CDCs (“CDCs”); (3) c‐kitDEP CDCs; (4) CD90DEP CDCs; or (5) DoubleDEP CDCs. Heart morphometry at 3 weeks showed severe LV chamber dilatation and infarct wall thinning in control hearts (Figures 3A and S2). In contrast, all the CDC‐treated groups exhibited some degree of attenuated LV remodeling (Figures 3A and S2). Compared to control (white bars, Figures 3B, 3C, and S2), injection of CDCs (black bars, Figures 3B, 3C, and S2) or c‐kitDEP CDCs (blue bars, Figures 3B, 3C, and S2) increased infarct wall thickness (Figure 3B) and viable tissue mass (Figures 3C and S2) 3 weeks after treatment. The salutary effects were even greater in CD90DEP CDC‐ or DoubleDEP CDC‐treated hearts, which had the thickest infarcted walls and the largest amount of viable tissue among all groups (green and red bars, Figures 3B, 3C, and S2). These findings confirm the notion that CD90− CDCs are superior to their CD90‐containing counterparts in attenuating LV remodeling and preserving heart morphology in post‐MI mice.
Figure 3.
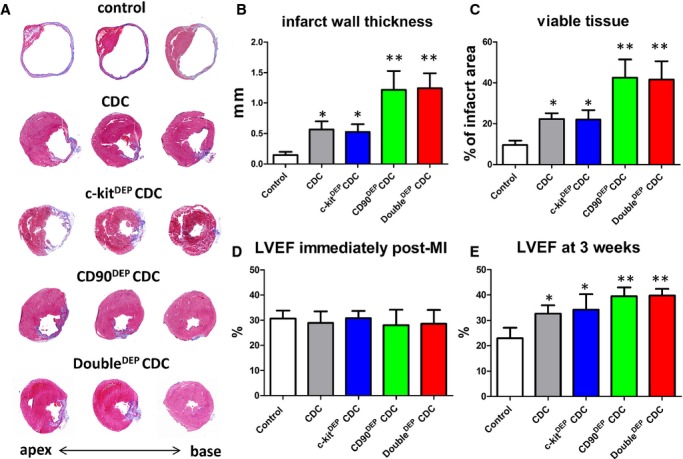
Therapeutic potencies of c‐kitDEP, CD90DEP, and DoubleDEP CDCs in a mouse model of myocardial infarction. A, Representative Masson's trichrome‐stained myocardial sections 3 weeks after treatment with control (vehicle only), unsorted CDCs, ckitDEP CDCs, CD90DEP CDCs, and DoubleDEP CDCs. Scar tissue and viable myocardium are identified by blue and red color, respectively. B and C, Quantitative analysis of infarct thickness and viable tissue size from the Masson's trichrome images (n=5 animals per group). D and E, Left ventricular ejection fraction (LVEF) was measured by echocardiography at baseline (4 hours post‐MI) (D) and 3 weeks afterward (E). *P<0.05, when compared to control; **P<0.05, when compared to any other group. Error bars=SDs. CDCs indicates cardiosphere‐derived cells; MI, myocardial infarction.
A consistent indicator of cell potency in this mouse model is the ability to produce functional benefit after transplantation.1,7,10 LVEF values at baseline (ie, 4 hours post‐MI) were comparable, indicating similar degrees of ischemic injury among groups (Figure 3D; Table S3). Over the following 3 weeks, LVEF deteriorated in the control group (white bar, Figure 3E; Table S3), but CDCs (consistent with previous studies7,10) or c‐kitDEP CDCs preserved LVEF. CD90DEP CDCs and DoubleDEP CDCs actually resulted in a sizable boost in LVEF (P<0.05 vs. all other groups; Figure 3E; Table S3). Taken together, these compound data sets show that depleting the c‐kit+ fraction from CDCs does not affect cardioprotective/regenerative potency, whereas depleting the CD90+ fraction enhances the structural and functional benefits of CDC therapy. Consistent with the latter conclusion, in a separate set of studies using the same mouse MI model, we found that unsorted CDCs outperformed the CD90+ fraction in augmenting cardiac function at 3 weeks (Figure S3).
Cardiomyocyte Proliferation and Apoptosis Assay
The in vivo therapeutic benefit of CDCs can be mimicked in vitro by coculturing neonatal rat cardiomyocytes with CDCs.9 Our previous work indicates that CDC therapy promotes cardiomyocyte cycling9 in vitro and in vivo. It is intriguing to ask whether depleting CD90+ cells increases the ability of CDCs to promote cardiomyocyte proliferation. Consistent with the previous findings, the percentage of NRCMs that have undergone DNA replication (Ki67+) is indeed higher when NRCMs are cocultured with human CDCs than in solitary NRCM culture (Figure 4A). However, depleting c‐kit+ and/or CD90+ cells had no effect on cardiomyocyte proliferation in vitro (Figure 4A) and in vivo (Figure S4). In contrast, cardiomyocyte apoptosis was inhibited by the presence of CDCs or c‐kitDEP CDCs in the culture (Figure 4B, gray and blue bars), an effect that was enhanced by the depletion of CD90+ cells (CD90DEP CDCs or DoubleDEP CDCs; Figure 4B, green and red bars). Furthermore, CD90DEP CDCs themselves are more resistant to oxidative stress than control CDCs (Figure S5). These results indicate that CD90 depletion enhances the antiapoptotic, but not the cardioproliferative, activity of CDCs.
Figure 4.
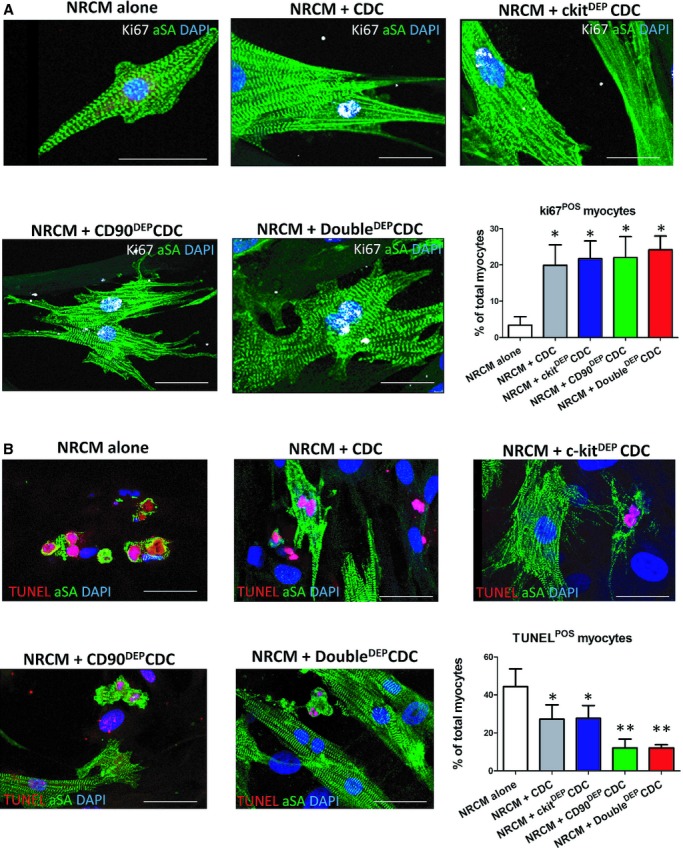
Cardiomyocyte proliferation and apoptosis assays. A, Representative fluorescent micrographs showing cycling (Ki67+ nuclei, white) NRCMs (αSA+, green). B, Representative fluorescent micrographs showing apoptotic (TUNEL+ nuclei, red) NRCMs (αSA+, green). *P<0.05, when compared to “NRCM alone”; **P<0.05, when compared to any other group. Data were obtained from 3 different CDC samples. Bars=10 μm. Error bars=SDs. CDCs indicates cardiosphere‐derived cells; NRCM, neonatal rat cardiomyocyte; SA, sarcomeric actin; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Paracrine Factor and Cytokine Secretion
Paracrine mechanisms underlie the majority of the beneficial effects of CDC transplantation.7–8,19 Consistent with our previous findings, CDCs secreted various growth factors (Figure S6A through E). Neither c‐kit depletion nor CD90 depletion changed the production of these factors. Also, no differences in metalloproteinase activities (MMP2/MMP9) were found among the 4 CDC groups (Figure S6F). On the other hand, inflammatory cytokine array analysis (Figure S7A) revealed that depleting CD90+ cells reduced the production of inflammatory cytokines (namely, interleukins (ILs) 1‐α and 1‐β, monocyte chemotactic protein‐3 [MCP], regulated on activation, normal T‐cell expressed and secreted [RANTES], granulocyte colony‐stimulating factor [G‐CSF], granulocyte‐macrophage colony‐stimulating factor [GM‐CSF], and chemokine (C‐C motif) ligand 1 [CCL‐1] by CDCs (Figure S7B). These results are consistent with the notion that the CD90+ fraction in CDCs is more proinflammatory than the CD90− fraction.
CD90– CDCs Can Differentiate Into Mature Cardiomyocytes
After 14 days of in vitro differentiation, both CDCs and CD90DEP CDCs started to express the cardiac‐specific marker, αSA (Figure 5A, red). CD90DEP CDC‐differentiated cardiomyocytes exhibited clear sarcomeric structures (Figure 5B), whereas most unsorted CDCs differentiated into an immature cardiomyocyte phenotype. Overall, more αSA+ cells were evident in CD90DEP CDC or DoubleDEP CDC cultures than in unsorted CDC or c‐kitDEP CDC cultures (Figure 5C), suggesting a superior cardiac differentiation potential from CD90 depletion. To confirm these results in vivo, SCID mice treated with various CDC groups were sacrificed 3 weeks after treatment and heart sections were stained for αSA and HNA (to identify engrafted cells; Figure 5D). More αSA+/HNA+ cells were detected in the CD90DEP CDC‐ and DoubleDEP CDCs‐treated hearts (Figure 5E), indicating that the CD90− CDCs are more likely to undergo cardiac differentiation in the post‐MI heart.
Figure 5.
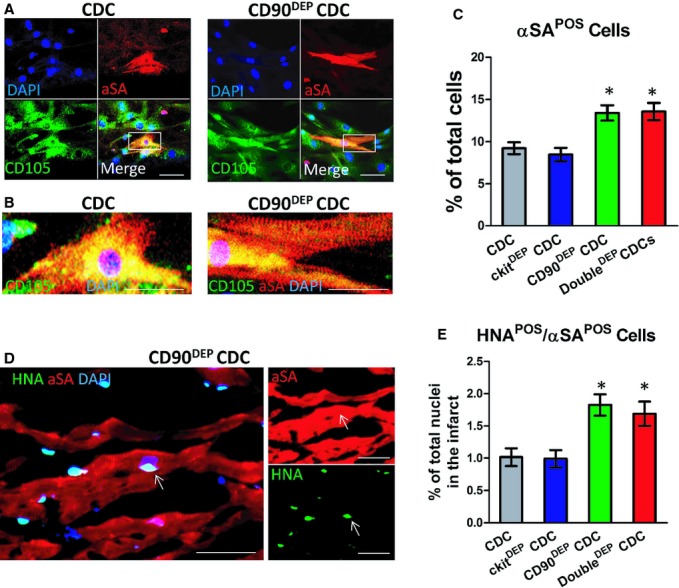
Enhanced cardiac differentiation of CD90DEP cardiosphere‐derived cells (CDCs). A, Representative confocal images showing unsorted CDCs and CD90DEP CDCs (expressing CD105; green) beginning to express α‐sarcomeric actin (αSA, red) after 14 days of differentiation. B, Magnified images of the cardiomyocytes shown in A. CD90DEP CDC‐differentiated cardiomyocytes exhibited clear sarcomeres. C, Differentiation potential was quantified by counting numbers of αSA+ cells in the CDC culture. D, Representative confocal images showing engrafted CD90DEP CDCs (positive for human nuclei antigen [HNA]; green) differentiated into cardiomyocytes (αSA+, red). E, Quantification of in vivo differentiation of transplanted CDCs and CD90DEP CDCs (αSA+/HNA+ cells) in the peri‐infarct area. *P<0.05, when compared to CDC. Bars=10 μm. In vitro data (C) were obtained with 6 experiments from 2 different CDC lines. In vivo data (D) were obtained from 5 animals injected with CDCs. Error bars=SDs.
Discussion
The last decade witnessed a burst of cell therapy trials for ischemic cardiomyopathy.20 The CADUCEUS trial11 and the SCIPIO trial15 have tested heart‐derived cells in human beings. The first study used unsorted CDCs from endomyocardial biopsies and the second study used purified c‐kit+ cells from surgical specimens (although concerns have been expressed regarding the cells used in SCIPIO21). CDCs contain a small, but highly variable, fraction of c‐kit+ cells (Figure 1C), which have been postulated to be cardiac stem cells.22 Therefore, we had initially hypothesized that c‐kit+ cells are the active principles in CDCs, whereas the remaining cells function to support the c‐kit+ fraction and increase their potency.10 Several years later, such a premise led to the purposeful admixture of cardiac c‐kit+ cells and marrow‐derived mesenchymal stem cells (MSCs) in a preclinical study.23 However, the present results disprove our original hypothesis: c‐kit expression was irrelevant for the therapeutic efficacy of CDCs in the CADUCEUS trial (Figure 1E), and c‐kit depletion did not affect functional or structural recovery in murine MI (Figure 3). Consistent with these results, it has been reported that unsorted human CDCs are functionally superior to c‐kit+ purified cells.7 Although the percentage of c‐kit+ cells in CDCs is low, the absolute number of c‐kit+ cells infused in a typical CDC treatment rivals the 1M that were transplanted in SCIPIO15 (and in the preclinical c‐kit+/MSCs admixture study23). Our results further support the conclusion that c‐kit+ cells are not important to the overall benefit of CDCs.
The observed irrelevance of c‐kit+ cells led us to test a second hypothesis: The CD90+ cells in CDCs are the active principles and are indispensable. CD90 (well known as Thy‐1) was originally discovered as a thymocyte antigen.24 CD90 is also widely used as a marker of a variety of stem cells (eg, MSCs, hepatic stem cells, keratinocyte stem cells, putative endometrial progenitor/stem cells, and hematopoietic stem cells).25–26 In humans, Thy‐1 is also expressed by endothelial cells, smooth muscle cells, a subset of CD34+ bone marrow cells, fibroblasts, and fetal liver‐derived hematopoietic cells. Strikingly, our results indicate that CD90 expression in CDCs undermines the functional benefit. In CADUCEUS, higher CD90 expression was associated with smaller therapeutic benefit (Figure 1F). Moreover, depleting the CD90+ fraction enhanced the functional benefit of CDCs in mice with MI (Figure 3). These findings support the previous report that CDCs are functionally superior to MSCs in a mouse model of MI7: >99% of bone‐marrow–derived MSCs and 85% of adipose‐derived MSCs expressed CD90, whereas only 18% of CDCs did so in that particular study. We further confirmed that the CD90+ cell fraction of CDCs secretes more inflammatory cytokines than the CD90− counterpart (Figure S4). Elevation of these inflammatory cytokines (eg, IL‐1) may lead to cardiomyocyte death and cardiac dysfunction.27 In any case, the overall benefit of CDCs is diminished by the presence of CD90+ cells. Our findings confirm and extend a recent report that CD90− CDCs are not only the majority fraction, but also crucial for the benefits of CDCs.28
Our study has several limitations. First, we did not include a CD90+ CDC group for direct comparison of its functional benefit with that of the CD90− subpopulation. However, we did confirm that unsorted CDCs are functionally superior to the CD90+ subpopulation of CDCs (Figure S2). Second, our data in support of enhanced in vivo cardiac differentiation by CD90DEP CDCs are limited in that we did not use confocal z‐stack imaging to confirm the location of HNA+ nuclei, nor did we perform ex vivo analysis of enzymatically isolated cardiomyocytes. However, the absolute number of HNA+/αSA+ cells is small and unlikely to explain the observed benefits of cell transplantation. Indeed, the overall differentiation rate in CDCs is marginal in both in vitro and in vivo experiments, consistent with the idea that the differential functional performance of the various groups may be dominated by indirect paracrine effects.7 Another limitation of our study is that a CDC‐specific positive marker has yet to be discovered. We have not found any markers unique to the CD90− population that are not expressed on the CD90+ population (or vice versa). Studies are ongoing to profile the CD90− CDCs using genomics approaches. Last, but not least, the lack of significance for some our data sets may be a result of low statistical power.
Nevertheless, our results point to a crucial, indispensable role of the c‐kit−/CD90− population for the regenerative potential of CDCs. In almost all CDC isolates from different human donors, this is the majority cell population (Figure 2).7,10 Future therapeutic trials of CDCs may benefit from CD90 depletion to enhance efficacy or, in allogeneic applications, selection of master cell banks that are naturally low in CD90 expression. Further studies are needed to fully elucidate the mechanisms for the observed benefit from CD90 depletion.
Supplementary Material
Appendix Supplementary Tables S1-S3 and Figures S1-S7.
Sources of Funding
This work was supported by the NIH (2R01HL083109) to Marbán, and American Heart Association (12BGIA12040477) to Cheng. The study was also partially supported by the National Center for Advancing Translational Sciences (UL1TR000124). Cheng is also supported by the North Carolina State University Chancellor's Faculty Excellence Program.
Disclosures
Dr Marbán is a founder, equity holder, and unpaid advisor to Capricor Inc. Dr Smith is an employee of Capricor Inc. Capricor provided no funding for the present study and had no approval rights over the manuscript. Drs Marbán and Cheng are inventors of a patent application covering the work described in the study.
Acknowledgments
The authors thank S. Chowdhury, T. Early, J. Doldron, and K. Wawrowsky for their technical assistance. The authors also thank J. Piedrahita for lab equipment support.
References
- 1.Zhang Y, Li TS, Lee ST, Wawrowsky KA, Cheng K, Galang G, Malliaras K, Abraham MR, Wang C, Marban E. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS One. 2010; 5:e12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng K, Malliaras K, Li TS, Sun B, Houde C, Galang G, Smith J, Matsushita N, Marban E. Magnetic enhancement of cell retention, engraftment and functional benefit after intracoronary delivery of cardiac‐derived stem cells in a rat model of ischemia/reperfusion. Cell Transplant. 2012; 21:1121-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng K, Shen D, Xie Y, Cingolani E, Malliaras K, Marban E. Brief report: mechanism of extravasation of infused stem cells. Stem Cells. 2012; 30:2835-2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis DR, Zhang Y, Smith RR, Cheng K, Terrovitis J, Malliaras K, Li TS, White A, Makkar R, Marban E. Validation of the cardiosphere method to culture cardiac progenitor cells from myocardial tissue. PLoS One. 2009; 4:e7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee ST, White AJ, Matsushita S, Malliaras K, Steenbergen C, Zhang Y, Li TS, Terrovitis J, Yee K, Simsir S, Makkar R, Marban E. Intramyocardial injection of autologous cardiospheres or cardiosphere‐derived cells preserves function and minimizes adverse ventricular remodeling in pigs with heart failure post‐myocardial infarction. J Am Coll Cardiol. 2011; 57:455-465. [DOI] [PubMed] [Google Scholar]
- 6.Li TS, Cheng K, Lee ST, Matsushita S, Davis D, Malliaras K, Zhang Y, Matsushita N, Smith RR, Marban E. Cardiospheres recapitulate a niche‐like microenvironment rich in stemness and cell‐matrix interactions, rationalizing their enhanced functional potency for myocardial repair. Stem Cells. 2010; 28:2088-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li TS, Cheng K, Malliaras K, Smith RR, Zhang Y, Sun B, Matsushita N, Blusztajn A, Terrovitis J, Kusuoka H, Marban L, Marban E. Direct comparison of different stem cell types and subpopulations reveals superior paracrine potency and myocardial repair efficacy with cardiosphere‐derived cells. J Am Coll Cardiol. 2012; 59:942-953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malliaras K, Li TS, Luthringer D, Terrovitis J, Cheng K, Chakravarty T, Galang G, Zhang Y, Schoenhoff F, Van Eyk J, Marban L, Marban E. Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere‐derived cells. Circulation. 2012; 125:100-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malliaras K, Zhang Y, Seinfeld J, Galang G, Tseliou E, Cheng K, Sun B, Aminzadeh M, Marbán E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol Med. 2013; 5:191-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marban E. Regenerative potential of cardiosphere‐derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007; 115:896-908. [DOI] [PubMed] [Google Scholar]
- 11.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marban L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marban E. Intracoronary cardiosphere‐derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012; 379:895-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malliaras K, Makkar RR, Smith RR, Cheng K, Wu E, Bonow RO, Marban L, Mendizabal A, Cingolani E, Johnston PV, Gerstenblith G, Schuleri KH, Lardo AC, Marban E. Intracoronary cardiosphere‐derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1‐year results of the CADUCEUS trial (cardiosphere‐derived autologous stem cells to reverse ventricular dysfunction). J Am Coll Cardiol. 2014; 63:110-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White AJ, Smith RR, Matsushita S, Chakravarty T, Czer LS, Burton K, Schwarz ER, Davis DR, Wang Q, Reinsmoen NL, Forrester JS, Marban E, Makkar R. Intrinsic cardiac origin of human cardiosphere‐derived cells. Eur Heart J. 2011; 34:68-75. [DOI] [PubMed] [Google Scholar]
- 14.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal‐Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003; 114:763-776. [DOI] [PubMed] [Google Scholar]
- 15.Bolli R, Chugh AR, D'Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK, Fahsah I, Rokosh DG, Slaughter MS, Kajstura J, Anversa P. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011; 378:1847-1857. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.da Silva Meirelles L, Chagastelles PC, Nardi NB. Mesenchymal stem cells reside in virtually all post‐natal organs and tissues. J Cell Sci. 2006; 119:2204-2213. [DOI] [PubMed] [Google Scholar]
- 17.Kapoor N, Galang G, Marban E, Cho HC. Transcriptional suppression of connexin43 by TBX18 undermines cell‐cell electrical coupling in postnatal cardiomyocytes. J Biol Chem. 2011; 286:14073-14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kapoor N, Liang W, Marban E, Cho HC. Direct conversion of quiescent cardiomyocytes to pacemaker cells by expression of TBX18. Nat Biotechnol. 2013; 31:54-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chimenti I, Smith RR, Li TS, Gerstenblith G, Messina E, Giacomello A, Marban E. Relative roles of direct regeneration versus paracrine effects of human cardiosphere‐derived cells transplanted into infarcted mice. Circ Res. 2010; 106:971-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malliaras K, Marban E. Cardiac cell therapy: where we've been, where we are, and where we should be headed. Br Med Bull. 2011; 98:161-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Lancet Editors. Expression of concern: the SCIPIO trial. Lancet. 2014; 383:1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anversa P, Kajstura J, Leri A, Bolli R. Life and death of cardiac stem cells: a paradigm shift in cardiac biology. Circulation. 2006; 113:1451-1463. [DOI] [PubMed] [Google Scholar]
- 23.Hatzistergos KE, Quevedo H, Oskouei BN, Hu Q, Feigenbaum GS, Margitich IS, Mazhari R, Boyle AJ, Zambrano JP, Rodriguez JE, Dulce R, Pattany PM, Valdes D, Revilla C, Heldman AW, McNiece I, Hare JM. Bone marrow mesenchymal stem cells stimulate cardiac stem cell proliferation and differentiation. Circ Res. 2010; 107:913-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ades EW, Zwerner RK, Acton RT, Balch CM. Isolation and partial characterization of the human homologue of Thy‐1. J Exp Med. 1980; 151:400-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carlyle JR, Zuniga‐Pflucker JC. Lineage commitment and differentiation of T and natural killer lymphocytes in the fetal mouse. Immunol Rev. 1998; 165:63-74. [DOI] [PubMed] [Google Scholar]
- 26.Saalbach A, Kraft R, Herrmann K, Haustein UF, Anderegg U. The monoclonal antibody AS02 recognizes a protein on human fibroblasts being highly homologous to Thy‐1. Arch Dermatol Res. 1998; 290:360-366. [DOI] [PubMed] [Google Scholar]
- 27.Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin‐1 in heart disease. Circulation. 2013; 128:1910-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gago‐Lopez N, Awaji O, Zhang Y, Ko C, Nsair A, Liem D, Stempien‐Otero A, MacLellan WR. THY‐1 receptor expression differentiates cardiosphere‐derived cells with divergent cardiogenic differentiation potential. Stem Cell Reports. 2014; 2:576-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix Supplementary Tables S1-S3 and Figures S1-S7.